
What Would a Knowledge Network and New Taxonomy Look Like?
In the previous chapter, the Committee outlined the reasons it concluded that the time is right to develop a Knowledge Network of Disease and New Taxonomy. But what would these resources look like and what implications would they have for disease classification, basic research, clinical care, and the health-care system? This chapter describes the Committee’s vision of a comprehensive Knowledge Network of Disease and New Taxonomy that would unite the biomedical-research, public-health, and health-care-delivery communities around the related goals of advancing our understanding of disease pathogenesis and improving health. The Committee envisions that the proposed resources would have several key features:
- They would drive development of a disease taxonomy that describes and defines diseases based on their intrinsic biology in addition to traditional physical “signs and symptoms”.
- They would go beyond description and be directly linked to a deeper understanding of disease mechanisms, pathogenesis, and treatments.
- They would be highly dynamic, continuously incorporating newly emerging disease information.
- They would be based on an Information Commons that draws upon as much disease-related information, from as large a number of individual patients, as possible.
- Much of the data that would populate the Information Commons would be generated during the ordinary course of clinical care.
Physical signs and symptoms are the overt manifestations of disease observed by physicians and patients. However, symptoms are not the best descriptors of disease. Symptoms are often non-specific and rarely identify a disease unambiguously. Physical signs and symptoms are generally also difficult to measure quantitatively. Furthermore, numerous diseases—including some of the most common ones such as cancer, cardiovascular disease, and HIV infection—are asymptomatic in early stages. Indeed, in a strict sense, all diseases are presumably asymptomatic for some “latent period” following the initiation of pathological processes. As a consequence, diagnosis based on traditional “signs and symptoms” alone carries the risk of missing opportunities for prevention, or early intervention can readily misdiagnose patients altogether. Even when histological analysis is performed, typically on tissue obtained after diseases become clinically evident, obtaining optimal diagnostic results can depend on supplementing standard histology with ancillary genetic or immunohistochemical testing to identify specific mutations or marker proteins.
Biology-based indicators of disease such as genetic mutations, marker-protein molecules, and other metabolites have the potential to be precise descriptors of disease. They can be measured accurately and precisely—be it in the form of a standardized biochemical assay or a genetic sequence—thus enabling comparison across datasets obtained from independent studies. Particularly when multiple molecular indicators are used in combination with conventional clinical, histological, and laboratory findings, they offer the opportunity for a more accurate and precise description and classification of disease.
Numerous molecularly-based disease markers are already available, and the number will grow rapidly in the future. Among the most prominent parameters of disease are an individual’s:
- Genome
- Transcriptome
- Proteome
- Metabolome
- Lipidome
- Epigenome
As discussed in Chapter 2, it is increasingly feasible to obtain substantial information about these biological features for each individual patient. The cost of sequencing an individual’s genome is rapidly dropping, and significant advances in the ability to globally and affordably characterize proteomes, me-
tabolomes, lipidomes, epigenomes, and microbiomes of individual subjects will continue, creating the potential for an increasingly rich molecular characterization of individuals in the future. Eventually, it is likely that extensive molecular characterization of individuals will occur routinely as a normal part of health care—even prior to appearance of disease, thereby allowing the collection of data on both sick and healthy individuals on a scale vastly exceeding current practice. In addition to providing a new resource for research on disease processes, these data would provide a far more flexible and useful definition of the “normal” state, in all its diversity, than now exists. The ability to make such measurements on both non-affected tissues and in sites altered by disease would allow monitoring of the development and natural history of many disorders about which even the most basic information is presently unavailable.
It is well recognized that health outcomes, disease phenotypes, and treatment response are determined by the individual and combined effects of various factors ranging from the molecular to the environmental (Collins 2004; IOM 2006; HealthyPeople.gov 2011). Gene-environment interactions have been implicated in a diverse group of diseases and pathological processes, including some psychological illnesses (Caspi et al. 2010), hypertension (Franks et al. 2004), tumor growth (J.B. Williams et al. 2009), HIV (Nunez et al. 2010), asthma (Chen et al. 2009), and cardiovascular reactivity (Williams et al. 2001; Snieder et al. 2002). Furthermore, the fact that numerous genome-wide association studies (GWASs) have revealed rather modest, albeit highly statistically significant, hazard ratios of disease risk highlights the need to investigate interactions among genetic and non-genetic factors to identify specific disease risk factors not found in conventional GWAS studies (Khoury and Wacholder 2009; Murcray et al. 2009; Cornelis et al 2010 ). Therefore, data added to the Information Commons should not be limited to molecular parameters as they are currently understood: patient-related data on environmental, behavioral, and socioeconomic factors will need to be considered as well in a thorough description of disease features1 (see Box 3-1).
Despite the focus on the individual patient in the creation of the Information Commons, the Committee expects that the inclusion of patients from diverse populations coupled with the incorporation of various types of infor-
_____________
1 As with all patient-related data in electronic medical records and contributed to the Information Commons, information in the exposome layer requires that attention be paid to data sharing, informed consent, and privacy issues; see discussion Chapter 4.
The exposome is a characterization of both exogenous and endogenous exposures that can have differential effects on disease predisposition at various stages during a person’s lifetime (Wild 2005; Rappaport 2011). The emerging science of exposomics is concerned with the application of innovative approaches to comprehensively measure a person’s exposure events, from conception to death, and determine how those exposures relate to health and disease (CDC 2010; NAS 2010; Rappaport 2011). A long-range goal is to ascertain the combined effects of these exposures by assessing the biomarkers and diseases they influence.
In its broadest definition, the exposome encompasses all exposures—internal (such as the microbiome, described elsewhere in this report) and external—across the lifespan. Physical environment (e.g., occupational hazards, exposure to industrial and household pollutants, water quality, climate, altitude, air pollution, and living conditions (Smith et al. 2008; Klecka et al. 2010; Alexeeff et al. 2011; Brookhart et al. 2011; Cutts et al. 2011; Yorifuji et al. 2011; Zanobetti et al. 2011; McMichael and Lindgren 2011) and lifestyle and behavior (e.g., diet, physical activity, cultural practices, and use of addictive substances [DHHS 2010; Hu and Malik 2010; Arem et al. 2011]), are some of the more apparent exogenous exposures. However, the concept of the exposome extends beyond these factors to include social factors, such as socioeconomic status, quality of housing, neighborhood, social relationships, access to services, and experience of discrimination that can contribute to psychological stress, poor health, and health inequities (Epel et al. 2004; Krieger et al. 2005; IOM 2006; Cole et al. 2007; Unnatural Causes 2008; Bruce et al. 2009; Gravlee 2009; Williams and Mohammed 2009; Cardarelli et al. 2010; Kim et al. 2010; Pollack et al. 2010; CDC 2011; Karelina and DeVries 2011; Sternthal et al. 2011; WHO 2011).
Despite the many practical and methodological challenges in characterizing and measuring these variables, rigorous evaluation of human exposures is needed. By incorporating data derived from multi-level assessments, a Knowledge Network of Disease could lead to better understanding of the variables and mechanisms underlying disease and health disparities, thereby helping to reveal a truer picture of the ecology of human health and facilitating a more holistic approach to health promotion and disease prevention.
mation contained in the exposome will result in a Knowledge Network that could also inform the identification of population-level interventions and the improvement of population health. For example, a better understanding of the impact of a sedentary lifestyle at the molecular level could conceivably facilitate the development of new approaches to physical education in early childhood. In addition, findings from the Knowledge Network and the New Taxonomy could reveal yet unidentified behavioral, social, and environmental factors that
are associated with particular diseases or subclassifications of diseases in certain populations and are amenable to public health interventions.
The Healthy People 2020 Initiative (Healthy People.gov. 2011) emphasizes an ecological approach to disease prevention and health promotion that focuses on both individual-level and population-level determinants of health and interventions. While molecular variables are often more easily measured and more directly tied to disease outcomes, if the modifiable factors that have contributed to the signature are known, we will be better able to prevent disease and to phenotype, genotype, and treat patients.
Asthma illustrates the interplay of social, behavioral, environmental, and genetic factors in disease classification. It is estimated that various types of asthma affect more than 300 million people worldwide. The term “asthma” is now used to refer to a set of “signs and symptoms” including reversible airway narrowing (“wheezing”), airway inflammation and remodeling, and airway hyper-reactivity. These various signs and symptoms likely reflect distinct etiologies in different patients. Many subjects with asthma have an allergic component, while in other cases, no clear allergic contributor can be defined (Hill et al. 2011; Lee et al. 2011). In some patients, asthma attacks are precipitated by exercise or aspirin (Cheong et al. 2011). Some patients, particularly those with severe asthma, may be resistant to treatment with corticosteroids (Searing et al. 2010). This phenomenological approach to asthma diagnosis has led to a plethora of asthma subtypes such as “allergic asthma,” “exercise-induced asthma,” and “steroid-resistant asthma” that may be clinically useful but provide little insight into underlying etiologies.
Over the years, linkage-analysis, candidate-gene, and genome-wide-association approaches have been applied to the study of the genetic underpinnings of asthma, leading to the identification of several associated genes and subphenotypes (Lee et al. 2011). However, these findings still leave most of the genetic influences of asthma unexplained (Li et al. 2010; Moffatt et al. 2010). Moreover, pediatric asthma research, in particular, has focused on a broad range of social and environmental, as well as genetic, contributors to the increased prevalence and severity of illness (Hill et al. 2011). Since the burden of asthma disproportionately affects children living in socioeconomically disadvantaged neighborhoods (D.R. Williams et al. 2009; Quinn et al. 2010), asthma may prove useful as a model for testing the Knowledge Network’s value in attaining a broader and deeper understanding of disease and health, in both the clinical and public-health policy domains. A knowledge-network-derived-taxonomy based on the biology of disease may help to divide patients with asthma—as well as many other diseases—into subtypes in which the different etiologies of the disorder can be better understood, and for which appropriate, subtype-specific approaches to treatment and prevention can be devised and tested.
Particularly because of advances in genomics, the proposed Knowledge Network of Disease has unprecedented potential to incorporate information about disease-causing and disease-associated microbial agents. Thousands of microbial genomes have been sequenced, providing a wealth of data on pathogenic and non-pathogenic organisms, and there has been an associated renaissance in studies of the molecular mechanisms of host-pathogen interactions. In parallel with these advances in microbiology, the analysis of human-genome sequences is enhancing the understanding of host responses and variation in individual susceptibility to microbial pathogens and infectious diseases. Today, sequence data, combined with other biochemical and microbiological information, are being used to understand microbial contribution to health, improve detection of pathogens, diagnose infectious diseases, and identify potential new targets for novel drugs and vaccines. In addition, comparing the sequences of different strains, species, and clinical isolates is crucial for identifying genetic polymorphisms that correlate with phenotypes such as drug resistance, morbidity, and infectivity. Combining this information with the molecular signature of the host will provide a more complete picture of an individual’s diseases allowing custom-tailoring of therapeutic interventions.
THE PROPOSED KNOWLEDGE NETWORK OF DISEASE WOULD GO BEYOND DESCRIPTION
A Knowledge Network of Disease would aspire to go far beyond disease description. It would seek to provide a unifying framework within which basic biology, clinical research, and patient care could co-evolve. The scope of the Knowledge Network’s influence would encompass:
Disease classification. The use of multiple molecular-based parameters to characterize disease may lead to more accurate and finer-grained classification of disease (see Box 3-2). Disease classification is not merely an academic exercise: more nuanced diagnostic accuracy and ability to recognize disease subtypes would undoubtedly have important therapeutic consequences, allowing treatment regimes to be customized based on the precise molecular features of a patient’s disease.
Disease-mechanism discovery. A Knowledge Network in which diseases are increasingly understood and defined in terms of molecular pathways has the potential to accelerate discovery of underlying disease mechanisms. In a molecularly-based Knowledge Network, a researcher could readily compare the
Box 3-2
Distinguishing Disease Types
Recent progress in the classification of lymphomas illustrates how a Knowledge Network could help distinguish diseases or disease states with similar symptoms and clinical presentations. Gene-expression profiling led to the discovery that B-cell lymphomas comprise two distinct subtypes of disease with different driver mutations and different prognoses (Alizadeh et al. 2000; Sweetenham 2011). One subtype bears a gene-expression profile similar to germinal center B-cells and has a good prognosis, while a second subtype bears a gene-expression profile similar to activated B-cells and has a poor prognosis. Recognition of these biological and clinical differences between subtypes of B-cell lymphomas makes it possible to predict patient prognosis more accurately and guide treatment decisions.
Similarly, leukemias are also now categorized based on differences in driver mutations, revealing subtypes with different prognoses and responses to particular treatment approaches. Acute myeloid leukemias with FLT3/ITD mutations have a poorer prognosis than acute myeloid leukemias with a normal FLT3 gene (Kiyoi et al. 1999; Kottaridis et al. 2001). As a consequence, patients bearing FLT3/ITD mutations are more likely to receive allogenic bone-marrow transplants or be offered experimental therapy with FTLs kinase inhibitors, while patients who do not have FLT3/ITD mutations are more likely to be treated only with chemotherapy. These are two of many known examples in which molecular data have been used to distinguish subtypes of malignancies with different prognoses and that benefit from different treatments. The proposed Knowledge Network of Disease could be expected to lead to many more insights of this type. By allowing any researcher to carry out analyses of this type on large numbers of patients, tracked over long periods of time, it is likely that insights such as the clinical relevance of FLT3 mutations in leukemia could be achieved for many other cancers and in situations where tumor behavior depends on a more complex interplay of influences.
molecular fingerprint (such as one defined by the transcriptome or proteome) of a disease with an unknown pathogenic mechanism to the information available for better understood diseases. Similarities between the molecular profiles of diseases with known and unknown pathogenic mechanisms might point directly to shared disease mechanisms, or at least serve as a starting point for directed molecular interrogation of cellular pathways likely to be involved in the pathogenesis of both diseases.
Disease detection and diagnosis. A Knowledge Network that integrates data from many different levels of disease determinants collected from individual subjects over time may reveal new opportunities for detection and early diagnosis. The availability of information on a multitude of diverse diseases should facilitate epidemiological research to identify novel diagnostic markers based
on correlations among diverse datasets (including clinical, social, economic, environmental, and lifestyle factors) and disease incidence, treatment decisions, and outcomes. In some instances, these advances would follow from the new insights into pathogenic mechanisms discussed above. The most robust early-detection tests—for example, assessment of an asymptomatic patient’s HIV status—are based on a clear understanding of pathogenic mechanism. In other cases, however, molecular profiles may prove sufficiently predictive of a patient’s future health to have substantial clinical utility long before the mechanistic rationale of the correlation is understood.
Disease predisposition. A Knowledge Network of Disease that links information from many levels of disease determinants, from genetic to environment and lifestyle, will improve our ability to predict and survey for diseases. Following outcomes in individual patients over time will allow the prognostic value of molecular-based classifications to be tested and, ideally, verified. Multi-parameter data across the entire spectrum of disease will become available. Obviously, the clinical utility of identifying disease predispositions depends on the availability of interventions that would either prevent or delay onset of disease or perhaps ameliorate disease severity.
Disease treatment. The ultimate goal of most clinical research is to improve disease treatments and health outcomes. There are many ways in which a Knowledge Network of Disease and its derived taxonomy may be expected to impact disease treatment and to contribute to improved health outcomes for patients. Accurate diagnosis is the foundation of all medical interventions. As many of the examples already discussed illustrate, finer-grained diagnoses often are the key to choosing optimal treatments. In some instances, a molecularly informed disease classification offers improved options for disease prevention or management even when different disease subtypes are treated identically (see Box 3-3). A Knowledge Network that integrates data from multiple levels of disease determinants will also facilitate the development of new therapies by identifying new therapeutic targets and may suggest off-label use of existing drugs. In other cases, the identification of links between environmental factors or lifestyle choices and disease incidence may make it possible to reduce disease incidence by lifestyle interventions.
Importantly, as discussed below, the Committee believes the Knowledge Network and its underlying Information Commons would enable the discovery of improved treatments by providing a powerful new research resource that would bring together researchers with diverse skills and integrate knowledge about disease processes in an unprecedented way. Indeed, it is quite possible that the transition to a modernized “discovery model” in which disease data generated during the course of normal health care and analyzed by a diverse set
Box 3-3
Information to Guide Treatment Decisions
The example of a patient such as Patient 1 with breast cancer, described in the Introduction, illustrates the potential of a Knowledge Network of Disease to provide patients with valuable information even when there is no difference in treatment for different diseases subtypes (e.g., sporadic vs. BRCA1/2-associated breast cancer). While mutations in the tumor-suppressor genes BRCA1 and BRCA2 strongly predispose women to breast and ovarian cancer, the extent to which particular germline mutations in these genes increase cancer risk often remains uncertain (Gayther et al. 1995). Consequently, patients and physicians must currently make decisions about whether to undertake more intensive cancer surveillance (for example, by breast magnetic resonance imaging or vaginal ultrasound) without being able clearly to assess the risks and benefits of such increased screening and the anxiety and potential morbidity that arises from inevitable false positives. Furthermore, some patients elect to undergo prophylactic mastectomies or oophorectomies without definitive information about the extent to which these drastic procedures actually would reduce their cancer risk.
Studies attempting to quantify these risks have largely focused on particular ethnic groups in which a limited set of mutations occur at high enough frequencies to allow reliable conclusions from analyses carried out on a practical scale. If BRCA1/2 genotypes and health histories could be compared across the large datasets currently segregated among different health-care organizations, it would become possible to assess accurately cancer risks for people with different mutations and genetic backgrounds. Such data would allow more rational recommendations regarding risk-reduction strategies, thereby creating enormous value for individual patients, health-care providers, and payers, by making it possible to avoid unnecessary screening and treatment while reducing cancer incidence and promoting early detection.
of researchers would ultimately prove to be a Knowledge Network of Disease’s greatest legacy for biomedical research.
Drug development. Molecular similarities among seemingly unrelated diseases would also be of direct relevance to drug discovery as it would lead to targeted investigation of disease-relevant pathways that are shared between molecularly related diseases. In addition, ongoing access to molecular profiles and health histories of large numbers of patients taking already-approved drugs would undoubtedly lead to improved drug safety by allowing identification of individuals at higher-than-normal risk of adverse drug reactions. Indeed, our limited understanding of—and lack of a robust system for studying—rare
adverse reactions is a major barrier to the introduction of new drugs in our increasingly risk-aversive and litigious society.
Health disparities. Major disparities in the health profiles of different “racial”, ethnic, and socio-economic groups within our diverse society have proven discouragingly refractory to amelioration. As discussed above, it is quite likely that key contributors to these disparities can be most effectively addressed through public-health measures and other public policies that have little to do with the molecular basis of disease, at least as we presently understand it. However, the Committee regards the Information Commons and Knowledge Network of Disease, as potentially powerful tools for understanding and addressing health disparities because they would be informed by data on the environmental and social factors that influence the health of individual patients. For the first time, these resources would bring together, in the same place, molecular profiles, health histories, and data on the many determinants of health and disease, thereby optimizing the ability to decipher the mechanisms through which exogenous factors give rise to endogenous, biological inputs, directly affecting health. Researchers and policy makers would then be better able to sort out the full diversity of possible reasons for observed individual and group differences in health and to devise effective strategies to prevent and combat them.
The establishment of a Knowledge Network, and its research and clinical applications, would depend on the availability of a hierarchy of large, well-integrated datasets describing what we know about human disease. These datasets would establish the foundation for the New Taxonomy and many other basic and applied activities throughout the health-care system. The Information Commons would contain the raw information about individual patients from which meaningful links and relationships could be derived. Recognizing that the Knowledge Network would need to be informed by vast amounts of information external to the network itself, the Committee envisions the need for substantial research in medical informatics directed at all steps of the creation and curation of the network, and, equally importantly, its use by individuals with diverse backgrounds and goals. The creation of the Knowledge Network and its underlying Information Commons would enable the continuous compilation and analysis of molecular, environmental, behavioral, social, and clinical data in a dynamic, shared platform. Such an information platform would need to be accessible by users across the entire spectrum of research and clinical care, including payers. Data would be continuously deposited by the research
community and extracted directly from the medical records of participating patients. The roles of the different datasets in this information resource are schematized in Figure 3-1.
The precise structures of both the Information Commons and Knowledge Network of Disease remain to be determined and would be informed by pilot studies, as discussed in Figure 1-2):
Multilayered. Given the inclusion of multiple parameters ranging from genomic to environmentally modulated disease factors, the Information Commons would likely have a multi-layered structure with each layer containing the information for one disease parameter, such as “signs and symptoms”, genetic mutations, epigenetic patterns, metabolic characteristics, or other risk factors (including social, behavioral, and environmental influences).
Individual-centric. The Information Commons should register all measurements with respect to individuals so that the multitude of influences on pathophysiological states can be viewed at scales that span all the way from the molecular to the social level. Only in this way could, for example, individual environmental exposures be matched to individual changes in molecular profiles. These data would need to be stored in an escrowed, encrypted depository that allows graded release of data depending on the questions asked, the access level of the individual making the inquiry, and other parameters that would undoubtedly emerge in the course of pilot studies. The Committee realizes that this is a radical approach and intense public education and outreach about the value of the Information Commons to the progress of medicine would be essential to harness informed volunteerism, the support of disease-specific advocacy groups, and the engagement of other stakeholders. The Committee regards careful handling of policies to ensure privacy as the central issue in its entire vision of the Information Commons, the Knowledge Network of Disease, and the New Taxonomy. Hence, this topic is discussed in more detail in Chapter 4.
The Knowledge Network of Disease, created by integrating data in the Information Commons with fundamental biological knowledge, drawn from the biomedical literature and existing community databases such as GenBank, would be the centerpiece of the informational resources underlying the New Taxonomy. The Committee envisions this network as:
Highly inter-connected. In order to extract relationship information between multiple parameters—for example, the transciptome and the exposome—the multiple data layers must be inter-connected (see Figure 3-1). Ideally, each information layer would be connected to every other layer: thus, “signs and symptoms” would be linked to mutations, mutations to metabolic defects, exposome
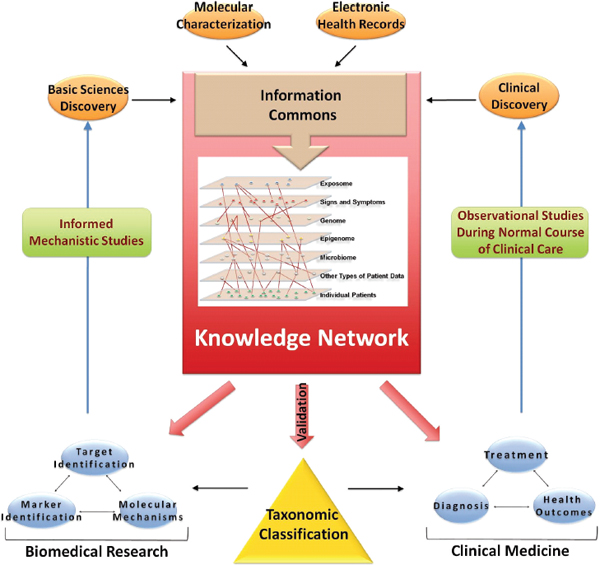
FIGURE 3-1 Building a biomedical Knowledge Network for basic discovery and Medicine.
At the center of a comprehensive biomedical information network is an Information Commons that contains current disease information linked to individual patients and is continuously updated by a wide set of new data emerging though observational studies during the course of normal health care. The data in the Information Commons and Knowledge Network serve three purposes: (1) they provide the basis to generate a dynamic, adaptive system that informs taxonomic classification of disease; (2) they provide the foundation for novel clinical approaches (diagnostics, treatments, strategies), and (3) they provide a resource for basic discovery. Validated findings that emerge from the Knowledge Network, such as those which define new diseases or subtypes of diseases that are clinically relevant (e.g., which have implications for patient prognosis or therapy) would be incorporated into the New Taxonomy to improve diagnosis (i.e., disease classification) and treatment. The fine-grained nature of the taxonomic classification would aid in clinical decision-making by more accurately defining disease.
SOURCE: Committee on A Framework for Developing a New Taxonomy of Disease.
to the epigenome, and so forth. The links could be one-to-one but most commonly would be many-to-one, and one-to-many (e.g., particular signs and symptoms arise when other parameters fall into many otherwise unrelated clusters). The interrelationships among such features within and between the layers could be characterized through a variety of representations that attempt to extract meaning from the Information Commons. For example, distinct transcriptomes may define several types of B-cell lymphomas. Meanwhile, different types of lymphomas, defined by transcriptome analysis, may have distinct metabolomic profiles. The similarities of multiple diseases could be discerned either from relationships among the networks of individual parameters (e.g., transcriptomes of multiple B-cell lymphomas) or from common patterns that emerge once multiple parameters are combined.
Flexible. A highly inter-connected Knowledge Network would link multiple individual networks of parameters in a flexible way. A user could chose to interrogate only a small part of the network by limiting his or her analysis to a single information layer, or even a small portion of this layer; alternatively, a user could interrogate the complex interrelationship of multiple parameters. High flexibility ensures easy cross-comparison and cross-correlation of any desired dataset, making it a versatile tool for a wide spectrum of applications ranging from basic research to clinical studies and healthy system administration.
Widely accessible. The Knowledge Network would need to be accessible and usable by a wide range of stakeholders from basic scientists to clinicians, health-care workers, and the public. Furthermore, the available information would need to be mineable in ways that are custom-tailored to the needs of different users, possibly by implementation of purpose-specific user interfaces.
While the Committee agreed upon the generalities listed above and illustrated in Figure 3-1 about the Information Commons and Knowledge Network—and their relationship to a New Taxonomy— specifics of implementation such as the detailed design of the Information Commons, the information technology platforms used to create it, questions about where key infrastructure should be physically housed, who would oversee it, and how the Information Commons would be financed, were considered beyond the scope of the Committee’s charge in a framework study. Nonetheless, dramatic developments in the fields of medical information technology—and other developments discussed in Chapter 2—give the Committee confidence that the creation and implementation of this ambitious and novel infrastructure is a feasible goal.
Immense progress has been made during the past 25 years in organizing our knowledge of basic biology, health, and disease, even as many components of this knowledge base have grown super-exponentially. The National Library of Medicine and its National Center for Biotechnology Information division (NCBI), created in 1988, maintains the closest current counterpart to the information infrastructure that the Committee envisions. The NCBI maintains a vast array of information about basic biology, health, and disease—ranging from the PubMed system for indexing the biomedical literature to GenBank, the primary depository for DNA-sequence data—and its databases are queried daily by nearly anyone involved in biomedical research. So, what is the difference between the Committee’s vision of the Information Commons and Knowledge Network of Disease and reasonable extrapolations of what the NCBI has already accomplished?
The key difference is that the Information Commons, which would underlie the other databases, would be “individual-centric.” The various databanks curated by NCBI generally only contain a single disease parameter and even if multiple pieces of information from an individual make it into multiple databanks—say a breast cancer patient’s transcriptome stored in the GeneOmnibus database of published microarray data and information about her chromosome translocations in the Cancer Chromosome databank—they are not linked between databases. An independent researcher, who was not involved in the study that contributed these entries, has no way of knowing that they are from the same individual. As a consequence, relationships between multiple parameters that determine disease status in a given individual are impossible to extract. However, motivated by the recent proliferation of GWAS studies, NCBI has developed an individual-centric database, dbGap (the database of Genotypes and Phenotypes). This database was “developed to archive and distribute the results of studies that have investigated the interaction of genotype and phenotype” (NCBI 2011b). The Committee considers NCBI’s success in doing so—despite severe current constraints on the sharing of phenotypic information about individuals—as evidence that the obstacles to creating an Information Commons can be overcome. This issue is discussed in more detail in Chapter 4. However, the important point is that little of the NCBI’s vast current store of information could, even in principle, be organized along the lines suggested for the Information Commons. This information was not collected in a way that allows the individual to be the central organizing principle, and no amount of redesign of the inter-connections between different entries in the current system could achieve the goals the Committee has outlined.
The Committee would like to emphasize the novelty and power of an
Information Commons that is “individual-centric.” As discussed in Figure 1-2). Following public access to the Global Positioning System (GPS) and dramatic improvements in database technology in many ways analogous to the driving forces current advances in data generation and handling in biomedicine, it became apparent to many users of geographically indexed information that a surprisingly high portion of the world’s information could be organized around GPS coordinates. Like the proposed Information Commons, GISs are layered data structures that inter-connect vast amounts of information and can be mined for information that is not readily apparent in the primary GPS of an object. For example, given the coordinates of a large number of, say, backyard barbecue grills, one can suddenly overlay a vast amount of socio-economic, ethnic, climatological, and other data on what—at the start of the investigation—appeared a peculiar, anecdotal inquiry. In some respects, this approach is counter-intuitive. The GPS coordinates of someone’s backyard barbecue grill may appear to take one away from useful generalizations about grills: it reveals more detail than one might want to know about an individual grill without laying any obvious foundations for developing an integrated perspective on the cultural practice of backyard-barbecuing. However, it is the precise GPS coordinates of an individual grill that are the key to inter-connecting whatever has been learned about this particular grill to a larger world of information.
Despite significant challenges to constructing an individual-centric Information Commons, the Committee concluded that this is a realistic undertaking and would be essential to the success of the Knowledge-Network/New Taxonomy initiative. The Committee is of the opinion that “precision medicine,” designed to provide the best accessible care for each individual, is not achievable without a massive reorientation of the information systems on which researchers and health-care providers depend: these systems, like the medicine they aspire to support, must be individualized. Generalizations must be built up from information on large numbers of individuals. Efforts to reverse this process will fail since indispensable information is lost when molecular profiles, data on other aspects of an individual’s circumstances, and health histories are abstracted away from the individual at the very beginning of investigations into the determinants of health and disease.
A KNOWLEDGE NETWORK OF DISEASE WOULD CONTINUOUSLY EVOLVE
Although knowledge of disease, and particularly molecular mechanisms of pathogenesis, is still limited, the pace of progress has never been greater. New insights into the biology of disease are emerging rapidly from a wealth of molecular approaches, as well as from new insights into the importance
of environmental factors. However, the system for updating current disease taxonomies, at intervals of many years does not permit the rapid incorporation of new information, thereby contributing to the delayed introduction of advances that have the potential, over time, to guide mainstream practice. The individual-centric nature of an Information Commons is an important means of ensuring that the data underlying the Knowledge Network, and its derived taxonomy, would be constantly updated. As participating patients undergo new tests and treatments, associated information would enter the Information Commons and, on the basis of these data, the taxonomies, such as the ICD, could be updated continuously. Such a dynamic system would not only accept new inputs for established disease parameters, it would also accommodate new types of information generated by newly developed technologies to identify, acquire, measure, and analyze new biological features of disease.
THE NEW TAXONOMY WOULD REQUIRE CONTINUOUS VALIDATION
Bad information is worse than no information. A key feature of a clinically useful taxonomy is the requirement for a validation system. The logic of the classification scheme, and especially its utility for practical applications, needs to be carefully and continuously tested. This is particularly important when patients and clinicians use the New Taxonomy to inform clinical decisions. The New Taxonomy should be routinely tested to provide all stakeholders with data indicating the extent to which decisions guided by it can be made with confidence. Clearly, some patients and clinicians will be more comfortable than others with making decisions that are based on clinical intuition rather than proven evidence. However, a physician should be able to interrogate the Knowledge Network that underlies the New Taxonomy to learn whether others have had to make a similar decision, and, if so, what the consequences were. For example, if a drug has been introduced to target a particular driver mutation in a cancer, a physician needs to know whether or not rigorous clinical testing has determined that the drug is safe and effective. Is the drug effective only in some patients who can be identified in some way, such as by analyzing variants of genes that affect cell growth or drug metabolism? Similarly, if a laboratory test is considered to be a candidate predictor for the later development of disease, has that hypothesis been rigorously validated? Is the candidate test valid in some patient groups but not others? Whether a given test is used to identify predictors of disease or the existence of disease, the test result must be interpreted in the context of knowledge about the “normal range” of results. This requirement is not a trivial consideration, especially for tests based on integration of vast amounts of data, such as the genome, transcriptome, and metabolome of the patient. Even with a conventional sequencing test, it is often difficult to ascertain with certainty whether a sequence change is disease-causing or insig-
nificant. This dilemma is multiplied many times over for genome-level testing. Some initial results from whole-human-genome-sequencing data indicate the scale of this problem: most individuals have dozens to hundreds of sequence variants that are readily recognizable, on biochemical grounds, as potentially pathogenic: examples include variants that cause premature-protein truncation or loss of normal stop codons (Ge et al. 2009; Pelak et al. 2010)—yet the clinical significance of nearly all such variants remains obscure. Defining and continuously refining our understanding of the normal “reference range” for such tests would require being able to access and effectively analyze biological and other relevant clinical data derived from large and ethnically diverse populations. Ultimately, the Knowledge Network that underlies the New Taxonomy will make it possible to develop decision-support tools that synthesize information and alert health-care providers to all validated insights that emerge from the Knowledge Network and that are relevant to clinical decisions under consideration.
THE NEW TAXONOMY WOULD DEVELOP IN PARALLEL WITH THE CONTINUED USE OF CURRENT TAXONOMIES
Existing disease taxonomies, such as ICD, clearly have utility and are likely to continue to be employed throughout the health-care system far into the future. The organizational and financial costs of systematically replacing these systems would be substantial. Moreover, as noted above, those responsible for revision of the ICD taxonomy are actively engaged in incorporating molecular characteristics of disease into that system. Hence, it is quite possible that the New Taxonomy could ultimately subsume the ICD system, with the latter comprising the most rigorously validated subset of disease classifications. Such issues must be addressed but, given the magnitude of the tasks associated with launching the creation of the Information Commons and the Knowledge Network of Disease, and seeing it through its formative stages, their consideration can safely be postponed for many years.
THE PROPOSED INFORMATIONAL INFRASTRUCTURE WOULD HAVE GLOBAL HEALTH IMPACT
A Knowledge Network of Disease should ultimately provide global benefits. Inevitably, the Knowledge Network initially would be devised primarily through data acquired, placed in the Information Commons, and analyzed by researchers and medical institutions in developed countries. However, a comprehensive and fully developed Knowledge Network of Disease must include the many diseases, including infectious diseases and disorders linked to geographically limited environmental exposures that are endemic in low- and middle-income settings throughout the world. Therefore, the Knowledge Net-
work effort should be extended to include analysis of data derived in these settings.
Improved precision in defining disease is of particular importance in regions of the world with under-developed health-care systems. Disease misdiagnosis in such settings has contributed to the improper use of therapy and the establishment of widespread drug resistance among disease-causing infectious agents. Malaria is one disease where misdiagnosis is common with dramatic consequences and costs (D’Acremont et al. 2009). In general, patients presenting with fever in regions where malaria is endemic are administered anti-malarial treatment without direct evidence that the patient actually has malaria. In part, this practice is due to limited resources—the state-of-the-art diagnostic test in most areas is a microscopy-based blood-smear diagnosis, which requires expert training. The lack of adequate point-of-care diagnostic tests to ascertain whether the patient has malaria represents a significant impediment to the selection of appropriate targeted therapy. As a consequence, major efforts are under way to develop molecular diagnostics for malaria and other major killers such as tubercuolosis (Boehme et al. 2010; Small and Pai 2010). Ultimately, such diagnostics will need to include tests that differentiate among various disease agents and also take into account genetic or molecular markers in the host that influence host responses to the infection or potential treatments. A globally relevant Information Commons and Knowledge Network could be useful in facilitating this process—for example, to distinguish between malaria caused by Plasmodium falciparum versus Plasmodium vivax, which are susceptible to different anti-malarial drugs (malERA Consultative Group on Diagnoses and Diagnostics 2011). The Knowledge Network and its associated taxonomy should not be designed exclusively to meet the needs of countries with advanced medical systems. Indeed, the individual-centric character of the Information Commons—and the inclusion of available data about contributing individuals, including information about where and in what circumstances they live—offers an unprecedented path toward a Knowledge Network of Disease that can meet global needs for health care and disease prevention.


















