
2
Evaluation of Current Animal Models
Session chair Stevin Zorn, executive vice president of neuroscience research at Lundbeck Research USA, opened the session with the following questions: What leads to poor translation of animal models of nervous system disorders to clinical practice? Is it the models themselves, researcher expectations of models, how models are used to make predictions and decisions, or perhaps the level of knowledge about underlying pathophysiology for any given disease? Finally, Zorn asked, what is the impact of generalizing animal model capabilities, of poor study design, or of publication bias?
To set the stage for discussion, Steven Paul provided background on the challenges of drug discovery for nervous system disorders and why animal models are useful. Mark Tricklebank followed with a discussion about validation of animal models for drug discovery and how translation of preclinical research can be enhanced through skillful study design, planning, and proper statistical analysis; these points were echoed by other speakers and many participants throughout the workshop. Next, Katrina Kelner described three forms of publication bias that can impact the success of animal models: (1) the tendency to publish positive findings; (2) the publication of poorly designed and executed animal studies that could contribute to incorrect conclusions; (3) and assumptions about what constitutes “good science.”
EXPECTATIONS FOR ANIMAL MODELS IN DRUG DEVELOPMENT
The past decade has been challenging for the biopharmaceutical industry. These challenges include the development and regulatory approval of innovative new medicines, said Steven Paul, director of the Helen and Robert Appel Alzheimer’s Disease Research Institute at Weill Cornell Medical College and former president of Lilly Research Laboratories. It has been a particularly difficult time for neuroscience research and development. There have been therapeutic successes in select areas, for example, multiple sclerosis and epilepsy. However, in other areas of research, such as schizophrenia and depression, new drugs are not significantly better than those developed 50 years ago. For the most part, there are still no disease-modifying drugs for diseases like Alzheimer’s and Parkinson’s. Many pharmaceutical companies are restructuring and/or deemphasizing research on nervous system diseases and disorders, Paul noted, while some have completely left the field.
A major problem in bringing a new therapy to market is attrition, Paul said. When a new drug target is discovered and validated, lead candidate molecules are then identified and optimized. Preclinical testing evaluates the pharmacology and toxicology of the lead compound in animal models and Phase I clinical studies establish safety and dosing in humans. The problem of attrition occurs in Phases II and III, during the testing of whether the drug “works” as a treatment for the particular disease.
A 2010 study by Paul and colleagues found that roughly 66 percent of compounds that entered Phase II development did not advance to Phase III and about 30 percent of those that did enter Phase III failed to move on to regulatory submission (Paul et al., 2010). A more recent analysis suggests that, from 2008 through 2010, 82 percent of compounds failed to advance from Phase II clinical trials, with more than half failing due to issues of efficacy (Figure 2-1). Equally concerning, Paul said, is data from 2007 through 2010 show that 50 percent of compounds failed in Phase III, 66 percent of which were due to efficacy concerns (Arrowsmith, 2011b). By Phase III, Paul suggested, there should be confidence in efficacy and compounds should really only fail infrequently or due to rare adverse events. The approval rate for new drug applications (NDAs) by the U.S. Food and Drug Administration (FDA) is currently about 70 percent. In other words, 30 percent of new drugs that make it through Phase III to regulatory filing will not gain regulatory approval.
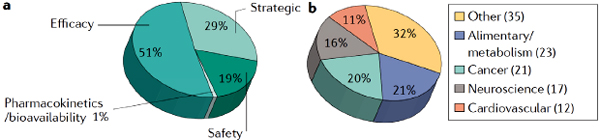
FIGURE 2-1 Failure of drugs in Phase II according to reason for failure (a) and therapeutic area (b).
SOURCE: Paul and Tricklebank presentations, March 28, 2012, from Arrowsmith, 2011a.
Animal models have long had an important role in the drug development process. As outlined by Paul, current expectations are that animal models can help researchers to
• Better understand the fundamental pathology and pathogenesis of a disease. An example is how genes and mutations result in observed disease phenotypes. This knowledge of underlying disease biology can aid in selecting and validating drug targets and defining pathways of intervention;
• Test drugs and treatments that could be effective for a specific disease or a particular symptom of a complex disease;
• Ascertain the safety of a drug or treatment, for example, toxicity or adverse events; and
• Establish the therapeutic index, or dose, that produces the desired effect compared to the dose that is toxic or lethal, for a given drug or treatment prior to testing in humans.
Paul concluded that although, animal models cannot solve all of the challenges of drug development, they are an important part of the solution. In particular, improvements in the ability of animal models to predict efficacy can help to reduce issues of attrition.
CHOICE AND VALIDATION OF ANIMAL MODELS FOR CENTRAL NERVOUS SYSTEM DRUG DISCOVERY
Mark Tricklebank, director of the Lilly Center for Cognitive Neuroscience at Eli Lilly and Company in the United Kingdom, said there are an unprecedented number of potential molecular targets in the nervous system, but there is little direct, clinically-based evidence to support a rational method for choosing one target over another. Selection of targets at the start of a drug discovery program would benefit from a solid biological hypothesis. This might be accomplished through manipulation of the target in vitro and development of a strong hypothesis of the expected in vivo results (Sarter and Tricklebank, 2012). Once a therapeutic target is selected, advances in in vitro screening technologies have made identification of potential drug molecules relatively easy. Tricklebank concurred with Paul that the system breaks down at the target validation stage, with too many false positives or false negatives.
Working Backward from Failure in the Clinic
Investigational new therapeutics are dismissed on the basis of the clinical result, most commonly, for lack of efficacy. However, Tricklebank cited a recent analysis suggesting that in many cases where the conclusion is lack of efficacy, the fact is that the hypothesis was not tested adequately and the results were not definitive (Morgan et al., 2012).
The way in which animal models are used may also contribute to failure in clinical trials, Tricklebank offered several possible reasons:
• Preclinical evidence supporting the hypothesis is given unrealistic weight in comparison to evidence against the hypothesis. Pressure on researchers to focus on positive results and ignore information that might need to be addressed and understood before deciding to move forward.
• Insufficient attention is paid to the design and analysis of experiments so that false positives or false negatives incorrectly influence decisions.
• Preclinical data collected to support the hypothesis are irrelevant to the mechanisms underlying the disease of interest. For example, not much is known about the pathophysiology of psychiatric diseases, and therefore it is difficult to make rational drug target choices.
• The preclinical data accurately conveys the drug responses in a very specific, genetically circumscribed population of animals maintained in highly controlled environments, but this does not necessarily hold true in the heterogeneous human clinical population.
• The measurements taken have, at best, only face validity for the variables that the experimenter would like to measure, and they need to be quantified in relation to the disease of interest. There is a need to understand in greater detail what researchers want to investigate, Tricklebank said.
• The measured effects are confounded by competing responses.
• There is a species gap, so the systems being manipulated in experimental animals might never fully predict outcomes in humans.
Expanding on the issue of experimental design and analysis, Tricklebank cited a study of 513 publications in top neuroscience journals that looked for appropriateness of statistical analysis (Nieuwenhuis et al., 2011). The study showed that 79 of these publications used an incorrect statistical approach. In a number of cases, the signal was so large that this error was irrelevant, but in two-thirds of the cases, this incorrect statistical analysis actually influenced the interpretation of the experimental results, Tricklebank explained.
Another issue is validation of the assay and model. Tricklebank outlined six types of validity to be considered in defining animal models (Box 2-1; see also Markou et al., 2009).1 There is some confusion, he noted, about what an “assay” is and what a “model” is. An assay is a means of quantifying a dependent variable. A model is a theoretical description of the way a system, process, or disease works. An animal
___________________________
1The types of validity described in Box 2-1 can be generalized under external validity or the “extent to which the results of an animal experiment provide a correct basis of generalizations to the human condition.” Not included in this list, but discussed throughout the workshop by participants, is the concept of internal validity or the “extent to which the design and conduct of the trial eliminate the possibility of bias” (van der Worp et al., 2010).
BOX 2-1
Types of Validity Relative to Neuroscience Animal Models
Construct validity: Ideally mimicking the molecular and/or structural basis of the disease
Convergent validity: Evidenced by high correlation among performance patterns across cognitive tasks designed to measure the same neurocognitive process
Criterion validity: The ability of performance in one task to predict performance on another, more ecologically valid test
Discriminant validity: Evidenced by low correlation among outcomes across tasks designed to measure distinct neurocognitive constructs
Face validity: Degree of similarity to disease-specific symptoms
Predictive validity: Based on currently available therapy
SOURCE: Tricklebank presentation (March 28, 2012).
model induces over- or under-expression of a biological variable which the assay quantifies.
Improving the Probability of Clinical Success Through Validation
Ensuring that preclinical models have some validity for the system of interest starts with a better definition of the animal behaviors to be measured across the domains of sensory-motor function, arousal, affect, motivation, cognition, and social processes. Assays to measure these aspects of animal behavior should be designed to be as similar as possible to the assays used in the clinical situation, Tricklebank said. Also, assays measuring these functions in the clinical study should be as close as possible to those used in animals. Focusing on measuring the right thing and measuring it accurately is important. This includes maximizing signal-to-noise, removing confounds, determining both intra- and inter-laboratory reproducibility, and testing compounds against the most appropriate baseline perturbation.
Consider the manipulation: Is that manipulation clinically relevant; is it engaging the circuitry that is thought to be dysfunctional in the clinical indication of interest? From a practical perspective, Tricklebank suggested that increasing the view of psychiatric disorders as aspects of disturbed brain circuitry will lead to more rational profiling of compounds. Validation of potential compounds delivered via local injection, for example, would require evidence of engagement of the neurocircuitry involved in the disease. Biomarkers can also serve as indicators of the circuitry involved in the measured behaviors. Improving animal models might occur through the use of clinically relevant pharmacological, environmental, neurodevelopmental, and genetic methods to perturb or impair normal function.
In discussion, Sharon Rosenzweig-Lipson of IVS Pharma Consulting added that in addition to establishing the validity of an animal model, it is important to understand how decisions are made based on the model. How much of failure to translate is attributable to novel animal models versus models that are well defined (e.g., standards of care, known mechanisms)?
Collaborative Partnerships and Cross-Disciplinary Research
The design and validation of preclinical assays and models for drug discovery is ideally served by precompetitive, collaborative approaches via industrial, academic, and clinical consortium, Tricklebank said.
The Lilly Center for Cognitive Neuroscience, he explained, has adopted a very detailed approach to profiling a molecule though assays, disease models, tools, targets, and biomarkers as the basis for Phase I clinical studies. To do this, Lilly is leveraging external capacity, capabilities, and innovation all along the development pathway through a consortium of academic and industry scientists.
On a much broader scale, the European Union (EU) through its Innovative Medicines Initiative (IMI) launched the Novel Methods leading to New Medications in Depression and Schizophrenia (NEWMEDS) project, an international consortium of academic and industry scientists.2 Tricklebank is involved in NEWMEDS Work Package 2 (WP2), which is focused on cognition assays and animal models. A key aspect of WP2 is multisite validation, establishing new assays in one laboratory and then running them in the partner laboratories to gauge reproducibility from
___________________________
2Discussed further by Steckler in Chapter 6.
methodological and pharmacological sensitivity perspectives. One example, Tricklebank mentioned, involves the development of a touchscreen-based translational assay of animal cognition and validation of the pharmacological sensitivity.3 Multiple laboratories within the consortium are using this touchscreen assay under standardized experimental conditions to compare the rate of task acquisition and response to drugs administered during task acquisition or posttraining.
Tricklebank noted that convergent approaches combining behavior and physiology are critical for the preclinical validation of drug targets. He concluded that cross-disciplinary approaches are essential for the preclinical validation of drug targets, and collaborative precompetitive approaches to verifying findings will pay off in the long run.
THE IMPACT OF PUBLICATION BIAS
The process of peer review and publication in established journals improves the quality of science and helps to filter out invalid or unimportant results. However, scientific literature is still subject to publication bias (Easterbrook et al., 1991; Sena et al., 2010; ter Riet et al., 2012). Katrina Kelner, editor of Science Translational Medicine, reviewed three types of publication bias.
Failure to Publish Negative Results
Sociological factors strongly influence what is published in the literature and what is not, Kelner said. A much larger fraction of papers that report positive findings are published than those reporting negative findings. The end result is that scientific literature can be distorted. Kelner illustrated this with a hypothetical scenario (see Box 2-2).
This problem has no easy solution. There have been several calls for the pharmaceutical industry to publish their preclinical data and results from failed clinical trials (Clozel, 2011; Rogawski and Federoff, 2011). Several new journals are dedicated to publishing negative results (e.g., Journal of Negative Results in Biomedicine, Journal of Negative Results—Ecology and Evolutionary Biology, Journal of Articles in Support of the Null Hypothesis). In addition, Kelner noted that PLoS ONE will publish studies with negative findings. To this point, one participant
___________________________
3Discussed further by Bussey in Chapter 4.
BOX 2-2
Hypothetical Publication Bias Scenario: Failure to Publish Negative Results
Twenty laboratories tested drug X and only one laboratory found that it lowers blood pressure and published their statistically significant results (p < 0.05). The rest of the laboratories did not observe any effect and did not publish their results. Because the p value was set at 0.05, there was a 5 percent chance of getting a positive result by chance alone. If researchers could look at the results of all 20 studies done all over the world together, they would have concluded that it is unlikely that drug X lowers blood pressure.
Therefore, based on the single positive result that was published, drug X appeared to have important clinical potential to control blood pressure. The results were recognized by peer/ service user reviewers and endorsed by publication in a top-tier journal. Shortly after, another laboratory that worked on drug X tried to replicate the published study but could not. Its work was published in a lower-tier journal and far fewer people saw it. Three other laboratories that had found negative results decided to publish their work, but no journal was interested.
SOURCE: Kelner presentation (March 28, 2012).
suggested that scientific quality and transparency would increase if there were a widespread effort by journal editors to publish a certain percentage of articles each year that contain negative results.
During the discussion, a participant suggested that top-tier journals establish sections for “replications and challenges” where one laboratory’s failure to replicate another’s work can reach the same level of attention as the original article. This could be a data-driven commentary on articles that have been published. Kelner supported the idea of a site that could publish negative results following review for rigorous experimental design and analysis. A participant from the National Institutes of Health (NIH) noted that the concept of a repository of negative data has been proposed many times. The NIH has, in essence, such a repository of data from its funded studies and perhaps that knowledge of what works and what does not could somehow be tapped to help address publication bias.
One participant noted that another pathway toward publication bias might arise if agencies disproportionately fund labs with publications of positive results in high-impact journals; this might lead to greater fund-
ing of fewer labs that are emphasizing singular approaches. A participant followed up that this could potentially lead to a push for postdoctoral fellows and graduate students to seek out training opportunities in highly recognized labs leading toward further scientific biases.
Poorly Designed and Executed Studies
Another type of publication bias is the publication of studies that are poorly designed, executed, and/or analyzed, in which the conclusions drawn are invalid or not meaningful.4 This results in proliferation of articles that are basically uninformative. Weeding these studies out of the submission pool can be difficult. Journals rely, by necessity, on the expertise of their peer reviewers and not all reviewers are aware of or appropriately trained in experimental design and data analysis. Kelner noted that medical journals have been ahead of preclinical journals in adopting specific, independent review criteria, for example, the Consolidated Standards of Reporting Trials or CONSORT guidelines (Schultz et al., 2010).
Journals can help address this, Kelner said, by enforcing quality standards for peer review and scientists can help journals by developing and disseminating relevant standards for their field. In addition, she said, funding agencies can require high-quality output from their funded scientists.
Cultural Assumptions About What Good Science Is
The third type of publication bias Kelner described results from cultural assumptions about what constitutes “good science.” A common assumption is that the best science increases fundamental knowledge, not practical application. This belief is part of the cultural fabric of the scientific community, including study sections, mentors, and some top-tier journals, she said.
___________________________
4The National Institute of Neurological Disorders and Stroke convened major stakeholders in June 2012 to discuss how to improve the methodological reporting of animal studies in grant applications and publications. The main workshop recommendation was that at a minimum studies should report on sample-size estimation, whether and how animals were randomized, whether investigators were blind to the treatment, and the handling of data (Landis et al., 2012).
Some journals, such as Science Translational Medicine, are embracing a new assumption, that is, the best science makes progress in improving the lives of human beings. From this new perspective, Kelner suggested, work that was previously thought to be dull becomes exciting, other studies become much less interesting, and the difficulties of doing research in humans might become worth tackling. This is not to say that science should stop doing fundamental research, she stressed, but rather, unexamined assumptions about what makes “good science” can introduce biases in what research is completed and which studies are published.
New Knowledge Calls for New Models
Neuroscience has changed significantly over the past few decades, moving from the study of simple systems, such as the squid axon, to imaging studies of complex systems such as humans. Scientists can conduct sophisticated, non-invasive studies in the living human brain to learn about diseases and the effects of treatments. In conclusion, Kelner challenged workshop participants to think about whether it is time to design a whole new set of animal models based on this knowledge, rather than trying to refine existing models. She suggested that resources be focused on advancing the understanding of neurological and psychiatric disease in people, and then, on the basis of that information, building new in vitro and animal models for drug development.
In response, Paul noted that most psychiatric drugs were discovered in humans, not in animals (Conn and Roth, 2008). Chlorpromazine, for example, was developed as an anesthetic sedative, but had a unique calming effect that was then tested and approved for use in psychotic schizophrenic patients. A participant added that if the mouse models for polygenic psychiatric disorders are really as poor as people think, perhaps it is time to ask under what circumstances it would be both worthwhile and ethical to go straight into human clinical trials after establishing safety.












