
The Clinical Trial Life Cycle and When to Share Data
During the course of a clinical trial, different types of data are collected, transformed into analyzable data sets to address specific research questions, and used to generate various publications and reports for different audiences (Drazen, 2002).
Publication in peer-reviewed scientific journals is currently the primary method for sharing clinical trial data with the scientific and medical communities, as well as the public (often through media coverage of published findings). These publications, however, contain only a small subset of the data collected, produced, and analyzed in the course of a trial (Doshi et al., 2013; Zarin, 2013).
Clinical trial sponsors seeking regulatory approval from authorities such as the U.S. Food and Administration (FDA) and the European Medicines Agency (EMA) must submit detailed clinical study reports (CSRs) and individual participant data, which form the basis for the marketing application for a product. In trials that are not part of a regulatory submission, detailed CSRs may or may not be prepared (Doshi et al., 2012; Teden, 2013).
Beyond the selected clinical trial data that are disclosed in journal publications, individual participant data and metadata (i.e., “data about the data”) have not been shared routinely with the broader scientific community or the public. As stated in previous chapters, however, an increasing number of organizations are taking the initiative to share their data more actively, and the committee believes the time is right to make recommendations for responsible sharing of clinical trial data that will
increase the availability and usefulness of the data while mitigating the risks outlined in Chapter 2. The committee acknowledges that no single body or authority in the global clinical trials ecosystem has the power to enforce the recommendation offered at the end of this chapter on what data to share and when. Moreover, the committee recognizes that no single solution will fit all clinical trials, and there will be exceptions to any proposed recommendations. The risks and benefits of sharing data may vary depending on the context and purpose of a trial and/or the particular type of data to be shared. Thus, there will be instances when it is reasonable for data to be shared either sooner or later than would generally be expected. The committee interpreted its charge as helping to establish professional standards and set expectations for responsible sharing of clinical trial data, together with requirements to be imposed by supporting organizations such as funders, medical journals, and professional societies, rather than suggesting new laws and regulations.
Many discussions of the sharing of clinical trial data to date have failed to specify which of the many clinical trial data elements or data sets might be shared at different time points in the life cycle of a trial. To address this shortcoming, this chapter briefly describes the major stages of the clinical trial life cycle and then offers specific definitions and descriptions of the individual participant data, metadata, and summary data that are generated at each stage. It then examines the benefits and risks associated with sharing the various types of data generated and presents the committee’s recommendation for when to share specific data packages in common scenarios such as after publication or regulatory application, as well as the case of sharing data with participants themselves.
Data are generated at nearly every stage of the clinical trial life cycle, from the initial protocol and statistical analysis plan prepared prior to registration, to the collection of baseline participant data at participant enrollment, to the analysis of the analyzable data set. To help frame the discussion of what data should be shared at what times, the committee conceptualized the clinical trial life cycle as consisting of five major stages (see Figure 4-1):
- Trial design and registration: Clinical trials are carefully designed; the protocol1 for conducting the trial and the statistical analysis
________________
1 The protocol is approved by a Research Ethics Committee (in the United States, an Institutional Review Board [IRB]) and explained to participants through the informed consent process.
-
plan (SAP) detailing the planned data analyses are developed well before the first participant is enrolled. The protocol and the SAP constitute some of the most important metadata of the trial. During the course of a trial, the protocols and the SAP generally undergo amendments, which should be explicitly documented within the protocol and the SAP. Since 2004, the International Committee of Medical Journal Editors (ICMJE) has required that all trials be registered prior to participant enrollment as a condition for consideration of publication (De Angelis et al., 2004). In 2006, the World Health Organization’s (WHO’s) International Clinical Trials Registry Platform identified a set of 20 data elements for all trials to include in registration. These 20 elements, along with narrative summaries of the protocol, make up the registration elements in Figure 4-1 (Sim et al., 2006).
- Participant enrollment: Clinical trial data originate from patients and healthy volunteers who participate in studies. Raw data are collected between the time of first participant enrollment and study completion. During the course of the trial, the raw data are abstracted, coded, and transcribed. After participant activities have ended, the data are cleaned into an analyzable data set. Both the raw data and the analyzable data set are individual participant data.
- Study completion: For the purposes of this report, study completion is defined as either “the study has concluded normally; participants are no longer being examined or treated,” or the study has been terminated;2 “recruiting or enrolling participants has halted prematurely and will not resume; participants are no longer being examined or treated” (ClinicalTrials.gov, 2012).3 Although study completion is often referred to as “last participant’s last visit” (European Union Clinical Trials Register, 2011), study completion may in fact include measurements that occur without face-to-face interaction between participants and investigators (e.g., data may be collected remotely using sensors or from medical records or reports by patients, or an endpoint such as death may occur after the last visit). After study completion, investigators and/or sponsors clean the data, derive additional variables, adjudicate endpoints, lock
________________
2 A study may be completed prematurely for various reasons: a Data Monitoring Committee/Data and Safety Monitoring Board may recommend termination to the sponsor on the basis of interim monitoring for ethical and scientific reasons, or the sponsor may decide to terminate the study for ethical, scientific, or business reasons.
3 “STUDY COMPLETION DATE: The date that the final data for a clinical study were collected because the last study participant has made the final visit to the study location (that is, ‘last subject, last visit’). The estimated study completion date is the date that researchers think will be the completion date for the study” (ClinicalTrials.gov, 2012).
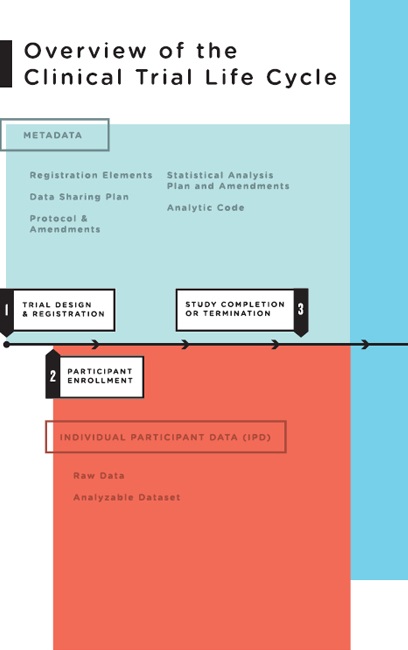
FIGURE 4-1 Overview of the clinical trial life cycle.
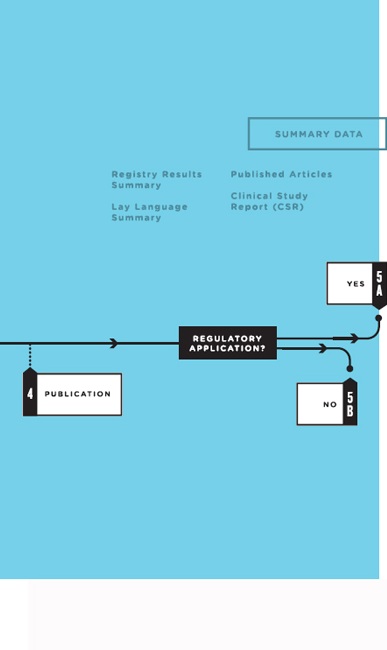
-
the data set to create the full analyzable data set, carry out prespecified and additional analyses using software programs (the analytic code), and prepare manuscripts for publication. Investigators also can use parts of the analyzable data set to prepare analyses for presentations, data exploration, and hypothesis generation. A biostatistics best practice is to freeze a copy of whatever data were used in an analysis so the results can be repeated later if necessary. It is also desirable to store the analytic code that generated the results (i.e., the computer program), especially for any derived data.
- Publication: A subset of the data from the analyzable data set is used to generate the tables, figures, and results for published articles. Publication may occur at any time during the course of the clinical trial life cycle, although it most often occurs after study completion. Several publications may result from one trial.
- Regulatory application: Clinical trials are required by regulatory authorities in countries around the world before a new medical product can be brought to market, or before a new indication, formulation, or target population can be approved for an intervention already on the market (ICH, 1995). Analyses prespecified in the SAP form the basis for the CSR, which includes a detailed analysis of the study efficacy data and the complete adverse event data. After a product’s introduction, additional clinical trials are commonly conducted by both industry and academia to further define the product’s efficacy and safety. Clinical trials also are used to study interventions that do not involve regulated medical products, such as surgical techniques, behavioral interventions, means of improving disease management practice, or changes to a health care system (Califf, 2013).4 Thus, as the committee examined what clinical trial data should be shared and when, it was useful to consider clinical trials in these two broad categories: (5a) those intended to support a regulatory application5 and (5b) those not intended to support a regulatory application.
This section further describes each of the above forms of clinical trial data and presents the committee’s analysis of the benefits and risks of sharing each.
________________
4 As noted in Chapter 1, the scope of this study is limited to interventional clinical trials, as defined in Box 1-1.
5 Whether or not the product or indication ultimately receives regulatory approval, is abandoned by the sponsor, or licensed to another entity.
Individual Participant Data
Raw Data
Raw data (sometimes called source data) are observations about individual participants used by the investigators. These data may be collected specifically for the study protocol or as part of routine care. At the source, these data may be in the form of measurements of participant characteristics such as weight, blood pressure, or heart rate and may be associated with the baseline (or initial) visit or subsequent follow-up visits. Raw data may also include a baseline description of the participant’s medical history, physical exam information, clinical laboratory results (e.g., serum lipid values, hemoglobin levels), whole exome or genome sequences, imaging results (e.g., X-ray, magnetic resonance imaging [MRI]), procedure results (e.g., electroencephalogram [EKG], endoscopy), or self-reported data (e.g., symptoms, quality of life). Some data are derived or calculated from raw data (e.g., change in weight from baseline). Other data must be abstracted (i.e., interpreted) according to rules set forth in the protocol—for example, reading the X-ray for tumor size or evaluating the electrocardiogram (ECG) for evidence of a heart attack. Depending on the study under consideration, demographics, clinical outcome data, and other appropriate raw data are entered into case report forms. Some observations (e.g., imaging studies) are interpreted by study investigators—a process referred to as abstraction—and are entered into case report forms as transcribed narrative data or as coded data according to study guidelines (e.g., men may be coded as “1” and women as “0”). Abstracted data may also include assessments by clinical study staff or adjudication committees to determine whether specific clinical endpoints or adverse events (e.g., heart attack, cardiac death) meet protocol-specified criteria. In addition to physiologic and clinical measurements, other types of health data are increasingly being collected in clinical trials, including quantified sensor data (e.g., readings from remote monitoring devices, including smartphone apps), consumer genomics data (e.g., 23andMe), and participant-reported outcomes (e.g., PatientsLikeMe).
After collection, as well as derivation, assessment, abstraction, or adjudication as appropriate, data must be entered into an organized data management system (i.e., database) for further evaluation and processing. Data typically undergo a process of cleaning, quality assurance, and quality control to detect inconsistent, incomplete, or inaccurate entries, and to confirm that the data were collected and evaluated according to the protocol and match the source data. This process of data transcription continues over the course of the trial as data are collected and afterward.
The argument for sharing raw data is that raw data most closely reflect the study observations. The analyzable data set, by contrast, is the
result of many decisions made by clinical trialists, as explained above. If there are errors, flaws, or biases in the processing of raw data, such problems will not necessarily be identified in the analyzable data set. Examples of the value of raw data include the detection of serious errors or biases as well as fraud uncovered by detailed and intense audits of raw data conducted by central statistical centers when inconsistencies or anomalies have been noted in analyzable data sets (Fisher et al., 1995; Soran et al., 2006; Temple and Pledger, 1980). Secondary researchers who have questions about the original analyses may therefore want the raw data to verify how the analytic data set was derived or to test alternative ways of deriving the variables in that data set. In other cases, sharing raw data can be extremely beneficial for additional research. For instance, the Alzheimer Disease Neuroimaging Initiative (ADNI) has published a number of important secondary analyses of a shared raw data set of images among a group of neuroscience investigators (ADNI, 2014; Bradshaw et al., 2013; Casanova et al., 2013; Haight et al., 2013).
However, there are strong arguments against sharing raw data routinely. Raw data sets are large and complex, include potentially sensitive individual participant data, and are not needed for most secondary analyses of shared clinical trial data. For example, raw data from MRI and computed tomography (CT) scans or genome sequences may be very large, require extensive documentation, and carry inherent privacy risks. The effort and cost of sharing such data routinely might better be spent on improved quality control and independent oversight of the data processing carried out during the course of a trial such that the analyzable data set can be shared with high levels of confidence in its integrity. These quality control processes should be documented and made available along with the analytic data set. If questions remained, investigators, sponsors, regulatory agencies, or external researchers could request “for cause” audits on a case-by-case basis.
Conclusion: For most trials sharing raw data would be overly burdensome and impractical; on a case-by-case basis, however, it would be beneficial to share raw data in response to reasonable requests.
Analyzable Data Set
Typically, after data have been entered in computerized form, new variables are generated mathematically to serve as the basis for later analyses. These variables are sometimes called “derived” variables. For example, patient age may not be entered directly but calculated by subtracting the birth date from the date of a given clinic visit, or “treatment response” may be entered as a mathematical comparison of lesion sizes
recorded from two images. After the trial has been declared complete and the editing and cleaning process is done, the data are moved into an analyzable data file and locked (i.e., no further changes may be made). If the study is blinded (or masked), the treatment code file is typically merged with the analyzable data file after the latter has been locked, and the data are unblinded to the investigators. Some or all of this now-unblinded analyzable data set is then used for data analyses.6 It is called analyzable rather than analyzed because a very large percentage of it is never used. The final cleaned and locked analyzable data set consists of various components (participant characteristics and primary outcome, prespecified secondary and tertiary outcomes, adverse event data, and exploratory data). A statistical analysis may involve a composite outcome using any of the various components. In addition, when data are missing, values may be imputed using this data set. Results are derived from data in the cleaned and locked analyzable data set, which have undergone statistical analysis.
The full analyzable data set is generally the most useful set of data to share from a trial, with large and likely important benefits to science and society. First, in most trials, as noted, large portions of the full analyzable data set are never analyzed and published. Sharing these data may increase the scientific return on the funder’s investment in the trial and the benefits to the public and future patients. Second, sharing the full analyzable data set allows other investigators to reproduce the original analyses and carry out alternative, scientifically valid analyses of the primary study aim. Such additional analyses help determine how robust the original analyses are. Third, meta-analytic syntheses of the results of similar trials increase the statistical power for detecting effects and maximize the evidentiary value of the clinical trial knowledge base. Finally, sharing analyzable data allows for further scientific discovery through additional secondary analyses, as well as the conduct of exploratory research to general hypotheses for additional studies.
Sharing the full analyzable data set also poses risks, as described in Chapter 2. One risk is invalid secondary analyses and use of statistical techniques that are not scientifically accepted. A particular concern is that a secondary user will carry out multiple statistical analyses on a complex data set without adjusting for the increased likelihood that one association will be found to be significant at a p level of 0.05 by chance alone. A second concern is that the clinical trial team often plans to publish a series of secondary analyses, particularly in large clinical trials. Often junior members of the team who are assigned to be lead author on one of these
________________
6 Best practice is to conduct the analyses on blinded data with dummy codes representing treatment arms.
secondary analyses regard this authorship as a key opportunity for career advancement and a quid pro quo for the effort they put into working on the trial. The clinical trialists may fear that sharing the full analyzable data set will give other investigators who did not contribute to the conduct of the study an opportunity to publish additional analyses before the members of the study team have done so. A third risk is that the more detailed the information is in a data set, the easier it is to re-identify an individual in that data set, particularly if “side information” is included (Dwork, 2014). For some situations in which the risk of identification is judged to be too high—for example, with sensitive health conditions or when harm may come to identified participants—sharing may not be advisable. Finally, the risk of re-identification of trial participants increases as the scope of data on each person and the number of participants increase. Using “big data” analytic techniques on de-identified data, researchers may re-identify participants by combining additional information, or they may determine whether an individual about whom they have independent information is a participant in the study and draw inferences about him or her, as discussed in Chapter 3.
Conclusion: Sharing the analyzable data set would benefit science and public health by allowing reanalysis, meta-analysis, and scientific discovery through hypothesis generation.
Conclusion: The risks of sharing individual participant data are significant and need to be mitigated in most cases through appropriate controls. In certain circumstances, the risks or burdens may be so great that sharing is not feasible or requires enhanced privacy protections.
Metadata and Additional Documentation
To use clinical trial data (e.g., to perform the primary analysis or carry out confirmatory or exploratory analyses), researchers need metadata (i.e., “data about the data”) and additional documentation beyond the individual participant data described above. Currently the trial registration data set of 20 items identified by WHO is publicly available for most trials (WHO, 2014). This data set includes the trial and investigator identification information, names of sponsors and funders, study type, participant enrollment information, and primary and secondary outcomes. These 20 items, however, are a small subset of the metadata and additional documentation developed for clinical trials, which include the data sharing plan, protocol and all amendments, the SAP and all amendments, analytic code, and other documents described in Box 4-1 and in the following pages.
BOX 4-1
Metadata and Additional Documentation
- Data sharing plan
- Clinical trial registration number and data set (available through Clinical Trials.gov and other World Health Organization [WHO] registries)
- Full trial protocol (e.g., all outcomes, study structure), including first version, final version, and all amendments
- Manual of operations describing how a trial is conducted (e.g., assay method) and standard operating procedures, including names of parties involved, specifically
- – names of persons on the clinical trial team, trial sponsor team, data management team, and data analysis team; and
- – names of members of the steering committee, Clinical Events Committee (CEC, which adjudicates endpoints), and Data and Safety Monitoring Board (DSMB)/Data Monitoring Committee (DMC), as well as committee charters
- Details of study execution (e.g., participant flow, deviations from protocol)
- Case report templates describing what measurements will be made and at what timepoints during the trial, as defined in the protocol
- Informed consent templates describing what participants agreed to, what hypotheses were included, and for what additional purposes participants’ data may be used
- Full statistical analysis plan (SAP), which includes all amendments and all documentation for additional work processes (including codes, software, and audit of the statistical workflow)
- Analytic code describing the clinical and statistical choices made during the clinical trial
Data Sharing Plan
The data sharing plan describes what specific types of data will be shared at various time points and how to seek access to the data. According to the U.S. National Institutes of Health (NIH):
The precise content of the data-sharing plan will vary, depending on the data being collected and how the investigator is planning to share the data. Applicants who are planning to share data may wish to describe briefly the expected schedule for data sharing, the format of the final data set, the documentation to be provided, whether or not any analytic tools also will be provided, whether or not a data-sharing agreement will be required and, if so, a brief description of such an agreement (including the criteria for deciding who can receive the data and whether or not any conditions will be placed on their use), and the mode of data sharing (e.g., under their own auspices by mailing a disk or posting
data on their institutional or personal website, through a data archive or enclave). (NIH, 2003)
Sharing the data sharing plan publicly before the first participant is enrolled allows the public, Research Ethics Committee, and potential participants to know what the sponsor and investigator are planning to do. As noted in Chapter 3 and Box 4-1, the data sharing plan should be discussed during the informed consent process. Disclosure of the data sharing plan also helps identify cases in which the sponsor changed its plan for data sharing after the trial began.
To be useful to potential secondary users of a clinical trial data set, the data sharing plan needs to be publicly available and readily accessible. One means of accomplishing this goal is to make data sharing plans a 21st element in the WHO trial registration data set.
In the interests of full transparency, the data sharing plan should include a comprehensive justification for any intent not to share data. In some situations, an exception to sharing the analyzable data set may be appropriate, or additional controls over access may be required. This may occur, for example, because of exceptional privacy risks. Such exceptions to usual data sharing plans should be made publicly available.
Trial Protocol
The trial protocol provides the overall experimental design. It describes the rationale for the trial, the eligibility and exclusion criteria for participants, the primary and secondary hypotheses and the corresponding primary and secondary outcome measures, the methods used to identify and adjudicate adverse events, and other measures intended for use in evaluating the intervention, as well as a full description of the entire study and all interventions and tests and how they are to be administered. A SAP should also be part of the protocol. When registering a trial, investigators and sponsors must provide both a brief summary and a detailed description of the protocol (see Box 4-2).
Over the life of a clinical trial, the protocol usually is modified a number of times. All amendments to the initial protocol should be dated and saved. To replicate or reproduce a study, other investigators need the version of the protocol in effect when the first participant was enrolled, as well as all modifications and the full protocol version in force when the data set was locked (and before unblinding). This should be the final protocol, as revisions made after the data set has been locked and unblinded are fraught with bias.
Sharing of protocols promotes protocol quality improvement efforts (e.g., Standard Protocol Items: Recommendations for Interventional Trials
BOX 4-2
Required Descriptions of Trial Protocols
According to ClinicalTrials.gov, the following descriptions of the protocol for a trial are required:
Brief Summary
Definition: Short description of the protocol intended for the lay public. Include a brief statement of the study hypothesis. (Limit: 5,000 characters)
Example: The purpose of this study is to determine whether prednisone, methotrexate, and cyclophosphamide are effective in the treatment of rapidly progressive hearing loss in both ears due to autoimmune inner ear disease.
Detailed Description
Definition: Extended description of the protocol, including more technical information (as compared to the Brief Summary) if desired. Do not include the entire protocol; do not duplicate information recorded in other data elements, such as eligibility criteria or outcome measures. (Limit: 32,000 characters)
SOURCE: ClinicalTrials.gov, 2012.
[SPIRIT]) (Chan et al., 2013), complements trial registration in identifying trials that were initiated, allows future auditing of data sharing, facilitates meta-analyses and systematic reviews, promotes greater standardization of protocol elements (e.g., interventions, outcomes), and may help reduce unnecessary duplication of studies. The full protocol with all amendments (see Figure 4-1) should be shared to help other investigators understand the original analysis, replicate or reproduce the study, and carry out additional analyses.
Statistical Analysis Plan
A SAP also is prespecified before the start of a trial. It may be amended during the trial while the data are blinded but is finalized before the analysis is completed and the data are unblinded. The SAP drives the primary analyses of the analyzable data set. It describes the analyses to be conducted and the statistical methods to be used, as determined by the protocol. The SAP includes, for example, plans for analysis of baseline descriptive data and adherence to the intervention, prespecified primary and secondary outcomes, definitions of adverse and serious
adverse events, and comparison of these outcomes across interventions for prespecified subgroups. These SAP-defined analyses are often extensive and can result in several dozen tables and figures.
For many clinical trials, the SAP-defined analyses may not use all of the data available in the analyzable data set. Moreover, as discussed earlier, publications of clinical trials in peer-reviewed journals generally draw on only part of the analyzable data set. Supplemental data often are collected to permit exploration of ancillary questions not directly related to the primary purpose of the protocol, and researchers may conduct exploratory and post hoc analyses not defined in the SAP to answer additional questions.
The full SAP describes how each data element was analyzed, what specific statistical method was used for each analysis, and how adjustments were made for testing multiple variables. The full SAP includes all amendments and all documentation for additional work processes from earlier versions. If some analysis methods require critical assumptions, data users will need to understand how those assumptions were verified.
Sharing the SAP enables additional scientific discoveries by allowing other investigators to carry out alternative or additional analyses to test the robustness of published findings, such as post hoc subgroup analyses or composite endpoints; to compare outcomes from other trials; to plan meta-analyses; and to carry out exploratory studies to generate new hypotheses. In addition, sharing the SAP allows other investigators to replicate or reproduce the original analysis. Reproducibility of results is discussed in more detail below.
Analytic Code
The analyzable data set from a clinical trial is transformed into scientific results through an analytic process that involves many steps and many clinical and statistical choices. These choices include but are not limited to which and how many analyses to conduct, which variables to include in an analysis, which statistical procedures to use, and whether and how to adjust analyses statistically for confounders. Some of these choices are detailed in the study’s SAP, while other choices are necessarily made during the process of data analysis. Regardless, these choices are reflected in the statistical programming code used to produce the study results. Another researcher attempting to reproduce the findings of a trial will need access to the statistical programming code, as well as the version of the software used.
To promote overall scientific validity and trust in the clinical research enterprise, there has been an increasing focus on ensuring the reproducibility of research (Collins and Tabak, 2014; IOM, 2014; Jasny, 2011). Repro-
duction requires access to the original analytic code, which reflects the investigators’ clinical and statistical choices, detailed earlier, that may not have been included in any data analysis plan or publication. Access to the analytic code also allows for verification of the correctness of the code and should be coincident with the availability of the data. Recent examples of coding errors in widely quoted economic reports have come to light because independent researchers had access to the data and the analytic code (Giles, 2014; Krugman, 2013). In a highly publicized example, external scientists raised concerns about the validity of gene-expression tests that were used to assign treatment arms in three NIH-sponsored cancer clinical trials. The study team did not respond to requests by external scientists to obtain the analytic code for the studies presenting the tests; when external biostatisticians obtained access, they identified serious errors (IOM, 2012). Arguably, had the analytic code and full analytic data set of the publications been shared, serious harms to participants in the cancer clinical trials—including misclassification of patients as being at low or high risk of recurrence—could have been averted. Such examples illustrate the importance of demonstrating statistical reproducibility. Sharing of the analytic code along with the analytic data set also aligns with the principles of transparency and accountability that underlie effective sharing of clinical trial data. At the same time, licensing models for reproducible research are needed to protect the rights of investigators over their code and associated products (IOM, 2012; Laine et al., 2007; Sandve et al., 2013; Stodden, 2009, 2013).
Conclusion: The clinical trial protocol, the SAP, amendments, and other metadata need to be shared along with the analyzable data set so that secondary investigators can plan and carry out analyses rigorously and efficiently.
Thus, a “full data package”—which the committee defines as the full analyzable data set, the full protocol (including first version, final version, and all amendments), the SAP (including all amendments and all documentation for additional work processes), and the analytic code—will allow for the majority of secondary analyses that other investigators may wish to carry out, including systematic reviews and meta-analyses of individual participant data.
Summary Data
Box 4-3 lists some of the documents that are that are commonly generated from analysis of the data from a clinical trial. These include publications describing the primary and major secondary outcomes specified
BOX 4-3
Documents Based on Analysis of Clinical Trial Data
- Publications (including those in peer-reviewed scientific journals)
- Summaries of results for registries (e.g., for ClinicalTrials.gov)
- Lay-language summaries
- Clinical study reports (CSRs)
- – Full CSR, with or without appendixes
- – CSR synopsis (executive summary)
- – Redacted CSR
- – Abbreviated CSR
in the protocol, summaries for registries, lay summaries, and CSRs for regulators.
Publications
Several scientific journal publications are commonly derived from the prespecified analyses driven by the SAP and from post hoc analyses. Typically, a primary publication will address the primary and possibly the leading secondary outcome measures specified in the protocol. The primary publication will also include the baseline measures to demonstrate the comparability of participants in the different intervention arms and comparisons across the intervention arms of any adverse events of major interest or frequency. Subsequent journal publications may address in greater detail a specific aspect of the primary analysis that was not included in the primary publication or analyze outcomes in particular prespecified subgroups of participants. Each journal publication is supported by a specific analytic data set corresponding exclusively to the data used to generate the tables and figures in the publication (which will be a subset of the full analyzable data set). A snapshot of each analytic data subset is typically stored in a separate set of data files to document the data used for each journal publication.
The committee appreciates that many clinical trialists plan to publish a number of papers from the data collected during a trial, particularly a long and complex trial. Other important secondary analyses may become apparent only after the trial team has become familiar with the data. As noted earlier, particularly for junior members of the clinical trial team, the prospect of being lead author on a secondary analysis is viewed as
their fair professional reward for their work in planning and carrying out the trial. Understandably, these investigators might feel distressed and demoralized by the possibility that other investigators who did not put effort into collecting the data would gain priority in publishing secondary analyses on the basis of shared data. On this issue, the committee shares the view of a previous National Research Council committee (addressing the more general context of life science research):
Community standards for sharing publication-related data and materials should flow from the general principle that the publication of scientific information is intended to move science forward. More specifically, the act of publishing is a quid pro quo in which authors receive credit and acknowledgment in exchange for disclosure of their scientific findings. An author’s obligation is not only to release data and materials to enable others to verify or replicate published findings (as journals already implicitly or explicitly require) but also to provide them in a form on which other scientists can build with further research. All members of the scientific community—whether working in academia, government, or a commercial enterprise—have equal responsibility for upholding community standards as participants in the publication system, and all should be equally able to derive benefits from it. (NRC, 2003, p. 4)
The committee agrees that sharing the analytic data set supporting a publication is an integral part of the process of communicating results through publication. Once a study finding has been published, the scientific process is best served by allowing other investigators to reproduce the findings and carry out additional analyses to test the robustness of the published conclusions. Robust conclusions increase confidence in the validity of the publication’s findings. For example, repeating the prespecified analysis and obtaining identical results will provide evidence that the results of the study, given the approach taken to data collection and analysis, are accurate. If alternative analyses addressing the research questions in the publication also yield the same findings, the reported findings can be considered robust. By contrast, if the published results do not hold when analytic approaches other than the prespecified approach are used, the report’s findings may need to be qualified, modified, or discussed further. In this case, readers of the publication will have reason to be cautious in applying the reported results to clinical practice or the design of future trials. Access to data that allow replication of the published results can therefore benefit the public by strengthening the evidence base on which physicians draw when making clinical recommendations.
Turning to what data should be shared after publication of a manuscript, the committee advocates sharing all the data that are needed to support the results reported in a manuscript, including those presented in tables, figures, and supplementary material. The committee refers to
this as the “post-publication data package,” which consists of the analytic data set and metadata, including the protocol, the SAP, and analytic code, supporting published results.
Conclusion: It is beneficial to share the analytic data set and appropriate metadata supporting published results.
Results Summary for Registries and Lay-Language Summary
Many clinical trials are subject to a requirement that their results be reported to one or more registries, in formats specified by the particular registry. These summaries, which are publicly available on the registry website, are generally limited to major outcomes and adverse events.7 As mentioned in Chapter 3, the Food and Drug Administration Amendments Act of 2007 (FDAAA) requires results of trials of FDA-regulated products to be reported to ClinicalTrials.gov within 12 months of study completion. However, a 2012 study found that the results of 30 percent of 400 clinical trials had neither been published nor reported on ClinicalTrials.gov 4 years after study completion (Saito and Gill, 2014). Following this study and others showing a similar dearth of reporting of results for registered trials, the U.S. Department of Health and Human Services (HHS) has proposed a new rule requiring that results of trials of unapproved products be reported to ClinicalTrials.gov within 12 months of study completion. NIH has also proposed a draft policy calling for all NIH-funded trials not subject to the FDAAA (e.g., trials of surgical or behavioral interventions and phase 1 trials) to also be reported to ClinicalTrials.gov within 12 months of study completion (Hudson and Collins, 2014). In support of this proposed rule, Drs. Hudson and Collins state in an editorial in the Journal of the American Medical Association (JAMA),
When research involves human volunteers who agree to participate in clinical trials to test new drugs, devices, or other interventions, this principle of data sharing properly assumes the role of an ethical man-
________________
7 Adverse events are “unfavorable changes in health, including abnormal laboratory findings, that occur in trial participants during the clinical trial or within a specified period following the trial” (ClinicalTrials.gov, 2014). Generally, two types of adverse event data sets are generated at the conclusion of a trial, described as follows by ClinicalTrials.gov:
Serious Adverse Events: A table of all anticipated and unanticipated serious adverse events, grouped by organ system, with number and frequency of such events in each arm of the clinical trial.
Other (Not Including Serious) Adverse Events: A table of anticipated and unanticipated events (not included in the serious adverse event table) that exceed a frequency threshold within any arm of the clinical trial, grouped by organ system, with number and frequency of such events in each arm of the clinical trial.
date. These participants are often informed that such research might not benefit them directly, but may affect the lives of others. If the clinical research community fails to share what is learned, allowing data to remain unpublished or unreported, researchers are reneging on the promise to clinical trial participants, are wasting time and resources, and are jeopardizing public trust. (Hudson and Collins, 2014)
The committee strongly endorses this reasoning and agrees that public reporting of clinical trial results is an ethical mandate. As noted above, although sharing of clinical trial results has been a requirement, not all investigators and sponsors have fulfilled it. In conjunction with the proposed new requirement, NIH recently announced a multipronged approach to increasing the reporting of public summaries of clinical trial results (NIH, 2014). As a positive incentive, ClinicalTrials.gov is increasing one-to-one support for the posting of these summaries. As an enforcement mechanism, NIH will begin to withhold funds from grantees who do not comply and will take reporting of results into account when reviewing future grant applications (Kaiser, 2014). The committee appreciates that, in a similar manner, adoption of recommendations for sharing clinical trial data (beyond public posting of summary results) may well face challenges in implementation and acceptance. The committee notes that, in support of the requirement to post summary results, ICMJE has modified its statement on registration of clinical trials to make clear that reporting summary results in tabular form in a registry or with a 500-word descriptive abstract is not considered prior publication (ICMJE, 2014).
Additionally, clinical trial participants are interested in knowing the aggregate results of the trial in lay language. A lay-language summary is a brief, nontechnical overview written for the general public and trial participants. Lay summaries of the clinical trial protocol are often required by Research Ethics Committees to assist their nonscientist members in the protocol review and approval process. Sponsors or investigators commonly prepare lay-language press releases after publishing articles or making presentations at professional meetings. However, sending lay-language summaries of clinical trial results to participants is uncommon (Getz et al., 2012). A recent study suggests that trial participants would value such summaries and that providing them is feasible (Getz et al., 2012).
As a matter of public transparency and respect for participants, the committee supports publicly sharing summaries of results for registries and lay-language summaries with participants after publication of an article, presentation at a professional meeting, issuance of a press release, or disclosure to the Securities and Exchange Commission, or no later than 1 year after completion of the trial.
Clinical Study Report
When a clinical trial is submitted to regulatory agencies as part of an application for marketing approval of an intervention or new indication, the trial sponsors usually submit a detailed CSR. Specifications for CSRs have been defined by the International Conference on Harmonisation (ICH) and adopted by the FDA, the EMA, and the Japanese Ministry of Health, Labor, and Welfare in an effort to simplify the application process for new interventions globally (ICH, 1995). According to the FDA guidance, the CSR is an
integrated full report of an individual study of any therapeutic, prophylactic or diagnostic agent … conducted in patients. The clinical and statistical description, presentation, and analyses are integrated into a single report incorporating tables and figures into the main text of the report or at the end of the text, with appendices containing such information as the protocol, sample case report forms, investigator-related information, information related to the test drugs/investigational products including active control/comparators, technical statistical documentation, related publications, patient data listings, and technical statistical details such as derivations, computations, analyses, and computer output. (FDA, 1996, p. 1)
Although a CSR contains mainly summary data and summary tables and graphs, it also usually contains considerable additional information (often thousands of pages), including, as described in the definition above, numerous large appendixes. Supplemental information can include detailed narratives describing individual participants. In some instances, the CSR and/or its appendixes may include identifiable participant information, commercially confidential information, or other protected health information or intellectual property. Currently sensitive information about sponsor strategy or manufacturing may be contained in the background, rationale, and interpretation sections of a CSR because the investigators and sponsor did not anticipate that anyone other than the sponsor, regulatory authorities, and the investigative team would have access to the CSR. A CSR synopsis (i.e., executive summary) is sometimes drafted to accompany a full CSR. Some of the supporting clinical trials included in a regulatory submission do not directly contribute to the evaluation of the effectiveness of the intervention; for these studies, sponsors may be permitted to submit an abbreviated CSR (FDA, 1999). CSRs need to be carefully reviewed and manually redacted of any personally identifiable and commercially confidential information before they are shared with persons other than regulatory authorities.
Sharing CSRs, which provide far more information than is contained in published articles, allows other investigators to carry out meta-analyses
and systematic reviews that combine data from a number of trials. Because a large proportion of clinical trial data is never published, critical reviews and meta-analyses that do not draw on unpublished data in CSRs may be biased (Doshi et al., 2013). Furthermore, there can be discrepancies between published results and CSRs (Doshi and Jefferson, 2013; Jefferson et al., 2014), so access to CSRs allows other investigators to correct errors or understand discrepancies in the published literature. In addition, sharing CSRs allows other investigators to gain new insights by analyzing the spectrum of data, including data on efficacy and safety, among different subgroups of patients. Therefore, a “post-regulatory data package” will consist of the full data package, including the full analyzable data set, the full protocol (including the initial and final versions and all amendments), the SAP (including all amendments and all documentation for additional work processes), the analytic code, and the CSR (redacted for commercially or personal confidential information).
Conclusion: Sharing the CSR will benefit science and public health by allowing a better understanding of regulatory decisions and facilitating use of the analyzable data set.
Conclusion: CSRs may contain sensitive information, including participant identifiers and commercially confidential information. The risks of sharing CSRs are significant and may need to be mitigated in most cases through appropriate controls.
Legacy Trials
The benefit of sharing data from legacy trials (i.e., trials initiated before this report was issued) is similar to the benefit of sharing data from future trials: Other researchers can reproduce the original findings and carry out secondary analyses and meta-analyses, and this additional knowledge benefits the public and patients. Many treatment decisions are based on evidence from past clinical trials, and thus the potential benefits from sharing those data should not be ignored.
However, sharing data from legacy clinical trials also presents particular risks and challenges, which need to be balanced against the potential benefits. One concern is the higher cost of preparing the data for sharing relative to future trials. Data collected during older trials may not be as easily redacted for sensitive information; for example, CSRs can contain commercially confidential information about further analyses, studies, or strategies for regulatory approval or data or narratives that allow participants to be identified. Such CSRs may need to be redacted by
hand to remove commercially confidential information8 and identifiable participant information—a laborious and expensive process. In contrast, future clinical trials can design CSRs to exclude such sensitive information prospectively.
A second concern is that after a trial has been completed staff who are most familiar with the data set generally move on to other research projects or organizations and therefore are not available to answer questions about the data set from secondary users. Even if the staff can be located and are available, they may have forgotten key features of the data set. Staff turnover and loss of familiarity are an issue after the conclusion of any clinical trial, but the problem becomes more serious as the time since study completion becomes longer.
A third challenge is the marked variability in how consent forms for older clinical trials address data sharing (O’Rourke and Forster, 2014). Some consent forms expressly exclude the type of data sharing that is being considered here, while others may be ambiguous or silent on the matter. Although it is legally acceptable to share the trial data without the consent of the trial participants (for example, if the data have been de-identified), Research Ethics Committees may or may not allow data sharing that is contrary to the consent form even if the data are de-identified. Moreover, in a multisite trial, consent forms from different sites may differ. If data from only some sites are available for sharing, secondary analyses will be carried out on a different data set from that used by the original clinical trialists, and inconsistencies between secondary and original analyses will result.
Finally, the results of some completed trials may be viewed as having little scientific or clinical import, for example, because the trial was poorly designed, the intervention was not approved, or if approved is not widely used in clinical care. For such legacy trials, the costs and effort of data sharing may better be spent on creating infrastructure and processes for data sharing for future clinical trials, when data collection can be designed with data sharing in mind. However, the committee believes great benefit would be gained from sharing data from legacy trials that continue to influence decisions about clinical care, and sponsors and investigators are encouraged to make every effort to share those data when requested to do so.
Conclusion: Sponsors and principal investigators will decide, on a case-by-case basis, whether they will share data from clinical trials initi-
________________
8 Some companies have committed to sharing CSRs without redaction for CCI; for example, GlaxoSmithKline has committed to posting CSRs from the year 2000 forward on its multi-sponsor data request system.
ated before the recommendations in this report are implemented. They are strongly urged to do so for major and significant clinical trials whose findings influence decisions about clinical care.
WHEN DATA PACKAGES SHOULD BE SHARED
As discussed previously, vast amounts of data from clinical trials currently are never made public or shared beyond the original investigator team or company, although, as noted in previous chapters, this situation is beginning to change. This section presents the committee’s findings and conclusions regarding when the various types of clinical trial data detailed above should be shared. As noted earlier, the committee acknowledges that currently no single body or authority is capable of enforcing its recommendations for all stakeholders; rather, the committee interpreted its charge as helping to establish professional standards and set expectations for responsible sharing of clinical trial data. Given the complexities of the issues surrounding data sharing, the committee found it helpful to separate the consideration of when data should be shared from that of how they should be shared. This chapter focuses on the former question; the terms, conditions, and operational strategies for how data should be shared—including whether data should be made available to the public or access should be controlled in some manner—are discussed in detail in Chapter 5.
The committee appreciates the wide variation in clinical trials with regard to the clinical conditions and interventions studied, trial designs, and populations of participants. Given this variation, the recommendations regarding the timing for sharing specific types of data detailed at the end of this chapter need to be implemented with discretion and a willingness to consider exceptions to general rules. Justifiable exceptions should be permitted, particularly when there are compelling public health reasons for doing so. The committee believes that agreement regarding common exceptions will develop over time.
The committee expects that standards for responsible sharing of clinical trial data will evolve. The implementation of data sharing will require new infrastructure and will be facilitated by changes in how clinical trials are carried out and analyzed, as well as changes in the culture of the clinical trials enterprise. As this occurs, the committee expects that data will be shared in a more timely manner than as currently recommended, particularly with respect to the analytic data set supporting a publication.
Finally, the committee hopes that the evolution of responsible sharing of clinical trial data will be guided by evidence. There are many unknowns, opportunities, and controversies entailed in sharing clinical trial data that could be clarified with empirical data. For example, it is
not yet known what actual benefits will flow from sharing clinical trial data and what adverse consequences will occur. Comparisons of different approaches to implementing data sharing will be enlightening, helping to identify challenges, ways of overcoming them, and best practices that could be more widely adopted. It would be desirable for stakeholders in clinical trials to convene after some experience with sharing clinical trial data has been gained, perhaps in 3 to 5 years, so they can reconsider, based on evidence, the timing of data sharing and the conditions under which various types of data should be shared.
In deliberating on when the various types of clinical trial data should be shared, the committee found it helpful to summarize the benefits and concerns, discussed in detail in Chapter 3, associated with the timing of data sharing from the perspectives of key stakeholders:
- Benefit patients and future research participants. An essential step in science is verification and replication of investigators’ claims. Once investigators have published their findings, as responsible scientists they should allow other researchers to subject those findings to scrutiny. Allowing timely verification and reproduction serves the public good by preventing other researchers or clinicians from building on findings whose validity cannot be established and by preventing patients from receiving recommendations for clinical care that are based on invalid information. Moreover, sharing clinical trial data benefits patients by allowing other investigators to carry out additional analyses that provide valid scientific information about the effectiveness and safety of the study intervention. In addition, sharing clinical trial data prevents participants in future research from being placed at unwarranted risk because the benefits of an intervention are smaller or the risks greater than claimed in a publication. Furthermore, sharing clinical trial data may increase public trust in the scientific and clinical trials ecosystem.
- Protect the professional interests of clinical trialists in gaining fair professional rewards for their intellectual effort and time by giving them reasonable time to analyze and publish data from a trial they have planned and carried out. In the long run, this protection of the interests of trialists will benefit the public and future patients by creating incentives for investigators to carry out clinical trials; conversely, failure to allow clinical trialists time to publish their findings will discourage investigators from proposing and conducting trials.
- Protect the commercial interests of the sponsors in gaining regulatory approval for a product or indication they have developed and tested, so that they can gain fair financial rewards for their investment of financial
-
and intellectual capital. If a sponsor shares extensive data before a product gains regulatory approval, follow-on developers may gain commercial and strategic advantages. In addition, as a matter of fairness, other companies should not use shared clinical trial data as the predominant basis for their own regulatory submissions—for example, seeking approval for a generic version of the product in a country that does not recognize data exclusivity. In the long run, such incentives to sponsors will benefit the public and future patients by incentivizing investors and companies to develop new medical products and bring basic science discoveries to patients; failure to do so will discourage investments in health care innovation and product development that would ultimately benefit future patients.
- Allow other researchers to access the data in order to reproduce the results of a published trial, synthesize results across trials, and carry out additional or exploratory analyses. Such secondary uses of the data will increase the benefits to society and future patients in terms of knowledge gained from the clinical trial and the contributions of the trial participants.
Policies regarding responsible sharing of specific clinical trial data at particular times in the life cycle of a clinical trial need to balance these countervailing goals and interests. To strike a reasonable balance, the committee proposes releasing specific types of clinical trial data “packages” at different timepoints in the life of a clinical trial (see Figure 4-2).
As discussed below, the committee determined that most clinical trial data should not be shared routinely before study completion. Sharing data before this time would jeopardize the integrity of the clinical trial process and risk the scientific validity of the results. As a matter of fairness, clinical trialists should have a moratorium that lasts long enough for them to gain fair professional rewards from their effort by publishing their work. Similarly, sponsors should have a “quiet period” of a reasonable length to allow for the regulatory process of seeking approval for a new product or indication. Giving too much weight to the interests of secondary users and competitor sponsors in gaining access to data would in the long run present strong disincentives for clinical trialists to design and carry out future trials and for sponsors and their investors to develop and test new products and indications.
Notwithstanding this general presumption that clinical trial data should not be shared before the conclusion of a trial, and allowing for a reasonable moratorium and quiet period, the committee recognizes that exceptions to this presumption are justified. For example, as discussed in more detail below, once a clinical trialist and sponsor publish the results
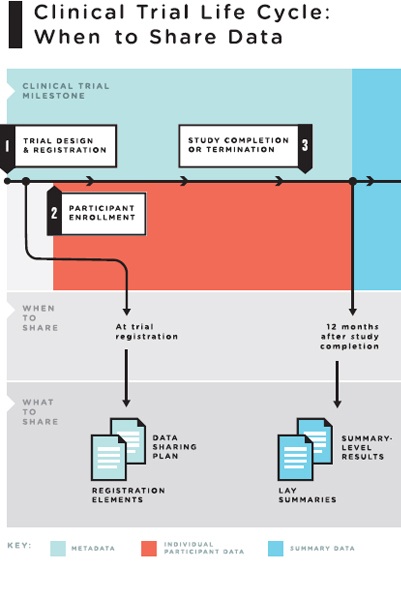
FIGURE 4-2 The clinical trial life cycle: When to share data.
NOTES: Full data package = the full analyzable data set, full protocol (including initial version, final version, and all amendments), full statistical analysis plan (including all amendments and all documentation for additional work processes), and analytic code. Post-publication data package = a subset of the full data package supporting the findings, tables, and figures in the publication. Post-regulatory
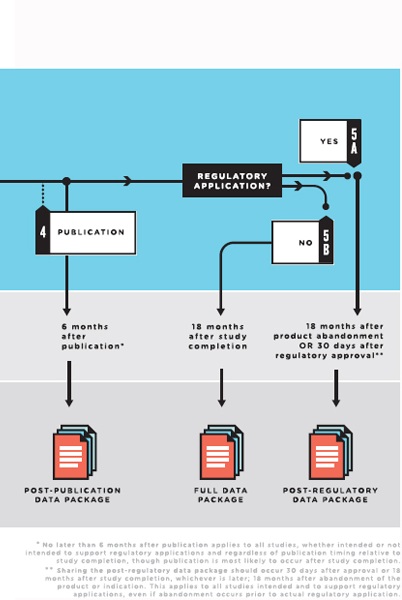
data package = the full data package plus the clinical study report (redacted for commercially or personal confidential information). Legacy trials: For trials initiated prior to the publication of these recommendations, sponsors and principal investigators should decide on a case-by-case basis whether to share data from the completed trials. They are strongly urged to do so for major and significant clinical trials whose findings will influence decisions about clinical care.
of a clinical trial, the goal of allowing verification and replication of a public claim regarding the study intervention takes on additional importance and changes how the countervailing goals described above should be balanced.
Publication
As noted earlier, a publication from a clinical trial is a public statement and discussion about the findings of a trial. Rapid publication commonly occurs with findings that are considered highly important scientifically or clinically. Thus, clinical trialists may publish the results of a trial shortly after its completion. For some trials, trialists may publish the primary trial endpoints despite ongoing longer-term participant follow-up; in this case, the last participant’s last visit may not occur for some time, and hence the full analyzable data set may not be complete at the time of the original publication. Regardless, once a study finding has been published, the scientific process is best served by allowing other investigators to reproduce the findings and carry out additional analyses to test the robustness of the published conclusions. Unless other investigators can reproduce the findings, claims could be made that might mislead other researchers working on similar products or create an impression among clinicians that a drug is safer or more effective than it really is. Even if a drug is not yet on the market, the “buzz” anticipating approval might set the stage for overly enthusiastic adoption.
As discussed previously, the committee appreciates that many clinical trialists feel strongly that, having put years of effort into carrying out a clinical trial, it is only fair that they have the opportunity to write a series of papers analyzing data collected during the trial before other investigators have access to the data. Although sharing data after the results of a trial have been published benefits the public and the scientific process, trial investigators face risks that competitors could publish additional trial findings before they can do so. When reporting the primary results of clinical trials, investigators routinely report a variety of participant characteristics. Reporting these characteristics enables readers to assess the types of patients to whom the results apply, and investigators sometimes use such characteristics to adjust analyses for differences among participants across the trial arms. These uses of participant characteristics augment the utility of the primary manuscript. On the one hand, sharing the individual participant analytic data set supporting the results reported in the publication will allow other researchers to scrutinize, verify, and reproduce the conclusions reported to determine their validity and robustness. On the other hand, the analytic data set supporting the publication, if shared, will also enable other investigators to carry out subgroup analyses, or assessments
of whether the effects of the intervention differ among different types of patients. Because such prespecified subgroup analyses are often the topic of a second paper planned by the trialists, they have an interest in maintaining a period of exclusive access to these data following publication of the initial manuscript.
The committee was mindful of these countervailing goals of sharing clinical trial data soon after publication and appreciates that the scientific community is divided over when the analytic data set supporting a publication should be shared. It is likely that some trialists believe they need 1 year to carry out secondary analyses they were planning. Moreover, clinical trialists may fear that preparing the analytic data set for sharing immediately upon publication would pose an undue administrative burden. On the other hand, other stakeholders believe that the analytic data set should be shared simultaneously with publication so that others can reproduce the findings and build on discoveries. As noted previously, for example, The Bill & Melinda Gates Foundation recently announced that it will require that “data underlying the published research results be immediately accessible and open” (The Bill & Melinda Gates Foundation, 2014). However, the Gates Foundation will allow a 1-year embargo on this requirement to allow investigators to transition to it (The Bill & Melinda Gates Foundation, 2014).
In an ideal clinical trials ecosystem, the committee would favor sharing of the analytic data set supporting a publication immediately upon publication. However, the committee recognizes that currently many practical constraints and challenges need to be addressed before this can be recommended. For the present, the committee recommends a pragmatic compromise time frame of no later than 6 months after publication, with the expectation that in several years the standard will become sharing simultaneously with publication. The committee believes that at the present time, an expectation of no later than 6 months after publication balances the public health benefits of facilitating rapid reanalysis of reported data with the interests of investigators in maintaining a deserved competitive advantage in generating subsequent manuscripts.
In its deliberations, the committee also considered the possibility that an expectation to share data within 6 months of publication may cause some investigators to delay publication to protect their competitive advantage. The committee believes this is unlikely because investigators are strongly motivated to publish important papers rapidly in order to gain credit and prestige. The committee also noted that for the vast majority of clinical trials, there is a time period in which a manuscript is under review and in press, during which the trial team can work on secondary analyses without competitors having access to any data.
The committee recognizes that, as with any guideline, there will be
justifiable exceptions to this 6-month time period; thus, it is not intended to be a hard-and-fast, inflexible rule. Case-by-case exceptions can and should be made with respect to the time period or what data need to be shared for trials that adjust for covariates at baseline and for which sharing the underlying data supporting the adjustments would allow other investigators to carry out subgroup analyses that the clinical trialists had preplanned.
For trials that are likely to have a major clinical, public health, or policy impact, the committee favors sharing the analytic data set sooner than the 6-month window. More rapid sharing will allow the results of important trials to be translated more promptly into improved clinical care, public health, and public policy after other investigators have scrutinized the data. The committee notes that for the majority of trials sharing the analytic data set will not allow other investigators to carry out secondary analyses but only to reproduce the published findings.
One situation that would justify a shorter time period between publishing the primary results and sharing the analytic data set is a publication showing that a drug already marketed is effective for preventing or treating an infection causing a public health crisis, such as pandemic influenza. In such a case, to enable public health officials to plan guidelines and decide whether to stockpile and distribute the drug, it would be desirable to have other investigators analyze data from this pivotal clinical trial to ascertain whether its findings were robust. In this situation, urgent public health considerations should override the clinical trialists’ interests in protecting their advantage in carrying out additional analyses. These additional analyses may supplement discussions among government agencies and sponsors around the world as new data are being generated.
Another example of justification for a shorter time period before sharing the analytic data set supporting a publication is a trial comparing standard medical practices or therapeutic targets in wide clinical use with no implications for regulatory approval of products or indications. If a well-designed, adequately powered trial showed that a widely used practice was less effective or less safe than another widely used alternative, and if the differences would have great clinical significance, the trial findings could strongly influence clinical practice. In such a case, shortening the time before clinical trial data are shared would be justified so that other investigators could reproduce the published findings or employ different valid analytic approaches. These additional analyses could establish more rapidly whether the trial findings were sufficiently robust to warrant prompt modifications in clinical practice. Shortening the period before data are shared would be particularly warranted if it
were highly unlikely that a second confirmatory trial of the same hypothesis would be carried out.
An example of a high-impact pivotal clinical trial comparing clinical interventions and therapeutic approaches widely used in practice is the ARDSNet studies showing “improved survival with lung protective ventilation and shortened duration of mechanical ventilation with conservative fluid management” (NHLBI ARDS Network, 2010). Additional trials suggested “no role for routine use of corticosteroids, beta agonists, [or] pulmonary artery catheterization.” These trials had an important impact on the clinical care of patients with acute respiratory distress syndrome (ARDS). In such a case, the public health interest in major improvements in patient outcomes should override the concerns of the clinical trialists that their advantage in carrying out secondary analyses might be compromised by sharing data on how outcomes were adjusted for covariates.
Trials Not Part of Regulatory Application
As noted previously, the results of many clinical trials remain unpublished long after the trial’s completion. Fewer than half (46 percent) of NIH-funded trials are published within 30 months of completion, and a Kaplan-Meier plot of the time to publication shows a continuous curve with no discontinuities (Gordon et al., 2013; Ross et al., 2012) (see Figure 4-3). According to Gordon and colleagues,
The NHLBI [National Heart, Lung, and Blood Institute], along with other stakeholders in the research enterprise, should seriously examine how best to comprehend and enhance the investment value of smaller trials with surrogate end points and should consider how best to facilitate the rapid publication of all funded randomized trials. (Gordon et al., 2013, p. 1933)
Similarly, Ross and colleagues (2012) found that one-third of trials remained unpublished a median of 51 months after study completion. The authors suggest that
Steps must be taken to ensure the timely dissemination of publicly funded research so that data from all those who volunteer are available to inform future research and practice. (Ross et al., 2012)
This problem exists not just for publicly funded trials but also for trials funded privately by industry and nonprofit sponsors, and across countries and sizes and phases of trials (Ross et al., 2009). Another study found that 29 percent of completed clinical trials had not been published or posted 4 years after completion (Saito and Gill, 2014). Further, a body of evidence reveals selective publication bias (i.e., publication of posi-
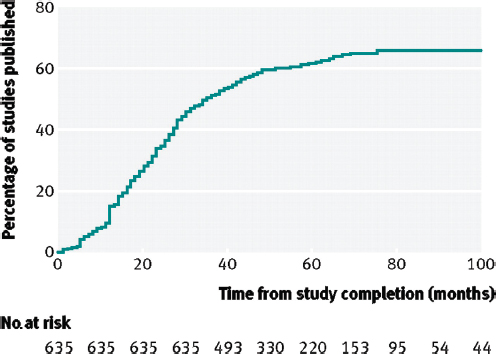
FIGURE 4-3 Cumulative percentage of studies published in a peer-reviewed biomedical journal indexed by Medline during 100 months after trial completion among all National Institutes of Health (NIH)–funded clinical trials registered in ClinicalTrials.gov.
SOURCE: Reproduced from [British Medical Journal, Joseph S. Ross, Tony Tse, Deborah A. Zarin, Hui Xu, Lei Zhou, and Harlan M. Krumholz, 344, d7292, 2012] with permission from BMJ Publishing Group Ltd.
tive results at a higher rate) (Bardy, 1998; Chan et al., 2004; Decullier et al., 2005; Dickersin and Min, 1993; Dickersin et al., 1992; Dwan et al., 2008; Easterbrook et al., 1991; Ioannidis, 1998; Krzyzanowska et al., 2003; Misakian and Bero, 1998; Stern and Simes, 1997; Turner et al., 2008; von Elm et al., 2008).
From these findings, the committee concluded that steps should be taken to encourage timely publication of clinical trial results and sharing of clinical trial data after the study investigators have had a fair opportunity to publish their findings. However, if the clinical trial team does not publish its findings in timely manner, other investigators should have the opportunity to access and analyze the trial data so the public can gain the benefit of knowledge produced by the trial and the contributions of participants who volunteered to participate. It is important that negative
as well as positive clinical trial results be made known and the underlying data shared. The committee rejected the option of sharing clinical trial data immediately upon the conclusion of a trial. Instead, the committee concluded that the moratorium discussed above should be provided, for several reasons. First, the primary investigators, who designed the trial, secured funding, implemented trial-related procedures, trouble-shot unexpected problems, and carried out data collection, should be given a fair opportunity to gain the rewards of publication and professional recognition for their intellectual contributions and efforts. Second, the primary investigators have unique insights into the strengths, weaknesses, and idiosyncrasies of the trial’s conduct and data, so they may be able to complete the planned analyses in the most rigorous and efficient fashion. Third, without having some competitive advantage in analyzing and publishing the results of a trial to which they devoted years of professional effort, highly trained clinical trialists might shift their careers toward other paths. In its public meetings, the committee heard clinical trialists declare that if other scientists could publish the results of a trial first, the trialists would have strong disincentives for undertaking the arduous process of organizing and conducting trials. If fewer scientists became clinical trialists, the production of clinical trial data would decline and the result in the long run could be fewer new therapies and less evidence for important clinical decisions. Fourth, junior members of a clinical trial team might expect to be first author on a secondary paper from the trial, which would be a major milestone in their careers.
The committee understands that the recommendations presented here apply for the current academic reward system. As discussed previously, fair sharing of clinical trial data necessitates that sharing be valued independently as a duty of scientific citizenship. Thus, as the academic reward system recognizes and affords fair credit for sharing data that enable other investigators to publish findings, the committee anticipates that the calculus for how much total credit can be obtained by sharing data earlier will evolve to favor more rapid sharing.
Conclusion: Once a clinical trial has been completed, a moratorium before the trial data are shared is generally appropriate to allow the trialists who have planned the trial and generated the data to complete their analyses.
After concluding that clinical trial data should be shared only after a moratorium following completion of the trial, the committee considered how long that moratorium should be. In addition to balancing the countervailing interests and goals discussed above, the committee weighed the following pragmatic considerations. First, as noted above, the avail-
able data on the time to publication after completion of a clinical trial suggest that the percentage of trials published increases continuously over time, with a leveling off between 40 and 60 months after completion. Second, the original investigators in the clinical trial have a head start relative to secondary users that is longer than the moratorium period because, even after obtaining access to clinical trial data, a secondary investigator will require time to become familiar with the data set, to plan and run data analyses, and to prepare a manuscript for submission to a medical journal. Third, changing the incentives and expectations for sharing data and publishing results of clinical trials may change investigator behaviors. If investigators know they will share data at a certain time after completion of a trial, they may alter their planning to attempt to obtain appropriate levels of funding and staffing or arrange to collaborate so as to publish the primary and some secondary analyses within the moratorium period. For junior investigators planning to be lead author in secondary analyses, it will be important to carry out those analyses as soon as possible so that they can be submitted shortly after the primary paper has been accepted.
How long the moratorium after trial completion should be is a matter of judgment. However, establishing a professional standard for the length of a moratorium is important to give all stakeholders in the clinical trials enterprise the same expectations. Sponsors and investigators who decide to release data sooner would be encouraged to do so for the sake of more rapid completion of secondary analyses. Other sponsors and investigators may have justifiable and predictable reasons to delay release beyond the moratorium period recommended here. If such circumstances can be anticipated when the trial is designed, it will be appropriate to include the expected delay as part of the data sharing plan at registration, and to provide the alternative expected time at which data will be shared for the trial.
The committee also recognizes that what stakeholders consider acceptable for a moratorium period may change over time as more experience is gained with sharing clinical trial data, particularly if data are collected prospectively on the outcomes of such a data sharing policy. Such outcomes might include the Kaplan-Meier curves on the percentage and types of trials published as a function of time since trial completion, as well as investigators’ willingness to lead or participate in other clinical trials. Taking all these considerations into account, the committee reached the following conclusion regarding the best balance of countervailing interests and goals, recognizing that some stakeholders will advocate a shorter and some a longer moratorium.
Conclusion: It is reasonable to expect clinical trial data that will not be part of a regulatory application to be available for sharing no later than 18 months after study completion.
Regulatory Application
This section addresses when the post-regulatory data package should be shared for those clinical trials that are intended to support a regulatory application for a new product or a new indication for a product already marketed. The committee considered when data should be shared for products and indications that are (1) granted regulatory approval or (2) abandoned by the sponsor (either before or after a regulatory submission) and/or licensed to another entity for further development.
Regulatory Approval
Regulatory authorities such as the EMA and the FDA have a more comprehensive view of clinical trial data than do other secondary users of shared data. These two agencies make extensive efforts to understand, challenge, and reanalyze submitted data. In addition, both have mechanisms for asking questions, requesting new analyses or data sets from the trial sponsors, and obtaining external expert advice. Moreover, they have the authority—which they often exercise—to audit sponsors as well as investigational sites. These audits include, for example, announced or unannounced visits to the sponsor to review the accuracy and veracity of the submitted files, interview staff, and test software and systems. Visits to investigational sites allow these agencies to review study documentation and procedures through interviews with study and hospital staff and review of administrative documents and patient and study records. Therefore, the agencies have the benefit of seeing how the data were collected and how they were handled and analyzed by the sponsor and of performing their own independent analysis of the data. They have the unique ability to evaluate individual study data in the context of all other studies conducted on the product, as well as other products—from other sponsors—in the same therapeutic area (especially those with similar mechanisms).
Regulatory authorities have public health responsibilities. The FDA, for example, is charged with “protecting the public health by assuring the safety, efficacy and security of human and veterinary drugs, biological products, medical devices,” and “is also responsible for advancing the public health by helping to speed innovations that make medicines more effective, safer and more affordable, and by helping the public get the accurate, science-based information they need to use medicines and foods
to maintain and improve their health” (FDA, 2014). Regulatory authorities also may have fewer conflicts of interest relative to sponsors and clinical trial investigators, by their very nature. The FDA is firewalled from the direct financial conflicts of interest that other secondary users of shared clinical trial data may have through research grants and contracts, although the FDA does receive funding through user fees. In addition, the FDA staff are free from the academic need to publish papers to advance their careers.
Given regulatory agencies’ broad perspective on the data, their public health mandate, and their accountability as government agencies, the committee believes, as noted earlier, that it is beneficial to allow regulators a “quiet period” as they carry out their review, during which clinical trial data need not be shared with other secondary users, even beyond the 18-month period after study completion. The committee concludes that a “post-regulatory data package”—consisting of the CSR (redacted for commercially or personal confidential information), the full analyzable data set, the full protocol (including the initial and final versions and all amendments), the SAP (including all amendments and all documentation for additional work processes), and the analytic code—should be shared either 18 months after study completion or 30 days after the FDA and/or the EMA approval, whichever occurs later. If regulatory approval is denied and the sponsor continues to pursue approval, the regulatory quiet period should continue so that competitors will not have the opportunity to use shared data for their own regulatory submissions even before the originator has obtained approval. Sponsors should have the opportunity to resubmit their application or have data and information requested by the regulatory authority evaluated during a quiet period. As discussed later, there should also be no disincentive for secondary sponsors that may want the opportunity to license and repurpose the product even after initial nonapproval from regulatory authorities (see the discussion below regarding abandonment).
An important exception to the quiet period arises if a sponsor chooses to publish a manuscript prior to regulatory approval, as commonly occurs with important trials and novel therapies. In this case, the sponsor has put the results out for scientific and medical discussion and therefore should share the analytic data set supporting the publication—in essence, negating the quiet period. Once the scientific and medical community has read a publication with the sponsor’s analysis and conclusions, the community should have the opportunity to scrutinize and reproduce the analysis and conclusions. Therefore, if the sponsor has elected to publish, the data supporting the publication—the post-publication data package—should be shared no later than 6 months after publication even if the product has not yet been approved.
Conclusion: It is beneficial to allow a “quiet period” while a product or indication is undergoing development for a regulatory application during which the full analyzable data set and metadata need not be shared unless the data are published.
Abandonment
In some cases, a clinical trial (whether terminated early or completed) may be part of a product development program that is abandoned. Products and indications may be abandoned either before or after regulatory submission and for a variety of reasons. Sponsors may decide to abandon products and indications undergoing development for regulatory application for scientific or medical reasons (e.g., a lack of efficacy for the indication of interest or a serious safety issue) or for administrative reasons (e.g., business considerations, drug supply) (Paul and Lewis-Hall, 2013; Psaty and Rennie, 2003). If a product development program is abandoned,
- the sponsor may continue to seek or already have regulatory approval for the product for a different indication, using other clinical trial data; or
- the sponsor may abandon the new product under development for all indications, in which case the sponsor may or may not decide to transfer intellectual property rights to another sponsor, either for-profit or nonprofit, that wants to develop the product.
If the sponsor transfers intellectual property rights to another sponsor, the new sponsor has an interest in having an exclusive period in which to conduct additional trials, seek additional patents, and prepare a regulatory submission (Rai and Rice, 2014). Lack of such an exclusive period would be a disincentive for another sponsor to develop the product and seek regulatory approval. Indeed, because of the potential importance of this approach to developing new therapies, the committee encourages making such decisions to transition abandoned products to other interested parties as expeditiously as possible.
The committee also considered the case in which a sponsor abandons a product or indication and does not transfer the intellectual property rights to develop the product to another sponsor. In such cases, sharing clinical trial data may help other researchers studying and other sponsors developing similar products. The design of trials on these other products may be modified by the results of the abandoned trial, for example, if the results suggest safety or efficacy endpoints. If a sponsor abandons a new indication for a marketed product, sharing the clinical trial data can benefit other researchers, clinicians, and the public. Sharing data on a product
approved for other uses will increase general knowledge of the product, including its efficacy and safety profile.
Taking the above considerations into account, the committee reached the following conclusions regarding abandoned trials (whether terminated early or completed) conducted for products or indications intended for regulatory application.
Conclusion: If a clinical trial has ended and the sponsor abandons development of a new product (and does not transfer rights to develop the product to another sponsor), it is appropriate to share the post-regulatory data package 18 months after the decision has been made definitively to abandon the product and not pursue further development.
In this case, the 18-month moratorium will allow the trial investigators to analyze the data from the trial and publish their findings. This 18-month moratorium is similar to the moratorium for completed trials, allowing investigators to analyze and publish their work. As with all trial data, the analytic data set supporting a publication should be available no later than 6 months after publication.
Conclusion: If a product will continue to be developed by the sponsor or if it is transitioned or licensed to a new sponsor that is pursuing development and approval, it is appropriate to share the post-regulatory data package 30 days after regulatory approval of the product or 18 months after study completion, whichever occurs later.
Conclusion: If a sponsor will not be seeking regulatory approval of the new indication for a marketed product for which a trial was intended to be part of a regulatory submission, it is appropriate to share the post-regulatory data package 18 months after the decision has been made definitively to abandon the indication.
Box 4-4 presents three case examples of the timeline for sharing clinical trial data.
Sharing Data with Participants
Clinical trial participants are interested in certain types of trial data, sometimes for purposes other than carrying out secondary analyses. Table 4-1 summarizes these types of data, when they should be shared, and the benefits/risks of sharing them. (Sharing of a summary of clinical trial results with participants was discussed earlier in the chapter.)
The committee next turned to the issue of sharing individual par-
BOX 4-4
Case Examples: Timeline for Sharing Clinical Trial Data
Case 1: Trial Not for Regulatory Application
University X conducts a comparative effectiveness trial that is not intended for regulatory approval. The trial starts January 1, 2015, and includes secondary outcomes that are 5 years out, with study completion anticipated January 1, 2020. On July 1, 2018, University X publishes a paper on early outcomes. It should then release the post-publication data package by December 1, 2018. The remainder of the data that constitute the full data package should be released by July 1, 2021.
Case 2: Regulatory Application—Approval
Sponsor Y runs a trial on a drug intended for regulatory approval. The trial is completed on July 1, 2014. Because this is a regulatory trial, the post-regulatory data package should be released 18 months after study completion (December 31, 2015) or 30 days after approval, whichever is later, if the product is approved, or 18 months after product abandonment if the product is abandoned. Sponsor Y publishes an article on the primary outcome of the trial on February 1, 2015. As recommended by the committee, the investigators should then release the post-publication data package no later than August 1, 2015 (6 months after publication). The product is approved on March 1, 2016. The remainder of the data that constitute the post-regulatory data package should be released by April 1, 2016.
Case 3: Regulatory Application—Abandonment
Sponsor Z runs a trial on a drug intended for regulatory approval. The trial is completed on July 1, 2014. Because this is a regulatory trial, the post-regulatory data package should be released 18 months after study completion (December 31, 2015) or 30 days after approval, whichever is later, if the product is approved, or 18 months after product abandonment if the product is abandoned. Although results of the initial phase 2 trial ending on July 1, 2014, are encouraging, final analyses of the phase 3 trials reveal new safety issues, and the product is abandoned on August 31, 2017. Sponsor Z publishes an article on December 1, 2017. Sponsor Z should then release the post-publication data package before June 1, 2018. The remainder of the data that constitute the post-regulatory data package should be released by February 28, 2019.
ticipant data with participants. Clinical trial participants are increasingly interested in obtaining their own data gathered during a trial. In accordance with the conceptual framework for this report, the committee considered what individual data might be shared with a clinical trial’s participants and at what points during the trial.
Baseline individual data. At baseline, data may be collected from physical examinations; blood tests; and other tests that are routinely available in clinical practice, such as urine analyses, ECGs, and chest X-rays. These data may have implications for participants’ own clinical care. For
TABLE 4-1 Timing and Benefits/Risks of Sharing Data with Participants
| Type of Data | When Shared | Benefits/Risks |
| Trial results in lay language | 1 year after study completion | Benefit: informing participants of the trial’s scientific benefits Benefit: better public understanding of clinical trial results |
| Individual participants’ own results of baseline clinical tests | As discussed during participant enrollment and the informed consent process | Benefit: potential benefit to participants through routine screening |
| Individual participants’ own results of clinical tests obtained during the trial | As discussed during participant enrollment and the informed consent process—typically after study completion | Benefit: potential personal benefit to participants or to inform participation in future research Risk: concern that if results are shared during the trial, the trial could be unblinded and its results potentially biased through differential dropout rates Risk: concern that the clinical significance of innovative tests may be unclear |
example, abnormal results for blood pressure or cholesterol may need to be followed up by a participant and his or her physician. If a participant has a chronic condition that requires periodic monitoring with clinical tests, sharing these data can obviate the need for duplicative tests. It is already good clinical trial practice to share such clinically actionable data with participants in real time.
Clinical data collected during the course of the trial. As in the case of baseline individual data, some data collected during a trial come from tests that are routinely available in clinical practice, such as tests ordered to assess endpoints or monitor adverse events. Abnormal findings may require follow-up by the participant and his or her physician in real time—for example, if there is evidence of cancer recurrence. In addition, it may be necessary to modify the dose of a drug, suspend its use, or carry out additional monitoring tests. The nature of the condition being studied
and the study intervention will determine the benefits to the patient of sharing data that are widely available in clinical care.
Sharing some of their individual participant data with participants in real time may increase the risk of unblinding the study. For instance, participants may infer which arm of a trial they were assigned to if certain laboratory abnormalities are known to be much more likely in one arm. (Of course, participants may also infer which arm they are in from clinical signs and symptoms, such as a local reaction to an active vaccination or a slower pulse rate from a study drug.) Unblinding may alter participants’ behavior, for example, causing higher dropout rates in the control arm compared with the active arm. If such behaviors are widespread and differ between the arms of a trial bias may result, compromising the scientific validity of the trial and thereby undermining the contributions of other participants and the potential benefits of the trial.
Participants are increasingly collaborating to pool their individual clinical trial data and experiences and using the pooled data. For example, participants may share their symptoms and signs, as well as results of laboratory tests obtained as part of or outside the trial. Furthermore, clinical trial participants may pool their individual data to analyze the outcomes of a trial before its prespecified completion. As a result of such interim analyses, participants may decide to drop out of a trial or take additional therapies outside the protocol and its restrictions. Although pooling and analyzing individual data may make sense from the perspective of an individual participant, dropouts and deviations from the protocol may compromise the power of the trial to detect a clinically meaningful difference between the two arms or cause bias in the trial results. As a result, the trial may not fulfill its objective of obtaining valid scientific information about the benefits and risks of the study intervention. Moreover, there is a risk that such interim analyses may be misleading if they do not take into account the variability of outcomes due to chance early in a trial and the need to correct statistical analyses for multiple interim analyses of the data before a trial’s prespecified conclusion.
Novel data that may not be clinically interpretable until after the trial is completed. For levels of new biomarkers for the study drug or for innovative tests not yet used in clinical care, individual results may be of uncertain significance and little direct clinical benefit until clinical trial data have been analyzed. For example, a trial may be assessing the predictive power of new biomarkers or the sensitivity of a new imaging technique. However, some participants may want to know their own data even if the clinical significance is unclear, and some clinical trial sponsors and investigators may choose to offer participants such data together with appropriate information about their clinical significance or lack thereof.
The committee believes the appropriateness of sharing individual
data collected in the course of a trial is highly dependent on the particular trial, the condition being studied, the study interventions, and the risk of altering adherence to the study protocol and/or unblinding the trial. As noted above, the sponsor and investigators often share standard baseline clinical data from routine laboratory tests with individual participants shortly after the samples have been collected and analyzed. Such sharing increases potential benefits to participants and may incentivize enrollment. Moreover, clinically actionable information clearly should be shared with participants in a timely manner as appropriate as a matter of exercising good clinical trial practice and acting to benefit participants. The protocol should specify what kinds of abnormal follow-up test results would be disclosed and how the study interventions would be modified. Additional disclosures should be made in the case of unexpected serious adverse events that require clinical follow-up.
Clinical trial sponsors and investigators may also benefit from reaching out to disease groups and community groups at various stages of a trial. In addition to strengthening the trial design, the consent process, and recruitment, these groups can help draft lay-language summaries of trial results, disseminate those summaries through their organizations, and provide information and support to trial participants who have received individual participant data. Such outreach and collaboration may also increase public understanding of and trust in the clinical trial process.
Conclusion: Investigators can help uphold public trust in clinical trials and adhere to current best practices and legal standards by
- explaining to trial participants what data will be shared with them and with other interested parties and when, as part of the informed consent process; and
- as appropriate, making individual participants’ own data collected during the course of a trial available to them following study completion and data analysis.
Drawing together the considerations detailed above, the committee formulated the following recommendation for what data should be shared after key points in a clinical trial. The committee believes that this recommendation will set as professional standards that clinical trial data should be shared while mitigating associated risks and concerns.
Recommendation 2: Sponsors and investigators should share the various types of clinical trial data no later than the times specified
below. Sponsors and investigators who decide to make data available for sharing before these times are encouraged to do so.
Trial registration:
- The data sharing plan for a clinical trial (i.e., what data will be shared when and under what conditions) should be publicly available at a third-party site that shares data with and meets the data requirements of WHO’s International Clinical Trials Registry Platform; this should occur before the first participant is enrolled.
Study completion:
- Summary-level results of clinical trials (including adverse event summaries) should be made publicly available no later than 12 months after study completion.
- Lay summaries of results should be made available to trial participants concurrently with the sharing of summary-level results, no later than 12 months after study completion.
- The full data package (including the full analyzable data set, the full protocol,9 the full statistical analysis plan, and the analytic code) should be shared no later than 18 months after study completion (unless the trial is in support of a regulatory application).
Publication:
- The post-publication data package (including the subset of the analyzable data set supporting the findings, tables, and figures in the publication and the full protocol, full statistical analysis plan, and analytic code that supports the published results) should be shared no later than 6 months after publication.
Regulatory application:
- For studies of products or new indications that are approved, the post-regulatory data package (including the full analyzable data set and clinical study report redacted for commercially or personal confidential information, together with the full protocol, full statistical analysis plan, and analytic code) should be shared 30 days after regulatory approval or 18 months after study completion, whichever occurs later.
- For studies of new products or new indications for a marketed product that are abandoned, the post-regulatory data package should be shared no later than 18 months after abandonment.
________________
9 Includes the protocol in place at the start of the trial, any modifications, and the final protocol.
However, if the product is licensed to another party for further development, these data need be shared only after publication, approval, or final abandonment.
ADNI (Alzheimer’s Disease Neuroimaging Initiative). 2014. ADNI publications. http://www.adni-info.org/scientists/adniscientistshome/adnipublications.aspx (accessed October 29, 2014).
Bardy, A. H. 1998. Bias in reporting clinical trials. British Journal of Clinical Pharmacology 46(2):147-150.
The Bill & Melinda Gates Foundation. 2014. Open access policy. http://www.gatesfoundation.org/how-we-work/general-information/open-access-policy (accessed December 10, 2014).
Bradshaw, E. M., L. B. Chibnik, B. T. Keenan, L. Ottoboni, T. Raj, A. Tang, L. L. Rosenkrantz, S. Imboywa, M. Lee, A. Von Korff, M. C. Morris, D. A. Evans, K. Johnson, R. A. Sperling, J. A. Schneider, D. A. Bennett, and P. L. De Jager. 2013. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nature Neuroscience 16(7):848-850.
Califf, R. M. 2013. Clinical trial data sharing and challenges to data sharing: Current and future. Paper presented at IOM Committee on Strategies for Responsible Sharing of Clinical Trial Data: Meeting One, October 22-23, Washington, DC.
Casanova, R., F. C. Hsu, K. M. Sink, S. R. Rapp, J. D. Williamson, S. M. Resnick, and M. A. Espeland. 2013. Alzheimer’s disease risk assessment using large-scale machine learning methods. PLoS ONE 8(11):e77949.
Chan, A. W., K. Krleza-Jeric, I. Schmid, and D. G. Altman. 2004. Outcome reporting bias in randomized trials funded by the Canadian Institutes of Health Research. Canadian Medical Association Journal 171(7):735-740.
Chan, A. W., J. M. Tetzlaff, D. G. Altman, K. Dickersin, and D. Moher. 2013. Spirit 2013: New guidance for content of clinical trial protocols. Lancet 381(9861):91-92.
ClinicalTrials.gov. 2012. Glossary of common site terms. http://clinicaltrials.gov/ct2/about-studies/glossary (accessed October 15, 2014).
ClinicalTrials.gov. 2014. “Basic results” data element definitions (draft). http://prsinfo.clinicaltrials.gov/results_definitions.html#AdverseEvents (accessed October 16, 2014).
Collins, F. S., and L. A. Tabak. 2014. NIH plans to enhance reproducibility. Nature 505(7485):612-613.
De Angelis, C., J. M. Drazen, F. A. Frizelle, C. Haug, J. Hoey, R. Horton, S. Kotzin, C. Laine, A. Marusic, A. J. P. M. Overbeke, T. V. Schroeder, H. C. Sox, and M. B. V. D. Weyden. 2004. Clinical trial registration: A statement from the International Committee of Medical Journal Editors. New England Journal of Medicine 351(12):1250-1251.
Decullier, E., V. Lheritier, and F. Chapuis. 2005. Fate of biomedical research protocols and publication bias in France: Retrospective cohort study. British Medical Journal 331(7507):19.
Dickersin, K., and Y. I. Min. 1993. NIH clinical trials and publication bias. The Online Journal of Current Clinical Trials 50.
Dickersin, K., Y. I. Min, and C. L. Meinert. 1992. Factors influencing publication of research results. Follow-up of applications submitted to two institutional review boards. Journal of the American Medical Association 267(3):374-378.
Doshi, P., and T. Jefferson. 2013. Clinical study reports of randomised controlled trials: An exploratory review of previously confidential industry reports. BMJ Open 3(2):e002496.
Doshi, P., T. Jefferson, and C. Del Mar. 2012. The imperative to share clinical study reports: Recommendations from the Tamiflu experience. PLoS Medicine 9(4):e1001201.
Doshi, P., K. Dickersin, D. Healy, S. S. Vedula, and T. Jefferson. 2013. Restoring invisible and abandoned trials: A call for people to publish the findings. British Medical Journal 346:f2865.
Drazen, J. M. 2002. Who owns the data in clinical trial? Science and Engineering Ethics 8(3):407-411.
Dwan, K., D. G. Altman, J. A. Arnaiz, J. Bloom, A. W. Chan, E. Cronin, E. Decullier, P. J. Easterbrook, E. Von Elm, C. Gamble, D. Ghersi, J. P. A. Ioannidis, J. Simes, and P. R. Williamson. 2008. Systematic review of the empirical evidence of study publication bias and outcome reporting bias. PLoS ONE 3(8):e3081.
Dwork, C. 2014. State of the art of privacy protection. Paper presented at Big Data Privacy Workshop: Co-hosted by The White House Office of Science and Technology Policy and MIT, March 3, Cambridge, MA.
Easterbrook, P. J., R. Gopalan, J. A. Berlin, and D. R. Matthews. 1991. Publication bias in clinical research. Lancet 337(8746):867-872.
European Union Clinical Trials Register. 2011. Clinical trials. https://www.clinicaltrialsregister.eu/ctr-search/trial/2010-020943-10/GB#P (accessed December 9, 2014).
FDA (U.S. Food and Drug Administration). 1996. Guideline for industry: Structure and content of clinical study reports. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073113.pdf (accessed October 17, 2014).
FDA. 1999. Guidance for industry: Submission of abbreviated reports and synopses in support of marketing applications. http://www.fda.gov/downloads/Drugs/Guidances/ucm072053.pdf (accessed October 16, 2014).
FDA. 2014. what we do. http://www.fda.gov/AboutFDA/WhatWeDo (accessed October 16, 2014).
Fisher, B., S. Anderson, C. K. Redmond, N. Wolmark, D. L. Wickerham, and W. M. Cronin. 1995. Reanalysis and results after 12 years of follow-up in a randomized clinical trial comparing total mastectomy with lumpectomy with or without irradiation in the treatment of breast cancer. New England Journal of Medicine 333(22):1456-1461.
Getz, K., Z. Hallinan, D. Simmons, M. J. Brickman, Z. Jumadilova, L. Pauer, M. Wilenzick, and B. Morrison. 2012. Meeting the obligation to communicate clinical trial results to study volunteers. Expert Review of Clinical Pharmacology 5(2):149-156.
Giles, C. 2014. Thomas Piketty’s exhaustive inequality data turn out to be flawed. Financial Times, http://www.ft.com/cms/s/0/c9ce1a54-e281-11e3-89fd-00144feabdc0.html#axzz3GR9NAHW5 (accessed October 17, 2014).
Gordon, D., W. Taddei-Peters, A. Mascette, M. Antman, P. G. Kaufmann, and M. S. Lauer. 2013. Publication of trials funded by the National Heart, Lung, and Blood Institute. New England Journal of Medicine 369(20):1926-1934.
Haight, T. J., S. M. Landau, O. Carmichael, C. Schwarz, C. DeCarli, and W. J. Jagust. 2013. Dissociable effects of Alzheimer disease and white matter hyperintensities on brain metabolism. JAMA Neurology 70(8):1039-1045.
Hudson, K. L., and F. S. Collins. 2014. Sharing and reporting the results of clinical trials. Journal of the American Medical Association. http://dx.doi.org/10.1001/jama.2014.10716 (accessed December 10, 2014).
ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use). 1995. Structure and content of clinical study reports (E3). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E3/E3_Guideline.pdf (accessed October 16, 2014).
ICMJE (International Committee of Medical Journal Editors). 2014. Clinical trials registration. http://www.icmje.org/about-icmje/faqs/clinical-trials-registration (accessed November 25, 2014).
Ioannidis, J. P. 1998. Effect of the statistical significance of results on the time to completion and publication of randomized efficacy trials. Journal of the American Medical Association 279(4):281-286.
IOM (Institute of Medicine). 2012. Evolution of translational omics: Lessons learned and the path forward. Washington, DC: The National Academies Press.
IOM. 2014. Workshop on principles and best practices for sharing data from environmental health research, March 19, Washington, DC.
Jasny, B. R. 2011. Again, and again, and again… Science 334(6060):1225.
Jefferson, T., M. A. Jones, P. Doshi, C. B. Del Mar, R. Hama, M. J. Thompson, I. Onakpoya, and C. J. Heneghan. 2014. Risk of bias in industry-funded oseltamivir trials: Comparison of core reports versus full clinical study reports. BMJ Open 4(9):e005253.
Kaiser, J. 2014. Proposed rules will vastly expand trove of clinical trial data reported in U.S. database. Science News, http://news.sciencemag.org/health/2014/11/proposed-rules-will-vastly-expand-trove-clinical-trial-data-reported-u-s-database (accessed December 9, 2014).
Krugman, P. 2013. The Excel depression. The New York Times, April 19, 2013, A31.
Krzyzanowska, M. K., M. Pintilie, and I. F. Tannock. 2003. Factors associated with failure to publish large randomized trials presented at an oncology meeting. Journal of the American Medical Association 290(4):495-501.
Laine, C., S. N. Goodman, M. E. Griswold, and H. C. Sox. 2007. Reproducible research: Moving toward research the public can really trust. Annals of Internal Medicine 146(6):450-453.
Misakian, A. L., and L. A. Bero. 1998. Publication bias and research on passive smoking: Comparison of published and unpublished studies. Journal of the American Medical Association 280(3):250-253.
NHLBI ARDS Network (National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Network). 2010. ARDSNet studies. http://www.ardsnet.org/clinicians/ studies (accessed October 24, 2014).
NIH (U.S. National Institutes of Health). 2003. NIH data sharing policy and implementation guidance. http://grants.nih.gov/grants/policy/data_sharing/data_sharing_guidance.htm (accessed December 9, 2014).
NIH. 2014. HHS and NIH take steps to enhance transparency of clinical trial results. http://www.nih.gov/news/health/nov2014/od-19.htm (accessed December 9, 2014).
NRC (National Research Council). 2003. Sharing publication-related data and materials: Responsibilities of authorship in the life sciences. Washington, DC: The National Academies Press.
O’Rourke, P. P., and D. Forster. 2014. Considerations of informed consent in the discussion of clinical trial data sharing—domestic issues. Paper presented at IOM Committee on Strategies for Responsible Sharing of Clinical Trial Data: Meeting Two, February 3-4, Washington, DC.
Paul, S. M., and F. Lewis-Hall. 2013. Drugs in search of diseases. Science Translational Medicine 5:186fs118.
Psaty, B. M., and D. Rennie. 2003. Stopping medical research to save money: A broken pact with researchers and patients. Journal of the American Medical Association 289(16):2128-2131.
Rai, A. K., and G. Rice. 2014. Use patents can be useful: The case of rescued drugs. Science Translational Medicine 6(248):248fs230.
Ross, J. S., G. K. Mulvey, E. M. Hines, S. E. Nissen, and H. M. Krumholz. 2009. Trial publication after registration in ClinicalTrials.gov: A cross-sectional analysis. PLoS Medicine/Public Library of Science 6(9):e1000144.
Ross, J. S., T. Tse, D. A. Zarin, H. Xu, L. Zhou, and H. M. Krumholz. 2012. Publication of NIH funded trials registered in ClinicalTrials.gov: Cross sectional analysis. British Medical Journal 344:d7292.
Saito, H., and C. J. Gill. 2014. How frequently do the results from completed US clinical trials enter the public domain? A statistical analysis of the ClinicalTrials.gov database. PLoS ONE 9(7):e101826.
Sandve, G. K., A. Nekrutenko, J. Taylor, and E. Hovig. 2013. Ten simple rules for reproducible computational research. PLOS Computational Biology 9(10):e1003285.
Sim, I., A. W. Chan, A. M. Gulmezoglu, T. Evans, and T. Pang. 2006. Clinical trial registration: Transparency is the watchword. Lancet 367(9523):1631-1633.
Soran, A., L. Nesbitt, E. P. Mamounas, B. Lembersky, J. Bryant, S. Anderson, A. Brown, and M. Passarello. 2006. Centralized medical monitoring in phase III clinical trials: The National Surgical Adjuvant Breast and Bowel Project (NSABP) experience. Clinical Trials 3(5):478-485.
Stern, J. M., and R. J. Simes. 1997. Publication bias: Evidence of delayed publication in a cohort study of clinical research projects. British Medical Journal 315(7109):640-645.
Stodden, V. 2009. Enabling reproducible research: Licensing for scientific innovation. International Journal of Communications Law and Policy 13:1-25.
Stodden, V. 2013. what scientific idea is ready for retirement? http://edge.org/annual-question/2014/response/25340 (accessed October 17, 2014).
Teden, P. 2013. Types of clinical trial data to be shared. Paper presented at IOM Committee on Strategies for Responsible Sharing of Clinical Trial Data: Meeting One, October 22-23, Washington, DC.
Temple, R., and G. W. Pledger. 1980. The FDA’s critique of the Anturane Reinfarction Trial. New England Journal of Medicine 303(25):1488-1492.
Turner, E. H., A. M. Matthews, E. Linardatos, R. A. Tell, and R. Rosenthal. 2008. Selective publication of antidepressant trials and its influence on apparent efficacy. New England Journal of Medicine 358(3):252-260.
von Elm, E., A. Rollin, A. Blumle, K. Huwiler, M. Witschi, and M. Egger. 2008. Publication and non-publication of clinical trials: Longitudinal study of applications submitted to a Research Ethics Committee. Swiss Medical weekly 138(13-14):197-203.
WHO (World Health Organization). 2014. wHO data set. http://www.who.int/ictrp/network/trds/en (accessed December 12, 2014).
Zarin, D. A. 2013. Participant-level data and the new frontier in trial transparency. New England Journal of Medicine 369(5):468-469.
















































