
7
Future Genetic-Engineering Technologies
This report has focused thus far on the “experiences” aspect of the committee’s statement of task. The purpose of the present chapter is to consider the “prospects,” that is, how genetic engineering might be used in the future in agricultural crops. That includes speculation about future genetic-engineering technologies.
To provide a context for genetic engineering in overall crop improvement, the chapter first provides a description of plant-breeding methods and of genomics approaches that enable rapid advances in basic knowledge related to crop genetics and plant breeding. It then discusses commonly used genetic-engineering technologies, examining the breadth and depth of current use and current limitations. Next, it scans the horizon for emerging genetic-engineering technologies, including synthetic biology and genome editing, and speculates about how they might shape the future of crops. The expected applications of genome editing and the technologies available for assessing associated nontarget effects are discussed in more detail.
Finally, “-omics” (genomics, transcriptomics, proteomics, metabolomics, and epigenomics) approaches are reviewed to evaluate their potential to assess intended and unintended effects of genetic engineering and conventional plant breeding. The committee concludes that advances in genetic engineering and -omics technologies have great potential to enhance crop improvement in the 21st century, especially when coupled with advanced conventional-breeding methods.
MODERN PLANT-BREEDING METHODS
Conventional plant-breeding approaches rely on the selection of plant germplasm with desirable agronomic and product characteristics (that is, phenotypes) from among individual plants created by using crosses and mutagenesis. Breeding used to be entirely phenotype-based; that is, plants were selected solely on the basis of features such as yield, without knowledge of the genetic composition of the plants. All plants of potential interest would be grown, phenotyped, and harvested, all of which are time-intensive and resource-intensive. The entry of molecular biology into breeding programs in the 1980s enabled knowledge of genetic determinants of phenotypes and marker-assisted selection (MAS) in which DNA-based molecular markers are used to screen germplasm for individual plants that have desired forms of genes, known as alleles. MAS reduced plant sample sizes needed to select desirable individual plants and has been used in many crops to reduce costs and increase efficiency. MAS allows the identification and elimination of an individual plant from a population on the basis of its genetic composition and, as a consequence, reduces the costs associated with both continued propagation and downstream phenotyping (Ru et al., 2015). For example, before MAS, each tree in a fruit-tree breeding program had to be grown for years before it would produce fruit that could be phenotyped. However, molecular markers associated with self-compatibility (in which fertilization does not require outside donor pollen) and fruit size have recently been used to eliminate seedlings that lack alleles favorable for these two critical market traits in a sweet cherry (Prunus avium) breeding program, resulting in substantial savings (Ru et al., 2015). MAS does not require knowledge of the specific genes that confer a trait; it only requires markers that are tightly associated with a trait, which may or may not be within the gene controlling the trait (see, for example, Box 7-1). MAS is not used in all plant-breeding programs, but its use might soon become universal as more genetic information is made available and screening costs are reduced.
As in all other disciplines of biology, plant breeding is now in the genomics era, in which paradigm-changing methods are being incorporated to accelerate and improve the efficiency of breeding. Incorporation of genomics into breeding and genetics research has resulted in an increased knowledge base on crop genetics, species diversity, the molecular basis of traits, and the evolutionary history of crop origins from primitive wild species. MAS and genomics greatly reduce the number of individual plants that need to be retained in the breeding pipeline for phenotyping. Genome-level datasets and genomic technologies have been used to identify causal genes, alleles, and loci important to relevant agronomic traits and have thereby become tools to accelerate breeding cycles (Box 7-1).
An array of genomic technologies can be used to generate large-scale genetic-diversity data on any species that can be used to breed improved crop varieties through such techniques as MAS. For example, genome sequencing and resequencing (in which all or part of the genome is sequenced) and single-nucleotide polymorphism (SNP) assays (in which hundreds to millions of individual loci are assayed for allelic diversity) are genomic methods that are used routinely in many crops. Several technological approaches can be applied to assay SNP loci, including platforms that use mass spectrometry, primer extension, or reduced-representation targeted resequencing to assay the polymorphism. Major considerations for the plant breeder in the choice of technological platform are marker density, sample throughput, cost, and number of loci to assay. Depending on the crop, a publicly or commercially available SNP platform is used, as are custom SNP arrays for specific applications.
The coupling of continual advancements in genomic technologies with increased throughput and decreasing costs means that conventional and genetic-engineering breeding programs now have access to a wealth of genetic-diversity data that can be used to link genes (and alleles) with phenotypes and agronomic traits. For example, large-scale, genome-diversity data on several major crops have been generated, including not only cultivated lines but related wild species and landraces. The information has provided insights into the genetic and molecular basis of
agronomic traits, genetic bottlenecks that restrict major improvements in breeding gains, and key genes and events in domestication and crop improvement, all of which lead to more efficient breeding (Huang et al., 2010; Lam et al., 2010; Chia et al., 2012; Hufford et al., 2012; Jiao et al., 2012; Li et al., 2014b; Lin et al., 2014).
At the time when the committee’s report was being written, a reference genome of nearly every major crop species was available. The quality of the reference genome sequences varies substantially, depending on technical and cost limitations. A complete genomic sequence with few or no gaps is the “gold standard” for a reference genome, but incompletely characterized genomes are also useful. For many crops, diversity panels1 with their associated genome and phenotype datasets have been or are being developed. However, the availability of large-scale genetic information is not a panacea for plant breeding. A reference genome derived from a single individual or genotype does not provide full representation of the genome information needed for crop improvement; as a consequence, multiple reference genomes for each species are needed to adequately capture the genome diversity. For various reasons—including lack of access to data, lack of computational tools, and insufficient analytical expertise in genomics—some researchers do not take full advantage of genomic data. However, as genomic technologies improve so that any individual plant’s genome can be sequenced and analyzed, as breeders acquire more expertise in using relevant genomic and bioinformatic technologies, and as genotyping methods improve in throughput and cost efficiency, those limitations will be overcome, and this will leave phenotyping as the major limitation of efficient breeding. Thus, it is likely that high-throughput, field-based phenotyping technologies will be developed to provide parallel datasets, increase efficiency, and reduce costs associated with breeding.
FINDING: Conventional and genetically engineered plant-breeding approaches in the 21st century have been enabled by increased knowledge of plant genomes, the genetic basis of agronomic traits, and genomic technologies to genotype germplasm.
FINDING: Continued improvements in genomic technologies and algorithm and software development in the coming decades will facilitate further improvements in the efficiency of plant breeding.
__________________
1 Diversity panels are collections of germplasm that represents a crop species. The panels include cultivars, landraces, and wild species related to the crop that collectively represent the genetic diversity of the crop or can be used to improve phenotypic traits in the crop.
FINDING: As genomic technologies increase in throughput and decrease in cost, thousands of genomes will be characterized per crop species.
COMMONLY USED GENETIC-ENGINEERING TECHNOLOGIES
This section reviews widely used genetic-engineering technologies that have been applied in the development of commercialized genetically engineered (GE) crops.
Expression of Added Genes
Since the application of recombinant-DNA technology to plants in the 1980s, most of the technology deployed commercially has consisted of constant or constitutive expression of transgenes in a few crop species in which Agrobacterium-mediated or gene gun-mediated transformation is used to insert a specific gene of interest into a random location in the plant nuclear genome. A single gene that endows a simple trait, such as the production of an enzyme that confers herbicide resistance or a Bt toxin for insect resistance, is incorporated into all the cells of the plant. In addition to the single transgene of interest, a selectable marker gene is sometimes included in the same DNA molecule that is transferred into the plants to facilitate identification of plants that have the transgene of interest. This type of “one-gene” genetic engineering was exemplified in most commercialized crops grown in 2015.
Transgenics versus Cisgenics versus Intragenics
Because of legislative, regulatory, marketing, and public-perception concerns, efforts have been made to develop GE crops with genes found in a crop species of interest or a plant species that can naturally interbreed with it (Rommens, 2004); this has become known as cisgenesis (Schouten et al., 2006). In the strictest definition of cisgenesis, an entire native gene would be cloned intact from a different variety of the crop or a sexually compatible relative and inserted into the genome of the crop of interest. In a related approach called intragenesis, the researcher recombines various plant DNAs, all of which came from the crop or its relatives into a single genetic construct to be introduced; for example, the promoter could come from one gene and the coding region from another gene (Holme et al., 2013). Several cisgenic–intragenic GE crops have been developed and field-tested. At the time the committee’s report was being written, only one intragenic crop species, the Innate™ potato developed by Simplot Plant Sciences, had been approved for commercialization.
Nuclear Genome Transformation versus Plastome Transformation
All plant cells have three separate genomes. The largest is in the nucleus, which contains hundreds of millions to billions of nucleotide bases.2 Much smaller genomes are in the mitochondria and plastids (hundreds of thousands of bases each). The plastid genome is known as the plastome.
As discussed above, almost all GE crops sold at the time the committee’s report was being written had been created with recombinant-DNA technology using Agrobacterium-mediated or gene gun-mediated nuclear genome transformation methods. Whereas gene gun-mediated transformation theoretically results in the possibility of transforming any of the three genomes, Agrobacterium favors gene transfer into the nuclear genome of plants (Zhang et al., 2007). As discussed in Chapter 3, nuclear genome transformation has proved to be successful in a wide array of plant taxa.
Plastome transformation was developed in the 1980s and was enabled by the invention of the gene gun (Svab et al., 1990). Plant mitochondrial genomes have not yet been transformed. As an alternative to nuclear genome transformation, plastome transformation has several advantages over nuclear transformation (Maliga, 2003; Jin and Daniell, 2015). First, transgenes are targeted to a specific locus in the plastome by using homologous recombination; thus, there are no “position effects” that influence gene expression. Second, extraordinarily high concentrations of recombinant protein can be produced; for example, Bt Cry2A accumulated to 46 percent of total soluble protein when the gene was localized in the plastome of tobacco (De Cosa et al., 2001). Third, most plant species are characterized by maternal inheritance of plastids, which could address transgene bioconfinement goals (Daniell, 2002); because most pollen does not contain plastids, concerns about gene flow are eliminated or reduced.
In spite of those advantages, some notable hurdles have prevented plastome transformation from being widely used in crops (Maliga, 2003). First, because there are thousands of plastomes per plant cell, it is not trivial to achieve homoplasmy, in which all plastomes in the cell have an identical GE change. An effective tissue-culture protocol is required to rid chloroplasts of native, untransformed plastomes. Thus, relatively few species (mostly in the Solanaceae family, such as tobacco, potato, and tomato) are amenable to tissue culture and have been routinely subjected to transplastomic methods. Second, few selectable marker genes and antibiotic combinations are effective for obtaining transgenic and homogeneous (transplastomic homoplasmic) cells. Nonetheless, there is tremendous
__________________
2 See Chapter 3 (section “The Development of Genetic Engineering in Agriculture”) for how the four nucleotide bases adenine, guanine, thymine, and cytosine form the structure of DNA.
interest in expanding the use of plastomes transformation in various crop species, especially when high transgene expression is desired.
Antisense or RNA Interference Approaches to Decrease Gene Expression
In delivering agronomic traits of interest by using genetics, the simple add-new-DNA-and-protein approach has been successful. However, silencing the expression of one or more native genes in plants or silencing the expression of pest genes, such as those found in pathogens or herbivorous insects, is sometimes desired.
The first method that was used to decrease the expression of genes in GE plants is termed antisense silencing. In an antisense gene construct delivered into the plant genome, the gene to be downregulated (that is, silenced) is essentially put in “backwards” into a plant transformation vector. When the backwards gene is transcribed, the messenger RNA (mRNA) produced from the transgene interferes with the translation of complementary mRNA of the gene to be silenced in the plant or pest into protein (or it can lead to RNA interference, described below). The FLAVR SAVR™ tomato is one of the first examples of the use of antisense technology to produce a GE crop; the FLAVR SAVR tomato had the desirable trait of altered fruit ripening and enhanced fruit quality owing to interference with the expression of the gene that encodes the polygalacturonase enzyme (Kramer and Redenbaugh, 1994).
A second method of silencing gene expression was developed in the late 1990s on the basis of fundamental biological research in plants: RNA interference (RNAi). RNAi started to be used extensively to genetically engineer plants in the 2000s as plasmid vectors and more plant-specific biological information became available. It had been known since the earliest days of plant biotechnology that post-transcriptional gene regulation (silencing) was an important process that could regulate the level of expression of plant genes. For example, it was thought that overexpression of a gene important in the anthocyanin pigment production pathway in petunias would produce flowers that had more rich purple pigmentation; instead, the resulting petunia flowers were white (Napoli et al., 1990). Unbeknownst to the researchers and to the producers of FLAVR SAVR tomato, the RNAi mechanism was the cause of gene silencing; that is, attempts at antisense silencing or overexpression triggered the RNAi mechanism (Krieger et al., 2008). Whereas the actual mechanism of RNAi was first mechanistically elucidated in nematodes (Fire et al., 1998, which resulted in the award of a 2006 Nobel prize to Craig Mello and Andrew Fire), RNAi is now known to be a natural molecular pathway that all higher organisms use to defend themselves against parasites and pathogens.
For uses in GE crops, noncoding RNA production in the form of double-stranded RNA (dsRNA) is an efficient way to set off a cascade
of molecular events within the cell to silence genes in the target plant gene or pest gene of interest, which can result in a new trait. Such traits can involve reduced lignin in plant cell walls, decreased browning in apples, or insect resistance (see Chapter 8). RNAi transformation vectors used for plant transformation are designed to allow the production of an mRNA molecule that will fold back on itself (hairpin RNA) to produce dsRNA. The RNAi machinery in the plant processes the dsRNA to produce 21- to 23-nucleotide small interfering RNA (siRNA) that ultimately targets the mRNA of interest for destruction. The degraded mRNA cannot be translated into protein, so a new trait is created. When the committee’s report was being written, RNAi was being used extensively as the primary tool in plant biology for silencing the expression of endogenous genes and was beginning to be used in commercial GE crops (see Chapter 8).
A notable use of RNAi, published in two papers in 2015 (Jin et al., 2015; Zhang et al., 2015), sought to engineer insect-pest resistance in crops. The researchers used a novel approach, which was to make dsRNA via transgenic chloroplast genomes (plastomes) in crops that were often subjected to damaging herbivory by insects. The dsRNA was designed to trigger the RNAi pathway in the insects that consume the GE plant. The method was effective for two reasons. First, the dsRNA cannot be processed within the plastids because the RNAi machinery does not exist in plastids, thereby ensuring the presence of intact dsRNA in the GE plant when the insect feeds on the plant. When the insect consumes intact dsRNA designed to target a vital insect gene, the gene’s expression in the insect is silenced, killing the insect. Second, plastome expression provides high expression of the dsRNA relative to that possible by nuclear transformation (discussed above). An important consideration in using this technique is to design dsRNAs and their component siRNAs highly specific to the target gene to avoid effects on nontarget genes. As with any genetic engineering-based insect-control strategy, potential nontarget effects need to be investigated.
Development of Non–Tissue-Culture Transformation Methods
As described in Chapter 3, the construction of GE plants commonly relies on in vitro plant tissue culture, transformation, and plant regeneration. Among the complications often associated with the regenerated plants is that they can be variable in phenotype and fertility because of somaclonal variation rather than the genetic-engineering event itself (see Chapter 3 for description of somaclonal variation). Many factors—including crop, culture media, length of time in tissue culture, and genotype—can affect the frequency and severity of somaclonal variation. Altered gene expression can result from changes in chromosome number or structure, in DNA sequence, in epigenetic status—for example, DNA methylation (see below)—or in all
the above (Jiang et al., 2011; Stroud et al., 2013). Because tissue-culture–derived GE plants, also known as transformants, are potentially subject to the effects of somaclonal variation on any gene or transgene and to positional effects on the degree and stability of transgene function, researchers routinely screen multiple independent transformants to select individual “events” that do not display aberrant phenotypes.
There are a few notable exceptions to the requirement of tissue culture for plant transformation. One is the floral-dip method. Some members of the Brassicaceae family, such as Arabidopsis thaliana and Camelina sativa, can be transformed with the floral-dip method (Clough and Bent, 1998; Liu et al., 2012), in which Agrobacterium tumefaciens delivers the transgene directly into the genome of egg cells, thereby permitting production of transgenic plants directly from seed. Numerous laboratories have attempted to adapt the floral-dip method to other species, but results have not been reliable or reproducible. Another is the use of particle bombardment to directly transform cells in plant organs that can be rapidly regenerated into plants; this avoids a prolonged cell-culture phase in which somaclonal variability can accumulate (Christou, 1992).
It is well known that, if a plant is grafted, RNAs and proteins can move between the rootstock and the scion; thus, in a grafted plant with a transgenic rootstock or a transgenic scion, there is the potential for GE-derived molecules to be transported to non-GE portions of the plant (Haroldsen et al., 2012). For example, if a rootstock were transgenic then fruits might have products of the transgene.
FINDING: Construction of GE plants commonly relies on in vitro plant tissue culture that can result in unintended, somaclonally induced genetic change. Development of transformation methods that minimize or bypass tissue culture for all crop species would reduce the frequency of tissue-culture–induced somaclonal variation.
EMERGING GENETIC-ENGINEERING TECHNOLOGIES
In addition to the technologies discussed above, new genetic-engineering approaches have been developed and are being refined and improved. Although they have not been applied to commercial products yet, they hold practical value for future GE crops. The technologies include genome editing, synthetic DNA components and artificial chromosomes, and targeted epigenetic modifications.
Genome Editing
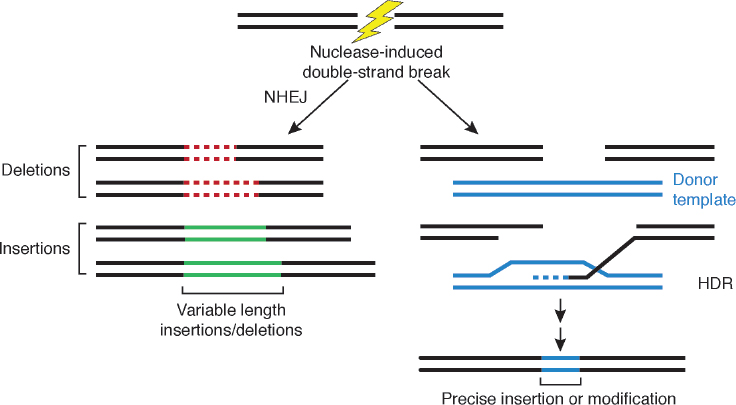
Genome editing uses site-directed nucleases (sequence-specific nucleases; SSNs) to mutate targeted DNA sequences in an organism. Using SSN systems, scientists can delete, add, or change specific bases at a designated locus. SSNs cleave DNA at specific sites and leave a single break (known as a nick) or a double-strand break. The DNA break can be repaired in two ways (Figure 7-1):
- Through the cell’s native nonhomologous end joining (NHEJ) process, which leads to a mutation at the site.
- If a donor DNA molecule is provided at the same time as the DNA is being edited by the nucleases, through the cell’s own native DNA repair machinery—known as homology directed repair (HDR)—which incorporates the donor molecule at the cleavage site.
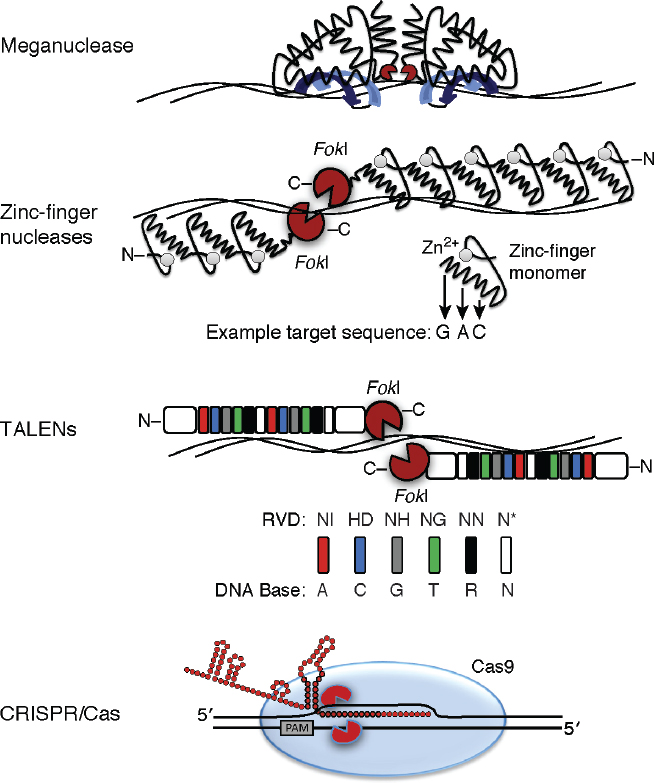
Four main classes of SSNs are used in plant genome editing (reviewed in Voytas and Gao, 2014): meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system (Figure 7-2). The field of genome-editing technologies and application has burgeoned, especially since the advent of the CRISPR/Cas9 system, and the committee expects additional discoveries to facilitate genome editing in the coming decade.
Meganucleases naturally occur in bacteria, archaea, and eukaryotes and were the first SSNs examined for genome editing. Meganucleases are single proteins that recognize a sequence in the DNA that is at least 12 nucleotides long and cleave the target DNA, leaving a double-strand break that can be repaired through NHEJ or HDR by using a donor molecule (reviewed in Silva et al., 2011). Meganuclease-mediated genome editing has been demonstrated in maize (Zea mays) and tobacco (Nicotiana spp.) (reviewed in Baltes and Voytas, 2014). It is difficult to change the target sequence specificity of meganucleases, so they are not widely used for genome editing.
Zinc finger–domain-containing proteins bind to DNA and are widespread in nature, often functioning as transcription factors (proteins that regulate gene expression by binding directly or indirectly to regulatory DNA sequences usually found in the promoter regions of genes3). The zinc finger domains can be manipulated to bind specific sequences of DNA; when fused to the DNA-cutting nuclease domain of the FokI protein, a ZFN is the resulting hybrid molecule. A pair of ZFNs functions in tandem
__________________
3 Examples of genetic engineering using transcription factors are given in Chapter 8.
SOURCE: Sander and Joung (2014).
NOTE: After cleavage of double-stranded DNA by sequence-specific nucleases such as meganucleases, zinc fingers, transcription activator-like effectors, and clustered regularly interspaced palindromic repeats/Cas9 (represented by the lightning bolt), the double-strand break in the DNA molecule can be repaired through native nonhomologous end joining (NHEJ) mechanisms of the cell, which leads to an altered DNA sequence with either a deletion (dotted red line) or an insertion (solid green line). If a donor DNA template is provided (see blue DNA fragment), the homology-directed repair (HDR) mechanisms within the cell will insert the donor molecule into the locus, and this leads to an edited gene or target region (blue region with a precise insertion or modification).
to cut DNA at the desired target site (reviewed in Urnov et al., 2010). As with meganucleases, ZFNs are used for introducing mutations through NHEJ and HDR. ZFNs have been used in engineering of numerous plant species (reviewed in Baltes and Voytas, 2014). ZFNs were the first widely used designer genome-editing tool in biology.
Transcription activator-like effectors (TALEs) were discovered in the bacterial plant pathogen Xanthomonas and could be engineered to bind to virtually any DNA sequence. Their ease of design for specific target DNA sequences revolutionized genome editing. In nature, Xanthomonas species secrete TALEs into plant cells to enable pathogenicity. TALEs bind to promoters in plant genes to suppress the plant’s resistance to the pathogen. The bacteria encode TALEs through a simple code or cipher that has been
SOURCE: Baltes and Voytas (2014).
exploited to engineer proteins with custom site specificity in any target genome (Boch et al., 2009; Moscou and Bogdanove, 2009). Like ZFNs, TALEs can be fused with the nuclease domain of FokI and are then referred to as TALENs. TALENs are used in pairs like ZFNs to affect targeted mutations. TALENs have been used to edit genomes in several plants, including rice (Oryza spp.), maize, wheat (Triticum spp.), and soybean (Glycine max) (reviewed in Baltes and Voytas, 2014).
CRISPR was the most recently developed genome-editing tool when the committee’s report was being written. Bacteria harbor CRISPR as an innate defense mechanism against viruses and plasmids that uses RNA-guided nucleases to target the cleavage of foreign DNA sequences. At the time the committee’s report was being written, the CRISPR/Cas system used in genome editing was primarily the Type II CRISPR/Cas9, from Streptococcus pyogenes, in which foreign DNA sequences are incorporated between repeat sequences at the CRISPR locus and then transcribed into an RNA molecule known as crRNA (reviewed by Sander and Joung, 2014). The crRNA then hybridizes with a second RNA, the tracrRNA, and the complex binds to the Cas9 nuclease. The crRNA guides the complex to the target DNA and, in the case of innate immunity in bacteria, binds to the complementary sequence in the target DNA that is then cleaved by the Cas9 nuclease. Scientists have dissected the innate CRISPR/Cas9 system and re-engineered it in such a way that a single RNA, the guide RNA, is needed for Cas9-mediated cleavage of a target sequence in a genome. Guide RNA design requirements are limited to a unique sequence of about 20 nucleotides in the genome (to prevent off-target effects) and are restricted near the protospacer adjacent motif sequence, which is specific for the CRISPR/Cas system. Newer applications of CRISPR include the use of two unique guide RNAs with a modified nuclease that “nicks” one strand of the DNA, providing greater specificity for targeted deletions. The ease of design, the specificity of the guide RNA, and the simplicity of the CRISPR/Cas9 system have resulted in rapid demonstration of the utility of this method of editing genomes in plants and other organisms (reviewed in Baltes and Voytas, 2014). Genome editing, especially CRISPR, is changing rapidly. At the time the committee’s report was being written, non-Cas9 endonucleases (for example, Cpf1) had been recently described for CRISPR genome editing (Zetsche et al., 2015).
When the committee was writing its report, applications of SSNs in genome editing had been used mostly to introduce mutations at the target locus through NHEJ to produce gene knockouts. As shown in Figure 7-1, nucleases can also be used for sequence replacement via HDR (or potentially NHEJ) if a donor DNA is co-introduced into the cell. Multiple types of donor molecules can be used. First, an alternative allele of the target locus can be introduced in such a way that the modified gene encodes a protein that confers a novel or enhanced trait. For example, modification of a specific single nucleotide in the acetolactate synthase (ALS) gene can confer resistance to herbicides that use ALS inhibition as their mode of action (Jander et al., 2003). Second, homologous recombination can be used to introduce a novel sequence at that locus. This “precision genome insertion” by engineering of a landing site would eliminate the semirandom insertion of transgenes in Agrobacterium-mediated and gene gun-mediated transformation methods. It would also permit combining of modified or
edited genes at a single locus, termed “trait landing pads,” in the genome rather than randomly in the genome, so it would be easier to add new traits to a GE crop that are physically linked in the genome (Ainley et al., 2013). This strategy would also enable the removal of inserted genes when desired.
Genome editing via CRISPR and other techniques might be performed in plants via transient expression of transgenes (Clasen et al., 2016) or without exogenous DNA at all (Woo et al., 2015) resulting in GE plants that lack the transgene. Clasen et al. (2016) used a genetically encoded TALEN pair to produce specific mutations in potato protoplasts. Potato plants recovered from the protoplasts with mutated targeted alleles contained a new quality trait, and seven of the 18 GE lines did not contain any TALEN transgene constructs in the potato genome. Woo et al. (2015) used in vitro–translated Cas9 protein coupled to guide RNA to mutate genes in Arabidopsis, tobacco, lettuce, and rice protoplasts, from which mutated plants were recovered. Thus, it appears that genome editing can be performed in crops without leaving any transgenic DNA footprint in the genome. In the absence of any off-target mutations, which is evident from deep targeted sequencing in the potato experiment (Woo et al., 2015), genome-editing reagents4 that do not leave a transgene in the genome would appear to be a valuable crop-breeding approach.
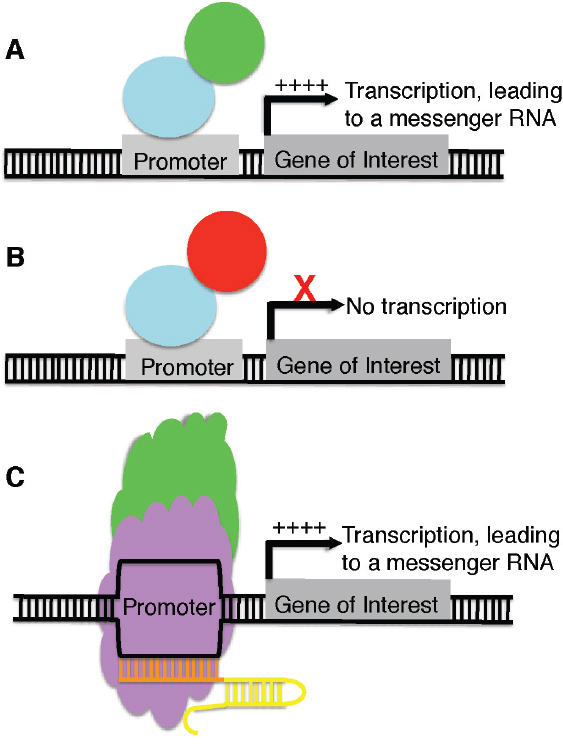
The above examples center on the use of SSNs to cleave DNA and introduce permanent changes in the sequence of the double-stranded DNA through either NHEJ or HDR. However, there are other applications by which proteins that bind to DNA in a sequence-specific manner can be exploited to modify genes and gene activity. They include fusion of the DNA-binding features of zinc finger proteins (ZFPs) or TALEs to activate domains to produce synthetic transcriptional activators. Without activator fusions, the ZFPs or TALEs can be used as transcriptional repressors. The design features of ZFPs and TALEs enable the production of synthetic transcription factors (TFs) to regulate the expression of practically any target gene (Figure 7-3). Indeed, TALE-TFs have been used in tandem to produce additive gene activation in transgenic plants (Liu et al., 2014). For the CRISPR/Cas9 system, a suite of molecules can be fused with a catalytically inactive Cas9 nuclease (dCas9) to permit a wide array of modifications of gene regulation, collectively known as CRISPR interference (reviewed in Doudna and Charpentier, 2014; Figure 7-3). Perhaps the simplest application is fusion of the dCas9 to a transcriptional activator or a transcriptional repressor; when introduced into a cell with a guide RNA, the transcriptional activator or repressor can modify the number of transcripts
__________________
4 Nucleases that have been customized to target a specific sequence are referred to as reagents.
of the target gene in a way similar to ZFPs and TALEs. The dCas9 genes could potentially be fused with chromatin modification genes and guided to a target region by the guide RNA to modify the epigenetic state of a locus and thereby affect transcription (Doudna and Charpentier, 2014).
FINDING: Exploitation of inherent biological processes—DNA binding-zinc finger proteins (ZFNs), pathogen-directed transcription of host genes (TALEs), and targeted degradation of DNA sequences (CRISPR/Cas)—now permit precise and versatile manipulation of DNA in plants.
Artificial and Synthetic Chromosomes
Increased knowledge of biological processes and advanced molecular-biology tools will not only facilitate the engineering of multiple genes into plants but will also make possible the insertion of complete and novel biochemical pathways or processes. DNA constructs used in plant genetic engineering have been small (less than 20–40 kilobases) and have used traditional molecular cloning techniques that are slow and laborious. However, new and inexpensive methods of synthesizing DNA molecules and assembling them into larger DNA molecules have been developed. The methods permit rapid, easy construction of multigene pathways on a single DNA molecule (for review, see Ellis et al., 2011). The feasibility of the techniques has been demonstrated by the synthesis of an entire bacterial genome (Gibson et al., 2010) and the creation of a synthetic yeast chromosome (Annaluru et al., 2014). Researchers would like to be able to introduce tens to hundreds of genes into plants. One method envisaged to accomplish such advances would be the use of artificial minichromosomes or synthetic chromosomes. An artificial minichromosome is a chromosome added into the plant’s natural composite of chromosomes in the nucleus. A synthetic chromosome (Annaluru et al., 2014) is a total synthesis of DNA to replace a natural chromosome or even an entire organelle genome, such as the plastome genome.
Artificial or synthetic chromosomes and genomes would allow the assembly of a large number of genes in a self-replicating molecule. In eukaryotes, chromosomes are linear DNA molecules with the proper sequences for replication (origins of replication), integrity (telomeres), and segregation in dividing cells (centromeres). The use of artificial or synthetic chromosomes would permit the introduction of genes into plants in a manner that does not have the potential to disrupt genes on native plant chromosomes at the sites of insertion.
No synthetic chromosomes or genomes have been created in plants, but the methods used to make a yeast synthetic chromosome (Annaluru et
SOURCE: Illustration provided by C. R. Buell.
NOTE: A, A zinc finger or transcription activator-like effector (TALE) (blue) can be fused to a transcriptional activator (green) to increase transcription of a gene of interest. B, A zinc finger or TALE (blue) can be fused to a transcriptional repressor (red) to suppress transcription of a gene of interest. C, A catalytically inactive Cas9 nuclease (purple) can be fused to a transcriptional activator (green) and, in the presence of a guide RNA (orange and yellow), guide the complex to the promoter of a gene of interest and increase transcription of the target gene. D, A catalytically
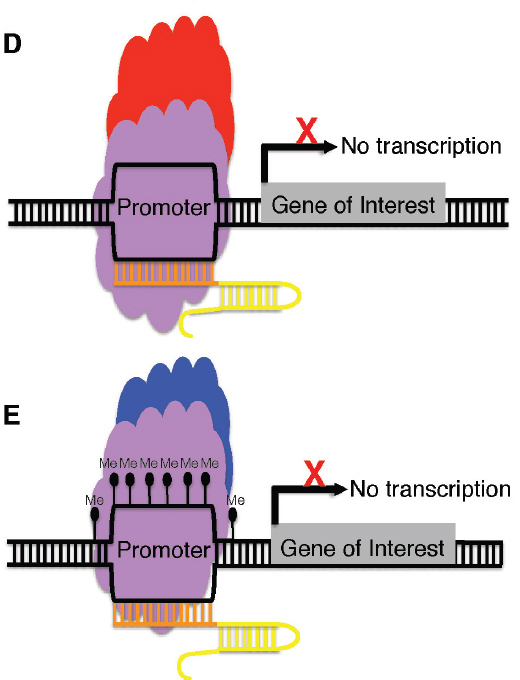
inactive Cas9 nuclease (purple) can be fused to a transcriptional repressor (red) and, in the presence of a guide RNA (orange and yellow), guide the complex to the promoter of a gene of interest and decrease transcription of the target gene. E, A catalytically inactive Cas9 nuclease (purple) can be fused to a DNA-methylating enzyme and, in the presence of a guide RNA (orange and yellow), guide the complex to the promoter of a gene of interest, targeting that gene for methylation and, as a consequence, suppressing transcription.
al., 2014) should be transferrable to plants. Yeast chromosome III, which is 316,617 bases, was replaced with a laboratory-synthesized chromosome of 272,871 bases. The chloroplast genome, which is one-half to one-third the size of yeast chromosome III, could foreseeably be synthesized and inserted into a plant, such as tobacco, that is already amenable to plastome transformation. The cost of DNA synthesis continues to decrease, so experimental development in this field would be economically feasible.
Research on artificial minichromosomes for plants has generally taken two approaches (reviewed in Gaeta et al., 2012; Birchler, 2015). In the “bottom-up” approach, the key parts of a chromosome—such as the centromere, telomere, origin of replication, and genes of interest—are assembled, and this results in a de novo minichromosome. Although progress has been made with this approach, additional characteristics, such as epigenetic modification of DNA nucleotides, can affect gene expression (see below) and the ability of the minichromosome to be replicated in a cell and have prevented the approach from being used routinely. The “top-down” approach essentially uses existing chromosomes—approximating the synthetic yeast chromosome approach—to build an artificial chromosome with an existing template. This approach has resulted in transmission of the minichromosome through meiosis but not as efficiently as that of native chromosomes. Neither approach has resulted in practically deployable minichromosomes. Thus, the use of a synthetic approach to replace DNA systematically, analogous to the approach in the yeast project, might be possible in the future (Birchler, 2015). It might also be possible that current bottom-up or top-down approaches could work in clonally propagated crops, such as potato, given that meiosis is not required (Birchler, 2015).
With a robust knowledge base of genes underlying plant biochemistry, precise constructs can be synthesized in vitro and introduced into a heterologous species through Agrobacterium-mediated or gene gun-mediated transformation or nuclease-targeted insertion at a single selected site. For example, sweet wormwood (Artemisia annua) produces artemisinin, an antimalarial compound. With five genes from yeast and from A. annua, tobacco was engineered to synthesize artemisinin (Farhi et al., 2011). Although the yield of artemisinin in tobacco was lower than that in A. annua, this proof-of-concept study demonstrated the feasibility of engineering heterologous biosynthetic pathways in plants. With further refinement of promoters, improved understanding of metabolite compartmentalization and transport and flux in cells and tissues, and development of such vectors as artificial chromosomes capable of transferring large segments of DNA to plant cells, genetic engineering of plants for complex traits, such as heterologous or novel biochemical pathways, and for specialized physiological
and developmental processes, such as C4 photosynthesis, may be possible (see Chapter 8 for detail).
Targeted Epigenetic Modifications
As outlined earlier in this chapter, relatively stable, heritable changes in the expression of specific genes can result from changes in the epigenome. Changes in the methylation of DNA in particular are most likely to result in heritable changes in gene expression in plants. As the understanding of the biochemistry of the systems that change DNA methylation patterns increases, the ability to target specific genes for epigenetic modifications also increases. Such targeting would involve expressing the proteins and perhaps also specific nucleic acids (for example, a guide RNA) that would direct DNA methylation to a specific locus. The targeting system could be removed after the epigenetic modification is accomplished—for example, by using conventional plant breeding to segregate the targeting system.
On the key issue of safety, changing the epigenetic modifications of particular plant genes presents no fundamental problems; the genomes of plants are replete with epigenetic modifications, and epigenetically modified DNA has been in the environment and consumed by humans for thousands of years. Thus, the safety issue is the same as that involved in any technique, whether genetic engineering or non-genetic engineering, that results in an increase or decrease in the expression of specific genes in a plant and in specific trait alterations.
It should be noted that, in addition to targeted approaches, there are several ways to broadly and randomly alter the epigenome of a plant, such as overexpression of an enzyme that alters DNA methylation. Of course, broad and random approaches to altering a plant genome are not new: mutagenesis is a broad and random approach to genome modification. The utility of broad and random approaches to genome or epigenome modification is that if the biochemistry or genetics of a process is not sufficiently understood to permit a targeted approach—whether a genetic-engineering or a non–genetic-engineering approach (such as marker-assisted breeding)—a random approach might produce a desirable result. As discussed above, broad and random approaches will result in more unknown changes than targeted approaches.
Consistency is typically a desirable feature of new plant varieties. Given that epigenetic changes can revert to the initial state at varied frequencies, epigenetic modification might not be an ideal approach to produce new plant varieties. However, if a new plant variety derived from an epigenetic change is superior to stable alternatives, the potential for some degree of reversion may not be an obstacle to commercialization.
FUTURE APPLICATIONS OF GENOME EDITING
Of the emerging genetic-engineering technologies reviewed above, genome editing is the closest to being used to modify commercially available crops. A key component of effective and efficient application of genome editing is understanding the biochemical, molecular, and physiological basis of agronomic traits such as plant architecture, photosynthesis, pathogen resistance, and stress tolerance. Given advances in knowledge, additional genome-editing targets will emerge, and they will probably involve the manipulation of multiple genes (see Chapter 8). The committee also expects advances in the ability to edit plant genes precisely—that is, to make precise changes in specific genes without disrupting other genes. Such advances often come from basic research, whose far-reaching applications are not anticipated. For example, TALENs resulted from a study of how some plant-pathogenic Xanthomonas species modify gene expression in host plant cells. The CRISPR/Cas9 system resulted from the surprising discovery that some bacteria have an adaptable “immune system” to resist infection by viruses. The TALEN and CRISPR/Cas9 systems ushered in rapid advances in not only the ease but the range of genome-editing possibilities. In this section, the committee provides an overview of some of the expected applications of this transformation technology.
Removal of Genome-Editing Reagents in Genetically Engineered Crops
It is envisioned that genome editing will be useful in most agricultural crops in generating modified alleles that are homozygous in the modified line for several reasons: to prevent segregation of the altered allele in derived progeny, to eliminate the production of the wild-type target mRNA and protein, and to increase the dosage of the modified allele as the level of transcript of a gene is correlated with numbers of alleles. As described earlier, DNA-free genome editing via CRISPR is possible (Woo et al., 2015). With TALEN and CRISPR/Cas9 reagents, both heterozygous and homozygous mutations can be generated in the first transformed generation. Simple molecular-biology screens can be performed to identify individuals homozygous for the modified allele. Alternatively, for sexually reproducing self-compatible species, individual heterozygous transformed plants can be crossed with their own pollen and homozygous progeny identified in the second transformed generation. An important consideration when generating some genome-edited plants is to ensure that the reagent (TALEN, Cas9) is not present in the selected progeny; additional mutations can be generated if the reagents remain active. In some situations, the continued presence of the reagent is desirable. For example, the retained editing reagents can act as a constant mutagen and generate various modified alleles
if sufficient gene family members that could be substrates for the reagents are present. However, that might not be desirable in an agronomic situation as stability of genetic material is essential for commercial production. Another example in which the continued presence of the reagent is necessary is gene-drive applications.
Gene Drive
In natural populations, genome-editing reagents can be used to create a gene-drive system, in which the frequency of a specific allele in the population is altered, affecting the likelihood that the desired allele will be inherited. A gene-drive system can be created by retaining the genome-editing reagents in the transgenic organism to enable continued editing of the target alleles throughout the population when the reagents are incorporated into the germline and passed to other members of the population through sexual reproduction; the use of CRISPR/Cas9 to create such a genome-editing system has been referred to as mutagenic chain reaction (Gantz and Bier, 2015). Gene drive has applications in the control of insect pests, such as mosquitoes, and various pests in crops (Esvelt et al, 2014). Inadvertent creation of a CRISPR/Cas9 gene-drive system can be avoided by ensuring that the gene constructs that encode the two cassettes5 for Cas9 and the guide RNAs are not present in the GE plant that is developed. There are many ways to ensure this (Akbari et al., 2015). One way is to genetically segregate away the constructs after editing occurs. Another way is to employ DNA-free or transgene-free genome editing (Woo et al., 2015) in which only proteins and RNAs are introduced into the plant to accomplish gene editing.
Traits Involving Gain versus Loss of Function
Most commercialized GE crops have gain-of-function traits, such as herbicide resistance or insect resistance. Only recently have loss-of-function traits been readied for commercial sale in GE crops. An example is the nonbrowning apple (see Chapters 3 and 8). Chapter 8 describes a number of complex traits that were in the research stage when the committee was writing its report; many of them (such as water-use efficiency, nitrogen fixation, and enhanced carbon-fixation efficiency) exhibit gain of function, or perhaps a combination of gain of function with loss of function, and will probably involve the introduction of multiple genes (Box 7-2).
Given the time and expense associated with the regulatory process, intellectual-property constraints, and consumer wariness (see Chapter 6),
__________________
5 A cassette is a DNA molecule that contains several genes that collectively function in a single process.
some firms and organizations seek to develop desired traits without genetic-engineering approaches. Loss-of-function traits, particularly if only a single gene has to be disabled, can be readily obtained with the non-GE approach of mutagenesis because random changes in DNA sequence are more likely to disrupt than to improve protein function. At the other end of the spectrum, traits that require introduction of novel genes, or perhaps complex redirecting of gene expression to different tissue or cell types, may be achievable only with genetic-engineering approaches.
Consider the following theoretical example that aims to reduce the concentration of a toxic compound in the leaves of a potentially new crop plant. All available germplasm of the plant contains the toxic compound at unacceptable concentrations. Proof-of-concept studies might first explore the efficacy of downregulating the target gene responsible for synthesis of the toxic compound by using RNAi or knocking it out via CRISPR/Cas 9 or TALENs. Once the target is identified, an approach that does not involve genetic engineering, such as targeting induced local lesions in genomes (TILLING), could be applied. TILLING relies on initial chemical mutagenesis of a plant population followed by molecular identification of the required mutant allele and then crossing to obtain plants homozygous in that allele (Henikoff et al., 2004). It does not require the target plant to be genetically transformable. Although setting up the initial mutant TILLING population is expensive and fixing the required allele can be complex in polyploid outcrossing species, the approach can be cost-effective if multiple traits are being sought in the same species. TILLING populations have been available for several years for many crop and model species (Perry et al., 2003; Comis, 2005; Weil, 2009) and are being used in agricultural biotechnology (Comis, 2005; Slade and Knauf, 2005). However, it is not clear that traits produced by TILLING would pose less risk of unintended effects to the environment or food safety than those introduced by genetic-engineering approaches, such as RNAi and genome editing. The chemical mutagenesis used in TILLING introduces random mutations into the plant genome; although most of the mutations can be removed by backcrossing in most crop species, the resulting modified crop plant might have more unknown changes than the same change in the target gene of interest brought about by use of CRISPR/Cas 9 (although somaclonal variation will not be an issue because TILLING does not require a tissue-culture step).
There are both conventional-breeding and genetic-engineering approaches for the selection of gain-of-function traits such as increasing the amount of a beneficial component (for example, a nutrient or useful pharmaceutical compound). If sufficient natural variation is present in the species, breeding for increased production can be advanced by using marker-based or genomics-based approaches and development of an im-
proved variety through marker-assisted introgression. A good example of this approach is the increase in production of the antimalarial compound artemisinin in wormwood (Graham et al., 2010). If natural variation is not present, as is the case in trying to introduce condensed tannins in foliage to improve forage quality in alfalfa (Lees, 1992), a genetic-engineering approach may be the only option available. Theoretically, it may be possible to introduce a gain-of-function trait through TILLING; if not, the trait can be introduced by overexpression of a key, rate-limiting biosynthetic enzyme or one or more positively regulating transcription factors. Examples of these approaches are presented in Chapter 8.
Editing Quantitative Trait Loci
Not every trait is governed by a single gene. Many agronomic traits are complex traits and are governed by multiple genetic loci that contribute to overall variation in the phenotype; the multiple genes involved in complex traits are known as quantitative trait loci (QTL). Scientists have identified a number of QTL for diverse agronomic and quality traits of a wide array of crop species. On the basis of high rates of coinheritance (linkage) with specific DNA sequences, specific progeny can be selected from conventional breeding to create combinations of QTL that are expected to perform well. They can be tested by field experimentation.
The use of QTL selection has limitations and impediments. First, for a number of crop species, backcrossing is difficult or impossible because of low sexual fertility, long reproduction cycles, inbreeding depression, or some combination thereof. Second, desirable QTL may be closely linked (coinherited) with genes that adversely affect other important traits, that is, “linkage drag;” this is common among genes in low-recombination regions of chromosomes and when desirable QTL are found in wild crop relatives whose genomes are less prone to recombination with the crop genome. Third, considerable effort is required to introgress a specific QTL into all plants of interest. Multiple backcrosses must be made to remove unlinked introgression events, which is expensive in terms of growing populations in the greenhouse or field for multiple generations. Thus, genome editing of QTL provides an alternative approach for developing elite varieties in species whose breeding cycles pose logistical challenges.
In the event that the specific nucleotides that govern a QTL are known, genome editing could be used to modify nucleotides in the QTL to the favorable alleles. Not all QTL have been defined at the gene or allele level; for most QTL, only a localized region of the genome has been defined as the QTL. Therefore, a region of the genome might be replaced by using genome-editing technologies, although current methods are inefficient and the restrictions on the length of DNA that can be edited are not yet known.
FINDING: Genome-editing methods can complement and extend contemporary methods of genetic improvement by modifying composition and expression of genes and by targeting insertion events.
FINDING: Current genome-editing methods and reagents are improving rapidly in precision and efficiency.
EMERGING TECHNOLOGIES TO ASSESS GENOME-EDITING SPECIFICITY
A highly touted feature of emerging genome-editing methods is their extreme specificity—ZFNs, TALENs, and the CRISPR/Cas9 nuclease system rely on recognition of a target sequence, so a single nucleotide in a genome can be targeted and modified. However, the extent of off-target effects is not well established, and off-target effects would potentially have unintended effects. The development of tools to assess such effects is important, and some emerging approaches are described below.
In Silico Prediction Methods
For ZFNs, TALENs, and CRISPR, computational programs have been developed that facilitate design of reagents to minimize off-target effects (for example, Fine et al., 2014; Heigwer et al., 2014; Naito et al., 2014). In general, the programs are tailored for each genome-editing reagent. The programs assess the homology of the target sequence to other sites in the genome; to address technical limitations or preferences such as nuclease specificity, binding energy, binding preferences, and maximal permitted mismatches, the user can modify various search parameters. As empirical data on off-target effects of genome-editing reagents accumulate (see below), the programs can be refined to improve their sensitivity and specificity.
Molecular-Based Methods
Multiple methods have been used to assess off-target effects of genome-editing reagents, from targeted amplification and assessment of one or a few candidate off-target loci to an unbiased approach in which all possible off-target effects are captured and examined by whole-genome DNA resequencing. For a few potential off-target loci, simple molecular-biology techniques can be used in which the locus is amplified and treated with a mismatch-specific nuclease that recognizes SNPs or insertions and deletions in heteroduplex DNA, and the products are then separated with gel electrophoresis. Alternatively, the polymerase chain reaction product can be sequenced and examined through sequence alignments. This inexpensive,
rapid method is sufficient for small numbers of loci but is not scalable and is limited in requiring a priori knowledge of candidate off-target loci. Recently developed unbiased approaches include detection of double-strand breaks through the capture of synthesized double-stranded oligonucleotides, which is then coupled with high-throughput sequencing. This technique—termed genome-wide, unbiased identification of double-strand breaks enabled by sequencing (GUIDE-seq)—was recently demonstrated in human cell lines with the CRISPR/Cas9 genome-editing platform (Tsai et al., 2015). The GUIDE-seq experiment was able to identify a substantially higher number of off-target loci for 13 different CRISPR RNA-guided nucleases than predicted through computational off-target prediction programs. Indeed, although the GUIDE-seq method captured all known, predicted off-target loci, most of them were not detected computationally, and this suggests substantial sensitivity limitations in off-target prediction software. The GUIDE-seq method detected double-strand breaks that were independent of the CRISPR RNA-guided nucleases, so there may be inherent breakage of chromosomes independent of the genome-editing reagent. What was most notable about the Tsai et al. study was that a substantial reduction in off-target effects was observed by using truncated guide RNAs; this suggests that improvements in the design of the CRISPR reagents have strong potential to affect the specificity of genome editing. The GUIDE-seq method could readily be applied to plants for testing and optimizing genome-editing reagents in protoplasts before construction of an engineered, regenerated plant.
DETECTION OF GENOME ALTERATIONS VIA -OMICS TECHNOLOGIES
In the last 15 years, various advanced technologies have been developed that permit accumulation and assessment of large-scale datasets of biological molecules, including DNA sequence (the genome), transcripts (the transcriptome; involving RNA), DNA modification (the epigenome), and, to lesser extents, proteins and their modifications (the proteome) and metabolites (the metabolome). Such datasets enable comparative analyses of non-GE and GE lines in such a way that effects on plant gene expression, metabolism, and composition can be assessed in a more informed manner. Access to the technologies also permits analysis of the extent of the natural variation in a crop species at the DNA, RNA, protein, metabolite, and epigenetic levels, enabling determination of whether variation in GE crops is within the range found naturally and among cultivars. As discussed below for each of the -omics data types, technologies to access the molecules were relatively recent as of 2015 but were advancing rapidly. Some technologies were ready to be deployed to generate datasets for assessment of the
effects of genetic-engineering events when the committee’s report was being written. Others will improve in precision and throughput in the coming decade and may someday be useful technologies for assessing effects of genetic-engineering events. The Precision Medicine Initiative announced by President Obama in January 20156 focuses on understanding how genetic differences between individuals and mutations present in cancer and diseased cells (versus healthy cells) affect human health. An analogous project that uses diverse -omics approaches in crop plants with genetic engineering and conventional breeding could provide in-depth improvements in the understanding of plant biological processes that in turn could be applied to assessing the effects of genetic modifications in crop plants.
Genomics
One way to ascertain whether genetic engineering has resulted in off-target effects (whether through nuclear transformation with Agrobacterium or gene guns, RNAi, or such emerging technologies as genome editing) is to compare the genome of the GE plant with an example—or reference—genome of the parent non-GE plant. The reference genome is like a blueprint for the species, revealing allelic diversity and identifying the genes associated with phenotype. Knowing the variation that occurs naturally in a species, one can compare the engineered genome with the reference genome to reveal whether genetic engineering has caused any changes—expected or unintended—and to gain context for assessing whether changes might have adverse effects. Because there is inherent DNA-sequence variation among plants within a species, and even between cultivars, any genetically engineered changes would need to be compared to the non-GE parent and the range of natural genomic variation. That is, changes made by genetic engineering must be placed in an appropriate context.
Background
In July 1995, the first genome sequence of a living organism, the bacterium Haemophilus influenza (1,830,137 base pairs), was reported (Fleischmann et al., 1995). This paradigm-changing technological achievement was possible because of the development of automated DNA-sequencing methods, improved computer-processing power, and the development of algorithms for reconstructing a full genome on the basis of fragmented, random DNA sequences. In October 1995, the genome of the
__________________
6 Fact Sheet: President Obama’s Precision Medicine Initiative. Available at https://www.whitehouse.gov/the-press-office/2015/01/30/fact-sheet-president-obama-s-precision-medicineinitiative. Accessed November 12, 2015.
bacterium Mycoplasma genitalium was released (Fraser et al., 1995); this solidified whole-genome shotgun sequencing and assembly as the method for obtaining genome sequences. In the next two decades, higher throughput and less expensive methods for genome sequencing and assembly emerged (for review, see McPherson, 2014) and enabled the sequencing of the genomes of hundreds of species, as well as thousands of individuals, in all kingdoms of life. For example, since the release of the draft sequence of the human reference genome in 2001 (Lander et al., 2001; Venter et al., 2001), thousands of individual human genomes have been sequenced, including such comparative genome-sequencing projects as: a deep catalog of human variation of thousands of individuals,7 normal versus tumor cells from a single individual, families with inherited genetic disorders, and diseased versus healthy populations. Those projects have focused on detecting the allelic diversity in a species and associating genes with phenotypes, such as the propensity for specific diseases.
Limitations in Current De Novo Genome Sequencing and Assembly Methods for Plants
Current methods to sequence a genome and assemble a genome de novo entail random fragmentation of DNA, generation of sequence reads, and reconstruction of the original genome sequence by using assembly algorithms. Although the methods are robust and continue to improve, it is important to note that they fail to deliver the full genome sequence of complex eukaryotes. Indeed, even the human genome sequence—for which billions of dollars have been spent to obtain a high-quality reference genome sequence that has provided a wealth of useful information in understanding of human biology, including cancer and other diseases—is still incomplete. For plants, the benchmark for a high-quality genome assembly is that of the model species Arabidopsis thaliana, which has an extremely small genome that was published in 2000 (Arabidopsis Genome Initiative, 2000). More than 15 years after the release of the A. thaliana reference genome sequence and with the availability of sequences from more than 800 additional accessions,8 an estimated 30–40 million nucleotides of sequence were still missing from the A. thaliana Col-0 reference genome assembly (Bennett et al., 2003). Most of the missing sequences are highly repetitive (such as ribosomal RNA genes and centromeric repeats), but some gene-containing regions are absent because of technical challenges.
__________________
7 1000 Genomes: A Deep Catalog of Human Genetic Variation. Available at http://www.1000genomes.org/. Accessed November 12, 2015.
8 1001 Genomes: A Catalog of Arabidopsis thaliana Genetic Variation. Available at http://1001genomes.org/. Accessed November 12, 2015.
With increased genome size and repetitive-sequence complexity, complete representation of the genome sequence becomes more challenging. Indeed, the genome assemblies of most major crop species (maize, wheat, barley, and potato) are all of only draft quality and have substantial gaps (Schnable et al., 2009; Potato Genome Sequencing Consortium, 2011; International Barley Genome Sequencing, 2012; Li et al., 2014a); none provides a complete, full representation of the genome.
In several major crops, when the committee was writing its report, projects equivalent to the human 10,000-genomes project were under way to determine the overall diversity of the species by documenting the “pan-genome” (Weigel and Mott, 2009). It has been surprising in several of these studies that there is substantial genomic diversity in some plant species not only in allelic composition but also in gene content (Lai et al., 2010; Hirsch et al., 2014; Li et al., 2014b). Thus, a single “reference” genome sequence derived from a single individual of a species will fail to represent the genetic composition and diversity of the overall population adequately and will therefore limit interpretations of directed changes in the genome (such as ones that can be delivered by emerging genome-editing methods that are being used to generate GE crops).
Resequencing: Assessing Differences Between the Reference and Query Genome
Once the DNA sequence of a crop’s genome is assembled well enough to serve as a reference genome, resequencing becomes a powerful and cost-effective method for detecting genomic differences among related accessions (individuals) or GE lines. Resequencing entails generating random-sequence reads of the query genome (the genome that is being compared with the reference genome), aligning those sequence reads with a reference genome, and using algorithms to determine differences between the query and the reference. The strengths of this approach are that it is inexpensive and permits many query genomes to be compared with the reference genome and thereby provides substantial data about similarities and differences between individuals in a species (Figure 7-5). However, limitations of the approach can affect determination of whether two genomes are different. First, sequence read quality will affect data interpretation in that read errors can be misinterpreted as sequence polymorphisms. Second, the coverage of sequence reads generated can limit interrogation of the whole genome because the sampling is random and some regions of the genome are underrepresented in the read pool. Third, library construction9 and sequencing
__________________
9 A library of DNA sequence is made by generating random fragments of the genome that collectively represent the full sequence of the genome.
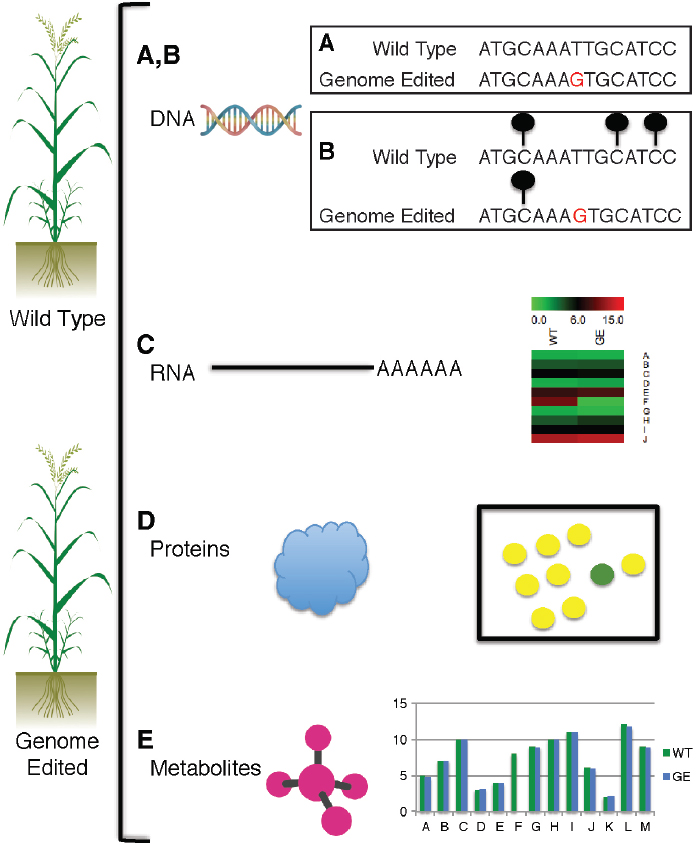
SOURCE: Illustration by C. R. Buell.
NOTE: To perform various -omics assessments of genome-edited plants, both the wild-type (unmodified) and the genome-edited plant are subjected to genome sequencing, epigenomic characterization, transcriptome profiling, proteome profiling, and metabolite profiling. A, genome sequencing is performed on both the wild-type and genome-edited accession, and differences in the DNA sequence (red G) are detected with bioinformatics methods. B, changes in the epigenome are assessed with bisulfite sequencing and chromatin immunoprecipitation with
bias will affect which sequences are present in the resequencing dataset and consequently available for alignment with the reference genome. Fourth, read-alignment algorithms fail to detect all polymorphisms if the query diverges too widely from the reference, especially with insertions and deletions or with SNPs near them. Fifth, read alignments and polymorphism detection are limited to nonrepetitive regions of the genome, so regions that are repetitive in the genome cannot be assessed for divergence. Although obstacles remain, resequencing is a powerful method for measuring differences in genome sequences between wild-type plants (normal untransformed individuals) and engineered plants. With expected improvements in technology, the resolution of resequencing to reveal differences between two genomes will improve.
Computational Approaches
Alternatives to resequencing approaches to identify polymorphisms in DNA sequence between two genomes were emerging when the committee was writing its report. The foundation of computational approaches to identify polymorphisms is algorithms that perform k-mer counting (a k-mer is a unique nucleotide sequence of a given length) in which unique k-mers are identified in two read pools (for example, wild type and mutant) and k-mers that differ between the two samples are then computationally identified. Those k-mers are then further analyzed to identify the nature of the polymorphism (SNP versus insertion or deletion) and to associate the polymorphism with a gene and potential phenotype (Nordstrom et al., 2013; Moncunill et al., 2014). The sensitivity and specificity of such programs are comparable with or better than the current methods that detect SNPs and
antibodies that target modified histones that are associated with chromatin; lollipops signify methylated cytosine residues. C, transcriptome sequencing is used to measure expression abundances in wild-type (WT) and genome-edited (GE) lines; in example shown, expression ranges from 0 to 15 for genes A through J; variance in all the genes is apparent, with only gene F showing substantial expression differences between the wild-type and the genome-edited line as would be expected in a knockout line. D, proteomics is used to measure differences in protein abundance in wild-type vs genome-edited line; all proteins are equally present in wild-type and genome-edited lines (yellow dots), whereas protein F is present only in wild-type line (green dot), as expected from a knockout line. E, levels of metabolites A through M in wild-type and genome-edited lines; levels of metabolite F are zero in contrast to the wild type, as would be expected in a knockout line.
insertions/deletions by using genome-sequencing methods and thus have the potential to identify more robustly genome variation introduced through genetic engineering. The committee expects the field to continue to develop rapidly and to enable researchers to read genomic DNA with increased sensitivity and specificity.
Utility of Transcriptomics, Proteomics, and Metabolomics in Assessing Biological Effects of Genetic Engineering
As stated in the 2004 National Research Council report Safety of Genetically Engineered Foods, understanding the composition of food at the RNA, protein, and metabolite levels is critical for determining whether genetic engineering results in a difference in substantial equivalence compared to RNA, protein, and metabolite levels in conventionally bred crops (NRC, 2004; see Chapter 5). Although the genome provides the “blueprint” for the cell, assessment of the transcriptome, proteome, and metabolome can provide information on the downstream consequences of genome changes that lead to altered phenotype. Methods used to assess transcripts, proteins, and metabolites in plants are described below with the committee’s commentary on limitations of the sensitivity and specificity of detection and interpretation that existed when this report was being written. One caveat in the use of any of these techniques is related to inherent biological variation regardless of genetic-engineering status. Even with identical genotypes grown under identical conditions, there is variation in the transcriptome, proteome, and metabolome. Scientists address such variation by using biologically replicated experiments and multiple -omics and molecular-biology approaches. In addition to biological variation, allelic variation results in different levels of transcripts, proteins, and metabolites in different accessions. To provide context to any observed changes in the transcriptome, proteome, or metabolome attributable to a genetic-engineering event, the broader range of variation in commercially grown cultivars of a crop species can be compared with that of a GE line to determine whether modified levels are outside the realm of variation in a crop. Thus, in assessment of GE crops, interpretation must be in the context of inherent biological and allelic variation of the specific crop. Assessment is also made difficult by the fact that scientists have little or no knowledge of what functions a substantial number of genes, transcripts, proteins, and metabolites perform in a plant cell.
Transcriptomics
Advancements in high-throughput sequencing technologies have enabled the development of robust methods for quantitatively measuring
the transcriptome, the expressed genes in a sample. One method, known as RNA sequencing (RNA-seq), entails isolation of RNA, conversion of the RNA to DNA, generation of sequence reads, and bioinformatic analyses to assess expression levels, alternative splicing, and alternative transcriptional initiation or termination sites (Wang et al., 2009; de Klerk et al., 2014). This method can be applied to mRNA, small RNAs (which include interfering RNAs involved in RNAi), total RNA, RNA bound to ribosomes, and RNA-protein complexes to gain a detailed assessment of RNAs in a cell. Methods to construct RNA-seq libraries, generate sequence reads, align to a reference genome, and determine expression abundances are fairly robust even with draft genome sequences if they provide nearly complete representation of the genes in the genome (Wang et al., 2009; de Klerk et al., 2014). Statistical methods to determine differential expression between any two samples, such as two plants with identical genotypes at different developmental stages, are continuing to mature but are limited by inherent biological variation in the transcriptome. Indeed, variation between independent biological replicates of wild-type tissues is well documented. For example, estimation of whole-transcriptome expression abundance in independent biological replicates of a given experimental treatment is considered to be highly reproducible if Pearson’s correlation values are more than 0.95; values greater than 0.98 are typically observed. However, even with high Pearson’s correlation values, numerous genes may exhibit different expression among biological replicates. Thus, differential gene expression in GE plants would need to be compared with the observed variation in gene expression in biological replicates of untransformed individuals to ensure the absence of major effects of the genetic-engineering event on the transcriptome.
Overshadowing any expression differences discovered between a wild-type plant and an engineered plant is the fact that little is known about the exact function of a substantial number of genes, transcripts, and proteins for any plant species. In maize, nearly one-third of the genes have no meaningful functional annotation; even when informative functional annotation is provided, the annotation was most likely assigned by using automated transitive annotation methods that depend heavily on sequence similarity. Thus, even if differentially expressed genes are detected between the wild-type and GE samples, interpreting them in the context of health or effects on the ecosystem may be challenging at best. For example, a study of the effects of expression of the antifungal protein in rice that was introduced with genetic engineering showed changes in about 0.4 percent of the transcriptome in the GE lines (Montero et al., 2011). Analysis of 20 percent of the changes indicated that 35 percent of the unintended effects could be attributed to the tissue-culture process used for plant transformation and regeneration, whereas 15 percent appeared to be event-specific and attributable to the presence of the
transgene. About 50 percent of the changes that were attributed to the presence of the transgene were in expression of genes that could be induced in the non-GE rice by wounding. It is impossible to determine whether the changes in transcript levels recorded in the study indicate that the GE rice might be worse than, equal to, or better than its non-GE counterpart as regards food safety. One way to assess the biological effects of genetic engineering on the transcriptome is to include a variety of conventionally bred cultivars in the study and determine whether the range of expression levels in the GE line falls within the range observed for the crop, but this method will not provide definitive evidence of food or ecosystem safety.
Proteomics
Several methods permit comparison of protein composition and post-translational protein modifications between samples (for review, see May et al., 2011). For example, two-dimensional difference in-gel electrophoresis permits quantitative comparison of two proteomes through differential labeling of the samples followed by separation and quantification (Figure 7-5 D). In mass spectrometry (MS), another method for examining the proteome, proteins are first broken into specific fragments (often by proteases, which are enzymes that catalyze the cleavage of proteins into peptides at specific sites) and fractionated with such techniques as liquid chromatography. Then the mass-to-charge ratios of the peptides are detected with MS. MS data typically provide a unique “signature” for each peptide, and the identity of the peptides is typically determined by using search algorithms to compare the signatures with databases of predicted peptides and proteins derived from genome or transcriptome sequence data. Differential isotope labeling can be used in the MS approach to determine quantitative differences in protein samples. One limitation of all current proteomic techniques is sensitivity; whole-proteome studies typically detect only the most abundant proteins (Baerenfaller et al., 2008). Furthermore, sample-preparation methods need to be modified to detect different fractions of the proteome (such as soluble versus membrane-bound and small versus large proteins) (Baerenfaller et al., 2008). Thus, to provide a broad assessment of the proteome, an array of sample-preparation methods must be used. Finally, as with the other -omics methods, interpretation of the significance of proteomic differences is made difficult by the fact that scientists have little knowledge of what a large number of proteins do in a plant cell.
Metabolomics
It is common practice in evaluating GE crops for regulatory approval to require targeted profiling of specific metabolites or classes of metabolites
that may be relevant to the trait being developed or that are known to be present in the target species and to be potentially toxic if present at excessive concentrations. Under current regulatory requirements, substantial metabolic equivalence is assessed on the basis of concentrations of gross macromolecules (for example, protein or fiber), such nutrients as amino acids and sugars, and specific secondary metabolites that might be predicted to cause concern.
As with genomics, transcriptomics, and proteomics, the approaches collectively known as metabolomics have been developed to determine the nature and concentrations of all metabolites in a particular organism or tissue. It has been argued that such information should be required before a GE crop clears regulatory requirements for commercialization. However, in contrast with genomic and transcriptomic approaches, with which it is now technically easy to assess DNA sequences and measure relative concentrations of most or all transcripts in an organism with current sequencing technologies respectively, metabolomics as currently performed can provide useful data only on a subset of metabolites. That is because each metabolite is chemically different, whereas DNA and RNA comprise different orderings of just four nucleotide bases. Metabolites have to be separated, usually with gas chromatography or high-performance liquid chromatography; their nature and concentrations are then determined, usually with MS. The mass spectra are compared with a standard library of chemicals run on the same analytical system. The major problem for this type of metabolomic analysis of plants is the possession in the plant kingdom of large numbers of genus-specific or even species-specific natural products (see section “Comparing Genetically Engineered Crops and Their Counterparts” in Chapter 5 for discussion of plant natural products). Advanced commercial platforms for plant metabolomics currently measure about 200 identified compounds, usually within primary metabolism, and less broadly distributed natural products are poorly represented (Clarke et al., 2013). However, these approaches can differentiate a much larger number of distinct but unidentified metabolites, and it is useful to know whether concentrations of a metabolite are specifically affected in a GE crop even if the identity of the particular metabolite is not known. For example, with a combination of separation platforms coupled to mass spectrometry, it was possible to resolve 175 unique identified metabolites and 1,460 peaks with no or imprecise metabolite annotation, together estimated to represent about 86 percent of the chemical diversity of tomato (Solanum lycopersicum) as listed in a publicly available database (Kusano et al., 2011). Although such an approach allows one to determine whether metabolite peaks are present in a GE crop but not in the non-GE counterpart or vice versa, metabolomics, in the absence of a completely defined metabolome for the target species in which the toxicity of all components is known, is not able to determine
with confidence that a GE or non-GE plant does not contain any chemically identified molecule that is unexpected or toxic.
An alternative approach to nontargeted analysis of metabolites is to perform metabolic fingerprinting and rely on statistical tools to compare GE and non-GE materials. That does not necessarily require prior separation of metabolites and can use flow-injection electrospray ionization mass spectrometry (Enot et al., 2007) or nuclear magnetic resonance (NMR) spectroscopy (Baker et al., 2006; Ward and Beale, 2006; Kim et al., 2011). NMR spectroscopy is rapid and requires no separation but depends heavily on computational and statistical approaches to interpret spectra and evaluate differences.
Generally, with a few exceptions, metabolomic studies have concluded that the metabolomes of crop plants are affected more by environment than by genetics and that modification of plants with genetic engineering typically does not bring about off-target changes in the metabolome that would fall outside natural variation in the species. Baseline studies of the metabolomes (representing 156 metabolites in grain and 185 metabolites in forage) of 50 genetically diverse non-GE DuPont Pioneer commercial maize hybrids grown at six locations in North America revealed that the environment had a much greater effect on the metabolome (affecting 50 percent of the metabolites) than did the genetic background (affecting only 2 percent of the metabolites); the difference was more striking in forage samples than in grain samples (Asiago et al., 2012). Environmental factors were also shown to play a greater role than genetic engineering on the concentrations of most metabolites identified in Bt rice (Chang et al., 2012). In soybean, nontargeted metabolomics was used to demonstrate the dynamic ranges of 169 metabolites from the seeds of a large number of conventionally bred soybean lines representing the current commercial genetic diversity (Clarke et al., 2013). Wide variations in concentrations of individual metabolites were observed, but the metabolome of a GE line engineered to be resistant to the triketone herbicide mesotrione (which targets the carotenoid pathway that leads to photobleaching of sensitive plants) did not deviate with statistical significance from the natural variation in the current genetic diversity except in the expected changes in the targeted carotenoid pathway. Similar metabolomic approaches led to the conclusion that a Monsanto Bt maize was substantially equivalent to conventionally bred maize if grown under the same environmental conditions (Vaclavik et al., 2013) and that carotenoid-fortified GE rice was more similar to its parental line than to other rice varieties (Kim et al., 2013). Those studies suggest that use of metabolomics for assessing substantial equivalence will require testing in multiple locations and careful analysis to differentiate genetic from environmental effects, especially because there will probably be effects of gene–environment interactions.
Some metabolomic and transcriptomic studies have suggested that transgene insertion or the tissue-culture process involved in regeneration of transformed plants can lead to “metabolic signatures” associated with the process itself (Kusano et al., 2011; Montero et al., 2011). That was reported for GE tomatoes with overproduction of the taste-modifying protein miraculin, although it was pointed out by the authors that, as in comparable studies with other GE crops, “the differences between the transgenic lines and the control were small compared to the differences observed between ripening stages and traditional cultivars” (Kusano et al., 2011).
For metabolomics to become a useful tool for providing enhanced safety assessment of a specific GE crop, it will be necessary to develop a chemical library that contains all potential metabolites present in the species under all possible environmental conditions. It is a daunting task that may be feasible for a few major commodity crops under currently occurring biotic and abiotic stresses, but even that would not necessarily cover future environmental conditions. Annotated libraries of metabolites are unlikely to be developed for minor crops in the near future.
The Epigenome
Background
Whereas the DNA sequence of a gene encodes the mRNA that is translated into the corresponding protein, the rate at which a gene in the nucleus of a eukaryotic cell is transcribed into mRNA can be heavily influenced by chemical modification of the DNA of the gene and by chemical modification of the proteins associated with the DNA. In plants and other eukaryotes, genomic nuclear DNA can be chemically modified and is bound to an array of proteins in a DNA–protein complex termed chromatin. The major proteins in chromatin are histone proteins, which have an important role in regulating the accessibility of the transcriptional machinery to the gene and its promoter (regulatory region) and thereby control synthesis of mRNAs and proteins. Multiple types of histone proteins are found in plants, each with an array of post-translational modification (for example, acetylation and methylation) that can affect transcriptional competence of a gene. DNA can also be covalently modified by methylation of cytosines that affect transcriptional competence. Collectively, those modifications, which influence the expression of genes and are inheritable over various time spans, are known as epigenetic marks.
Epigenetic marks are determinants of transcriptional competence, and alteration of the epigenetic state (which occurs naturally but infrequently) can alter expression profiles or patterns of target genes. For example, when a transposable element inserts in or near a gene, the gene can be “silenced”
as regions near a transposon become highly methylated and transcription-ally suppressed owing to the activity of the cell’s native RNA-mediated DNA methylation machinery. Different epigenetic marks occur naturally in crop species; examples of transposable element-mediated gene silencing include allelic variation at the tomato 2-methyl-6-phytylquinol methyltransferase gene involved in vitamin E biosynthesis (Quadrana et al., 2014) and imprinting as seen in endosperm tissue, in which differential insertion of transposable elements occurs in the maternal and paternal parents (Gehring et al., 2009).
Methods of Characterizing the Epigenome
Methods of characterizing the epigenome are available and improving rapidly. For DNA methylation, high-throughput, single-nucleotide resolution can be obtained through bisulfite sequencing (BS-seq; for review, see Feng et al., 2011; Krueger et al., 2012). BS-seq methods mirror that of genome resequencing except that the genomic DNA is first treated with bisulfite, which converts cytosines to uracils but does not affect 5-methyl-cytosine residues. As a consequence, nonmethylated cytosines will be detected as thymidines after the polymerase chain reaction step during epigenome-library construction. After sequencing, reads are aligned with a reference genome sequence, and nonmethylated cytosines are detected as SNPs and compared with a parallel library constructed from untreated DNA (see section above “Resequencing: Assessing Differences Between the Reference and Query Genome”; Figure 7-5). There are limitations of BS-seq approaches, such as incomplete conversion of cytosines, degradation of DNA, and an inability to assess the full methylome because of read mapping limitations, sequencing depth, and sequencing errors, as described above for resequencing. Another limitation is the dynamic nature of plant genome cytosine methylation. Plants derived from an identical parent that have not been subject to any traditional selection or GE transformation can have different epigenomes—an example of “epigenetic drift” (Becker et al., 2011). Thus, determining the epigenome of a plant at one specific point in time will not necessarily indicate the future epigenome of offspring of that plant.
Histone marks can be detected through chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-Seq; for review see Yamaguchi et al., 2014; Zentner and Henikoff, 2014). First, chromatin is isolated so that the proteins remain bound to the DNA. Then the DNA is sheared, and the DNA that is bound to specific histone proteins is selectively removed by using antibodies specific to each histone mark. The DNA bound to an antibody is then used to construct a library that is sequenced and aligned with a reference genome, and an algorithm is used to define the
regions of the genome in which the histone mark is found. Sensitivity and specificity of ChIP-Seq depend heavily on the specificity of the histone-mark antibodies, on technical limitations in alignment of sequence reads with the reference genome, and on the overall quality of the reference genome itself. Also, the present state of understanding does not permit robust prediction of the effects of many epigenetic modifications on gene expression, and gene expression can be more thoroughly and readily assessed by transcriptomics.
Evaluation of Crop Plants Using -Omics Technologies
The -omics evaluation methods described above hold great promise for assessment of new crop varieties, both GE and non-GE. In a tiered regulatory approach (see Chapter 9), -omics evaluation methods could play an important role in a rational regulatory framework. For example, consider the introduction of a previously approved GE trait such as a Bt protein in a new variety of the same species. Having an -omics profile in a new GE variety that is comparable to the profile of a variety already in use should be sufficient to establish substantial equivalence (Figure 7-6, Tier 1). Furthermore, -omics analyses that reveal a difference that is understood to have no adverse health effects (for example, increased carotenoid content) should be sufficient for substantial equivalence (Figure 7-6, Tier 2).
The approach described above could also be used across species. For example, once it is established that production of a protein (such as a Bt protein) in one plant species poses no health risk, then the only potential health risk of Bt expression in another species is unintended off-target effects. -Omics analyses that reveal no differences (Figure 7-6, Tier 1) or in which revealed differences present no adverse health effects (Figure 7-6, Tier 2) in comparison with the previously deregulated GE crop or the range of variation found in cultivated, non-GE varieties of the same species provide evidence for substantial equivalence. As discussed in Chapter 5 (see section “Newer Methods for Assessing Substantial Equivalence”), there have been more than 60 studies in which -omics approaches were used to compare GE and non-GE varieties, and none of these studies found differences that were cause for concern.
There are also scenarios for which -omics analyses could indicate that further safety testing is warranted, such as if -omics analyses reveal a difference that is understood to have potential adverse health effects (for example, increased expression of genes responsible for glycoalkaloid synthesis) (Figure 7-6, Tier 3). Another scenario is if -omics analyses reveal a change of a protein or metabolite for which the consequences cannot be interpreted and are outside the range observed in GE and non-GE varieties of the crop (Figure 7-6, Tier 4). It is important to note that a Tier 4 scenario is not in and of itself an indication of a safety issue. The functions
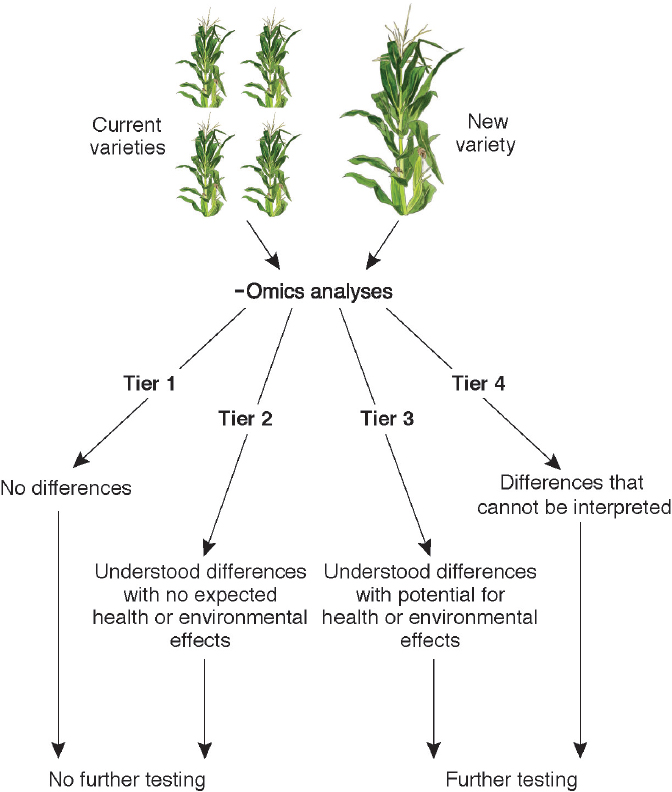
SOURCE: Illustration by R. Amasino.
NOTE: A tiered set of paths can be taken depending on the outcome of the various -omics technologies. In Tier 1, there are no differences between the variety under consideration and a set of conventionally bred varieties that represent the range of genetic and phenotypic diversity in the species. In Tier 2, differences are detected that are well understood to have no expected adverse health or environmental effects. In Tiers 3 and 4, differences are detected that may have potential health or environmental effects and thus require further safety testing.
or health effects of consumption of many genes and corresponding RNAs, proteins, and metabolites in non-GE plants are not known. Furthermore, the chemical structure of many metabolites in plants that can be detected as “peaks” in various analytical systems is not known. Substantially more basic knowledge is needed before -omics datasets can be fully interpreted.
The state of the art of the different -omics approaches varies considerably. Advances in the efficiency of DNA-sequencing technology enable a complete genome or transcriptome to be sequenced at a cost that is modest on the scale of regulatory costs. Transcriptomics could play an important role in evaluation of substantial equivalence because it is relatively straightforward to generate and compare extensive transcriptomic data from multiple biological replicates of a new crop variety versus its already-in-use progenitor. As noted above, if no unexpected differences are found, this is evidence of substantial equivalence. It is possible that two varieties with equivalent transcriptomes have a difference in the level of a metabolite due to an effect of the product of a transgene on translation of a particular mRNA or on activity of a particular protein, but these are unlikely scenarios.
It is also straightforward and relatively low in cost to generate genome-sequence data from many individuals from a new GE or non-GE variety to determine which lineage has the fewest nontarget changes to its genome. As noted earlier in the chapter, mutagenesis, although currently classified as conventional breeding, can result in extensive changes to the genome; thus generating DNA sequence data will be useful in evaluating varieties produced by this method.
Metabolomic and proteomic techniques cannot presently provide a complete catalog of the metabolome or proteome. Nevertheless, these -omics approaches can play a role in assessment. For example, a similar metabolome or proteome in a new variety compared to an existing variety provides supporting evidence of substantial equivalence, whereas a difference can indicate that further evaluation may be warranted.
The most thorough evidence of substantial equivalence would result from a complete knowledge of the biochemical constituents of one crop variety compared to other varieties. As noted above, that is not possible with present techniques for the proteome and metabolome. However, looking to the future, an increasing knowledge base of plant biochemistry will translate into fewer analyses that result in a Tier 4 situation, and basic research in plant biochemistry will continue to expand the knowledge base that will enable the thorough and rational evaluation of new crop varieties; basic research will also expand fundamental understanding of basic biological processes in plants and thus enable advances in molecular plant breeding.
FINDING: Application of -omics technologies has the potential to reveal the extent of modifications of the genome, the transcriptome, the epigenome, the proteome, and the metabolome that are attributable to conventional breeding, somaclonal variation, and genetic engineering. Full realization of the potential of -omics technologies to assess substantial equivalence would require the development of extensive species-specific databases, such as the range of variation in the transcriptome, proteome, and metabolome in a number of genotypes grown in diverse environmental conditions. Although it is not yet technically feasible to develop extensive species-specific metabolome or proteome databases, genome sequencing and transcriptome characterization can be performed.
RECOMMENDATION: To realize the potential of -omics technologies to assess intended and unintended effects of new crop varieties on human health and the environment and to improve the production and quality of crop plants, a more comprehensive knowledge base of plant biology at the systems level (DNA, RNA, protein, and metabolites) should be constructed for the range of variation inherent in both conventionally bred and genetically engineered crop species.
CONCLUSIONS
Modern plant breeding and genetic engineering are complementary methods for improving crop yield, production efficiency, and composition. Anti-sense and RNAi technologies offer capabilities that differ from simple transgene overexpression strategies. The emerging genetic-engineering technologies—such as genome editing and synthetic biology—when coupled with knowledge of the genetic basis of phenotypes enable construction of not only improved varieties of crops but crops with novel traits.
Both genetic engineering and conventional breeding have been greatly enhanced by increases in basic knowledge about plant biology and by technological innovations. Emerging genetic-engineering technologies have the potential to substantially change future crop production because of paradigm-shifting capabilities in precision, complexity, and diversity (see examples in Chapter 8). They have also developed markedly in ease of application and are poised to extend the range of individual improvements in crops—that is, endowing them with new traits (sometimes single traits but also multiple traits) and capabilities. Since the 1990s, there has been a revolution in DNA-sequencing technologies and associated technologies for detecting major molecules in cells, especially DNA and transcripts but also proteins, metabolites, and epigenetic marks. Plant breeders, geneticists, and other scientists have used -omics technologies to understand biological
function better. The breadth of understanding has improved the efficiency of conventional plant breeding and increased scientists’ ability to “read” DNA, which will be a harbinger of parallel developments in the ability to “write” DNA into organisms by using various advances in genome editing and synthetic biology. It is important that basic knowledge regarding biology continues to grow and that technology continues to improve.
In addition to contributing to crop improvement, emerging -omics methods could provide a rational pathway to developing a tiered approach for assessing health and environmental effects of new crop varieties produced by conventional breeding and genetic engineering.
REFERENCES
Ainley, W.M., L. Sastry-Dent, M.E. Welter, M.G. Murray, B. Zeitler, R. Amora, D.R. Corbin, R.R. Miles, N.L. Arnold, T.L. Strange, M.A. Simpson, Z. Cao, C. Carroll, K.S. Pawelczak, R. Blue, K. West, L.M. Rowland, D. Perkins, P. Samuel, C.M. Dewes, L. Shen, S. Sriram, S.L. Evans, E.J. Rebar, L. Zhang, P.D. Gregory, F.D. Urnov, S.R. Webb, and J.F. Petolino. 2013. Trait stacking via targeted genome editing. Plant Biotechnology Journal 11:1126–1134.
Akbari, B.O.S., H.J. Bellen, E. Bier, S.L. Bullock, A. Burt, G.M. Church, K.R. Cook, P. Duchek, O.R. Edwards, K.M. Esvelt, V.M. Gantz, K.G. Golic, S.J. Gratz, M.M. Harrison, K.R. Hayes, A.A. James, T.C. Kaufman, J. Knoblich, H.S. Malik, K.A. Matthews, K.M. O’Connor-Giles, A.L. Parks, N. Perrimon, F.Port, S. Russell, R. Ueda, and J. Wildonger. 2015. Safeguarding gene drive experiments in the laboratory. Science 349:927–929.
Annaluru, N., H. Muller, L.A. Mitchell, S. Ramalingam, G. Stracquadanio, S.M. Richardson, J.S. Dymond, Z. Kuang, L.Z. Scheifele, E.M. Cooper, Y. Cai, K. Zeller, N. Agmon, J.S. Han, M. Hadjithomas, J. Tullman, K. Caravelli, K. Cirelli, Z. Guo, V. London, A. Yeluru, S. Murugan, K. Kandevlou, N. Agier, G. Fischer, K. Yang, J.A. Martin, M. Bilgel, P. Bohutskyi, K.M. Boulier, B.J. Capaldo, J. Chang, K. Charoen, W.J. Choi, P. Deng, J.E. DiCarlo, J. Doong, J. Dunn, J.I. Feinberg, C. Fernandez, C.E. Floria, D. Gladowski, P. Hadidi, I. Ishizuka, J. Jabbari, C.Y.L. Lau, P.A. Lee, S. Li, D. Lin, M.E. Linder, J. Ling, J. Liu, M. London, H. Ma, J. Mao, J.E. McDade, A. McMillan, A.M. Moore, W.C. Oh, Y. Ouyang, R. Patel, M. Paul, L.C. Paulsen, J. Qiu, A. Rhee, M.G. Rubashkin, I.Y. Soh, N.E. Sotuyo, A. Srinivas, A. Suarez, A. Wong, R. Wong, W.R. Xie, Y. Xu, A.T. Yu, R. Koszul, J.S. Bader, J.D. Boeke, and S. Chandrasegaran. 2014. Total synthesis of a functional designer eukaryotic chromosome. Science 344:55–58.
Arabidopsis Genome Initiative. 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815.
Asiago, V.M., J. Hazebroek, T. Harp, and C. Zhong. 2012. Effects of genetics and environment on the metabolome of commercial maize hybrids: A multisite study. Journal of Agricultural and Food Chemistry 60:11498–11508.
Baerenfaller, K., J. Grossmann, M.A. Grobei, R. Hull, M. Hirsch-Hoffmann, S. Yalovsky, P. Zimmermann, U. Grossniklaus, W. Gruissem, and S. Baginsky. 2008. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science 320:938–941.
Baker, J.M., N.D. Hawkins, J.L. Ward, A. Lovegrove, J.A. Napier, P.R. Shewry, and M.H. Beale. 2006. A metabolomic study of substantial equivalence of field-grown genetically modified wheat. Plant Biotechnology Journal 4:381–392.
Baltes, N.J., and D.F. Voytas. 2014. Enabling plant synthetic biology through genome engineering. Trends in Biotechnology 33:120–131.
Becker, C., J. Hagmann, J. Muller, D. Koenig, O. Stegle, K. Borgwardt, and D. Weigel. 2011. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480:245–249.
Bennett, M.D., I.J. Leitch, H.J. Price, and J.S. Johnston. 2003. Comparisons with Caenorhabditis (approximately 100 Mb) and Drosophila (approximately 175 Mb) using flow cytometry show genome size in Arabidopsis to be approximately 157 Mb and thus approximately 25% larger than the Arabidopsis genome initiative estimate of approximately 125 Mb. Annals of Botany 91:547–557.
Birchler, J.A. 2015. Promises and pitfalls of synthetic chromosomes in plants. Trends in Biotechnology 33:189–194.
Boch, J., H. Scholze, S. Schornack, A. Landgraf, S. Hahn, S. Kay, T. Lahaye, A. Nickstadt, and U. Bonas. 2009. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512.
Büschges, R., K. Hollricher, R. Panstruga, G. Simons, M. Wolter, A. Frijters, R. van Daelen, T. van der Lee, P. Diergaarde, J. Groenendijk, S. Töpsch, P. Vos, F. Salamini, and R. Schulze-Lefert. 1997. The barley Mlo gene: A novel control element of plant pathogen resistance. Cell 88:695–705.
Chang, Y., C. Zhao, Z. Zhu, Z. Wu, J. Zhou, Y. Zhao, X. Lu, and G. Xu. 2012. Metabolic profiling based on LC/MS to evaluate unintended effects of transgenic rice with cry1Ac and sck genes. Plant Molecular Biology 78:477–487.
Chia, J.M., C. Song, P.J. Bradbury, D. Costich, N. de Leon, J. Doebley, R.J. Elshire, B. Gaut, L. Geller, J.C. Glaubitz, M. Gore, K.E. Guill, J. Holland, M.B. Hufford, J. Lai, M. Li, X. Liu, Y. Lu, R. McCombie, R. Nelson, J. Poland, B.M. Prasanna, T. Pyhäjärvi, T. Rong, R.S. Sekhon, Q. Sun, M.I. Tenaillon, F. Tian, J. Wang, X. Xu, Z. Zhang, S.M. Kaeppler, J. Ross-Ibarra, M.D. McMullen, E.S. Buckler, G. Zhang, Y. Xu, and D. Ware. 2012. Maize HapMap2 identifies extant variation from a genome in flux. Nature Genetics 44:803–807.
Christou, P. 1992. Genetic transformation of crop plants using microprojectile bombardment. Plant Journal 2:275–281.
Clarke, J.D., D.C. Alexander, D.P. Ward, J.A. Ryals, M.W. Mitchell, J.E. Wulff, and L. Guo. 2013. Assessment of genetically modified soybean in relation to natural variation in the soybean seed metabolome. Scientific Reports 3:3082.
Clasen, B.M., T.J. Stoddard, S. Luo, Z.L. Demorest, J. Li, F. Cedrone, R. Tibebu, S. Davison, E.E. Ray, A. Daulhac, A. Coffman, A. Yabandith, A. Retterath, W. Haun, N.J. Baltes, L. Mathis, D.F. Voytas, and F. Zheng. 2016. Improved cold storage and processing traits in potato through targeted gene knockout. Plant Biotechnology Journal 14:169–176.
Clough, S.J., and A.F. Bent. 1998. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant Journal 16:735–743.
Comis, D. 2005. TILLING genes to improve soybeans. Agricultural Research 53:4–5.
Daniell, H. 2002. Molecular strategies for gene containment in transgenic crops. Nature Biotechnology 20:581–586.
De Cosa, B., W. Moar, S.B. Lee, M. Miller, and H. Daniell. 2001. Overexpression of the Bt cry2Aa2 operon in chloroplasts leads to formation of insecticidal crystals. Nature Biotechnology 19:71–74.
de Klerk, E., J.T. den Dunnen, and P.A.C. ‘t Hoen. 2014. RNA sequencing: From tag-based profiling to resolving complete transcript structure. Cellular and Molecular Life Sciences 71:3537–3551.
Doudna, J.A., and E. Charpentier. 2014. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096.
Ellis, T., T. Adie, and G.S. Baldwin. 2011. DNA assembly for synthetic biology: From parts to pathways and beyond. Integrative Biology: Quantitative Biosciences from Nano to Macro 3:109–118.
Enot, D., M. Beckmann, and J. Draper. 2007. Detecting a difference—assessing generalisability when modelling metabolome fingerprint data in longer term studies of genetically modified plants. Metabolomics 3:335–347.
Esvelt, K.M., A.L. Smidler, F. Catteruccia, and G.M. Church. 2014. Emerging technology: Concerning RNA-guided gene drives for the alteration of wild populations. eLife 3:e03401.
Farhi, M., E. Marhevka, J. Ben-Ari, A. Algamas-Dimantov, Z. Liang, V. Zeevi, O. Edelbaum, B. Spitzer-Rimon, H. Abeliovich, B. Schwartz, T. Tzfira, and A. Vainstein. 2011. Generation of the potent anti-malarial drug artemisinin in tobacco. Nature Biotechnology 29:1072–1074.
Feng, S., L. Rubbi, S.E. Jacobsen, and M. Pellegrini. 2011. Determining DNA methylation profiles using sequencing. Methods in Molecular Biology 733:223–238.
Fine, E.J., T.J. Cradick, C.L. Zhao, Y. Lin, and G. Bao. 2014. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Research 42:e42.
Fire, A., S. Xu, M.K. Montgomery, S.A. Kostas, S.E. Driver, and C.C. Mello. 1998. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811.
Fleischmann, R.D., M.D. Adams, O. White, R.A. Clayton, E.F. Kirkness, A.R. Kerlavage, C.J. Bult, J.F. Tomb, B.A. Dougherty, J.M. Merrick, K. McKenney, G. Sutton, W. Fitzhugh, C. Fields, J.D. Gocayne, J. Scott, R. Shirley, L.I. Liu, A. Glodeck, J.M. Kelley, J.F. Weidman, C.A. Phillips, T. Spriggs, E. Hedblom, M.D. Cotton, T.R. Utterback, M.C. Hanna, D.T. Nguyen, D.M. Saudek, R.C. Brandon, L.D. Fine, J.L. Fritchman, J.L. Fuhrmann, N.S.M. Geoghagen, C.L. Gnehn, L.A. McDonald, K.V. Small, C.M. Fraser, H.O. Smith, and J.C. Venter. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512.
Fraser, C.M., J.D. Gocayne, O. White, M.D. Adams, R.A. Clayton, R.D. Fleischmann, C.J. Butt, A.R. Kerlavge, G. Sutton, J.M. Kelley, J.L. Fritchman, J.F. Weidman, K.V. Small, M. Sandusky, J. Fuhrmann, D. Nguyen, T.R. Utterback, D.M. Saudek, C.A. Phillips, J.M. Merrick, J.F. Tomb, B.A. Dougherty, K.F. Bott, P.C. Hu, T.S. Lucier, S.N. Peterson, H.O. Smith, C.A. Hutchison, and J.C. Venter. 1995. The minimal gene complement of Mycoplasma genitalium. Science 270:397–403.
Gaeta, R.T., R.E. Masonbrink, L. Krishnaswamy, C. Zhao, and J.A. Birchler. 2012. Synthetic chromosome platforms in plants. Annual Review of Plant Biology 63:307–330.
Gantz, V.M., and E. Bier. 2015. The mutagenic chain reaction: A method for converting heterozygous to homozygous mutations. Science 348:442–444.
Gehring, M., K.L. Bubb, and S. Henikoff. 2009. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324:1447–1451.
Gibson, D.G., J.I. Glass, C. Lartigue, V.N. Noskov, R.Y. Chuang, M.A. Algire, G.A. Benders, M.G. Montague, L. Ma, M.M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N.Assad-Garcia, C. Andrews-Pfannkoch, E.A. Denisova, L. Young, Z.-Q. Qi, T.H. Segall-Shapiro, C.H. Calvey, P.P. Parmar, C.A. Hutchinson III, H.O. Smith, and J.C. Venter. 2010. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329:52–56.
Graham, I.A., K. Besser, S. Blumer, C.A. Branigan, T. Czechowski, L. Elias, I. Guterman, D. Harvey, P.G. Isaac, A.M. Khan, T.R. Larson, Y. Li, T. Pawson, T. Penfield, A.M. Rae, D.A. Rathbone, S. Reid, J. Ross, M.F. Smallwood, V. Segura, T. Townsend, D. Vyas, T. Winzer, and D. Bowles. 2010. The genetic map of Artemisia annua L. identifies loci affecting yield of the antimalarial drug artemisinin. Science 327:328–331.
Haroldsen, V.M., M.W. Szcerba, H. Aktas, J. Lopez-Baltazar, M.J. Odias, C.L. Chi-Ham, J.M. Labavitch, A.B. Bennett, and A.L.T. Powell. 2012. Mobility of transgenic nucleic acids and proteins within grafted rootstocks for agricultural improvement. Frontiers in Plant Science 3:39.
Heigwer, F., G. Kerr, and M. Boutros. 2014. E-CRISP: Fast CRISPR target site identification. Nature Methods 11:122–123.
Henikoff, S., B.J. Till, and L. Comai. 2004. TILLING. Traditional mutagenesis meets functional genomics. Plant Physiology 135:630–636.
Hirsch, C.N., J.M. Foerster, J.M. Johnson, R.S. Sekhon, G. Muttoni, B. Vaillancourt, F. Penagaricano, E. Lindquist, M.A. Pedraza, K. Barry, N. de Leon, S.M. Kaeppler, and C.R. Buell. 2014. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 26:121–135.
Holme, I.B., T. Wendt, and P.B. Holm. 2013. Intragenesis and cisgenesis as alternatives to crop development. Plant Biotechnology Journal 11:395–407.
Huang, X., X. Wei, T. Sang, Q. Zhao, Q. Feng, Y. Zhao, C. Li, C. Zhu, T. Lu, Z. Zhang, M. Li, D. Fan, Y. Guo, A. Wang, L. Wang, L. Deng, W. Li, Y. Lu, Q. Weng, K. Liu, T. Huang, T. Zhou, Y. Jing, W. Li, Z. Lin, E.S. Buckler, Q. Qian, Q.-F. Zhang, J. Li, and B. Han. 2010. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics 42:961–967.
Hufford, M.B., X. Xu, J. van Heerwaarden, T. Pyhäjärvi, J.M. Chia, R.A. Cartwright, R.J. Elshire, J.C. Glaubitz, K.E. Guill, S.M. Kaeppler, J. Lai, P.L. Morrell, L.M. Shannon, C. Song, N.M. Spring, R.A. Swanson-Wagner, P. Tiffin, J. Wang, G. Zhang, J. Doebley, M.D. McMullen, D. Ware, E.S. Buckler, S. Yang, and J. Ross-Ibarra. 2012. Comparative population genomics of maize domestication and improvement. Nature Genetics 44:808–811.
International Barley Genome Sequencing. 2012. A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716.
Jander, G., S.R. Baerson, J.A. Hudak, K.A. Gonzalez, K.J. Gruys, and R.L. Last. 2003. Ethylmethanesulfonate saturation mutagenesis in Arabidopsis to determine frequency of herbicide resistance. Plant Physiology 131:139–146.
Jiang, C., A. Mithani, X. Gan, E.J. Belfield, J.P. Klingler, J.K. Zhu, J. Ragoussis, R. Mott, and N.P. Harberd. 2011. Regenerant Arabidopsis lineages display a distinct genome-wide spectrum of mutations conferring variant phenotypes. Current Biology 21:1385–1390.
Jiao, Y., H. Zhao, L. Ren, W. Song, B. Zeng, J. Guo, B. Wang, Z. Liu, J. Chen, W. Li, M. Zhang, S. Xie, and J. Lai. 2012. Genome-wide genetic changes during modern breeding of maize. Nature Genetics 44:812–815.
Jin, S., and H. Daniell. 2015. Engineered chloroplast genome just got smarter. Trends in Plant Science 20:622–640.
Jin, S., N.D. Singh, L. Li, X. Zhang, and H. Daniell. 2015. Engineered chloroplast dsRNA silences cytochrome p450 monooxygenase, V-ATPase and chitin synthase genes in the insect gut and disrupts Helicoverpa armigera larval development and pupation. Plant Biotechnology Journal 13:435–446.
Kim, H.K., Y.H. Choi, and R. Verpoorte. 2011. NMR-based plant metabolomics: Where do we stand, where do we go? Trends in Biotechnology 29:267–275.
Kim, J., S.-Y. Park, S. Lee, S.-H. Lim, H. Kim, S.-D. Oh, Y. Yeo, H. Cho, and S.-H. Ha. 2013. Unintended polar metabolite profiling of carotenoid-biofortified transgenic rice reveals substantial equivalence to its non-transgenic counterpart. Plant Biotechnology Reports 7:121–128.
Kloosterman, B., J.A. Abelenda, M. Gomez Mdel, M. Oortwijn, J.M. de Boer, K. Kowitwanich, B.M. Horvath, H.J. van Eck, C. Smaczniak, S. Prat, R.G.F. Visser, and C.W.B. Bachem. 2013. Naturally occurring allele diversity allows potato cultivation in northern latitudes. Nature 495:246–250.
Kramer, M.G., and K. Redenbaugh. 1994. Commercialization of a tomato with an antisense polygalacturonase gene: The FLAVR SAVR™ tomato story. Euphytica 79:293–297.
Krieger, E.K., E. Allen, L.A. Gilbertson, J.K. Roberts, W. Hiatt, and R.A. Sanders. 2008. The Flavr Savr tomato, an early example of RNAi technology. HortScience 43:962–964.
Krueger, F., B. Kreck, A. Franke, and S.R. Andrews. 2012. DNA methylome analysis using short bisulfite sequencing data. Nature Methods 9:145–151.
Kusano, M., H. Redestig, T. Hirai, A. Oikawa, F. Matsuda, A. Fukushima, M. Arita, S. Watanabe, M. Yano, K. Hiwasa-Tanase, H. Ezura, and K. Saito. 2011. Covering chemical diversity of genetically-modified tomatoes using metabolomics for objective substantial equivalence assessment. PLoS ONE 6:e16989.
Lai, J., R. Li, X. Xu, W. Jin, M. Xu, H. Zhao, Z. Xiang, W. Song, K. Ying, M. Zhang, Y. Jiao, P. Ni, J. Zhang, D. Li, X. Guo, K. Ye, M. Kian, B. Wang, H. Zheng, H. Liang, X. Zhang, S. Wang, S. Chen, J. Li, Y. Fu, N.M. Springer, H. Yang, J. Wang, J. Dai. P.S. Schnable, and J. Wang. 2010. Genome-wide patterns of genetic variation among elite maize inbred lines. Nature Genetics 42:1027–1030.
Lam, H.-M., X. Xu, X. Liu, W. Chen, G. Yang, F.-L. Wong, M.-W. Li, W. He, N. Qin, B. Wang, J. Li, M. Jian, J. Wang, G. Shao, J. Wang, S.S.-M. Sun, and G. Zhang. 2010. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nature Genetics 42:1053–1059.
Lander, E.S., L.M. Linton, B. Birren, C. Nusbaum, M.C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J.P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J.C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R.H. Waterston, R.K. Wilson, L.D. Hillier, J.D. McPherson, M.A. Marra, E.R. Mardis, L.A. Fulton, A.T. Chinwalla, K.H. Pepin, W.R. Gish, S.L. Chissoe, M.C. Wendl, K.D. Delehaunty, T.L. Miner, A. Delehaunty, J.B. Kramer, L.L. Cook, R.S. Fulton, D.L. Johnson, P.J. Minx, S.W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J.-F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R.A. Gibbs, D.M. Muzny, S.E. Scherer, J.B. Bouck, E.J. Sodergren, K.C. Worley, C.M. Rives, J.H. Gorrell, M.L. Metzker, S.L. Naylor, R.S. Kucherlapati, D.L. Nelson, G.M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, D.R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H.M. Lee, J. Dubois, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R.W. Davis, N.A. Federspiel, A.P. Abola, M.J. Proctor, B.A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W.R. McCombie, M. de la Bastide, N. Dedhia, H. Blöcker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J.A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D.G. Brown, C.B. Burge, L. Cerutti, H.-C. Chen, D. Church, M. Clamp, R.R. Copley, T. Doerks, S.R. Eddy, E.E. Eichler, T.S. Furey, J. Galagan, J.G.R. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L.S. Johnson, T.A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W.J. Kent, P. Kitts, E.V. Koonin, I. Korf, D. Kulp, D. Lancet, T.M. Lowe, A. McLysaght, T. Mikkelsen, J.V. Moran, N. Mulder, V.J. Pollara, C.P. Ponting, G. Schuler, J. Schultz, G. Slater, A.F.A. Smit, E. Stupka, J. Szustakowki, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y.I. Wolf,
K.H. Wolfe, S.-P. Yang, R.-F. Yeh, F. Collins, M.S. Guyer, J. Peterson, A. Felsenfeld, K.A. Wetterstrand, R.M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D.R. Cox, M.V. Olson, R. Kaul, C. Raymond, N. Shimizu, K. Kawasaki, S. Minoshima, G.A. Evans, M. Athanasiou, R. Schultz, A. Patrinos, and M.J. Morgan. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921.
Lees, G.L. 1992. Condensed tannins in some forage legumes: Their role in the prevention of ruminant pasture bloat. Pp. 915–934 in Plant Polyphenols, R.W. Hemingway and P.E. Laks, eds. New York: Plenum Press.
Li, F., G. Fan, K. Wang, F. Sun, Y. Yuan, G. Song, Q. Li, Z. Ma, C. Lu, C. Zou, W. Chen, X. Liang, H. Shang, W. Liu, C. Shi, G. Xiao, G. Gou, W. Ye, X. Xu, X. Zhang, H. Wei, Z. Li, G. Zhang, J. Wang, K. Liu, R.J. Kohel, R.G. Percy, J.Z. Yu, Y.-X. Zhu, J. Wang, and S. Yu. 2014a. Genome sequence of the cultivated cotton Gossypium arboreum. Nature Genetics 46:567–572.
Li, Y.-H., G. Zhou, J. Ma, W. Jiang, L.-G. Jin, Z. Zhang, Y. Guo, J. Zhang, Y. Sui, L. Zheng, S.-S. Zhang, Q. Zuo. X.-H. Shi, Y.-F. Li, W.-K. Zhang, Y. Hu, G. Kong, H-L. Hong, B. Tan, J. Song, Z.-X. Liu, Y. Wang, H. Ruan, C.K.L. Yeung, J. Liu, H. Wang, L.-J. Zhang, R.-X. Guan, K.-J. Wang, W.-B. Li, S.-Y. Chen, R.-Z. Chang, Z. Jiang, S.A. Jackson, R. Li, and L.-J. Qiu. 2014b. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nature Biotechnology 32:1045–1052.
Lin, T., G. Zhu, J. Zhang, X. Xu, Q. Yu, Z. Zheng, Z. Zhang, Y. Lun, S. Li, X. Wang, Z. Huang, J. Li, C. Zhang, T. Wang, Y. Zhang, A. Wang, Y. Zhang, K. Lin, C. Li, G. Xiong, Y. Xue, A. Mazzucato, M. Causse, Z. Fei, J.J. Giovannoni, R.T. Chetelat, D. Zamir, T. Städler, J. Li, Z. Ye, Y. Du, and S. Huang. 2014. Genomic analyses provide insights into the history of tomato breeding. Nature Genetics 46:1220–1226.
Liu, W., M.R. Rudis, Y. Peng, M. Mazarei, R.J. Millwood, J.P. Yang, W. Xu, J.D. Chesnut, and C.N. Stewart, Jr. 2014. Synthetic TAL effectors for targeted enhancement of transgene expression in plants. Plant Biotechnology Journal 12:436–446.
Liu, X., J. Brost, C. Hutcheon, R. Guilfoil, A.K. Wilson, S. Leung, C.K. Shewmaker, S. Rooke, T. Nguyen, J. Kiser, and J. De Rocher. 2012. Transformation of the oilseed crop Camelina sativa by Agrobacterium-mediated floral dip and simple large-scale screening of transformants. In Vitro Cellular & Development Biology-Plant 48:462–468.
Maliga, P. 2003. Progress towards commercialization of plastid transformation technology. Trends in Biotechnology 21:20–28.
May, C., F. Brosseron, P. Chartowski, C. Schumbrutzki, B. Schoenebeck, and K. Marcus. 2011. Instruments and methods in proteomics. Methods in Molecular Biology 696:3–26.
McPherson, J.D. 2014. A defining decade in DNA sequencing. Nature Methods 11:1003–1005.
Moncunill, V., S. Gonzalez, S. Bea, L.O. Andrieux, I. Salaverria, C. Royo, L. Martinez, M. Puiggros, M. Segura-Wang, A.M. Stutz, A. Navarro. R. Royo, J.L. Gelpí, I.G. Gut, C. López-Otín, J.O. Korbel, E. Campo, X.S. Puente, and D. Torrents. 2014. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nature Biotechnology 32:1106–1112.
Montero, M., A. Coll, A. Nadal, J. Messeguer, and M. Pla. 2011. Only half the transcriptomic differences between resistant genetically modified and conventional rice are associated with the transgene. Plant Biotechnology Journal 9:693–702.
Moscou, M.J., and A.J. Bogdanove. 2009. A simple cipher governs DNA recognition by TAL effectors. Science 326:1501.
Naito, Y., K. Hino, H. Bono, and K. Ui-Tei. 2014. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 31:1120–1123.
Napoli, C., C. Lemieux, and R. Jorgensen. 1990. Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 2:279–289.
Nordstrom, K.J., M.C. Albani, G.V. James, C. Gutjahr, B. Hartwig, F. Turck, U. Paszkowski, G. Coupland, and K. Schneeberger. 2013. Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nature Biotechnology 31:325–330.
NRC (National Research Council). 2004. Safety of Genetically Engineered Foods: Approaches to Assessing Unintended Health Effects. Washington, DC: National Academies Press.
Perry, J.A., T.L. Wang, T.J. Welham, S. Gardner, J.M. Pike, S. Yoshida, and M. Parniske. 2003. A TILLING reverse gentics tool and a web-accessible collection of mutants of the legume Lotus japonicus. Plant Physiology 131:866–871.
Potato Genome Sequencing Consortium. 2011. Genome sequence and analysis of the tuber crop potato. Nature 475:189–195.
Quadrana, L., J. Almeida, R. Asis, T. Duffy, P.G. Dominguez, L. Bermudez, G. Conti, J.V. Correa da Silva, I.E. Peralta, V. Colot, S. Asurmendi, A.R. Fernie, M. Rossi, and F. Carrari. 2014. Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nature Communications 5:3027.
Rommens, C. 2004. All-native DNA transformation: A new approach to plant genetic engineering. Trends in Plant Science 9:457–464.
Ru, S., D. Main, and C. Peace. 2015. Current applications, challenges, and perspectives of marker-assisted seedling selection in Rosaceae tree fruit breeding. Tree Genetics & Genomics 11:8.
Sander, J.D., and J.K. Joung. 2014 CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology 32:347–355.
Schnable, P.S., D. Ware, R.S. Fulton, J.C. Stein, F.S. Wei, S. Pasternak, C.Z. Liang, J.W. Zhang, L. Fulton, T.A. Graves, P. Minx, A.D. Reily, L. Courtney, S.S. Kruchowski, C. Tomlinson, C. Strong, K. Delehaunty, C. Fronick, B. Courtney, S.M. Rock, E. Belter, F. Du, K. Kim, R.M. Abbott, M. Cotton, A. Levy, P. Marchetto, K. Ochoa, S.M. Jackson, B. Gillam, W. Chen, L. Yan, J. Higginbotham, M. Cardenas, J. Waligorski, E. Applebaum, L. Phelps, J. Falcone, K. Kanchi, T. Thane, A. Scimone, N. Thane, J. Henke, T. Wang, J. Ruppert, N. Shah, K. Rotter, J. Hodges, E. Ingenthron, M. Cordes, S. Kohlberg, J. Sgro, B. Delgado, K. Mead, A. Chinwalla, S. Leonard, K. Crouse, K. Collura, D. Kudrna, J. Currie, R. He, A. Angelova, S. Rajasekar, T. Mueller, R. Lomeli, G. Scara, A. Ko, K. Delaney, M. Wissotski, G. Lopez, D. Campos, M. Braidotti, E. Ashley, W. Golser, H.R. Kim, S. Lee, J. Lin, Z. Dujmic, W. Kim, J. Talag, A. Zuccolo, C. Fan, A. Sebastian, M. Kramer, L. Spiegel, L. Nascimento, T. Zutavern, B. Miller, C. Ambroise, S. Muller, W. Spooner, A. Narechania, L. Ren, S. Wei, S. Kumari, B. Faga, M.J. Levy, L. McMahan, P. Van Buren, M.W. Vaughn, K. Ying, C.-T. Yeh, S.J. Emrich, Y. Jia, A. Kalyanaraman, A.-P. Hsia, W.B. Barbazuk, R.S. Baucom, T.P. Brutnell, N.C. Carpita, C. Chaparro, J.-M. Chia, J.-M. Deragon, J.C. Estill, Y. Fu, J.A. Jeddeloh, Y. Han, H. Lee, P. Li, D.R. Lisch, S. Liu, Z. Liu, D. Holligan Nagel, M.C. McCann, P. SanMiguel, A.M. Myers, D. Nettleton, J. Nguyen, B.W. Penning, L. Ponnala, K.L. Schneider, D.C. Schwartz, A. Sharma, C. Soderlund, N.M. Springer, Q. Sun, H. Wang, M. Waterman, R. Westerman, T.K. Wolfgruber, L. Yang, Y. Yu, L. Zhang, S. Zhou, Q. Zhu, J.L. Bennetzen, R.K. Dawe, J. Jiang, N. Jiang, G.G. Presting, S.R. Wessler, S. Aluru, R.A. Martienssen, S.W. Clifton, W.R. McCombie, R.A. Wing, and R.K. Wilson. 2009. The B73 maize genome: Complexity, diversity, and dynamics. Science 326:1112–1115.
Schouten, H.J., F.A. Krens, and E. Jacobsen. 2006. Cisgenic plants are similar to traditionally bred plants. EMBO Reports 7:750–753.
Silva, G., L. Poirot, R. Galetto, J. Smith, G. Montoya, P. Duchateau, and F. Paques. 2011. Meganucleases and other tools for targeted genome engineering: Perspectives and challenges for gene therapy. Current Gene Therapy 11:11–27.
Slade, A.J., and V.C. Knauf. 2005. TILLING moves beyond functional genomics into crop improvement. Transgenic Research 14:109–115.
Stroud, H., B. Ding, S.A. Simon, S. Feng, M. Bellizzi, M. Pellegrini, G.L. Wang, B.C. Meyers, and S.E. Jacobsen. 2013. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2:e00354.
Svab, Z., P. Hajdukiewicz, and P. Maliga. 1990. Stable transformation of plastids in higher plants. Proceedings of the National Academy of Sciences of the United States of America 87:8526–8530.
Tsai, S.Q., Z. Zheng, N.T. Nguyen, M. Liebers, V.V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A.J. Iafrate, L.P. Le, M.J. Aryee, and J.K. Joung. 2015. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology 33:187–197.
Urnov, F.D., E.J. Rebar, M.C. Holmes, H.S. Zhang, and P.D. Gregory. 2010. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics 11:636–646.
Vaclavik, L., J. Ovesna, L. Kucera, J. Hodek, K. Demnerova, and J. Hajslova. 2013. Application of ultra-high performance liquid chromatography-mass spectrometry (UHPLC-MS) metabolomic fingerprinting to characterise GM and conventional maize varieties. Czech Journal of Food Science 31:368–375.
Venter, J.C., M.D. Adams, E.W. Myers, P.W. Li, R.J. Mural, G.G. Sutton, H.O. Smith, M. Yandell, C.A. Evans, R.A. Holt, J.D. Gocayne, P. Amanatides, R.M. Ballew, D.H. Huson, J.R. Wortman, Q. Zhang, C.D. Kodira, X.Q.H. Zheng, L. Chen, M. Skupski, G. Subramanian, P.D. Thomas, Z.H. Zhang, G.L.G. Miklos, C. Nelson, S. Broder, A.G. Clark, C. Nadeau, V.A. McKusick, N. Zinder, A.J. Levine, R.J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z.M. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A.E. Gabrielian, W. Gan, W.M. Ge, F.C. Gong, Z.P. Gu, P. Guan, T.J. Heiman, M.E. Higgins, R.R. Ji, Z.X. Ke, K.A. Ketchum, Z.W. Lai, Y.D. Lei, Z.Y. Li, J.Y. Li, Y. Liang, X.Y. Lin, F. Lu, G.V. Merkulov, N. Milshina, H.M. Moore, A.K. Naik, V.A. Narayan, B. Neelam, D. Nusskern, D.B. Rusch, S. Salzberg, W. Shao, B.X. Shue, J.T. Sun, Z.Y. Wang, A.H. Wang, X. Wang, J. Wang, M.H. Wei, R. Wides, C.L. Xiao, C.H. Yan, A. Yao, J. Ye, M. Zhan, W. Q. Zhang, H.Y. Zhang, Q. Zhao, L.S. Zheng, F. Zhong, W.Y. Zhong, S.P.C. Zhu, S.Y. Zhao, D. Gilbert, S. Baumhueter, G. Spier, C. Carter, A. Cravchik, T. Woodage, F. Ali, H.J. An, A. Awe, B. Baldwin, H. Baden, M. Barnstead, I. Barrow, K. Beeson, D. Busam, A. Carver, A. Center, M.L. Cheng, L. Curry, S. Danaher, L. Davenport, R. Desilets, S. Dietz, K. Dodson, L. Doup, S. Ferriera, N. Garg, A. Gluecksmann, B. Hart, J. Haynes, C. Haynes, C. Heiner, S. Hladun, D. Hostin, J. Houck, T. Howland, C. Ibegwam, J. Johnson, F. Kalush, L. Kline, S. Koduru, A. Love, F. Mann, D. May, S. McCawley, T. McIntosh, I. McMullen, M. Moy, L. Moy, B. Murphy, K. Nelson, C. Pfannkoch, E. Pratts, V. Puri, H. Qureshi, M. Reardon, R. Rodriguez, Y.H. Rogers, D. Romblad, B. Ruhfel, R. Scott, C. Sitter, M. Smallwood, E. Stewart, R. Strong, E. Suh, R. Thomas, N.N. Tint, S. Tse, C. Vech, G. Wang, J. Wetter, S. Williams, M. Williams, S. Windsor, E. Winn-Deen, K. Wolfe, J. Zaveri, K. Zaveri, J.F. Abril, R. Guigo, M.J. Campbell, K.V. Sjolander, B. Karlak, A. Kejariwal, H.Y. Mi, B. Lazareva, T. Hatton, A. Narechania, K. Diemer, A. Muruganujan, N. Guo, S. Sato, V. Bafna, S. Istrail, R. Lippert, R. Schwartz, B. Walenz, S. Yooseph, D. Allen, A. Basu, J. Baxendale, L. Blick, M. Caminha, J. Carnes-Stine, P. Caulk, Y.H. Chiang, M. Coyne, C. Dahlke, A.D. Mays, M. Dombroski, M. Donnelly, D. Ely, S. Esparham, C. Fosler, H. Gire, S. Glanowski, K. Glasser, A. Glodek, M. Gorokhov, K. Graham, B. Gropman, M. Harris, J. Heil, S. Henderson, J. Hoover, D. Jennings, C. Jordan, J. Jordan, J. Kasha, L. Kagan, C. Kraft, A.
Levitsky, M. Lewis, X.J. Liu, J. Lopez, D. Ma, W. Majoros, J. McDaniel, S. Murphy, M. Newman, T. Nguyen, N. Nguyen, M. Nodell, S. Pan, J. Peck, M. Peterson, W. Rowe, R. Sanders, J. Scott, M. Simpson, T. Smith, A. Sprague, T. Stockwell, R. Turner, E. Venter, M. Wang, M.Y. Wen, D. Wu, M. Wu, A. Xia, A. Zandieh, and X.H. Zhu. 2001. The sequence of the human genome. Science 291:1304–1351.
Visker, M.H., L.C. Keizer, H.J. Van Eck, E. Jacobsen, L.T. Colon, and P.C. Struik. 2003. Can the QTL for late blight resistance on potato chromosome 5 be attributed to foliage maturity type? Theoretical and Applied Genetics 106:317–325.
Voytas, D.F., and C. Gao. 2014. Precision genome engineering and agriculture: Opportunities and regulatory challenges. PLoS Biology 12:e1001877.
Wang, Y., X. Cheng, Q. Shan, Y. Zhang, J. Liu, C. Gao, and J.L. Qiu. 2014. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nature Biotechnology 32:947–951.
Wang, Z., M. Gerstein, and M. Snyder. 2009. RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics 10:57–63.
Ward, J.L., and M.H. Beale. 2006. NMR spectroscopy in plant metabolomics. Pp. 81–91 in Plant Metabolomics, K. Saito, R.A. Dixon, and L. Willmitzer, eds. Berlin: Springer-Verlag.
Weigel, D., and R. Mott. 2009. The 1001 genomes project for Arabidopsis thaliana. Genome Biology 10:107.
Weil, C.F. 2009. TILLING in grass species. Plant Physiology 149:158–164.
Woo, J.W., J. Kim S.I. Kwon, C. Corvalán, S.W. Cho, H. Kim, S.-G. Kim, S.-T. Kim, S. Choe, and J.-S.Kim. 2015. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology 33:1159–1161.
Yamaguchi, N., C.M. Winter, M.F. Wu, C.S. Kwon, D.A. William, and D. Wagner. 2014. PROTOCOLS: Chromatin immunoprecipitation from Arabidopsis tissues. The Arabidopsis Book/American Society of Plant Biologists 12:e0170.
Zentner, G.E., and S. Henikoff. 2014. High-resolution digital profiling of the epigenome. Nature Reviews Genetics 15:814–827.
Zetsche, B., J.S. Gootenberg, O.O. Abudayyeh, I.M. Slaymaker, K.S. Makarova, P. Essletzbichler, S.E. Volz, J. Joung, J. van der Oost, A. Regev, and E.V. Koonin. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771.
Zhang, J., D. Guo, Y. Chang, C. You, X. Li, X. Dai, Q. Weng, J. Zhang, G. Chen, X. Li, H. Liu, B. Han, Q. Zhang, and C. Wu. 2007. Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analyzing 13 804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant Journal 49:947–959.
Zhang, J., S.A. Khan, C. Hasse, S. Ruf, D.G. Heckel, and R. Bock. 2015. Pest control. Full crop protection from an insect pest by expression of long double-stranded RNAs in plastids. Science 347:991–994.




















































