
As noted earlier (Chapter 3), assessment of human health hazards should be included in each alternatives assessment. Human health hazard assessment of chemical alternatives is very similar to the hazard identification step of a traditional risk assessment. They are similar in that the types of adverse health end points and the sources of data for decisions are largely identical. Chemical alternatives assessments, however, typically use a comparative approach and are not meant to emulate formal dose-response or weight-of-evidence mode-of-action evaluations found in other chemical hazard assessments.
As shown in Figure 8-1, human health assessment is Step 6.1 of the committee’s overall framework. Box 8-1 provides the elements of the committee’s suggested approach.
TYPES OF DATA FOR HUMAN HEALTH ASSESSMENT
As illustrated in Figure 8-2, an implicit hierarchy exists with respect to the sources of data that are used in chemical assessments. Knowledge obtained from controlled clinical studies in humans is arguably the most desirable data for decisions on the potential for human health hazard. With the exception of pharmaceuticals, very few chemicals have this type of data available. Epidemiological studies of various designs are the next most useful data source because they examine whether there is an association between chemical exposure and human health.
The main advantage of these studies is that they involve humans; however, they are difficult to conduct and human evidence of chemical-induced effects, especially chronic effects, is rarely available. Data from experimental animal studies are often used to draw inferences about the potential hazard to humans when no adequate human data are available, yet the uncertainties associated with extrapolating the results from traditional toxicity studies in animals to humans are frequently poorly characterized. Other types of data, including results from studies in invertebrates, microorganisms, or in vitro experiments in animal or human cells, are also useful for traditional risk assessments. However, such data are most frequently used to determine the chemical mode of action. Traditionally, they have not been widely used to identify human health hazards beyond predicting specific hazard end points, such as genotoxicity, skin irritation, and eye irritation. Data from in vitro and in silico models, however, are, or will be, available for a far larger number of chemicals than experimental or epidemiological data will be (Collins et al. 2008). Thus, it is likely that at some point in the future, most decisions about environmental health protection will be made with in vitro and in silico33 data and models, rather than traditional data (NRC 2007).
There are several different approaches for using the various levels of human health related data in an alternatives assessment:
- a. Using traditional data, such as those that can be classified by the United Nations Globally Harmonized System of Classification and Labelling of Chemicals34 (GHS) criteria. This is the typical approach used by alternatives assessments and is illustrated in the DecaDBE example in Chapter 12.
- b. Using traditional data in combination with the use of new types of in vitro screening and in silico data as another type of primary data (for end points where this is deemed appropriate) or to fill data gaps.
_____________
33 The term in silico is used here to describe prediction and modeling (typically computational modeling) of effects based on information about a chemical’s structure or physicochemical characteristics, including but not limited to structural alerts and structure activity relationship analysis.
34 GHS health hazards are agreed upon internationally for characterizing chemical hazards (76 Fed. Reg. 40850 2011). The Organisation for Economic Co-operation and Development High Production Volume Screening Information Data Set endpoints (OECD 2005), EU’s Classification and Labeling and Packaging of Products regulation (EC 2011) and the U.S Occupational Safety and Health Hazard Communication Standard (77. Fed. Reg.17574 2012) are aligned with GHS. GHS criteria have been established for a number of human health end points.
- c. A hybrid of a and b, which uses traditional data along with screening chemicals of concern with in vitro screening data and in silico modeling. This approach is illustrated in the glitazone example in Chapter 12.
There is interplay between health concerns, available data streams, and expertise that will contribute to the type of approach used. In any case, the type of approach used should be described in Step 2, as part of formulation and scoping. That said, the committee strongly supports a movement toward using in vitro screening and in silico data to fill data gaps when the necessary information is not available in the more traditional epidemiological and animal testing data. The committee points out later in this chapter that many high throughput in vitro assays may still have only limited applicability as primary data for predicting in vivo chemical hazards. The committee does, however, believe that the science will continue to advance in this area and that even today, there are opportunities to fill data gaps or screen for unexpected consequences using high throughput in vitro assays and in silico approaches.
To build on existing approaches, this chapter first describes how human health has been considered in existing alternatives assessment approaches and then describes the committee’s framework for evaluating chemicals using traditional human health data in alternatives assessments. Second, the chapter provides more information on the state-of-the science of in vitro and in silico data, by health end point—showing where the science is in terms of predictivity and where the challenges remain. Third, the committee describes three scenarios of how novel in vitro and in silico data can be used in the context of chemical alternatives assessments and illustrates how visualization tools can inform the stakeholders about information available to help them make a decision about alternatives. Lastly, the committee offers advice on the path forward in using existing health data, as well as in vitro and in silico data, in chemical alternatives assessments.
BOX 8-1
HUMAN HEALTH ASSESSMENT AT A GLANCE
Phase 1. Evaluate the required health end points (identified in Steps 2) that must be addressed in the alternatives assessment
- Gather available data on health hazards associated with the chemical of interest and alternatives.
- Use authoritative lists to record previously identified health concerns.
- Use GHS criteria and hazard descriptors to the fullest extent possible to assess data, including potential effects on vulnerable populations, and classify the hazard data as indicating high (H), medium (M), or low (L) hazard. Indicate whether certainty of this classification is high, medium, or low.
- When conducting assessments of chemicals for reproductive toxicity and other health end points that require expert judgment to apply GHS criteria, use existing health hazard assessment guidance to ensure consistency and transparency.
- Where appropriate (e.g., for genotoxicity), use in vitro and in silico as primary data for an end point of concern (e.g., mutagenicity).
- Identify data gaps.
Phase 2. Develop strategies to address data gaps
- Use in vitro and in silico data and models to fill data gaps for an end point of concern (e.g., endocrine toxicity).
- Remaining data gaps should be classified as “No Data.”
Phase 3. Develop a graphic or tabular display of health hazards associated with the chemical of interest and alternatives.
- Tabulation should include a placeholder for the full range of health end points typically considered in alternatives assessment, indication of which end points were considered, which end points warranted a H, M, L hazard level, which end points were based on novel in vitro or in silico approaches, and the certainty associated with each end point.
- ToxPi and similar approaches may be useful for visualizing novel high throughput data sets.
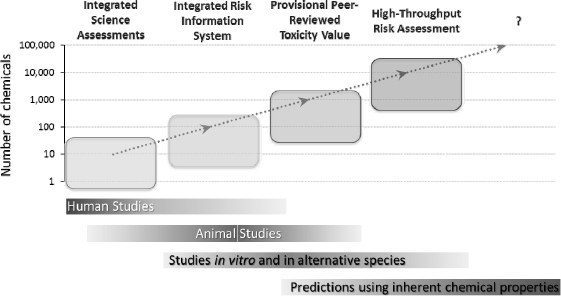
FIGURE 8-2 Decision contexts, data type, and data availability determine the type of human health assessment that can be performed on chemicals. The examples shown illustrate assessments performed by the EPA (EPA/NCEA).
HOW HUMAN HEALTH IS CONSIDERED IN EXISTING FRAMEWORKS
End Points Considered in Existing Frameworks
The committee considered the human health end points described in eight existing frameworks to compare current practices related to evaluating health hazards in chemical alternatives assessments and to inform the development of the committee’s framework. Table 8-1 shows specific health end points; prioritized end points; the criteria and information sources the reviewed frameworks use to evaluate chemicals based on specific end points; and the types of data (e.g., human, animal, in vitro) upon which the criteria and source information are based. Appendix D provides more details on health end points and their evaluation in existing frameworks.
While the existing alternatives assessment frameworks are not identical, they contain common end points of concern, including carcinogenicity, mutagenicity, reproductive and developmental toxicity, endocrine disruption, acute and chronic or repeat dose toxicity, dermal and eye irritation, and dermal and respiratory sensitization. Several frameworks go further by identifying priority end points (e.g., carcinogenicity, mutagenicity/genotoxicity, reproductive toxicity, developmental toxicity, and endocrine toxicity). In determining which are “priority” end points, many of the frameworks use essentially the same rationale or basis—serious or irreversible health effects, or effects that may be transferred between generations and caused by low exposures to toxicants.
With two exceptions (endocrine activity/toxicity and epigenetic toxicity), the health end points in Table 8-1 align closely with the health hazards identified in the GHS. For example, the GHS defines acute mammalian toxicity as “adverse effects occurring following oral or dermal administration of a single dose of a substance, or multiple doses given within 24 hours, or an inhalation exposure of 4 hours” (UNECE 2013c). Regarding their acute toxicity, chemicals are classified into five hazard categories based on animal LD50 (oral, dermal) or LC50 (inhalation) values.
Endocrine toxicity is not included as a health hazard in the GHS. However, several frameworks identify endocrine-related health effects as an end point of concern. The criteria used in the frameworks vary because endocrine effects are not defined uniformly across frameworks. Data and authoritative lists are used to provide evidence of endocrine activity and/or disruption. For example, the DfE framework evaluates endocrine activity of chemicals, but does not characterize hazard in terms of endocrine disruption. On the other hand, the IC2 and BizNGO frameworks use criteria developed by GreenScreen® to evaluate chemicals for endocrine activity and assign hazard values based on adverse
TABLE 8-1 Health End Points Established by Other Frameworks
| Human Health End Point (more detail in Appendix D) | Criteria and Information Sources Frameworks Use to Establish Evidence of the Human Health End Points | Types of Data Used to Establish the Health End Points | ||||
| Human | Animal | Human & Animal (WOE) | In Vitro | |||
| Acute Mammalian Toxicity1,2,4,5,6,7,8 Priority7 |
GHS criteria 1,2,4,5,6,8 Authoritative lists/databases2,4 EU Risk phrases, Hazard statements2,4,8 NIOSH7 | MSDS7 IDLH7; LD50/LC507 OELs (NIOSH, OSHA, ACGIH)7 HSDB7; RTECS7 | OELs | OELs GHS 1-5; LD50/LC50; IDLH | OELs | |
| Carcinogenicity1,2,3,4,5,6, 7,8 Priority2,3,4,5,7 | GHS criteria1,2,3,4,5,6,7,8 Authoritative lists 2,3,4,6,7,8 | EU Risk phrases, Hazard statements2,3,4,8 | GHS 1A | GHS 1B | GHS 1B, 2 | |
| Mutagenicity/Genotoxicity1,2,3,4,5,6,7,8 Priority 2,3,4,5, | GHS criteria 1,2,3,4,5,6, 7,,8 Authoritative lists 2,3,4,6,7,,8 | EU R-phrases, Hazard statements2,3,4,5,6,7,,8 | GHS 1A | GHS 1B | GHS 1B; 2 | |
| Reproductive Toxicity1,2,3,4,5,6,7,8 Priority 2,3,4,5, | GHS criteria2,3,4,5,6,7,8 EPA OPPT criteria (HPV)1 REACH criteria (Annex IV)1 | Authoritative lists2,3,4,6,7,8 EU Risk phrases, Hazard statements2,3,4,6,7,8 RTECS7 | GHS 1A | GHS 1B | GHS 1B; 2 | |
| Developmental Toxicity1,2,3,4,5,6,7,8 Priority 2,3,4,5, | GHS criteria2,3,4,5,6,7,8 Authoritative lists2,3,4,6,7,8 EPA OPPT criteria (HPV)1 | EU Risk phrases, Hazard statements2,3,4,6,7,8 REACH criteria (Annex IV)1 | GHS 1A | GHS 1B | GHS 1B; 2 | |
| Neurotoxicity3 †Single Exposure (SE)2,4 † †Repeated Exposure (RE)1,2,4 Priority3 |
GHS criteria1,2,4 Neurotoxicants (ATSDR; EPA IRIS)3 | EU Risk phrases, Hazard statements2,4 Authoritative lists/databases2,4, | GHS 1(a); 3; ATSDR; IRIS (SE) GHS 1(a) ATSDR; IRIS (RE) | GHS 1(b); 2 ATSDR; IRIS (SE) GHS 1(b); 2 ATSDR; IRIS (RE) |
ATSDR; IRIS (SE) ATSDR; IRIS (RE) |
|
| ††Repeated Dose Toxicity1,5,7 | GHS criteria1,2,4 Authoritative lists 2,4 | EU Risk phrases, Hazard statements2,4 EPA IRIS Reference Doses (RfDs)7 | GHS 1(a) EPA RfDs | GHS 1(b) EPA RfDs | EPA IRIS RfDs | |
| Systemic Toxicity/Organ Effects2,4,6,7,8 †Single Exposure (SE) ††Repeated Exposure (RE) | GHS criteria2,4,6 Authoritative lists2,4 | EU Risk phrases, Hazard statements8 EPA IRIS Reference Doses (RfDs)7 | GHS 1(a); 3 (SE) GHS 1(a) (RE) | GHS 1(b); 2 (SE) GHS 1(b); 2 (RE) | EPA RfDs | |
| Human Health End Point (more detail in Appendix D) | Criteria and Information Sources Frameworks Use to Establish Evidence of the Human Health End Points | Types of Data Used to Establish the Health End Points | ||||
| Human | Animal | Human & Animal (WOE) | In Vitro | |||
| Respiratory Sensitization1,2,3,4,5, Priority3,5 |
GHS criteria 1,2,4,5 Authoritative lists /databases2,4 | EU Risk phrases, Hazard-statements2,4 EU Annex VI Category 13 | GHS 1A; 1B | |||
| Skin Sensitization1,2,4,7,8 | GHS criteria1,2,4, Authoritative lists2,4 | EU Risk phrases, Hazard-statements2,4,8 HSDB7; Sax7; MSDSs7 | GHS 1A;1B | GHS 1A; 1B | ||
| Skin & Eye Irritation /Corrosivity 1,2,4,5,7,8 | GHS criteria2,4,5,8 Authoritative lists 2,4,8 NIOSH7 | EU Risk phrases, Hazard-statements2,4,8 | HSDB; MSDSs; NIOSH | GHS 1, 2A, 2B | REACH skin irritation | |
| Respiratory Irritation5,7 | EPA Office of Pesticide Programs 1 | HSDB7 MSDSs7 | & corrosion tests | |||
| Endocrine Activity1,2,4,7/Toxicity3,5,6,7,8 Priority 2,3,4,5 | All available data2,4,7 Authoritative lists2,3,4,6,8 | |||||
| Epigenetic Toxicity 6 | No information provided | NA | NA | NA | NA | |
1DfE; 2 IC2; 3CA SCP; 4 BizNGO; 5REACH; 6UCLA MCDA (Malloy et al. 2013); 7TURI; 8German Guide; †GHS Specific Target Organ Toxicity –Single Exposure (see Appendix D); ††GHS Specific Target Organ Toxicity –Repeated Exposure (see Appendix D).
Acronyms: OELs= Occupational Exposure Limits. AOEC=Association of Occupational and Environmental Clinics database of asthmagens. HSDB= Hazardous Substances Data Bank. CLP= ECHA’s Classification and Labelling Inventory database. GHS 1A and 1B refer to GHS categories, which are explained in Appendix D. ACGIH = American Conference of Industrial Hygienist, ATSDR = Agency for Toxic Substances and Disease Registry, GHS = Globally Harmonized System of Classification and Labelling of Chemicals, HPV = High Production Volume, HSDB = Hazardous Substances Data Bank, IDLH = Immediately Dangerous to Life or Health, IRIS = Integrated Risk Information System, LC50 = Lethal Concentration that kills 50% of population, LD50 = Lethal Dose that kills 50% of population, MSDS = Material Safety Data Sheets, NIOSH = National Institute for Occupational Safety and Health, OELs = Occupational Exposure Limits, OPPT = Office of Pollution Prevention and Toxics, OSHA = Occupational Safety and Health Administration, RfDs = Reference Doses, RTECS = Registry of Toxic Effects of Chemical Substances.
Information in the table was obtained from a review of guidance documents, regulations, and other available information on the frameworks. Specifically, the guidance document for the hazard assessment tool, GreenScreen® (Clean Production Action 2013), is the source for information on the IC2 and BizNGO health end points. The California Safer Consumer Products (CA SCP) framework’s health end points are the hazard traits that are used for listing a chemical as a “Candidate Chemical” or a potential “Chemical of Concern” in a priority consumer product (CA DTSC 2013c). The health end points for the UCLA Multi-Criteria Decision Analysis (MCDA) framework are the eight measures linked to the toxicity sub-criterion (associated with human health impacts) in a generic alternatives assessment model (Malloy et al. 2013). The REACH health end points are those specified in the REACH legislation (EC 2007). The health end points for the German Guide on Sustainable Chemicals framework (Reihlen et al. 2011) are based on the risk phrases and hazard statements used to identify high- priority chemicals for substitution (“red color code”). Two additional frameworks, the Lowell Center Alternatives Assessment Framework (Rossi et al. 2006) and the UNEP Persistent Organic Pollutants Review Committee General Guidance on Alternatives (UNEP 2009) framework, which are reviewed in other sections of the report, do not identify human health end points and are not included here.
endocrine-related health effects. Additional information about criteria the frameworks use to provide evidence of endocrine-related health effects is presented in Appendix D. Appendix D also discusses how other end points are characterized by the GHS classification scheme and their application in the GreenScreen® tool and DfE framework. Although GHS is widely used, different approaches have also been used to inform other alternatives assessment frameworks (e.g., TURI).
Information Sources Used by Existing Frameworks
Table 8-1 shows that frameworks use a variety of information sources, including authoritative lists and databases, to establish evidence of health end points when evaluating chemicals. Some of the frameworks specify review of all available, relevant information, including information obtained from searches of the scientific literature. For example, the EPA DfE framework uses primary data sources, public and confidential business information, expert predictive models, and other forms of expert judgment to characterize health hazards (Lavoie et al. 2010).
Authoritative lists are used extensively as the basis for alternatives assessments (i.e., as a reason for entry into Step 1 of the committee’s framework). Authoritative lists, databases, and risk phrases35 are also used to assess the health impacts of potential substitute chemicals. Several of the examined frameworks (e.g., IC2 and BizNGO) rely on the GreenScreen® chemical hazard assessment tool, which uses authoritative lists and databases to classify chemical health hazards. GreenScreen® defines “authoritative list” as “those lists developed by governmental bodies or government recognized expert bodies and include chemicals that are listed based on results from expert review of test data and scientific literature”. The hazard lists that are required for a full GreenScreen® are called GreenScreen® Specified Lists (Clean Production Action 2012) and also include screening lists. According to GreenScreen®, “lists are identified as Screening Lists if they were developed using a less comprehensive review; or if they have been compiled by an organization that is not considered to be authoritative; or if they are developed using exclusively estimated data; or if the chemicals are listed because they have been selected for further review and/or testing, and result in a classification with a lower level of confidence.”36 Table 8-2 provides an example of how authoritative lists are used by the DfE framework and the GreenScreen® tool.
The IC2 and BizNGO frameworks also use authoritative lists (with GreenScreen®-assigned hazard levels) to establish evidence of the reproductive toxicity health end point.
Below are some of the lists included in their frameworks:
- High Hazard = NTP-OHAT (Clear Evidence of Adverse Effects-Reproductive);
- CA Prop 65 (known to the state to cause reproductive effects--male or female);
- H360F (may impair fertility);
- EU H360FD (may damage fertility); and
- EU 360Fd (may damage fertility).
Authoritative lists used by GreenScreen® are divided into A37 and B lists. The A and B lists are
_____________
35 Risk phrases were developed in the European Union (prior to adoption of the GHS Classification and Labelling System) to communicate risk. They are based on criteria that are essentially the same as GHS criteria and are being replaced by hazard statements based on GHS criteria. Both risk phrases and hazard statements are examples of authoritative lists.
36 Although the types of lists the frameworks use are defined and explained in the GreenScreen guidance document (Clean Production Action 2013), some questions and issues remain. For example, it is not clear: (a) how lists are selected; (b) why some lists are used and others are not; (c) to what extent scientific rigor determines the level of confidence in lists; and (d) how the level of confidence in lists impacts the selection of safer alternative chemicals. The use of authoritative lists is discussed further in Appendix D.
37 Authoritative A lists include: IARC Group 1 or 2A chemicals (carcinogenicity); EU CMR Category 1 or 2 chemicals [mutagenicity/genotoxicity]; and chemicals classified as H360F (may damage fertility), H360FD (may damage fertility; may damage the unborn child) and 360df (may damage the unborn child, suspected of damaging fertility) [reproductive and developmental toxicity]. Authoritative B lists include: IARC Group 3 chemicals (carcinogenicity); MAK Germ cell mutagens 1, 2, or 3a chemicals (mutagenicity/genotoxicity); chemicals classified as EU H334 (respiratory sensitization); and DOT Class 2,3 Group B chemicals (acute mammalian toxicity). Screening A lists include, predominantly, GHS lists of various countries, including Korea, Japan, Indonesia, and Australia for several end points (e.g., carcinogenicity, developmental toxicity, acute mammalian toxicity). Screening B lists include: WHMIS D1B chemicals (acute mammalian toxicity); OSPAR (endocrine disruptor); and MAK Pregnancy Risk Group D chemicals.
TABLE 8-2 Use of Authoritative Lists by the DfE Framework and GreenScreen® Tool
| End Point/List Classification | DfE Classification | GreenScreen® Tool |
| Carcinogenicity | ||
| NTP—Known to be human carcinogen | Very High Hazard | High Hazard |
| NTP—Reasonably Anticipated to be a Human Carcinogen | ||
| IARC Group 1—Carcinogenic to Humans | ||
| IARC Group 2A—Probably Carcinogenic to Humans | ||
| GHS H350—May Cause Cancer | ||
| GHS H350i—May cause cancer by inhalation | ||
| IARC 2B—Possibly carcinogenic to humans | High Hazard | Moderate Hazard |
| EU CMR List Category 3—Cause for concern for humans owing to possible carcinogenic effects | ||
| EU 351—Suspected of causing cancer | ||
| Mutagenicity/genotoxicity | ||
| EU CMR Category 1—Substances known to be mutagenic to man | Very High Hazard | |
| Category 2—Substances which should be regarded as if they are mutagenic to man | ||
| EU H340—May cause genetic defects | ||
distinguished based on whether categories in the list translate directly into a single level of concern for a single GreenScreen® health end point or a single benchmark. In addition, the assigned health hazard level of an Authoritative A list cannot be modified using additional data; Authoritative B lists, however, can be modified. The confidence level is “high” for Authoritative A lists (Clean Production Action 2012, 2013). For Authoritative B lists, the confidence level is “low” in the current guidance document (Clean Production Action 2013), but is listed as “high” on the Specified List (Clean Production Action 2012).
The TURI framework also uses material safety data sheets (MSDSs), which in the future must be based on GHS criteria, as a source of information to compare the toxicity of chemicals 38 Another data source is the Hazardous Substances Data Bank (HSDB).39 This database focuses on the toxicology of potentially hazardous chemicals and includes up-to-date abstracts of animal and human studies, including studies on the acute and chronic toxicity of chemicals. The abstracts undergo peer review before they are added to the database; however, the studies are not evaluated, and expert judgment is required to determine their relevance in providing evidence of chemical toxicity.
Use of Hazard Classification Levels in Existing Frameworks
To facilitate comparison of hazard levels across chemicals, some frameworks use hazard classification levels to describe information about the severity of the effect. Hazard classification levels are used most extensively in the DfE framework (Davies et al. 2013) and the GreenScreen® tool (Heine and Franjevic 2013).
For each human health end point considered in DfE and GreenScreen®, a descriptor is assigned based on criteria that constitute High, Moderate, or Low and, in some cases, Very High or Very Low (Davies et al. 2013). As discussed earlier, these
_____________
38 Under the revised Hazard Communication Standard (77 Fed. Reg. 17574 2012), MSDSs will be renamed Safety Data Sheets, or SDS, and based on GHS criteria, which should make them a good information source. As of now, however, the committee’s comparison of harmonized and un-harmonized chemical classifications for the acute toxicity end point in the European Chemical Agency (ECHA) Classification and Labeling Inventory Database, using the H330 and H311 hazard statements, showed a ten-fold difference in the number of classified chemicals (ECHA 2014e). This indicates that un-harmonized chemical classifications (by individual manufacturers and other safety data sheet preparers) may be inconsistent or inaccurate.
39 Available at http://toxnet.nlm.nih.gov/cgibin/sis/htmlgen?HSDB
descriptors are usually based on GHS criteria. Table 8-3 shows acute mammalian toxicity levels by different exposure routes. It indicates that the hazard classification levels described by DfE can range from Very High Hazard = (Category 1 or 2) to Low (Category 5). Appendix D has a more detailed description of the hazard level classification systems in DfE and GreenScreen® for various end points. The hazard profile and assigned concern levels are ultimately reviewed by a group of experts before they are used in decision-making.
GreenScreen® uses a similar overall process of assigning High, Moderate, and Low classification levels. It groups human health hazard end points in the following way:
- Group I hazards can lead to chronic or life-threatening effects or adverse impacts that are potentially induced at low doses and transferred between generations. Group I end points include carcinogenicity, mutagenicity/genotoxicity, reproductive toxicity, developmental toxicity (including neurodevelopmental), and endocrine activity.
- Group II/II* hazards are additional end points that are necessary for understanding and classifying hazards (Heine and Franjevic 2013). Group II end points are acute mammalian toxicity, systemic toxicity/organ effects (single exposure), neurotoxicity (single exposure), and irritation/corrosivity for eyes and skin. Group II* end points include systemic toxicity/organ effects (repeated exposure), neurotoxicity (repeated exposure), and respiratory and skin sensitization.
Approaches to Handling Data Gaps in Existing Frameworks
Comparative chemical alternatives assessments, similar to more traditional human health risk assessments, are only as good as the data and information available. Identifying and addressing the potential impacts of health hazard data gaps in alternatives assessments is an important issue because it can help ensure that what are thought to be safer chemical substitutes do not subsequently pose health concerns. The extent to which data gaps and related issues are discussed in the frameworks varies widely.
The CA SCP framework does not require new data collection to address data gaps. Gaps in toxicity and health effects information are acknowledged in the German and TURI frameworks and noted in health hazard summaries, but are not discussed further. When primary data are not available or deemed inadequate, DfE has explicit procedures in place to assign a hazard concern level based on structure-activity relationship (SAR) considerations and professional judgment. These procedures ensure that all end points are covered so that the hazard profile can be completed. Similarly, GreenScreen® specifies review of at least one readily available suitable analog for each hazard end point for which data are missing on the parent compound, stating that expert judgment and estimated data from analog and SAR analyses may be used in lieu of measured data. If information is still deemed insufficient to provide any classification for a hazard end point, as is frequently the case, the end point is assigned a “data gap” or “no data” designation. For example, GreenScreen® states that a data gap exists when measured data and authoritative screening lists have been reviewed, and expert judgment and estimation such as modeling and analog data have been applied, and there is still insufficient information to assign a hazard level.
With regard to how any remaining data gaps are handled in the final analyses, a range of possibilities is described in Chapter 9. The UCLA MCDA framework evaluates the impact of data gaps in an alternatives assessment using multi-attribute utility theory and outranking. The GreenScreen® tool (and by extension, the IC2 and BizNGO frameworks) is the only example found to describe how data gaps are handled in the analysis. GreenScreen®’s procedure defines the minimum data requirements to achieve a given benchmark and describes the required data and permissible data gaps for each hazard end point category (Group I Human and Group II and II* are specified). The treatment of gaps, or failure to meet minimum data requirements, is negative as opposed to neutral and is benchmark-specific. For example, if a chemical meets Benchmark 2 based on hazard analysis but fails to meet the minimum requirements for this benchmark because of gaps in data, it is assigned an “unspecified” designation. If a chemical fails to meet the minimum data requirements for Benchmark 3 in the gap analysis, it is downgraded to 2DG. No data gaps are allowed for Benchmark 4.
TABLE 8-3 Acute Mammalian Toxicity
| Very High | High | Moderate | Low | |
| Oral LD50 (mg/kg) | ≤ 50 | < 50-300 | > 300-2000 | > 2000 |
| Dermal LD50 (mg/kg) | ≤ 200 | > 200-1000 | > 1000-2000 | > 2000 |
| Inhalation LC50 (vapor/gas) (mg/L) | ≤ 2 | > 2-10 | > 10-20 | > 20 |
| Inhalation LC50 (dust/mist/fume) (mg/L/day) | ≤ 0.5 | > 0.5-1.0 | > 1.0-5 | > 5 |
SOURCE: EPA 2011a
HUMAN HEALTH IN THE COMMITTEE’S FRAMEWORK
For its recommended framework, the committee suggests that the assessment of human health hazards follow a similar process as that used by the frameworks described above, with these two modifications included:
- Consider the full range of scientific information to fill data gaps (see below).
- Continued focus on hazard as opposed to risk, except when directed otherwise by comparative exposure or decision rules.
The committee’s framework for human health assessment would begin with the following end points, which are GHS health hazards, supplemented with endocrine activity that is not included in GHS at this point in time.40
- Acute toxicity
- Carcinogenicity
- Mutagenicity/genotoxicity
- Reproductive toxicity
- Development toxicity
- Respiratory sensitization
- Skin sensitization
- Specific target organ toxicity (single exposure):
- Neurotoxicity
- Respiratory irritation
- Specific target organ toxicity (repeated exposure):
- Neurotoxicity
- Skin and eye corrosion/irritation
- Endocrine activity
The committee did not strictly define the above list as a minimum set of adverse health end points to be considered in alternatives assessment, but suggests that this list be used as the initial list for selecting end points in the problem formulation exercise in Step 2, with clear documentation of which end points were not considered in Step 6.1 of the assessment. Additional end points considered in the assessment also should be specified and clearly documented.
The committee advises using GHS criteria and hazard descriptors to the fullest extent possible in evaluating human health hazards, which is consistent with what is described in Chapter 7 for ecotoxicity. This approach is also consistent with several existing approaches that use GHS as their ultimate common denominator in human health assessment. The use of health end points that are aligned with health hazards identified in GHS ensures that assessments address internationally recognized chemical hazards. In addition, using GHS criteria enables alternatives assessments to use toxicity information on chemicals submitted as Screening Information Data Sets (SIDS) for the OECD SIDS program because GHS criteria include SIDS end points. EPA’s Office of Pollution Prevention and Toxics is making non-confidential information submitted to this program publicly available (EPA 2007), enabling assessment of unpublished data, which is especially important for assessments of chemicals for which there are no published toxicity studies. This information should reduce data gaps. In addition, GHS alignment enables information from other resources, such as the ECHA Classification and Labeling Inventory Database and guidance documents, to be used in alternatives
_____________
40 The descriptors for the Mutagenicity/Genotoxicity and Skin and Eye Corrosion/Irritation end points listed for the committee’s framework do not conform to the GHS health hazard descriptors. The rationales for this are in Appendix D and would be included in the problem formulation section of the framework.
assessments. Another advantage of GHS alignment is that it links safer alternatives directly to workplace chemical hazards identified under the GHS-based OSHA Hazard Communication Standard.
While supporting the use of GHS criteria, the committee suggests the following refinements to the reliance on GHS criteria and hazard descriptors:
- Use available human data when GHS criteria indicate that they should be used.41
- Describe criteria used. When non-GHS criteria are used, explain the rationale.
- Align the description of GHS hazards to the GHS criteria. If there is a rationale for using a hazard description that is different from the ones used by GHS, explain this rationale and how to apply the criteria. An example of misalignment between GHS hazards and GHS criteria is use of the GHS criteria for “Specific Target Organ Toxicity (Repeated Exposure)” when referring to the health end point as “Systemic Organ Toxicity/Organ Effects (Repeated Exposure).”
The committee’s framework advises using authoritative lists, as has been done by a number of existing approaches. The rationale for using such health end point-specific authoritative lists to compare chemical hazards is that it maximizes the use of existing evaluations of scientific information and helps ensure that alternatives assessments are efficient and based on consistent science. Assessments following the committee’s framework would:
- Define “authoritative” lists.
- Describe criteria for which authoritative lists are used or not used in the framework.
- Include end point-specific authoritative lists of toxicants developed by government agencies that use human or animal data. For example, Agency for Toxic Substances and Disease Registry Minimal Risk Levels (ATSDR 2013), California Environmental Protection Agency Office of Environmental Health Hazard Assessment acute and chronic reference exposure levels (OEHHA 2014), and respiratory irritants identified by the National Institute for Occupational Safety and Health (NIOSH 2005), and others that include human data in the assessments.
- Ensure that the listing criteria are transparent, understood by the assessor, and consistent with the criteria used to establish evidence of the health end point that the list is addressing.
- Use authoritative lists only to identify hazards
Assessments following the committee’s framework would consider existing health hazard assessment guidance to classify chemicals based on their end point effects, when GHS criteria require the use of expert judgment to establish a health end point. It is not clear whether some existing frameworks do this, but assessments following the committee’s framework would do so in a transparent way when conducting de novo assessment to classify chemicals for reproductive toxicity and other end points. Box 8-2 describes examples of existing health hazard assessment guidance and includes EPA risk assessment guidelines for reproductive toxicity, neurotoxicity, and developmental toxicity.
Notably different from several existing frameworks, the output of the committee’s framework would not include a “score” integrating human health data across health end points. Instead, the committee’s framework would tabulate (a potential format is shown in Table 8-4) health end points, noting: which end points were considered; which of the typically assessed end points were not considered; indication of the hazard level suggested by the data (H,M,L); and an indication of the certainty of the data (known, limited certainty, highly uncertain). Gaps in data at this point would be clearly indicated.
The resulting tabulation makes no attempt to integrate information across health end point domains for three primary reasons: 1) there is no established consensus on which end points are of greater concern; 2) doing so unnecessarily carries forward the impact of benchmarking cutoffs; and 3) it is important to carry forward the certainty and the level of the hazard into the integration of other data in the decision-making step (Step 7). This approach is in contrast to the common approach of creating scores that integrate information and account for data gaps and uncertainty at this point in the process. While such approaches may be easy to use, they obscure information that should be considered across domains. Ideally, gaps would be addressed using novel high throughput in vitro data and in silico modeling, as described in the rest of the chapter.
_____________
41 In some existing frameworks, it is not clear if human data are used when prescribed by GHS criteria.
Note: More specifics about minimal evidence requirements are described in the risk assessment guidelines.
- EPA’s Risk Assessment Guidelines for Neurotoxicants (EPA 1998)
Sufficient Experimental Animal Evidence/Limited Human Data
The minimum evidence necessary to judge that a potential hazard exists would be data demonstrating an adverse neurotoxic effect in a single appropriate, well-executed study in a single experimental animal species.The minimum evidence needed to judge that a potential hazard does not exist would include data from an appropriate number of end points from more than one study, and two species showing no adverse neurotoxic effects at doses that were minimally toxic in terms of producing an adverse effect. Information on pharmacokinetics, mechanisms, or known properties of the chemical class may also strengthen the evidence.
- EPA’s Risk Assessment Guidelines for Developmental Toxicants (EPA 1991)
Sufficient Experimental Animal Evidence/Limited Human Data
The minimum evidence necessary to judge that a potential hazard exists generally would be data demonstrating an adverse developmental effect in a single appropriate, well-conducted study in a single experimental animal species.The minimum evidence needed to judge that a potential hazard does not exist would include data from appropriate, well-conducted laboratory animal studies in several species (at least two) that evaluated a variety of the potential manifestations of developmental toxicity and showed no developmental effects at doses that were minimally toxic to the adult.
- EPA’s Risk Assessment Guidelines for Reproductive Toxicants (EPA 1996)
Sufficient Experimental Animal Evidence/Limited Human Data
The minimum evidence necessary to determine if a potential hazard exists would be data demonstrating an adverse reproductive effect in a single appropriate, well-executed study in a single test species.The minimum evidence needed to determine that a potential hazard does not exist would include data on an adequate array of endpoints from more than one study, with two species that showed no adverse reproductive effects at doses that were minimally toxic in terms of inducing an adverse effect. Information on pharmacokinetics, mechanisms, or known properties of the chemical class may also strengthen the evidence.
IN VITRO DATA AND IN SILICO MODELS FOR CHEMICAL ALTERNATIVES ASSESSMENTS
Released in 2007 by the National Research Council (NRC), the report, “Toxicity Testing in the 21st Century (TT21C): A Vision and a Strategy” (NRC 2007), described the promise of high throughput in vitro approaches and in silico models in evaluating chemical safety. The idea that these novel approaches could replace animals in toxicity testing has been treated by some with skepticism and claims of unrealistic overreaching (Bus and Becker 2009; Meek and Doull 2009). But significant research investments have revealed numerous advantages to using high throughput methods in toxicology (Krewski et al. 2011; Kavlock et al. 2012) and led to the generation of a vast amount of data (Table 8-5) that are in the public domain and available for analysis and evaluation in hazard identification and dose-response assessments (Tice et al. 2013). Advances in molecular, cell, and systems biology, together with advanced analytical methods in biostatistics, bioinformatics, and computational biology, have led to toxicity testing now being routinely conducted in vitro by evaluating cellular responses in a suite of toxicity pathway-centric assays.
In silico approaches for predicting adverse effects have existed for more than 30 years, but research and development in this area has increased exponentially in recent years. Most in silico methods for toxicity prediction have focused on hazard identification; for example, determining whether a compound has properties associated with liver injury. The concept of chemical similarity has been used to develop a variety of methods to predict
TABLE 8-4 Hypothetical Tabulation Evaluations of Human Health Impact: One Potential Format.*
| Human Health Impacts | |||||||||||||||||||||||||||||||||||
| Alternatives | Acute Toxicity | Carcinogenicity | Mutagenicity/Genotoxicity | Reproductive Toxicity | Developmental Toxicity | Specific Target Organ Toxicity (Single Exposure)—Neurotoxicity | Specific Target Organ Toxicity (Single Exposure)—Respirator Irritation | Specific Target Organ Toxicity (Repeated Exposure) - Neurotoxicity | Specific Target Organ Toxicity (Repeated Exposure)—Respiratory Irritation | Respiratory Sensitivity | Endocrine Activity | Skin Sensitivity | Skin and Eye Corrosion/Irritation | ||||||||||||||||||||||
| H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | H | M | L | |||
| C | N C |
N C |
|||||||||||||||||||||||||||||||||
| A | |||||||||||||||||||||||||||||||||||
| B | |||||||||||||||||||||||||||||||||||
*High, medium, and low indicate health impact and relative uncertainty of each finding is depicted by colors (dark blue = known, light blue = limited certainty, pink = highly uncertain, gray = unknown (data gap), NC = not considered). C = chemical of concern.
chemical-induced responses based only on chemical structure. Both the simpler read-across analysis (Enoch et al. 2008; Hewitt et al. 2010) and more complex machine learning-based approaches (Voutchkova et al. 2010) can be easily adopted for the purpose of chemical alternatives assessment.
It is crucial that the next generation of alternatives assessment frameworks incorporate the use of in vitro and other high throughput assays—toxicity pathway-centric assays—into the assessment process. The question of how various types of human health assessments of chemicals, including chemical alternatives assessment, will be conducted once a proper suite of in vitro assays, in silico models, and other novel data streams become available has come to the forefront of the debate in the environmental health community, largely because the feasibility of obtaining complex data on hundreds, if not thousands, of chemicals became a reality in the past several years. A number of broadly applicable opinions have been voiced that, while unanimous in the overall conclusion that human and environmental health decisions will be made with new data, are somewhat different in how this information should be used and for what type of decisions (e.g., relative ranking/prioritization to select candidates for further traditional testing, or making choices about alternatives in the context of alternatives assessment).
Several approaches to using in vitro data in human health assessments impact the thinking about how to incorporate these data in alternatives assessment:
- Using in vitro data and in silico predictions in ways similar to current practices that rely on human and animal health end points, with the use of additional uncertainty or safety factors to account for in vitro to in vivo extrapolation (Crump et al. 2010). Crump et al (2010) reasoned that toxicity pathway-based models are unlikely to contribute quantitatively to decision making for several reasons, including that the statistical variability inherent in such complex models will hinder their ultimate utility for estimating small changes in response, and that such models will likely continue to involve empirical modeling of dose-response relationships.
- Using in vitro data and in silico models, coupled with estimates of population variability and uncertainty, to estimate the human dose at which a chemical may significantly alter a biological pathway in vivo. This dose is referred to as a biological pathway altering dose (BPAD) (Judson et al. 2011). This approach draws parallels between a chemical-associated perturbation of a pathway as observed in in vitro assays and a key event in the chemical’s
| ToxCast (Knudsen et al. 2013) |
Quantitative (in most cases concentration-response based) information from a suite of diverse HTS cellular (viability, proliferation, or reporter gene), biochemical (enzymes and receptors), and zebrafish assays. | Phases I and II: 293 and 767 chemicals screened in 600+ assays Phase III: 1K+ chemicals screened in 100+ assays |
| Tox21 (Collins et al. 2008) |
Ultra-qHTS (all data are collected for 8-15 concentrations of each agent) molecular, biochemical, and cell-based assays from a consortium of U.S. federal agencies. | 8K+ environmental chemicals and drugs 50+ assays and 120+ endpoints |
| HTS Zebrafish (Truong et al. 2014) |
HTS (concentration-response) of embryonic zebrafish for developmental, morphological, and behavioral end points | 8K chemicals, including those screened by ToxCast and Tox21 |
| PubChem (Wang et al. 2014) |
A database of biological tests of small molecules generated through high throughput screening experiments, medicinal chemistry studies, chemical biology research, and drug discovery programs. | 10K chemicals screened in up to 10K cellular, molecular or biochemical assays |
| Drug Matrix (Fostel 2008) |
A large compendium of microarray data from in vivo (rat) exposures to various drugs and chemicals; profiling was performed on 9 organs. | 658 drugs and chemicals 4.3K studies (dose, time, organ, etc.)/13K arrays |
| TG-Gates (Uehara et al. 2010) |
Gene expression data from liver (rat) and cultured hepatocytes (rat and human) in dose- and time-dependent study design. Matching toxicity (pathology and clinical chemistry) data is also available. | 170 drugs and chemicals 33K+ microarrays |
| CTD (Davis et al. 2013) |
Manually curated chemical-gene/protein interactions, chemical-disease relationships, and exposure relationships (stressors, receptors, events, and outcomes) from published literature. | 28K genes (change in expression information) 886K chemical-gene interactions |
| CEBS (Waters et al. 2008) |
The Chemical Effects in Biological Systems (CEBS) database houses several types of study data from academic, industrial, and governmental laboratories. | 6K chemicals and drugs 800+ molecular (mostly microarray) datasets |
| ToxRefDB (Martin et al. 2009) |
Manually curated chronic toxicity data for a variety of organ systems in experimental animals from regulatory submissions to EPA. | 474 environmental chemicals tested in guideline studies |
| NTP Toxicity data | Detailed toxicity data from bioassay (rat and mouse sub- and chronic regulation toxicity studies), CHO Cell Cytogenesis, Drosophila, Micronucleus, Mouse Lymphoma, Rodent Cytogenetics, and Salmonella assays on hundreds of environmental chemicals. | Close to 1K environmental chemicals Multiple doses, tissues, and end points |
-
mode of action that may lead to an adverse health outcome. It offers an opportunity to not only compare alternatives with regard to the potential of human health hazard, but also take into account the quantitative and variability aspects of the underlying adverse effects.
- A step-wise decision tree that incorporates structure-activity relationship models, in vitro assays, toxicokinetic modeling, and short-term animal data into toxicity testing and risk assessment in an integrated fashion (Thomas et al. 2013). Tier 1 of this approach uses in vitro assays to rank chemicals based on their relative selectivity in interacting with biological targets that have been associated with known toxicity outcomes and to identify the concentration at which these effects occur. Reverse toxicokinetic modeling and in vitro to in vivo extrapolations (IVIVE) (Rotroff et al. 2010; Wetmore et al. 2012; Wetmore et al. 2013) are used to convert in vitro concentrations into external dose for derivation of the point-of-departure values. The latter can be compared to human exposure data or estimates (Wambaugh et al. 2013) to yield a margin of exposure (MOE).
BOX 8-3
IN VITRO TESTING BY END POINT
Genotoxicity - Direct
A battery of well-defined tests to assess a number of genotoxicity end points induced by direct-acting chemicals, such as point mutations, aneuploidy and chromosomal fragmentation, is necessary for regulatory consideration of drugs and other chemicals (Doak et al. 2012). Many OECD guideline protocols for genotoxicity assessment have been established and include the in vitro bacterial reverse gene mutation test (Ames; OECD 471), an in vitro mammalian cell gene mutation test (e.g., HPRT forward mutation assay, mouse lymphoma TK assay; OECD 476), and an in vitro mammalian cell chromosome aberration (OECD 473) or micronucleus (OECD 487) assays (Pfuhler et al. 2007). Despite concerns that such a battery of tests may result in a large number of false positives, it was shown recently that a combination of the Ames test and in vitro micronucleus assay can identify 78% of compounds known to be genotoxic in vivo (Kirkland et al. 2011). Standard OECD-approved assays for direct-acting genotoxicity are not meant for high throughput testing. Additional assays that can be used for screening of large chemical libraries are under evaluation. Additional assays in which large numbers of chemicals have been evaluated, without the advantage of established formal sensitivity or specificity of these assays, include a cell-based quantitative high throughput ATAD5-luciferase assay (Fox et al. 2012) and the induction of increased cytotoxicity in isogenic chicken DT40 cell lines deficient in DNA repair pathways (Yamamoto et al. 2011). While it is yet difficult to reach firm conclusion on the genotoxicity and potential tumorigenicity of a chemical using novel assays (Mahadevan et al. 2011; Benigni 2013), these experimental tools may be used in the context of a comparative assessment to provide a relative notion of safety among the alternative compounds being considered.
Genotoxicity-Indirect
A number of high throughput approaches are being considered in ToxCast and Tox21 programs. Despite concerns raised about the predictive nature of these in vitro (and rodent in vivo) approaches (Kleinstreuer et al. 2013; Corton et al. 2014; Rusyn et al. 2014), these assays likely will also prove useful for relative ranking of a chemical of concern and its alternatives. For example, a classification model that utilized in vitro screening data of 309 environmental chemicals in human constitutive androstane receptor (CAR/NR1I3), pregnane X receptor (PXR/NR1I2), aryl hydrocarbon receptor (AhR), peroxisome proliferator-activated receptors (PPAR/NR1C), liver X receptors (LXR/NR1H), retinoic X receptors (RXR/NR2B), and steroid receptors (SR/NR3) has been developed (Shah et al. 2011).
Endocrine Disruption
Endocrine disrupting chemicals have received heightened attention because of concerns that they may cause delayed reproductive and developmental effects in the general population (Birnbaum 2013). Concerns related to endocrine disrupting chemicals led to the EPA’s development of an endocrine disruptor screening program (EDSP) and identification of chemicals that require screening. EDSP requires an initial screening battery (Tier 1) for the estrogen, androgen, and thyroid hormones, as well as steroidogenesis (EATS) pathways consisting of five in vitro (estrogen receptor binding—rat uterine cytosol, androgen receptor binding—rat prostate cytosol, estrogen receptor α transcriptional activation, recombinant aromatase, and steroidogenesis in H295R cell line), and six in vivo (rodent, fish and amphibian models) assays to evaluate a chemical’s potential to interact with the endocrine system. High throughput screening assays that are not part of the Tier 1 panel in EDSP may have the potential for providing in vitro biological activity indicative of the potential to disrupt the endocrine pathways, as there are a number of assays that are highly relevant to EATS pathways (Martin et al. 2010; Huang et al. 2011) or nuclear receptor activation (see above). It has been suggested that such assays may assist in developing a prioritized list of chemicals for evaluation in the current Tier 1 battery or possibly replace in vivo assays in the long term. For example, a comparison of the results of in vitro screening of chemicals in a growth assay in the estrogen-responsive human mammary ductal carcinoma cell line T-47D with data from estrogen receptor binding and transactivation assays demonstrated that chemicals detected as active in both types of assays showed potencies that were highly correlated (Rotroff et al. 2013a). A follow-up study used high throughput screening assays for estrogen, androgen, steroidogenic, and thyroid-disrupting mechanisms to classify compounds and compare the results to in vitro and in vivo data from EDSP Tier 1 (Rotroff et al. 2013b). While it was reported that ToxCast estrogen receptor and androgen receptor assays showed significantly high concordance with the results of relevant in vitro and in vivo EDSP Tier 1 assays, no classification model could be developed for steroidogenic and thyroid hormone-related effects with the currently available ToxCast data.
Reproductive and Developmental Toxicity
Several recent studies have taken advantage of the available in vitro and in vivo information in the ToxCast Phase I chemical library and animal studies in the Toxicity Reference Database (Martin et al. 2009) to evaluate the utility of toxicity screening for predicting reproductive and developmental toxicity. These studies showed that ToxCast in vitro assay-derived information on steroidal and nonsteroidal nuclear receptors, cytochrome P450 enzyme inhibition, G protein-coupled receptors,
and disturbances in cell signaling pathways could identify rodent reproductive toxicants with about 75% accuracy (Martin et al. 2011). Similarly, ToxCast in vitro assay-derived information on transforming growth factor beta, receptor signaling in the rat, and inflammatory signals in the rabbit can be used to classify compounds as developmental toxicants in the rat or rabbit with greater than 70% accuracy (Sipes et al. 2011).
Studies of chemical effects on zebrafish are also useful for predicting rodent developmental toxicity (60%-70% concordance) (Padilla et al. 2012; Truong et al. 2014). (While not technically an “in vitro” assay, zebrafish are high throughput animal models for development effects.)
Acute, Chronic, and Repeat Dose Toxicity
A variety of biochemical, molecular, and cellular assays are used in drug safety evaluation to identify potential unintended “off-target” effects that may result in adverse drug reactions (Kola and Landis 2004). A comprehensive profiling of compounds through a large-scale battery of experimental assays and in silico models is usually conducted, and many publications suggest that straightforward in vitro cytotoxicity assays are very informative of in vivo health hazard (Benbow et al. 2010; Greene et al. 2010a). The utility of a large-scale inference on the potential adverse drug reactions was recently demonstrated using prediction and testing of the reactivity of drug candidates toward a panel of 73 “receptors” that are known as side-effect targets (Lounkine et al. 2012). Importantly, human health hazard evaluation through these pipelines is not limited to a qualitative binary prediction of the potential to cause adverse drug reaction, but must also be accompanied by a quantitative prediction of the dose at which such effects may be seen. The latter is as, or even more, important for the estimation of the “safety margin” between the desired (i.e., therapeutic) and side effect (i.e., adverse) health effects.
Dermal Irritation/Sensitization
Predictive identification of skin sensitizers is now highly reliant on a range of in vitro approaches. A 2013 European prohibition on animal testing (Adler et al. 2011) of ingredients in cosmetics has led to a novel in vitro strategy that can reliably identify sensitizing chemicals and predict their relative sensitizing potential. Numerous in vitro approaches address key parameters of the sensitizing process (Gerberick et al. 2008; Vocanson et al. 2013). These include testing for the ability of chemicals to modify skin protein (e.g., by covalent binding), activate innate skin immunity, and promote skin emigration or surface/intracellular changes in dendritic cell phenotypes, or T-cell priming. Several recent tests have proven successful in correctly detecting large numbers of reference chemicals, sometimes with >85% of correlation to standard in vivo animal assays. Importantly, it has been found that none of these methods alone is able to detect all the sensitizers, and some of them are more likely to detect certain classes of chemicals (Vocanson et al. 2013). Nevertheless, in combination, they hold the promise that in vitro assays can detect chemicals with sensitizing properties. However, while appropriate in vitro solutions for the hazard identification step appear to be within reach, the field is now faced with the challenge of obtaining robust in vitro data on the potency of identified skin-sensitizing chemicals. The availability of such quantitative information may be crucial for an alternatives assessment, if the potential for human exposure varies widely among the chemicals being evaluated.
- Modification of methods under development by the EPA’s Advancing the Next Generation of Risk Assessment program NexGen (EPA 2013e). The agency’s draft approach to using in vitro data is based on the recognition that EPA deals with various decision contexts and that a “toolbox” of various NexGen methodologies could provide information and knowledge to support each of these decision contexts, from screening and prioritization, to limited and major scope assessments.
- Using the Adverse Outcome Pathway (AOP) concept to link molecular screening and mechanistic toxicology data to adverse effects of interest in assessments (OECD 2013b). The goal here is to reduce uncertainty by identifying key intermediate events and quantitatively linking them to adverse outcomes. An AOP should describe a sequential progression from the molecular initiating event to the cellular, organ, and organism response that underlies the in vivo outcome of interest (OECD 2013b). If an AOP accurately describes a sequence of events through the different levels of biological organization, it may be possible to determine which in vitro assays may be useful in identifying chemical effects or molecular initiating events. Conceptually, the AOP concept may therefore be very useful if a defined set of “adverse outcomes” to avoid in alternative selection are identified. Several AOPs for human health effects have emerged, including a well-developed one for skin sensitization (MacKay et al. 2013; Maxwell et al. 2014). Several additional AOPs are under development for mutagenicity, nuclear receptor-mediated non-genotoxic liver carcinogenesis, neurodevelopmental effects and
thyroid disruption, hematotoxicity, hepatotoxicity, and liver fibrosis (OECD 2013b).
Based on the above proposals for how in vitro data may be used to evaluate the potential for human health hazard, the committee suggests the following potential uses of high throughput in vitro data in alternatives assessment:
- Using in vitro data as primary evidence for an end point of concern (e.g., mutagenicity);
- Using in vitro data to fill data gaps for an end point of concern (e.g., endocrine toxicity);
- Using in vitro data to screen out possible unintended consequences of data-poor chemicals
These uses are consistent with an emerging structure for how in vitro and other high throughput assays, as well as in silico model-based predictions, may be used in the broader context of risk assessment (EPA 2013e,f; Thomas et al. 2013). In vitro and in silico approaches that address human health hazard are described in the next section.
In Vitro Approaches for Evaluation of Human Health Hazards
Using in vitro data in an alternatives assessment is conceptually similar to using in vivo animal data. For example, animal studies are used to make predictions of the potential for health hazards in humans, whereas in vitro assays are used to assess whether chemicals may perturb certain biological pathways. The committee did not undertake a comprehensive review of which health end points can now be assessed using novel in vitro data as primary data in the same way more traditional types of data are used. Similarly, a complete discussion of the strengths, limitations, and predictive value of in vitro tests is beyond the scope of this committee. The committee acknowledges that scientific input will be necessary to determine the breadth of assays that may be required to adequately assess a chemical of concern and alternatives. This could involve the identification of one or more assays to assess different endpoints of interest. In Box 8-3, the committee describes the state of the science of high throughput in vitro toxicity assays for several end points. Box 8-4 describes the committee’s thinking on how to consider the relationship between the in vitro concentration used in such assays and the in vivo dose that elicits adverse health effects.
In Silico Approaches for Evaluation of Human Health Hazards
In silico models exist for a variety of human health end points, but the accuracy of these predictions can vary dramatically. The accuracy of in silico toxicity predictions is typically measured through internal and external validation of the model using data sets of known experimental activity. Internal validation is used during development to show that statistically derived models are robust, but this type of validation provides little information about the ability of the model to predict the activity of compounds outside the training set (Tropsha et al. 2003; Gramatica 2007). External or prospective validation is the gold standard method for evaluating model performance, but results have proved to be context dependent and difficult to generalize beyond the data training set. Furthermore, in silico prediction of a variety of toxicity end points has been limited by the quantity and quality of data available in the public domain for model development. In addition, most in silico approaches do not identify the dose at which effects are likely to happen.
In Box 8-5, the models and approaches available for predicting genotoxicity, carcinogenicity, skin sensitization, reproductive and developmental toxicity, and hepatotoxicity are discussed briefly. There are many commercial systems available, and a discussion of their strengths and weaknesses is beyond the scope of this chapter. Rather, the committee provides a survey of approaches and, where appropriate, provides illustrations of their use in toxicology. While Box 8-5 looks at approaches by end point, Box 8-6 discusses specific chemical structure and physicochemical properties that influence toxicity.
Most, if not all, novel high throughput toxicity screening assays evaluate the relationship between dose of the chemical and response of the assay. Dose-response relationship data are becoming a source of increasingly accessible information for evaluating the potential human health hazard. This information may be useful even in the context of chemical alternatives assessment. Potential dose metrics or points of departure that can be applied to high throughput toxicity data include:
- Chemical concentration that elicits a 50% effect (EC50) in the assay (Neubig et al. 2003; Huang et al. 2008; Xia et al. 2008). The limitations of the EC50 approach in the analysis of in vitro screening data have been addressed by (Sand et al. 2012).
- Binary “active/inactive” classification of responses in each assay (Shukla et al. 2010). This approach facilitates concordance analysis with in vivo toxicity outcomes that are also frequently binary.
- The use of logistic curve modeling to fit the concentration-response relationships that may not reach the maximum effect (Sirenko et al. 2013).
- One standard deviation-based benchmark concentrations (BMCs) (Sirenko et al. 2013).
- Benchmark dose-transition (BMDT), which represents the dose where the slope of the dose-effect curve changes the most (per unit log-dose) in the low dose region (Sand et al. 2012).
- Lowest dose at which the signal can be reliably detected (Sand et al. 2011).
Several of these methods rely on statistically based approaches. Human health risk assessments, including alternatives assessments, will likely be improved if these approaches also consider inter and intraspecies adjustments and biological considerations relating to the assessed in vitro end points (Chiu et al. 2012).
Even though concentration-response data are routinely collected in most in vitro assays, it has been repeatedly noted that in vitro toxicity screening-derived points-of-departure are not directly useable in assessment decisions (e.g., comparative analysis) unless they are converted to in vivo dose equivalents (Blaauboer 2010; Basketter et al. 2012; Blaauboer et al. 2012; Thomas et al. 2013; Yoon et al. 2014; Groothuis et al. in press). The relationship between in vitro concentrations and the concentration of the chemical in the blood/target tissue in vivo, however, can be complex and dependent on variables that are not captured in screening assays. The high throughput screening data do not account for pharmacokinetic factors, such as bioavailability, clearance, and protein binding, which can significantly influence in vivo toxicity and, depending on the assay, may not account for metabolism.
Computational in vitro-to-in vivo extrapolations (IVIVE) use data generated within in vitro assays to estimate in vivo drug or chemical fate. IVIVE is increasingly being used to predict the in vivo pharmacokinetic behavior of environmental and industrial chemicals (Basketter et al. 2012). A combination of IVIVE and reverse dosimetry can be used to estimate the daily human oral dose, called the oral equivalent dose, necessary to achieve steady state in vivo blood concentrations equivalent to the point-of-departure values derived from the in vitro assays (Rotroff et al. 2010; Wetmore et al. 2012,2013). Incorporation of pharmacokinetic and exposure information enhances the use of high throughput in vitro screening data by providing a risk context (Judson et al. 2011; Thomas et al. 2013), but more research is needed to produce such information. Consideration of alternative dose metrics instead of nominal concentrations is needed to reduce effect concentration variability between in vitro assays and between in vitro and in vivo assays in toxicology (Groothuis et al. in press). The quantitative IVIVE efforts will add information critical to interpreting the biological relevance of exposure scenarios (Wetmore et al. 2012).
BOX 8-5
IN SILICO PREDICTION BY END POINT
Genotoxicity
In silico prediction of genotoxicity has been a major research focus since the initial publication of structural alerts for DNA reactivity (Ashby and Tennant 1991). Access to large public domain data sets has helped stimulate progress and has resulted in a fair degree of success in the prediction of genotoxicity, particularly in the prediction of the Ames salmonella assay for mutagenicity by in silico models (Naven et al. 2010; Lynch et al. 2011). The overall concordance between the predictive tools and the assays they are designed to predict ranges between 70% and 85%. It is worth noting that these values are close to the inter- and intra-laboratory reproducibility of the Ames assay, reported as 87% (Kamber et al. 2009). However, the sensitivity of the in silico model—its ability to accurately predict an Ames positive compound—can vary much more dramatically, from up to 85% for public domain data sets to just 17% for some proprietary (e.g., pharmaceutical) data sets (Hillebrecht et al. 2011). This variability in sensitivity may result from the fact that few active pharmaceutical ingredients contain the classical DNA-reactive functionsal groups that are a common cause of genotoxicity.
Commercially available software packages for conducting in silico predictions—such as Derek for Windows55 (DfW; Marchant et al. 2008), MC4PC (Saiahhov and Klopman 2010), and Leadscope Model Applier (LSMA; Valerio and Cross (2012))—are commonly used in the pharmaceutical industry for the prediction of genotoxicity and other toxicological end points. Other readily available systems like Toxtree (Benigni et al. 2010) are also being evaluated for their usefulness. Their comparative performances have been extensively reviewed and published (Hillebrecht et al. 2011; Sutter et al. 2013), but it is clear that no single system performs significantly better than any of the others. Although other models exist for the prediction of chromosomal aberrations, such as clastogenicity and anugenicity, these systems are generally less accurate than the other modeling tools and are not commonly used in industry settings.
Carcinogencity
Various methods for structure-based prediction of carcinogenicity have been developed over the last several decades, including some commercial applications, such as Derek, Case Ultra, Leadscope Model Applier, ToxTree, and OncoLogic. The value of these methods for predicting carcinogens has been limited by lack of public data availability and the complexity of the end point itself. Carcinogenicity can occur through genotoxic and non-genotoxic mechanisms. Most structure-based approaches are able to predict DNA-reactive genotoxic compounds (as discussed above). Some systems, such as Derek, contain structural alerts specifically targeting certain classes of non-genotoxic carcinogens. Other predictive packages, such as Case Ultra, do not always differentiate between these two classes in their predictions.
Two prospective exercises conducted by the National Toxicology Program (NTP) in the 1990s evaluated the performance of computational models for carcinogenicity. In these exercises, the NTP invited interested parties to publish model predictions on a set of chemicals that were scheduled for testing in the NTP’s two-year rodent bioassay. Once the tests were completed, the in vivo results were compared to the predictions. The carcinogenicity of the first set of 65 chemicals was reasonably well predicted with computational models, achieving between 50%-65% accuracy. However, the second set consisting of only 30 chemicals was not predicted as well by the in silico systems and tended to over-predict non-carcinogens as carcinogenic (Benigni and Giuliani 2003). Ongoing effort to predict carcinogenicity through structure-based approaches continues with some recent examples from Fjodorova et al. (2010) and Kar et al. (2012).
Reproductive and Developmental Toxicity
“Developmental and Reproductive Toxicity (DART) occurs through many different mechanisms and involves a number of different target sites, making it very difficult to predict this end point” (Wu et al. 2013). Most of the published QSAR development has been done through collaborative projects with the computational toxicology group within the U.S. Food and Drug Administration (FDA), using data collected from preclinical and clinical data submitted by pharmaceutical companies. Matthews et al. (2007) reported the use of computational QSAR approaches to predict male and female reproductive toxicity, fetal dysmorphogenesis, functional toxicity, mortality, growth, and newborn behavioral toxicity. Matthews reported high specificity (i.e., the number of correctly predicted negatives) and positive predictive value (i.e., the number of correct positive predictions when compared to the total number of positive predictions) of greater than 80%. However, the sensitivity (i.e., the number of correctly identified positive compounds) was often less than 50%. Unlike the NTP carcinogenicity exercises, to date there have been no published prospective tests of performance of these DART models, so their accuracy compared to a set of novel compounds cannot be ascertained. Published models are available in commercial packages such as Case Ultra and Leadscope Model Applier. In addition, Derek Nexus also contains some structural alerts for DART effects that have been
developed as part of acollaboration with Pfizer Inc., although these alerts and their respective performance have not been formally published.
Wu et al. (2013) recently published “an empirically based decision tree for determining whether or not a chemical has receptor-binding properties and structural features that are consistent with chemical structures known to have toxicity for DART end points.” As with the above models and structural alerts, the performance of this decision tree has not been independently assessed, so its performance for truly novel chemical series that have not been previously tested may well be limited.
Skin Sensitization
Skin sensitization is primarily driven through hapten reactivity, which supports a central role for chemical reactivity in allergic sensitization (Vocanson et al. 2013), as well as skin permeability and metabolic activation. This requirement for chemical reactivity makes the prediction of skin sensitizers more feasible, and there has been substantial progress in this area. Structural alerts for skin sensitization have been implemented in Derek, ToxTree, and other systems; the relative performance of these approaches has not been extensively reviewed, but external validation studies do point out limitations in applicability and low external predictivity (Teubner et al. 2013).
Predictive tests for allergic contact dermatitis (ACD) have also been developed. ACD depends on the intrinsic capability of the chemical to cause skin sensitization and the ability of a chemical to penetrate viable epidermis. Numerous QSAR methods that predict ACD for specific chemical classes or non-congeneric data sets have been published (Deardon 2002; Guha and Jurs 2004; Sutherland et al. 2004). Factors that affect the ability of chemical to be absorbed into the epidermis are discussed in more detail in Chapter 5.
Respiratory Sensitization
Respiratory sensitization is an important disease (Mekenyan et al. 2014), but there are “no validated or widely accepted models for characterizing the potential of a chemical to induce respiratory sensitization.” While efforts to model respiratory sensitization in silico have been hampered by an incomplete understanding of immunological mechanisms, structural alerts for this end point have been developed (Agius et al. 1991, 1994; Enoch et al. 2012). Typical structural alerts “have been encoded into the Derek Nexus knowledge based expert system developed by LHASA Ltd. Other efforts have focused on establishing statistical QSAR models; examples include those first derived by the developers of MCASE, Jarvis et al. (2005) and more recently by Warne et al. (2009), who investigated the use of pattern recognition methods to discriminate between skin and respiratory sensitizers” (Mekenyan et al. 2014).
Despite the lack of a universally accepted test method, REACH regulations and others still require the assessment of respiratory sensitization as part of a risk assessment. The REACH guidance describes an integrated evaluation strategy that includes a consideration of well-established structural alerts and existing read-across, QSAR, and in vivo data. As with many other toxicological end points, there has been no published comparison of these methods for prediction, so it is difficult to draw conclusions on the relative merits and accuracy of the models.
Hepatoxicity
Drug-inducted liver injury (DILI) is a major concern in the pharmaceutical industry and has led to the withdrawal of a significant number of marketed drugs (Holt and Ju 2006; Kaplowitz 2005). Adverse effects range from hepatic enzyme elevations to liver failure (Zimmerman 1999; Williams 2006) and are often difficult to predict in the preclinical stages. As a result of this interest, numerous in silico approaches for predicting hepatotoxicity have been developed. These approaches range from structural alerts associated with DILI (Hewitt et al. 2013) to QSAR methods (Chen et al. 2013). The mechanisms involved in DILI are often complex, making accurate prediction of this end point using QSAR and other computational approaches challenging. Most of these methods claim to have a sensitivity and specificity between 65%-70% depending on the method and data test set; however, no independent evaluation has been published, so true head-to-head performances are difficult to ascertain.
BOX 8-6
OTHER CHEMICAL STRUCTURE AND PHYSICOCHEMICAL PROPERTIES THAT INFLUENCE TOXICITY
While structures can certainly not predict all chemical activity, physicochemical and structural characteristics have been used for predicting toxicity end points. Changes in key physicochemical properties, such as pKa, lipophilicity, and polar surface area, can lead to dramatic effects on the toxicity of a chemical, either through influencing pharmacokinetic properties, such as clearance of the compound, or its ability to interact with a biological system in the form of pharmacological interactions and/or non-specific protein binding.
The effect of physicochemical properties on bioavailability is discussed in more detail in Chapter 5. Other physicochemical and structural characteristics that have been developed for predicting toxicity endpoints include:
- pKa and LogP (or calculated LogP, cLogP), which correlate with mitochondrial uncoupling for certain classes of chemicals (Naven et al. 2013).
- The energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), which is associated with a chemical’s ability to absorb light and phototoxicity (Peukert et al. 2011).
- A basic center and one or more lipophilic chains in a compound is associated with a higher likelihood of inhibiting an ion channel important to cardiac cells action potentials (Schmid et al. 2003).
- Amphiphilicity, pKa, and lipophilicity (LogP), which have been used to predict the likelihood of phospholipidosis (Goracci et al. 2013).
- Structures indicating the potential for generating reactive metabolites (Stepan et al. 2011).
- Acidity or basicity of the molecule as an indication of propensity to interact with different classes of receptors such as cyclooxygenases and the nuclear hormone receptors (acidic molecules) or the aminergic G-protein coupled receptors (basic molecules) (Parker at al. 2014).
- High lipophilicity, low polar surface area and low passive permeability are associated with induction of endoplasmic reticulum stress, which has been linked to drug-induced toxicity (Koslov-Davino et al. 2013).
- Structures predicted to have endocrine effects, using various QSAR methods to predict chemical docking with cellular targets (Vuorinen et al. 2013).
- LogP and topological polar surface area may be related to a chemical’s ability to cause in vivo toxicity at low plasma exposures, cytotoxicity, and off-target pharmacological effects (Benbow et al. 2010, Greene et al. 2010b; Wang and Greene 2012).
- Higher volume of distribution, when combined with in vitro indications of greater cytotoxicity in hepatocytes, is associated with a reduction in the lowest observable adverse effect level (LOAEL) (Sutherland et al. 2012). LogD and pKa predictions based on structure are useful in predicting volume of distribution and plasma protein binding (Lombardo et al. 2002). Other structural predictors of the steady state volume of distribution and clearance (Gombar and Hall 2013) are also helpful.
- Lipophilicity (or LogP) and polar surface area descriptors are strongly correlated with chemical clearance (Hsiao et al. 2013).
- Higher LogP, low polar surface area, and pKa have been associated with higher target promiscuity (a wider area of nonselective interaction with biological targets) (Seidler et al. 2003).
Use of Novel In Vitro Data and In Silico Models as Primary Evidence
The NTP Report on Carcinogens identifies that among the most crucial information considered in the evaluation of human cancer hazard of chemicals is that obtained from “studies on genotoxicity [ability to damage genes]” (NTP 2011). Genotoxicity may be either the result of interaction between a chemical and DNA (direct genotoxicity) or action between a chemical and DNA regulatory elements (indirect genotoxicity). A number of in vitro assays are available to test for the potential of a chemical to be genotoxic. Due to the complexity and diversity of mechanisms that may lead to DNA damage (and the mutagenic events that follow), a battery of in vitro tests is needed to establish a chemical’s genotoxic potential. In vitro mutagenicity data is included in
GHS as a primary data type (UNECE 2013c). More details are in Appendix D, but primary data are sufficient for considering a chemical as a GHS Category 2 Suspected/Possible Germ Cell Mutagenicity. Other (i.e., not mutagenicity specific) in vitro testing for genotoxicity is described in Box 8-3.
Use of Novel In Vitro Data and In Silico Models to Fill Data Gaps
High throughput in vitro data can also be used to fill certain primary data gaps for particular health end points. For example, ToxCast in vitro assay-derived information on steroidal and non-steroidal nuclear receptors, cytochrome P450 enzyme inhibition, G protein-coupled receptors, and disturbances in cell signaling pathways may identify rodent reproductive and developmental toxicants (Martin et al. 2011; Sipes et al. 2011), potentially showing a path toward replacement of more traditional (Clode 2006) uterotrophic assays and the Hershberger assays found in tiered endocrine assays.
How novel in vitro data can be used to address gaps in the human health data in an alternatives assessment was illustrated by Russell Thomas of EPA’s National Center for Computational Toxicology (Thomas 2014). Thomas used the information from two case studies by Martin et al. that analyzed the potential use of ToxCast data (Martin et al 2012) for tiered testing. While these case studies were designed to examine the use of in vitro data to determine which chemicals warranted further reproductive toxicity testing in animals, Thomas showed how the concept applies to alternatives assessment as well, in that those chemicals with higher or lower probability of exerting reproductive effects can be identified. This concept is explained in further detail in Box 8-7.
Use of Novel Data to Screen out Possible Unintended Consequences in Data-Poor Chemicals
The third use of novel high throughput in vitro data suggested by the committee is to screen out unintended consequences. Some companies in the pharmaceutical industry are using this information in this way. In vitro high throughput screening and toxicogenomics appear promising in the screening of data-poor chemical alternatives for biological activity that may contribute to hazard identification. The information about potential modes of action can support the transition toward an integrated testing and assessment strategy. The pharmaceutical industry routinely uses mechanistic in vitro tests and high throughput screening to look for unexpected safety issues with potential drug candidates. As a result, the industry brings valuable experience tempered with some caution about setting overly high expectations for in vitro toxicity testing technologies (MacDonald and Robertson 2009).
In addition to screening for unintended and unexpected consequences by looking at mode- of-action information, it is also possible to use high throughput in vitro screening to look for evidence of nonselective chemical activity at low concentrations. That is, use the data for screening out chemicals that have general indicators of potential toxicity, even if the specific toxicities and their modes of action are not identified. This concept is based on the observation that while the batteries of in vitro toxicity assays utilized in the Tox21 and ToxCast programs provide a broad biological profile of the potential proximal biochemical and cellular targets for a chemical, the majority of environmental chemicals being tested likely act via nonselective interactions with cellular macromolecules (Thomas et al. 2013). Because most high throughput assays incorporate an extensive concentration range usually spanning several orders of magnitude, it is frequently observed that many biological targets are “engaged” at, or near, concentrations that result in cytotoxicity (Martin et al. 2010). Thus, one use of high throughput assay-derived information may be for separating chemicals into either those that cause toxicity primarily through nonselective interactions with cells and cellular macromolecules or those that act through more selective interactions (e.g., receptor-mediated chemicals). Specifically, it has been shown that promiscuity across multiple pharmacological targets at a concentration of 10 µM can lead to an increased likelihood of observing toxicity in vivo at low exposures (Hughes et al. 2008; Wang and Greene 2012). In the context of alternatives assessment, if alternatives under consideration exhibit varying levels of “selectivity,” a compound with higher “selectivity” may be considered as lower risk for additional “off target” effects and thus be assigned a higher relative rank. Clearly, if a chemical is considered to be selectively active against a pharmacological target, whether intended or otherwise, the implications of this specific pharmacological activity in relation to potential safety concerns should be considered. For example, agonism of the 5HT2b receptor has been implicated in causing cardiac valvulopathy.
It is also possible that high throughput in vitro data could be used to screen out unintended consequences associated with particularly susceptible subpopulations of people. In general, existing alternatives assessment frameworks do not consider human variability in their analysis, but because this variability underlies differences in how people respond, addressing these differences is a key consideration in human health assessments for chemicals (NRC 2009). By using and expanding upon current analytical methods, these assessments may take advantage of novel in vitro data to better characterize and quantify variability in susceptibility (Zeise et al. 2013). Approaches that are now possible include using large-scale in vitro screening (Rusyn et al. 2010) in human cell lines obtained from genetically diverse subjects (Durbin et al. 2010; Welsh et al. 2009; Wheeler and Dolan 2012). The utility of such in vitro models to toxicology, especially for exploring the extent and nature of genetic components of inter-individual variability in pharmacodynamics, was recently demonstrated (O’Shea et al. 2011; Lock et al. 2012). The extent of inter-individual variability in response that was observed for different chemicals in in vitro assays could also be compared with previously collected sets of in vivo human pharmacodynamics variability data.
Limitations of Using In Vitro Data in Alternatives Assessments
High throughput in vitro screening to identify chemical hazards and prioritize chemicals for additional in vivo testing is an area of heightened scientific inquiry and regulatory scrutiny. While the promise of the novel in vitro assays and statistical methods is difficult to underestimate, their predictive power or classification accuracy is still not clear. It has been observed that the findings from in vitro assays may not provide more information than that of the chemical structural descriptors (see below), and aggregating the assays based on genes or pathways may even lead to reduced predictive performance (Thomas et al. 2012). Significant potential biases in the estimates of the performance of the classification models have been noted, though this point is still a subject of active debate (Dix et al. 2012; Knudsen et al. 2013). Because the current high throughput in vitro assays may still have only limited applicability for predicting in vivo chemical hazards, the committee believes that high throughput in vitro assays with limited or uncertain predictivity should generally only be used in alternatives assessments to fill data gaps or screen for unexpected consequences, except as described earlier for certain mutagenicity and endocrine/reproductive toxicity assays.
Limitations of Using In Silico Approaches in Alternatives Assessments
When using in silico methods or read-across approaches to infer toxicological activity, there are two main limitations that need careful consideration in the assessment:
- Measures of chemical similarity and their appropriate application to the effect being predicted; and
- The reported applicability domain of a prediction and hence the reliability of the prediction being made.
These aspects of computational models are briefly summarized in the following sections. Recent reviews by Patlewicz et al. (2013) and Modi et al. (2012) are available for more information.
Limitations of Chemical Similarity and Read-Across Approaches
Similarity in chemical structure is often used in read-across and QSAR models to identify chemical structures with known activities that could be used to infer the activity of a molecule with unknown activity. In other words, the idea is to infer that the less understood chemical will produce an equivalent test result as the more well-understood one (OECD 2014b). This approach presents the dilemma of how to define what is similar and what is not. Defining chemical similarity has been debated for several decades, and no one method for applying or presenting read- across concepts has been agreed upon, despite the frequent use42 of grouping and read across to satisfy information requirements under REACH, legislation that has been enacted in Europe. The method used is often case dependent; for example, in genetic toxicology, when a chemical bears the same structural alert as an Ames negative comparison compound (and no other alert) in the same position and environment and has a similar molecular weight, then the chemical is considered
_____________
42 For example, more than 20% of high production volume chemicals submitted for the first REACH deadline relied on read-across for hazard information on a number of toxicity end points necessary for registration (ECHA 2011).
In this example, high throughput in vitro screening data (36 assays) from the ToxCast database were compared to the in vivo data from the ToxRef database to identify the in vitro assays that best predict in vivo results. The information about how activity of 56 chemicals (ToxCast Phase I and II chemicals, including plasticizers or other industrial chemicals) in these 36 in vitro assays was mapped onto the 8 pathways most predictive of in vivo assays identified (pathways illustrated here as pie slices for PPARα, AR, etc.). Strategies for predicting in vivo toxicity outcomes from in vitro data are detailed in Figure A and in Martin et al. (2012).
Based on the available data, there are numerous ways to visualize and compare the profiles of the chemicals. No one way is considered the preferred method. The Toxicological Priority Index (ToxPi) software (described in detail in Appendix C) is a prioritization support tool that incorporates diverse chemicals’ bioactivity profiles and other relevant data (Reif et al. 2013). It is used here to visualize the outcome of the analysis and then rank the chemicals according to their impact on the eight pathways. ToxPi is a dimensionless index score that enables integration of multiple sources of evidence on exposure and/or safety, which is then transformed into transparent visual rankings to facilitate decision-making. The rankings and associated graphic profiles can be used to prioritize resources in various decision contexts, such as testing chemical toxicity or assessing the similarity of predicted compound activity profiles.
Figure B shows these results as ToxPi for 12 of 52 chemicals analyzed, where each slice represents multiple assays associated with a given gene pathway, and the chemicals are ordered based on the ToxPi score. For each slice, distance from
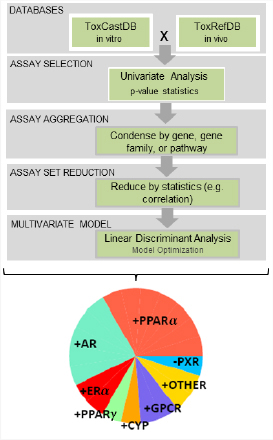
FIGURE A General workflow for developing statistical classification models for in vivo toxicity outcomes based on in vitro toxicity data. The example of the classification model for reproductive toxicity is for the rat (Martin et al., 2011). SOURCE: Thomas 2014. Reprinted with permission of the author.
the origin (center) is proportional to the normalized value (e.g., assay potency) of the component data points. The width (in radians) shows the relative weight of that slice in the overall ToxPi score calculation. Values closer to the unit score (equal to 1) translate to higher potency, or greater pathway perturbation relative to other chemicals in the analysis. Conversely, values closer to the origin (equal to 0) translate to lower potency and lesser pathway perturbation across the corresponding domain Values at zero (i.e., slices not extending at all from the origin) translate to “inactive/no activity.”
A judgment is made about which gene pathways should contribute most to the ToxPi score. These weighting factors, as well as the assays used, are described in Martin et al. (2011). The model shown here, however, used data from human cytochrome P450 assays instead of rat cytochrome P450 assays, and the Bioseek assays were removed because the data were not publicly available at the time of press. For each chemical, weighted combinations of data were combined from multiple data streams, with relative scores shown in ToxPi profiles as slices based on one or more components. Martin et al (2011) explains this process in more detail.
In this analysis, the chemicals with high ToxPi scores may be considered as representing a higher degree of reproductive health hazard. Indeed, a number of phthalates and other chemicals that have been associated with adverse reproductive health effects are at the top of the list. Among “alternatives,” some compounds demonstrate low relative scores, at least for the type of information used to develop the classification model. In an alternatives analysis, the chemicals with higher ToxPi scores would be assumed to represent a greater hazard.
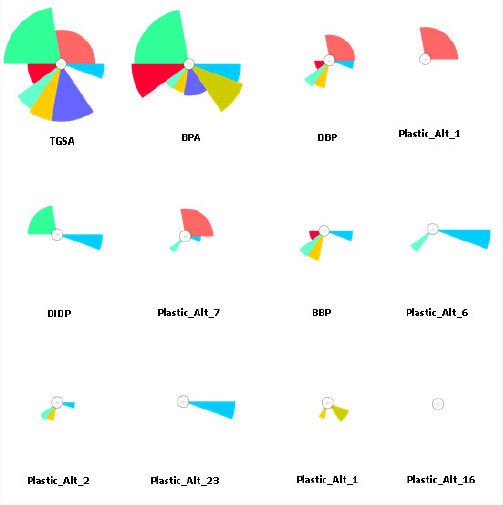
FIGURE B ToxPi profiles (ToxPi GUI v. 1.3) were used to prioritize chemical alternatives using 36 assays and pathways associated with reproductive toxicity (Reif et al. 2013). A subset of these prioritized chemicals is shown here. Some of the compounds were evaluated as potential alternatives and labeled “Plastic_Alt_n.”
likely to be negative in the Ames test as well. In this case, no further testing is generally needed (EMA 2014).
In a read-across assessment, a chemically defined category of known adverse activity is represented by compounds that have common structural features and exhibit similar trends in their physicochemical properties. Generally, the presence of a common biological or chemical behavior is associated with a common underlying mechanism of action (e.g. alkylating compounds). This categorical approach provides the basis for identifying trends in properties across the category of compounds, resulting in the possibility of extending the use of measured data to similar untested chemicals. These estimates of biological activity may be considered adequate, without further testing, for regulatory purposes (e.g., classification and labeling and/or impurity hazard assessment for classification with respect to toxicity potential). Enoch et al (2011) have provided a description of chemical category function (Sutter et al. 2013). However, the standardization of this approach for defining structural similarity on the basis of a chemically-defined class of known biological actives is much more difficult when the mechanisms of action are both diverse and complex.
In QSAR approaches, the definition of structural similarity is crucial to the final result of an in silico prediction (Naven et al. 2012). The assessment of chemical similarity usually begins with a quantitative description of the molecular structure or fingerprint” (Sutter et al. 2013). Comparisons between structures are then performed using one of a variety of indices that have been developed; for example, Euclidean distance measures or maximum common substructures. However, similarity is a multidimensional concept, and the similarity between two compounds can be difficult to determine or set guidelines for. For instance, compounds (1) and (2) in Figure 8-3 are similar in that they both have the same molecular formulas (C6H5NO2), yet their chemical structures bear little resemblance. They have different electron delocalization properties or aromatic behavior, physicochemical properties, and most importantly, probably dissimilar biological properties. Likewise, glucose (3) and galactose (4) share structural similarities but have very different pharmacologic properties. Many methods to measure the structural similarity between two compounds have been developed, but the more relevant question to consider is whether structural similarity an important factor for the toxicological end point being studied.
This is because minor modifications to the structural alert can significantly influence toxicological activity, yet major modifications to the periphery of the chemical structure may have little impact on activity so long as the structural alert remains intact. When assessing the relevance of a prediction of activity, it is not enough to ask how similar the query compound is to other inactive compounds. It is also important to identify the features of structurally alerting, active compounds that would attenuate the activity and to assess if these features can be adequately extrapolated to the compound being studied.
Limitations of Defining Applicability Domains
OECD guidelines currently recommend that QSAR models should define the domain within which the predictions of a model can be deemed reliable—the applicability domain (AD). Many methods exist for defining the AD of a QSAR model, and they have been extensively reviewed (Dragos et al. 2009; Hewitt and Ellison. 2010; Ellison et al. 2011). The AD of a model can be broadly described using two non-exclusive terms: (a) the region of chemical or response space relating to the model training set and (b) the region of a chemical or response space where a model makes an acceptable prediction error. For toxicological end points like mutagenicity or the uncoupling of oxidative phosphorylation, which are dependent on the presence or absence of structural alerts, the less applicable the concept of similarity becomes.
In the first definition (a), the underlying assumption is that those predictions based on interpolation from data in the training set are generally more reliable than those based on extrapolation. The second definition (b) is based on the assumption that by assessing where compounds are predicted most accurately, we can gain valuable information. The thinking is that inevitably, a subset of the training set will be incorrectly classified, casting doubt on the reliability of the predictions based on similarity to these compounds. In addition, definition (b) does not automatically assume that predictions for compounds that are considered dissimilar to the training set are unreliable (Dragos et al. 2009).
FIGURE 8-3 Selected chemical compounds to illustrate structural similarities and differences.
Defining the AD of any model is difficult and presents challenges to the end user about whether a prediction is reliable or not. In addition, although the scope of structural alerts can be used to define their AD, this provides little information to a user when alerts are not matched to the compound in question. Expert systems that rely on structural alerts do not have a model training set per se, as the alerts are often based on disparate data sources, such as toxicity data, information pertaining to the biological mechanism, and knowledge of chemistry and reactivity, which are synthesized into the development of an alert. Furthermore, not all data are publicly available; thus, current approaches cannot reflect this expert knowledge and often require a complete model training set.
Most of the methods for defining ADs have been trained to reduce the error in continuous output QSARs where the assay data provides homogeneous responses; for example, for LogP values or an experimentally derived IC50 for protein inhibition. It should be noted that there is a distinct gap between the applicability of ADs of chemicals producing homogenous responses to categorical models based on assays that generate a more diverse range of outputs, such as carcinogenicity or reproductive effects. Because a large number of available QSARs are categorical, and their use in alternatives assessment is likely even if the prediction outcome is of active/inactive type, the assessors should have confidence that the alternatives they are evaluating can be classified using a particular QSAR, or fall outside of the AD of that model. There are a few exceptions, but generally it has been shown that there is only value in using an AD to qualify confidence in a positive response, rather than as a prediction for absence of activity.
IN VITRO AND IN SILICO DATA INTEGRATION:
AN OPPORTUNITY FOR NEW INSIGHTS
Most of the current computational tools used in toxicity assessments, including chemical alternatives assessments, rely either on chemical or biological data. Specifically, structure activity-based computational approaches attempt to predict the toxicity for a particular health end point from chemical structure alone, whereas novel biological data-based bioinformatics approaches do not usually take full advantage of the inherent structural features of chemical molecules. Incorporating these features may improve the accuracy of the prediction or increase the confidence of the assessor. Integrative chemical-biological modeling may both improve the prediction accuracy and uncover insights previously invisible to either informatics discipline alone.
Using only chemical or biological modeling is unlikely to take full advantage of the richness of the modern data streams, which effectively capture chemical-biological interactions. Few integrative studies, however, have been reported; their paucity is stemming from the lack of both suitable data and integrative methods. Several recent reviews (Rusyn et al. 2012; Valerio and Choudhuri 2012) have proposed general schemes to integrate cheminformatics and bioinformatics for improved understanding of chemical effects on biological systems. A simple means of integrating the disparate data streams that may be available for an alternatives assessment is to apply existing statistical methods to both chemical and biological types of molecular features. Another way is to merge chemical models with biological models. Other approaches may be less straightforward. These include strategically combining chemical structures and biological assays such that the two data sources compensate for each
other’s shortcomings and the complementary information between them is maximally used.
While the approaches to integrative analyses of chemical and novel biological data streams are still maturing, it is clear that a multidisciplinary systems approach is the best available solution for translating molecular and preclinical insights into practice and guarding against unwanted outcomes of chemical use. In addition to addressing the issue of data quality, further gains in methodological innovations and cohesive integration of the various disciplines will be necessary. Starting such multidisciplinary efforts is unlikely to occur organically and will require deliberate efforts to foster a collaborative environment. As more data come online and advances in assay technologies reduce experimental variability, we expect integrative approaches to play a greater role in toxicology.
IMPLEMENTATION OF STEP 6.1: HUMAN HEALTH ASSESSMENT IN THE COMMITTEE’S FRAMEWORK
Box 8-1 at the beginning of this chapter provides a summary of how Step 6.1 in the committee’s framework should be implemented. Additional illustrations of the committee’s approach are also presented as two case studies in Chapter 12.
Specific advice for the completion of Step 6.1 includes:
- The assessment should focus on health hazards as opposed to health risks. Risk includes exposure, which is considered elsewhere in the framework.
- The health end points to be addressed in an alternatives assessment should be specified in Step 2. The set of human health hazard end points discussed in this chapter is the suggested initial list. there should be clear documentation of which end points were not considered, and which points, if any, were added.
- Use health end point-specific authoritative lists to identify previously identified health concerns.
- Collect available data, including by conducting scientific literature searches on health hazards associated with the chemical of interest and alternatives. Use the GHS criteria and hazard descriptions, when available, to assess available information for the required end points.
- Existing hazard identification guidance (e.g., as described in EPA risk assessment guidelines) should be used to ensure consistency and transparency when conducting de novo assessments of chemicals for reproductive toxicity and other health end points that require use of expert judgment.
- Assign hazard designations to criteria and authoritative lists to facilitate their use in comparing the health hazards of chemicals and selecting safer alternatives.
- Gaps in data for required end points should be addressed with qualitative and quantitative predictions based on high throughput in vitro data and in silico modeling, when available. The large-scale governmental efforts to collect novel data streams through Tox21 and ToxCast programs will provide critically important information that should inform gap analysis.
- Equally important is to evaluate which computational modeling approaches may serve as acceptable substitutes for alternatives assessment in lieu of additional data collection. In this regard, a closer examination of the read-across assessment framework in development by the European Chemicals Agency may provide additional guidance to inform user implementation of these approaches in alternatives assessments.
Implementation of the committee’s framework will require the ongoing development of tools and methods and regulatory guidance by the scientific community. This need is especially acute with the use of emerging in vitro and in silico data from high throughput toxicology programs (e.g., Tox21 and ToxCast programs). The committee anticipates that these data streams will provide critically important information that can be used to fill data gaps when traditional data from human and experimental animal studies are lacking. In addition, there is a need for scientists and regulatory agencies to determine which high throughput toxicology assays, end points, and model systems are most informative in assessing the human health hazard types used in chemical alternatives assessment. Once these decisions are made, then development of well-accepted classification schemes for these high throughput and in silico data, analogous to the GHS system, would enhance the use of this information.
In the case of the Tox21 and ToxCast programs, there is also a need for the development of data mining tools (e.g., user-friendly dashboards and
software) that will enable stakeholders to access the novel data types that have been already collected and support comparative analyses in a transparent and statistically rigorous way. The EPA is uniquely positioned to demonstrate leadership in incorporating novel data streams and modeling outcomes on human health into alternatives assessment. Specifically, EPA can help develop best practices by determining which information available through the Tox21 and ToxCast programs are most informative in assessing human health hazards and how and when such data may be incorporated in alternatives assessment. Such model efforts could demonstrate how decisions based in part on high throughput data could be formulated and communicated to the stakeholders. In addition, EPA may want to consider assisting parties interested in alternatives assessment in their interpretation and use of results from innovative toxicity testing methods.
Alternatives assessment of human health hazards is critical not only within the committee’s framework, but also in most other frameworks because it is central to determining whether an alternative meets the criteria for being considered safer than a chemical of concern. Identification of human health hazards will require evaluation of multiple data streams, including human epidemiologic or experimental studies in animals, and will be increasingly dependent on the use of novel in vitro and in silico approaches. It is important to keep in mind that in keeping with the committee’s approach to the task (see Chapter 1), required tools were not specified as part of this step. The committee found that most frameworks rely heavily on GHS criteria for evaluation of human health hazard, which are generally thought to be acceptable for this purpose. However, the committee strongly encourages users with adequate scientific resources to move beyond relying solely on traditional types of data associated with GHS or other benchmarking approaches toward data from novel in vitro and in silico approaches. This is especially true as the development and application of tools to assess and integrate novel data streams into the alternatives assessment process evolves.
In Chapter 12, the committee provides one case example in which these novel types of data were used to evaluate and rank several alternatives. As can quickly be discerned from this case study, the application of novel data streams in particular will require expertise in computational modeling, molecular toxicology, and other scientific disciplines that may go beyond the capabilities of some existing assessment teams. This dilemma is not unique to the application of high throughput data. Indeed, the committee found that interpretation of traditional toxicology data even when using the GHS and GreenScreen® tools can remain a challenge even for experienced toxicologists. For example, the committee found that some differences in end points and descriptor language exist between the GHS hazard categories and the reviewed frameworks. In addition, cutoffs in classification tools used in some frameworks could result in the assignment of alternatives to different hazard categories (e.g., High vs. Moderate) when the actual difference in response can be toxicologically insignificant. Users of the committee’s framework will need to exercise professional judgment so that they do not discount possible beneficial alternatives or adopt others that may have unintended health consequences.






























