
Appendix D
Impact of the Regulatory Framework on Medical Device Development and Innovation
David W. Feigal, Jr., MD, MPH
The pace of innovation for regulated products is the combination of the speed of the development of science and engineering needed to make science-based regulatory decisions. Just as innovative medical products have a life cycle, from concept to obsolescence, new scientific and public health challenges have a life cycle. The two are intertwined and the lack of speed in scientific developments and the lack of a responsive science-based regulatory decision-making process can both slow progress. The US Food and Drug Administration (FDA) Critical Path initiative focused on the science of development: better toxicology, biomarkers, improved clinical trials, and personalized medicine, but that initiative, like others including the re-engineering initiatives of the 1990s and legislative changes that accompanied device user fees, did not examine the regulatory structure of the approval process itself and how the regulatory structure determines the choices in the science of development.
SCIENTIFIC AND PRODUCT LIFE CYCLES
In 2003, severe acute respiratory syndrome (SARS), a coronavirus causing severe sometimes fatal pulmonary infections, emerged in China and rapidly spread around the world (Anderson et al., 2004). Pandemics have a life cycle characterized by rapid worldwide spread which grows as long as each new infected person on average transmits the infection to more than one other person. An effective public health response in the life cycle of a new potential pandemic threat begins with identification of cases (see Figure D-1). Sometimes the first case in a community can be identified,
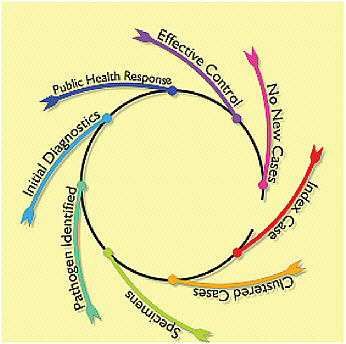
FIGURE D-1 The life cycle of the public health response to a new infection.
but more often the threat is recognized as clusters and then communities develop disease.
Collections of body fluids or tissue specimens are needed to identify the pathogen. Diagnostic devices are often an important element in an effective public health response to break the cycle of spread and prevent the emergence of new cases. The World Health Organization estimated that there were 8,096 cases with 774 deaths in 2002 and 2003 (Anderson et al., 2004; WHO, 2004). But by 2006 this SARS pandemic threat had been completely contained.
Parallel to the scientific and public health life cycle is the in vitro diagnostic product life cycle (see Figure D-2). As the clinical specimens from infected patients become available, the search for a pathogen begins. As infectious candidates are identified, analytes, the active ingredients of in vitro diagnostics, are created to develop investigational diagnostic devices. In 2003 SARS-infected clinical specimens were scarce, but with the pathogen identified “spiked” samples could be developed to refine diagnostic methods. Investigational diagnostics for emerging infections are initially tested against available specimens from sporadic cases and epidemiologic studies. As the test develops it may be prospectively studied to evaluate new cases and make decisions about clinical treatment or quarantine. Patient research
protocols and informed consent are often required at this time. As the test matures the quality system manufacturing processes prepare for commercial scale production. Once introduced into the market and in widespread use it isn’t long before the development life cycle repeats as the next generation tests are developed and improve the diagnostic test.
The scientific and product life cycles are interconnected and involve coordinated efforts by clinicians, academic research groups from many scientific disciplines, local, national and international public health organizations, and the regulatory oversight of research by institutional review boards (IRBs), clinical laboratory licensing agencies, such as the Clinical Laboratory Improvement Amendments (CLIA) program in the United States, and last but not least, the regulatory oversight of investigators and the device manufacturers by the FDA and other national device regulatory authorities.
While the effectiveness of the public health measures to control SARS forestalled the need to develop a commercial widely available diagnostic, the tools for innovation provided by molecular methods allowed public health laboratories and multiple in vitro diagnostic (IVD) manufacturers to have the investigational diagnostics available to help in the public health response. Another example of rapid development, approval and use of a new diagnostic, still in use, is the blood screening test for West Nile disease available since 2003.
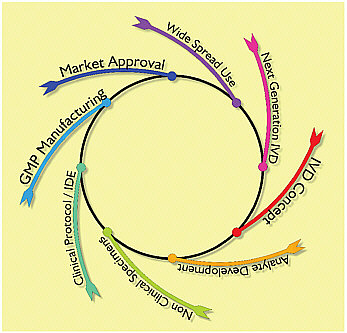
FIGURE D-2 The scientific life cycle of a new in vitro diagnostic device in vitro diagnostic device.
THE TOTAL PRODUCT LIFE CYCLE AND FDA’S DEVICE REGULATION
For the last decade the Center for Devices and Radiological Health (CDRH) at FDA has promoted the total product life cycle as a framework for medical device regulation. The origins of the US medical device laws were embedded in the US drug laws, and before the 1976 Device Amendments to the Food, Drug, and Cosmetic Act, some medical devices were approved with new drug applications (NDAs).1 As it began, device regulation largely reflected drug regulation. The initial medical device good manufacturing practices (GMPs) borrowed heavily from drug GMPs, the investigational device exemptions (IDEs) were patterned on the investigational new drug exemptions (INDs), and the premarket authorization (PMA) process borrowed advisory committees, preapproval inspections and clinical trial evidence standards from the NDA process. Drug regulation’s most powerful tool—the authority to approve drug marketing—became a key milestone in device development. Much of drug regulation is framed in terms of premarket and postmarket requirements. But device regulation does not fit as well into this simple premarket–postmarket regulatory framework.
While digoxin has always been and always will be digoxin and penicillin always will be penicillin, medical devices which are not as much discovered as they are designed, are iteratively developed technology. Devices have physical properties and performance characteristics that can be tailored for use, directly observed, and modified. Designs evolve throughout early development from prototype to bench testing to the clinical experience and use in IDEs and even once in the marketplace. Individual device failures provide opportunities for iterative improvements. The more complex the design, the more likely the product will rapidly evolve. All the phases of the device life cycle are interconnected and include connections across generations of a device.
Device consumer protections since the device amendments have recognized that wide range of potential device hazards and that the extent of regulation should be proportional to the level of risk. One of FDA’s first tasks in the 1970s was to classify devices and determine risk levels. While the basic regulatory framework put in place in the 1970s remain, the requirements began to evolve away from the drug regulatory paradigms. In the middle 1990s the device quality-system manufacturing requirements replaced the more drug-like GMPs. In 2000 CDRH began to create programs that combined the regulatory programs across the product life cycle and were tailored to the specific risks and clinical uses of particular products. The Division of In Vitro Diagnostics within the Office of New Devices became
the first TPLC (total product life cycle) program combining together all the regulatory teams with IVD responsibilities from premarket and postmarket surveillance to compliance activities.
Without accounting for iterative innovation it is difficult to design long term postmarket studies for a product which will be replaced in a year by the next generation of that manufacturer’s product. The best source of safety information about a product not yet cleared or approved for marketing may not be the preclinical testing of the new product, but the postmarket experiences from previous generations of the same product. Judicious device design changes during investigational or marketed use encourages innovation and optimization, and CDRH provided guidance on how much a product can be changed before it is a new product (FDA, 1997). A product designer or regulatory reviewer will have more insight into design challenges when considered along with failure mode analyses done after the recall of a similar product or in light of the product specific problems identified on compliance inspections. The science is all interconnected across the life cycle, not just for the innovative product developers, but also within the FDA and other regulatory bodies.
An interconnected life-cycle vision of medical device innovation emphasizes the need to link the diverse scientific disciplines and the regulatory mechanisms (see Figure D-3). Early in the process the device engineers develop prototypes and begin bench testing different designs to evaluate biocompatibility, or strength or flexibility, for example. FDA provides guidance to determine if the product will be regulated as a device, and if so, at which risk classification and product group. Specific guidances have been developed for many products on specific standards and regulatory requirements. Primary mode of action and intended use both guide the design and shape the regulatory path.
When products require clinical testing both the manufacturer and FDA will involve clinical scientists and statisticians and IRBs. Regulatory meetings may produce agreements and provide advice on the scientific evidence needed for regulatory decisions leading to market clearance or approval. Formal advisory panels may become part of the process for external scientific advice and CDRH calls on panel members for advice between meetings. Later in the life cycle the scientific disciplines become less experimental and more observational as medical device reporting (MDR) provides signals of potential new problems. Failed devices are analyzed and compliance inspections ensure appropriate quality systems. Some products need to be recalled, others grow old gracefully and are replaced by newer products over time. Devices frequently are improved by considering human factors engineering, training, and the diversity of clinical use. As illustrated in Figure D-3, both the scientific and regulatory processes are intertwined throughout the product life cycle. Just as different parts of the science life cycle are intercon-
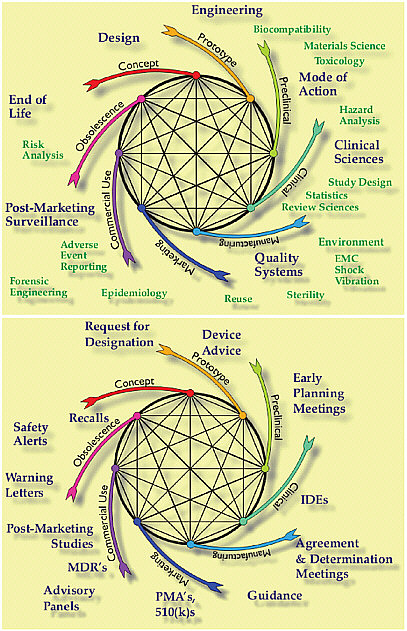
nected, the science and the regulatory requirements are intertwined, each informing and determining the other. There is an opportunity to build the connections, both at FDA and in manufacturers, so parts of the life cycle do not risk only being considered in isolation. For example, it is not uncommon for a premarket application to be reviewed without considering postmarket experience of similar products.
RISK CLASSIFICATION AND INNOVATION
Medical device regulations, around the world, begin by assessing the risk of the device and more specifically, the risk of the intended use of the device. The regulatory requirements (or “burdens,” as the US Congress implies when the statute refers to the “least burdensome regulatory path”) are proportional to risk. The lowest risk devices are required to meet general controls, register their facilities and list their devices but in large part are only regulated by FDA “for cause” when problems are identified. The highest risk products, the class III PMA products have development paths that are very similar to new drug development, as described briefly above. There have been as few as a dozen and never more than a hundred new PMAs in a year although there are also several hundred supplements and IDE protocols to review. The class III products occupy a disproportionate share of the review and inspectional resources at FDA. The ten to twenty new PMAs and the several hundred supplements, by CDRH’s estimates, occupy as much review time as the 3,500 501(k) applications. While PMAs require a preapproval inspection, 510(k) inspections are worked into the schedule of routine inspections, and the PMA products are far more likely to be topics at advisory panel meetings. The flexibility of the supplemental PMA process, the process to modify an approved PMA product, includes regulatory innovations such as real-time review and accelerates the innovation life cycle for these products, once initially approved.
CDRH summarizes the number of applications received and the review performance every year in an annual report that can be found on the FDA Web site (FDA, 2008). The approximately 3,500 class II 510(k) notifications each year represent the bulk of new devices with active FDA oversight. To achieve a steady state CDRH must clear approximately 15 notifications each business day. The review time to final action is accomplished more than half the time within the 90 day review clock. Historically CDRH has estimated that about 25 percent of its staff devotes their time to 510(k) activities which is not more than 20 days review time per application. By contrast the review of NDAs, biologic license applications, and even PMAs is measured in person-years not person-days. 510(k) notifications, however, do not have supplements or annual reports and these estimates do not count the regulatory hours spent on the rest of the product life cycle such as postmarketing
surveillance and compliance activities. Even if all of CDRH’s resources were dedicated to class II products, at current staffing, there would be no more than 0.25 person-year per application for review. Class II review cannot be modeled on NDA or PMA review, and because requirements should be proportionate to risk, the design of the class II regulations must be streamlined, as they are now, compared to PMA reviews.
The large difference in regulatory burden between class II and class III applications allows the development of smaller companies and more rapid product innovation cycles. While small companies have completed PMAs, the majority of class III products are developed by the larger medical device companies. The two factors that have the biggest impact on regulated product development are review cycle time and level of evidence requirements. The impact of the short review cycle can be illustrated when comparing US PMA products to European class III products. Because the US review cycle for some products is twice as long as the European cycle, not only are innovative products introduced later to the US markets, but the US markets miss every other new model when the PMA supplement review cycle is twice as long as the innovation and approval cycle outside the United States.
Risk reclassification is a difficult process for CDRH. Innovation is hampered when a product is classified in too high a risk class. The infrequency of reclassifications and the ponderousness of rule making which can add years of delay to implement reclassification decisions combine to slow the pace of implementing scientific change. The statutory default which assumes novel products, that is, products without a predicate, are class III products hampers innovation.
DEVICE CLASSIFICATION AND INNOVATION
By page volume, the largest part of the US FDA device regulations are devoted to describing approximately one thousand different medical device classifications. Along with risk classification, device classification is a strong determinant of the regulatory requirements for specific products. Each device classification has its own section in the regulations, and approximately half of all products approved also have an FDA guidance document that provides FDA’s best advice on regulatory requirements necessary for clearance or approval. The products with guidances have both a more rapid review cycle and a higher probability of first cycle approval. Products with guidances were also the first products that FDA allowed to be reviewed by third-party reviewers. Guidances advise not only the innovators but also FDA review scientists, which may account for the predictably faster review cycles.
LEVEL OF EVIDENCE AND INNOVATION
Finally, once risk classified and product classified that last determinant of the regulatory requirements is the product’s intended use. 510(k) clearance requires demonstrating substantial equivalence to one or more predicate devices with the same intended use. It is often the case that the predicate will be an earlier version of the same device from the same manufacturer, but a predicate can be any medical device marketed before 1976 or legally marketed 510(k) since. FDA has weak authorities to remove obsolete products and critics of the 510(k) process worry that comparison to any available predicate is a low bar. While guidances and standards can recommend more stringent requirements FDA cannot require them.
The extent and type of evidence are determined by the intended use. The same product can have more than one intended use. Different intended uses can even result in different risk classifications. To illustrate, a diagnostic device for cancer prognosis in someone already known to have breast cancer is a class II risk device. The same diagnostic, if solely relied on to make a specific treatment decision for breast cancer, would be a class III risk device. The level of evidence for prognosis could be based on retrospective observational data while the evidence for a treatment decision would likely require a prospective clinical trial.
“Tool claims” have less burdensome evidence requirements than specific clinical benefit claims. CT scanners are class II products. The quality of the images is assessed in light of the risks of the radiation exposure. Producing images is a “tool claim.” NIH is conducting a large trial to assess whether CT scan detected pulmonary abnormalities results in clinical benefit from earlier detection of lung cancer. If established, FDA could allow “detection and prevention of morbidity from lung cancer” as a claim or intended use. Not having explicitly established a clinical benefit from CT images has not kept the products from the market. Innovation in image quality and speed of image acquisition would likely not have occurred if product innovations required demonstration of clinical benefit.
Innovation is often established in research and practice outside of the regulatory framework. Laparoscopic cholecystectomy was first conducted in 1987 using laparoscopic tools developed for gynecologic surgery (preamendment devices). Adoption by general surgeons was rapid and by 1992 an NIH consensus conference recommended it as the treatment of choice (NIH, 1993). Randomized clinical trials were never done. “Tool claims” are often controversial, particularly for surgical materials such as patches or artificial membranes, as illustrated by public discussions of the concerns around surgical materials for meniscus repair, or surgical mesh for pelvic surgery.
STANDARDS AND INNOVATION
Embedded in the evidence requirements for approval and clearance are many standards. There are thousands of useful standards documents including biocompatibility testing standards, physical characteristics of biomaterials, metallurgy standards for implants or surgical equipment, electromagnetic shielding requirements, aseptic and sterile manufacturing standards to name a few. CDRH has recognized the use of approximately 500 standards. One special set of standards are the harmonization documents of the Global Harmonization Task Force, with the goal of harmonizing regulatory bodies. CDRH scientists participate in committees of the standards organizations and have a formal recognition process for standards which can be used in the 510(k) process. Just as FDA guidances promote innovation, standards provide a predictable regulatory framework. Predictability promotes innovation.
CLASS II PRODUCTS “WITH CLINICAL” AND INNOVATION
Many countries have four risk levels, most by splitting the class II products into those which require clinical testing as part of their special controls vs those which can be evaluated at the bench. Approximately ten percent of US 510(k) notifications rely on clinical testing as part of the evidence for substantial equivalence to a predicate device. Sometimes the clinical experience is integral to evaluating the performance standards required to assess the device and other times the clinical experience is required to develop training materials or to do human factors studies to ensure safe use.
Class II implants are a sometimes controversial example of these products. Since 510(k) applications have no annual reports or supplements the manufacturer decides whether changes in the product require a new 510(k). An implant with a specific design that had been clinically tested could be changed and introduced to the market without additional testing if the manufacturer concluded the change was allowed without a new 510(k). The product on the market would not match the product in FDA’s records. Unintended consequences of the changes may be difficult to detect. By not having a FDA review, the opportunity is lost for FDA to detect a problem, unknown to the applicant, that had been seen in a similar product from a different applicant. The 510(k) process can be criticized for creating opportunities to not conduct trials.
EXCLUSIVITY, TRANSPARENCY, AND INNOVATION
FDA approved pharmaceutical and biologics products can obtain exclusivity when they are new molecular entities, obtain new indications with
clinical trials, are orphan products,2 or have been the subject of certain pediatric studies. The progress made in orphan drugs and the development of pediatric pharmaceutical products after exclusivity was provided in new laws are examples cited of the role that incentives play in innovation.
There are no provisions for medical device exclusivity, even for the humanitarian device exemption products. It is not that the medical device innovators do not benefit from patents and intellectual property protections. The nature of medical devices themselves, often complex products with multiple components and rapid market cycles, makes exclusivity more difficult to define. Other than a rule which specifies a time limit during which FDA cannot rely on evidence from other applications, exclusivity is not a feature of device regulation.
More important to innovation is the transparency of the FDA processes. FDA is unique in the breadth and scope of its medical device regulatory decisions. In Europe, the early development process is divided between approximately 50 third-party notified bodies which have the sole delegated authority for premarket review in the European Union. Their review information is not shared. European postmarket surveillance is divided between the member states. No other regulatory body has as many medical device applications, public meetings, or guidances or has the small business assistance programs of FDA’s CDRH. Innovation is greatly fostered by this transparency.
BIOMATERIALS, COMPONENTS, PARTS, AND INNOVATION
The regulatory framework for innovation is more uncertain for components or accessories that are not approved in their own right. The components are not without oversight. If the biomaterial is part of an implant, standards such as the ISO biocompatibility testing standard specify safety assessment requirements. If the material is on the surface of the implant there are testing standards for wear and durability. If it changes the clinical risk to benefit ratio, for example by preventing lead fracture or infection, scientific evidence would be required if the manufacturer would like to base a new claim on that innovation.
There are many components that are difficult to individually assess in any scientific testing process. In a complex device this may be true of most of the parts of the device. But the answer is not to try and solve this in the development and design phase and the 510(k) clearance review alone. The quality of the device needs attention throughout the product life cycle. Innovations in medical devices are not just big fixes and big improvements.
Devices are products that can be continually improved. The regulatory framework becomes a problem when it creates disincentives to improve and innovate. An option to modify and change class II products would need to be less burdensome than the PMA supplement process, but it could create a clear path to innovation.
CLASS II LABELING AND INNOVATION
At first look, it would appear that the 510(k) process discourages innovative use since clearance is based on showing substantial equivalence to a predicate device in order to make the same labeling claims. If the claims are changed, a new 510(k) is required to make the same claims as some other predicate. For example, a replacement knee joint that is glued in place and the same prosthesis intended for use without glue require a different 510(k). There are separate device classifications for the two intended uses. How then is the 510(k) process able to foster innovation? Arguably with adjectives and adverbs and a flexible interpretation of the word predicate by FDA. Substantial equivalence is not the device counterpart to generic drug bioequivalence. In the latter the products are expected to be interchangeably alike, but the devices have to be “as good as” or better. A rapid pregnancy test can be substantially equivalent to a slow pregnancy test but a rapidly absorbed generic would not be bioequivalent to a slowly absorbed reference product. The effect of 510(k) labeling constrains innovation to the framework of the predicates’ labels.
HUMAN FACTORS AND INNOVATION
Medical devices usually require an operator, sometimes with considerable training and skill. One important component of labeling for many products is the IFU, the instructions for use. User errors are a common safety problem for medical devices, even when the device is not defective and is functioning as intended. Training programs and human factors engineering not only improve safe use but contribute useful information throughout the product life cycle to improve the design to minimize errors. Designing medical gas delivery devices so that a vacuum hose can’t be hooked up where the oxygen hose was supposed to be is a simple example of human engineering.3 A product with technology that has human factors innovations is particularly important for high risk devices and medical products designed for home use.
“LEAST BURDENSOME” AND INNOVATION
The 2007 FDA Modernization Act (FDAMA) required CDRH to use the “least burdensome path to market” for medical devices. The legislation deregulating dietary supplements (the Dietary Supplement Health and Education Act of 1994) provides ample evidence that claim creativity, for better or worse, is greatly enhanced by reduction of regulatory oversight. More examples of the impact of regulation and innovation are found in the gaps between FDA and other consumer protection regulations.
Laboratory developed tests4 fall in such a gap between FDA’s regulation of in vitro diagnostic devices manufactures, the CLIA regulation of clinical laboratories, and the state medical boards’ regulation of the practice of laboratory medicine. The widespread use of FDA cleared general purpose laboratory equipment, such as the polymerase chain reaction, to detect specific nucleic acid sequences has allowed laboratories to offer hundreds of different genetic tests, few of which have been evaluated by FDA.
CLIA has standards for the quality of laboratory processes but does not evaluate individual tests. Many of these diagnostics are used to evaluate reproductive risks for genetic disease or diagnose genetic diseases. FDA would clearly classify many of these devices as class III or class II products and require PMAs or 510(k)s if applications were submitted. Of these genetic tests, only about a dozen have been approved by FDA. Aside from the interesting legal questions about FDA jurisdiction,5 the fact remains that IVD manufacturers have not brought forward innovative genetic diagnostic tests through either the 510(k) or PMA processes. Some argue that these small markets do not support the cost of the regulatory requirements to get to the market. On the other hand, the state of New York requires review and approval by its own state health department reviewers, and requires payment of a user fee and many of the lab-based tests (LBTs) meet those regulatory requirements. Perhaps the low regulatory hurdle at the front end of the product development life cycle promotes innovation.6 Several public advisory committees, such as the Secretary’s Advisory Committee on Genetics, Health, and Society, have expressed concerns about the lack of FDA oversight. Innovation with biomarkers is even more complicated since the Center for Drug Evaluation and Research has expressed a strong preference for only approving indications that are guided by a diagnostic when the diagnostic is FDA approved.
RECOMMENDATIONS TO PROMOTE INNOVATION IN CLASS II PRODUCTS
With innovation in mind, what changes could be made to improve innovation for class II products?
Remove the requirement that novel products are class III (PMA) products by default. Although the de novo process is a work around, the assumption that novel innovations are synonymous with high risk is not correct.
Create a class II approval process. The 510(k) procedure deserves its own home outside of the historical work around of cobbling together a “clearance” (aka approval) process with registration and listing.
Remove reference to 1976. No other country in the world assumes that all class II products legally marketed before 1976 are suitable as reference products for performance standards.
Harmonize EU ISO 134857 requirements with the US quality system regulations. FDA does not have the resources to do biannual inspections of all class II manufacturers and lacks many authorities outside the United States. Use required third party manufacturing quality systems to supplement FDA inspectional authorities. Compliance predictability removes potential impediments to innovation implementation.
Do not require comparisons to predicate devices where performance standards are a better alternative. In those cases predicates should not be allowed and clearance should rely on accepted standards. Confidence in the evidence for clearance fosters acceptance of innovation.
Risk reclassification should be re-engineered to become a process that is applied routinely to keep the regulatory requirements up to date with current science.
Humanitarian device exemptions (HDEs) should be split into class II and class III products. Orphan unmet medical need are not synonymous with high risk. The potential for HDEs to foster innovation has not been reached because of the regulatory burdens of the program.
Refine the methodology for collecting clinical performance data across the product life cycle and across product generations. Unlike drugs where large, blinded placebo controlled trials are often needed to detect drug effects, device performance, and failures are often more directly observed. Health care providers and users benefit from precision in the relevant estimates relevant to the safe and effective use tracked over the product’s life cycle.
Refine the safety information collected about devices and tailor the information to the specific performance characteristics of specific devices.
Although class II “tool claim” products do not have clear off-label use (since there is barely a description of on-label use), collect the information about clinical outcomes and safety across the uses. Do not clear a surgical mesh just because it is “fit for use” without a plan to find out how the product is used and where it performs well and poorly.
SUMMARY AND CONCLUSIONS
There are many things in the current life-cycle regulatory framework that FDA “gets right.” Device marketing requirements are and should be risk based. Class II reviews are multidisciplinary and science-based and assess the requirements determined by the use of the product in the clinic. Manufacturing quality systems for a given product are tailored to the complexity and risk of that product. Attention to corrective and prevention action programs creates iterative product improvements. Recognized standards and product guidances are integral parts of the device regulatory framework. CDRH is effective in assisting small businesses and promoting innovation.
Changes in the 510(k) process potentially would better foster innovation and ensure confidence that the process results in safe and effective medical devices. Class II approvals should no longer reference preamendments (1976) products. Class II approvals should be based on objective performance criteria that ensure safe and effective use, when appropriate based on comparison to predicate devices recognized as meeting those standards. “Tool claims” are essential for the practice of medicine but some of these products would be more safely used if their actual use were better understood. Manufacturers should still be able to modify their devices when a new 510(k) is not required, but they should send the device equivalent of a PMA CBE (changes being effected) to the file at FDA so that FDA could request more information when appropriate. Class II device manufactures should be required to provide ISO 13485 certification for their manufacturing quality systems.
No review of FDA performance is complete without a comment on FDA resources. CDRH should have the resources to make classification reviews an on-going and dynamic process, complete a guidance document for every product classification, create an oversight process for a mandatory ISO 13485 certification by third party inspectors, develop a gap-closing process with CLIA for oversight of lab based tests that is streamlined and risk based, and continually seek proposals on how to maintain a vibrant and innovative medical device development community.
REFERENCES
Anderson, R.M., Fraser, C., Ghani, A.C., Donnelly, C.A., Riley, S., Ferguson, N.M., Leung, G.M., Lam, T.H., and Hedley, A.J. 2004. Epidemiology, transmission dynamics and control of SARS: The 2002–2003 epidemic. Philos Trans R Soc Lond B Biol Sci. 359(1447):1091–1105.
FDA (US Food and Drug Administration). 1997. Deciding When to Submit a 510(k) for a Change to an Existing Device (K97-1). http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080235.htm (accessed June 1, 2010).
FDA. 2008. CDRH Annual Report. http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM199014.pdf (accessed June 1, 2010).
NIH Consensus conference. 1993. Gallstones and laparoscopic cholecystectomy. JAMA. 269(8):1018–1024.
WHO (World Health Organization). 2004. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. http://www.who.int/csr/sars/country/table2004_04_21/en/index.html (accessed June 1, 2010).
















