
This chapter focuses on a critical part of the systematic-review process: the assessment of the individual studies that are selected for inclusion in a review. As depicted in Figure 5-1, this step comes after the comprehensive search for and identification of relevant studies and precedes the integration of human, animal, and mechanistic evidence from the systematic reviews. The chapter first reviews the recommendations on evidence evaluation from the National Research Council (NRC) formaldehyde report and the US Environmental Protection Agency (EPA) responses to them. Best practices for evaluating clinical and epidemiologic studies, animal toxicology studies, and mechanistic studies in the systematic-review process are then discussed. Drawing on approaches developed for systematic reviews in clinical practice and public health, the committee emphasizes the need for EPA to assess the “risk of bias” in individual studies. Accordingly, the best-practice section defines the terms, notes the possibility that bias could arise throughout the conduct and reporting of a study, and discusses how to review studies within a risk-of-bias framework. Needs for further developing best practices are identified, and the committee’s findings and recommendations are provided at the conclusion of the chapter.
RECOMMENDATIONS ON EVIDENCE EVALUATION FROM THE NATIONAL RESEARCH COUNCIL FORMALDEYDE REPORT
The earlier formaldehyde report observed that “ultimately, the quality of the studies reviewed and the strength of evidence provided by the studies for deriving RfCs [reference concentrations] and unit risks need to be clearly presented” (NRC 2011a, p. 155). To that end, the report provided several recommendations for evaluating the evidence; these are provided in Box 5-1. Briefly, the recommendations focus on standardizing the presentation of the reviewed studies and evaluating the studies with standardized approaches that consider confounding and other biases.
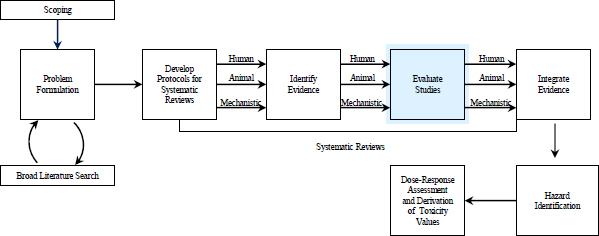
FIGURE 5-1 The IRIS process; the evidence-evaluation step is highlighted. The committee views public input and peer review as integral parts of the IRIS process, although they are not specifically noted in the figure.
• All critical studies need to be thoroughly evaluated with standardized approaches that are clearly formulated and based on the type of research, for example, observational epidemiologic or animal bioassays. The findings of the reviews might be presented in tables to ensure transparency.
• Standardize the presentation of reviewed studies in tabular or graphic form to capture the key dimensions of study characteristics, weight of evidence, and utility as a basis for deriving reference values and unit risks.
• Standardized evidence tables for all health outcomes need to be developed. If there were appropriate tables, long text descriptions of studies could be moved to an appendix or deleted.
• Develop templates for evidence tables, forest plots, or other displays.
• Establish protocols for review of major types of studies, such as epidemiologic and bioassay.
Source: NRC 2011a, pp. 152 and 165.
As discussed in Chapter 1 (see Table 1-1), the committee used the EPA reports Status of Implementation of Recommendations (EPA 2013a) and Chemical-Specific Examples (EPA 2013b) to evaluate EPA’s progress in implementing the recommendations in the NRC formaldehyde report (NRC 2011a). The committee also reviewed the draft IRIS assessment for benzo[a]pyrene (EPA 2013c) to gauge EPA’s progress in implementing recommendations for evidence evaluation. The draft preamble for IRIS assessments (EPA 2013a, Appendix B) provides a checklist that EPA will use to assess the quality of epidemiologic and experimental studies (see Box 5-2). EPA (2013a, p. F-44) also states that those and other considerations are “consistent with guidelines for systematic reviews that evaluate the quality and weight of evidence.” On the basis of those comments, the committee assumes that EPA intends to adopt principles associated with systematic review and with analysis of studies for risk of bias.
EPA (2013a, Appendix F) also includes a draft handbook for IRIS assessment development. The handbook mentions the following approach for epidemiologic studies: “to the extent possible, you want to assess not just the ‘risk of bias,’ but also the likelihood, direction, and magnitude of bias” (EPA 2013a, p. F-23). The draft handbook provides a table that outlines general considerations for evaluating features of epidemiologic studies. Features that would be assessed for quality and potential risk of bias include study design, study population and target-population setting, participation rate and follow-up, comparability between exposed and control populations, exposure-assessment methods, outcome measures, and data presentation, such as statistical analyses. The handbook also develops similar criteria for assessing the quality of animal studies. Animal-study features that would be assessed for quality and potential risk of bias include study design, exposure quality, test animals, end-point evaluation, data presentation and analysis, and reporting.
Several factors, including blinding and sampling bias, are discussed in the handbook, but several important risk-of-bias and quality-control elements, including attrition and statistical-power calculations, are not explicitly considered. The handbook emphasizes evaluation of how a study is reported (as opposed to how it is conducted). EPA describes the use of tables to present
Epidemiologic Studies
• Documentation of study design, methods, population characteristics, and results.
• Definition and selection of the study group and comparison group.
• Ascertainment of exposure to the chemical or mixture.
• Ascertainment of disease or health effect.
• Duration of exposure and follow-up and adequacy for assessing the occurrence of effects.
• Characterization of exposure during critical periods.
• Participation rates and potential for selection bias as a result of the achieved participation rates.
• Measurement error…and other types of information bias.
• Potential confounding and other sources of bias addressed in the study design or in the analysis of results.
Experimental Studies
• Documentation of study design, animals or study population, methods, basic data, and results.
• Nature of the assay and validity for its intended purpose.
• Characterization of the nature and extent of impurities and contaminants of the administered chemical or mixture.
• Characterization of dose and dosing regimen (including age at exposure) and their adequacy to elicit adverse effects, including latent effects.
• Sample sizes and statistical power to detect dose-related differences or trends.
• Ascertainment of survival, vital signs, disease or effects, and cause of death.
• Control of other variables that could influence the occurrence of effects.
Source: EPA 2013a, Appendix B.
the results of the study-quality evaluations (if there are robust datasets). Some examples provide qualitative descriptors for quality factors being assessed, for example, robust, moderate, or poor; or + to indicate that “criteria [are] not completely met or potential issues identified, but [they are] unlikely to directly affect study interpretation”; and ++ to indicate that “criteria [are] determined to be completely met” (EPA 2013a, Appendix F, p. F-32). Evidence tables presented in the draft IRIS assessment for benzo[a]pyrene (EPA 2013c, Tables 1-1 through 1-9 and 1-11 through 1-16) describe the populations, exposures, and outcomes of each study and the results. However, there is no assessment of the risk of bias in the studies evaluated, so it is unclear how EPA will meet its goal of assessing the direction and magnitude of bias in epidemiologic or animal studies. There is also no description of quality-assurance measures for the collection of assessment data.
Overall, EPA (2013a) has identified relevant study attributes to consider in evaluating study quality and indicates its intention to adopt risk-of-bias analyses. However, its considerations ignore some important elements that should be covered; accordingly, the following section focuses heavily on best practices for evaluating risk of bias in individual studies.
BEST PRACTICES FOR EVALUATING EVIDENCE FROM INDIVIDUAL STUDIES
Best practices for evaluating evidence from individual studies have developed in a number of fields. Here, following the general recommendation of the NRC formaldehyde report and the direction that EPA is taking with its revisions, the committee focuses on the best practices as developed for systematic reviews in clinical medicine and public health. As described above, although EPA has identified and is assessing important characteristics of the quality of human and animal studies, it has not historically conducted the assessments in a consistent and standardized way for studies included in IRIS assessments.
The report Finding What Works in Health Care: Standards for Systematic Reviews (IOM 2011) provides a useful roadmap for conducting systematic reviews, including the process of evaluating individual studies (see Chapter 4). Key elements of the Institute of Medicine (IOM) standards for study evaluation include an assessment of the relevance of the study’s populations, interventions, and outcomes for the systematic-review questions. After studies and outcomes are assessed for relevance, the IOM standards recommend a systematic assessment of the risk of bias (see below) according to predefined criteria. The potential biases must be assessed to determine how confident one can be in the conclusions drawn from the data.
The IOM standards are directed primarily toward the evaluation of evidence that compares the benefits and harms associated with alternative methods for preventing, diagnosing, treating for, and monitoring a clinical condition or for improving the delivery of care (that is, comparative-effectiveness research). Although the reviews conducted by the IRIS program have a distinctly different objective, many of the guiding principles identified by IOM can be applied to the IRIS program. For example, a systematic evaluation of research evidence for selection bias, dose-response associations, plausible confounders that could bias an estimated effect, and the strength of observed associations is relevant for both comparative-effectiveness research and toxicologic assessment.
Evaluation of Risk of Bias and Study Quality in Human Clinical and Epidemiologic Research
The validity of scientific evidence from a particular study has multiple determinants, from the initial formulation of a study hypothesis to the reporting of findings. In its assessment of findings that will be used from one or more studies in an IRIS assessment, EPA needs to address potential threats to the validity of evidence. The threats are generally well recognized by researchers but are referred to in different terms among fields. Here, the committee adopts the terminology that is used in systematic reviews of the medical literature.
The concept of risk of bias is central to the evaluation of studies for systematic reviews of clinical evidence (Higgins and Green 2008). The term is now widely used by those conducting such reviews; for example, it is extensively discussed in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins and Green 2008). The term, which might be unfamiliar to those in the field of toxicology, is defined by IOM as “the extent to which flaws in the design and execution of a collection of studies could bias the estimate of effect for each outcome under study” (IOM 2011, p. 165). Bias is generally defined as error that reduces validity, and risk of bias refers to the potential for bias to have occurred. A study might be in compliance with regulatory standards but still have an important risk of bias. For example, a study might adhere to all rules regarding the ethical treatment of animals (for example, appropriate housing, diet, and pain management) but have a bias due to lack of blinding of outcome measurement. The committee notes that risk of bias is not the same as imprecision (Higgins and Green 2008). Bias refers to systematic error, and imprecision refers to random error; smaller studies are less precise, but they might be less biased than larger ones.
Risk of bias has been empirically linked to biased effect estimates, but the direction (overestimate or underestimate) of the effect cannot be determined a priori on the basis of the specific type of risk of bias. For example, inadequate randomization (a risk of bias) in a drug study has been associated with overestimates of efficacy measures and underestimates of harm measures. In controlled human clinical trials that test drug efficacy, studies that have a high risk of bias—such as those lacking randomization, allocation concealment, or blinding of participants, personnel, and outcome assessors—tend to produce larger treatment-effect sizes and thus falsely inflate the efficacy of the drug being evaluated compared with studies that include those design features (Schulz et al. 1995; Schulz and Grimes 2002a,b).1 Biased human studies that assess the harm of drugs are less likely than nonbiased studies to report statistically significant adverse effects (Nieto et al. 2007). Those results reflect the need to consider bias related to funding source that might arise from systematic influences on the design and conduct of a study and the extent to which the full results and analyses of the study are published (Lundh et al. 2012).
Study quality and risk of bias are not equivalent concepts, and there is a difference between assessing risk of bias and assessing other methodologic characteristics of a particular study (Higgins and Green 2008). Assessment of study quality involves an investigation of the extent to which study authors conducted their research to high standards—for example, by following a well-documented protocol with trained study staff, by conducting an animal study according to good laboratory practices (GLP), or by complying with human-subjects guidelines for a clinical study. Assessment of study quality also evaluates how a study is reported—for example, whether the study population is described sufficiently. Risk of bias involves the internal validity of a study and reflects study-design characteristics that can introduce a systematic error or deviation from a true effect that might affect the magnitude and even the direction of the apparent effect (Higgins and Green 2008). 2
Table 5-1 identifies for the present report the various forms of bias within a study that underlie risk-of-bias assessments. The committee recognizes that terminology related to bias is not standardized and varies among fields and textbooks. It has not proposed specific definitions, but to ensure consistency throughout this report, it uses the terminology and descriptions of how biases arise that are presented in Table 5-1. The terms in the table are broad but cover the major types of bias that are relevant to IRIS assessments. EPA will need to give consideration to the terminology and definitions that it will use.
The Cochrane Handbook provides a classification scheme that includes selection, performance, detection, attrition, and reporting bias (Higgins and Green 2008). That scheme is oriented toward randomized clinical trials. In observational studies, the potential for confounding is a critical aspect of study design, data collection, and data analysis that needs to be assessed; in randomized clinical trials, confounding is addressed through randomization. Selection bias reflects differences in participant characteristics at baseline or that arise during follow-up and reflects patterns of participation that distort the results from those that would be found if the full
_____________________________
1The committee defines randomization, allocation concealment, and blinding as follows: randomization is a process that ensures that test subjects are randomly assigned to treatment groups, allocation concealment is a process that ensures that the person allocating subjects to treatment groups is unaware of the treatment groups to which the subjects are being assigned, and blinding (or masking) is a set of procedures that keeps the people who perform an experiment, collect the data, or assess the outcome measures unaware of the treatment allocation.
2The committee distinguishes internal validity, which is related to the bias of an individual study design, from external validity, which is the degree to which the results of the study can be generalized to settings or groups other than those used in the given study. Although external validity is relevant for determining whether a study should be included in a systematic review, it is not relevant for assessing bias within a study.
TABLE 5-1 Types of Biases and Their Sources
| Type of Bias | Sources |
| Randomized Studies | |
| Selection | Systematic differences between exposed and control groups in baseline characteristics that result from how subjects are assigned to groups |
| Performance | Systematic differences between exposed and control groups with regard to how the groups are handled |
| Detectiona | Systematic differences between exposed and control groups with regard to how outcomes are assessed |
| Attrition or exclusion | Systematic difference between exposed and control groups in withdrawal from the study or exclusion from analysis |
| Observational Studies | |
| Confounding and selection | Differences in the distribution of risk factors between exposed and nonexposed groups—can occur at baseline or during follow-up |
| Measurement | Mismeasurement of exposures, outcomes, or confounders—can occur at any time during the study |
| Randomized or Observational Studies | |
| Reporting | Selective reporting of entire studies, outcomes, or analyses |
aDetection bias includes measurement errors.
Source: Adapted from Higgins and Green 2008, Table 8.4, p. 8.7.
population could be observed. Problems with measurements in observational studies might also affect outcomes (detection bias in randomized clinical trials), exposures, potential confounders, and modifying factors. Such measurement problems might be systematic or random.
A number of methods can be used to minimize bias. The biases—and methods used to reduce them—are the same for human and animal studies. For example, randomized studies of humans or rodents could be at risk of selection or exclusion bias. There is empirical evidence that some methodologic characteristics can protect against specific biases. For example, in randomized clinical trials, selection bias can be minimized by randomization and concealment of allocation. In observational studies, confounding that arises from differences between exposed and nonexposed groups at enrollment or from differences that develop during follow-up in a prospective study can sometimes be addressed by using statistical techniques to adjust for group differences that can be measured. Selection bias that arises from difference at baseline or patterns of dropout during follow-up is not readily addressed by modeling.
The NRC formaldehyde report (NRC 2011a) noted a number of study characteristics that should be included in a template for evaluating observational epidemiologic studies (see Box 5-3). The recommended evaluation template includes items for assessing bias in design (the internal validity of a study) and the generalizability of the study (the external validity). As noted above, a number of assessment tools have been developed to assess the different types of biases for different study designs and data streams and are discussed in the following sections.
Cochrane Approach
The Cochrane Handbook for Systematic Reviews of Interventions includes a tool for systematically assessing the risk of bias in individual studies of the causal efficacy of a health intervention (Higgins and Green 2011). The tool is designed to handle randomized trials that assign human exposures at random and asks reviewers to assess whether the treatment and control groups are comparable (random allocation and allocation concealment), whether the participants or subjects were blind to their treatment, whether there was detection bias (whether knowledge of exposure condition affected the measurement of outcome), and whether there was attrition bias (whether dropout was associated with treatment), reporting bias, or “other sources of bias.”
BOX 5-3 Considerations for a Template for Evaluating an Epidemiologic Study
• Approach used to identify the study population and the potential for selection bias.
• Study population characteristics and the generalizability of findings to other populations.
• Approach used for exposure assessment and the potential for information bias, whether differential (nonrandom) or nondifferential (random).
• Approach used for outcome identification and any potential bias.
• Appropriateness of analytic methods used.
• Potential for confounding to have influenced the findings.
• Precision of estimates of effect.
• Availability of an exposure metric that is used to model the severity of adverse response associated with a gradient of exposures.
Source: NRC 2011a, p. 158.
The Cochrane treatment of risk of bias in nonrandomized studies (Higgins and Green 2011, Section 13.5.1.1) is extremely limited, but a new tool is being developed. It states that the sources of bias remain the same but that in some cases statistical techniques are used to control or adjust for potential confounding. It provides little help in systematic review of observational studies, which predominate in human research on the risks posed by chemicals.
The Cochrane Handbook (Higgins and Green 2011, Section 8.3.1) states that “the Collaboration’s recommended tool for assessing risk of bias is neither a scale nor a checklist. It is a domain-based evaluation, in which critical assessments are made separately for different domains.” Cochrane discourages using a numerical scale because calculating a score involves choosing a weighting for the subcomponents, and such scaling generally is nearly impossible to justify (Juni et al. 1999). Furthermore, a study might be well designed to eliminate bias, but because the study failed to report details in the publication under review, it will receive a low score. Most scoring systems mix criteria that assess risk of bias and reporting. However, there is no empirical basis for weighting the different criteria in the scores. Reliability and validity of the scores often are not measured. Furthermore, quality scores have been shown to be invalid for assessing risk of bias in clinical research (Juni et al. 1999). The current standard in evaluation of clinical research calls for reporting each component of the assessment tool separately and not calculating an overall numeric score (Higgins and Green 2008).
The Cochrane tool does include sources of risk of bias that are empirically based. For example, empirical studies of clinical trials show that inadequate or unclear concealment of allocation results in greater heterogeneity of results and effect sizes that are up to 40% larger than those of studies that contain adequate concealment of allocation (Schulz et al. 1995; Schulz and Grimes 2002b). In an empirical evaluation of the literature, Odgaard-Jensen et al. (2011) also found that both randomization and concealment of allocation can be associated with the estimated effect; results of randomized and nonrandomized studies differed, although variably, and nonblinded studies tended to provide larger effect estimates.
Ottawa-Newcastle Tool
The Ottawa-Newcastle tool for observational studies is an alternative to the Cochrane tool and, because it is intended for epidemiologic studies, is more relevant to the human studies eval-
uated in IRIS assessments.3 Developed primarily for case-control and cohort studies, the tool contains separate coding manuals for each. It is relatively short and easy to implement and has been used to evaluate risk of bias in studies of the association between coronary heart disease and hormone-replacement therapy in postmenopausal women. However, the Ottawa-Newcastle tool focuses on only three dimensions of a study: selection of the sample, comparability of the study groups, and outcome assessment (for cohort studies) or exposure assessment (for case-control studies).
National Toxicology Program Tool
The National Toxicology Program (NTP) in collaboration with the Office of Health Assessment and Translation (OHAT) of the National Institute of Environmental Health Sciences has recently constructed a method for systematic reviews to “assess the evidence that environmental chemicals, physical substances, or mixtures…cause adverse health effects and [provide] opinions on whether these substances may be of concern given what is known about current human exposure levels” (NTP 2013, p. 1). NTP assesses the risk of bias in individual studies by using questions related to five categories—selection, performance, detection, attrition or exclusion, and selective reporting bias (the latter term, used by OHAT, is equivalent to reporting bias)—that are similar to those used by the Cochrane Collaboration and adapted for environmental studies by the Navigation Guide Work Group (Woodruff and Sutton 2011). For each study outcome, risk of bias is assessed on a four-point scale: definitely low, probably low, probably high, or definitely high.
Evaluation of Tools for Assessing Risk of Bias in Human Studies
All the existing tools applicable to assessing risk of bias in a study of human risks require a substantial amount of expert judgment to rate a study’s effectiveness in controlling or adjusting for potential bias with methods other than randomization, allocation concealment, or blinding. One challenge in adapting systematic-review methods for environmental-epidemiology studies is the formal consideration of potential confounding and its consequences. Standard reviews and meta-analyses of epidemiologic studies typically include identification of key confounding variables; the failure to adjust for which is thought to result in bias. For example, a review of epidemiologic studies of maternal caffeine consumption and low birth weight would usually exclude (or stratify) studies that did not adequately adjust for maternal smoking as a confounding variable because maternal smoking is a risk factor for low birth weight and a known correlate of caffeine consumption.
Confounding is such an important bias in environmental epidemiology that it is often the primary consideration in assessing a study. Statistical techniques can be used to adjust for confounding in observational studies,4 but they require expert knowledge to identify, measure, and model the confounders correctly and resources to measure the confounders. It is often helpful to represent one’s understanding of potential confounding in a causal diagram (Greenland et al. 1999). In practice, however, it is unlikely that the etiology of any disease is sufficiently understood to create a causal diagram that fully and correctly accounts for all possible confounding factors. A reviewer of observational studies of a particular chemical and adverse health outcome might start by drawing several possible causal diagrams, including all known and suspected etio-
_____________________________
3The Ottawa-Newcastle tool (Wells et al. 2013) was developed at the Ottawa Hospital Research Institute and is available at http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp.
4For example, see Hernan and Robins 2008; Hernan et al. 2009; Hernan and Cole 2009; Hernan and Robins 2013; Robins and Hernan 2008; Robins 2000; Robins et al. 2000; Greenland 1989; Greenland et al. 1999; Greenland 2000.
logic factors. Each reviewed study could then be compared with each causal diagram to determine whether control for confounding was adequate given the present state of knowledge regarding the etiology of the health outcome.
Measurement error is another common source of bias in epidemiologic research; it can be systematic or random and can affect data related to exposures, outcomes, confounders, or modifiers (Armstrong 1998). Indeed, chemical-exposure assessment can be so difficult that one environmental-epidemiology textbook devotes an entire chapter to the topic of exposure-measurement error and states that “the quality of exposure data is a major factor in the validity of epidemiological studies” (Baker and Nieuwenhuijsen 2008, p. 258). Difficulties in exposure assessment arise from the time variability of personal exposures to chemicals, and it is often not feasible to assess exposures of each participant in an epidemiologic study. In addition, lifetime exposure is often relevant for cancer, and various estimation approaches are needed to fill gaps in the context of an epidemiologic study; the error that results from the estimation approaches might lead to measurement bias. Thus, many environmental epidemiology studies rely on complex exposure assignments based on a few environmental measurements and occupational records, residential histories, or time-activity surveys. Exposure biomarkers are increasingly used for exposure assessment but are not necessarily superior to other methods, depending on the period covered and their sensitivity and specificity for the exposure of interest. Although biomarkers might provide an accurate snapshot of personal biologic exposure at the time of sampling, their usefulness for epidemiologic-exposure assignment decreases with decreasing biologic half-life and increasing temporal variation in exposure (Handy et al. 2003; Bartell et al. 2004). Exposure biomarkers are also potentially susceptible to reverse causation—whereby an adverse health outcome or a risk factor for it induces a change in the exposure biomarker rather than reverse— and confounding by unmeasured physiologic differences.
Evaluation of Risk of Bias and Study Quality in Animal Toxicologic Research
Animal toxicology studies encompass a variety of experimental designs and end points. The studies can include short- to long-term bioassays that evaluate a wide array of clinical outcomes and more focused studies that evaluate one or more end points of interest. Results from animal toxicology studies are a critical stream of evidence and often the only data available for an IRIS assessment. Despite their importance, how to use animal studies in risk assessments and in regulatory decision-making is a subject of continuing debate (Guzelian et al. 2005; Weed 2005; ECETOC 2009; Adami et al. 2011; Woodruff and Sutton 2011). The NRC formaldehyde report concluded that EPA should develop a template for evaluation of toxicology studies in laboratory animals that would “consider the species and sex of animals studied, dosing information (dose spacing, dose duration, and route of exposure), end points considered, and the relevance of the end points to human end points of concern” (NRC 2011a, p. 159). Experience gained from randomized clinical trials in human and veterinary medicine suggests that systematic reviews that assess animal toxicology studies for quality and risk of bias would improve the quality of IRIS reviews. The committee notes that efforts have been made over the last decade to apply principles of systematic review, such as those made by the Evidence-based Toxicology Collaboration (EBTC 2012).
When the body of evidence is considered collectively, an evaluation of risk of bias in data derived from toxicologic studies can help to determine sources of inconsistency. Broad sources of bias considered earlier for human studies—including selection, performance, attrition, detection, and reporting bias—apply equally well to animal studies. A priori determination of suitable risk-of-bias questions tailored for animal studies and a variety of acceptable responses can help to guide the review process.
Because empirical studies of risk of bias in animal toxicology studies are few, the committee’s recommendations are based primarily on information derived from other types of research. On the basis of experience with risk of bias in controlled human clinical trials (Schulz et al. 1995;
Schulz and Grimes 2002a,b), one can anticipate that estimates of treatment effect might be influenced in animal studies that lack randomization, blinding, and allocation concealment and that such animal studies would therefore have a high risk of bias. Reviews of human clinical studies have shown that study funding sources and financial ties of investigators are associated with research outcomes that are favorable for the sponsors (Lundh et al. 2012). Favorable research outcomes were defined as increased effect sizes in drug-efficacy studies and decreased effect sizes in studies of drug harm. One study (Krauth et al. 2014) has demonstrated funding bias in preclinical studies of statins. Although selective reporting of outcomes is considered an important source of bias in clinical studies (Rising et al. 2008; Hart et al. 2012) and one study (Tsilidis et al. 2013) suggests that there is selective outcome reporting in animal studies of neurologic disease, further research is needed to determine the importance of biases—for example, related to funding and selective reporting—in the animal-toxicology literature.
Some empirical evidence shows that several risk-of-bias criteria are applicable to animal studies. For example, lack of randomization, blinding, specification of inclusion and exclusion criteria, statistical power, and use of clinically relevant animals has been shown to be associated with a risk of inflated estimates of the effects of an intervention (Bebarta et al. 2003; Crossley et al. 2008; Minnerup et al. 2010; Sena et al. 2010; Vesterinen et al. 2011). Because a detailed discussion of all possible risk-of-bias criteria was beyond the scope of the present report, the committee focused its attention on sources of bias in animal studies that could affect the scientific rigor of a systematic review that uses animal toxicology studies as an evidence stream. The committee briefly discusses the importance of randomization, allocation concealment, and blinding. Although GLPs contain those practices, many high-quality studies are conducted outside the regulatory framework and hence not directly subject to GLPs. Moreover, a recent systematic review of tools for assessing animal toxicology studies identified 30 distinct tools for which the number of assessed criteria ranged from two to 25 (Krauth et al. 2013). The most common criteria were randomization (25 of 30, 83%) and investigator blinding (23 of 30, 77%).
Regarding randomization, several approaches are used in toxicology to assign animals to treatment groups randomly, including computer-based algorithms, random-number tables, and card assignment (Martin et al. 1986). Conversely, having study staff select animals from their cages “at random” poses a risk of conscious or unconscious manipulation and does not lead to true randomization (van der Worp et al. 2010). Proper randomization includes not only generating a truly random-number sequence but ensuring that the person who is allocating animals is unaware of whether the next animal will be assigned to the control group or the treatment group. If allocation is not concealed, animals can be differentially assigned to control and treatment groups on the basis of characteristics other than their random-number assignment. Allocation concealment therefore helps to prevent selection bias, and it has been shown empirically that lack of randomization or of allocation concealment in animal studies biases research outcomes by altering effect sizes (Bebarta et al. 2003; Sena et al. 2007; Macleod et al. 2008; Vesterinen et al. 2011). For example, Macleod et al. (2008) published a systematic review of the effects of the free-radical scavenger disufenton sodium in animal experiments on focal cerebral ischemia. Studies that included methods for randomization and allocation concealment and studies that used a more clinically relevant animal model (spontaneously hypertensive rats) reported lower efficacy than other studies. However, a similar meta-analysis of experimental studies of stroke performed by Crossley et al. (2008) did not find that lack of randomization was associated with a bias although lack of concealment was so associated.
As noted earlier, blinding or masking is a set of procedures that keeps the people who perform an experiment, collect the data, or assess the outcome measures unaware of the treatment allocation. If treatment allocation is known, that knowledge could affect decisions regarding the supply of additional care and the withdrawal of animals from an experiment and affect how outcomes are assessed. In blinded studies, those involved in the study will not be influenced by knowing treatment allocation, so performance, detection, and attrition bias will be prevented (Sargeant et al. 2010). Thus, blinding is used to prevent bias toward fulfilling the expectations of
the investigator on the basis of knowledge of the treatment (Kaptchuk 2003). There is substantial evidence that lack of blinding in a variety of types of animal studies is associated with exaggerated effect sizes (Bebarta et al. 2003; Sena et al. 2007; Vesterinen et al. 2011). Crossley et al. (2008) found that studies that did not use blinded investigators and studies that used healthy animals instead of animals that had relevant clinical comorbidities reported greater effect sizes. The review of animal drug interventions by Bebarta et al. (2003) showed that studies that lacked blinding or randomization in their design were more likely to find positive outcomes than studies that included blinding and randomization.
In contrast with allocation concealment, blinding might not always be possible throughout a toxicology experiment, for example, when the agent being tested imparts a visible change in the outward appearance of an animal. However, blinding of outcome assessment is almost always possible, and there are many ways to achieve it, such as providing study personnel with coded animal numbers (which blinds the study personnel to treatment assignment). Coded data can be analyzed by a statistician who is independent of the rest of the research team. The blinding of study personnel to treatment groups is not without controversy. For example, several groups and individuals have recommended blinded evaluation of histopathology slides in animal toxicology studies (EFSA 2011; Holland and Holland 2011) whereas others have argued against this approach (Neef et al. 2012). The committee notes that the terms single-blinded, double-blinded, and triple-blinded are often used to describe blinding in human clinical trials, but such terms can be ambiguous (Devereaux et al. 2001), and, unlike human study subjects, animals cannot be blinded to treatment assignment. Therefore, it is preferable to state which members of the study team were blinded and how. In general, it is insufficient to state that staff members were blinded to treatment groups. The method of blinding should be described in publications or in an accessible protocol to allow readers to assess the validity of the blinding procedures.
Another type of bias can occur in animal studies and can be reduced with proper procedures. Exclusion bias refers to the systematic difference between treatment and control groups in the number of animals that were included in and completed the study. Data on whether all animals in a study are accounted for and use of intention-to-treat analysis (analyzing animals in the groups to which they were assigned) can reduce exclusion bias (Marshall et al. 2005).
Several studies have shown that assessing risk of bias in animal studies is challenging because of inconsistent standards for reporting procedures in preclinical animal experiments (Lamontagne et al. 2010; Faggion et al. 2011). Similarly, several systematic reviews related to disease in animals have noted “a lack of reporting of group-allocation methods, blinding, and details related to intervention protocols, outcome assessments, and statistical analysis methods in some published veterinary clinical trials” (O’Connor et al. 2006; Wellman and O’Connor 2007; Burns and O’Connor 2008; Sargeant et al. 2010, p. 580). Those deficiencies have led to the development of reporting guidelines for randomized controlled trials in livestock and food safety (Sargeant et al. 2010). The reporting guidelines have been endorsed by several veterinary journals, and their adoption is expected to improve the quality of reporting of livestock-based randomized clinical trials (Sargeant et al. 2010). Because of the similarities between many animal toxicology studies and livestock clinical trials, the committee also considered those guidelines during its deliberations. Common deficiencies noted in many animal studies included lack of sample-size calculations, sufficient sample sizes, appropriate animal models, randomized treatment assignment, conflict-of-interest statements , and blinded procedures for drug administration, induction of injury, and outcome assessment (Knight 2008; Sargeant et al. 2010).
Because of the importance of many of those factors for study quality, they are included in GLPs that apply to animal studies as required by EPA or the US Food and Drug Administration (FDA) for product registration. The GLPs form a framework for study design, conduct, and oversight that reduces the risk of bias that can be associated with the adequacy of temperature, humidity, and other environmental conditions; experimental equipment and facilities; animal care; health status of animals; animal identification; separation from other test systems; and presence of contaminants in feed, soil, water, or bedding. The GLP regulations also require that “the test,
control, and reference substances be analyzed for identity, strength, purity, and composition, as appropriate for the type of study” (EPA 1999) and that the solubility and stability of the substances be determined. The GLP regulations also consider the need for blinding and randomization of the animal test system. In some tools used to assess study quality (Klimisch et al. 1997), tests conducted and reported according to accepted test guidelines (of the European Union, EPA, FDA, the Organisation for Economic Co-operation and Development) and in compliance with GLP principles have the highest grade of reliability.
Other considerations of quality of animal toxicology studies, such as the choice of the species for study, are important. Commonly used mammalian orders include rodents (such as mice, rats, hamsters, and guinea pigs), carnivores (such as dogs), lagomorphs (such as rabbits), primates (such as monkeys), and artiodactyla (such as pigs and sheep). Other vertebrates—including birds, amphibians, and fish—are also widely used in toxicologic research. Invertebrate models, including such flies as Drosophila melanogaster and such nematodes as Caenorhabditis elegans, are increasingly used, especially for mechanistic toxicology studies. Not all animal models are relevant for assessing human health effects, so understanding of the toxicologic end point of interest, concordance between animal and human responses, pharmacokinetic data, and mechanistic information is often key to evaluating the importance of results of an animal study for human health. Animal models that are irrelevant to the end point being studied are often excluded a priori from the review.
One of the most important elements in the design and interpretation of animal toxicology studies is selection of the appropriate doses for investigation (Rhomberg et al. 2007). Details of the test chemical and its administration are also quality concerns. For toxicologic treatments, the investigators should at least provide the chemical name, the concentration, and the delivery matrix and describe how the doses were selected and administered. Additional information, such as particle size and distribution, is often needed for inhalation studies. High-quality studies should include multiple dose groups, including a high-dose group that increases a study's statistical power to detect effects that might be rare and other groups to allow characterization of the dose-response relationship for observed adverse effects (Rhomberg et al. 2007).
The committee recognizes that many toxicologists are unfamiliar with the concept of risk of bias and that underreporting of its determinants probably affects the toxicology literature. Recent calls for reporting criteria for animal studies (NC3Rs 2010; NRC 2011b; Landis et al. 2012) recognize the need for improved reporting of animal research. Reporting of animal research is likely to improve if risk-of-bias assessments become more common, particularly after use in IRIS assessments.
Evaluation of Risk of Bias and Study Quality in Mechanistic Research
As described in Chapter 3, EPA often relies on a third evidence stream in reaching conclusions about the risk associated with a chemical hazard. In the broadest terms, mechanistic studies can include animal or human pharmacokinetic studies, nonanimal alternatives to hazard identification, and cell-based in vitro assays that evaluate responses of interest. Mechanistic studies can also include high-throughput assays that exploit technologic advances in molecular biology and bioinformatics. Those studies often evaluate cellular pathways that are thought to lead to such adverse health effects as carcinogenicity, genotoxicity, reproductive and developmental toxicity, neurotoxicity, and immunotoxicity in humans. Data are increasingly generated from mechanistic toxicology studies, so it is important for EPA to develop a framework that will facilitate regulatory acceptance of the results of such studies.
As in other experiments, risk of bias should be considered in evaluating mechanistic toxicology data. One challenge that EPA faces is the lack of critically evaluated risk-of-bias assessment tools. Empirically, many of the same risk-of-bias considerations that apply to bioassay studies could apply equally well to animal-based pharmacokinetic studies. Pharmacokinetic data can be used to extrapolate dose among species and treatment groups and to quantify interindivid-
ual variability. They are also used to assess the quality of individual studies, consistency among outcomes, and differences among population groups, such as susceptible populations. Like bioassays, pharmacokinetic studies need to specify test-chemical stability, chemical exposure route, and exposure-measurement methods. A recent review of mechanistic studies used a systematic approach to identify studies, extract information, and summarize study findings but fell short of assessing risk of bias in the studies (Kushman et al. 2013).
The committee encourages EPA to advance its methods for using in vitro studies in hazard assessment. Several criteria should be considered in assessing in vitro toxicology studies for risk of bias and toxicologic relevance. Relevance should be determined in several domains, including cell systems used, exposure concentrations, metabolic capacity, and the relationship between a measured in vitro response and a clinically relevant outcome measure. Few tools are available for assessing risk of bias in in vitro studies. Because of the nascent status of this field, the committee can provide only provisional recommendations for EPA to consider.
FDA’s Good Manufacturing Practice (GMP) regulations describe how to ensure performance and consistency of in vitro methods when they are approved for commercial use by FDA, including criteria for setting performance standards for each assay that falls under FDA authority as a device or kit for medical purposes.5 The provisions in the regulations can also be applied to many in vitro tests that are used in toxicology (Gupta et al. 2005). Likewise, GLP principles discussed earlier could be applied to in vitro tests (OECD 2004; Gupta et al. 2005). Useful guidelines, such as Good Cell Culture Practice (Bal-Price and Coecke 2011), have also been developed to define minimum standards in cell and tissue culture.
Another approach that could be used by EPA is to consider criteria developed by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) to validate in vitro and other alternative (nonanimal) toxicity assays. Test methods validated by ICCVAM for regulatory risk-assessment purposes generally include (a) a scientific and regulatory rationale for the test method, (b) an understanding of the relationship of the test method’s end points to the biologic effect of interest, (c) a description of what is measured and how, (d) test performance criteria (for example, the use of positive and negative controls), (e) a description of data analysis, (f) a list of the species to which the test results can be applied, (g) and a description of test limitations, including classes of materials that the test cannot accurately assess. Joint efforts between ICCVAM and the European Centre for the Validation of Alternative Methods (ECVAM) are evaluating criteria for the validation of toxicogenomics-based test systems (Corvi et al. 2006). Potential sources of bias that have been empirically identified during the joint ICCVAM-ECVAM effort include data quality, cross-platform and interlaboratory variability, lack of dose- and time-dependent measurements that examine the range of biologic variability of gene responses for a given test system, unknown concordance with known toxicologic outcomes (phenotypic anchoring), and poor microarray and instrumentation quality. The following items were deemed necessary for the use of microarray-based toxicogenomics in regulatory assessments: (a) microarrays should be made according to GMP principles; (b) “specifications and performance criteria for all instrumentation and method components should be available”; (c) “all quality-assurance and quality-control…procedures should be transparent, consistent, comparable, and reported”; (d) “arrays should have undergone sequence verification, and the sequences should be publicly available”; and (e) all data should be exportable in a microarray and gene expression compatible format (Corvi et al. 2006, p. 423). The extent of bias associated with each of those items is not known.
The test chemicals used in in vitro systems should also include known positive and known negative agents. Whenever possible, chemicals or test agents should be coded to reduce bias. Other problems that might be associated with study quality and risk of bias involve the character-
_____________________________
5See 21 CFR 210 and 211 [2003] Good Manufacturing Practices and 21 CFR 800 [2003] Good Manufacturing Practices Device.
ization of the source, the growing conditions of the cultured cell, and the genetic materials used in microarrays; documentation of cell-culture practices, such as incubator temperatures; and definition of reagents and equipment. The limitations of the test systems should be understood and described. Important limitations that could affect how study results are interpreted include the inability to replicate the metabolic processes relevant to chemical toxicity that occur in vivo in an in vitro test system.
Faggion (2012) recently assessed existing guidelines for reporting in vitro studies in dentistry and presented a method for reporting risk of bias that was based on the Consolidated Standards of Reporting Trials (CONSORT) checklist for reporting randomized clinical trials. Important elements in the checklist included (a) the scientific background and rationale for the study; (b) the interventions used for each group, including how and when they were administered, with sufficient detail to enable replication; (c) outcome measures, including how and when they were assessed; (d) sample-size calculations; (e) how the random-allocation sequence was generated and implemented; (g) blinding of investigators responsible for treatment and others responsible for outcome assessment; (h) statistical methods; and (i) trial limitations, such as sources of potential bias or imprecision.
Reporting inadequacies similar to those mentioned earlier have been noted in the literature describing results of in vitro studies. Watters and Goodman (1999) compared the rigor with which tissue-culture and cell-culture in vitro studies were reported and randomly selected clinical studies published in the same anesthesia journals. They focused on basic aspects of study design and reporting that might lead to bias, including sample size, randomization, and reporting of exclusions and withdrawals. They found that reporting of those aspects in the in vitro studies occurred at much lower rate than that of clinical studies and thus hindered interpretation of reported tissue-culture and cell-culture studies.
EVALUATING EVIDENCE FROM INDIVIDUAL STUDIES
Assessment of the risk of bias and other methodologic characteristics of relevant studies is a critical part of the systematic-review process. The committee acknowledges that these assessments will take time and effort; the resources required will depend on the complexity and amount of data available on a given chemical. However, the use of standard risk-of-bias criteria by trained coders can be efficient. Koustas et al. (in press) and Johnson et al. (in press) found that risk-of-bias assessment, data analysis, and evaluation of quality and strength of evidence took about 2-3 additional months.
The risk-of-bias assessment can be used to exclude studies from a systematic review or can be incorporated qualitatively or quantitatively into the review results. The plan for incorporating the risk-of-bias assessment into a systematic review should be specified a priori in the review protocol. Various options are discussed in the sections that follow.
Exclusion of Studies
Individual studies are most commonly excluded from a systematic review if they do not meet the inclusion criteria for the review with respect to the population examined, the exposures measured, or the outcomes assessed. Less often, individual studies are excluded because they did not meet a particular threshold of risk of bias or other methodologic criteria. Some studies that entail a substantial risk of bias or that have severe methodologic shortcomings (“fatal flaws”) could be excluded from consideration. Examples of such exclusion criteria include instability of the test compound, inappropriate animal models, inadequate or no controls (or comparison group), or invalid measures of exposure or outcome. EPA needs a transparent process and clear criteria for excluding certain studies that have important deficiencies. If studies are excluded because of risk of bias, the reasons for the exclusion should be described.
Incorporating the Assessment of a Risk of Bias in the Review
There are two main options for presenting an assessment of the risk of bias and methodologic characteristics of studies that will be included in the IRIS assessment. First, if a meta-analysis has been conducted, a sensitivity analysis can be conducted to determine the effects of risk of bias on the meta-analytic result. For example, a sensitivity analysis could be conducted to determine whether a meta-analysis result is affected by excluding studies that have a high risk of bias. All studies would be included in the meta-analysis, and then the meta-analysis would be recalculated by including only studies that meet a threshold for low risk of bias. If the summary estimate does not change, the estimate is not sensitive to the risk of bias of the studies. If the included studies are very heterogeneous or if all included studies have a high risk of bias, it might be inappropriate to calculate a quantitative summary statistic.
The second option is to describe the results of individual study evaluations qualitatively. Qualitative presentation of individual study evaluations can include narrative descriptions or graphic presentations. Tables summarizing the evidence could include a column that summarizes the risk-of-bias assessment in text and provides a ranking (for example, one, two, or three stars). Graphic displays could show the evaluation of each individual item assessed for each study. Two displays from a Cochrane review are shown in Figures 5-2 and 5-3. As described in later chapters of the present report, the risk-of-bias assessment can be included in the process for selecting studies for calculating toxicity values or in the uncertainty analysis.
Finding: The checklist developed by EPA that is presented in the preamble and detailed in the draft handbook addresses many of the concerns raised by the NRC formaldehyde report. EPA has also developed broad guidance for the assessment of the quality of observational studies of exposed human populations and, to a smaller extent, animal toxicology studies. It has not developed criteria for the evaluation of mechanistic toxicology studies. Still lacking is a clear picture of the assessment tools that EPA will develop to assess risk of bias and of how existing assessment tools will be adapted.
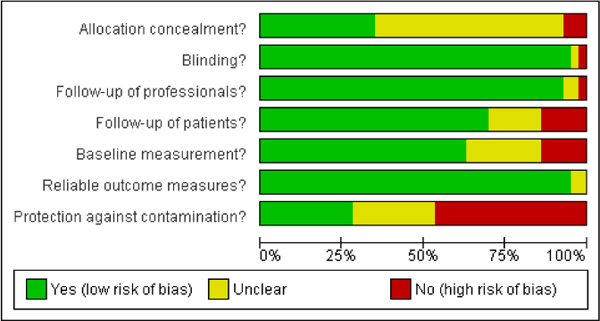
FIGURE 5-2 Sample graphic display of risk-of-bias evaluations. Source: Nkansah et al. 2010. Reprinted with permission; copyright 2010, Cochrane Database of Systematic Reviews.
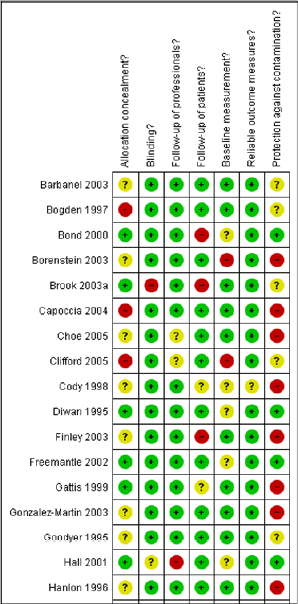
FIGURE 5-3 Truncated graph of the risk-of-bias summary that shows review authors’ judgments about each risk-of-bias item for each included study. Green: yes (low risk of bias); yellow: unclear; red: no (high risk of bias). Source: Nkansah et al. 2010. Reprinted with permission; copyright 2010, Cochrane Database of Systematic Reviews.
Recommendation: To advance the development of tools for assessing risk of bias in different types of studies (human, animal, and mechanistic) used in IRIS assessments, EPA should explicitly identify factors, in addition to those discussed in this chapter, that can lead to bias in animal studies—such as control for litter effects, dosing, and methods for exposure assessment—so that these factors are consistently evaluated for experimental studies. Likewise, EPA should consider a tool for assessing risk of bias in in vitro studies.
Finding: The development of standards for evaluating individual studies for risk of bias is most advanced in human clinical research. Even in that setting, the evidence base to support the standards is modest, and expert guidance varies. Furthermore, many of the individual criteria included in risk-of-bias assessment tools, particularly for animal studies and epidemiologic studies, have not been empirically tested to determine how the various sources of bias influence the results of individual studies. The validity and reliability of the tools have also not been tested.
Finding: Thus, the committee acknowledges that incorporating risk-of-bias assessments into the IRIS process might take additional time; the ability to do so will vary with the complexity and extent of data on each chemical and with the resources available to EPA. However, the use of standard risk-of-bias criteria by trained coders has been shown to be efficient.
Recommendation: When considering any method for evaluating individual studies, EPA should select a method that is transparent, reproducible, and scientifically defensible. Whenever possible, there should be empirical evidence that the methodologic characteristics that are being assessed in the IRIS protocol have systematic effects on the direction or magnitude of the outcome. The methodologic characteristics that are known to be associated with a risk of bias should be included in the assessment tool. Additional quality-assessment items relevant to a particular systematic-review question could also be included in the EPA assessment tool.
Recommendation: EPA should carry out, support, or encourage research on the development and evaluation of empirically based instruments for assessing bias in human, animal, and mechanistic studies relevant to chemical-hazard identification. Specifically, there is a need to test existing animal-research assessment tools on other animal models of chemical exposures to ensure their relevance and generalizability to chemical-hazard identification. Furthermore, EPA might consider pooling data collected for IRIS assessment to determine whether, among various contexts, candidate risk-of-bias items are associated with overestimates or underestimates of effect.
Recommendation: Although additional methodologic work might be needed to establish empirically supported criteria for animal or mechanistic studies, an IRIS assessment needs to include a transparent evaluation of the risk of bias of studies used by EPA as a primary source of data for the hazard assessment. EPA should specify the empirically based criteria it will use to assess risk of bias for each type of study design in each type of data stream.
Recommendation: To maintain transparency, EPA should publish its risk-of-bias assessments as part of its IRIS assessments. It could add tables that describe the assessment of each risk-of-bias criterion for each study and provide a summary of the extent of the risk of bias in the descriptions of each study in the evidence tables.
Finding: The nomenclature of the various factors that are considered in evaluating risk of bias is variable and not well standardized among the scientific fields relevant to IRIS assessments. Such terminology has not been standardized for IRIS assessments.
Recommendation: EPA should develop terminology for potential sources of bias with definitions that can be applied during systematic reviews.
Finding: Although reviews of human clinical studies have shown that study funding sources and financial ties of investigators are associated with research outcomes that are favorable for the sponsors, less is known about the extent of funding bias in animal research.
Recommendation: Funding sources should be considered in the risk-of-bias assessment conducted for systematic reviews that are part of an IRIS assessment.
Finding: An important weakness of all existing tools for assessing methodologic characteristics of published research is that assessment requires full reporting of the research methods. EPA might be hampered by differences in traditions of reporting risk of bias among fields in the scientific literature.
Recommendation: EPA should contact investigators to obtain missing information that is needed for the evaluation of risk of bias and other quality characteristics of included studies. The committee expects that, as happened in the clinical literature in which additional reporting standards for journals were implemented (Turner et al. 2012), the reporting of toxicologic research will eventually improve as risk-of-bias assessments are incorporated into the IRIS program.
However, a coordinated approach by government agencies, researchers, publishers, and professional societies will be needed to improve the completeness and accuracy of the reporting of toxicology studies in the near future.
Finding: EPA has not developed procedures that describe how the evidence evaluation for individual studies will be incorporated, either qualitatively or quantitatively, into an overall assessment.
Recommendation: The risk-of-bias assessment of individual studies should be carried forward and incorporated into the evaluation of evidence among data streams.
Adami, H.O., S.C. Berry, C.B. Breckenridge, L.L. Smith, J.A. Swenberg, D. Trichopoulos, N.S. Weiss, and T.P. Pastoor. 2011. Toxicology and epidemiology: Improving the science with a framework for combining toxicological and epidemiological evidence to establish causal inference. Toxicol. Sci. 122(2):223-234.
Armstrong, B.G. 1998. Effect of measurement error on epidemiological studies of environmental and occupational exposures. Occup. Environ. Med. 55(10):651–656.
Baker, D., and M.J. Nieuwenhuijsen. 2008. Environmental Epidemiology. Study Methods and Application. New York: Oxford University Press.
Bal-Price, A., and S. Coecke. 2011. Guidance on Good Cell Culture Practice (GCCP). Pp. 1-25 in Cell Culture Techniques, M. Aschner, C. Sunol, and A. Bal-Price, eds. Springer Protocols, Neuromethods Vol. 56. New York: Humana Press.
Bartell, S.M., W.C. Griffith, and E.M. Faustman. 2004. Temporal error in biomarker based mean exposure estimates for individuals. J. Expo. Anal. Environ. Epidemiol. 14(2):173-179.
Bebarta, V., D. Luyten, and K. Heard. 2003. Emergency medicine animal research: Does use of randomization and blinding affect the results? Acad. Emerg. Med. 10(6):684-687.
Burns, M.J., and A.M. O’Connor. 2008. Assessment of methodological quality and sources of variation in the magnitude of vaccine efficacy: A systematic review of studies from 1960 to 2005 reporting immunization with Moraxella bovis vaccines in young cattle. Vaccine 26(2):144-152.
Corvi, R., H.J. Ahr, S. Albertini, D.H. Blakey, L. Clerici, S. Coecke, G.R. Douglas, L. Gribaldo, J.P. Groten, B. Haase, K. Hamernik, T. Hartung, T. Inoue, I. Indans, D. Maurici, G. Orphanides, D. Rembges, S.A. Sansone, J.R. Snape, E. Toda, W. Tong, J.H. van Delft, B. Weis, and L.M. Schechtman. 2006. Meeting report: Validation of toxicogenomics-based test systems: ECVAM-ICCVAM/NICEATM considerations for regulatory use. Environ. Health Perspect. 114(3):420-429.
Crossley, N.A., E. Sena, J. Goehler, J. Horn, B. van der Worp, P.M. Bath, M. Macleod, and U. Dirnagl. 2008. Empirical evidence of bias in the design of experimental stroke studies: A metaepidemiologic approach. Stroke 39(3):929-934.
Devereaux, P.J., B.J. Manns, W.A. Ghali, H. Quan, C. Lacchetti, V.M. Montori, M. Bhandari, and G.H. Guyatt. 2001. Physician interpretations and textbook definitions of blinding terminology in randomized controlled trials. JAMA 285(15):2000-2003.
EBTC (Evidence–based Toxicology Collaboration). 2012. Methods Work Group [online]. Available: http://www.ebtox.com/methods-work-group/ [accessed Feb. 14, 2014].
ECETOC (European Centre for Ecotoxicology and Toxicology of Chemicals). 2009. Framework for the Integration of Human and Animal Data in Chemical Risk Assessment. Technical Report No. 104. ECETOC, Brussels, Belgium [online]. Available: http://www.ecetoc.org/uploads/Publications/documents/TR%20104.pdf [accessed December 6, 2013].
EFSA (European Food Safety Authority). 2011. Scientific Opinion EFSA Guidance on Repeated-Dose 90-Day Oral Toxicity Study on Whole Food/Feed in Rodents. Draft for public consultation. Scientific Committee, EFSA, Parma, Italy [online]. Available: http://www.efsa.europa.eu/en/consultations/call/110707.pdf [accessed December 6, 2013].
EPA (U.S. Environmental Protection Agency). 2013a. Part 1. Status of Implementation of Recommendations. Materials Submitted to the National Research Council, by Integrated Risk Information System Program, U.S. Environmental Protection Agency, January 30, 2013 [online]. Available: http://www.
epa.gov/iris/pdfs/IRIS%20Program%20Materials%20to%20NRC_Part%201.pdf [accessed Nov. 13,2013].
EPA (U.S. Environmental Protection Agency). 2013b. Part 2. Chemical-Specific Examples. Materials Submitted to the National Research Council, by Integrated Risk Information System Program, U.S. Environmental Protection Agency, January 30, 2013 [online]. Available: http://www.epa.gov/iris/pdfs/IRIS%20Program%20Materials%20to%20NRC_Part%202.pdf [accessed October 22, 2013].
EPA (U.S. Environmental Protection Agency). 2013c. Toxicological Review of Benzo[a]pyrene (CAS No. 50-32-8) in Support of Summary Information on the Integrated Risk Information System (IRIS), Public Comment Draft. EPA/635/R13/138a. National Center for Environmental Assessment, Office of Research and Development, U.S. Environmental Protection Agency, Washington, DC. August 2013 [online]. Available: http://cfpub.epa.gov/ncea/iris_drafts/recordisplay.cfm?deid=66193 [accessed Nov. 13, 2013].
EPA (U.S. Environmental Protection Agency). 1999. Audit for Determining Compliance of Studies with GLP Standards Requirements. Standard Operating Procedure SOP No. GLP-C-02 [online]. Available: http://www.epa.gov/compliance/resources/policies/monitoring/fifra/sop/glp-c-02.pdf [accessed December 11, 2013].
Faggion, C.M., Jr. 2012. Guidelines for reporting pre-clinical in vitro studies on dental materials. J. Evid. Based Dent. Pract. 12(4):182-189.
Faggion, C.M., Jr., N.N. Giannakopoulos, and S. Listl. 2011. Risk of bias of animal studies on regenerative procedures for periodontal and peri-implant bone defects - a systematic review. J. Clin. Periodontol. 38(12):1154-1160.
Finley, P.R., H.R. Rens, J.T. Pont, S.L. Gess, C. Louie, S.A. Bull, J.Y. Lee, and L.A. Bero. 2003. Impact of a collaborative care model on depression in a primary care setting: A randomized controlled trial. Pharmacotherapy 23(9):1175-1185.
Greenland, S. 1989. Modeling and variable selection in epidemiologic analysis. Am. J. Public Health 79(3):340-349.
Greenland, S. 2000. An introduction to instrumental variables for epidemiologists. Int. J. Epidemiol. 29(4):722-729.
Greenland, S., J. Pearl, and J.M. Robins. 1999. Causal diagrams for epidemiologic research. Epidemiology 10(1):37-48.
Gupta, K., A. Rispin, K. Stitzel, S. Coecke, and J. Harbell. 2005. Ensuring quality of in vitro alternative test methods: Issues and answers. Regul. Toxicol. Pharmacol. 43(3):219-224.
Guzelian, P.S., M.S. Victoroff, N.C. Halmes, R.C. James, and C.P. Guzelian. 2005. Evidence-based toxicology: A comprehensive framework for causation. Hum. Exp. Toxicol. 24(4):161-201.Hall, L., M. Eccles, R. Barton, N. Steen, and M. Campbell. 2001. Is untargeted outreach visiting in primary care effective? A pragmatic randomized controlled trial. J. Public Health Med. 23(2):109-113.
Handy, R.D., T.S. Galloway, and M.H. Depledge. 2003. A proposal for the use of biomarkers for the assessment of chronic pollution and in regulatory toxicology. Ecotoxicology 12(1-4):331-343.
Hart, B., A. Lundh, and L. Bero. 2012. Effect of reporting bias on meta-analyses of drug trials: Reanalysis of meta-analyses. BMJ 344:d7202.
Hernán, M.A., and S.R. Cole. 2009. Causal diagrams and measurement bias. Am. J. Epidemiol. 170(8):959-962.
Hernán, M.A., and J.M. Robins. 2008. Observational studies analyzed like randomized experiments: Best of both worlds. Epidemiology 19(6):789-792.
Hernán, M.A., and J.M. Robins. 2013. Causal Inference. New York: Chapman & Hall/CRC [online]. Available: http://www.hsph.harvard.edu/miguel-hernan/causal-inference-book/ [accessed December 6, 2013].
Hernán, M.A., M. McAdams, N. McGrath, E. Lanoy, and D. Costagliola. 2009. Observation plans in longitudinal studies with time-varying treatments. Stat. Methods Med. Res. 18(1):27-52.
Higgins, J.P.T., and S. Green, eds. 2008. Cochrane Handbook for Systematic Reviews of Interventions. Chichester, UK: John Wiley & Sons.
Higgins, J.P.T., and S. Green, eds. 2011. Cochrane Handbook for Systematic Reviews of Interventions, Version 5.1.0. The Cochrane Collaboration [online]. Available: http://handbook.cochrane.org/ [accessed December 11, 2013].
Holland, T., and C. Holland. 2011. Analysis of unbiased histopathology data from rodent toxicology studies or, are these groups different enough to ascribe it to treatment? Toxicol. Pathol. 39(4):569-575.
IOM (Institute of Medicine). 2011. Finding What Works in Health Care: Standards for Systematic Reviews. Washington, DC: National Academies Press.
Johnson, P.I., P. Sutton, D.S. Atchley, E. Koustas, J. Lam, S. Sen, K.A. Robinson, D.A. Axelrad, and T.J. Woodruff. In press. The Navigation Guide - Evidence-based medicine meets environmental health: Systematic review of human evidence for PFOA effects on fetal growth. Environmental Health Perspectives.
Juni, P., A. Witschi, R. Bloch, and M. Egger. 1999. The hazards of scoring the quality of clinical trials for meta-analysis. JAMA 282(11):1054-1060.
Kaptchuk, T.J. 2003. Effect of interpretive bias on research evidence. BMJ 326(7404):1453-1455.
Klimisch, H.J., M. Andreae, and U. Tillmann. 1997. A systematic approach for evaluating the quality of experimental toxicological and ecotoxicological data. Regul. Toxicol. Pharmacol. 25(1):1-5.
Knight, A. 2008. Systematic reviews of animal experiments demonstrate poor contributions toward human healthcare. Rev. Recent Clin. Trials 3(2):89-96.
Koustas, E., J. Lam, P. Sutton, P.I. Johnson, D.S. Atchley, S. Sen, K.A. Robinson, D.A. Axelrad, and T.J. Woodruff. In press. The Navigation Guide - Evidence-based medicine meets environmental health: Systematic review of non-human evidence for PFOA effects on fetal growth. Environmental Health Perspectives.
Krauth, D., T. Woodruff, and L. Bero. 2013. A systematic review of instruments for assessing risk of bias and other methodological criteria of published animal studies: A systematic review. Environ. Health Perspect. 121(9):985-992.
Krauth, D., A. Anglemyer, R. Philipps, and L. Bero. 2014. Nonindustry-sponsored preclinical study on statins yield greater efficacy estimates than industry-sponsored studies: A meta-analysis. PLOS Biol. 12(1):e1001770.
Kushman, M.E., A.D. Kraft, K.Z. Guyton, W.A. Chiu, S.L. Makris, and I. Rusyn. 2013. A systematic approach for identifying and presenting mechanistic evidence in human health assessments. Regul. Toxicol. Pharmacol. 67(2):266-277.
Lamontagne, F., M. Briel, M. Duffett, A. Fox-Robichaud, D.J. Cook, G. Guyatt, O. Lesur, and M.O. Meade. 2010. Systematic review of reviews including animal studies addressing therapeutic interventions for sepsis. Crit. Care Med. 38(12):2401-2408.
Landis, S.C., S.G. Amara, K. Asadullah, C.P. Austin, R. Blumenstein, E.W. Bradley, R.G. Crystal, R.B. Darnell, R.J. Ferrante, H. Fillit, R. Finkelstein, M. Fisher, H.E. Gendelman, R.M. Golub, J.L. Goudreau, R.A. Gross, A.K. Gubitz, S.E. Hesterlee, D.W. Howells, J. Huguenard, K. Kelner, W. Koroshetz, D. Krainc, S.E. Lazic, M.S. Levine, M.R. Macleod, J.M. McCall, R.T. Moxley, III, K. Narasimhan, L.J. Noble, S. Perrin, J.D. Porter, O. Steward, E. Unger, U. Utz, and S.D. Silberberg. 2012. A call for transparent reporting to optimize the predictive value of preclinical research. Nature 490(7419):187-191.
Lundh, A, S. Sismondo, J. Lexchin, O.A. Busuioc, and L. Bero. 2012. Industry sponsorship and research outcome. Cochrane Database Syst. Rev. 12:Art. MR000033. doi: 10.1002/14651858.MR000033.pub2.
Macleod, M.R., H.B. van der Worp, E.S. Sena, D.W. Howells, U. Dirnagl, and G.A. Donnan. 2008. Evidence for the efficacy of NXY-059 in experimental focal cerebral ischaemia is confounded by study quality. Stroke 39(10):2824-2829.
Marshall, J.C., E. Deitch, L.L. Moldawer, S. Opal, H. Redl, and T. van der Poll. 2005. Preclinical models of shock and sepsis: What can they tell us? Shock 24(suppl. 1):1-6.
Martin, R.A., A. Daly, C.J. DiFonzo, and F.A. de la Iglesia. 1986. Randomization of animals by computer program for toxicity studies. J. Environ. Pathol. Toxicol. Oncol. 6(5-6):143-152.
Minnerup, J., H. Wersching, K. Diederich, M. Schilling, E.B. Ringelstein, J. Wellmann, and W.R. Schäbitz. 2010. Methodological quality of preclinical stroke studies is not required for publication in high-impact journals. J. Cereb. Blood Flow Metab. 30(9):1619-1624.
NC3Rs (National Centre for the Replacement, Refinement and Reduction of Animals in Research). 2010. The ARRIVE Guidelines. Animal Research: Reporting of in Vivo Experiments [online]. Available: http://www.nc3rs.org.uk/downloaddoc.asp?id=1206&page=1357&skin=0 [accessed Feb. 14, 2014].
Neef, N., K.J. Nikula, S. Francke-Carroll, and L. Boone. 2012. Regulatory forum opinion piece: Blind reading of histopathology slides in general toxicology studies. Toxicol. Pathol. 40(4):697-699.
Nieto, A., A. Mazon, R. Pamies, J.J. Linana, A. Lanuza, F.O. Jimenez, A. Medina-Hernandez, and F.J. Nieto. 2007. Adverse effects of inhaled corticosteroids in funded and nonfunded studies. Arch. Intern. Med. 167(19):2047-2053.
Nkansah, N., O. Mostovetsky, C. Yu, T. Chheng, J. Beney, C.M. Bond, and L. Bero. 2010. Effects of outpatient pharmacists’ non-dispensing roles on patient outcomes and prescribing patterns. Cochrane Database Syst. Rev. 2010(7):Art. No. CD000336. doi: 10.1002/14651858.CD000336.pub2.
NRC (National Research Council). 2011a. Review of the Environmental Protection Agency's Draft IRIS Assessment of Formaldehyde. Washington, DC: National Academies Press.
NRC (National Research Council). 2011b. Guidance for the Description of Animal Research in Scientific Publications. Washington, DC: National Academies Press.
NTP (National Toxicology Program). 2013. Draft OHAT Approach for Systematic Review and Evidence Integration for Literature-Based Health Assessments – February 2013. U.S. Department of Health and Human Services, National Institute of Health, National Institute of Environmental Health Sciences, Division of the National Toxicology Program [online]. Available: http://ntp.niehs.nih.gov/NTP/OHAT/EvaluationProcess/DraftOHATApproach_February2013.pdf [accessed December 11, 2013].
O’Connor, A.M., N.G. Wellman, R.B. Evans, and D.R. Roth. 2006. A review of randomized clinical trials reporting antibiotic treatment of infectious bovine keratoconjunctivitis in cattle. Anim. Health Res. Rev. 7(1-2):119-127.
Odgaard-Jensen, J., G.E. Vist, A. Timmer, R. Kunz, E.A. Akl, H. Schuemann, M. Briel, A.J. Nordmann, S. Pregno, and A.D. Oxman. 2011. Randomization to protect against selection bias in healthcare trials. Cochrane Database Syst. Rev. 2011(4):Art. No. MR0000012. doi: 10,1002/14651858. MR000012. pub3.
OECD (Organization for Economic Cooperation and Development). 2004. Advisory Document of the Working Group on Good Laboratory Practice: The Application of the Principles of GLP to In vitro Studies. OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring No. 14. ENV/JM/MONO(2004)26. OECD, Paris, France [online]. Available: http://www.bfr.bund.de/cm/349/Glp14.pdf [accessed December 6, 2013].
Rhomberg, L.R., K. Baetcke, J. Blancato, J. Bus, S. Cohen, R. Conolly, R. Dixit, J. Doe, K. Ekelman, P. Fenner-Crisp, P. Harvey, D. Hattis, A. Jacobs, D. Jacobson-Kram, T. Lewandowski, R. Liteplo, O. Pelkonen, J. Rice, D. Somers, A. Turturro, W. West, and S. Olin. 2007. Issues in the design and interpretation of chronic toxicity and carcinogenicity studies in rodents: Approaches to dose selection. Crit. Rev. Toxicol. 37(9):729-837.
Rising, K., P. Bacchetti, and L. Bero. 2008. Reporting bias in drug trials submitted to the Food and Drug Administration: Review of publication and presentation. PLoS Med. 5(11):e217.
Robins J.M. 2000. Marginal structural models versus structural nested models as tools for causal inference. Pp. 95-134 in Statistical Models in Epidemiology: The Environment and Clinical Trials, M.E. Halloran, and D. Berry, eds. The IMA Volume in Mathematics and its Applications No. 116. New York: Springer.
Robins, J.M., and M.A. Hernán. 2008. Estimation of the causal effects of time-varying exposures. Pp. 553-600 in Advances in Longitudinal Data Analysis, G. Fitzmaurice, M. Davidian, G. Verbeke, and G. Molenberghs, eds. New York: Chapman and Hall/CRC Press.
Robins, J.M., M.A. Hernán, and B. Brumback. 2000. Marginal structural models and causal inference in epidemiology. Epidemiology 11(5):550-560.
Sargeant, J.M, A.M. O'Connor, I.A. Gardner, J.S. Dickson, M.E. Torrence, I.R. Dohoo, S.L. Lefebvre, P.S. Morley, A. Ramirez, and K. Snedeker. 2010. The REFLECT statement: Reporting guidelines for randomized controlled trials in livestock and food safety: Explanation and elaboration. J. Food Prot. 73(3):579-603.
Schulz, K.F., and D.A. Grimes. 2002a. Blinding in randomised trials: Hiding who got what. Lancet 359(9307):696-700.
Schulz, K.F., and D.A. Grimes. 2002b. Allocation concealment in randomised trials: Defending against deciphering. Lancet 359(9306):614-618.
Schulz, K.F., I. Chalmers, R.J. Hayes, D.G. Altman. 1995. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 273(5):408-412.
Sena, E., H.B. van der Worp, D. Howells, and M. Macleod. 2007. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci. 30(9):433-439.
Sena, E.S., C.L. Briscoe, D.W. Howells, G.A. Donnan, P.A. Sandercock, and M.R. Macleod. 2010. Factors affecting the apparent efficacy and safety of tissue plasminogen activator in thrombotic occlusion models of stroke: Systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 30(12):1905-1913.
Turner, L., L. Shamseer, D.G. Altman, K.F. Schulz, and D. Moher. 2012. Does use of the CONSORT statement impact the completeness of reporting of randomised controlled trials published in medical journals? A Cochrane Review. Syst. Rev. 1:60. doi:10.1186/2046-4053-1-60.
Tsilidis, K.K., O.A. Panagiotou, E.S. Sena, E. Aretouli, E. Evangelou, D.W. Howells, R. Al-Shahi Salman, M.R. Macleod, and J.P. Ioannidis. 2013. Evaluation of excess significance bias in animal studies of neurological diseases. PLOS Biol. 11(7):e1001609.
van der Worp, H.B., D.W. Howells, E.S. Sena, M.J. Porritt, S. Rewell, V. O'Collins, and M.R. Macleod.
2010. Can animal models of disease reliably inform human studies? PLoS Med. 7(3):e1000245.
Vesterinen, H.M., K. Egan, A. Deister, P. Schlattmann, M.R. Macleod, and U. Dirnagl. 2011. Systematic survey of the design, statistical analysis, and reporting of studies published in the 2008 volume of the Journal of Cerebral Blood Flow and Metabolism. J. Cereb. Blood Flow Metab. 31(4):1064-1072.
Watters, M.P., and N.W. Goodman. 1999. Comparison of basic methods in clinical studies and in vitro tissue and cell culture studies reported in three anaesthesia journals. Br. J. Anaesth. 82(2):295-298.
Weed, D.L. 2005. Weight of evidence: A review of concept and methods. Risk Anal. 25(6):1545-1557.
Wellman, N.G., and A.M. O’Connor. 2007. Meta-analysis of treatment of cattle with bovine respiratory disease with tulathromycin. J. Vet. Pharmacol. Ther. 30(3):234-241.
Wells, G.A., B. Shea, D. O'Connell, J. Petersen, V. Welch, M. Losos, and P. Tugwell. 2013. The Newcastle-Ottawa Scale (NOS) for Assessing the Quality of Nonrandomized Studies in Meta-Analyses [online]. Available: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm [accessed December 9, 2013].
Woodruff, T.J., and P. Sutton. 2011. An evidence-based medicine methodology to bridge the gap between clinical and environmental health sciences. Health Aff. (Millwood) 30(5):931-937.






















