
4
The Medical Device Industry Innovation Ecosystem
Part of the committee’s charge is to consider how the medical device industry innovation ecosystem is helped or hindered by the 510(k) statute and regulation. Two speakers reviewed the current environment of medical device innovation, including the effect of the current regulatory framework on device development. Panelists then discussed issues related to the balancing of patient safety and innovation
STRUCTURE OF THE MEDICAL DEVICE INDUSTRY INNOVATION ECOSYSTEM
Pain, suffering, and death from disease still plague patients worldwide. Even where solutions exist, many are suboptimal, and there is much room for improvement. Fortunately, the US economic system has created incentives and resources to promote and reward innovation, said Josh Makower, consulting associate professor of medicine at Stanford University Medical School and founder and CEO of ExploraMed Development, a medical device incubator. That has created a medical device (also called medical technology) innovation ecosystem in which ideas can become realities that can affect health care.
Many innovations in technology and procedure come from practicing physicians who have firsthand experience with what works and what does not. Their inspirations can become products. Makower cited a 1988 Institute of Medicine (IOM) and National Academy of Engineering (NAE) report on new medical devices and noted that not much has changed in medical
device development over the last 20 years—many of the challenges identified by IOM/NAE in 1988 persist today (IOM/NAE, 1988).
How Innovations Are Brought to Patients
The medical device innovation ecosystem has multiple components:
-
“Fuelers”—venture capitalists, investors, and public markets that support the process and invest in the innovators.
-
Innovation catalysts—small startups, large companies, incubators, and other entrepreneurs that invent the technology or take a concept through to commercialization.
-
Regulators—the Food and Drug Administration (FDA), the Centers for Medicare and Medicaid Services (CMS), third-party payers, and professional societies (which play a substantial role in patients’ access to new technologies).
-
Consumers—patients, physicians, and hospitals.
Innovation catalysts with ideas need resources if they are to advance their innovations to the product stage. Those resources come from the fuelers. Products then enter the regulatory system in the hope that they will leave it to be delivered to consumers (that is, patients, physicians, and hospitals). Marketed products produce revenue that is returned to the innovation catalyst and rewards the fuelers, consumers generate new ideas on the basis of experience with the products, and the cycle continues. All the players in the system are responding to their own sets of risks and rewards. As the risks and rewards change, the player’s behavior changes.
The primary fuel for device innovation comes from venture capital. However, little of the total pool of available investment capital is invested in medical device innovation. When venture capital underperforms, or when the total public market is compressed, venture capital for device innovation is reduced. From 2008 to 2009, for example, venture investment in medical technology declined by nearly $1 billion. As the global economy struggles, companies that have valuable technologies for patients are struggling to find capital. Only when venture capital outperforms does more money flow in.
The survival of small companies is critical for delivering innovation to patients, Makower said. Most of the ideas that really change the practice of medicine come from small companies or individual inventors. Department of Commerce statistics show that in 2002, 3,725 of the 6,007 US medical device firms being regulated by FDA had fewer than 20 employees, and only 150 had more than 500 employees. Large companies commonly acquire small companies. That provides a larger company with the innovation that it needs to grow and provides a small company with capital and with access to
the large company’s expertise in scaling up production and delivering patient solutions to a broader community. Public market success excites investors, who help to fuel the next round of innovation.
From a regulatory perspective, it is important to recognize that patients’ access to new health technologies is affected not only by FDA marketing approval or clearance but by the reimbursement process, which is also difficult to navigate.
Ensuring Safety in New Technologies
Patient safety is delivered primarily through good quality systems, and the vast majority of problems in the field are related to quality. In a well-run company, quality systems are integrated into the design process and follow a product through its life cycle, from concept through manufacturing and into the field (Figure 4-1). What is key for patients and for advancing
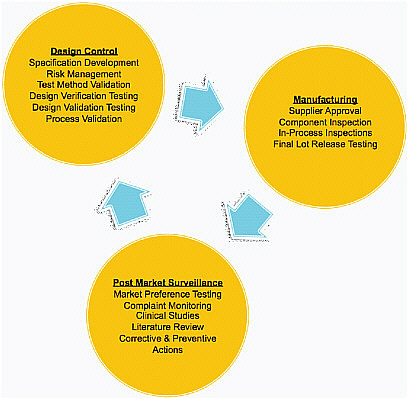
FIGURE 4-1 Patient safety is delivered primarily through good quality systems. Design control designs quality into a product from the beginning of development. Quality systems follow the product through its life cycle into the market.
technology is a system that permits rapid iteration, because it is impossible to model perfectly all the ways that devices and technologies are used in the field. Companies need to be able to make improvements quickly on the basis of feedback from the field.
There are substantial differences between how drugs and devices are developed and how they are used in practice. Devices span from low-technology tools, such as tongue depressors, to complex devices, such as implantable defibrillators. One size does not fit all with regard to evidence requirements. Some devices that are cleared through the 510(k) process undergo clinical trials, but many do not require clinical trials to establish safety. In fact, many of the structures of clinical trials that are used for drugs would be unethical and inappropriate to apply to devices (for example, blinding or sham groups for a dramatic surgical therapy). And devices rarely have distant systemic effects.
The standard device product life cycle is 18–24 months; that is, a product is replaced by a new or improved product within 2 years. Such a fast life cycle occurs, however, only when the reimbursement and approval pathways have already been pioneered. It often is not until the third or fourth generation of a medical device that clinical significance and cost savings start to become apparent; this is because of the time needed for adoption of the technology.
The Costs of Bringing Devices to Market Today
Before any funding is expended on pursuing a 510(k) clearance or premarket approval (PMA), there is a basic burn rate, the amount of money that a company has to spend every month to continue to exist. In addition, for any given product, costs are associated with concept development, proof of concept (for example, bench testing and animal testing), clinical unit development, obtaining an investigational device exemption (IDE), safety and feasibility studies (for example, small-group human trials), pivotal trials, the 510(k) or PMA process, and securing reimbursement. Today, navigating a device through the 510(k) process from concept through reimbursement will cost an average of $73 million for overhead and development. The cost to deliver new technologies to patients via the PMA path has historically been 2 to 5 times as much as the cost for 510(k) products (especially more novel products), averaging $136 million.
As noted earlier, iteration is the key to improving patient outcomes. The use of predicates allows innovators to build on established clinical and scientific data and bring incremental innovations to market quickly. Generally, little new science comes into play for 510(k) products. However, incremental technology innovation does not equate to incremental clinical value. Transformational leaps are created through a series of small steps.
One 510(k) product that delivered important outcomes was the delivery of insulin via pump vs multiple daily injections.
Time is money, and delays along the pathway from concept to market can be financially too much for a company to bear. If it takes a year to get an IDE approved, rather than 1–3 months, that can add $10 million to the overall cost. If later in the pathway the product is reassigned to the PMA track, there may be another $28 million in costs. Added time and expense at any step can become severe in the aggregate.
The Current State of the Device Innovation Ecosystem
In the marketplace, physicians are the natural gatekeepers for new-product implementation. They are cautious adopters, interested in both clinical data and the potential for reimbursement for their services. Device innovation is patient-driven (not technology-driven), and only technologies that address important patient needs can succeed, Makower said.
If the regulatory process is too difficult, it will deter even the most talented and creative innovators from entering the system. Similarly, most venture capitalists and entrepreneurs will avoid investing in projects that will require a PMA (although these are often the ones that have the greatest potential to affect human health). Over the last 10 years the number, of original PMAs has been declining; overall, there has been a disturbing compression in innovation in this country, Makower said. Companies’ expenses are increasing but not their returns. That reduces the financial returns to venture capitalists and decreases the likelihood that they are going to invest further in this sector. That, in turn, drives innovators out of device development. The net effect is that many valuable ideas and technologies never reach patients.
The medical device innovation ecosystem is fragile and extremely sensitive to changes in the cost of innovation, which is substantial, Makower concluded. The system is already under immense economic pressure. Innovation is driven by physicians and companies working together for the benefit of patients. The process is and must be iterative. The 510(k) process encourages multiple iterations, which can have a revolutionary effect on patient care. To ensure that safe and effective innovations sustain and improve patient health, regulatory systems must be predictable and reasonable.
Makower noted there has been much misunderstanding in the public press about the 510(k) process, some calling it a fast-track process and others believing that no 510(k)s involve clinical trials. Overall, the 510(k) system works well, and we should be looking at specific cases in which it did not work well—in which patients were harmed in some way—and ask what could have been done in those situations. The question is whether those are unique situations or require a global response.
Makower asked the committee to consider carefully whether the system needs to be fundamentally changed or whether it is only a question of opportunities for better management—for example, more resources, more and better-trained reviewers who have clinical expertise, a better process for resolving disputes fairly and promptly; synchronization of requirements between FDA and CMS that allows the reimbursement process to start earlier; greater investment in review of quality systems and less in premarket requirements for class I and II devices; and consideration of postmarket opportunities, such as unique device identification. Overall, Makower said, any recommendations should sustain innovation, improve predictability of the process, and not substantially increase cost or time to market.
EFFECT OF THE REGULATORY FRAMEWORK ON MEDICAL DEVICE DEVELOPMENT AND INNOVATION
David Feigal, vice president for regulatory affairs at Amgen and former director of FDA’s Center for Devices and Radiological Health (CDRH), presented an overview of a commissioned paper that he prepared for the committee on the regulatory burdens required to bring innovative medical technologies to market.1 FDA, Feigal said, is the nation’s oldest consumer-protection agency. The public health goals of the agency include safe human experimentation, marketing of products that have demonstrated effectiveness relative to known risks, manufacturing quality, truthful claims, prompt response to hazards, and prompt response to unmet medical needs. The question at hand is how well those goals are met in the regulation of medical devices, specifically, class II devices.
The Overlapping Life Cycles of Scientific Innovation and Product Regulation
Drugs and devices are different in many ways, but there are enough similarities for the device regulatory pathway to have borrowed some of its framework from that of drugs. For example, the biocompatibility testing of devices is based heavily on toxicology testing of drugs. Drugs have interactions; devices have malfunctions. Patients may receive the wrong dose of a drug; there may be user error with a device. Most drugs are clinically studied, whereas most devices (which are in class II) are bench-studied. Drugs rely on good manufacturing practices, and devices on quality systems. Those parallels often tempt people to say that devices should be subject to more drug-like regulation, Feigal said.
FDA’s most important regulatory tool, certainly on the drug and bio-
|
1 |
The complete commissioned paper is available as Appendix D. |
logic side, is market authorization. Consumer protections can be stratified as predominantly in the premarket part of the life cycle (safety experiments, premarket safety and effectiveness studies, and inspections focused on premarket research) as in the postmarket part of the life cycle (truthful promotion, adverse-event reporting, postmarket studies, and manufacturing inspection). When there are concerns, one’s instinct often is to require more evidence before marketing (for example, larger clinical trials before approval).
Some scientific problems, such as emerging public health threats, have life cycles of their own. Severe acute respiratory syndrome (SARS), for example, emerged rapidly and unexpectedly in 2003. The life cycle of the science of an infectious public health emergency starts with an index case (which is usually missed). Evidence of community wide infection begins to appear, and scientists begin to collect specimens (blood, saliva, and urine) and to try to isolate the infecting organism. Initial diagnostics are developed, usually in public health laboratories as laboratory-based tests, and begin to be used in the epidemic. The public health response (for example, quarantines and case tracking), is aimed at effective control and avoidance of new cases. As happened with SARS, by 2006 there were no new cases, at least for this initial cycle.
Similar to the cycle of public health response to a new infection are a product development cycle and a regulatory cycle. Development of a diagnostic device begins with identification of the concept, which is followed by development of the diagnostic active ingredient (that is, the analyte specific for the infection). It is often necessary to work with nonclinical specimens. Then there is a period of clinical investigation, which is followed by manufacturing scaleup of the device, market approval, and widespread use. Not long after that, there will be a next-generation diagnostic device, and the cycle will continue. The discussion at the time of SARS was about how to develop a rapid diagnostic that could be used in airports as people get off airplanes to determine whether to quarantine them.
The public health response cycle and the product development cycle have to be well synchronized, and they are inextricably connected to the regulatory cycle. Diagnostics, among the most regulated devices, are not only overseen by FDA in the United States but regulated under the Clinical Laboratory Improvement Amendments.
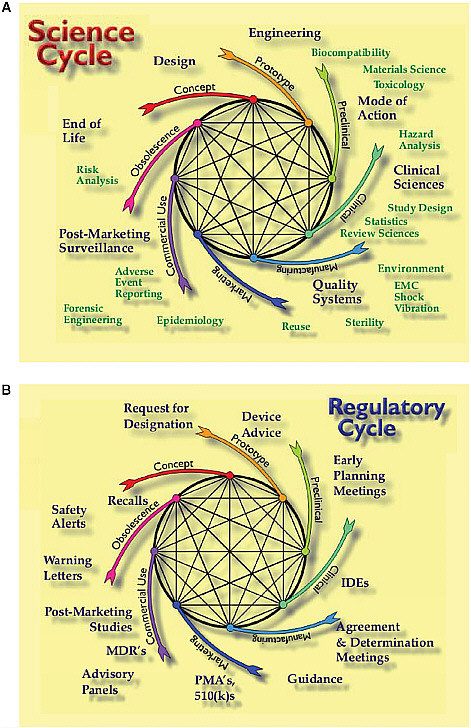
Instead of thinking only about devices as premarket and postmarket, CDRH looks at device products as a core cycle of concept, prototype, preclinical or bench testing, clinical evaluation, manufacturing, marketing, consumer use, and obsolescence. Every part of the cycle informs another part. An noted earlier, these products are iterative, and it does not make sense to think of postmarket requirements for a product that may be off the market—replaced by the next-generation product—before postmarket
studies could start. It requires a different kind of thinking, Feigal said. The next generation’s premarket studies are actually the postmarket studies for the current generation, and it is not yet clear how to manage this.
The science cycle that overlays the product development cycle is a multidisciplinary process that involves, for example, engineering, clinical science, statistics, quality systems, manufacturing, epidemiology, and postmarket surveillance (Figure 4-2a). The corresponding regulatory cycle incorporates, for example, requests for designation, device advice, early planning meetings, IDE discussions, pre-IDE meetings, determination guidance, the applications themselves, advisory committees, and medical device reporting (Figure 4-2b). When considering changing part of that cycle, Feigal noted, the committee needs to consider how the whole cycle may be affected.
Regulation of Class II Medical Devices
Risk Classification
Product risk classification can foster innovation if the regulatory requirements are proportional to risk, Feigal said. That is, if a less risky product has to go through less to get to the market, that creates a drive for innovation. The regulatory-review cycle length should be proportional to risk. The target cycle time for class II reviews is 90 days, compared with the PMA target cycle time of 180 days. Innovation is inhibited when the risk classification becomes uncertain, when it creates burdens that were not anticipated, or when the review cycle becomes long.
Feigal noted that FDA needs to clear 15 new 510(k) submissions on each business day to keep up with the 3,000–3,500 submissions that it receives per year; 5 or 6 years ago, he said, there were about 250 staff dedicated to reviewing 510(k) submissions. Simple mathematics shows that staff can spend no more than a few weeks in reviewing an application, assuming that they review full-time and do nothing else. (Staff effort for approval of a drug application, approval of a biologic license application, and the PMA process is measured in person-years, not person-days.)
Device Classification
Device classification depends on the device technology and the product claims. Guidance documents for many of the classifications help to make the process more rapid and predictable and to foster innovation. But over 1,000 medical device classification groups need guidance documents, and it is especially challenging to write guidance that will continue to be relevant to rapidly changing science.
Evidence Requirements
Evidence requirements are proportional to risk. The quantity and type of evidence required depend on the intended use of a product, and the 510(k) process of comparison with a previously approved product allows incremental improvements. A challenge for FDA is to manage functional or “tool” claims vs clinical claims. A functional claim requires less evidence, describing what a device does but not specifically which patient population it should be used for. There is concern that this approach creates a backdoor for technologies to be cleared without enough information about what they are going to be used for.
In addition to the evidence requirements, there are regulatory standards that help to simplify the review process. Evidence can be based on objective nonclinical performance criteria. In many cases, a performance assessment by an engineer or a physicist can provide more useful information about a device than a clinical trial. Although FDA makes a substantial investment in standards development, it is challenging to keep up. Standards are an important part of the assurance of the effectiveness of class II devices, Feigal said, and this function needs to be supported by adequate resources.
Clinical Evidence
Clinical evidence is needed more often for class II products than one might expect. Although a clinical trial may not be needed to establish safety and effectiveness, substantial equivalence sometimes can be evaluated only in the clinic. In other cases, there may be a need for clinical experience to address the human factors associated with the use and performance of a product; that is, clinical experience may be necessary to write effective training materials. Clinical evidence takes longer to obtain, and one approach could be to collect evidence across the entire class II product life cycle, not only before clearance.
Regulatory Incentives
For drugs and biologics, regulatory incentives, such as marketing exclusivity, encourage innovation. That approach is ill suited for the rapid changes and complexity of medical devices, Feigal said. Transparency is an important regulatory principle that promotes innovation and the adoption of safe and effective products. Transparency of the advisory-committees process and the transparency provided by guidance documents are strong development tools for innovators. One of the (somewhat controversial) proposals for increased transparency is that FDA make rejection letters public,
but there is some discomfort about the possibility that that would disclose trade secrets prematurely.
One of the principles of regulation is that claims are based on what is known. There is an incentive to know more, to have a better claim, and to secure a stronger market presence. The challenge is when to require that more be known as opposed to providing incentives to know more. That is, which information is important for the development of the medical knowledge base as opposed to essential to ensure safe and effective use? It is important to preserve the incentives and rewards that cultivate knowledge and to be cautions about requirements that might stifle innovation.
Biomaterials, Components, and Accessories
FDA struggles with the numbers and variety of biomaterials, components, and accessories in Class II products. There are often so many components in a device that it is impossible, for example, to remove components one at a time to see whether the product still works and what each component’s unique contribution is. The final manufacturer is responsible for the whole device although components have many sources. FDA’s challenge is to set priorities for oversight of manufacturers and their supply chains. It is important that standards not be so rigorous that we lock ourselves in to an existing technology and freeze out new technology in biomaterials and components, Feigal said.
Labeling
Class II devices are cleared on the basis of substantial equivalence to a predicate and so should have essentially the same labeling. That is counter to innovation. However, a class II device has to be “at least as good as …”, so there is some ability to modify labeling to indicate the improvement over the predicate; for example, a new diagnostic device may be a “rapid” version of the predicate. In considering the 510(k) process, the labeling limitations for new class II devices should be taken into account.
Opportunities for Improvement
Feigal offered several suggestions for changes in the 510(k) process that would foster innovation in class II devices. First, he said, it is confusing to the public to have “cleared” products and “approved” products. FDA is approving class II products on the basis of a set of standards relevant to the class. There should be a separate class II approval process based on objective performance standards, clinical safety and effectiveness, and predicates that meet appropriate standards. Feigal recommended removing the reference to
pre-1976 devices from the statute. In handling class II products, he said, there are times when it would be better to rely on absolute performance standards rather than a predicate. Feigal also supported harmonizing the US quality-system regulations with the International Organization for Standardization Standard 13485 requirements and allowing mutual recognition.
Feigal suggested a variety of opportunities for FDA to foster innovation. The agency can work toward streamlining the risk-classification process to keep up with science. There is a need for guidance on all product classifications. It is also important to use evidence from the whole life cycle in decision-making. Regulatory decisions need to be science-based, not legislation-based. The law sets the framework, and the decisions are based on science. Peer-review regulatory decisions could enhance consistency and quality. These opportunities could be incorporated into the agency’s review practices. Some lags in transparency, particularly in connection with Freedom of Information requests, can be fixed.
A part of the life-cycle map that is inadequate for 510(k) products is the postmarket period. More information is collected on PMA products than 510(k) products once they are on the market. Postmarket information is collected for PMA products, for example, in annual reports, periodic safety reports, and tracking reports. Some version of those could be used for 510(k) products. In addition, when a company decides to make a change in a product that does not require a new 510(k), it could submit general information about the change to FDA. It would not submit the whole set of changes that it documents internally but would keep FDA informed that the product has changed—for example, in a surface coating. If FDA sees a change in postmarket event reporting, it will know if there was a change in the product. Currently, FDA can learn about such minor changes on inspection, but it does not have enough resources for inspection. That would be one way to achieve better information flow. In addition, FDA does not know which products are in use and which ones have been withdrawn. Collecting information in this part of the life cycle is challenging. Notification of some events, such as withdrawal from the market, should be required.
In summary, Feigal said, risk-based regulation tailored to the specific nature of different class II devices is an appropriate way to protect the health of the public while encouraging innovation. Changes in the 510(k) process should strive to foster innovation, ensure confidence that the process has integrity, and bring to market tools and technologies that offer benefit with well-understood risks.
Panel Discussion: Balancing Patient Safety and Innovation
After the presentations, Makower and Feigal were joined by Hutt, Ulatowski, Phillips, and three other panelists: Amy Allina, program and policy director of the National Women’s Health Network (NWHN); Bruce Burlington, an independent consultant, former executive vice president for regulatory affairs and human safety and quality at Wyeth, and former director of CDRH; and William Vaughan, a consultant to Consumer’s Union on FDA issues, formerly staff of the House of Representatives Committee on Ways and Means, and staff director for the minority on the House Subcommittee on Health.
There was much discussion of the evidence base for device decisions. Panelists discussed how much evidence is enough for using products that are cleared through the 510(k) process and how such evidence should be obtained. Panelists also discussed how in vitro diagnostics fit into the medical devices structure, the need for consistent decision-making in classifying devices, and FDA’s role as an enforcement agency.
Evidence Base
Allina described the mission of the NWHN as working to bring the concerns and needs of women consumers to the health-policy and regulatory discussion. In addition to safety and effectiveness, consumer advocates are concerned with innovation, seeking development of better products and sometimes of products that are already approved outside the United States. One question raised during a presentation was, How high should the regulatory bar for evidence be set without risking the blocking of patient access to innovative products? A parallel question, Allina said, is, What are the effects in patient harm and dollars wasted on ineffective products if the bar is set too low?
What does it mean for a product to be effective? Allina offered the example of home uterine-activity monitors, which some pregnant women are instructed to use if they are at risk for preterm birth. The manufacturer did not have to show that using a home uterine-activity monitor would make a difference in preventing preterm birth, but only that it worked as it was intended to work. Ultimately, a study by the National Institute of Child Health and Human Development found that the monitors are not useful in predicting or preventing preterm birth, the American College of Obstetrics and Gynecology concluded that they should not be part of standard care, and the Agency for Healthcare Research and Quality advised against using the products because they confer no maternal, fetal, or neonatal benefits. FDA needs the authority, Allina said, to require companies to provide relevant efficacy data. Otherwise, health-care dollars are being wasted, and
patient harm can result. The FDA needs to know that it can use its flexibility to meet patient safety demands, not only to respond to the concerns of commercial sponsors, she said.
Makower responded that physicians should be using clinical data to drive their decision making and that FDA should not be preventing them access to a device if they decide that it is good for their patients. The question, he said, is, Where is the line between when there are enough data to allow a product onto the market and when the responsibility of the physician begins? Information is the key. If FDA said that “this is commercially available, but there is no evidence to support X, Y, and Z,” that would have a powerful effect on a product’s utility.
Hutt added that the laws do allow FDA to require clinical utility; that is part of “substantial equivalence.” With home uterine monitoring, he said, there was a requirement for a clinical trial, but the end point that was chosen was whether the device allows a doctor to obtain information earlier and therefore be able to intervene to prevent preterm birth. It may well have been the wrong end point to choose, but there was no lack of statutory authority or of clinical trials in this case.
In review of 510(k)s, Ulatowski said, much time is spent in trying to figure out what questions need to be answered; for example, Is it materials or clinical utility? What is necessary in the end point?
Hutt referred to the Cooper Committee report, which strongly concluded that the type, quality, and quantity of evidence required for devices were different from those required for drugs. As a result, the 1976 device statute contains different language regarding the safety and effectiveness of devices from the 1962 statute addressing the requirements for drugs.
Economics come into play in evidence requirements. Hutt asked whether a device company could afford a $50 million or $100 million controlled clinical trial for a medical device that does not have nearly the market value of a blockbuster new drug. High economic barriers harm innovation. The corollary question, Burlington said, is whether we can afford to have products on the market on which there is not enough information about proper use or even on whether use of the device provides more benefit than risk. Allina added that although NWHN advocates for new alternatives to existing products, no one is helped by approving or clearing more products on which there is not enough information about use. The challenge for FDA is to identify the middle ground.
A committee member asked whether Hutt, if he were to rewrite the 1976 amendments today, would consider requiring more scientific evidence regarding the safety of medical devices. Hutt responded that structurally, he does not see a need to change anything in the current statute. FDA can require whatever data are needed to show safety and effectiveness. The amount of data required is, and should be, a matter for FDA discretion.
Trying to legislate the level of evidence required would inevitably set the bar at the wrong place. It cannot be accomplished by statute, regulations, or guidance but only by individual reviewers case by case. Allina said that it is important to empower the agency to make those decisions on the basis of science and noted that FDA could use more direction in setting that bar.
In considering changes that could be made in the 510(k) process, Feigal encouraged the committee to look for ways to create incentives and rewards for accruing information as a product evolves and the science evolves over the course of the product life cycle. For example, digital mammography was first allowed onto the market for use in patients that were referred because of an abnormality. It was not clear at that time whether the technology would also be good for screening. Four companies agreed to conduct a 40,000-patient study funded through a public partnership with the National Institutes of Health. FDA created an incentive for the study by saying that unless one of the devices was a particular outlier, it would make a single decision for the group of products. The companies would not be allowed to compare each other’s products on the basis of data from the trial; the clinical utility of digital mammography for screening would be established as a group. When the study was completed, the data revealed findings that would not have been apparent in a small study, for example, that there were advantages for some groups of women with respect to detection. Those types of studies cannot be done every time, but the example shows that some questions can be answered only with large, multisponsor trials.
Burlington asked whether, as a result of the current financial situation, FDA is being forced into a system in which, to get companies to innovate, it needs to allow products to enter the market quickly and easily. Are products being developed in the marketplace rather than before they are put onto the market? Makower said that device companies are constantly solving problems and in the process learning new science and discovering new problems to solve. Feigal noted that the first blood-screening test for hepatitis C had only had about 60 percent sensitivity. The blood advisory committee recommended not telling blood donors their test results but using the test only to screen donor blood and destroying any blood that was probably infected. That was before treatments were available. Eventually, knowledge about hepatitis C infection accumulated, and treatments were developed. But it had been right to try to prevent transmission of hepatitis C through blood products even though the tools were less than ideal. That is the iterative nature of the development of medical products.
In Vitro Diagnostic Devices
Does it make sense, Burlington asked, to use the same regulatory framework for both diagnostic and therapeutic devices? If so, why is it appropri-
ate for FDA not to regulate the central laboratory–based high–information-content diagnostics?
Feigal said that inaccurate information from in vitro diagnostic tests can be as dangerous as a faulty medical device. If a diagnostic product yields an erroneous cancer diagnosis and the diagnosis is acted on, it is the information that has caused harm. One of FDA’s problems occurs when regulatory systems overlap. In this case, there is a gap between supervision of laboratory processes under the Clinical Laboratory Improvement Amendments and FDA supervision of in vitro diagnostics.
Phillips said that the FDA Office of In Vitro Diagnostics regulates diagnostic tests a bit differently from other products because the concept of substantial equivalence has evolved differently in relation to the two. There is less emphasis on substantial equivalence and more emphasis on characterizing the performance of products (for example, sensitivity, specificity, and accuracy), and there is a standardized regimen for assessing how products perform. In addition, a specific labeling regulation that governs in vitro diagnostics does not apply to other products.
Diagnostics are somewhat different, Feigal agreed, but the fundamental problem of having enough information is the same.
Consistent Criteria for Assigning Device Class
One particular issue of concern to NWHN is the lack of consistent criteria for determining what goes through the 510(k) process. Companies that were developing female condoms, for example, initially sought to use the 510(k) process, with the male condom as a predicate device. Of course, there are some obvious differences, and a female condom raises questions that would not have been asked in approving a male condom, Allina said. NWHN argued, and FDA agreed, that female condoms should have to go through PMA. But there was a great deal of confusion, controversy, and delay. The lack of clarity was a problem for the company, for FDA staff, and for women. As a result, FDA convened a group of product developers, consumers, and scientific experts to help them to develop guidelines for contraceptive-device approval. The new guidelines were helpful but did not solve the problem of inconsistent decision-making across the board. Later, when a female-condom manufacturer wanted to return to FDA with what appeared to be a simple materials change, it was required to go through PMA. In light of the existing vagueness and flexibility, as things change environmentally there is no consistency within the agency.
No one would disagree that greater consistency is needed in everything that FDA does, Hutt said. But he noted that achieving that goal is extremely difficult in what is now an over 12,000-person government agency that makes numerous decisions daily.
Enforcement
Burlington raised the question of whether the current enforcement regimen is an efficient or effective way to ensure to compliance. The agency has the authority to issue civil financial penalties, but the process is cumbersome and inefficient. Ulatowski responded that FDA has lost the efficiency to take actions quickly (for example, product seizures), and he supported the commissioner’s current efforts to revitalize the enforcement program. The tools for enforcement exist, but there could be greater efficiency in using them. It would also involve training staff to be more effective. Hutt added that although FDA is built on science, it is not a science agency. Its true mission is law enforcement, and the failure to bring strong enforcement action is a signal that the agency does not stand behind all its requirements to the degree that it should.
Additional authorities and resources are much needed for antifraud efforts, Vaughan said. Those resources will probably come from user fees, and this presents a conflict because an industry that funds an agency can have influence on the agency. User fees should not be associated with specific agency performance requirements, he said.
Participants discussed the extended length of time between inspections of foreign plants that manufacture class II devices and the need for more resources to tighten oversight. Burlington noted that public firms have to disclose financial information quarterly and annually and must have their disclosures attested to by an independent accounting firm. Perhaps a similar system for device compliance might be effective: disclosure by the company reinforced by an independent third-party audit and backed up by FDA, which would oversee the auditors and introduce sanctions when they are needed. That is essentially the system that is used in countries that require International Organization for Standardization Standard 13485 certification, Feigal said. Companies provide compliance information to the government before they file some kinds of applications, or they have independent audits of their quality systems. Such an audit is one way to keep up to date and avoid the problem of the 5- to 7-year inspection cycle. Feigal noted, however, that he sensed a strong preference in FDA for conducting inspections itself rather than through third parties and self-certification, in part because FDA, as a law-enforcement agency, has the responsibility.
REFERENCE
IOM/NAE (Institute of Medicine/National Academy of Engineering). 1988. New Medical Devices: Invention, Development, and Use. Washington, DC: National Academy Press.


















