
2
Skin and Musculoskeletal Tissues
Autologous and allogeneic skin therapies have a long history, from skin grafts that were performed more than 4,000 years ago to modern-day hair transplants, said Anthony Oro, a professor of dermatology at Stanford University. Remarkable advances have been made in the ability to grow healthy skin and musculoskeletal tissues for potential use in the
treatment of chronic wounds; neuromuscular diseases; trauma to bone, cartilage, tendons, or ligaments; and bone tumors. While promising, many of these approaches have faced roadblocks throughout the course of their research and development, and they still have not become routine in clinical care. Speakers in this panel discussed the significant clinical needs for patients with musculoskeletal and skin diseases, outlined the challenges facing research and patients, and described potential ways forward for the field.
CELL THERAPIES IN SKIN
Skin is composed of many types of cells, including keratinocytes, hair follicle cells, melanocytes, and fat, among numerous others, which together act to provide a barrier to the outside world. Because skin is externally accessible, it is fairly straightforward to perform autologous transplants from one part of the body to another, Oro said. However, if there is not sufficient tissue from the patient’s body, autologous tissue must be scaled up, presenting a challenge.
Patients with recessive dystrophic epidermolysis bullosa (DEB) have a defect in one of the keratin proteins that adhere the epidermis to the underlying dermis, resulting in impaired adherence, and patients suffer from severe blistering, scarring, deformity, squamous cell carcinoma, and often early death, Oro said. He and his team have been working on the regeneration of autologous skin tissue in order to treat DEB. In 2014 Oro’s team conducted Phase I clinical trials on an autologous retroviral-corrected keratinocyte sheet product called LEAES (for LZRSE-Col7A1 engineered autologous epidermal sheet). The sheet product is made from affected keratinocytes taken from a patient’s unscarred skin, and, using gene therapy, the genetic mutation is corrected in the keratinocytes, and the edited cells are cultured in sheets, which are then transplanted back to a patient’s wound sites. In a recent study of the application of this technique to treat DEB, researchers created these sheets by taking a patient’s unscarred skin tissue, using a murine leukemia virus (MLV) to correct the mutated COL7A1 gene, and culturing the tissue to make six skin grafts. Although Phase I trials are not meant to test the effectiveness of a treatment, the trials did successfully demonstrate safety and the presence of collagen in the four patients who participated, Oro said (Siprashvili et al., 2016).
A number of challenges have arisen during the process of scaling up autologous cells for regenerative therapies for DEB, Oro said. First, DEB patients have low keratinocyte stem cell numbers because of chronic wounding, which makes it difficult to produce sufficient skin grafts for the whole body. Second, gene transfer is ineffective for forms of the disease caused by dominant negative mutations. Finally, many patients’ skin cells have preclinical premalignant lesions, which will eventually result in squamous cell carcinoma, Oro said.
In order to address these issues, Oro and his collaborators used a new process called therapeutic reprogramming, in which iPS cells are created from the patient’s skin cells and genome editing is used to correct the mutation that causes DEB. The edited cells are screened to select for those that do not have mutations for squamous cell carcinoma, and then downstream differentiation techniques are used to make human skin cells for transplantation (Sebastiano et al., 2014). A schematic of this process is found in Figure 2-1. Oro noted a number of benefits of using therapeutic reprogramming to treat DEB. First, Oro’s patients are highly susceptible to squamous cell carcinoma, and the technique allows for the screening and selection of cells that do not have mutations that predispose them to squamous cell carcinoma. Second, the genome editing techniques such as clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 allow researchers to correct the mutation that is responsible for DEB. However, Oro noted, there remains the questions of whether CRISPR/Cas9 should be used to make changes to other genes, and if so, which ones. There are also issues associated with therapeutic reprogramming with regard to safety and efficacy, Oro said. Individual iPS cell lines vary in their ability to make the same keratinocytes, which can affect therapeutic efficacy. A detailed map of the differentiation pathway is needed to understand how to manufacture a defined product from different clones, he said.
Consortia are critical to the success of these new therapies, Oro said. He noted that the development of LEAES took 18 years, partially because of a lack of collaboration and sharing within and across groups. To further develop the use of iPS cell–derived keratinocytes, Oro and others have formed a DEB consortium to establish a coordinated process.1 In addition, the Stanford University Center for Definitive and Curative Medicine brings together groups working on different cell therapies to share best practices in order to improve and accelerate the practice.
___________________
1 For more information about the Epidermolysis Bullosa Research Consortium Study, see http://med.stanford.edu/dermatology/resources/gsdc/eb_clinic/trials/eb-ebcrc.html (accessed December 12, 2016).
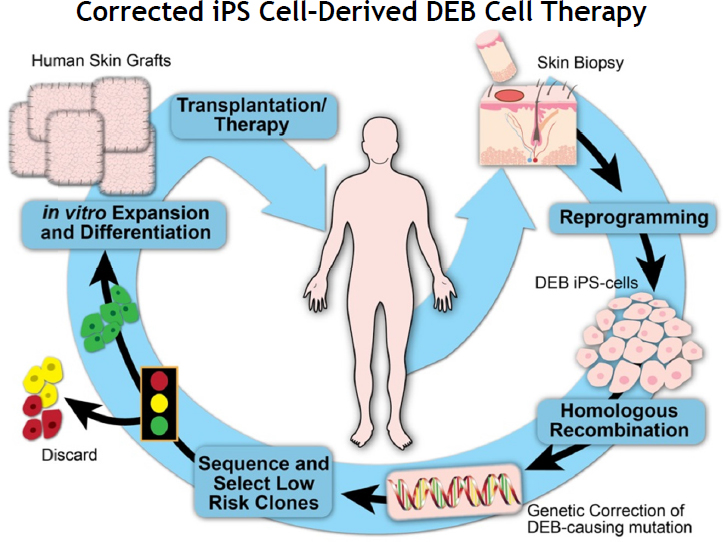
SOURCES: Anthony Oro, National Academies of Sciences, Engineering, and Medicine workshop presentation, October 13, 2016. From an internal report based on Sebastiano et al. (2014).
TRANSLATION OF RESEARCH DISCOVERIES INTO CLINICAL CARE
There are numerous opportunities for the use of cell-based therapies in the treatment of skin and musculoskeletal tissues. Despite the wealth of opportunities, there are several roadblocks that prevent advances from reaching the market, said Anthony Ratcliffe, the president and chief executive officer of Synthasome, Inc. There are challenges to the translation of research discoveries into products, Ratcliffe said, noting that in order to be successful, a product must not only be safe and effective, but must also be profitable.
The regulatory pathway from discovery to approval for musculoskeletal products takes years and millions of dollars, Ratcliffe said. A 510(k)
Food and Drug Administration (FDA) submission, which is for medical devices that are “substantially equivalent” to devices legally already on the market, can cost between $5 and $20 million and can take more than 3 years. The approval of a new biologic or drug can take more than 8 years and cost up to $300 million (Ratcliffe, 2004). There must be a positive return on investment (ROI) to make the development of a product worth the time and money, Ratcliffe said. In other words, the income generated by a product, taking into consideration the cost of manufacturing and selling it, must be greater than the costs of development (Ratcliffe, 2004).
There are challenges involved in making a product profitable, including technical difficulties such as sourcing and developing the product, manufacturing, and predicting market opportunity. Ratcliffe noted that scientific and technical advances may improve profitability. Innovative manufacturing processes may make safe, efficient, and consistent production possible; for example, Aesculap Biologics has developed a system for manufacturing cartilage that takes cartilage biopsy tissue, isolates and expands the cells, and then seeds them onto a scaffold to grow.
There are various roadblocks in the research and development process, including challenges with cell sourcing that could be mitigated by the development and implementation of clear standards and quality measures, Ratcliffe said. He also addressed the challenges faced when carrying out clinical trials, including the uncertainty about what data already exist and how a lack of data sharing can lead to inefficiencies and make it difficult to complete safety and efficacy studies. Ratcliffe mentioned several other challenges and proposed potential ways to mitigate them (see Table 2-1).
TABLE 2-1 Challenges and Opportunities to Profitability of Cell-Based Therapies
| Challenge | Opportunity |
|---|---|
| Cost | Increase efficiencies, standards |
| Time | Streamline studies required |
| Technical difficulty | Identify standards for cell sourcing |
| Clinical uncertainty | Improve clinical databases |
| Regulatory uncertainty | Develop standards |
| Manufacturing | Improve scalability, standards |
| Predicted market opportunity | Increase availability of data |
| International opportunities | Improve harmonization of standards |
SOURCE: Anthony Ratcliffe, National Academies of Sciences, Engineering, and Medicine workshop presentation, October 13, 2016.
PATIENT PERSPECTIVE: DUCHENNE MUSCULAR DYSTROPHY
Duchenne muscular dystrophy (DMD) is a rare disorder that affects 1 in 4,600 live male births, said Patricia Furlong, the chief executive officer of Parent Project Muscular Dystrophy (PPMD). Patients are usually diagnosed between the ages of 3 and 5, and the disorder causes weakness and wasting of muscles, beginning with trunk muscles and eventually affecting the heart and gut. The life expectancy of patients is around 25 years.2 Furlong’s two sons were diagnosed with DMD in 1984, and she immediately became involved in the search for a treatment or cure. After working independently for several years, Furlong founded PPMD in 1994. The organization’s mission is to accelerate DMD research, advocate for DMD causes, demand optimal care for all young men with DMD, educate the global community, and, ultimately, end DMD. PPMD was instrumental in passing the Muscular Dystrophy Care Act in 2001,3 which has delivered $700 million into muscular dystrophy research and development.
One of the first potential therapies for DMD was myoblast transfer, in which allogeneic nondystrophic muscle cells are injected into the patient’s muscles (Karpati et al., 1993). Furlong’s sons underwent this experimental therapy, but they experienced issues with the techniques used for cell delivery and migration, and the cells were rejected. Similar problems were encountered in the 2000s, when an Italian clinical trial using human mesenchymal cells was unsuccessful and even resulted in severe negative side effects for one child (Maffioletti et al., 2014). Noting the challenges that remain with cell delivery and engraftment, Furlong said that the following questions should be asked with regard to work using myloblast transfer as a therapy for patients:
- What kinds of cells are being used, and how do you know they will have the intended effect?
- How will the cells be delivered?
- Will the cells migrate?
___________________
2 See Parent Project Muscular Dystrophy at http://www.parentprojectmd.org/site/PageServer?pagename=Understand_about (accessed December 12, 2016).
3 The Muscular Dystrophy Community Assistance, Research and Education Amendments of 2001 is available at https://www.congress.gov/bill/107th-congress/house-bill/717 (accessed May 15, 2017).
The cause of death for most DMD patients is cardiac failure, although little is known about the mechanism behind it, Furlong said. Currently, a company called Capricor4 is pursuing a promising approach in which allogeneic cardiospheres are delivered into the patient’s myocardium using a cardiac catheter; the hope is that these cells will coax cardiac stem cells to regenerate normal cardiac cells. Phase I studies have been completed, with 24 boys over 12 years of age participating.5 The study is not without its challenges, Furlong said; for instance, the fact that so little is known about DMD-related heart failure has made it difficult to develop outcome measurements to indicate success. In addition, if the therapy proves beneficial, patients who receive it may need repeated doses of cardiospheres delivered on a regular basis, which will present a risk for a fatal arrhythmia. Early studies in mice by Capricor have demonstrated that the delivery of cardiosphere exosomes also results in the growth of new heart cells. However, mouse models may be of minimal value, Furlong said, commenting that “the mouse has been treated and cured many, many times; the boys not very many times.” Furlong described several other avenues that are being pursued, including the use of iPS cells to create more effective disease models to study cardiomyopathy and skeletal muscle pathologies.
While there has been success in the muscular dystrophy field over the past 30 years, much remains to be learned about how cell-based therapies may or may not be useful in treating the disease and creating better models to study DMD, Furlong said. She reiterated that there are significant challenges in identifying the right cells to use, delivering them to the right target, and ensuring that the cells have a positive effect on the targeted muscle tissue.
PANEL DISCUSSION
Commercialization
Workshop speakers and participants discussed the commercialization of these cellular therapies and whether and how regulatory requirements should or could be relaxed for potential treatments of rare diseases. The recent approval of Sarepta Therapeutics’ drug for DMD demonstrates the
___________________
4 For more information on Capricor, see Chapter 5.
5 For more information on the HOPE–Duchenne Study, see http://capricor.com/hope (accessed December 15, 2016).
difficulty of approving and paying for these types of therapies, Furlong said. FDA debated the approval of the drug, with some officials opposed to approval because the increase in dystrophin production was quite small, while those who wanted to approve the drug argued that while the production was small, it was statistically significant. Ultimately, FDA approved the drug under the accelerated approval pathway for rare diseases. While the increase in dystrophin in skeletal muscle was small, the fact that there was any effect indicated that the drug hit its target, Furlong said. She argued that in a “rare disease with a high unmet medical need and no options,” therapies that are safe and potentially beneficial need to be “out there on the market” so that patients can use them and researchers can learn more about how and whether the therapies work.
One insurance company has already announced that it will not pay for the Sarepta therapy, indicating that even therapies with regulatory approval may encounter challenges in entering the market.6 This potential roadblock could prevent companies or investors from initially investing in therapies that are not guaranteed commercial success. The traditional model of funding is one in which investors are motivated by the desire to make money, rather than to cure disease, Ratcliffe observed. He added, however, that there is “no reason why that has to be the only option.” If a product is developed for an application with a high unmet medical need and few existing therapeutic options, the product may require an alternative type of funding mechanism that is driven by a desire to get the product into the hands of those who need it most, he added. Dunbar noted that there may be similarities between the commercial structure for these skin or musculoskeletal therapies and the structure for vaccine development, because the potentially far-reaching benefits of these products may change the equation.
One workshop participant commented that part of the appeal of regenerative medicine is the “hope of a cure for diseases that are currently either untreatable or managed with chronic therapy,” but current U.S. payment systems are geared toward chronic therapy rather than one-time therapies, which may compromise commercial viability for some therapies. While one-time therapies may be cost-effective, the cost savings may not be manifest for years. Ratcliffe agreed, noting that payers are reluctant to foot the bill for an expensive treatment when the
___________________
6 The Anthem Medical Policy Statement on Eteplirsen can be found here: https://www.anthem.com/medicalpolicies/policies/mp_pw_c192386.htm (accessed December 15, 2016).
patient may switch insurance companies at any time, taking any future cost-savings with him or her. When there is the potential that a one-time therapy will not be paid for, the incentive to initially invest in a therapy is lessened, the participant suggested.
State of the Science
When considering the state of the science for skin and musculoskeletal cell–based therapies, Dunbar said that there are two important questions to ask: What types of models are used to study potential therapies? Is there enough information to understand whether the models are effective at representing the disease and how the treatment might work? For DMD, the mouse model has traditionally been used because researchers were able to produce a mouse with the mutation for DMD, Furlong said, but the more expensive models of dogs and pigs are used once the mouse model shows promise. Unfortunately, she said, even these models cannot represent the complexity of the multisystem disease as seen in humans with DMD. The lack of a perfect animal model means that tough decisions will have to be made about how much evidence is needed before moving a potential therapy into humans, Furlong said. Oro concurred, noting that mouse and human skin are quite different, so clinical trials must move forward with only partial results from the animal model. While animal models can be helpful in determining some level of safety and efficacy, it is important for researchers to understand the questions that these models are not capable of answering and to move into human trials when appropriate, Ratcliffe added.
Meeting Quality and Production Standards
One workshop participant asked how, during the manufacturing process, one can define and measure the critical quality attributes for a cell therapy product in order to ensure that it is safe, effective, and the same from batch to batch. This is an important issue because manufacturing and regulatory standards for biologics, such as cell therapies, are unclear, Oro said. Developing quality attributes begins with characterizing and understanding the cell that will be manufactured, he said. It is then possible to develop a GMP pathway and assays to measure the quality and safety of those cells. Ratcliffe said that he starts by applying a large number of assays and measurements and gradually
determines which assays are critical for identifying his unique cell population. Once those assays are identified, he said, he uses them to assess the quality of subsequent cells.
The topic of scaling up and expanding cell populations for use in cellular therapies was brought up by a workshop participant. How can scientists and manufacturers make sure that their expanded cell populations retain purity and potency? For example, fibroblasts are easily expanded, but may not retain the desired characteristics. This issue remains a significant challenge in the field of regenerative medicine, Ratcliffe said. In many cases, scientists can easily identify a defined cell type, but they do not know how to expand that cell population. Ratcliffe said that there is an opportunity for the field to study potential solutions to this problem.
Communication
How, asked one workshop participant, can research findings be best translated and communicated so that patients and other stakeholders can understand the science and manage expectations about product development timelines and what clinical outcomes are possible? Families approach researchers with money in hand to ask for a cure, and researchers have told the families, “Within 2 years, I will be giving you a prescription for this therapy,” Furlong said. These types of promises and highly publicized experimental therapies make it challenging to effectively communicate with families and patients who are desperate for help. Scientists need to be realistic about the time that it takes to move from the laboratory to helping a patient, Oro added. It is critical not only to educate patients on how to engage with researchers, but also to educate investigators about how to speak with patients about the research process and the possibilities, Furlong said.










