
Overview of Conclusions and Recommendations
Advances in biomedical research have produced significant opportunities to improve cancer prevention, detection, and treatment. Insights about the genomic and molecular mechanisms of disease have enabled basic scientists to identify new therapeutic targets and develop new agents that are changing the paradigm of cancer research from nonspecific, broadly toxic chemotherapies to highly targeted combinations of therapies. However, the ability to translate biomedical discoveries into advances in care for patients with cancer remains dependent on the clinical trials system. Clinical trials provide an essential link between scientific discovery and clinical practice. These trials are crucial to the translation of new knowledge into tangible benefits for patients, and the knowledge gained in a clinical trial can also inform and guide further research into the biology of the disease.
Many clinical trials are undertaken by the pharmaceutical and biotechnology industries, whose primary objectives are to develop novel therapeutic agents and gain Food and Drug Administration (FDA) approval for clinical use. These research and development efforts entail enormous costs (hundreds of millions of dollars) and are critical to progress in cancer treatment. Publicly funded clinical trials also play a vital role and are complimentary to industry trials in advancing science and patient care, particularly by addressing questions that are important to patients but are less likely to be top priorities of industry. For example, companies may have less incentive to
-
conduct clinical trials to compare the effectiveness of different treatment options that are already approved for clinical use,
-
combine novel therapies developed by different sponsors,
-
develop therapies for rare diseases,
-
determine optimal duration and dose of treatment with drugs in clinical use,
-
test multimodality therapies, such as radiation therapy, surgery, or devices in combination with drugs,
-
study screening and prevention strategies, or
-
focus on rehabilitation and quality of life following therapy.
Publication of negative research findings about the therapies used in practice, which are underreported in the literature but which are essential in setting the standard of care, is also an important aspect of publicly funded research.
To address these needs, the National Cancer Institute (NCI) supports the largest U.S. network of clinical trials of any type through several different funding mechanisms. The largest component of that network is the Clinical Trials Cooperative Group Program (informally known as the Cooperative Group Program), which comprises 10 Groups that involve more than 3,100 institutions and 14,000 investigators who enroll more than 25,000 patients in clinical trials each year. Most Cooperative Group trials are either moderate-scale Phase II or large-scale Phase III clinical trials that may have practice-changing implications directly relevant to patient care. In contrast, many single-institution, investigator-sponsored trials are relatively small, nonrandomized Phase II trials that are less likely to have a major impact on the standard of care.
Since its inception in the 1950s, the Clinical Trials Cooperative Group Program has been instrumental in establishing the standards for cancer patient care and clinical research methods. The research undertaken by the Cooperative Groups has contributed to significant advances in cancer treatment and prevention, including the introduction of new treatments or new drug indications that have led to improved survival and increased cure rates, particularly for pediatric cancers and some early-stage cancers in adults. Furthermore, the role of the Cooperative Group Program is growing in importance as industry trials are increasingly being conducted outside of the United States. The Cooperative Group Program provides a primary mechanism by which the value of therapeutic agents can be assessed within the medical milieu of the U.S. health care system.
One of the Program’s strengths is the extensive involvement of physicians and patients from the community setting. Participation by the diverse patient populations treated in the community setting helps to ensure that the results of clinical trials are meaningful to a broad segment of the U.S. population and provides these patients with access to promising, innovative therapies as they are developed and tested. In addition, Cooperative
Group trials have contributed high-quality, annotated biospecimens that have aided preclinical and translational research activities, providing critical prognostic and predictive markers of response to therapy. The Cooperative Groups also provide data that support initial or expanded FDA labeling on the basis of clinical trial results supporting new indications for cancer therapeutics. The Cooperative Groups also provide a valuable training ground for clinical investigators, offering opportunities for mentorship, collaboration, and career advancement.
However, despite these important contributions and a long record of accomplishments, the public clinical trials system in the United States is at a critical juncture. The Cooperative Group Program in particular is facing numerous challenges that threaten its ability to continue to undertake large-scale, innovative clinical trials that benefit patient care. Funding for the Program has never covered the full cost of the trials that the Groups undertake. Stagnant and declining funding, inefficient processes, extensive and complex government oversight, and a lack of resources to accommodate the new targeted and personalized approach to the development and evaluation of cancer therapy contribute to the Cooperative Group Program’s current difficulties in efficiently and effectively translating research discoveries into timely clinical applications.
Recognizing the importance of maintaining an effective publicly funded clinical trials system, the director of NCI, John Niederhuber, requested that the Institute of Medicine (IOM) conduct a consensus study of cancer clinical trials and the Clinical Trials Cooperative Group Program and develop recommendations for how to improve the current system. To address the charge, the IOM appointed a 17-member committee with a broad range of expertise and experience, including experts in biomedical and clinical investigations in academia and community practice, statistics, radiology, research and development in the biotechnology and pharmaceutical industries, management research, systems engineering, the health insurance industry, and patient advocacy.
Because the environment in which clinical trials are conducted influences the pace of clinical advances, the committee took a broad view of the clinical trials process rather than simply focusing on NCI’s role. The committee concluded that the academic, government, and commercial sectors must join with the public to develop a 21st-century clinical trials system to more effectively leverage scientific advancements and translate them into public health benefits by improving the science; technology; efficiency; and timely creation, launch, and completion of the very best cancer clinical trials. The committee began by describing the needs of an ideal cancer clinical trials system of the near future (Box O-1). Then, on the basis of a review of the available published literature along with input from experts in the field and interested individuals, the committee developed a set of goals and
|
BOX O-1 Needs for Cancer Clinical Trials in 2015 Rapid translation of scientific discoveries into public health benefits
A strong publicly supported clinical trials system in the United States that complements industry trials to develop drugs and devices
A robust, standardized, and accessible clinical trials infrastructure
|
strategies that aim to enhance the value of national Cooperative Group clinical trials in cancer.
The committee concluded that a robust, standing cancer clinical trials network is essential to effectively translate discoveries into clinical benefits for patients. Multi-institutional collaborations are necessary to conduct large Phase III trials for indications such as adjuvant therapy, first-line therapy of metastatic disease, and prevention; single institutions are not capable of undertaking such large-scale trials. For research on some other diseases, the National Institutes of Health (NIH) supports large trials on a case-by-case basis, aggregating appropriate institutions for a particular study and then disbanding the group on completion of the study. However, cancer encompasses more than 100 different diseases, the treatment regimens are complex and diverse (and becoming more so), and hundreds of experimental therapies for cancer are in development. Thus, there is a
Harmonized and synchronized rules and guidelines across federal regulatory agencies
Support for clinical investigators
Broad patient involvement in clinical trials
|
continuous need for the design and implementation of new trials, and it would be highly inefficient to fund and develop infrastructures and research teams separately for each new clinical trial.
If NCI is to achieve the goal of improving outcomes for patients with cancer, it is imperative to preserve and strengthen the unique capabilities of the NCI Clinical Trials Cooperative Group Program as a critical component of NCI’s translational continuum. Given its long and impressive history of accomplishment, the Cooperative Group Program should ideally provide an established infrastructure for the rapid and efficient translation of scientific knowledge into practical therapeutic solutions that incorporate targeted agents matched to the characteristics of the patient and tumor and routinely achieve change in clinical practice, as well as FDA approval, where appropriate.
However, although a strong and adequately funded clinical trials coop-
erative network is essential for addressing questions of national importance, the current structure and operating processes of the entire trials system need to be reevaluated to improve value by reducing redundancy and improving effectiveness and efficiency. Numerous changes, as further delineated throughout this report, are needed to fully achieve that ideal, including an evaluation and justification of the unique role of each Cooperative Group, as well as an evaluation of the key roles that NCI has in administering and overseeing the Cooperative Group Program. Novel, multidisciplinary solutions are needed for currently intractable problems in cancer clinical trials. Redesigning a more effective and efficient clinical trials system would likely speed the pace of advances in cancer patient care.
The committee’s recommendations are organized under four broad goals: Goal I, improve the speed and efficiency of the design, launch, and conduct of clinical trials; Goal II, incorporate innovative science and trial design into cancer clinical trials; Goal III, improve the prioritization, support, and completion of cancer clinical trials; and Goal IV, incentivize the participation of patients and physicians in clinical trials (Box O-2). Taken together, these recommendations would alter the entire clinical trials system, including the functions of NCI as well as those of the Cooperative Groups.
GOAL I.
IMPROVE THE SPEED AND EFFICIENCY OF THE DESIGN, LAUNCH, AND CONDUCT OF CLINICAL TRIALS
Background
A clinical trial is a highly complex endeavor. It comprises hundreds of steps that must be taken, numerous decision points, and multilayered and iterative review processes because multiple oversight bodies with different objectives and responsibilities have jurisdiction over clinical trials. Inefficiencies in the processes used to develop, launch, and complete cancer clinical trials lead to lengthy delays in each step. Recent studies indicate that the time needed to transit from concept approval to activation of a Phase III Cooperative Group trial often exceeds 2 years. Given the rapid pace at which new scientific findings from basic or preclinical studies accumulate, a trial concept may lose relevance or become outdated in that 2-year time period. Moreover, evidence indicates that trials with lengthy activation times are statistically less likely to accrue the targeted number of patients required to draw valid scientific conclusions. Thus, process improvements are essential to achieve the rapid translation of scientific discoveries into public health benefits.
The current structure and organization of the Cooperative Groups did not result from any kind of strategic planning with regard to what might be optimal with respect to trial design and execution. Each Group
|
BOX O-2 Summary of the Committee’s Goals and Recommendations Goal I. Improve the speed and efficiency of the design, launch, and conduct of clinical trials
Goal II. Incorporate innovative science and trial design into cancer clinical trials
Goal III. Improve prioritization, selection, support, and completion of cancer clinical trials
Goal IV. Incentivize the participation of patients and physicians in clinical trials
|
operates independently, with its own administrative structures and operating procedures, committees, and statistical and data management centers. Although the Groups were originally organized by geographic area or, in some cases, by type of disease or therapeutic modality, today there is considerable overlap in the interests of the existing Groups, most of which conduct clinical trials in medical oncology, radiation, and surgery and thus compete for similar trial strategies and funding. Although some overlap generates competition for trial ideas, and some replication is necessary to serve as validation, too much redundancy in the Groups and in individual activities can lead to an unnecessary duplication of efforts, which wastes limited resources. The recent voluntary consolidation of the pediatric oncology Groups, which was done to pool and conserve limited resources, serves as an informative precedent for how the system could change. Some consolidation of the Cooperative Groups and common activities, along
with a focus on best practices, could increase operational efficiencies and conserve resources, ease the workload of the Cooperative Groups, and lead to more consistency for providers who would like to enroll patients in trials launched by different Cooperative Groups. At the same time, maintaining a robust competition for innovative trial concepts is essential.
Large-scale clinical trials also necessitate interactions among numerous stakeholders, including governmental agencies, academic medical centers, community practices, patients, and industry. However, effective communication and collaboration among stakeholders has been challenging. Thus, meaningful change to the cancer clinical trials system will require actions by the numerous stakeholders. Although NCI should play a leading role in instituting the necessary changes, other agencies within the U.S. Department of Health and Human Services (HHS), such as FDA, as well as academic centers, community practices, and others, will need to be actively involved in improving the system. Because of the complexity of the system and the interconnected roles that these stakeholders play, changing only one or a few steps is unlikely to achieve the desired improvements.
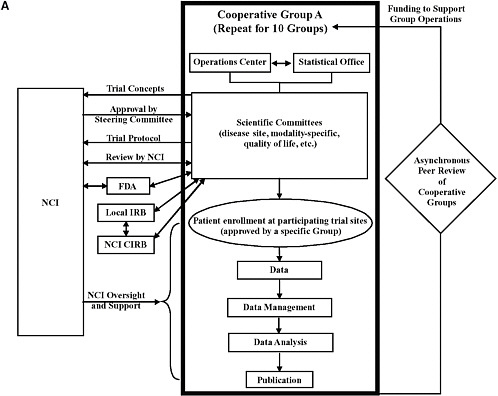
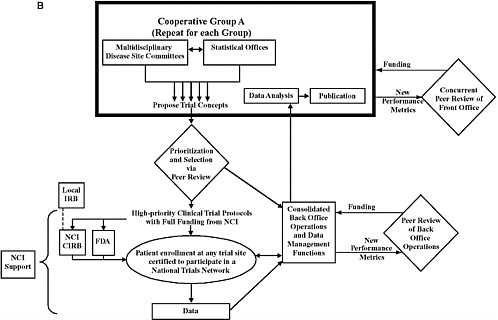
Recommendations 1 to 4 provide strategies to achieve the goal of improving the efficiency and the average time for the design and launch of innovative clinical trials by consolidating Groups, committees, and functions, by enhancing collaboration, and by streamlining and standardizing data collection and analysis. The first two recommendations, in particular, would significantly alter the definition, structure, and operations of Cooperative Groups (Figure O-1). The current system entails 10 independent Groups that generate ideas for clinical trials and then conduct trials using their own infrastructures for trial operations and data management. Cooperation primarily occurs among the members within each Group. The revised system that would result from implementation of the committee’s recommendations would go beyond cooperation to integration for many functions. Each Cooperative Group would consist of multidisciplinary committees focused on particular disease sites (referred to as disease site committees) and statistical offices that generate trial concepts and provide leadership for the conduct of trials selected as high priorities. The number of Groups and committees should be reduced on the basis of peer review. Most of the infrastructures used to support clinical trial operations and management would be consolidated to achieve greater consistency and efficiency under the new system. The committee’s recommendations also aim to streamline and coordinate the many iterative oversight processes to achieve further gains in the speed and efficiency of trial launch and conduct. NCI’s role would shift from a primary focus on oversight to a greater focus on supporting high-priority trials conducted through the national cancer trials network. Trial sites would be certified to participate in a national trials network and could enroll patients in any high-priority
FIGURE O-1 (a) Overview of the current structure and function of the Cancer Clinical Trials Cooperative Group Program. The components within the bold box are unique to each Cooperative Group. Each of the 10 Cooperative Groups has an operations center and a statistical center, scientific committees, and data management infrastructure. NCI is involved at multiple points throughout the process of trial concept design, protocol development, and trial implementation and conduct. Patient enrollment occurs at sites affiliated with and approved by specific Cooperative Groups to carry out clinical trials. The basis of peer review includes the scientific accomplishments and future plans of the Group, the organization of Group resources, the organization of data management and statistical centers, overall leadership, the Group’s publication record, and the effectiveness of disease- and modality-specific committees (collectively and, to some extent, individually). All of these factors are reviewed against a general standard for Cooperative Groups. FDA oversight is not required for all trials; FDA is involved any time a trial is conducted under an investigational new drug (IND) application. In general, any trial designed to provide information that may be used to change the drug label is conducted under an IND application, but this is not always the case. Double-sided arrows indicate repetitive interactions between organizations; changes in response to one review can trigger a re-review by another body.
(b) Overview of the proposed structure and function of the Cancer Clinical Trials Cooperative Group Program, as described in the committee’s recommendations. The components within the bold box would be unique to each Cooperative Group, while the other components would be consolidated and shared across the Groups. The Cooperative Groups would primarily consist of multidisciplinary disease site committees and statistical offices that generate concepts for clinical trials. These concepts would be prioritized through external peer review, with only high-priority trials being selected for implementation. Patient enrollment would be at sites certified to participate in a National Trials Network that comprises cancer centers, Community Clinical Oncology Programs, and community practices. Most of the infrastructure used to support trial operations and management would be consolidated to facilitate greater consistency and efficiency, but leadership for each trial would still be provided by the originating cooperative Group and principal investigators. The basis of front office peer review would include the success of the multidisciplinary disease site committee in innovation, winning study approvals, completing high-impact studies, mentorship of younger clinical investigators, and the publication of findings. Each multidisciplinary disease site committee will be reviewed against others in its peer group (i.e., committees focused on the same disease site will be reviewed against each other at the same time). Back office operations will also be evaluated through the use of new performance metrics and will be funded accordingly. In cases in which NCI is the IND holder in a clinical trial, the type of NCI oversight would be similar to that used for the current Cooperative Group Program. For other studies, NCI’s role would be limited to facilitating the launch and completion of the trial.
NOTE: CIRB = central institutional review board; FDA = Food and Drug Administration; IRB = institutional review board; NCI = National Cancer Institute.
trial launched under the new system, regardless of the origin of the trial concept.
Recommendation 1: NCI should facilitate some consolidation of Cooperative Group front office operations by reviewing and ranking the Groups with defined metrics on a similar timetable and by linking funding to review scores.
-
Key planning and scientific evaluations should be at the disease site committee level. The focus should be on the quality and success of the clinical trial concepts developed and the committee’s record of development of new investigators.
-
Committees that do well in review should be funded, and committees with low review scores should be eliminated.
-
Committees should be organized with a multidisciplinary focus on disease sites, and Group leaders should consolidate disease site committees from different Groups to strengthen their productivity and review scores.
Rationale
Some consolidation of the current Cooperative Group front offices,1 which entail the Groups’ disease site committees and statistical offices that generate and vet potential concepts and statistical designs for trials, would reduce redundancy in the Program, enable the pooling of resources, and reduce competition for enrollment in trials on the basis of Group-specific priorities. At present, the Groups are evaluated separately, on different schedules, and the score derived from that evaluation has no real impact on the amount of funding awarded to a particular group. Changing the timeline and focus of the review process to facilitate direct comparisons of the front office operations would ensure that only the most innovative and successful disease site committees would thrive, expand their membership, and maintain a sense of community.
The logical extension of the proposed consolidations will be a reduction in the number of Cooperative Groups. Cooperative Group leaders would have an incentive to work together to merge disease site committees
across the existing Groups to enhance productivity and review scores. For example, Groups focused on a single disease site or modality would likely need to merge with multidisciplinary Groups under this system. Such a system would ideally maintain strong competition for trial concepts among a smaller number of Cooperative Groups and their disease site committees and thus help to ensure that only the highest-priority trials are undertaken. A reduction in the number of trials competing for patient enrollment would help to align patient and clinician incentives, as providers would focus on finding the best possible trial for each patient and his/her particular disease, regardless of where the trial originated.
A similar rationale was behind the recent consolidation of the four Cooperative Groups focused on pediatric cancers into a single new Children’s Oncology Group (COG). Because of the relatively small number of children with cancer, greater collaboration was deemed essential to achieving adequate enrollment in trials and further progress in the cure rate for this disease. Operations of the consolidated Group are now streamlined, and pediatric patients are assured that they will be offered the best possible trial of the Program rather than the trial preferred by a particular Group. Other types of cancer that are rare or that have consistently had suboptimal accrual in trials (e.g., lymphoma) would also likely benefit from such a consolidation.
Recommendation 2: NCI should require and facilitate the consolidation of administration and data management operations across all of the Cooperative Groups (the back office operations) and, working with the extramural community, make process improvement in the operational and organizational management of clinical trials a priority. For example, NCI should
-
facilitate the consolidation of offices and personnel for such activities as data collection and management, data queries and reviews to ensure that the data collected are complete and accurate, patient registration, audit functions, submission of case report forms, training of clinical research associates, image storage and retrieval, drug distribution, credentialing of sites, and funding and reimbursement for patient accrual;
-
work with governmental and nongovernmental agencies with relevant expertise to facilitate the identification of best practices in the management of clinical research logistics and develop, publish, and use performance, process, and timing standards and metrics to assess the efficiency and operational quality of clinical trials;
-
coordinate and streamline the protocol development process, as recommended by the Operational Efficiency Working Group;
-
devote more funds to drug distribution;
-
provide resources and technical assistance to facilitate the rapid adoption of a common patient registration system as well as a common remote data capture system;
-
facilitate more efficient and timely methods for ensuring that trial data are complete and accurate; and
-
develop standardized case report forms that meet regulatory requirements.
Rationale
Each Cooperative Group devotes significant resources to support similar administrative structures and activities in what is defined in the operations management literature as back office operations. Although the ways in which the Groups accomplish these administrative and data management functions vary, there is little technical rationale for why they must be unique to the scientific focus of each Group. Consolidated back office operations work very successfully in other industries. The consolidation of offices and personnel to conduct these information-based activities across all the Cooperative Groups would streamline the operations, reduce redundancy, conserve resources, and offer greater consistency to providers enrolling patients in trials launched by different Cooperative Groups. The consolidation of COG again provides a model for such a transition, but NCI should work with the Cooperative Group leaders to develop a mechanism by which this transition can be accomplished efficiently and smoothly. It will be imperative to ensure high service quality and responsiveness to the principal investigators and Cooperative Groups leading the trials, through periodic peer review of formal metrics of performance.
In addition, the operational processes used to conduct clinical trials are idiosyncratic to individual institutions or Cooperative Groups, with little sharing of best practices or lessons learned. Although good clinical practice guidelines provide an international ethical and scientific quality standard for the design, conduct, recording, and reporting of the findings of clinical trials that involve the participation of human subjects, at present there is no mechanism for the systematic collection of best management and administrative practices that can be used as benchmarks by a clinical trials office in a cancer center or a Cooperative Group. Furthermore, few standard processes or metrics of what constitutes operational “quality” in the development or management of clinical trials exist. Thus, the operational performance metrics used to evaluate cancer centers and Cooperative Groups need to be enhanced and redefined to include quality, outcome, and timing metrics for clinical trials.
Because these operational issues can significantly delay clinical trials
and the evaluation of innovative therapies for all types of cancer, there is a need for NCI to work with other agencies and to make novel research on these topics a priority. In addition, a transparent process that could be used to measure and reward Cooperative Groups, cancer centers, and individual investigators not only for doing meaningful clinical research (as determined by an assessment of the essential scientific elements of the trial) but also for how rapidly and efficiently they conduct such research and for the impact of that research would greatly facilitate the adoption and use of best practices and metrics.
One of the most time-consuming and complex activities in the clinical trials process is the development of a scientific concept into a viable and approvable clinical trial protocol. NCI’s Operational Efficiency Working Group, which was charged with identifying ways to reduce the study activation time for Cooperative Group and cancer center trials by 50 percent, has recently put forth specific, measurable goals that the IOM committee endorses. These include reducing the time from protocol submission to final protocol approval to 300 workdays for Phase III trials and 210 work days for Phase II trials and eliminating trials that do not open and accrue patients within 18 calendar months for Phase II trials or 2 years for Phase III trials. To achieve those goals, the working group recommended staffing changes, more coordinated, parallel reviews, improved project management, and better tracking of the trial protocol.
More active and consistent support from NCI to facilitate trial operations would also be beneficial. For example, more resources for the rapid implementation and adoption of a common electronic registration and data capture system would increase consistency across trials, conserve resources by reducing the workload associated with patient enrollment and followup, allow for the more timely review of the data from a trial, and enhance the knowledge gained from a trial. Standardized case report forms would ease the burden of regulatory oversight and lead to better compliance.
However, all these activities will require additional NCI staff and resources to support the Cooperative Group Program, as noted in the discussion of Goal III.
Recommendation 3: The U.S. Department of Health and Human Services should lead a transagency effort to streamline and harmonize government oversight and regulation of cancer clinical trials. For example,
-
All review bodies should distinguish between major review concerns (regarding patient safety and critical scientific flaws, which must be addressed) and minor concerns (which should be considered, but are not obligatory).
-
NCI should coordinate with FDA for the review and oversight of trials involving an investigational new drug or investigational device exemption to eliminate iterative review steps.
-
FDA should establish a coordinated Cancer Program across its centers that regulate oncology products.
-
FDA should update its regulatory guidelines for the minimum data required to establish the safety and efficacy of experimental therapies (including combinations of products) and eliminate requirements for nonessential data, particularly for supplemental new drug and biologic license applications.
-
The Office for Human Research Protections should develop guidance that clearly establishes the accountability of the NCI central institutional review board, to encourage its wider use and acceptance by local institutions.
-
Federal oversight should be more flexible in allowing minor amendments to the protocol or consent form to fast-track the chain of reapprovals.
-
Patient consent forms should include a shortened and simplified summary to enhance the provision of informed consent.
Rationale
Compliance with regulatory requirements for the conduct of clinical trials is a major challenge for investigators in all fields of medicine. Multiple agencies and institutional bodies of HHS review and provide oversight for cancer clinical trials, including NCI, FDA, the Office for Human Research Protections (OHRP), the Office for Civil Rights (OCR), and institutional review boards (IRBs). The many oversight bodies have different objectives and responsibilities and thus seek similar, overlapping, but not identical information and action for compliance. Moreover, the review processes are serial and iterative. A change made in response to one review or in response to new findings can trigger a loop or re-review among the other review bodies as well. This delays the trial process and increases the burdens on investigators. Parallel, concurrent, or ideally, joint reviews and a clear distinction between major and minor reviewer concerns would help to reduce iterative reviews.
A departmental effort by HHS, with strong leadership from the HHS secretary and agency heads, to strengthen the scientific underpinnings of regulations, as well as to harmonize, coordinate, and streamline the oversight and review processes, could significantly improve the speed and efficiency of clinical trials, ease the burden on investigators, and better protect patients. For example, the HHS Secretary’s Advisory Committee on Human Research Protections and the IOM have recommended harmonization of
the regulatory language as well as the guidance and policies associated with the Common Rule2 and the Health Insurance Portability and Accountability Act Privacy Rule3 because of the difficulties that investigators and IRBs encounter in trying to reconcile the discrepancies between the two. Improved communication and coordination of reviews would also improve the process. For instance, NCI coordination with FDA for oversight of NCI-funded trials involving an investigational new drug application (IND) or investigational device exemption would ensure appropriate protocol design early in the process and thus reduce the number of revisions and re-reviews that may be required.
Changes within individual agencies would also be beneficial. For example, FDA may have multiple centers with jurisdiction over trials testing combination products, such as drug-biologic combinations or therapeutic-diagnostic combinations. A coordinated Cancer Program across the centers that regulate oncology products could avoid the conflicting expectations that may arise when sponsors seek approval through multiple centers. FDA committed in principle to the formation of such a Cancer Program in 2004 to “facilitate cross agency expert consultation,” but it has yet to follow through on that commitment. In addition, for trials intended to support product registration, FDA has extensive data collection requirements that could likely be reduced, especially for agents currently on the market that are being tested for new indications, as significant amounts of data on the safety of such agents often exist. Defining a core set of data elements, along with guidance on how those elements could be modified under certain circumstances, would speed the FDA review process and lead to greater uniformity in data requirements. Eliminating unnecessary and onerous data requirements would also conserve resources and result in the testing of more combination therapies in particular.
A major challenge unique to large multi-institutional studies is the involvement of many local IRBs. Regulatory language is often complex and subject to interpretation, so decisions by IRBs can be highly variable, which can cause delays and lead to protocol variations at different sites. Local IRBs can defer to a central IRB (CIRB), but in practice, many institutions are reluctant to rely on decisions made by the NCI CIRB, in large part because of concerns about being held accountable for the decisions that the CIRB makes. Guidance and policies from OHRP that address that concern would encourage the wider use of CIRBs and would thus increase
the efficiency and reduce the costs of clinical trials, as well as increase consistency in patient protections across sites. Guidance from OHRP and OCR to allow the use of simplified summaries of consent forms, which have become very lengthy and complex, would also improve patient communication and decision making.
Recommendation 4: NCI should take steps to facilitate more collaboration among the various stakeholders in cancer clinical trials. For example, NCI should
-
develop standard licensing language and contract templates for material and data transfer and for intellectual property ownership in biospecimen-based studies and trials that combine intellectual property from multiple sources;
-
facilitate the creation of more public-private partnerships and precompetitive consortia, guided in part by successful models;
-
facilitate the development of appropriate hybrid funding models, in which NCI and industry support clearly defined components of trials that are of mutual interest;
-
facilitate a process by which stakeholders (NCI, NIH, FDA, industry, investigators, and patients) can define an effective mechanism for the development of targeted cancer therapies, with particular emphasis on combinations of products; and
-
implement a highly visible grand challenge competition to engage experts in cancer and noncancer fields (e.g., engineering, social science, management, and marketing) and to reward significant innovation leading to increased efficiency in clinical trials processes.
Rationale
Cancer clinical trials often necessitate effective collaboration among diverse stakeholders, but there are numerous challenges to achieving such collaborations. For example, negotiations to reach contract and licensing agreements to transfer or share materials, data, and intellectual property (IP) are complex and can cause lengthy and costly delays in the launch of clinical trials. Pharmaceutical companies in particular may be reluctant to share IP or data and patient samples with academic collaborators and may require IP rights that are unacceptable to collaborators. However, valuable insights and discoveries may be lost and progress toward clinical advances may be slowed if important data or samples are withheld from collaborating institutions that could explore novel, additional hypotheses with those resources. Standard IP licensing language and contract templates, similar to the standardized trial contract language that was developed in conjunction
with the CEO Roundtable on Cancer, could reduce the delays due to these negotiations and facilitate important new research.
Given the limited funding capacity of NCI, it would also be beneficial to leverage the resources of industry to support the work of the Cooperative Groups in a transparent way to benefit patients, for example, in comparison trials or for secondary indications. Two recent reports from the President’s Council of Advisors on Science and Technology acknowledge the importance and value of strengthening public-private collaborations to enhance innovation, particularly for discovery and translational research in personalized medicine. However, industry funding for Cooperative Group trials has been limited for a variety of reasons, including concern about the inherent inefficiencies in the Program, the Groups’ concern about maintaining independence in study design and execution, and concerns about conflicts of interest. These concerns may contribute to the increasing tendency of pharmaceutical and biotechnology companies to conduct trials in other countries.
Commercial firms might be more interested in collaborations with the Cooperative Groups if the review and operational procedures of the Program were streamlined, as recommended in this report. However, novel hybrid funding mechanisms, as well as new efforts to establish public-private partnerships and precompetitive consortia would further aid progress toward effective collaboration, to the benefit of patients, who desire access to new and promising cancer therapies. Maintaining a critical mass of clinical trials in the United States via appropriate collaborations is important to ensure that patients in this country gain access to promising therapies as they develop, that trials address questions and generate data that are relevant and meaningful to patients in the United States, and that the nation retains a sufficient number of properly trained clinical trial specialists.
Effective collaboration among stakeholders will be particularly important for combination therapies, which may hold the key to successful personalized medicine. Traditionally, most cancer drugs have been broadly cytotoxic and nonselective in their mechanisms of action, resulting in significant toxicity to healthy tissues. In recent years, research has elucidated many of the molecular changes underpinning the initiation and progression of cancer (e.g., molecular changes affecting signaling pathways; cell death mechanisms; cancer spread; and DNA synthesis, repair, and modification). Because most cancers have multiple abnormalities, combination therapies that target multiple key cellular pathways and activities should benefit patients by increasing the efficacy of cancer treatments and reducing the likelihood that resistance will develop. Preclinical models of cancer support that hypothesis, and clinical studies also indicate that some targeted therapeutics that work effectively in concert with other agents may not induce significant responses when they are used as a single agent. This circum-
stance creates a significant challenge for the design of clinical trials and for FDA oversight. For example, traditional FDA standards require sponsors to “isolate the contribution of each agent.” Companies may also be reluctant to work with competitors to test promising combinations at an early and risky stage of development. To date, most combinations tested in Phase III trials have involved at least one agent currently approved by FDA.
The progress of clinical oncology research is also impeded by numerous obstacles that are well known but have eluded solution, despite decades of discussion and multiple reports by review panels. The low proportion (~3 to 5 percent) of adult cancer patients who enroll in clinical trials is a prime example. In recent years, NCI used the traditional request for application mechanism to solicit proposals that aimed to increase patient accrual, but that effort achieved little meaningful gains in accrual.
Well-run R&D organizations devote a portion of their resources to improve how they do research, not just doing research. A new and novel approach is required to solve these well-known intractable problems, with application of the best minds in multiple disciplines (engineering, social science, management, marketing, etc.). The potential for impact can often be a stronger motivator to good science than money per se, and competition can foster rapid and innovative solutions, much like what occurred with the sequencing of the human genome. Thus, one promising novel approach would be to develop a major, influential grand challenge for those well-known problems in oncology research.
Models for the development of such grand challenges exist and have shown some successes. A widely known example of such an approach is the X PRIZE, a multimillion-dollar award given to the first team to achieve a specific goal that has the potential to benefit humanity. A recent report on such incentive prizes, which spur innovation by tapping into competitive and entrepreneurial spirits rather than directly funding research, concluded that they are unique and powerful tools that can produce change not only by identifying new levels of excellence and by encouraging specific innovations but also by changing wider perceptions, improving the performance of communities of problem solvers, building the skills of individuals, and mobilizing new talent or capital.
GOAL II.
INCORPORATE INNOVATIVE SCIENCE AND TRIAL DESIGN INTO CANCER CLINICAL TRIALS
Background
Progress in the treatment of cancer patients depends on the effective incorporation of scientific advances into clinical trials. For example, to achieve the goals of targeted cancer therapy, the use of validated biomark-
ers will be essential. Cooperative Group clinical trials provide a unique opportunity to enable the emerging science of molecular markers through retrospective analyses of archived samples and prospective evaluations of biomarkers. High-quality annotated biorepositories are needed to gain useful knowledge about the biology of cancer and biomarkers from the analysis of patient samples archived from past trials, but the maintenance of tissue banks and the analysis of stored samples are costly activities that are not fully covered by the core funding that NCI provides to the Cooperative Groups. Access to stored samples can also be problematic. The increasing complexity of cancer clinical trials, along with the great expense and high failure rate of late-stage clinical trials, has spurred innovation in trial design as well, with the aim of conducting clinical trials more efficiently and with a greater likelihood of success. However, when new methods or technologies are incorporated into clinical trials, standards to ensure that the results collected at the various trial sites are consistent enough to attain accurate and meaningful conclusions from a study are often lacking.
Recommendations 5 to 7 aim to facilitate the incorporation of innovation in cancer clinical trial design and conduct.
Recommendation 5: NCI should mandate the submission of annotated biospecimens to high-quality, standardized central biorepositories when samples are collected from patients in the course of Cooperative Group trials and should implement new funding mechanisms and policies to support the management and use of those resources for retrospective correlative science. For example,
-
All data, including biomarker data from serum, tissue, and imaging analyses should be considered precompetitive, unencumbered by intellectual property restrictions, and made widely available.
-
The accompanying clinical data should be reported on standardized forms.
-
NCI should establish a national inventory of samples held in the central repositories and have a defined process for access by researchers that includes a single scientific peer review linked to funding.
Rationale
The Cooperative Groups have a history of collecting biospecimens from the diverse populations of patients who participate in their clinical trials and maintaining them in repositories with detailed information about patient characteristics, treatment, and outcome. These resources have proven immensely valuable in the development of molecular-based classifi-
cation schemes and diagnostic tests that now guide decisions on the most appropriate therapy for numerous types of cancer. However, high levels of evidence are needed to validate and qualify biomarkers for specific uses, and current funding is inadequate to support the research needed to generate that evidence. Although current NCI policies and funding do support a portion of the costs involved in the collection and storage of samples, the Groups must routinely seek supplemental funding to manage and maintain the repositories. Moreover, little funding is available to conduct retrospective studies of samples that have been collected in previous trials.
Furthermore, current NCI policies require research studies that propose to use specimens collected from intergroup protocols to undergo scientific review by a scientific steering committee before specimens are made available. However, such a review is not linked to funding, so investigators must often seek funding through other mechanisms. This process creates many review loops, time delays, and significant double jeopardy, in that each proposal requires at least two scientific reviews (one to receive specimens and one to receive funding) that are conducted at different times by different review groups. The availability of a consistent and adequate funding source devoted to correlative studies with stored samples and with appropriate peer review that includes direct input from the Group that collected the samples is imperative. The broader use of high-quality, standardized repositories would speed the pace of scientific and clinical advances at a much lower expense than would be required if new clinical samples had to be collected to study each new concept.
In addition, access to biospecimens for research is inconsistent and can entail complex negotiations with the various custodians of the samples. Policies regarding ownership and access vary across different institutions, and this impedes progress. Furthermore, many hospitals discard samples after a period of time, so valuable resources are lost to research. Because the Cooperative Groups have a long history of responsible stewardship of repositories, they are a logical choice to play a central role in the ongoing efforts of NCI to establish consistent policies on ownership and access. The creation of a national inventory of samples held by the Cooperative Groups would also greatly facilitate important research in correlative science.
Recommendation 6: Cooperative Groups should lead the development and assessment of innovative designs for clinical trials that evaluate cancer therapeutics and biomarkers (including combinations of therapies).
Rationale
Cooperative Groups are in a unique position to develop innovative designs for clinical trials and to demonstrate the feasibility and utility of
using innovative, efficient designs in their clinical trials. The development and use of innovative trial designs could speed progress in clinical trials in numerous ways. For example, prospective clinical trial designs that randomize patients on the basis of biomarkers or treatments, or both, should be explored and evaluated. For targeted therapies, a predictive hypothesis for a biomarker should be put forward in the preclinical phase and tested in early-phase clinical trials (Phase I and II trials). Better Phase II trial designs are needed to more accurately assess which patients benefit from a particular therapy, and thus guide the decisions about whether to move into Phase III trials. Improved designs for Phase III trials, which are the most costly and lengthy trials and entail the majority of Cooperative Group trials, could lead to faster, more accurate conclusions about new therapeutics and in the process reduce costs and conserve resources. For example, recent innovations, such as the use of adaptive designs for Phase II trials that assess response endpoints during trial accrual in real time, suggest that relevant clinical questions might be addressed more efficiently, with fewer patients required, with less time needed to show differences between Groups, and with enhanced confidence in the clinically (and statistically) meaningful differences that are observed between Groups. These or related designs may be particularly amenable for the comparison of treatment effects in patients with different biomarker profiles and could hasten the identification of the most promising predictive biomarkers that could be validated in a Phase III trial setting.
Recommendation 7: NCI, in cooperation with other agencies, should establish a consistent, dynamic process to oversee the development of national unified standards as needed for oncology research. This process should
-
be used by NCI when standards are required for any important new technology, tool, or breakthrough method (e.g., biomedical imaging and other biomarkers and biospecimens);
-
replicate successful aspects of standards development by other standard-setting bodies, both governmental and nongovernmental (e.g., the American Society for Testing and Materials, the National Standards Foundation, the National Institute for Standards and Technology, the International Organization for Standardization, and professional societies);
-
utilize the input of experts in both subject matter and standards design in developing standards;
-
include consistent operating procedures for developing standards (e.g., representation of stakeholders in committee composition, decision making, and voting rules); and
-
publish and update the standards in a timely manner such that they are useful to those performing clinical trials.
Rationale
As new scientific methods and technologies develop and mature, standards are needed to ensure appropriate and consistent use. The current approach to standards development is often ad hoc, with the processes and rules for such things as committee composition and voting rules being reinvented on a case-by-case basis. This can lead to heterogeneous and delayed results. A more systematic, multidisciplinary, and dynamic approach to standards development fostered by NIH and NCI would be advantageous for the rapid and consistent setting of unified national standards as the need arises. NCI could further assist by facilitating the creation of systems and software to aid the process of standards implementation.
This need for standards will become increasingly important as the science of cancer research becomes more complex and more dependent on technologies such as imaging and on molecular tools such as biomarkers. In the case of biomedical imaging, many technologies and imaging reagents, both those in current use and those under development, have the potential to provide information that can aid drug development and clinical decision making by providing improved means of diagnosis and monitoring. However, the lack of standards for image acquisition and quantification of results compromises the validity of the results and the interpretation of those results. In addition, the lack of harmonization of methods among the different vendors of imaging equipment compromises the quality and consistency of results. The consistent development of standard methodologies for established tumor-imaging modalities (e.g., computed tomography, fluorodeoxyglucose positron emission tomography, and conventional magnetic resonance imaging) by expert panels, along with a requirement that manufacturers meet those standards, could significantly improve the accuracy and value of those tests. Validation standards are also needed to continuously evaluate novel imaging methods and modalities to determine their merit and appropriate use.
Similarly, expert panels are needed to establish validation and qualification standards for the development and use of in vitro biomarker tests, to ensure that the results of those tests are consistent and accurate, and for the appropriate interpretation and use of those results. Such standards could also inform FDA guidance for the codevelopment of diagnostic-therapeutic combinations or for the inclusion of a biomarker test on the label for a drug or biologic that is already FDA approved.
GOAL III.
IMPROVE PRIORITIZATION, SELECTION, SUPPORT, AND COMPLETION OF CANCER CLINICAL TRIALS
Background
Clinical oncology research has changed a great deal since the early days of the Cooperative Group Program in the 1950s. The process of conducting large-scale trials has become highly complex, with the incorporation of new technologies and trial designs, the increasing number of therapeutic agents to be tested, the increase in the number of Cooperative Groups, and the evolving regulatory environment. Many of these issues are addressed in the recommendations in the preceding sections, but it is also necessary to examine the contributions of and interactions between NCI and the Cooperative Groups in developing and implementing large-scale cancer clinical trials. NCI’s coordination role within the current environment is quite challenging, and inefficient interactions between NCI and the Groups contribute to delays in the system. To improve the speed of advances in oncology care, streamlined processes are needed for the prioritization, selection, and support of trials and for the enrollment (accrual) of patients quickly after a trial is launched.
A major challenge that the Cooperative Group Program faces is the prioritization and selection of trial concepts before a trial is launched. The effective prioritization and selection of trial concepts is critical to ensure that limited public funds are used in ways that are likely to have the greatest impact on patient care. A previous report by the Clinical Trials Working Group (CTWG) recommended that NCI establish scientific steering committees that leverage intergroup, Cooperative Group, SPORE (Specialized Programs of Research Excellence), and cancer center structures to work with NCI in the design, evaluation, and prioritization of Phase III trials. The goal is to better allocate resources, increase scientific quality, and reduce duplication in trials proposed by Cooperative Group committees focused on a particular disease site, SPOREs, and other sources. However, the disease-specific steering committees set up in response to that recommendation do not appear to have fully achieved that goal.
Moreover, prioritization alone is not sufficient. At present, only about 60 percent of cancer clinical trials supported by NCI are completed and published. Inefficiencies in the system, including the time needed to respond to iterative reviews (as described in section I), can delay the launch of trials, and the longer it takes to open a trial, the less likely it is that a trial will meet its accrual goals. This represents a tremendous waste of very limited resources, including time, effort, and money. Once a priority trial has been selected, resources and effective procedures are needed to ensure rapid launch, patient accrual, and completion of the study.
The NCI Clinical Trials Cooperative Group Program has been chronically underfunded for the work that it performs, as noted in a 1997 review of the Program commissioned by the NCI director, and current funding does not cover the cost of the clinical trials undertaken. For the past 3 years, the annual budget for the Program has been held at about $145 million, but in real dollars it has declined to less than the 1999 funding level of $119 million, when the funding is adjusted for inflation. Despite this decrease in funding, the Cooperative Group Program has maintained patient accrual, with several hundred clinical trials ongoing at any given point. This level of funding, which represents approximately 3 percent of the total NCI budget, is simply not sufficient to support the number of trials that the Groups undertake. As a result, the Cooperative Group Program is highly dependent on the voluntary efforts of participating investigators and on supplemental funding from other sources, such as foundations, the pharmaceutical industry, and the institutional contributions of Cooperative Group members. Especially in light of the new focus on targeted therapy and personalized medicine, which raises the complexity and cost of clinical trials, the Cooperative Group funding process is becoming increasingly unsustainable.
Recommendations 8 to 10 aim to improve prioritization, selection, and support for clinical trials that have the greatest possibility of improving survival and quality of life for cancer patients and, along with Recommendations 11 and 12, aim to substantially increase the proportion of initiated clinical trials that are completed and published.
Recommendation 8: NCI should reevaluate its role in the clinical trials system. For example,
-
NCI should file more investigational new drug applications for agents to be tested in high-priority trials and provide a leadership role to ensure the success of those studies.
-
In cases in which NCI does not hold the investigational new drug application, the primary focus of NCI should be on supporting high-priority trials, with less emphasis on oversight of the selection and implementation process and greater focus on facilitating the launch and execution of the trial.
-
The process of peer review for trial concepts should be strengthened and streamlined and should entail the evaluation of concise proposals (including the intended statistical design) that are ranked against each other. The emphasis should be on scientific strength and opportunity, innovation, feasibility, and the importance to improving patient outcomes.
-
Steering committees administered by NCI should operate independently of NCI staff and should focus on the prioritization of
-
clinical needs and scientific opportunities, selection of trial concepts proposed by the Cooperative Group disease site committees, and facilitation of communication and cooperation among the Groups.
Since the funding mechanism for the Cooperative Group Program was changed from grants to cooperative agreements in 1980, NCI has exercised oversight of every aspect of the clinical trials process, including trial selection, protocol development, and trial operations. NCI has crucial responsibilities in the clinical trials system, for example, by providing a framework for both cooperatively and competitively organized interactions between Groups and their committees and in the management of IND sponsorship. As already noted in Recommendation 2, there are numerous steps that NCI could take to further improve the support and facilitation of high-priority trials. Helping Group investigators gain access to more experimental therapeutic agents for high-priority trials by filing an IND application would reduce the time that the Groups spend in negotiations with industry to acquire agents before a trial is launched and also ensure the availability of the agent during the trial.
At the same time, it is necessary to reassess NCI’s role and interaction with the Groups, which has evolved over the past 50 years and has become quite complex. NCI has leadership and legal obligations associated with holding an IND, but in cases in which NCI does not hold the IND, NCI should shift its limited resources from oversight to support of the trials process. A Cooperative Group whose trial concept has scored well in peer review should be able to request assistance from NCI as needed to develop and implement the protocol, but it should have the necessary expertise to develop and run the trial without extensive oversight by NCI, which can delay the process. Specific research projects funded through other grant mechanisms on the basis of peer review (the bulk of NCI extramural funding) are not subjected to such oversight.
The role of the steering committees should also be reevaluated. The historical CTEP approval rate for trial concepts before implementation of the steering committees was about 65 percent. As of January 1, 2010, the approval rate under the new system was not substantially different, with 62 percent of the concepts reviewed by these committees being approved. The length of concept proposals has also increased substantially (now about 20 to 25 pages compared with 10 to 12 pages in the past), making the review process more arduous. Moreover, multiple layers of review still slow the process, and trial concepts are still not ranked against each other with consistent criteria, as is usually done in peer review. Steering committees review and vote up or down on trial concepts as they are submitted and NCI staff actively participate in the review process, unlike other NCI
peer review groups. In addition, there is little interaction among the disease-specific steering committees to determine trial priorities across disease categories, nor do they consider how to balance the inclusion of Phase II or Phase III trials in the trial portfolio, although the steering committees are charged with “guiding the development of strategic priorities.” A possible alternative approach might be for the steering committees to identify research priorities and then issue requests for proposals to address them. If the steering committees continue to function as peer-review bodies, then NCI should have a more traditional role of facilitating the review process rather than actively participating in it.
In any case, it is imperative to strengthen the process for selecting high-priority trials. Launching only the highest-ranked trials would improve quality, speed advances, and ensure that patients are enrolling in the most meaningful and potentially beneficial trials.
Recommendation 9: NCI, Cooperative Groups, and physicians should take steps to increase the speed, volume, and diversity of patient accrual and to ensure high-quality performance at all sites participating in Cooperative Group trials. For example, they should
-
develop electronic tools that cue physicians practicing oncology via electronic medical record systems about trials for which a particular patient is eligible;
-
encourage patient eligibility criteria that allow the broadest participation possible;
-
encourage greater enrollment in high-priority trials, regardless of where the trial originates;
-
establish a centralized credentialing system for participating sites;
-
eliminate investigators and sites with low rates of accrual or inadequate data management skills or quality;
-
strive to make participation in clinical trials a key component of clinical practice and to achieve the exemplary attributes of the American Society of Clinical Oncology for academic and community clinical trial sites, including high accrual rates4 of 10 percent or more; and
-
encourage greater participation of patient advocates in trial concept development and accrual planning, and partnerships with patient advocacy organizations to support accrual efforts.
Rationale
Surveys indicate that the most important factor affecting patient accrual in clinical trials is whether a care provider offers participation in a trial to his or her patients. The majority of patients who participate in clinical trials are enrolled by a small percentage of participating sites, while many sites enroll only a few patients in trials to maintain their status as investigators. These circumstances can contribute to the underrepresentation in clinical trials of minority and medically underserved populations. Given the importance of trials in generating the evidence needed to make the best treatment decisions, more physicians should be encouraged to include trial participation in their clinical practice.
As noted in Recommendation 10, providing adequate case reimbursement would help to align physician and patient incentives and facilitate higher accruals at participating sites. However, another obstacle to increasing patient enrollment is that physicians may lack timely and easy-to-access information about clinical trials that would be appropriate for their patients. Some public databases with information about clinical trials exist, but in their current form, they may not adequately serve the information needs of physicians and patients as they are not part of the normal work flow of a busy clinical practice. User-friendly electronic tools, available with the right features for a physician’s work flow, would increase awareness of trials and make it easier for physicians and patients to enroll in the most appropriate studies.
Eligibility criteria present another challenge to increasing enrollment. Historically, stringent eligibility criteria have excluded many patients, including, for example, those with prior cancers or certain prior treatments. However, it has been argued that the adoption of less restrictive eligibility criteria for most studies would permit more rapid accrual and would also allow broader generalizations to be made, would better mimic the situation as it occurs in medical practice, and reduce the complexity and costs of clinical trials without compromising patient safety or requiring major increases in sample size. Greater involvement by patient advocates could help facilitate this change. Patient advocates also provide valuable input to study design and procedures, safety and confidentiality issues, feasibility, informed-consent processes, and other factors important to potential research participants to help facilitate the development, implementation, and recruitment processes.
Ensuring consistent quality at participating trial sites is also important. Site credentialing requirements vary among the Cooperative Groups, making the credentialing process onerous for sites that wish to engage with multiple Groups. A centralized credentialing system, perhaps outsourced to an independent entity, would increase consistency and quality across
sites and eliminate the burden of recredentialing. Such a system would also facilitate higher levels of enrollment in high-priority trials, regardless of where the trial originates, because sites would be credentialed to participate in any Cooperative Group trial. Moreover, elimination of sites with low rates of accrual would reduce costs and improve the efficiency of the clinical trial system.
Recommendation 10: NCI should allocate a larger portion of its research portfolio to the Clinical Trial Cooperative Group Program to ensure that the Program has sufficient resources to achieve its unique mission.
-
NCI should increase the per case reimbursement rate and adequately fund highly ranked trials to cover the costs of the trial, including the costs of biomedical imaging and other biomarker tests that are integral to the trial design.
-
To ensure sufficient funding for high-priority trials, the total number of NCI-funded trials undertaken by the Cooperative Groups should be reduced to a quantity that can be adequately supported.
-
External advisory boards, such as the National Cancer Advisory Board and the Board of Scientific Advisors, should have a greater roles in advising NCI on how it allocates its funds to support a national clinical trials program.
Rationale
High-priority trials must be adequately funded to efficiently and effectively attain results that can move the field forward. Compromising the science to launch more trials than the available funding can support is detrimental to progress. Innovative approaches to leveraging funding from sources other than NCI, as described in Recommendation 4, could also strengthen the Program, but NCI has an obligation to adequately fund trials identified as being of high priority. NCI should increase the total funding allocation for the Cooperative Group Program to ensure the effective translation of discoveries made with public funding to improved clinical care.
A first important step will be to raise the per case reimbursement, which has been set at $2,000 since 1999, although the median costs are estimated to be from $3,500 to $6,000 per patient. The many duties required of physicians and other key research staff, such as research nurses and clinical research associates, to participate in clinical trials are costly in terms of both time and resources. For example, before a trial can be opened at a particular site, much work must be done to ensure compliance with federal regulations
governing human subjects research. Once a trial is opened, a significant amount of time is spent discussing potential trial options with patients. If a patient enrolls, the data collection and documentation requirements are substantially more onerous than they are for patients receiving standard therapy outside of a trial. These voluntary contributions of clinicians who participate in the Cooperative Group Program constitute a substantial value and strength of the Program. However, when the discrepancy between the per case reimbursement and the actual cost of participation is excessive, as it is now, it becomes a major disincentive to participation. A substantial increase in the NCI per case reimbursement rate would constitute a major step toward aligning the incentives of physicians with those of their patients who wish to participate in clinical trials. Even in the absence of a substantial increase in the overall funding of the Program, the funds saved by launching fewer but higher-priority trials could be allocated for increased per case reimbursements to trial sites.
The existing system also often does not provide the resources required to thoroughly characterize each patient’s tumor and carefully match that profile to targeted therapeutics. Biomedical imaging and other biomarker tests are commonly becoming integral components of modern cancer clinical trials, but supplemental funding for these tests must be obtained by the Cooperative Groups through other support mechanisms.
The allocation of NCI funds among the competing needs of its various programs is a major challenge for the NCI director, who must take many factors into consideration. Greater input from the broad expertise and experience of external advisory boards, such as the National Cancer Advisory Board and Board of Scientific Advisors, would be helpful to ensure the most rational distribution of funds across the major NCI programs, in light of such factors as scientific opportunity and clinical need. These high-level boards should not be involved in the oversight of individual trials or in concept review, which would further slow the process, but rather, they should have a greater influence on how much funding is allocated to the overall Cooperative Group Program.
GOAL IV.
INCENTIVIZE THE PARTICIPATION OF PATIENTS AND PHYSICIANS IN CLINICAL TRIALS
Background
A robust clinical trials infrastructure is largely dependent on a critical mass of patients and physicians willing to participate in clinical trials. However, current indications suggest that participation in clinical trials is the exception rather than the rule, both for patients and for physicians. For clinical investigators, concerns about reimbursement, extensive regulatory
burdens, and academic procedures regarding tenure, promotion, and career development can all deter participation in trials. Investigator participation in trials requires substantial resources and staff. Given the limits in funding and capacity of the system, it is unrealistic to expect all or most clinicians to participate in trials, but those who are motivated to do so should be supported and encouraged.
Patient access to clinical trials is also an import issue to consider. Even if patients are eligible for trials and are informed about the option by their physicians (as discussed in the section describing Goal III), they may decline because of financial concerns, as coverage of patient care costs in clinical trials by health insurers is not consistent.
Recommendations 11 and 12 provide strategies to achieve the goals of increased participation by physicians and patients in Cooperative Group clinical trials.
Recommendation 11: All stakeholders, including academic medical centers, community practices, professional societies, and NCI, should work to ensure that clinical investigators have adequate training and mentoring, paid protected research time, the necessary resources, and recognition. For example,
-
NCI should recognize and reward Cooperative Group efforts in Cancer Center Support Grant (CCSG; P30) site visits, and allow the CCSG research base to include the federal per case funding received by cancer centers that participate in Cooperative Group trials.
-
NCI should provide funding to site and trial principal investigators to cover the time that they need to develop and oversee approved trials.
-
Academic medical centers should develop policies and evaluation metrics that recognize and reward clinical and team research in promotion and tenure decisions.
-
NCI should work with a nonprofit foundation to develop a certification program and registry, as recommended by the Clinical Trials Working Group.
Rationale
Multiple stakeholders need to take steps to support the recruitment and retention of clinical investigators both in community practice and in academia. The large-scale, multi-institutional trials that are the hallmark of the Cooperative Group Program require a team approach to research. However, career advancement in the field has traditionally focused on individual accomplishment. The current system does not adequately recognize,
reward, or support collaborative work. Furthermore, clinical investigation is often accorded less value than either basic research or patient care. This must change if the goal is to have talented individuals embark on a career that entails active participation in clinical trials for cancer as well as other diseases. Clinical research is a complex endeavor that requires training, mentoring, and paid time set aside for research to master and apply the skills needed to undertake innovative trials.
For example, the provision of funds for principal investigators to cover the time that they need to develop and oversee approved trials could improve the speed and quality of those trials. Recognizing the per case reimbursements for Cooperative Group trials in the CCSG assessment of a cancer center’s funding base would acknowledge the importance of patient accrual in these trials and encourage broader participation at those centers. A certification program for all research staff (including physicians, nurses, clinical research associates, pharmacists, etc.) would recognize the valuable contributions that these professionals make to the improvement of patient care and treatment.
Ultimately, the inability to recruit, train, and retain a sufficient number of talented clinical investigators will compromise the ability to conduct clinical trials in the United States, to the detriment of the U.S. biomedical research enterprise and to patients, those who participate in clinical trials as well as those who do not. Clinical trials help to raise the standard of care in the community by setting examples, and they have educational and training value for the oncologists involved, as physicians gain early knowledge of new drugs and gain experience with delivering complex therapies.
Recommendation 12: Health care payment policies should value the care provided to patients in clinical trials and adequately compensate that care. For example, the Centers for Medicare & Medicaid Services (via a national coverage decision), federal and state health benefits plans, and private health insurers should
-
establish consistent payment policies to cover all patient care costs (except for study-related costs, such as study drugs, devices, and tests, which should be paid for by the manufacturer) in clinical trials approved through the NCI prioritization mechanism, without having to pay for experimental therapies administered to patients outside of a clinical trial (any such limitation in coverage should not affect off-label use that is backed by evidence from clinical trials published in the scientific literature, as evidence-based off-label use constitutes the standard of care for many cancer therapies and is therefore not experimental) and
-
work with health care providers to educate patients more effectively about the availability, payment coverage, and value of clinical trials.
In addition,
-
The American Medical Association should establish new Current Procedural Terminology codes, reimbursed by the Centers for Medicare & Medicaid Services, private insurers, and other third-party payors, to pay an enhanced reimbursement for offering, enrolling, managing, and following a patient in a clinical trial.
-
The U.S. Congress should amend the Employee Retirement Income Security Act of 1974 to prohibit health plans from denying (or from limiting or imposing additional conditions on) coverage for the routine care associated with clinical trial participation.5
Rationale
Inadequate health care coverage is a major deterrent to participation in clinical trials for patients as well as physicians. Health care insurers traditionally have not paid for experimental therapies. However, much of the care provided to cancer patients is similar regardless of whether the patient is receiving a standard of care or an experimental drug. Some insurers and states acknowledge this and provide reimbursement for the routine clinical care of patients enrolled in trials, whereas others do not. The policies of the Centers for Medicare & Medicaid Services (CMS) regarding coverage of care in clinical trials have recently been in flux and, absent national coverage
decisions, may not be nationally uniform because fiscal intermediaries and carriers have some discretion on coverage, which can cause variations and inconsistencies by geographical region. Furthermore, the provisions of the Employee Retirement Income Security Act of 1974 (ERISA), which places the regulation of employee benefit plans (including health plans) primarily under federal jurisdiction for about 131 million people, preempts state laws governing such things as access to care and mandated coverage.
Thus, coverage of care in clinical trials is variable and may be uncertain, and patients who are interested and willing to enroll in a trial may decline because of an inability to pay for care that is not or may not be covered. Others might still enroll but may then experience significant financial hardship as a result. If such patients drop out of the trial, the scientific integrity of the trial can be compromised because of inferential problems that result from missing data. If cancer care is to be evidence based and relevant to the diverse population of patients with cancer, it is important for coverage policies to encourage rather than deter patient enrollment in trials. However, as a quid pro quo for improved coverage of care in clinical trials, insurers should be able to eliminate coverage of experimental therapies delivered outside of the clinical trial setting. Currently, many patients who are not enrolled in trials receive experimental therapy and expect coverage for it. The committee’s recommended approach is analogous to the “coverage with evidence development” mechanism that CMS has occasionally used, in which coverage is provided only within the context of a clinical trial. However, any such limitation in coverage should not affect off-label indications backed by evidence from clinical trials published in the scientific literature, as off-label use constitutes the standard of care for many cancer therapies and is therefore not experimental.
For physicians, even in cases in which routine patient care in a clinical trial is covered by health insurers, the current payment policies do not reflect the additional time needed to enroll and follow patients in a trial. For example, if a patient receiving off-protocol chemotherapy reports an adverse event or unanticipated problem, the physician can respond however he or she thinks is clinically the most appropriate. However, if a patient on a protocol who receives the same therapy reports the same adverse event, the physician must grade the severity, assess the attribution, document the event, consult the protocol, and make treatment modifications as required by the protocol. New codes in the Current Procedural Terminology, with higher reimbursement rates that acknowledge the additional time and resources needed to counsel and care for a patient in a clinical trial would address an important deterrent to physician participation in clinical trials. With a proper definition of eligible trials, use of such a code could easily be audited.
However, taking steps to align the incentives of patients and providers to participate in clinical trials may not be effective unless more is done to
educate patients about the availability and value of clinical trials. Educational efforts should focus on making the general population more aware of clinical trials. One reason is that it can be difficult for patients to sort through a large volume of new information and make complex decisions just after they have received a diagnosis of a life-threatening illness. Patients often lack comprehensive and reliable information about clinical trials and may not be able to identify the trials for which they might be eligible. Patients value reliable information from trusted sources, including family members, so appropriate education efforts could provide useful information that would allow patients to make informed choices about participation in a clinical trial. In addition, as noted in more detail in Recommendation 9, user-friendly electronic tools would increase awareness of clinical trials and make it easier for physicians and patients to enroll in the most appropriate studies.
SUMMARY
Collectively, the implementation of these recommendations would reinvigorate the Clinical Trials Cooperative Group Program for the 21st century and strengthen its position as a critical component of the translational pathway from scientific discovery to improved treatment outcomes for patients with cancer. Modifying any particular element of the Program or the clinical trials process will not suffice; changes across the board are urgently needed. All participants and stakeholders, including physicians, patients, and health care insurers, as well as NCI, other federal agencies, academia, foundations, and industry, must reevaluate their current roles and responsibilities in cancer clinical trials and work together to develop a more effective and efficient multidisciplinary trials system.
The Cooperative Group Program is beset by serious problems, but they are not intractable. The committee envisions a system that retains the current strengths, but moves beyond collaboration to integration, with reorganized structures and operations in a truly national clinical trials network and with sufficient funding and support to enable the rapid completion of well-designed, high-priority cancer clinical trials that advance patient care.




































