
Responsible sharing of clinical trial data is in the public interest. It maximizes the contributions made by clinical trial participants to scientific knowledge that benefits future patients and society as a whole. Results from many clinical trials are not published in peer-reviewed journals in a timely manner. Even when findings are published, large amounts of data remain unanalyzed. Data sharing makes data from clinical trials available to other investigators for secondary uses, which include carrying out additional analyses, analyzing unpublished data, reproducing published findings, and conducting exploratory analyses to generate new research hypotheses. In several highly publicized cases, independent investigators who have reanalyzed the data underlying published results of clinical trials have challenged the published results as invalid or incomplete. These allegations have sparked debates, additional analyses, and new clinical trials. Further, they have caused regulators to limit marketing of the products or led sponsors to withdraw them. This back-and-forth discussion, while complex and perhaps confusing to the public, is how scientific knowledge progresses, and it has resulted in a broader evidence base for regulatory and clinical decisions.
Taken together, these are compelling justifications for sharing clinical trial data to benefit society and future patients. The challenge is to set clear expectations that clinical trial data should be shared and to agree on how to do so in a responsible manner that mitigates the risks involved. Stakeholders have concerns about data sharing. Clinical trial participants want assurance that data will be shared in a way that protects privacy
and is consistent with informed consent. Sponsors want a quiet period for regulators to evaluate the entire body of evidence submitted to them, appropriate safeguards for intellectual property and commercially confidential information, and protections from invalid secondary analyses. Academic clinical trialists want time to analyze and publish the data they have collected and thereby gain appropriate professional recognition for planning, organizing, and running clinical trials whose data are subsequently used by other investigators. Research institutions fear that requirements for sharing clinical trial data will be unfunded mandates. Participant and patient advocates want clinical trial data to be widely available in order to advance the development of new treatments. If data sharing is to be broadly accepted and fulfill its promise, these concerns of key stakeholders will need to be acknowledged and addressed. Moreover, the sharing of clinical trial data needs to be carried out in a way that maintains incentives for sponsors and researchers to develop new therapies and carry out future clinical trials and that sustains participants’ willingness to participate in trials.
In addressing its statement of task (see Box S-1), the committee that conducted this study worked to craft a report that would be useful well into the future, as well as addressing specific issues that need attention in the short term. The committee acknowledges that no body or authority currently is capable of enforcing the recommendations offered in this report for all stakeholders; rather, the committee interpreted its charge as helping to establish professional standards and set expectations for responsible sharing of clinical trial data.
GUIDING PRINCIPLES FOR SHARING CLINICAL TRIAL DATA
The goal of responsible sharing of clinical trial data should be to increase scientific knowledge that leads to better therapies for patients. The committee formulated the following guiding principles for responsible sharing of clinical trial data:
- Maximize the benefits of clinical trials while minimizing the risks of sharing clinical trial data.
- Respect individual participants whose data are shared.
- Increase public trust in clinical trials and the sharing of trial data.
- Conduct the sharing of clinical trial data in a fair manner.
These guiding principles need to be specified and balanced in the context of specific issues associated with the sharing of clinical trial data. The committee determined that the public should benefit from the sharing of clinical trial data in the form of valid scientific knowledge and
BOX S-1
Statement of Task for This Study
An ad hoc committee of the Institute of Medicine will conduct a study to develop guiding principles and a framework (activities and strategies) for the responsible sharing of clinical trial data. For the purposes of the study, the scope will be limited to interventional clinical trials, and “data sharing” will include the responsible entity (data generator) making the data available via open or restricted access, or exchanged among parties. For the purposes of this study, data generator will include industry sponsors, data repositories, and researchers conducting clinical trials. Specifically, the committee will:
- Articulate guiding principles that underpin the responsible sharing of clinical trial data.
- Describe a selected set of data and data sharing activities, including but not limited to:
- − Types of data (e.g., summary, participant).
- − Provider(s) and recipient(s) of shared data.
- − Whether and when data are disclosed publicly, with or without restrictions, or exchanged privately among parties.
- For each data sharing activity, the committee will:
- − Identify key benefits of sharing and risks of not sharing for research sponsors and investigators, study participants, regulatory agencies, patient groups, and the public.
- − Address key challenges and risks of sharing (e.g., resource constraints, implementation, disincentives in the academic research model, changing norms, protection of human subjects and patient privacy, intellectual property/legal issues, preservation of scientific standards and data quality).
- − Outline strategies and suggest practical approaches to facilitate responsible data sharing.
- Make recommendations to enhance responsible sharing of clinical trial data. The committee will identify guiding principles and characteristics for the optimal infrastructure and governance for sharing clinical trial data, taking into consideration a variety of approaches (e.g., a distributed/federated data system).
In developing the principles and framework and in defining the rights, responsibilities, and limitations underpinning the responsible sharing of clinical trial data, the committee will take into account the benefits of data sharing, the potential adverse consequences of both sharing and not sharing data, and the landscape of regulations and policies under which data sharing occurs. Focused consideration will also be given to the ethical standards and to integrating core principles and values, including privacy. The committee is not expected to develop or define specific technical data standards.
A discussion framework will be released for public comment, which will include tentative findings regarding (a) guiding principles and (b) a selected set of data sharing activities. Based on the public comments received and further deliberations, the committee will prepare a final report with its findings and recommendations.
improved clinical practice and public health; at the same time, however, the legitimate interests of stakeholders—particularly their concerns about the potential harms and costs of data sharing—need to be recognized and addressed in a fair manner.
KEY STAKEHOLDERS INVOLVED IN DATA SHARING
In Chapter 3, the committee analyzes the perspectives of key stakeholders regarding the sharing of clinical trial data and their assessment of the associated benefits, risks, and challenges.
No stakeholder can single-handedly create a clinical trial ecosystem in which sharing data is expected and the risks of sharing are minimized. But all stakeholders have crucial roles and responsibilities in creating a culture of responsible sharing of clinical trial data and in providing effective incentives for such sharing. The committee envisaged that different approaches to sharing clinical trial data will be developed and urges learning from experience with these approaches.
Recommendation 1: Stakeholders in clinical trials should foster a culture in which data sharing is the expected norm, and should commit to responsible strategies aimed at maximizing the benefits, minimizing the risks, and overcoming the challenges of sharing clinical trial data for all parties.
Funders and sponsors should
- promote the development of a sustainable infrastructure and mechanism by which data can be shared, in accordance with the terms and conditions of grants and contracts;
- provide funding to investigators for sharing of clinical trial data as a line item in grants and contracts;
- include prior data sharing as a measure of impact when deciding about future funding;
- include and enforce requirements in the terms and conditions of grants and contracts that investigators will make clinical trial data available for sharing under the conditions recommended in this report; and
- fund and promote the development and adoption of common data elements.
Disease advocacy organizations should
- require data sharing plans as part of protocol reviews and criteria for funding grants;
- provide guidance and educational programs on data sharing for clinical trial participants;
- require data sharing plans as a condition for promoting clinical trials to their constituents; and
- contribute funding to enable data sharing.
Regulatory and research oversight bodies should
- work with industry and other stakeholders to develop and harmonize new clinical study report (CSR) templates that do not include commercially confidential information or personally identifiable data;
- work with regulatory authorities around the world to harmonize requirements and practices to support the responsible sharing of clinical trial data; and
- issue clear guidance that the sharing of clinical trial data is expected and that the role of Research Ethics Committees or Institutional Review Boards (IRBs) is to encourage and facilitate the responsible and ethical conduct of data sharing through the adoption of protections such as those recommended by this committee and the emerging best practices of clinical trial data sharing initiatives.
Research Ethics Committees or IRBs should
- provide guidance for clinical trialists and templates for informed consent for participants that enable responsible data sharing;
- consider data sharing plans when assessing the benefits and risks of clinical trials; and
- adopt protections for participants as recommended by this committee and the emerging best practices of clinical trial data sharing initiatives.
Investigators and sponsors should
- design clinical trials and manage trial data with the expectation that data will be shared;
- adopt common data elements in new clinical trial protocols unless there is a compelling scientific reason not to do so;
- explain to participants during the informed consent process
- − what data will (and will not) be shared with the individual participants during and after the trial,
- − the potential risks to privacy associated with the collection and sharing of data during and after the trial and a summary of the types of protections employed to mitigate this risk, and
- − under what conditions the trial data may be shared (with regulators, investigators, etc.) beyond the trial team; and
- make clinical trial data available at the times and under the conditions recommended in this report.
Research institutions and universities should
- ensure that investigators from their institutions share data from clinical trials in accordance with the recommendations in this report and the terms and conditions of grants and contracts;
- promote the development of a sustainable infrastructure and mechanisms for data sharing;
- make sharing of clinical trial data a consideration in promotion of faculty members and assessment of programs; and
- provide training for data science and quantitative scientists to facilitate sharing and analysis of clinical trial data.
Journals should
- require authors of both primary and secondary analyses of clinical trial data to
- − document that they have submitted a data sharing plan at a site that shares data with and meets the data requirements of the World Health Organization’s International Clinical Trials Registry Platform before enrolling participants, and
- − commit to releasing the analytic data set underlying published analyses, tables, figures, and results no later than the times specified in this report;
- require that submitted manuscripts using existing data sets from clinical trials, in whole or in part, cite these data appropriately; and
- require that any published secondary analyses provide the data and metadata at the same level as in the original publication.
Membership and professional societies should
- establish as policy that members should participate in sharing clinical trial data as part of their professional responsibilities;
- require as a condition of submitting abstracts to a meeting of the society and manuscripts to the journal of the society that clinical trial data will be shared in accordance with the recommendations in this report; and
- collaborate on and promote the development and use of common data elements relevant to their members.
WHAT DATA SHOULD BE SHARED AND WHEN IN THE LIFE CYCLE OF A CLINICAL TRIAL
In Chapter 4, the committee analyzes the benefits, risks, and challenges of sharing the various types of clinical trial data that are generated at different times during the clinical trial life cycle.
Data sharing can refer to various types of data, including individual participant data (i.e., raw data and the analyzable data set); metadata, or “data about the data” (e.g., protocol, statistical analysis plan, and analytic code); and summary-level data (e.g., summary-level results posted on registries, lay summaries, publications, and CSRs). Therefore, a clinical trial data sharing policy needs to specify what data will be shared, when, and under what conditions.
The committee recognizes that sharing the various types of data presents different benefits and risks.
- Sharing summary results helps protect against publication bias but does not enable new analysis.
- Sharing the analyzable data set allows for reanalysis, meta-analysis, and scientific discovery through hypothesis generation, but this data set needs to be accompanied by metadata in order for secondary analyses to be rigorous and efficient, and sharing it could also lead to privacy risks and inappropriate analyses.
- Sharing the CSR allows for better understanding of regulatory decisions and facilitates use of the analyzable data set, but the CSR may contain commercially confidential information or be used for unfair commercial purposes.
- Sharing raw data is useful in certain circumstances but is overly burdensome in most cases and also presents risks to privacy.
- Summary results may be posted on public websites with few risks, but the risks of sharing individual participant data and CSRs are significant and may need to be mitigated in most cases through appropriate controls.
- Explaining to trial participants what data will be shared during the informed consent process and making their own data available to them following study completion and data analysis help uphold public trust in clinical trials.
The committee applied these considerations to clinical trial data for trials initiated after this report only, recognizing that sharing data from legacy trials may present greater risks and burdens and thus needs to be deliberated on a case-by-case basis. Sponsors and investigators are strongly urged to give priority to sharing of data from legacy trials whose findings influence decisions about clinical care.
Next, the committee considered the timing of data sharing. The committee sought to balance several goals: (1) providing to trial investigators a fair opportunity to publish their analyses; (2) allowing other investigators to analyze and use data that are otherwise not being published in a timely manner and to reproduce the findings of a published paper; and (3) reducing the risks of data sharing, including risks to participants and sponsors and the risk of invalid analyses of shared data. The committee appreciated that many clinical trialists feel strongly that, after years of effort carrying out a clinical trial, they should have the opportunity to write a series of papers analyzing the collected data before other investigators have access to the data. The committee concluded that after completion of a clinical trial, a moratorium of 18 months is generally appropriate before data are shared to allow trialists to carry out their analyses.
The committee paid particular attention to sharing of the analytic data set that supports a published paper reporting results of a clinical trial. Once the results of a study have been published, the scientific process is best served by allowing other investigators immediate access to the analytic data set supporting the publication so they can reproduce the published findings and carry out additional analyses to test the robustness of the conclusions.
In an ideal clinical trials ecosystem, the committee would favor sharing the analytic data set supporting a publication immediately upon publication. However, the committee recognized that there currently are many associated practical constraints and challenges that need to be addressed. The committee therefore has recommended a pragmatic compromise time frame of 6 months after publication at this time, with the expectation that the standard ultimately will become sharing the analytic data set simultaneously with publication. The committee hopes that the evolution of responsible sharing of clinical trial data will be guided by empirical evidence.
At the same time, the committee recognized that there will be justifiable exceptions to its recommendations in light of the wide variation in clinical trials. The recommended time periods at which specific data are to be shared are not intended to be hard-and-fast, inflexible rules. For trials that are likely to have a major clinical, public health, or policy impact, the committee favors sharing the analytic data set supporting a publication sooner than the 6-month guideline.
The committee next considered clinical trials submitted to a regulatory agency. Regulatory agencies review data from many trials and may carry out further analyses or require additional data. There are advantages to allowing regulatory agencies a “quiet period” to review the totality of evidence without being influenced by multiple analyses of just a portion of the data under review. However, if the sponsor publishes results from
a trial prior to regulatory approval, the analytic data set supporting the publication should be shared as recommended, even if that occurs before the end of the regulators’ quiet period.
Turning to clinical trials of products that are abandoned, the committee distinguished situations in which the sponsor transfers rights to develop the product to another company from situations in which it does not. The committee also considered sharing of data with trial participants and the public, distinguishing summary results of a trial from results of measurements on individual participants made during the trial.
Drawing together these considerations, the committee formulated the following recommendation for what data should be shared after key points in a clinical trial. The committee believes that this recommendation will set professional standards and establish expectations that clinical trial data should be shared (see also Figure S-1):
Recommendation 2: Sponsors and investigators should share the various types of clinical trial data no later than the times specified below. Sponsors and investigators who decide to make data available for sharing before these times are encouraged to do so.
Trial registration:
- The data sharing plan for a clinical trial (i.e., what data will be shared when and under what conditions) should be publicly available at a third-party site that shares data with and meets the data requirements of the World Health Organization’s International Clinical Trials Registry Platform; this should occur before the first participant is enrolled.
Study completion:
- Summary-level results of clinical trials (including adverse event summaries) should be made publicly available no later than 12 months after study completion.
- Lay summaries of results should be made available to trial participants concurrently with the sharing of summary-level results, no later than 12 months after study completion.
- The “full data package”1 should be shared no later than 18 months after study completion (unless the trial is in support of a regulatory application).
________________
1 See the notes to Figure S-1 for definitions of “full data package,” “post-publication data package,” and “post-regulatory data package.”
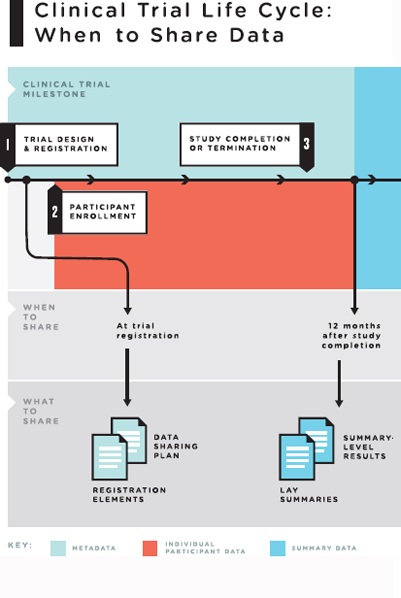
FIGURE S-1 The clinical trial life cycle: When to share data.
NOTES: Full data package = the full analyzable data set, full protocol (including initial version, final version, and all amendments), full statistical analysis plan (including all amendments and all documentation for additional work processes), and analytic code. Post-publication data package = a subset of the full data package supporting the findings, tables, and figures in the publication. Post-regulatory
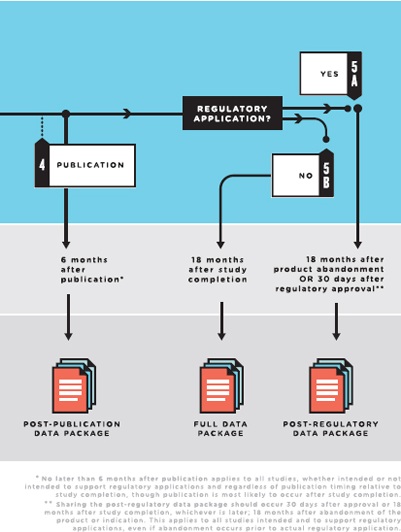
data package = the full data package plus the clinical study report (redacted for commercially or personal confidential information). Legacy trials: For trials initiated prior to the publication of these recommendations, sponsors and principal investigators should decide on a case-by-case basis whether to share data from the completed trials. They are strongly urged to do so for major and significant clinical trials whose findings will influence decisions about clinical care.
Publication:
- The “post-publication data package” should be shared no later than 6 months after publication.
Regulatory application:
- For studies of products or new indications that are approved, the “post-regulatory data package” should be shared 30 days after regulatory approval or 18 months after study completion, whichever occurs later.
- For studies of new products or new indications for a marketed product that are abandoned, the “post-regulatory data package” should be shared no later than 18 months after abandonment. However, if the product is licensed to another party for further development, these data need be shared only after publication, approval, or final abandonment.
ACCESS TO CLINICAL TRIAL DATA
In Chapter 5, the committee analyzes how several risks associated with sharing clinical trial data (in particular individual participant data and CSRs) might be addressed through controls on data access (i.e., with whom the data are shared and under what conditions) without compromising the usefulness of data sharing for the generation of additional scientific knowledge. Arrangements for determining access to clinical trial data need to balance several goals: protecting the privacy of research participants, reducing the likelihood of invalid analyses or misuse of the shared data, avoiding undue burdens on secondary users seeking access, avoiding undue harms to investigators and sponsors that share data, and enhancing public trust in the sharing of clinical trial data.
The committee noted support for open and free access to scientific publications immediately upon publication, as well as the requirement of the U.S. Food and Drug Administration (FDA) to make a summary of clinical trial results available to the public. The committee believes that open access (to the public with no controls) is appropriate and desirable for clinical trial results, and, in some cases, no or few controls on sharing other types of clinical trial data may be the preferred approach when all stakeholders involved in a trial (i.e., sponsors, investigators, and participants) are comfortable with this approach and believe the benefits outweigh the risks. In many cases, however, sponsors, investigators, and/or participants may have concerns about an open access model for certain clinical trial data and may wish to place some conditions on access to or uses of the data.
In reviewing protections for privacy, the committee noted that while
de-identification is commonly used to protect privacy, it has limitations. Different jurisdictions have different de-identification standards. Moreover, the risk of re-identification depends on the context in which data are released, the type of data, and the additional information that might be combined with the shared data. In the case of genome-wide sequencing data and “big data” analyses, for example, de-identification and data security alone may not provide adequate protection; additional privacy and security techniques are being developed for these cases.
The committee determined that data use agreements are a promising vehicle for reducing these risks and related disincentives for sharing clinical trial data. The committee reviewed a variety of provisions in existing data use agreements aimed at reducing risks to various parties, enhancing the scientific value of secondary analyses, and protecting the public health. The committee does not endorse all the specific provisions that were reviewed but believes they should be considered as potential options. Although it is unclear whether and how data use agreements will be enforced, the committee believes these agreements have significant normative, symbolic, and deterrent value, setting professional expectations and standards for responsible behavior.
The committee considered the review of requests for data access and reached several conclusions. Access restrictions based on the composition of the secondary analysis team—for example, requiring a biostatistician with particular qualifications or excluding lawyers—would not further the goals of responsible sharing of clinical trial data. Review of research proposals could mitigate risks, but overly restrictive controls would inhibit valid secondary analyses and innovative scientific proposals. If the trial sponsor or investigator, rather than an independent review panel, reviewed data requests and made decisions regarding access, concerns about conflicts of interest could lead to mistrust. Representatives of communities and patient and disease advocacy groups could serve as useful members of such review panels. Furthermore, making the policies and procedures regarding access to clinical trial data transparent would enhance the trustworthiness of data sharing programs.
Finally, the committee concluded that the experience of early adopters of the sharing of clinical trial data will undoubtedly offer lessons and best practices from which others can learn. As sponsors try different approaches to data sharing, collecting empirical data that allow comparison of different approaches will provide crucial information on what does and does not work in various contexts.
In light of the above considerations, the committee formulated the following recommendation regarding data access:
Recommendation 3: Holders of clinical trial data should mitigate the risks and enhance the benefits of sharing sensitive clinical trial data by implementing operational strategies that include employing data use agreements, designating an independent review panel, including members of the lay public in governance, and making access to clinical trial data transparent. Specifically, they should take the following actions:
- Employ data use agreements that include provisions aimed at protecting clinical trial participants, advancing the goal of producing scientifically valid secondary analyses, giving credit to the investigators who collected the clinical trial data, protecting the intellectual property interests of sponsors, and ultimately improving patient care.
- Employ other appropriate techniques for protecting privacy, in addition to de-identification and data security.
- Designate an independent review panel—in lieu of the sponsor or investigator of a clinical trial—if requests for access to clinical trial data will be reviewed for approval.
- Include lay representatives (e.g., patients, members of the public, and/or representatives of disease advocacy groups) on the independent review panel that reviews and approves data access requests.
- Make access to clinical trial data transparent by publicly reporting
- − the organizational structure, policies, procedures (e.g., criteria for determining access and conditions of use), and membership of the independent review panel that makes decisions about access to clinical trial data; and
- − a summary of the decisions regarding requests for data access, including the number of requests and approvals and the reasons for disapprovals.
- Learn from experience by collecting data on the outcomes of data sharing policies, procedures, and technical approaches (including the benefits, risks, and costs), and share information and lessons learned with clinical trial sponsors, the public, and other organizations sharing clinical trial data.
THE FUTURE OF CLINICAL TRIAL DATA SHARING IN A CHANGING LANDSCAPE
Chapter 6 presents the committee’s vision for responsible sharing of clinical trial data in the future. In this vision, all stakeholders are com-
mitted to sharing data responsibly, have modified their work processes to facilitate data sharing, and possess the resources and tools necessary to do so.
- A culture of sharing clinical trial data with effective incentives for sharing emerges.
- There are more platforms for sharing clinical trial data, with different data access models and with sufficient total capacity to meet demand. The different platforms are interoperable: data obtained from various platforms can easily be searched and combined to allow further analyses.
- There is adequate financial support for sharing clinical trial data, and costs are fairly allocated among stakeholders.
- Protections are in place to minimize the risks of data sharing and to reduce disincentives for sharing.
- Best practices for sharing clinical trial data are identified and modified in response to ongoing experience and feedback. The sharing of clinical trial data forms a “learning” ecosystem in which data on data sharing outcomes are routinely collected and continually used to improve how data sharing is conducted.
Next the committee identified remaining key challenges to responsible sharing of clinical trial data, which include the following:
- Infrastructure challenges—Currently there are insufficient platforms to store and manage clinical trial data under a variety of access models.
- Technological challenges—Current data sharing platforms are not consistently discoverable, searchable, and interoperable. Special attention is needed to the development and adoption of common protocol data models and common data elements to ensure meaningful computation across disparate trials and databases. A federated query system of “bringing the data to the question” may offer effective ways of achieving the benefits of sharing clinical trial data while mitigating its risks.
- Workforce challenges—A sufficient workforce with the skills and knowledge to manage the operational and technical aspects of data sharing needs to be developed.
- Sustainability challenges—Currently the costs of data sharing are borne by a small subset of sponsors, funders, and clinical trialists; for data sharing to be sustainable, costs will need to be distributed equitably across both data generators and users.
The committee gave particular attention to the need for a sustainable and equitable business model for responsible sharing of clinical trial data and developed the following conceptual framework:
- Responsible sharing of clinical trial data benefits the public and multiple stakeholders, including payers of health care as well as patients, their physicians, and researchers.
- As a matter of fairness, those who benefit from responsible sharing of clinical trial data, including the users of shared data, should also bear some of the costs of sharing. Additional sources of funding for responsible sharing of clinical trial data, such as private philanthropies, need to be identified.
- Policies on equitable distribution of the costs of responsible sharing of clinical trial data among stakeholders should be based on accurate information on the costs of data sharing for various kinds of clinical trials.
- The costs of responsible sharing of clinical trial data will decrease in the future if data collection and management are designed to facilitate sharing.
The committee concluded that a market analysis of the costs of sharing clinical trial data and an economic analysis of options for funding data sharing would provide an evidence base for developing sustainable and equitable models for responsible sharing of clinical trial data.
Finally, the committee considered the ecosystem of responsible sharing of clinical trial data. Individual sponsors and trusted intermediaries can do a great deal to make sharing clinical trial data more responsible, effective, and efficient. For responsible sharing of clinical trial data to become pervasive, sustained, and rooted as a professional norm, however, many challenges need to be addressed in collaboration with other institutions and stakeholders. The committee recommends a next step to promote discussion and exchange of ideas among a wide range of stakeholders in order to forge agreement on best practices, standards, and incentives:
Recommendation 4: The sponsors of this study should take the lead, together with or via a trusted impartial organization(s), to convene a multistakeholder body with global reach and broad representation to address, in an ongoing process, the key infrastructure, technological, sustainability, and workforce challenges associated with the sharing of clinical trial data.
















