
5
Examining Mechanisms of
Breast Cancer Over the Life Course:
Implications for Risk
The preceding chapters have summarized the available evidence for the relationship between environmental exposures and breast cancer, as well as the many challenges inherent in studying this issue. Although there is strong evidence of a modest role for a handful of modifiable environmental exposures as risk factors for breast cancer, many unanswered questions remain. These unanswered questions require new research approaches, which are discussed in Chapter 7.
Meanwhile, remarkable progress has been made in understanding the fundamentals of carcinogenesis, manifested in mechanisms at the genetic, epigenetic, cellular, and tissue levels. Scientific advances are revealing complex potential pathways and factors involved in cancer development. The committee sees the need for a continued and intensified focus on understanding the basic biology of breast carcinogenesis in order to gain better fundamental appreciation of the environmental factors with potential roles in the etiology of this disease.
As a crucial dimension of this research, the committee notes a growing appreciation among researchers of the important role that the timing of exposure plays in effecting changes that alter the likelihood for cancer and other diseases later in life. Observations in human studies of the effects of exposure to ionizing radiation and diethylstilbestrol (DES) in early life, as well as mechanistic and animal studies of other environmental exposures, suggest that existing assessments of the role of certain environmental factors derived from studies in adult women, such as those reviewed in Chapter 3, may have been negative or uninformative because they failed to consider critical periods of life stage and exposure—essentially asking the wrong
question. As understanding grows of how the genetic, epigenetic, cellular, and tissue changes in the breast during development and over the life course influence susceptibility to breast carcinogenesis, researchers have continuing and increased appreciation for the potential for timing of exposure to make a difference in the effects of environmental agents on breast cancer risk. The committee sees the need to direct attention to the accumulating evidence that environmental exposures may have a differential impact, depending on their timing during the life course.
ENVIRONMENTAL EXPOSURES OVER THE LIFE COURSE AS DETERMINANTS OF BREAST CANCER RISK
The female breast is not static; it changes in structure and function over the life course. Breast development begins in utero and continues into adulthood, with further differentiation occurring with pregnancy and lactation and involution occurring with menopause (Russo and Russo, 2004; Polyak and Kalluri, 2010). Most breast cancers arise in and spread from the ducts or the lobules, which are the breast’s main functional components (Figure 5-1).
The sections that follow consider the major life stages for women and the state of breast tissue during each stage, with indications of the potential for exposures during each stage to alter risk for breast cancer. Although evidence from human studies is limited, studies in animal models strongly indicate the potential for timing of environmental exposures to alter risk for developing cancer. Box 5-1 lists the life stages discussed by the committee and mechanisms of carcinogenesis likely to be of particular relevance or importance to breast cancer.
Early Life Exposures and Breast Cancer Risk
Preconceptional and Periconceptional Exposure
Preconception studies focus on parental exposure to environmental agents before the conception of offspring. There is no standard definition for the preconceptional period, and the term is used rather loosely. Some studies combine the time before conception with early pregnancy as the periconceptional period (Van Maele-Fabry et al., 2010). Studies examining pre- or periconceptional exposures may consider paternal or maternal exposure, or both.
To date, epidemiologic studies have not addressed parental exposure before conception and subsequent risk of breast cancer in offspring. However, childhood cancers, such as leukemia and brain tumors, have been linked to prenatal exposures such as maternal smoking, ionizing radiation,
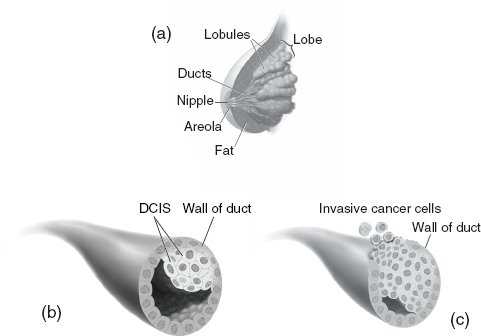
FIGURE 5-1 Schematic representation of (a) the breast, showing lobules and ducts, (b) ductal carcinoma in situ (DCIS), and (c) invasive ductal cancer.
SOURCE: NCI (2009).
and pesticides (Shim et al., 2009; Van Maele-Fabry et al., 2010). Preconceptional parental exposure to ionizing radiation was not associated with an increased risk of childhood leukemia (Wakeford, 1995), and parental exposure electromagnetic fields before conception has not been demonstrated to increase the risks of childhood cancers of any kind (Sorahan et al., 1999).
It is difficult for studies to track offspring of parents whose exposures before the child’s conception are known until the children reach the ages in which breast cancer tends to manifest itself, making this a potential area for future research.
In Utero and Neonatal Exposure
The human breast begins to develop once the embryo reaches 4.5–6 mm in length (Hughes, 1950). Embryonic epidermal cells proliferate to create a breast bud, which responds to cues from the embryonic mesenchyme (Anbazhagan et al., 1998). In the newborn human, the breast is characterized by “very primitive structures, composed of ducts ending in short duct-
Some mechanisms are likely to be more relevant or important to breast carcinogenesis at particular life stages (e.g., epigenetic reprogramming during the in utero/perinatal period).
Life Stages as Potential Windows of Susceptibility
• Preconception
• In utero/perinatal
• Early childhood
• Prepuberty
• Puberty
• Reproductive years
• Menopause
• Postmenopausal years
Hypothesized Mechanisms of Carcinogenesis
• Mutagenesis
• Nuclear hormone receptor signaling
• Mitogenic signaling leading to cell proliferation
• Epigenetic and developmental reprogramming
• Modulation of immune function, escape from immune surveillance
• Alterations of tissue microenvironment
ules lined by one to two layers of epithelial and one of myoepithelial cells” (Russo and Russo, 2004, p. 3). The breast in the newborn also contains a population of stem cells that are the undifferentiated precursors of the cellular expansion that occurs as the structures of the breast develop during puberty, pregnancy, and lactation.
Strong evidence indicates that aspects of fetal growth, such as birth weight, are associated with breast cancer risk as an adult (Potischman and Troisi, 1999; Michels and Xue, 2006; dos Santos Silva et al., 2008; Park et al., 2008b). According to one postulation advanced by Trichopoulos and others (2005), the number of mammary stem cells is determined during in utero or immediate postnatal life and is under the influence of estrogens and components of the insulin-like growth factor system during pregnancy. They hold that the number of mammary tissue-specific stem cells is the core determinant of breast cancer risk. Thus, an increase in mammary stem cells associated with higher birth weight would provide more target cells for breast carcinogenesis.
This postulation also includes recognition of the increased risk of breast cancer associated with increased breast density and mass of glandular tissue (Boyd et al., 1998, 2010), which is itself likely linked to the number of stem
cells (Trichopoulos et al., 2005). While this hypothesis focuses on the effects of endogenous estrogens and growth factors, it also provides a potential mechanism by which exogenous environmental agents might increase (or decrease) breast cancer risk if prenatal exposure to them promotes the formation of greater numbers of breast stem cells. There is growing evidence from rodent models to indicate that exposure to xenoestrogens during the prenatal and neonatal periods may affect mammary gland development and alter risk for cancer later in life (Soto et al., 2008). Some of these changes are thought to possibly result from developmental reprogramming as described below, but some of the mechanisms are just beginning to be elucidated.
During gestation, while maternal levels of the pregnancy hormones progesterone and estrogen soar, the developing fetus is protected from endogenous maternal hormones by steroid hormone binding proteins. Steroid hormones such as progesterone and estrogen circulate in the blood stream bound to proteins such as serum albumin and sex hormone binding globulin (SHBG), which is a glycoprotein that specifically binds testosterone and estradiol. Only a small fraction of steroid hormones is present in the circulation in the unbound “free” form. In utero, the fetal liver synthesizes sufficient amounts of steroid hormone binding proteins, including steroid-binding β-globulin (SBβG) and alpha fetoprotein (AFP), to protect the developing organism from rising levels of maternal hormones. Thus, even when maternal estrogen levels are extremely high, increased expression of AFP and other steroid hormone binding proteins (Mizejewski et al., 2004) reduces the unbound fraction of estradiol in the human fetus to 1.0 to 4.5 percent (i.e., greater than 95 percent is bound) (Pasqualini et al., 1985; vom Saal and Timms, 1999; Witorsch, 2002, citing Tulchinsky, 1973). In primates, fetal hormone levels actually decline in the face of increasing maternal hormones during pregnancy (Thau et al., 1976). Production of steroid hormone binding proteins by the fetal liver decreases dramatically after birth. Thus, some free endogenous steroid hormone is likely to come in contact with developing tissues of the fetus. In the context of environmental exposures, however, in vitro studies with human serum (e.g., Milligan et al., 1998; Jury et al., 2000) and in vivo studies in mice (Welshons et al., 1999) have found that various xenoestrogens are generally not recognized by these steroid hormone binding proteins. As a result, the mechanism that limits fetal exposure to endogenous estrogens may offer less protection from these xenoestrogens.
One of the best known examples in humans of an in utero exposure altering risk for cancer later in life is that of DES. Between 1938 and 1971, this estrogenic compound was used to prevent miscarriages, but it was taken off the market when daughters of women who took it during pregnancy developed adenocarcinomas of the vagina (Herbst et al., 1971).
Long-term follow-up studies of both mothers and their offspring have found that daughters who were exposed to pharmacological levels of DES in utero experienced important and devastating effects on adult health many years later, including an elevated risk of breast cancer (Palmer et al., 2006; Troisi et al., 2007). These studies have graphically demonstrated that the period of organogenesis during fetal life is a period when humans may be particularly sensitive to the effects of environmental agents—in this case synthetic estrogens. From an epidemiologic standpoint, these DES studies have provided a unique illustration of early development as a window of susceptibility to environmental exposures in terms of the clarity of the dose and timing of the exposure and the extremely long and costly period of follow-up.
Studies in Sprague Dawley rats exposed perinatally to 1.2 µg of DES by subcutaneous injection have demonstrated increased susceptibility to mammary carcinogenesis after postnatal treatment with the carcinogen 7,12-dimethylbenz(a)anthracene (DMBA) (Boylan and Calhoon, 1979, 1983). A higher incidence of palpable tumors and decreased tumor latency were observed compared with DMBA-treated rats without prior hormone exposure (Boylan and Calhoon, 1979, 1983); however, major differences in reproductive tracts or mammary gland structure were not observed at the DES doses used (Boylan and Calhoon, 1983). Kawaguchi et al. (2009a) also used Sprague Dawley rats to demonstrate that rats that had prenatal exposure to DES (their mothers had been fed DES [0.1 ppm] throughout pregnancy or from day 13 of pregnancy through to birth) and were then exposed to DMBA at 50 days after birth developed more mammary carcinomas than controls.
Experiments in ACI rats without additional carcinogen dosing demonstrated that mammary tumors can be induced in female rats by prenatal (0.8 µg or 8.0 µg s.c. to the pregnant mother, equivalent to 4.28 µg/kg or 42.8 µg/kg of body weight) or postnatal (2.5 mg via implanted pellet) DES exposure alone, and that prenatal and postnatal DES exposure combined yielded significantly greater tumor multiplicity and decreased tumor latency (Rothschild et al., 1987). The morphology of peripubertal mammary glands of a significant proportion of female ACI rats exposed to DES in utero were atypical; approximately 25 percent displayed hypodifferentiation and about 5 percent had hyperproliferation (Vassilacopoulou and Boylan, 1993).
In utero DES exposure is theorized to increase mammary cancer susceptibility by slowing mammary gland maturation and development (Jenkins et al., 2011).The most mature structures of the mammary gland, lobules, appear to be most resistant to carcinogenic transformation by chemical carcinogens, while terminal end buds are more susceptible (Russo and Russo, 1978; Russo et al., 1982). Prenatal DES exposure has been reported to increase the number of terminal end buds, increasing susceptibility to
chemical carcinogenesis (Ninomiya et al., 2007). In contrast to the effects from fetal exposure to DES, exposure to DES at other time windows has been shown to manifest different effects (Lamartiniere and Holland, 1992; Hovey et al., 2005; Kawaguchi et al., 2009b).
Studies carried out to explore the mechanisms of uterine estrogenic effects from perinatal exposure to DES in the CD-1 mouse model might prove helpful for understanding effects in the mammary gland. Estrogenic effects include persistent expression of the lactoferrin and c-fos genes (Newbold et al., 1997; Li et al., 2003) together with a high incidence of uterine adenocarcinoma (Newbold et al., 1990). DES exposure also causes changes in the expression of several uterine genes responsible for directing tissue architecture and morphology, resulting in altered tissue structures (Ma et al., 1998; Miller et al., 1998; Block et al., 2000). Thus, altered gene expression, likely as a result of epigenetic reprogramming, may also be a contributor to increased susceptibility to mammary carcinogenesis in DES-exposed animals. The body of animal studies carried out with DES demonstrates that the in utero and neonatal periods are especially vulnerable to inappropriate xenoestrogen exposure, with these exposures inducing developmental reprogramming and potentially altering risk for cancer later in life.
Although intentional human exposures to pharmacologic doses of estrogen compounds such as DES are hopefully unlikely to recur, exposure to environmental chemicals with estrogen-like activity is common. Analysis of data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 found a large range of chemicals present in blood or urine in women who were and were not pregnant (Woodruff et al., 2011). Concentrations tended to be similar or lower in pregnant women compared to those in women who were not pregnant. The analysis demonstrates the potential opportunity for exposure of the developing fetus and the pregnant mother’s breast to a wide range of chemical compounds. Whether or not the presence of these chemicals is associated with an increased risk of breast cancer in either child or parent requires additional investigation.
Early Childhood and Prepuberty
The rudimentary ductal system present in the breast at birth is under the influence of maternal hormones (Anbazhagan et al., 1991). But by age 2, the breast undergoes involution, and it has only a primitive ductal system without alveoli until the onset of puberty (Howard and Gusterson, 2000). During this period, however, the body is preparing for puberty and the next stage of mammary gland development. Various exposures during childhood appear to influence the timing of puberty. Because the timing of puberty is associated with the risk of breast cancer later in life, better understanding
of the factors in childhood that influence the timing of puberty may add to understanding of pathways that can contribute to breast cancer.
Diet and the nutritional content of food consumed in childhood help set the stage for puberty by developing adequate body mass. However, prepubertal overweight and obesity are positively associated with early puberty (Kaplowitz, 2008) and late pubertal peak height velocity and early menarche (Hauspie et al., 1997). Recent national health surveillance data for the United States show that 35 percent of girls ages 6–11 are at or above the 85th percentile of body mass index (BMI) standards for their age, and 18 percent are at or over the 95th percentile (Ogden et al., 2010). Biro and colleagues (2003, 2010) have shown that girls with higher BMIs are likely to begin puberty at an earlier age. However, the mechanism underlying the association between obesity and earlier puberty is not yet clear (Jasik and Lustig, 2008). The impact of decreased physical activity, independent of energy intake, and its effects in different subgroups of children in this prepubertal period may act through decreased insulin sensitivity and are also of concern (Sorensen et al., 2009). Further investigation is needed to understand the relation between activity levels in childhood and the timing of puberty or other factors that may influence breast cancer risk in later life.
Psychosocial factors have also been found to influence pubertal timing among girls (Graber et al., 1997; Ellis and Garber, 2000; Bogaert, 2005). They are likely to operate through pathways other than diet, physical activity, and obesity. In stressful family contexts, characterized by low-quality parental investment, high levels of stress, negative relationships, and prolonged distress, reproductive maturation appears to accelerate (Romans et al., 2003). Family relationships characterized by warmth, cohesion, and stability, on the other hand, consistently predict later pubertal onset (Graber et al., 1995). One particular manifestation of disrupted family relationships exists in households with the absence of a biological father. Studies have shown an association between early pubertal maturation in both boy and girl twins in homes where the father was absent when the children were age 14. Girls were about twice as likely to experience menarche before age 12 in such households, although the mechanisms for this observation are not clear (Quinlan, 2003; Mustanski et al., 2004). One study has found this relationship confined to white girls in families with higher socioeconomic status, as measured by household income (Deardorff et al., 2011). Overall, a review of the father-absence literature suggests that girls in father-absent homes experience menarche 2 to 5 months earlier than those in homes where the father is present (Ellis, 2004). The absence of the mother or the presence of a stepfather does not appear to be related to pubertal timing (Bogaert, 2005).
Animal studies suggest that endocrine-disrupting chemicals (EDCs) may alter hormone synthesis and metabolism during this prepubertal period
(Crain et al., 2008) and may advance the onset of puberty. Individual nutrients and dietary intake of substances such as phytoestrogens, which are considered xenoestrogens, may also be factors in pubertal development. In a study of pubertal development in 9-year-old girls, higher urinary concentrations of phytoestrogens, and in particular daidzein and genistein, were associated with later age of breast development, and this effect was stronger in girls with lower BMIs (Wolff et al., 2008). The delay in pubertal development is considered protective in terms of risk for breast cancer. A limited set of case–control studies provides some support for an association between higher consumption of soy products during childhood and lower risk of breast cancer (reviewed in Hilakivi-Clarke et al., 2010).
Studies in animal models of genistein exposure have investigated the importance of the timing of exposures in altering mammary tumor susceptibility. The animal data regarding postnatal, prepubertal exposure to genistein are described as “very consistent” in showing a reduction in mammary cancer risk (Warri et al., 2008). In early studies, Lamartiniere and coworkers (1995) observed that rats treated with genistein during the early postnatal period displayed increased mammary tumor latency and decreased tumor multiplicity in classic chemical carcinogenesis models. In contrast, animal studies of in utero genistein exposure have produced conflicting results (reviewed in Warri et al., 2008).
Differences in the impact of pre- and postnatal exposures in these animal models highlight the potential for the timing of human exposure to genistein, and likely other xenoestrogens, to have differing impacts on breast cancer risk.
Puberty and Adolescence
The onset of puberty, the pubertal period, and adolescence comprise another life stage during which environmental factors may influence the development of breast cancer in adult life in unique ways. This stage spans the period from the first signs of sexual development and the external appearance of the breast to sexual maturity (Russo and Russo, 2004). Breast development in adolescent girls is characterized by branching of terminal end buds in response to hormonal cues (Russo and Russo, 2004).
The onset of puberty is clinically defined by the first signs of breast development, pubic hair, and other secondary sex characteristics (Grumbach and Styne, 2002). It coincides with the activation of the hypothalamic– pituitary–gonadal (HPG) axis, or thelarche, and the activation of the hypothalamic–pituitary–adrenal (HPA) axis, or adrenarche, which are independent events. HPG axis activation is associated with a surge of pituitary follicle-stimulating hormone (FSH), which results in the stimulation of primordial ovarian follicles to secrete estrogen. Circulating estrogen then
has several important effects, including the release of pituitary luteinizing hormone (LH) and vascular proliferation and growth in the breast with the further development of the mammary ducts and the mammary stromal connective tissue (Rogol, 1998). The activation of the HPA axis stimulates the adrenal production of dehydroepiandrosterone, dehydroepiandrosterone sulfate, and androstenedione, which lead to the development of secondary sexual characteristics, including pubic hair and changing body proportions. The relative timing of thelarche and adrenache may differentially determine the onset of menarche (Biro et al., 2006).
Early age at menarche is an established risk factor for breast cancer (Kelsey and Bernstein, 1996), but its use as a marker of pubertal onset can be misleading because the relationship between the onset of puberty and menarche has not been constant over time (Euling et al., 2008; Mouritsen et al., 2010). In the United States, the correlation between the onset of puberty and menarche was greater than 0.9 for women born in the 1930s, 0.5–0.7 for those born in the 1950s, and 0.38–0.39 for those born in the 1970s (Biro et al., 2006). These results suggest that factors contributing to the onset of puberty and menarche were more similar in the past than in more recent years. Clear differentiation between the time of onset of puberty and menarche and the interval between them (“tempo”) is important in studies of pubertal development (Euling et al., 2008).
It is clear that both the age when girls begin puberty and their age of menarche have declined over the past century (Euling et al., 2008). However, historical data and more recent detailed epidemiologic studies have concluded that the age of menarche declined in industrialized countries over the course of the past century, whereas the decline in the age of onset of puberty has been rapid and observed since just since the early 1990s (de Muinck Keizer-Schrama and Mul, 2001; Euling et al., 2008; Aksglaede et al., 2009; Mouritsen et al., 2010). This latter decline has not been associated with development and socioeconomic conditions and has thus raised concerns about the possible role of environmental factors such as endocrine disrupting chemicals (Mouritsen et al., 2010). Genetic regulation of puberty is unlikely to explain these rapid secular trends, but genetic factors do influence the age of pubertal onset in individual girls (Parent et al., 2005). Supporting evidence comes from studies documenting a correlation between a mother’s and daughter’s ages at puberty, from twin correlation studies that suggest that most (70–80 percent) of the variance between twins is explained by genetic influences, and from the observation of marked differences in pubertal timing among racial and ethnic groups (Parent et al., 2003, 2005).
At the onset of puberty, the ratio of FSH to LH favors FSH, which inhibits ovulation, and even with the onset of menarche, ovarian function can continue to be anovulatory for a time (MacMahon et al., 1982b).
The duration of anovulatory menstrual cycles after the onset of menarche varies from 1 to more than 6 years, with longer intervals to ovulation in girls with a late menarche (MacMahon et al., 1982a; Clavel-Chapelon, 2002). The shorter period of anovulation for girls with earlier menarche would suggest that the increased risk of breast cancer that is associated with earlier menarche may be related to earlier and more frequent exposure to the hormones produced during the menstrual cycle. Moreover, acceleration of menarche without a concomitant acceleration in the timing of menopause increases the duration of estrogen exposure over a lifetime, which it is widely thought to promote the development of breast cancer (de Waard and Thijssen, 2005). For each additional year of delay in menarche, the risk of breast cancer is decreased by approximately 9 percent for premenopausal cases and by approximately 4 percent for postmenopausalcases (Hsieh et al., 1990; Clavel-Chapelon and Gerber, 2002). Among women in the Nurses’ Health Study II, who were followed between 1989 and 1993, a 1-year increase in age at menarche was associated with reduction in risk of 10 percent (RR = 0.90, 95% CI, 0.83–0.99) (Garland et al., 1998). In a comparison between women with an age of menarche of 13 versus 15 years or older, an older age of menarche was associated with a statistically significantly reduced risk for premenopausal breast cancer (OR = 0.72, 95% CI, 0.57–0.91), but the reduction in risk for postmenopausal breast cancer was not statistically significant (OR = 0.90, 95% CI, 0.80–1.03) (Titus-Ernstoff et al., 1998).
The contribution of timing of puberty and onset of menarche to increased breast cancer risk may be related to estrogen receptor signaling or to other mechanisms discussed later in this chapter. It is also possible that the rapidly duplicating cells of the breast during pubertal development are more susceptible to environmental insults. Studies in the rodent model show that the highest number and the greatest proliferative activity of the terminal duct lobular units (TDLUs) occurs during puberty (Rudland, 1993), and it has been suggested that this may be related to the apparent susceptibility of the breast to carcinogens during puberty (Colditz and Frazier, 1995; Knight and Sorensen, 2001).
Some of the best evidence for susceptibility of breast tissue during early-life exposures is derived from investigation of the effects of ionizing radiation from nuclear explosions and from medical diagnostic and treatment procedures. An increased risk for breast cancer has been documented among atomic bomb survivors in Japan (Tokunaga et al., 1991), and this increased risk has been related to younger age at exposure, especially during the period of puberty (Land et al., 2003). In an ecological study of the Chernobyl accident in Belarus, the areas with the highest levels of radiation contamination (estimated average cumulative doses ≥ 40 mSv) were associated with elevated breast cancer risk about 10 years after the incident,
especially among women who were younger than age 45 at the time of the event (Pukkala et al., 2006).
More recently, additional follow-up of the atomic bomb survivors and analysis of both excess relative risk and excess attributable risk suggest that excess relative risks are similar across ages of exposure, for example, at ages 10, 30, or 50 (Preston et al., 2007). However, models examining excess attributable risk show a large difference by age at exposure, which the authors suggest reflects differences in factors such as reproductive history that have changed across birth cohorts and that may act multiplicatively with age at radiation exposure (Preston et al., 2007).
The potential implications of these findings are important in terms of recommendations for earlier radiographic screening among high-risk populations such as women at increased genetic risk of breast cancer. Their risks with exposure to mammographic X-rays may vary according to whether they have completed a pregnancy or other factors. Research findings such as these may influence the age at which mammographic screening is begun, reliance on other screening techniques that do not use ionizing radiation, and issues to be covered by consent documents.
An increased risk of breast cancer has also been consistently reported for exposure to ionizing radiation at young ages in conjunction with medical treatments, such as radiotherapy for Hodgkin’s disease and childhood cancer, ankylosing spondylitis, tinea capitis, enlarged thymus (Shore et al., 1993), and skin hemangioma (John and Kelsey, 1993). Later in life into the reproductive years, radiation has been associated with breast cancer among women receiving radiation for postpartum mastitis (Shore et al., 1986) and during tuberculosis treatments (Boice et al., 1991). Exposures among radiologic technologists have been studied, but little impact has been found on risk for breast cancer among those employed over the past 40 years.
Reproductive Years
The reproductive period for women spans the time from sexual maturity at the end of puberty to menopause, providing an expansive time window in which exposures may influence the risk of breast cancer development. Established risk factors for breast cancer encountered during this period include later age at first full-term pregnancy and later age at menopause. The neutral or inverse association between weight or BMI and breast cancer during the reproductive period differs from the increased risk found during the years following menopause. Smoking, hormone therapy, and radiation are examples of other risk factors that may have differential effects over the life course and across the reproductive period. Indeed, pregnancy itself is associated with a short-term increased risk of breast cancer, making it a period of vulnerability of the breast for both the
mother and the developing infant exposed to the in utero environment, as discussed above.
Breast tissue continues to evolve and differentiate before and during pregnancy (Russo et al., 2006), and the differentiation that occurs during pregnancy may be a factor in the reduced risk of breast cancer that is associated with childbearing. During pregnancy, the breast “attains its state of maximum development” in two distinct stages—early and late in pregnancy (Russo and Russo, 2004, p. 7). The early stage involves differentiation of ductal trees and an increase by the third month of pregnancy in the number of well-formed lobules. In the later stages of pregnancy, the continued changes in the breast are related in large part to the secretory functions that the breast tissues will perform during lactation (Russo and Russo, 2004). During pregnancy, significant changes also occur in the mammary stroma, the connective tissue in the breast. This remodeling includes changes that result in increased angiogenesis, infiltration of immune cells, and fibroblast reorganization, all of which help supply nutrients and cues to the expanding ductal and lobular structures (McCready et al., 2010). An increase in circulating hormones during pregnancy also plays a role in breast development.
Studies in rats have shown that pregnancy leads to the maximum mammary gland development. This process includes alterations in mammary stem cells (breast progenitor cells) that make them more resistant to carcinogens by virtue of primed mechanisms for metabolism of carcinogens and improved DNA repair mechanisms (Russo et al., 2006). This type of process may explain the association in humans between young age at first pregnancy and reduced breast cancer risk, and the increased risk associated with nulliparity and late age at first pregnancy. Nulliparity or late age at first pregnancy would leave the breast more vulnerable to carcinogens because of the predominance of unaltered stem cells. With late age at first pregnancy, the differentiation and proliferation of breast tissue would be more likely to involve compromised stem cells that have suffered DNA damage, which would provide a favorable environment for progression of cancer cells. Some studies have found that the increase in risk associated with a later age at first full-term birth (age 30 and older) is greater than for nulliparity (e.g., Kotsopoulos et al., 2010; Newcomb et al., 2011).
A review of the literature on breast cancer and characteristics of pregnancy found conflicting evidence for factors such as weight gain during pregnancy, fetal growth, gestational age, and gestational diabetes (Nechuta et al., 2010). Findings were more consistent that multiple births and preeclampsia were associated with modest reductions in risk for breast cancer (Nechuta et al., 2010). In an example of the effects of an exogenous hormone exposure during pregnancy, follow-up of women who took DES to prevent pregnancy complications from 1940 through the 1960s has found a modest association between their adult DES exposure while pregnant
and subsequent breast cancer risk (RR = 1.27, 95% CI, 1.07–1.52) (Titus-Ernstoff et al., 2001), supporting the influence of hormonal factors during pregnancy and its period of rapid breast proliferation.
Other evidence that pregnancy can modify breast cancer risk comes from the literature on smoking and breast cancer. This literature is often characterized as mixed, with some studies finding associations and others not. However, a more nuanced picture emerges when the risks from smoking are analyzed separately for women who started to smoke before a first pregnancy and for those who began to smoke after their first child was born. As noted in Chapter 3, a meta-analysis of 23 studies found a weak association with increased risk of breast cancer for women who began smoking before a first pregnancy (DeRoo et al., 2011). The summary risk ratio was 1.10 (95% CI, 1.07–1.14), compared with a risk ratio 1.07 (95% CI, 0.99–1.15) for women who began smoking later (DeRoo et al., 2011).
Reynolds and colleagues (2004), for example, observed an elevated risk for women who smoked for 5 or more years before a first full-term pregnancy compared with never smokers with children (HR = 1.13, 95% CI, 1.00–1.28). Risk was not increased among the small group of women (42 cases) who began smoking after a first pregnancy (HR = 0.89, 95% CI, 0.65–1.21). This study also found no excess risk for women who initiated smoking at age 20 or later (HR = 1.03, 95% CI, 0.90–1.17), but a statistically significant increased risk for those who began before age 20 (HR = 1.17, 95% CI, 1.05–1.30). Consistent with these patterns, Ha and colleagues (2007) observed no association between breast cancer risk and pack-years of smoking after first childbirth, and no significant trend with cumulative smoking exposure. However, there was a significant trend (p <.0001) in the hazard ratios for pack-years of smoking before the birth of the first child, with the highest exposure group having a statistically significant increased risk (HR = 1.78, 95% CI, 1.27–2.49) (Figure 5-2).
As also noted in Chapter 3, recent reports from both the Nurses’ Health Study (Xue et al., 2011) and the observational component of the Women’s Health Initiative (Luo et al., 2011) appear to support this pattern. Xue and colleagues (2011) found that more pack-years of smoking from menarche to a first birth were associated with a statistically significant increase in risk (p for trend <.001). Luo et al. (2011) found that initiation of smoking before first full-term pregnancy was associated with a statistically significant increase in risk (HR = 1.28, 95% CI, 1.06–1.55). The risk with initiation after a first pregnancy was elevated but not statistically significant (HR = 1.17, 95% CI, 0.90–1.52).
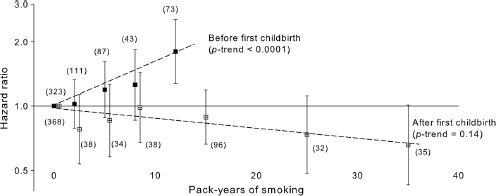
FIGURE 5-2 Breast cancer risk by pack-years of smoking before and after first childbirth among parous women, U.S. Radiologic Technologists Health Study, 1983–1998. Hazard ratios and 95 percent confidence intervals were estimated from one multivariate proportional hazards model including separate variables for pack-years of smoking before and after first childbirth, with age as the time scale, stratified for birth cohort in 5-year intervals and adjusted for alcohol intake, age at menarche, age at first childbirth, parity, family history of breast cancer, hormone replacement therapy, year that a woman first worked as a radiologic technologist, body mass index, and time-dependent menopausal status. The numbers of breast cancer cases in each category are provided in parentheses.
SOURCE: Ha et al. (2007, p. 58). Reproduced with permission from American Journal of Epidemiology.
Menopause and Older Ages
Most breast cancers are diagnosed in menopausal women. Menopause is characterized by a cessation of ovarian hormone production, and subsequently, the end of a woman’s menstrual cycles (McCready et al., 2010). The loss of estrogen and progesterone production coincides with a process known as lobular involution, or the reduction of both number and size of lobules in the breast (Russo et al., 2001). The number of epithelial cells in the breast expressing estrogen receptors increases significantly (Shoker et al., 1999), and the interlobular stroma of the breast is increasingly replaced by adipose tissue (Howard and Gusterson, 2000). At this stage of life, breast tissue has also been influenced by the cumulative opportunity for previous exposures to endogenous and exogenous factors to have generated compromised cells.
Clear evidence from studies of menopausal hormone therapy also shows that breast cancer risks can be influenced by exposures during this life stage. Postmenopausal use of hormonal therapy that combines estrogen and progestin increases both the incidence of and mortality from breast
cancer (Writing Group for the Women’s Health Initiative Investigators, 2002; Million Women Study Collaborators, 2003; Chlebowski et al., 2010). The risks decline rapidly on withdrawal of combined hormone therapy in the postmenopausal period (Collaborative Group on Hormonal Factors in Breast Cancer, 1997; Clarke et al., 2006; Chlebowski et al., 2009; Beral et al., 2011). While the mechanism for the increased risk is not known in detail, it is thought that hormone therapy may be causing proliferation of cancer stem cells (Eden, 2010, 2011). In the absence of hormonal stimulus, other regulatory processes in the body may be able to inhibit the progression of tumorigenesis or expansion of an existing tumor.
Studies have also examined the risk associated with timing of initiation of hormone therapy relative to onset of menopause (Prentice et al., 2009; Beral et al., 2011). From both the Women’s Health Initiative in the United States and the Million Women Study in the United Kingdom, evidence has emerged that the risk of invasive breast cancer from combination hormone therapy decreased with increasing gap time (time from menopause to initiating hormone therapy) for combined estrogen–progestin hormone therapy (Prentice et al., 2009; Beral et al., 2011; LaCroix et al., 2011). Risk was higher among women taking combined estrogen–progestin therapy within 5 years of menopause compared to those whose first use was 5 or more years after menopause (Prentice et al., 2009), and additional analyses demonstrated the risk of breast cancer increased with use within 2 years since menopause.
The Women’s Health Initiative found that estrogen-only hormone therapy, which is appropriate only for women who have had hysterectomies, was not associated with increased risk of breast cancer (Women’s Health Initiative Steering Committee, 2004; LaCroix et al., 2011). The Million Women Study, however, observed a small but statistically significant risk associated with estrogen-only therapy, but only when initiated before menopause or within 5 years after menopause (RR = 1.49, 95% CI, 1.40–1.58 and RR = 1.36, 95% CI, 1.27–1.46, respectively). Among estrogen-only users, there was no association with breast cancer risk if use was initiated 5 or more years after menopause (RR = 1.05, 95% CI, 0.90–1.24) (Beral et al., 2011). The findings from these studies are also discussed in Chapters 3 and 6.
MECHANISMS OF BREAST CANCER DEVELOPMENT
Several potential mechanisms may be operating in the development of breast cancer, and they may play out during a particular phase or across multiple phases of life. Researchers do not yet know the details of all of the potential mechanisms through which normal breast tissue changes to become cancerous tissue. Such changes, termed “transformation,” occur
through a multistage process of carcinogenesis. Multistage carcinogenesis encompasses both spontaneous and environmentally induced events that contribute to altering the cells from normal, healthy tissue into tumor tissue. Spontaneous events occur in cells by chance, or stochastically, as a by-product of normal processes. For example, reactive oxygen species (ROS) produced as a by-product of cellular respiration can cause damage to DNA, as can errors in DNA replication that result in spontaneous mutations. Environmentally induced events are those caused by an external exposure, for example, ultraviolet (UV)-induced damage from sunlight or exposure to tobacco carcinogens. In addition to physical and chemical insults, environmental contributors to cancer, as defined broadly by this committee (see Chapter 2), can include pathophysiological conditions, such as obesity.
Accumulated damage from both internal and external carcinogenic events or risk factors drives cancer development through multiple stages from normal cells through preneoplasia to metastatic disease, with many of these stages recognizable as distinct entities in the process of tumor development at the cellular (e.g., lobular hyperplasia) and/or molecular level (e.g., aberrant p16 expression). There is also a growing appreciation for the role that the tissue microenvironment can play in limiting, permitting, or potentiating the carcinogenesis. Thus, the development of breast cancer, like other adult cancers, occurs as a result of accumulated damage occurring over a person’s entire life. However, susceptibility to different types of damage induced by either endogenous or exogenous carcinogens may change over the life course, so that humans may be more or less vulnerable to the effects of any given environmental agent at different stages of life.
In the remainder of this chapter, critical mechanisms of carcinogenesis are reviewed, with the recognition that mechanistic explanations of the origins of breast cancer have not commonly been situated within the life course perspective that the committee recommends.
Mutagenesis
Damage to DNA was the earliest recognized mechanism for development of cancer from environmental causes. Changes to the sequence of base pairs that make up the genome (DNA) are called mutations, and they can be caused by internal cellular processes or outside effectors such as radiation, viruses, or certain chemicals. DNA mutations can result in changes in gene (and microRNA) expression, function, or regulation that may lead to the development of tumors. Many environmental agents can induce DNA damage, which, if not properly repaired, can give rise to mutations that contribute to multistage carcinogenesis. Induction of DNA damage can be direct—for example, induction of DNA double-strand breaks by gamma irradiation, or the creation of pyrimidine dimers by UV light. DNA dam-
age can also occur indirectly—for example, via oxidative damage resulting from depletion of ROS scavengers, or increased ROS production in the cell. The scientific community’s increasingly sophisticated understanding of “mutagens as carcinogens” now includes appreciation that not all agents that induce DNA damage cause cancer. Nevertheless, environmental agents that induce mutations are still considered among the most effective types of carcinogens.
Exposure to environmental mutagens may have very different effects, depending on the target cell1 and the extent of cell proliferation that occurs before and after repair of environmentally induced damage. If the target cells have insufficient time to repair DNA damage before they divide, mutations can be passed on to daughter cells. Therefore, those stages of life when breast epithelial cells are most proliferative (i.e., puberty and pregnancy) may be times when the breast is especially vulnerable to the effects of mutagens. If these mutations occur in genes that participate in tumor development, such exposures can be transforming and contribute to the accumulation of the necessary alterations for multistage tumorigenesis.
Estrogen, in the form of 17b-estradiol, is generally recognized as potentially carcinogenic through its promotion of cell proliferation via interactions with estrogen receptors (see below), and it may also be linked to mutagenesis through its metabolites (Yager and Davidson, 2006). In particular, a metabolite of 17b-estradiol (2,4-dihydroxy-17b-estradiol) has been found to be DNA-reactive (Cavalieri et al., 2006; Bolton and Thatcher, 2008). It is plausible for this estradiol metabolite to be produced in breast tissue, where it could contribute to the formation of DNA adducts prone to lead to changes in the gene sequence (Belous et al., 2007).
Although most studies focused on the potential mutagenic role of estradiol have been conducted in animal models, or with human tissues in vitro, there is some evidence that such estradiol-DNA adducts can occur in humans. As discussed in the International Agency for Research on Cancer (IARC) monograph update on the carcinogenicity of estrogen-related pharmaceuticals, stable DNA adducts were found in a small set of breast tissue samples from women with and without breast cancer who had used an estrogen–progestin form of hormone therapy (IARC, 2011).
Nuclear Hormone Receptor Signaling
The steroid hormones, which include estrogens and progestogens, are involved in the development and maintenance of female reproductive characteristics, via their physiological effects on a broad range of tissues (Björnström and Sjöberg, 2005). Like other hormones, the steroid
![]()
1A target cell or target tissue is the site at which an agent acts.
hormones function as signaling molecules by traveling through the blood stream to interact with cells in a variety of target tissues to affect cellular behavior. A principal point of interaction is through binding with receptors that are located within cells and referred to as nuclear hormone receptors. The receptors for the steroid hormones are part of a larger family of nuclear hormone receptors (Aranda and Pascual, 2001).
Physiologically, estrogen usually occurs in the form of estradiol, 17β-estradiol being the dominant and most potent form (Björnström and Sjöberg, 2005), or estrone. Estradiol and estrone are produced by the ovaries, and estrone can also be produced by peripheral tissues such as adipose tissue and the adrenal gland. Estrogen is especially important in the functioning of female reproductive tissues such as the uterus and breast. Estrogens are able to act on target tissues by binding to estrogen receptors. Receptors act by recognizing a molecule’s structure, thereby enabling only estrogen, or molecules that closely resemble estrogen in structure, to interact with receptors and promote receptor-mediated behavior.2
The fact that most breast cancers express estrogen receptor(s), coupled with estrogen’s potential to stimulate cell proliferation (mitogenesis), has led to the hypothesis that estrogen receptor signaling can ultimately increase susceptibility to breast cancer (Weinberg, 2007). In its classical signal transduction pathway, once estrogen is bound to its receptor, it is able to regulate gene expression, which, in turn, alters cellular function. Estrogen’s biological effects are regulated by two types of estrogen receptors, ERα and ERβ (Björnström and Sjöberg, 2005). These receptors act as ligand-activated transcription factors (Björnström and Sjöberg, 2005); when no ligand (in this case, estradiol or estrone) is bound to the receptor, the receptor’s ability to transactivate gene expression is greatly attenuated, although these receptors are capable of ligand-independent activity. Once receptors are activated (“turned on”), they bind to specific DNA response elements known as estrogen response elements (EREs) of target genes (Björnström and Sjöberg, 2005). Estrogen bound to its receptor also induces a change in the receptor’s structure that allows it to recruit coactivator proteins (NCI, 2010). By binding to the EREs and recruiting coactivator proteins, estrogen is able to influence the transcription of messenger RNA, which allows for the production of specific proteins that can, in turn, influence cellular behavior (NCI, 2010).
One such change in cellular behavior is increased cellular proliferation,
![]()
2Receptor-mediated cellular behavior is of particular importance when considering the effects of certain environmental exposures. As discussed later in this chapter, compounds that closely resemble hormones, such as EDCs, are able to activate or block receptor function. These compounds have the potential to subsequently alter the physiological status of the whole organism, especially if it is exposed during developmental stages (Caserta et al., 2008).
a result of estrogen’s mitogenic actions. Growth promotion can increase susceptibility to breast cancer in several ways: increasing the population of cells that are targets for transformation, increasing the potential for mutagenesis by shortening the cell cycle and decreasing time available for DNA repair; and selectively promoting the growth of preneoplastic and neoplastic cells of nascent tumors. As a result of the ability of 17β-estradiol and several other endogenous estrogens to promote tumor growth in the breast, estrogen is classified as a known human carcinogen by IARC (2011).
After menopause, ovarian production of 17β-estradiol diminishes, affording some protection against development of hormone-dependent tumors. Relatively little is known about how the effects of other mitogens are modulated during the life course to increase or decrease their carcinogenic potential. Some existing data suggest that promotion of growth by mitogens early in life may modulate breast cancer risk later in life.
Some evidence also indicates that progesterone has a mitogenic role in breast carcinogenesis (Bernstein, 2002; Kariagina et al., 2010). Recent studies in rats have shown that in both the normal mammary gland and mammary tumors, proliferation is stimulated more by a combination of estrogen and progesterone than by treatment with estrogen alone (Kariagina et al., 2010). This proliferation is mediated by amphiregulin, an epidermal growth factor receptor (EGFR) ligand, and intracellular signaling pathways downstream of EGFR that induce proliferation. Recently, these pathways have also received interest as potential targets for hormone-dependent cancer therapy (Kariagina et al., 2010).
Ligand-activated estrogen receptors can also affect cell growth via what is termed nongenomic signaling, which involves estrogen receptor activation of phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) and other cell signaling pathways in the cytoplasm rather than the nucleus. Research in animal models indicates that during development of the mammary gland, inappropriate activation of genomic or non-genomic ER signaling by estrogens can cause changes in the developing mammary gland (see Box 5-2). For example, a study examined the effects of a single subcutaneous injection (doses of 0.0125, 0.125, 12.5, 25, or 50 µg) of DES or tamoxifen given to female BALB/c mice within 36 hours of birth (Hovey et al., 2005). At 33 days, mice with DES exposure at 12.5 ìg had greater growth of the mammary ducts compared with controls. At maturity, all the mice that had received 12.5 ìg of DES and remained nulliparous showed abnormalities in the ducts and alveoli of the mammary gland compared to controls. In contrast, exposure to 25 ìg of tamoxifen, a selective estrogen receptor modulator, resulted in reduced ductal outgrowth relative to controls at 33 days. At 12 weeks, mice with exposure to 12.5 ìg to 50 ìg of tamoxifen had regressed and atrophic ducts (Hovey et al. 2005). Work by Couse et al. (2001) indicates that DES-induced developmental reprogram-
BOX 5-2
Nongenomic Estrogen Receptor Signaling by Environmental Estrogens
The classical effects of steroid hormones are mediated by nuclear hormone receptors functioning as ligand-activated transcription factors. However, evidence now indicates that steroid hormones manifest effects by other means as well (Castoria et al., 1999; Cato et al., 2002; Losel and Wehling, 2003; Cheskis, 2004; Björnström and Sjöberg, 2005; Edwards, 2005; Levin, 2005). These other effects have been termed “nongenomic” to distinguish them from the direct, or genomic, effects of the transcription factors in the nucleus. The nongenomic effects of steroid hormones involve a subpopulation of classical receptors that associate with signaling complexes in either the cytoplasm or the plasma cell membrane (Weinberg, 2007).
Estrogens stimulate mitosis in ER-positive (ER+) breast cells, yet the precise mechanism by which estrogen is able to drive the proliferation of ER+ breast cancer cells remains unknown (Weinberg, 2007). When estrogen is added to ER+ breast cancer cells, a rapid signaling cascade ensues unrelated to ER action at a nuclear receptor (Weinberg, 2007). Genomic receptor-mediated physiological responses, which may take up to several hours, cannot explain the rapid physiological changes that occur within seconds or minutes following the administration of estrogen (Hewitt et al., 2005).
Various cell or plasma-membrane-associated-receptor signal transduction pathways have been studied to understand the non-genomic path that estrogen may take. These pathways include association with and activation of insulin-like growth factor receptors (IGF-1Rs) (Kahlert et al., 2000) or direct association with Src and Shc proteins (Migliaccio et al., 1996, 2000; Castoria et al., 2001; Kousteni et al., 2001; Song et al., 2002a,b, 2004, 2005; Wong et al., 2002), which are known to be involved in the activation of PI3K and MAPK signaling. PI3K signaling is involved in many cellular processes, including survival, proliferation, growth, and motility (Vivanco and Sawyers, 2002; Sulis and Parsons, 2003). Association of estrogen with G-protein coupled receptors such as GPCR30, which is expressed in the membranes of breast cancer cells, has also been observed (Hewitt et al., 2005; Revankar et al., 2005; Thomas et al., 2005). With further study this may explain some of estrogen’s mitogenic effects (Hewitt et al., 2005). Interpreting the biological role of estrogen as a mitogen is challenging, given the myriad of proposed signal transduction pathways, and researchers urge further in vivo to assess the effects in mammary tissue (Hewitt et al., 2005).
ming of the murine reproductive tract requires the ERa, suggesting that this receptor has a crucial role in mediating the imprint. Further investigation is needed to assess the impact of morphologic changes in the developing mammary gland on the risk for mammary cancer later in life.
Epigenetic Reprogramming
During gestation, and in some cases postnatally, growth and differentiation of cells and tissues occurs via a well-orchestrated series of developmental processes directed by the genetic, epigenetic, and environmental milieu of the developing organism. The genetic contribution is shaped by the individual’s DNA sequence. Epigenetic contributions derive from processes that modify gene expression, but do not alter the DNA sequence. Feinberg and Tycko (2004) have described three principal epigenetic mechanisms: cytosine methylation at CpG sites in the DNA (termed “DNA methylation”), genomic imprinting (silencing of an allele based on whether it was of maternal or paternal origin), and histone modifications (affecting the proteins around which DNA strands are wound).
Epigenetic methyl “marks,” such as DNA methylation and histone methylation, are transmitted to daughter cells and are thus epigenetically “inherited” from the parent generation of cells (Feinberg and Tycko, 2004). Epigenetic methyl marks are the result of the addition of a methyl (–CH3) group to either cytosine nucleotides in the DNA or lysine or arginine residues of histones. Methylation of cytosine residues in DNA (hypermethylation) regulates gene expression largely via gene silencing, and hypomethylation of genes is permissive for gene expression. Methylation of arginine and lysine residues of histones creates binding sites for several regulators of gene expression that recognize these site-specific marks.
During gestation, epigenomic programs that are installed in cells direct patterns of differentiation crucial for tissue and organ specification. This programming also determines how cells and tissues will respond to physiological and environmental signals throughout the life of the organism. The “developmental reprogramming” hypothesis proposes that at critical times during development, exposure of developing tissues to an adverse stimulus can result in permanent epigenetic reprogramming of normal physiological responses, and so alter risk for metabolic and hormonal disorders later in life (Frankel et al., 1996; Hattersley and Tooke, 1999; Barker, 2002; Couzin, 2002). Studies in animal models have demonstrated that perinatal exposure to xenoestrogens can reprogram the development of the mammary gland and reproductive tract, causing alterations in tissue morphology and gene expression (Muñoz-de-Toro et al., 2005; Durando et al., 2007; Murray et al., 2007; Vandenberg et al., 2008; Rudel et al., 2011).
Methylation of DNA and of histones are both thought to be targets
for developmental reprogramming. A direct link between environmental exposures and changes in DNA methylation is lacking, but a direct mechanism whereby environmental exposures can interact with the epigenetic machinery and perturb histone methylation during development has been demonstrated (Bredfeldt et al., 2010; Doherty et al., 2010). Environmental estrogens such as DES can inappropriately activate nongenomic (or more aptly “pregenomic”) ER signaling to perturb patterns of epigenetic histone methyl marks being installed during development. For example, DES activation of pregenomic signaling can initiate a process that lowers levels of histone H3 methylation at lysine (K) 27 (Bredfeldt et al., 2010). Reduced levels of histone H3K27 methylation, a repressive mark for gene expression, can result in increased expression of estrogen-responsive genes in response to normal physiological levels of this hormone, driving development of hormone-dependent tumors.
Although epigenetic alterations are heritable and stably maintained as cells replicate, the alterations are also potentially reversible. This has been done pharmacologically with the use of 5-azacytadine and valproic acid to modulate epigenetic methyl marks as part of cancer treatment (Yoo and Jones, 2006). It has also been found in studies in mice that maternal exposure to bisphenol A (BPA) can result in DNA hypomethylation in offspring, but that maternal folate and genistein supplementation blocked the BPA-induced DNA hypomethylation (Dolinoy et al., 2007). Elucidating the role of environmental agents in inducing epigenetic alterations and the potential contribution of these alterations in the development of breast cancer will broaden understanding of the mechanisms of action of these agents, and potentially open avenues to reverse or prevent their adverse health effects.
Modulation of Immune Function
“Immunoediting” is a term used to describe the process by which the immune system influences cancer development and progression (Bui and Schreiber, 2007). The immune system functions to eliminate cancer cells by immunosurveillance, that is, controlling the growth of transformed cells by mechanisms of adaptive immunity. However, cancer cells may escape detection by the body’s immune cells (immune escape), and thus evade being killed by the immune system.
Environmental exposures may modulate the effectiveness of the immune system in detecting or eliminating cancer cells. For example, the environmental estrogen genistein, present at levels associated with soy consumption, has been shown in studies using human breast cancer cell lines grown in mice to block the ability of cytotoxic T-cells and natural killer cells to recognize and destroy breast cancer cells during immunosurveillance (Jiang et al., 2008). Chemical immunotoxicity is an active area of research, with
investigation of effects from lead, cigarette smoke, endocrine disruptors, and ambient air pollution, among others (Svensson et al., 1994; Weisglas-Kuperus et al., 2000; Bunn et al., 2001; Hertz-Picciotto et al., 2005; Ng et al., 2006; Park et al., 2008a). Conversely, pregnancy can increase the expression of immunosurveillance genes in breast epithelial cells, suggesting that one of the protective effects of pregnancy may be to enhance elimination of nascent tumor cells by immunosurveillance (Balogh et al., 2007). More needs to be learned about how environmental factors influence immunoediting and how modulation of immune function by environmental exposures over the life course influences breast cancer risk.
The Microenvironment Model of Carcinogenesis
In the multistage model of carcinogenesis, cancer progression results from the stimulation of abnormal growth of an initiated cell with altered genetic material. However, mutations that may initiate carcinogenesis can occur frequently in the body and do not necessarily result in cancer. Rodent studies have demonstrated that the number of initiated cells far exceeds the number of tumors that develop in vivo (as reviewed in Barcellos-Hoff and Ravani, 2000). The multistage model of carcinogenesis alone does not fully explain the complex series of transitions from mutation to a cancerous state. It is now appreciated that in addition to the genetic and epigenetic alterations that occur within transformed cells, the process of carcinogenesis and tumor progression includes alterations of the dialog between cells and their surroundings (Bissell and Hines, 2011).
Cells do not exist as isolated units, but are part of a complex environment of various cell types and tissues, as well as extracellular space. The extracellular space is filled with a network of macromolecules called the extracellular matrix (ECM), which serves as a structural anchor for cells. Once viewed as an inert scaffold, it is now accepted that the ECM is dynamic and influences cellular processes through initiation of intracellular signal transduction (Divoux and Clement, 2011). A sheet-like deposition of ECM, called the basement membrane, separates epithelial cells from supportive connective tissue cells called the stroma. The stroma consists of fibroblasts, immune cells, fat cells, and blood vessel cells (Mueller and Fusenig, 2004). Because all cells in the body have the same genetic code, molecular signals are necessary to direct the cells destined to become an eye to become an eye, and the nose to become a nose, and so on.
The stroma is responsible for some of the regulatory signals necessary to determine proper organ development and function in an embryo, and later to maintain homeostasis in an adult organism (Shekhar et al., 2003). Occasionally, such regulatory signals are disturbed, resulting in an alteration of normal external context for cells. Usually the effects are transient,
but sustained inflammation may result in an up-regulation of enzymes such as matrix metalloproteinases (Bissell and Radisky, 2001). These enzymes normally promote healing, but their prolonged presence can contribute to the degradation of the ECM and allow for invading immune cells to overproduce factors that may lead to unregulated cellular proliferation. In this way, it is thought that the cellular environment, including the extracellular matrix and stroma, may play an important role in progression from normalcy to neoplasia. It has been proposed that disruption of the stromal–cell interactions in early stages of carcinogenesis can “provide the stimulus for initiated cells to move further down the neoplastic pathway” (Barcellos-Hoff and Ravani, 2000, p. 1254).
Microenvironments are specialized systems, composed of the ECM and soluble growth factors. They mediate epithelial–stromal interactions and thereby play an important role in normal growth and tissue development (Barcellos-Hoff and Ravani, 2000). The microenvironment theory complements the multistage model of carcinogenesis, with the microenvironment and stroma in the breast interacting with transformed epithelial cells to constrain or permit invasive tumor growth (Shekhar et al., 2003; Bissell, 2007). Based on this model, the destruction of tissue itself could be a carcinogenic event; an event such as wounding or tissue damage could trigger an inflammatory cascade leading to unregulated proliferation.
Alternatively, exposure to a carcinogen may disrupt the interactions between a cell and its external environment in a manner that permits transformation. Ionizing radiation has carcinogenic potential in both the breast and the rodent mammary gland, and it has been demonstrated to elicit rapid remodeling of the mammary gland ECM (Barcellos-Hoff, 1993). When genomically unstable cells harboring mutations in both alleles of p53 (COMMA-D cells) were transplanted into epithelial-free mammary stroma in irradiated host mice, tumors arose more quickly and were able to grow larger than in unirradiated hosts (Barcellos-Hoff and Ravani, 2000).
Since carcinogenic environments are not necessarily mutagenic or mitogenic, it is likely that changes in cell–cell contact or cell–ECM interactions promote a malignant phenotype (Barcellos-Hoff and Ravani, 2000). Studies in a mouse mammary chimera model showed that when an irradiated host received oncogenic tissue, the development of aggressive tumors was accelerated and the molecular signatures of the tumors differed from those arising in nonirradiated hosts (Nguyen et al., 2011). Maffini et al. (2004) also demonstrated the stroma to be a critical target for carcinogenesis using a rat mammary tissue recombination model and the chemical carcinogen N-methyl-N-nitrosourea (MNU). They found that mammary epithelial cells were neoplastically transformed only when the stroma was exposed to MNU, regardless of MNU exposure of the epithelial cells.
The microenvironment model is especially relevant to breast cancer
because of both the nature of the disease and the tissue structure of the organ. Most breast cancers originate from the epithelial cells of the terminal lobular ducts, and stroma accounts for more than 80 percent of breast volume (Shekhar et al., 2003). Furthermore, the breast is an organ that undergoes substantial development after birth and is a dynamic tissue that continually undergoes changes throughout a woman’s lifetime, most notably during puberty, pregnancy, and menopause (McCready et al., 2010). Changes in the stromal and hormonal environments are part of these age- and event-related processes.
One reflection of stromal differences is mammographic density. Breasts with a greater proportion of fat produce a darker radiographic image that is described as less dense, and breasts with greater amounts of connective and epithelial tissue produce lighter images that are considered more dense (Boyd et al., 2010). Greater breast density is strongly associated with a higher risk of breast cancer (reviewed in Boyd et al., 2010). Women with higher mammographic density have been shown to have an altered ECM composition, including an increase in fibrillar collagen (McCready et al., 2010). As discussed above, it is hypothesized that changes to ECM architecture may play an early role in the path of tumorigenesis (e.g., McCready et al., 2010) or may reflect the effects of processes promoting tumorigenesis (e.g., Martin and Boyd, 2008). Further research is required to elucidate the mechanisms underlying the association between mammographic density and breast cancer risk, including the influence of environmental factors in altering breast tissue composition.
POSSIBLE BIOLOGIC MECHANISMS FOR ALTERATIONS IN BREAST CANCER RISK ASSOCIATED WITH OBESITY
As summarized in Chapter 3, obesity or excess body fatness has become a major public health issue in the United States. Obesity is associated with a myriad of adverse health outcomes, including many forms of cancer (ACS, 2011).3 The picture for breast cancer is mixed. Obesity is associated with a lower risk for premenopausal breast cancer, while it is associated with an increased risk for postmenopausal breast cancer (Carmichael and Bates, 2004; WCRF/AICR, 2007). Pooled analysis of cohort and case–control studies suggests that greater abdominal fatness (measured by waist circumference or waist-to-hip ratio) is a “probable cause” of breast cancer diagnosed in postmenopausal women (WCRF/AICR, 2007, 2010).
![]()
3Overweight and obesity are commonly defined on the basis of body mass index (BMI), which is an approximate measure of body fat based on height and weight. BMI is calculated as body weight (kilograms) divided by height (meters) squared. The following BMI categories are used: Underweight, <18.5; Normal weight, 18.5–24.9; Overweight, 25.0–29.0; and Obese, ≥30.0.
Being obese is associated with a poorer prognosis and higher mortality rates for women diagnosed with breast cancer (Calle et al., 2003; Carmichael, 2006; Protani et al., 2010). The American Cancer Society’s Cancer Prevention Study II, a prospective cohort study that included 2,852 breast cancer deaths, indicated that breast cancer mortality rates increased continually with increasing BMI at entry into the study. For women with the highest BMI (.40) compared to women with normal weight (BMI 18.5–20.49), the relative risk (RR) of death from breast cancer was 3.08 (95% CI, 2.09–4.51) (Petrelli et al., 2002).
Overweight and obesity have been found to be associated with diagnosis of advanced-stage disease; however, the experience of African American women differs from that of white women (Hunter et al., 1993; Jones et al., 1997). Jones et al. (1997), in a retrospective study in Connecticut, found that a greater prevalence of severe obesity (BMI ≥ 32.3) among black women could account for approximately 33 percent of their excess risk of diagnosis of later-stage cancer compared to white women. In the National Cancer Institute survival study by Hunter et al. (1993), no differences between blacks and whites were observed.
Obesity probably affects breast cancer risk through several different and overlapping pathways (Fletcher et al., 2005; Slattery et al., 2007; WCRF/AICR, 2007; Cleary and Grossmann, 2009). Obesity-associated carcinogenesis has been explained by three main candidate mechanisms involving (1) insulin and insulin-like growth factor axis, (2) steroid hormones, and (3) circulating levels of cytokines and adipokines (Roberts et al., 2010).
Insulin plays an integral role in short-term metabolism, signaling muscle, liver, and adipose tissues to convert the glucose in the bloodstream into glycogen for storage. Circulating insulin levels have been shown to correlate positively with an increasing BMI (Roberts et al., 2010). Insulin-like growth factors (IGFs) mediate metabolism in the longer term and affect cell growth. Insulin contributes to inhibiting IGF by reducing hepatic secretion of IGF binding proteins (IGFBPs) that bind IGFs with high affinity (Giovanucci, 2001).
Obese individuals are often insulin resistant, a state of reduced responsiveness of muscle, liver, and adipose tissues to insulin. As a result, obese individuals are likely to have higher blood glucose and circulating insulin levels. The insulin resistance–cancer hypothesis is based on hyperinsulinemia, or elevated insulin levels, reducing production of IGFBPs and leaving free “bioactive” IGFs, which are then able to promote cell proliferation (Roberts et al., 2010).
Insulin itself has also been shown to suppress apoptosis (programmed cell death) and contribute to cell proliferation by acting as a mitogen for breast epithelial cells via insulin and IGF-1 receptors (Ish-Shalom et al., 1997; Chappell et al., 2001). In animal models, administration of insu-
lin promotes mammary tumor growth (Heuson et al., 1972; Shafie and Grantham, 1981; Shafie and Hilf, 1981). Insulin also has the ability to affect sex hormone levels (Poretsky and Kalin, 1987) and to lower levels of SHBG, which binds sex hormones and renders them temporarily biologically “inactive” (Pugeat et al., 1991).
Several clinical trials are under way to evaluate potential benefits of the common diabetes drug metformin in treatment of women who have breast cancer (NIH, 2010). Early studies also suggested that metformin, but not other diabetes drugs, decreased the incidence of breast and other cancers in diabetics (Bodmer et al., 2010; Zakikhani et al., 2010). The mechanism of the protective effect of metformin is thought to be mediated through the action of this drug on AMP-activated protein kinase (AMPK), an enzyme that acts as a calorie restriction mimetic and that inhibits mitogenic signaling via the PI3K pathway in tumor cells (Zakikhani et al., 2006; Wysocki and Wierusz-Wysocka, 2010), as well as perhaps from direct reduction in levels of HER2 protein levels through inhibition of specific kinase activity (Vazquez-Martin et al., 2009).
Obesity may also be linked with carcinogenesis through its impact on estrogen and other steroid hormones and circulating growth factors. Adipose tissue is an important source of the estrogen estrone in postmenopausal women, and production of estrone by aromatization of androstenedione in the breast is thought to be one of the major factors in obesity’s association with increased risk of breast cancer in postmenopausal women. Estrogens such as estrone can induce IGF-1 and increase cellular proliferation via IGF-1 (Suenson et al., 1984; Stewart et al., 1990; Owens et al., 1993; Ruan et al., 1995). As noted, the IGFs are key mitogens in regulating cell proliferation and differentiation, and they are anti-apoptotic (Pollak, 1998; Lee et al., 1999; Yu and Rohan, 2000). Studies have produced conflicting results regarding the association between higher IGF-1 levels and increased breast cancer risk (Yu and Rohan, 2000; Renehan et al., 2004; Shi et al., 2004; Sugumar et al., 2004; Fletcher et al., 2005). A collaborative group formed to carry out pooled analyses of individual data from prospective studies explored this relationship (Endogenous Hormones and Breast Cancer Collaborative Group, 2010). The group’s analysis of data from 17 studies showed that IGF-1 concentrations, adjusted for age, were higher in moderately overweight women than in other women. A positive association was found between circulating IGF-1 and breast cancer risk; the odds ratio for breast cancer for women in the highest versus the lowest fifth of IGF-1 concentration was 1.28 (95% CI, 1.14–1.44). The association was not influenced by menopausal status at the time blood samples were collected or by adjustment for other risk factors. In addition, it seemed to be limited to estrogen receptor–positive tumors (Endogenous Hormones and Breast Cancer Collaborative Group, 2010).
Another aspect of obesity that may relate to carcinogenesis is its induction of a chronic state of low-grade inflammation. Inflammation promotes an increase in cell proliferation and differentiation, inhibition of apoptosis, generation of new blood vessels, and induction of epigenetic events (Federico et al., 2007; WCRF/AICR, 2007). Obesity is characterized by an increased production of inflammatory factors by adipocytes, or fat cells; depending on the degree of obesity, up to 40 percent of fat tissue may be composed of macrophages, a type of white blood cell that plays a role in adaptive immunity and acts as a chemoattractant (WCRF/AICR, 2007, p. 39). These macrophages in turn recruit proinflammatory cells such as monocytes (McCready et al., 2010). Compared with lean people, obese individuals also have elevated concentrations of circulating leptin, which can function as an inflammatory cytokine (Zhang et al., 2002); tumor necrosis factor (TNF)-alpha (Jarvinen et al., 2001); interleukin (IL)-6; and C-reactive protein (Lunn et al., 1997). Human studies of obesity, inflammation, and breast cancer are limited (Kundu and Surh, 2008; Aggarwal and Gehlot, 2009), but studies in animal models suggest a connection between chronic inflammation and mammary tumors (McCready et al., 2010).
The mechanisms proposed to explain obesity-related carcinogenesis have many notable shortcomings, including, but not limited to, a focus on endocrine mechanisms that may ignore the role of paracrine signaling; complex isoforms of insulin, and IGF receptors that may alter their roles in carcinogenesis; and some inconsistency between animal and epidemiologic evidence (e.g., IGF-1 increases with increasing fatness in mice but declines in humans above a BMI of about 27) (Roberts et al., 2010). Other proposed mechanisms of obesity-induced carcinogenesis include obesity-related hypoxia, migrating stromal cells, and shared genetic susceptibility (Roberts et al., 2010). Adipose tissue hypoxia (ATH), or reduced levels of oxygen in adipose tissue, is seen in the white adipose tissue of obese mice in comparison to lean mice (Ye et al., 2007). ATH also plays a role in increased insulin resistance and chronic inflammation, potentially leading to an increased cancer risk (Roberts et al., 2010).
Migrating stromal cells have also been proposed to play a role in obesity-induced carcinogenesis. Studies have shown that adipose stromal cells from fat deposits in obese mice can promote tumor vascularization by migrating to the tumor site upon recruitment (Zhang et al., 2009). Collagen, a component of connective tissue, is also up-regulated in the adipose tissue of mice during the progression of mammary tumors, due to promotion from adipocyte-derived factors (Iyengar et al., 2003).
Shared genetic susceptibility and “obesity genetics” are a third novel area of research, in which genetic maps are used to explore overlapping “mutual candidate genes” for both obesity and cancer. Such approaches
have found promising patterns of polymorphisms for diseases such as colorectal cancers (Roberts et al., 2010).
A NEED TO CONSIDER TIMING OF EXPOSURE ACROSS THE LIFE COURSE
Thus, substantial evidence supports varying effects of environmental exposures at different stages along the life course. As science in this field has matured, it now seems clear that to examine the effect of exposure to an environmental chemical or other factor without consideration for the stage in life when that factor may be acting is shortsighted. The currently modest amount of evidence on the impact of environmental factors on breast cancer may actually stem from a lack of clear, biologically based hypotheses about the relevant timing of exposures and implementation of designs and protocols that address such hypotheses. Although studies that take into account the timing of exposure will be challenging to conduct, the committee strongly recommends that both epidemiologic and laboratory investigations of breast cancer risk factors specify the stage of the life course being studied (see Chapter 7).
ACS (American Cancer Society). 2011. Cancer facts and figures 2011. http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-figures-2011 (accessed June 22, 2011).
Aggarwal, B. B., and P. Gehlot. 2009. Inflammation and cancer: How friendly is the relationship for cancer patients? Curr Opin Pharmacol 9(4):351–369.
Aksglaede, L., K. Sorensen, J. H. Petersen, N. E. Skakkebaek, and A. Juul. 2009. Recent decline in age at breast development: The Copenhagen Puberty Study. Pediatrics 123(5): e932–e939.
Anbazhagan, R., J. Bartek, P. Monaghan, and B. A. Gusterson. 1991. Growth and development of the human infant breast. Am J Anat 192(4):407–417.
Anbazhagan, R., P. P. Osin, J. Bartkova, B. Nathan, E. B. Lane, and B. A. Gusterson. 1998. The development of epithelial phenotypes in the human fetal and infant breast. J Pathol 184(2):197–206.
Aranda, A., and A. Pascual. 2001. Nuclear hormone receptors and gene expression. Physiol Rev 81(3):1269–1304.
Balogh, G. A., I. H. Russo, C. Spittle, R. Heulings, and J. Russo. 2007. Immune-surveillance and programmed cell death-related genes are significantly overexpressed in the normal breast epithelium of postmenopausal parous women. Int J Oncol 31(2):303–312.
Barcellos-Hoff, M. H. 1993. Radiation-induced transforming growth factor β and subsequent extracellular matrix reorganization in murine mammary gland. Cancer Res 53(17):3880–3886.
Barcellos-Hoff, M. H., and S. A. Ravani. 2000. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res 60(5):1254–1260.
Barker, D. J. 2002. Fetal programming of coronary heart disease. Trends Endocrinol Metab 13(9):364–368.
Belous, A. R., D. L. Hachey, S. Dawling, N. Roodi, and F. F. Parl. 2007. Cytochrome P450 1B1-mediated estrogen metabolism results in estrogen-deoxyribonucleoside adduct formation. Cancer Res 67(2):812–817.
Beral, V., G. Reeves, D. Bull, and J. Green. 2011. Breast cancer risk in relation to the interval between menopause and starting hormone therapy. J Natl Cancer Inst 103(4):296–305.
Bernstein, L. 2002. Epidemiology of endocrine-related risk factors for breast cancer. J Mammary Gland Biol Neoplasia 7(1):3–15.
Biro, F. M., A. W. Lucky, L. A. Simbartl, B. A. Barton, S. R. Daniels, R. Striegel-Moore, S. S. Kronsberg, and J. A. Morrison. 2003. Pubertal maturation in girls and the relationship to anthropometric changes: Pathways through puberty. J Pediatr 142(6):643–646.
Biro, F. M., P. Khoury, and J. A. Morrison. 2006. Influence of obesity on timing of puberty. Int J Androl 29(1):272–277; discussion 286–290.
Biro, F. M., M. P. Galvez, L. C. Greenspan, P. A. Succop, N. Vangeepuram, S. M. Pinney, S. Teitelbaum, G. C. Windham, et al. 2010. Pubertal assessment method and baseline characteristics in a mixed longitudinal study of girls. Pediatrics 126(3):e583–e590.
Bissell, M. J. 2007. Modelling molecular mechanisms of breast cancer and invasion: Lessons from the normal gland. Biochem Soc Trans 35(Pt 1):18–22.
Bissell, M. J., and W. C. Hines. 2011. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17(3):320–329.
Bissell, M. J., and D. Radisky. 2001. Putting tumours in context. Nat Rev Cancer 1(1):46–54.
Björnström, L., and M. Sjöberg. 2005. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 19(4):833–842.
Block, K., A. Kardana, P. Igarashi, and H. S. Taylor. 2000. In utero diethylstilbestrol (DES) exposure alters Hox gene expression in the developing mullerian system. FASEB J 14(9):1101–1108.
Bodmer, M., C. Meier, S. Krahenbuhl, S. S. Jick, and C. R. Meier. 2010. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care 33(6):1304–1308.
Bogaert, A. F. 2005. Age at puberty and father absence in a national probability sample. J Adolesc 28(4):541–546.
Boice, J. D., Jr., D. Preston, F. G. Davis, and R. R. Monson. 1991. Frequent chest X-ray fluoroscopy and breast cancer incidence among tuberculosis patients in Massachusetts. Radiat Res 125(2):214–222.
Bolton, J. L., and G. R. Thatcher. 2008. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol 21(1):93–101.
Boyd, N. F., G. A. Lockwood, J. W. Byng, D. L. Tritchler, and M. J. Yaffe. 1998. Mammographic densities and breast cancer risk. Cancer Epidemiol Biomarkers Prev 7(12): 1133–1144.
Boyd, N. F., L. J. Martin, M. Bronskill, M. J. Yaffe, N. Duric, and S. Minkin. 2010. Breast tissue composition and susceptibility to breast cancer. J Natl Cancer Inst 102(16): 1224–1237.
Boylan, E. S., and R. E. Calhoon. 1979. Mammary tumorigenesis in the rat following prenatal exposure to diethylstilbestrol and postnatal treatment with 7,12-dimethylbenz[a]anthracene. J Toxicol Environ Health 5(6):1059–1071.
Boylan, E. S., and R. E. Calhoon. 1983. Transplacental action of diethylstilbestrol on mammary carcinogenesis in female rats given one or two doses of 7,12-dimethylbenz(a) anthracene. Cancer Res 43(10):4879–4884.
Bredfeldt, T. G., K. L. Greathouse, S. H. Safe, M. C. Hung, M. T. Bedford, and C. L. Walker. 2010. Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol 24(5):993–1006.
Bui, J. D., and R. D. Schreiber. 2007. Cancer immunosurveillance, immunoediting and inflammation: Independent or interdependent processes? Curr Opin Immunol 19(2):203–208.
Bunn, T. L., P. J. Parsons, E. Kao, and R. R. Dietert. 2001. Exposure to lead during critical windows of embryonic development: Differential immunotoxic outcome based on stage of exposure and gender. Toxicol Sci 64(1):57–66.
Calle, E. E., C. Rodriguez, K. Walker-Thurmond, and M. J. Thun. 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348(17):1625–1638.
Carmichael, A. R. 2006. Obesity and prognosis of breast cancer. Obes Rev 7(4):333–340.
Carmichael, A. R., and T. Bates. 2004. Obesity and breast cancer: A review of the literature. Breast 13(2):85–92.
Caserta, D., L. Maranghi, A. Mantovani, R. Marci, F. Maranghi, and M. Moscarini. 2008. Impact of endocrine disruptor chemicals in gynaecology. Hum Reprod Update 14(1):59–72.
Castoria, G., M. V. Barone, M. Di Domenico, A. Bilancio, D. Ametrano, A. Migliaccio, and F. Auricchio. 1999. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J 18(9):2500–2510.
Castoria, G., A. Migliaccio, A. Bilancio, M. Di Domenico, A. de Falco, M. Lombardi, R. Fiorentino, L. Varricchio, et al. 2001. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J 20(21):6050–6059.
Cato, A. C., A. Nestl, and S. Mink. 2002. Rapid actions of steroid receptors in cellular signaling pathways. Sci STKE 2002(138):re9.
Cavalieri, E., D. Chakravarti, J. Guttenplan, E. Hart, J. Ingle, R. Jankowiak, P. Muti, E. Rogan, et al. 2006. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta 1766(1):63–78.
Chappell, J., J. W. Leitner, S. Solomon, I. Golovchenko, M. L. Goalstone, and B. Draznin. 2001. Effect of insulin on cell cycle progression in MCF-7 breast cancer cells: Direct and potentiating influence. J Biol Chem 276(41):38023–38028.
Cheskis, B. J. 2004. Regulation of cell signalling cascades by steroid hormones. J Cell Biochem 93(1):20–27.
Chlebowski, R. T., L. H. Kuller, R. L. Prentice, M. L. Stefanick, J. E. Manson, M. Gass, A. K. Aragaki, J. K. Ockene, et al. 2009. Breast cancer after use of estrogen plus progestin in postmenopausal women. N Engl J Med 360(6):573–587.
Chlebowski, R. T., G. L. Anderson, M. Gass, D. S. Lane, A. K. Aragaki, L. H. Kuller, J. E. Manson, M. L. Stefanick, et al. 2010. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 304(15):1684–1692.
Clarke, C. A., S. L. Glaser, C. S. Uratsu, J. V. Selby, L. H. Kushi, and L. J. Herrinton. 2006. Recent declines in hormone therapy utilization and breast cancer incidence: Clinical and population-based evidence. J Clin Oncol 24(33):e49–e50.
Clavel-Chapelon, F. 2002. Evolution of age at menarche and at onset of regular cycling in a large cohort of French women. Hum Reprod 17(1):228–232.
Clavel-Chapelon, F., and M. Gerber. 2002. Reproductive factors and breast cancer risk. Do they differ according to age at diagnosis? Breast Cancer Res Treat 72(2):107–115.
Cleary, M. P., and M. E. Grossmann. 2009. Minireview: Obesity and breast cancer: The estrogen connection. Endocrinology 150(6):2537–2542.
Colditz, G. A., and A. L. Frazier. 1995. Models of breast cancer show that risk is set by events of early life: Prevention efforts must shift focus. Cancer Epidemiol Biomarkers Prev 4(5):567–571.
Collaborative Group on Hormonal Factors in Breast Cancer. 1997. Breast cancer and hormone replacement therapy: Collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Lancet 350(9084):1047–1059.
Couse, J. F., D. Dixon, M. Yates, A. B. Moore, L. Ma, R. Maas, and K. S. Korach. 2001. Estrogen receptor-alpha knockout mice exhibit resistance to the developmental effects of neonatal diethylstilbestrol exposure on the female reproductive tract. Dev Biol 238(2):224–238.
Couzin, J. 2002. Quirks of fetal environment felt decades later. Science 296(5576):2167–2169.
Crain, D. A., S. J. Janssen, T. M. Edwards, J. Heindel, S. M. Ho, P. Hunt, T. Iguchi, A. Juul, et al. 2008. Female reproductive disorders: The roles of endocrine-disrupting compounds and developmental timing. Fertil Steril 90(4):911–940.
de Muinck Keizer-Schrama, S. M., and D. Mul. 2001. Trends in pubertal development in Europe. Hum Reprod Update 7(3):287–291.
de Waard, F., and J. H. Thijssen. 2005. Hormonal aspects in the causation of human breast cancer: Epidemiological hypotheses reviewed, with special reference to nutritional status and first pregnancy. J Steroid Biochem Mol Biol 97(5):451–458.
Deardorff, J., J. P. Ekwaru, L. H. Kushi, B. J. Ellis, L. C. Greenspan, A. Mirabedi, E. G. Landaverde, and R. A. Hiatt. 2011. Father absence, body mass index, and pubertal timing in girls: Differential effects by family income and ethnicity. J Adolesc Health 48(5):441–447.
DeRoo, L. A., P. Cummings, and B. A. Mueller. 2011. Smoking before the first pregnancy and the risk of breast cancer: A meta-analysis. Am J Epidemiol 174(4):390–402.
Divoux, A., and K. Clement. 2011. Architecture and the extracellular matrix: The still unappreciated components of the adipose tissue. Obes Rev 12(5):e494–e503.
Doherty, L. F., J. G. Bromer, Y. Zhou, T. S. Aldad, and H. S. Taylor. 2010. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer 1(3):146–155.
Dolinoy, D. C., D. Huang, and R. L. Jirtle. 2007. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 104(32):13056–13061.
dos Santos Silva, I., B. De Stavola, and V. McCormack. 2008. Birth size and breast cancer risk: Reanalysis of individual participant data from 32 studies. PLoS Med 5(9):e193.
Durando, M., L. Kass, J. Piva, C. Sonnenschein, A. M. Soto, E. H. Luque, and M. Muñoz-de-Toro. 2007. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect 115(1):80–86.
Eden, J. A. 2010. Breast cancer, stem cells and sex hormones. Part 2: The impact of the reproductive years and pregnancy. Maturitas 67(3):215–218.
Eden, J. A. 2011. Breast cancer, stem cells and sex hormones. Part 3: The impact of the menopause and hormone replacement. Maturitas 68(2):129–136.
Edwards, D. P. 2005. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol 67:335–376.
Ellis, B. J. 2004. Timing of pubertal maturation in girls: An integrated life history approach. Psychol Bull 130(6):920–958.
Ellis, B. J., and J. Garber. 2000. Psychosocial antecedents of variation in girls’ pubertal timing: Maternal depression, stepfather presence, and marital and family stress. Child Dev 71(2):485–501.
Endogenous Hormones and Breast Cancer Collaborative Group. 2010. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: Pooled individual data analysis of 17 prospective studies. Lancet Oncol 11(6):530–542.
Euling, S. Y., M. E. Herman-Giddens, P. A. Lee, S. G. Selevan, A. Juul, T. I. Sorensen, L. Dunkel, J. H. Himes, et al. 2008. Examination of U.S. puberty timing data from 1940 to 1994 for secular trends: Panel findings. Pediatrics 121(Suppl 3):S172–S191.
Federico, A., F. Morgillo, C. Tuccillo, F. Ciardiello, and C. Loguercio. 2007. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer 121(11):2381–2386.
Feinberg, A. P., and B. Tycko. 2004. The history of cancer epigenetics. Nat Rev Cancer 4(2):143–153.
Fletcher, O., L. Gibson, N. Johnson, D. R. Altmann, J. M. Holly, A. Ashworth, J. Peto, and S. Silva Idos. 2005. Polymorphisms and circulating levels in the insulin-like growth factor system and risk of breast cancer: A systematic review. Cancer Epidemiol Biomarkers Prev 14(1):2–19.
Frankel, S., P. Elwood, P. Sweetnam, J. Yarnell, and G. D. Smith. 1996. Birthweight, body-mass index in middle age, and incident coronary heart disease. Lancet 348(9040):1478–1480.
Garland, M., D. J. Hunter, G. A. Colditz, J. E. Manson, M. J. Stampfer, D. Spiegelman, F. Speizer, and W. C. Willett. 1998. Menstrual cycle characteristics and history of ovulatory infertility in relation to breast cancer risk in a large cohort of U.S. women. Am J Epidemiol 147(7):636–643.
Giovannucci, E. 2001. Insulin, insulin-like growth factors and colon cancer: A review of the evidence. J Nutr 131(11 Suppl):3109S–3120S.
Graber, J. A., J. Brooks-Gunn, and M. P. Warren. 1995. The antecedents of menarcheal age: Heredity, family environment, and stressful life events. Child Dev 66(2):346–359.
Graber, J. A., P. M. Lewinsohn, J. R. Seeley, and J. Brooks-Gunn. 1997. Is psychopathology associated with the timing of pubertal development? J Am Acad Child Adolesc Psychiatry 36(12):1768–1776.
Grumbach, M. M., and D. M. Styne. 2002. Puberty: Ontogeny, neuroendocrinology, physiology, and disorders. In Williams textbook of endocrinology, 10th ed. Edited by P. R. Larsen, H. M. Kronenberg, S. Melmed, and K. S. Polonsky. Philadelphia, PA: Saunders. Pp. 1115–1286.
Ha, M., K. Mabuchi, A. J. Sigurdson, D. M. Freedman, M. S. Linet, M. M. Doody, and M. Hauptmann. 2007. Smoking cigarettes before first childbirth and risk of breast cancer. Am J Epidemiol 166(1):55–61.
Hattersley, A. T., and J. E. Tooke. 1999. The fetal insulin hypothesis: An alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet 353(9166):1789–1792.
Hauspie, R. C., M. Vercauteren, and C. Susanne. 1997. Secular changes in growth and maturation: An update. Acta Paediatr Suppl 423:20–27.
Herbst, A. L., H. Ulfelder, and D. C. Poskanzer. 1971. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med 284(15):878–881.
Hertz-Picciotto, I., C. E. Herr, P. S. Yap, M. Dostal, R. H. Shumway, P. Ashwood, M. Lipsett, J. P. Joad, et al. 2005. Air pollution and lymphocyte phenotype proportions in cord blood. Environ Health Perspect 113(10):1391–1398.
Heuson, J. C., N. Legros, and R. Heimann. 1972. Influence of insulin administration on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in intact, oophorectomized, and hypophysectomized rats. Cancer Res 32(2):233–238.
Hewitt, S. C., J. C. Harrell, and K. S. Korach. 2005. Lessons in estrogen biology from knockout and transgenic animals. Annu Rev Physiol 67:285–308.
Hilakivi-Clarke, L., J. E. Andrade, and W. Helferich. 2010. Is soy consumption good or bad for the breast? J Nutr 140(12):2326S–2334S.
Hovey, R. C., M. Asai-Sato, A. Warri, B. Terry-Koroma, N. Colyn, E. Ginsburg, and B. K. Vonderhaar. 2005. Effects of neonatal exposure to diethylstilbestrol, tamoxifen, and toremifene on the BALB/c mouse mammary gland. Biol Reprod 72(2):423–435.
Howard, B. A., and B. A. Gusterson. 2000. Human breast development. J Mammary Gland Biol Neoplasia 5(2):119–137.
Hsieh, C. C., D. Trichopoulos, K. Katsouyanni, and S. Yuasa. 1990. Age at menarche, age at menopause, height and obesity as risk factors for breast cancer: Associations and interactions in an international case–control study. Int J Cancer 46(5):796–800.
Hughes, E. S. 1950. The development of the mammary gland: Arris and Gale Lecture, delivered at the Royal College of Surgeons of England on 25 October, 1949. Ann R Coll Surg Engl 6(2):99–119.
Hunter, C. P., C. K. Redmond, V. W. Chen, D. F. Austin, R. S. Greenberg, P. Correa, H. B. Muss, M. R. Forman, et al. 1993. Breast cancer: Factors associated with stage at diagnosis in black and white women. Black/White Cancer Survival Study Group. J Natl Cancer Inst 85(14):1129–1137.
IARC (International Agency for Research on Cancer). 2011. IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 100, Part A: Pharmaceuticals. Lyon, France: IARC.
Ish-Shalom, D., C. T. Christoffersen, P. Vorwerk, N. Sacerdoti-Sierra, R. M. Shymko, D. Naor, and P. De Meyts. 1997. Mitogenic properties of insulin and insulin analogues mediated by the insulin receptor. Diabetologia 40(Suppl 2):S25–S31.
Iyengar, P., T. P. Combs, S. J. Shah, V. Gouon-Evans, J. W. Pollard, C. Albanese, L. Flanagan, M. P. Tenniswood, et al. 2003. Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 22(41):6408–6423.
Jarvinen, R., P. Knekt, T. Hakulinen, H. Rissanen, and M. Heliovaara. 2001. Dietary fat, cholesterol and colorectal cancer in a prospective study. Br J Cancer 85(3):357–361.
Jasik, C. B., and R. H. Lustig. 2008. Adolescent obesity and puberty: The “perfect storm.” Ann N Y Acad Sci 1135:265–279.
Jenkins, S., A. M. Betancourt, J. Wang, and C. A. Lamartiniere. 2011. Endocrine-active chemicals in mammary cancer causation and prevention. J Steroid Biochem Mol Biol June 23. [Epub ahead of print]
Jiang, X., N. M. Patterson, Y. Ling, J. Xie, W. G. Helferich, and D. J. Shapiro. 2008. Low concentrations of the soy phytoestrogen genistein induce proteinase inhibitor 9 and block killing of breast cancer cells by immune cells. Endocrinology 149(11):5366–5373.
John, E. M., and J. L. Kelsey. 1993. Radiation and other environmental exposures and breast cancer. Epidemiol Rev 15(1):157–162.
Jones, B. A., S. V. Kasi, M. G. Curnen, P. H. Owens, and R. Dubrow. 1997. Severe obesity as an explanatory factor for the black/white difference in stage at diagnosis of breast cancer. Am J Epidemiol 146(5):394–404.
Jury, H. H., T. R. Zachzrewski, and G. L. Hammond. 2000. Interactions between human plasma sex hormone-binding globulin and xenobiotic ligands. J Steroid Biochem Mol Biol 75:167–176.
Kahlert, S., S. Nuedling, M. van Eickels, H. Vetter, R. Meyer, and C. Grohe. 2000. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem 275(24):18447–18453.
Kaplowitz, P. B. 2008. Link between body fat and the timing of puberty. Pediatrics 121(Suppl 3):S208–S217.
Kariagina, A., J. Xie, J. R. Leipprandt, and S. Z. Haslam. 2010. Amphiregulin mediates estrogen, progesterone, and EGFR signaling in the normal rat mammary gland and in hormone-dependent rat mammary cancers. Horm Cancer 1(5):229–244.
Kawaguchi, H., N. Miyoshi, Y. Miyamoto, M. Souda, Y. Umekita, N. Yasuda, and H. Yoshida. 2009a. Effects of fetal exposure to diethylstilbestrol on mammary tumorigenesis in rats. J Vet Med Sci 71(12):1599–1608.
Kawaguchi, H., Y. Umekita, M. Souda, K. Gejima, H. Kawashima, T. Yoshikawa, and H. Yoshida. 2009b. Effects of neonatally administered high-dose diethylstilbestrol on the induction of mammary tumors induced by 7,12-dimethylbenz[a]anthracene in female rats. Vet Pathol 46(1):142–150.
Kelsey, J. L., and L. Bernstein. 1996. Epidemiology and prevention of breast cancer. Annu Rev Public Health 17:47–67.
Knight, C. H., and A. Sorensen. 2001. Windows in early mammary development: Critical or not? Reproduction 122(3):337–345.
Kotsopoulos, J., W. Y. Chen, M. A. Gates, S. S. Tworoger, S. E. Hankinson, and B. A. Rosner. 2010. Risk factors for ductal and lobular breast cancer: Results from the Nurses’ Health Study. Breast Cancer Res 12(6):R106.
Kousteni, S., T. Bellido, L. I. Plotkin, C. A. O’Brien, D. L. Bodenner, L. Han, K. Han, G. B. DiGregorio, et al. 2001. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: Dissociation from transcriptional activity. Cell 104(5):719–730.
Kundu, J. K., and Y. J. Surh. 2008. Inflammation: Gearing the journey to cancer. Mutat Res 659(1–2):15–30.
LaCroix, A. Z., R. T. Chlebowski, J. E. Manson, A. K. Aragaki, K. C. Johnson, L. Martin, K. L. Margolis, M. L. Stefanick, et al. 2011. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: A randomized controlled trial. JAMA 305(13):1305–1314.
Lamartiniere, C. A., and M. B. Holland. 1992. Neonatal diethylstilbestrol prevents spontaneously developing mammary tumors. In Hormonal carcinogenesis: Proceedings of the first international symposium. Edited by J. Li, S. Nandi, and S. A. Li. New York: Springer-Verlag.
Lamartiniere, C. A., J. B. Moore, N. M. Brown, R. Thompson, M. J. Hardin, and S. Barnes. 1995. Genistein suppresses mammary cancer in rats. Carcinogenesis 16(11):2833–2840.
Land, C. E., M. Tokunaga, K. Koyama, M. Soda, D. L. Preston, I. Nishimori, and S. Tokuoka. 2003. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950–1990. Radiat Res 160(6):707–717.
Lee, A. V., J. G. Jackson, J. L. Gooch, S. G. Hilsenbeck, E. Coronado-Heinsohn, C. K. Osborne, and D. Yee. 1999. Enhancement of insulin-like growth factor signaling in human breast cancer: Estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol 13(5):787–796.
Levin, E. R. 2005. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol 19(8):1951–1959.
Li, S., R. Hansman, R. Newbold, B. Davis, J. A. McLachlan, and J. C. Barrett. 2003. Neonatal diethylstilbestrol exposure induces persistent elevation of c-fos expression and hypomethylation in its exon-4 in mouse uterus. Mol Carcinog 38(2):78–84.
Losel, R., and M. Wehling. 2003. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol 4(1):46–56.
Lunn, R. M., Y. J. Zhang, L. Y. Wang, C. J. Chen, P. H. Lee, C. S. Lee, W. Y. Tsai, and R. M. Santella. 1997. p53 mutations, chronic hepatitis B virus infection, and aflatoxin exposure in hepatocellular carcinoma in Taiwan. Cancer Res 57(16):3471–3477.
Luo, J., K. L. Margolis, J. Wactawski-Wende, K. Horn, C. Messina, M. L. Stefanick, H. A. Tindle, E. Tong, et al. 2011. Association of active and passive smoking with risk of breast cancer among postmenopausal women: A prospective cohort study. BMJ 342:d1016.
Ma, L., G. V. Benson, H. Lim, S. K. Dey, and R. L. Maas. 1998. Abdominal B (AbdB) Hoxa genes: Regulation in adult uterus by estrogen and progesterone and repression in mullerian duct by the synthetic estrogen diethylstilbestrol (DES). Dev Biol 197(2):141–154.
MacMahon, B., D. Trichopoulos, J. Brown, A. P. Andersen, K. Aoki, P. Cole, F. deWaard, T. Kauraniemi, et al. 1982a. Age at menarche, probability of ovulation and breast cancer risk. Int J Cancer 29(1):13–16.
MacMahon, B., D. Trichopoulos, J. Brown, A. P. Andersen, P. Cole, F. deWaard, T. Kauraniemi, A. Polychronopoulou, et al. 1982b. Age at menarche, urine estrogens and breast cancer risk. Int J Cancer 30(4):427–431.
Maffini, M. V., A. M. Soto, J. M. Calabro, A. A. Ucci, and C. Sonnenschein. 2004. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci 117(Pt 8):1495–1502.
Martin, L. J., and N. F. Boyd. 2008. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: Hypotheses based on epidemiological evidence. Breast Cancer Res 10(1):201.
McCready, J., L. M. Arendt, J. A. Rudnick, and C. Kuperwasser. 2010. The contribution of dynamic stromal remodeling during mammary development to breast carcinogenesis. Breast Cancer Res 12(3):205.
Michels, K. B., and F. Xue. 2006. Role of birthweight in the etiology of breast cancer. Int J Cancer 119(9):2007–2025.
Migliaccio, A., M. Di Domenico, G. Castoria, A. de Falco, P. Bontempo, E. Nola, and F. Auricchio. 1996. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 15(6):1292–1300.
Migliaccio, A., G. Castoria, M. Di Domenico, A. de Falco, A. Bilancio, M. Lombardi, M. V. Barone, D. Ametrano, et al. 2000. Steroid-induced androgen receptor–oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J 19(20):5406–5417.
Miller, C., K. Degenhardt, and D. A. Sassoon. 1998. Fetal exposure to DES results in de-regulation of Wnt7a during uterine morphogenesis. Nat Genet 20(3):228–230.
Milligan, S. R., O. Khan, and M. Nash. 1998. Competitive binding of xenobiotic oestrogens to rat alpha-fetoprotein and to sex steroid binding proteins in human and rainbow trout (Oncorhynchus mykiss) plasma. Gen Comp Endocrinol 112(1):89–95.
Million Women Study Collaborators. 2003. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362(9382):419–427.
Mizejewski, G., G. Smith, and G. Butterstein. 2004. Review and proposed action of alpha-fetoprotein growth inhibitory peptides as estrogen and cytoskeleton-associated factors. Cell Biol Int 28(12):913–933.
Mouritsen, A., L. Aksglaede, K. Sorensen, S. S. Mogensen, H. Leffers, K. M. Main, H. Frederiksen, A. M. Andersson, et al. 2010. Hypothesis: Exposure to endocrine-disrupting chemicals may interfere with timing of puberty. Int J Androl 33(2):346–359.
Mueller, M. M., and N. E. Fusenig. 2004. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 4(11):839–849.
Muñoz-de-Toro, M., C. M. Markey, P. R. Wadia, E. H. Luque, B. S. Rubin, C. Sonnenschein, and A. M. Soto. 2005. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology 146(9):4138–4147.
Murray, T. J., M. V. Maffini, A. A. Ucci, C. Sonnenschein, and A. M. Soto. 2007. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod Toxicol 23(3):383–390.
Mustanski, B. S., R. J. Viken, J. Kaprio, L. Pulkkinen, and R. J. Rose. 2004. Genetic and environmental influences on pubertal development: Longitudinal data from Finnish twins at ages 11 and 14. Dev Psychol 40(6):1188–1198.
NCI (National Cancer Institute). 2009. What you need to know about™ breast cancer. http://www.cancer.gov/cancertopics/wyntk/breast/page2 (accessed August 1, 2011).
NCI. 2010. Understanding cancer series: Estrogen receptors/SERMs. http://www.cancer.gov/cancertopics/understandingcancer/estrogenreceptors (accessed November 2, 2011).
Nechuta, S., N. Paneth, and E. M. Velie. 2010. Pregnancy characteristics and maternal breast cancer risk: A review of the epidemiologic literature. Cancer Causes Control 21(7):967–989.
Newbold, R. R., B. C. Bullock, and J. A. McLachlan. 1990. Uterine adenocarcinoma in mice following developmental treatment with estrogens: A model for hormonal carcinogenesis. Cancer Res 50(23):7677–7681.
Newbold, R. R., R. B. Hanson, and W. N. Jefferson. 1997. Ontogeny of lactoferrin in the developing mouse uterus: A marker of early hormone response. Biol Reprod 56(5):1147–1157.
Newcomb, P. A., A. Trentham-Dietz, J. M. Hampton, K. M. Egan, L. Titus-Ernstoff, S. Warren Andersen, E. R. Greenberg, and W. C. Willett. 2011. Late age at first full term birth is strongly associated with lobular breast cancer. Cancer 117(9):1946–1956.
Ng, S. P., A. E. Silverstone, Z. W. Lai, and J. T. Zelikoff. 2006. Effects of prenatal exposure to cigarette smoke on offspring tumor susceptibility and associated immune mechanisms. Toxicol Sci 89(1):135–144.
Nguyen, D. H., H. Martinez-Ruiz, and M. H. Barcellos-Hoff. 2011. Consequences of epithelial or stromal TGFβ1 depletion in the mammary gland. J Mammary Gland Biol Neoplasia 16(2):147–155.
NIH (National Institutes of Health). 2011. Clinical trials.gov. http://clinicaltrials.gov/ct2/results?term=breast+cancer+and+metformin (accessed November 4, 2011).
Ninomiya, K., H. Kawaguchi, M. Souda, S. Taguchi, M. Funato, Y. Umekita, and H. Yoshida. 2007. Effects of neonatally administered diethylstilbestrol on induction of mammary carcinomas induced by 7,12-dimethylbenz(a)anthracene in female rats. Toxicol Pathol 35(6):813–818.
Ogden, C. L., M. D. Carroll, L. R. Curtin, M. M. Lamb, and K. M. Flegal. 2010. Prevalence of high body mass index in U.S. children and adolescents, 2007–2008. JAMA 303(3):242–249.
Owens, P. C., P. G. Gill, N. J. De Young, M. A. Weger, S. E. Knowles, and K. J. Moyse. 1993. Estrogen and progesterone regulate secretion of insulin-like growth factor binding proteins by human breast cancer cells. Biochem Biophys Res Commun 193(2):467–473.
Palmer, J. R., L. A. Wise, E. E. Hatch, R. Troisi, L. Titus-Ernstoff, W. Strohsnitter, R. Kaufman, A. L. Herbst, et al. 2006. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev 15(8):1509–1514.
Parent, A. S., G. Teilmann, A. Juul, N. E. Skakkebaek, J. Toppari, and J. P. Bourguignon. 2003. The timing of normal puberty and the age limits of sexual precocity: Variations around the world, secular trends, and changes after migration. Endocr Rev 24(5):668–693.
Parent, A. S., G. Rasier, A. Gerard, S. Heger, C. Roth, C. Mastronardi, H. Jung, S. R. Ojeda, et al. 2005. Early onset of puberty: Tracking genetic and environmental factors. Horm Res 64 (Suppl 2):41–47.
Park, H. Y., I. Hertz-Picciotto, J. Petrik, L. Palkovicova, A. Kocan, and T. Trnovec. 2008a. Prenatal PCB exposure and thymus size at birth in neonates in Eastern Slovakia. Environ Health Perspect 116(1):104–109.
Park, S. K., D. Kang, K. A. McGlynn, M. Garcia-Closas, Y. Kim, K. Y. Yoo, and L. A. Brinton. 2008b. Intrauterine environments and breast cancer risk: Meta-analysis and systematic review. Breast Cancer Res 10(1):R8.
Pasqualini, J. R., F. A. Kincl, and C. Sumida. 1985. Hormones and the fetus. New York: Pergamon Press.
Petrelli, J. M., E. E. Calle, C. Rodriguez, and M. J. Thun. 2002. Body mass index, height, and postmenopausal breast cancer mortality in a prospective cohort of U.S. women. Cancer Causes Control 13(4):325–332.
Pollak, M. N. 1998. Endocrine effects of IGF-I on normal and transformed breast epithelial cells: Potential relevance to strategies for breast cancer treatment and prevention. Breast Cancer Res Treat 47(3):209–217.
Polyak, K., and R. Kalluri. 2010. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb Perspect Biol 2(11):a003244.
Poretsky, L., and M. F. Kalin. 1987. The gonadotropic function of insulin. Endocr Rev 8(2):132–141.
Potischman, N., and R. Troisi. 1999. In-utero and early life exposures in relation to risk of breast cancer. Cancer Causes Control 10(6):561–573.
Prentice, R. L., J. E. Manson, R. D. Langer, G. L. Anderson, M. Pettinger, R. D. Jackson, K. C. Johnson, L. H. Kuller, et al. 2009. Benefits and risks of postmenopausal hormone therapy when it is initiated soon after menopause. Am J Epidemiol 170(1):12–23.
Preston, D. L., E. Ron, S. Tokuoka, S. Funamoto, N. Nishi, M. Soda, K. Mabuchi, and K. Kodama. 2007. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat Res 168(1):1–64.
Protani, M., M. Coory, and J. H. Martin. 2010. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res Treat 123(3): 627–635.
Pugeat, M., J. C. Crave, M. Elmidani, M. H. Nicolas, M. Garoscio-Cholet, H. Lejeune, H. Dechaud, and J. Tourniaire. 1991. Pathophysiology of sex hormone binding globulin (SHBG): Relation to insulin. J Steroid Biochem Mol Biol 40(4–6):841–849.
Pukkala, E., A. Kesminiene, S. Poliakov, A. Ryzhov, V. Drozdovitch, L. Kovgan, P. Kyyronen, I. V. Malakhova, et al. 2006. Breast cancer in Belarus and Ukraine after the Chernobyl accident. Int J Cancer 119(3):651–658.
Quinlan, R. J. 2003. Father absence, parental care, and female reproductive development. Evol Hum Behav 24(6):376–390.
Renehan, A. G., M. Zwahlen, C. Minder, S. T. O’Dwyer, S. M. Shalet, and M. Egger. 2004. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 363(9418):1346–1353.
Revankar, C. M., D. F. Cimino, L. A. Sklar, J. B. Arterburn, and E. R. Prossnitz. 2005. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307(5715):1625–1630.
Reynolds, P., S. E. Hurley, K. Hoggatt, H. Anton-Culver, L. Bernstein, D. Deapen, D. Peel, R. Pinder, et al. 2004. Correlates of active and passive smoking in the California Teachers Study cohort. J Womens Health (Larchmt) 13(7):778–790.
Roberts, D. L., C. Dive, and A. G. Renehan. 2010. Biological mechanisms linking obesity and cancer risk: New perspectives. Annu Rev Med 61:301–316.
Rogol, A. D. 1998. Leptin and puberty. J Clin Endocrinol Metab 83(4):1089–1090.
Romans, S. E., J. M. Martin, K. Gendall, and G. P. Herbison. 2003. Age of menarche: The role of some psychosocial factors. Psychol Med 33(5):933–939.
Rothschild, T. C., E. S. Boylan, R. E. Calhoon, and B. K. Vonderhaar. 1987. Transplacental effects of diethylstilbestrol on mammary development and tumorigenesis in female ACI rats. Cancer Res 47(16):4508–4516.
Ruan, W., V. Catanese, R. Wieczorek, M. Feldman, and D. L. Kleinberg. 1995. Estradiol enhances the stimulatory effect of insulin-like growth factor-I (IGF-I) on mammary development and growth hormone-induced IGF-I messenger ribonucleic acid. Endocrinology 136(3):1296–1302.
Rudel, R. A., S. E. Fenton, J. M. Ackerman, S. Y. Euling, and S. L. Makris. 2011. Environmental exposures and mammary gland development: State of the science, public health implications, and research recommendations. Environ Health Perspect 119(8):1053–1061.
Rudland, P. S. 1993. Epithelial stem cells and their possible role in the development of the normal and diseased human breast. Histol Histopathol 8(2):385–404.
Russo, J., and I. H. Russo. 1978. DNA labeling index and structure of the rat mammary gland as determinants of its susceptibility to carcinogenesis. J Natl Cancer Inst 61(6): 1451–1459.
Russo, J., and I. H. Russo. 2004. Development of the human breast. Maturitas 49(1):2–15.
Russo, J., L. K. Tay, and I. H. Russo. 1982. Differentiation of the mammary gland and susceptibility to carcinogenesis. Breast Cancer Res Treat 2 (1):5–73.
Russo, J., H. Lynch, and I. H. Russo. 2001. Mammary gland architecture as a determining factor in the susceptibility of the human breast to cancer. Breast J 7(5):278–291.
Russo, J., G. A. Balogh, J. Chen, S. V. Fernandez, R. Fernbaugh, R. Heulings, D. A. Mailo, R. Moral, et al. 2006. The concept of stem cell in the mammary gland and its implication in morphogenesis, cancer and prevention. Front Biosci 11:151–172.
Shafie, S. M., and F. H. Grantham. 1981. Role of hormones in the growth and regression of human breast cancer cells (MCF-7) transplanted into athymic nude mice. J Natl Cancer Inst 67(1):51–56.
Shafie, S. M., and R. Hilf. 1981. Insulin receptor levels and magnitude of insulin-induced responses in 7,12-dimethylbenz(a)anthracene-induced mammary tumors in rats. Cancer Res 41(3):826–829.
Shekhar, M. P., R. Pauley, and G. Heppner. 2003. Host microenvironment in breast cancer development: Extracellular matrix-stromal cell contribution to neoplastic phenotype of epithelial cells in the breast. Breast Cancer Res 5(3):130–135.
Shi, R., H. Yu, J. McLarty, and J. Glass. 2004. IGF-I and breast cancer: A meta-analysis. Int J Cancer 111(3):418–423.
Shim, Y. K., S. P. Mlynarek, and E. van Wijngaarden. 2009. Parental exposure to pesticides and childhood brain cancer: U.S. Atlantic Coast Childhood Brain Cancer Study. Environ Health Perspect 117(6):1002–1006.
Shoker, B. S., C. Jarvis, R. B. Clarke, E. Anderson, J. Hewlett, M. P. Davies, D. R. Sibson, and J. P. Sloane. 1999. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am J Pathol 155(6):1811–1815.
Shore, R. E., N. Hildreth, E. Woodard, P. Dvoretsky, L. Hempelmann, and B. Pasternack. 1986. Breast cancer among women given X-ray therapy for acute postpartum mastitis. J Natl Cancer Inst 77(3):689–696.
Shore, R. E., N. Hildreth, P. Dvoretsky, E. Andresen, M. Moseson, and B. Pasternack. 1993. Thyroid cancer among persons given X-ray treatment in infancy for an enlarged thymus gland. Am J Epidemiol 137(10):1068–1080.
Slattery, M. L., C. Sweeney, R. Wolff, J. Herrick, K. Baumgartner, A. Giuliano, and T. Byers. 2007. Genetic variation in IGF1, IGFBP3, IRS1, IRS2 and risk of breast cancer in women living in Southwestern United States. Breast Cancer Res Treat 104(2):197–209.
Song, R. X., R. A. McPherson, L. Adam, Y. Bao, M. Shupnik, R. Kumar, and R. J. Santen. 2002a. Linkage of rapid estrogen action to MAPK activation by ERα-Shc association and Shc pathway activation. Mol Endocrinol 16(1):116–127.
Song, R. X., R. J. Santen, R. Kumar, L. Adam, M. H. Jeng, S. Masamura, and W. Yue. 2002b. Adaptive mechanisms induced by long-term estrogen deprivation in breast cancer cells. Mol Cell Endocrinol 193(1–2):29–42.
Song, R. X., C. J. Barnes, Z. Zhang, Y. Bao, R. Kumar, and R. J. Santen. 2004. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma membrane. Proc Natl Acad Sci U S A 101(7):2076–2081.
Song, R. X., Z. Zhang, and R. J. Santen. 2005. Estrogen rapid action via protein complex formation involving ERα and Src. Trends Endocrinol Metab 16(8):347–353.
Sorahan, T., L. Hamilton, K. Gardiner, J. T. Hodgson, and J. M. Harrington. 1999. Maternal occupational exposure to electromagnetic fields before, during, and after pregnancy in relation to risks of childhood cancers: Findings from the Oxford Survey of Childhood Cancers, 1953–1981 deaths. Am J Ind Med 35(4):348–357.
Sorensen, K., L. Aksglaede, T. Munch-Andersen, N. J. Aachmann-Andersen, J. H. Petersen, L. Hilsted, J. W. Helge, and A. Juul. 2009. Sex hormone-binding globulin levels predict insulin sensitivity, disposition index, and cardiovascular risk during puberty. Diabetes Care 32(5):909–914.
Soto, A. M., L. N. Vandenberg, M. V. Maffini, and C. Sonnenschein. 2008. Does breast cancer start in the womb? Basic Clin Pharmacol Toxicol 102(2):125–133.
Stewart, A. J., M. D. Johnson, F. E. May, and B. R. Westley. 1990. Role of insulin-like growth factors and the type I insulin-like growth factor receptor in the estrogen-stimulated proliferation of human breast cancer cells. J Biol Chem 265(34):21172–21178.
Suenson, E., O. Lutzen, and S. Thorsen. 1984. Initial plasmin-degradation of fibrin as the basis of a positive feed-back mechanism in fibrinolysis. Eur J Biochem 140(3):513–522.
Sugumar, A., Y. C. Liu, Q. Xia, Y. S. Koh, and K. Matsuo. 2004. Insulin-like growth factor (IGF)-I and IGF-binding protein 3 and the risk of premenopausal breast cancer: A meta-analysis of literature. Int J Cancer 111(2):293–297.
Sulis, M. L., and R. Parsons. 2003. PTEN: From pathology to biology. Trends Cell Biol 13(9):478–483.
Svensson, B. G., T. Hallberg, A. Nilsson, A. Schutz, and L. Hagmar. 1994. Parameters of immunological competence in subjects with high consumption of fish contaminated with persistent organochlorine compounds. Int Arch Occup Environ Health 65(6):351–358.
Thau, R., J. T. Lanman, and A. Brinson. 1976. Declining plasma progesterone concentration with advancing gestation in blood from umbilical and uterine veins and fetal heart in monkeys. Biol Reprod 14(4):507–509.
Thomas, P., Y. Pang, E. J. Filardo, and J. Dong. 2005. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146(2):624–632.
Titus-Ernstoff, L., M. P. Longnecker, P. A. Newcomb, B. Dain, E. R. Greenberg, R. Mittendorf, M. Stampfer, and W. Willett. 1998. Menstrual factors in relation to breast cancer risk. Cancer Epidemiol Biomarkers Prev 7(9):783–789.
Titus-Ernstoff, L., E. E. Hatch, R. N. Hoover, J. Palmer, E. R. Greenberg, W. Ricker, R. Kaufman, K. Noller, et al. 2001. Long-term cancer risk in women given diethylstilbestrol (DES) during pregnancy. Br J Cancer 84(1):126–133.
Tokunaga, M., C. E. Land, and S. Tokuoka. 1991. Follow-up studies of breast cancer incidence among atomic bomb survivors. J Radiat Res (Tokyo) 32 (Suppl):201–211.
Trichopoulos, D., P. Lagiou, and H. O. Adami. 2005. Towards an integrated model for breast cancer etiology: The crucial role of the number of mammary tissue-specific stem cells. Breast Cancer Res 7(1):13–17.
Troisi, R., E. E. Hatch, L. Titus-Ernstoff, M. Hyer, J. R. Palmer, S. J. Robboy, W. C. Strohsnitter, R. Kaufman, et al. 2007. Cancer risk in women prenatally exposed to diethylstilbestrol. Int J Cancer 121(2):356–360.
Tulchinsky, D. 1973. Placental secretion of unconjugated estrone, estradiol and estriol into the maternal and the fetal circulation. J Clin Endocrinol Metab 36(6):1079–1087.
Van Maele-Fabry, G., A. C. Lantin, P. Hoet, and D. Lison. 2010. Childhood leukaemia and parental occupational exposure to pesticides: A systematic review and meta-analysis. Cancer Causes Control 21(6):787–809.
Vandenberg, L. N., M. V. Maffini, C. M. Schaeberle, A. A. Ucci, C. Sonnenschein, B. S. Rubin, and A. M. Soto. 2008. Perinatal exposure to the xenoestrogen bisphenol-A induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol 26(3–4):210–219.
Vassilacopoulou, D., and E. S. Boylan. 1993. Mammary gland morphology and responsiveness to regulatory molecules following prenatal exposure to diethylstilbestrol. Teratog Carcinog Mutagen 13(2):59–74.
Vazquez-Martin, A., C. Oliveras-Ferraros, and J. A. Menendez. 2009. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 8(1):88–96.
Vivanco, I., and C. L. Sawyers. 2002. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2(7):489–501.
vom Saal, F. S., and B. G. Timms. 1999. The role of natural and manmade estrogens in prostate development. In Endocrine disruptors: Effects on male and female reproductive systems. Edited by R. K. Naz. Boca Raton, FL: CRC Press. Pp. 307–327.
Wakeford, R. 1995. The risk of childhood cancer from intrauterine and preconceptional exposure to ionizing radiation. Environ Health Perspect 103(11):1018–1025.
Warri, A., N. M. Saarinen, S. Makela, and L. Hilakivi-Clarke. 2008. The role of early life genistein exposures in modifying breast cancer risk. Br J Cancer 98(9):1485–1493.
WCRF/AICR (World Cancer Research Fund/American Institute for Cancer Research). 2007. Food, nutrition, physical activity, and the prevention of cancer: A global perspective. Washington, DC: AICR.
WCRF/AICR. 2010. Food, nutrition and physical activity and the prevention of breast cancer. WCRF/AICR Continuous update report summary. http://dietandcancerreport.org/downloads/cu/cu_breast_cancer_summary_2008.pdf (accessed October 29, 2011).
Weinberg, R. A. 2007. The biology of cancer. New York: Garland Science.
Weisglas-Kuperus, N., S. Patandin, G. A. Berbers, T. C. Sas, P. G. Mulder, P. J. Sauer, and H. Hooijkaas. 2000. Immunologic effects of background exposure to polychlorinated biphenyls and dioxins in Dutch preschool children. Environ Health Perspect 108(12):1203–1207.
Welshons, W. V., S. C. Nagel, K. A. Thayer, B. M. Judy, and F. S. vom Saal. 1999. Low-dose bioactivity of xenoestrogens in animals: Fetal exposure to low doses of methoxychlor and other xenoestrogens increases adult prostate size in mice. Toxicol Ind Health 15(1–2):12–25.
Witorsch, R. J. 2002. Low-dose in utero effects of xenoestrogens in mice and their relevance to humans: An analytical review of the literature. Food Chem Toxicol 40(7):905–912.
Wolff, M. S., J. A. Britton, L. Boguski, S. Hochman, N. Maloney, N. Serra, Z. Liu, G. Berkowitz, et al. 2008. Environmental exposures and puberty in inner-city girls. Environ Res 107(3):393–400.
Women’s Health Initiative Steering Committee. 2004. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 291(14):1701–1712.
Wong, C. W., C. McNally, E. Nickbarg, B. S. Komm, and B. J. Cheskis. 2002. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci U S A 99(23):14783–14788.
Woodruff, T. J., A. R. Zota, and J. M. Schwartz. 2011. Environmental chemicals in pregnant women in the United States: NHANES 2003–2004. Environ Health Perspect 119(6): 878–885.
Writing Group for the Women’s Health Initiative Investigators. 2002. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288(3):321–333.
Wysocki, P. J., and B. Wierusz-Wysocka. 2010. Obesity, hyperinsulinemia and breast cancer: Novel targets and a novel role for metformin. Expert Rev Mol Diagn 10(4):509–519.
Xue, F., W. C. Willett, B. A. Rosner, S. E. Hankinson, and K. B. Michels. 2011. Cigarette smoking and the incidence of breast cancer. Arch Intern Med 171(2):125–133.
Yager, J. D., and N. E. Davidson. 2006. Estrogen carcinogenesis in breast cancer. N Engl J Med 354(3):270–282.
Ye, J., Z. Gao, J. Yin, and Q. He. 2007. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 293(4):E1118–E1128.
Yoo, C. B., and P. A. Jones. 2006. Epigenetic therapy of cancer: Past, present and future. Nat Rev Drug Discov 5(1):37–50.
Yu, H., and T. Rohan. 2000. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst 92(18):1472–1489.
Zakikhani, M., R. Dowling, I. G. Fantus, N. Sonenberg, and M. Pollak. 2006. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66(21):10269–10273.
Zakikhani, M., M. J. Blouin, E. Piura, and M. N. Pollak. 2010. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat 123(1):271–279.
Zhang, Y., M. Matheny, S. Zolotukhin, N. Tumer, and P. J. Scarpace. 2002. Regulation of adiponectin and leptin gene expression in white and brown adipose tissues: Influence of β3-adrenergic agonists, retinoic acid, leptin and fasting. Biochim Biophys Acta 1584(2–3):115–122.
Zhang, Y., A. Daquinag, D. O. Traktuev, F. Amaya-Manzanares, P. J. Simmons, K. L. March, R. Pasqualini, W. Arap, et al. 2009. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer Res 69(12):5259–5266.
This page intentionally left blank.












































