
Appendix C
Commissioned Paper: Topical Dosage Form Development and Evaluation
CONTENTS
Potential Drug Absorption Pathways Across the Stratum Corneum
Physicochemical Properties of Active Pharmaceutical Ingredients and Dermal Absorption
The Formulation Characteristics and Dermal Drug Delivery
Pharmaceutical Excipients and Dermal Drug Delivery
Drugs Used in Compounded Pain Creams
Permeation Studies of Pain Molecules
THE SKIN
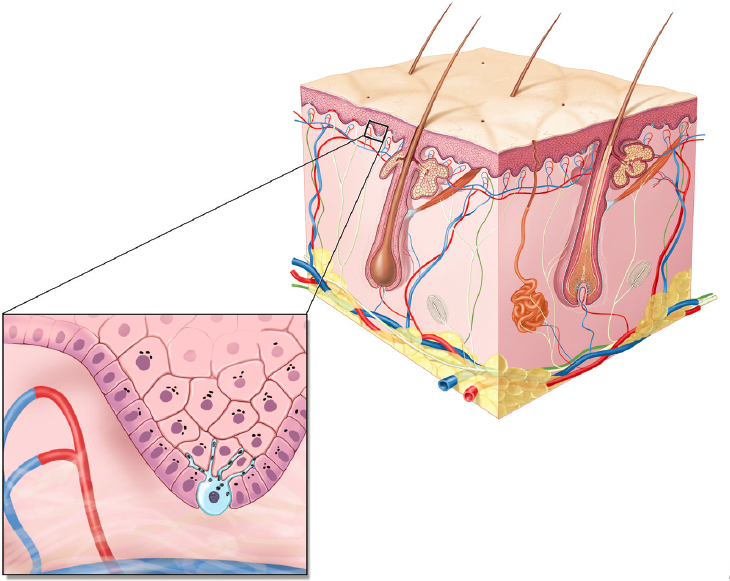
The skin is the largest organ and a protective barrier. The skin consists of several heterogeneous layers of cells, hair follicles, and glands. To understand the key mechanisms of topical drug delivery, the skin must be viewed as having two major layers: the outer epidermis and the inner dermis. The epidermis lacks any blood vessels whereas the dermis has a rich network of blood vessels (see Figure C-1). The epidermis consists of two distinct layers: the outermost stratum corneum and the underlying viable
SOURCE: Copyright 2008 Terese Winslow, U.S. government has certain rights.
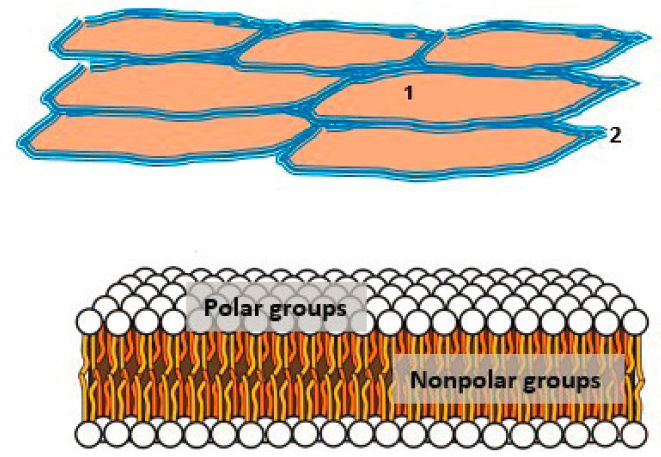
epidermis. The stratum corneum is constituted of tightly packed dead cells filled with keratin (see Figure C-2). The intercellular space is occupied by bilayer lamellar lipid structures (Elias, 2005). These lipid domains consist of fatty acids, cholesterol, and saturated fats called ceramides. Together, the tightly packed keratinocyte layers and intercellular lipids constitute the main barrier and keep external agents from penetrating into deeper layers of the skin. Melanin is the pigment responsible for skin color, and it protects the skin from intense ultraviolet (UV) radiation. Additionally, the skin is also associated with a great immune system to address any exposure to pathogens while also reacting to allergens. The skin surface is acidic in nature, while the inner layers are at physiological pH (Schmid-Wendtner and Korting, 2005). The pH gradient across the different layers prevents microorganisms from penetrating into deeper layers of skin. The skin tissue also has significant amounts of esterase, protease, and other enzymes that constitute a metabolic barrier (Martin and Axelrod, 1957; Park et al., 2011).
SOURCE: Phospholipids aqueous solution structures by Mariana Ruiz Villarreal, LadyofHats is licensed under Creative Commons CC0.
A water gradient exists from the inside to the outside layers of the skin; it is responsible for the constant evaporation of water across the stratum corneum. Any damage caused to the stratum corneum barrier will increase the transepidermal water evaporation rate (Sotoodian and Maibach, 2012).
Over the past several decades, research has clearly shown that macromolecules could not diffuse across the skin. The carrier-medicated delivery systems, nanoparticles, micron size surfactant, and polymeric carriers cannot penetrate the skin either (Campbell et al., 2012). There are some reports showing penetration of particulate drug delivery systems owing to their charge, shape, elasticity, and other characteristics via the hair follicles. However, there is not enough strong evidence to dispute the fact that large molecules and particles would not be able to penetrate the skin. Overall, there is a strong physical, metabolic, and physicochemical barrier embedded in the skin that limits the number of drugs that can be delivered topically in effective amounts. The criteria for the molecules to diffuse into and across the skin is discussed in later sections of this article.
TOPICAL PRODUCTS
Topical products are generally available in various forms such as liquids, ointments, gels, creams, and foams. In terms of complexity, the product can be a simple homogenous viscous solution or semisolid solution, or a suspension of active pharmaceutical ingredients (APIs) in the semisolid vehicle. The microstructure of the product becomes increasingly complex with the increasing heterogeneity of the product. For example, creams are heterogeneous and are considered complex products. The complexity increases when the API or microcarrier system is dispersed in the cream base.
In compounding pharmacies, the pharmacists generally incorporate the drug into ready semisolid bases. These bases are premixed and readily available for the pharmacist, into which the API is generally incorporated with the help of an ointment spatula or triturated using a mortar and pestle. Some of the semisolid bases that are commonly used in compounding pain creams include premium lecithin organogel (PLO), oleaginous base, Vanicream base, Aquaphor base, and Eucerin base. The excipients present would determine the type of semisolid base. The gel bases are made up of hydrogel polymers such as carbomers, celluloses, and pluronics. The organogels for the delivery of lipophilic compounds are made of lipids such as lecithin. The cream bases are emulsions in which an oil and aqueous phases are mixed together to form either an oil in water (o/w) or a water in oil (w/o) emulsion. Oleaginous bases are made of hydrocarbons, oils, fats, and waxes. Water-soluble bases are generally formulated using polyethylene glycols. The consideration of a particular type of base should be based on factors such as stability, site of delivery, and target site of action. The type of base chosen could largely influence the overall performance of the product (Padula et al., 2018).
The phrase “topical products” encompasses all of the products that are intended for application on the skin, mucous membranes, and cavities. In the case of dermatological products, the products may be intended just for the purpose of protection or hydration of skin. Such products generally may or may not contain any active ingredients and they are intended to spread well across the entire applied area, forming a thin layer. In products that are intended for local activity, such as antibacterial and antifungal products, the drug must be available on the surface of the skin in its active form, and it is not required to be absorbed into the skin. In the case of products intended for regional activity, such as local anesthetic products and pain creams, the drug must be retained in the skin. However, the drug would eventually be cleared by dermal circulation. In the case of transdermal drug delivery products, the skin is only used as a port of administration for systemic delivery of drugs. For such APIs, the target site of action is located elsewhere in the
body. For example, the antianginal drug nitroglycerine is designed to be absorbed into the systemic circulation from the site of application on the skin.
POTENTIAL DRUG ABSORPTION PATHWAYS ACROSS THE STRATUM CORNEUM
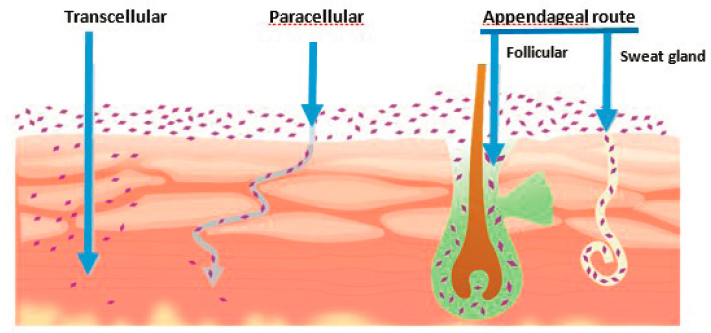
There are three potential pathways for the absorption of drugs across the stratum corneum (see Figure C-3). The drug could get absorbed via the transcellular pathway in which the drug would need to traverse from one cell to another. The paracellular pathway is diffusion through the intercellular lipid domains. The third potential pathway would be the transappendageal pathway, which involves the pilosebaceous routes (hair follicle and sebaceous glands) and sweat glands (Lauer et al., 1996; Meidan et al., 2005). Because a dense capillary network surrounds the sweat ducts and hair follicles, any small fraction of a drug that makes its way into the skin via the appendageal pathway will be quickly absorbed into the circulatory system, hence explaining why it is not available in effective amounts in the skin. Therefore, the contribution of the appendageal pathway to overall dermal drug absorption is generally negligible. The most important pathways are intercellular and intracellular pathways. Although paracellular volume is relatively smaller than the transcellular route, it can still contribute significantly, provided the drug molecule meets the required criteria to propagate through the lipid pathway.
SOURCE: Shaker et al., 2019.
PASSIVE DIFFUSION OF DRUG
The drug undergoes passive diffusion across the skin layers along the concentration gradient, which is thermodynamically a spontaneous process. A simple approach to mathematically explain the passive diffusion of drugs across the skin layers is through Fick’s first law:
![]()
Where J is the flux (rate of drug transfer per unit surface expressed mass/unit time/unit area), Cd is the concentration of drug in the formulation, and Cr is the concentration of drug in the receiver, which is generally considered as skin interstitial fluid or systemic circulation. Cd–Cr represents the concentration gradient across the barrier where h is thickness of the membrane. D is the diffusion coefficient of the drug in the stratum corneum (cm2/sec). K is the partition coefficient of drug between formulation and stratum corneum. (Generally, the partition coefficient of drug in octanol and water system is considered in this case.)
PHYSICOCHEMICAL PROPERTIES OF ACTIVE PHARMACEUTICAL INGREDIENTS AND DERMAL ABSORPTION
The most critical physicochemical characteristics that would influence the permeation of drugs across the skin are molecular weight, partition coefficient, melting point, and charge of the penetrant molecule.
The molecules greater than 600 da are known to be poorly permeable across the skin (Barry, 2001). The larger the molecule, the less its diffusivity is in the vehicle as well as in the skin layers. Therefore, smaller molecules are preferred for passive topical delivery over larger molecules. This is the primary criteria to be fulfilled by the penetrating molecule. Most pain medications satisfy this criterion unless they are modified by association, complexation, or structural modification.
The predominant pathway for drug penetration into the stratum corneum (paracellular or transcellular pathway) is lipid in nature. Unlike the stratum corneum, the viable epidermis and dermal layers are aqueous in nature. To make its way across the stratum corneum, the API must be able to partition into the lipid domains adequately; to penetrate across the viable epidermis and dermis, the molecule should possess significant water solubility as well. Therefore, the drugs that are adequately both lipid and
water soluble are known to permeate across the skin in significant amounts. If the drug is extremely hydrophilic in nature, then it would not be able to partition into the stratum corneum. On the other hand, the drugs that are extremely lipophilic (log Poct/water > 3) might be successful in localizing in the stratum corneum but will fail to get absorbed into the deeper layers in effective amounts. Therefore, an optimal log Poct/water partition coefficient value for a drug to be a good candidate for dermal product is reported to be 1–3 (Naik et al., 2000; Wiedersberg and Guy, 2014).
The melting point and the skin permeability have been known to be inversely related to each other. Generally, drugs with a melting point of less than 200ºC are known to permeate relatively better than the higher melting drugs (Naik et al., 2000). Therefore, approaches to decrease the melting point such as the formation of a eutectic mixture and modification of chemical structure are considered. In a study reported by Ki and Choi, the authors observed greater permeation of the pain medication meloxicam ethanolamine compared to meloxicam, owing to the lesser melting point of the ethanolamine form (Ki and Choi, 2007).
The charge-selective nature of the stratum corneum has been well investigated. The skin has a pI1 of 4–5. At a pH less than its pI, the surface of the skin is predominantly positively charged, whereas at a pH above pI, the skin’s surface is predominantly negatively charged. Evidently, the skin is selective to negatively charged drugs at pH < pI and vice versa (Hatanaka et al., 1996).
THE FORMULATION CHARACTERISTICS AND DERMAL DRUG DELIVERY
The quality attributes and microstructure of the topical products could potentially have a significant effect on their performance and sensorial characteristics.
Some of the critical quality attributes that are considered important are drug concentration, pH, amount of drug in the suspended form, particle size, polymorphic form, shape of particles, globule size, viscosity, and texture properties (Otto et al., 2009).
Generally, the drug permeation flux increases with an increase in the concentration of the drug in the formulation vehicle. Most appropriate would be to relate the permeation flux to the thermodynamic activity of the drug. The thermodynamic activity of the drug at saturation is the unity at which a maximum transdermal flux is achieved. When supersaturated systems are formed at the applied site, the absorption flux increases further (Moser et al., 2001b). If the drug is distributed in both dispersed as well as
___________________
1 Isoelectric point or the pH at which the charge is a net neutral.
continuous phases in a cream product, then the thermodynamic activity of the drug in both phases contributes to the performance of the topical product. Often the drug incorporated in the cream base gets localized only in one of the phases. If the drug predominantly concentrates in the dispersed phase, then the globule size of the dispersed phase becomes an important factor in determining the performance of the product (Ktistis and Niopas, 1998; Schwarz et al., 1995). If the drug is highly soluble in the continuous phase, then the globule size of the dispersed phase is not likely to have a significant influence on the drug permeation (Izquierdo et al., 2007). Friedman and coworkers demonstrated a superior permeation of steroidal and nonsteroidal anti-inflammatory drugs when delivered using a submicron emulsion vehicle as compared to conventional cream formulations indicating that reducing the size of the dispersed phase could influence the drug permeation and thus enhance the pharmacological activity of the drug significantly (Friedman et al., 1995). The efficacy of the nonsteroidal drugs, diclofenac and indomethacin, was enhanced by 40–50 percent, whereas the efficacy of steroidal drugs, which are relatively more lipophilic, was found to have been enhanced by four fold.
In topical products incorporated with poorly soluble drugs, an excess amount of drug is dispersed in the formulation due to saturation of the vehicle. The suspended drug would act like a reservoir during absorption process. The factors such as the amount of drug suspended, its particle size and distribution, shape of the particles, polymorphic form, and melting point influence the rate of dissolution of the drug in the remnant vehicle at the applied site. The smaller the diameter, the higher the specific surface area and the faster the dissolution of the particles in the formulation vehicle. As the polymorphic form of drugs could differ in their physicochemical characteristics, choosing the most appropriate form that dissolves rapidly and remains stable in the formulation is important while formulating a topical product. The X-ray diffraction studies and differential scanning calorimetric studies would shed some light on the solid-state nature of the drug.
Release of the drug is the first step in the dermal absorption process. The viscosity of a topical product is one of the major factors that determines the rate of release of the drug from the formulation (Binder et al., 2019). Indeed, it is the overall rheological behavior of the product that is known to play a significant role in determining the performance of the formulation. Generally, the topical semisolid products are shear thinning systems. This means their viscosity decreases as it is rubbed on the skin. When allowed to stand, the viscosity may or may not be recovered depending on the extent of disruption of the microstructure of the formulation. Yield stress is a property of some of the semisolids that represents their rigidity. The yield stress value determines the extrudability of the formulation from collapsible tubes and also the spreadability on the skin surface. In a study
by Binder and coworkers, the authors evaluated the effect of viscosity on drug penetration into the skin from cellulose ether-based hydrogels. It was observed that the drug penetration depth decreased as viscosity increased, suggesting a slower drug release due to an increasingly dense gel network (Binder et al., 2019).
Texture properties such as stringiness, adhesiveness, and spreadability influence the sensorial characteristics of formulation. Sensorial characteristics determine the acceptability of the formulation. Although the role of the work of adhesion has not been systematically investigated, fundamentally, it is understood that the work of adhesion represents the extent of interaction between the formulation and skin surface. There should be adequate interaction between the topical product and the skin for the product to spread and adhere to the surface of the skin so that the diffusion process continues uninterrupted.
The drying rate of the formulation is critical in determining the extent of drug delivery from a topical product. When applied in a clinically relevant dose on the skin surface, the products would start losing the volatile components immediately after application. The evaporative metamorphosis process leads to dynamic change in the viscosity of the formulation. The solvent evaporation also affects the thermodynamic activity of the API, as mentioned earlier.
The quality attributes of the product are highly dependent on the composition of the product and the manufacturing process implemented during the product’s preparation. A cream product prepared using different equipment could vary in its characteristics. Despite having the same composition and process of preparation, products that are prepared using different manufacturing variables could end up with different quality attributes and thus exhibit different levels of performance.
PHARMACEUTICAL EXCIPIENTS AND DERMAL DRUG DELIVERY
The excipients play different functional roles, which in turn determine the type, structure, and stability of the semisolid base. Often, the excipients also affect the absorption of drugs. The commonly used excipients in semisolid products include solvents, viscosifying agents, emulsifying agents, penetration enhancers, stabilizers, preservatives, and organoleptic agents. The excipients used in topical products should be inert and should comply with the quality standards specified in the pharmacopoeia. The ingredients should not cause any irritation, sensitization, or irreversible perturbation of the stratum corneum barrier. Therefore, the potential physiological interaction of excipients needs to be well understood. The excipients influence the quality attributes of the topical products, physicochemical characteristics of the drug, and sensorial characteristics of the formulation as well.
Solvents play multiple roles in a topical product. Solvents help enhance the solubility of the API in the product and also facilitate drug absorption. The solvents used in the formulation could enhance drug absorption through several mechanisms. The solvent could carry the drug into the skin via solvent transport pathways (Osborne and Musakhanian, 2018). At the site of application, the solvent evaporates, leading to enhanced drug absorption owing to increased concentration. In the case of products that are already saturated with drugs (Moser et al., 2001a), incorporation of solvents with relatively higher boiling points may help to keep the drug from precipitating over a long period of time at the site of application, facilitating the absorption process. Often, the solvents are also incorporated to dissolve some of the excipients like coloring agents, preservatives, and stabilizers in the formulation.
Within the formulation, one of the potential interactions of the excipient with the API that could enhance drug absorption is through the formation of an eutectic substance. For example, the transdermal permeation of meloxicam was enhanced significantly by incorporating thymol in the formulation, owing to the decrease in the melting point. The extent of decrease in the melting point was directly dependent on the concentration of thymol (Mohammadi-Samani et al., 2013). The excipients that are present in an ionized state could also form ion pairs with the API, leading to a relatively more lipophilic drug form that could enhance the drug penetration. In one of the studies, Green and Hadgraft (1987) reported the ability of oleic acid and lauric acid to form ion pairs with cationic drugs and enhanced their transdermal permeation.
SKIN PERMEABILITY ENHANCERS
The excipients used in the topical formulations are often intended to influence the skin permeability by interacting with the stratum corneum barrier. Some excipients are known to lead to hydration and swelling of the stratum corneum to enhance the drug delivery (examples include propylene glycol, urea, and polyethylene glycol). Some excipients would enhance the skin permeability of inherently poorly permeable polar molecules by fluidizing the rigid lipid lamellar structures. The chemical penetration enhancers can be classified into different categories such as solvents, surfactants, fatty alcohols, fatty acid esters, lipids, and terpenes. Most of these enhancers have been known to enhance the permeation by fluidizing lipids in the stratum corneum and to increase the diffusivity of molecules across the stratum corneum (Williams and Barry, 2012). Azones and dimethyl sulfoxide (DMSO) are also known to disrupt the lipid domains and improve the partitioning of drugs into the stratum corneum. Solvents such as propylene glycol and transcutol enhance the solubility of the drug in
the stratum corneum. Oleic acid is known to induce phase separation in the stratum corneum lipid domains. For example, amitriptyline, one of the topical pain medications formulated in combination with other drugs, was found to permeate 4- and 5-fold more in the presence of fatty acids such as oleic acid and linoleic acid (Jain and Panchagnula, 2003). The permeability enhancers should be used in optimal concentration. When used in higher amounts, the permeability enhancers could affect the formulation properties as well as potentially cause skin irritation (Haque and Talukder, 2018).
In Vitro Evaluation of Topical Products
In Vitro Release Testing
The topical products applied on the skin are required to release the drug from the formulation so that it is available for absorption at the site of application. The release rate of the drug is governed by the viscosity and heterogeneity of the topical product. The drug release rate would be significantly affected in the case of any thermodynamic instability in the formulation. A drastic drop in the viscosity of the formulation due to phase separation of the cream could lead to the rapid release of the drug concerning dose dumping. Therefore, in vitro release testing is considered an excellent tool to test the product’s performance, stability, batch-to-batch variability, and influence of changes in the composition (Nallagundla et al., 2004; Tiffner et al., 2018; USP, 2019).
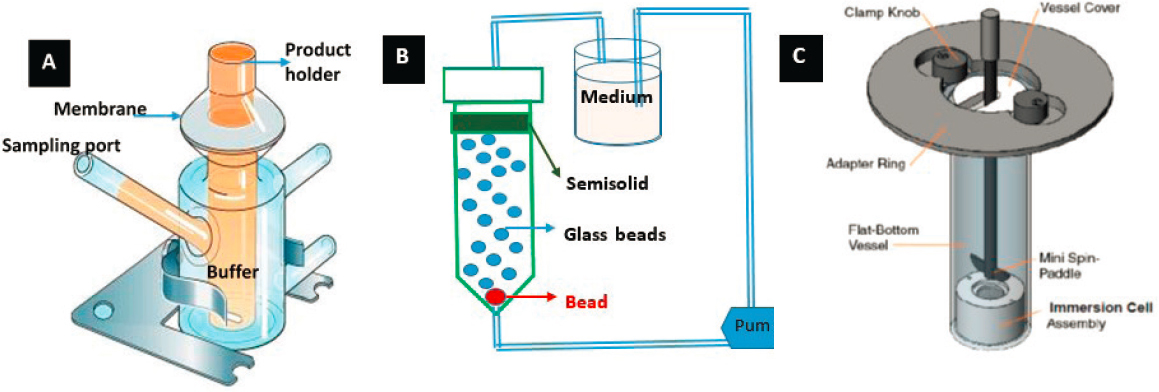
The in vitro release testing is generally performed using the USP II dissolution apparatus or Franz diffusion cells. In some cases, the use of flow through the apparatus has been demonstrated too (Chattaraj and Kanfer, 1996). The various apparatus that is used for in vitro testing of dermatological products are shown in Figure C-4. In principle, the in vitro release testing involves separation of the formulation from the receiver fluid using a nonrate controlling, nonbiological membrane. Generally, a filter membrane or a dialysis membrane would be used so the rate of release of the drug from the formulation is governed only by the quality attributes of the product (FDA’s SUPAC-SS guidance) (FDA, 1997). The release rate of drug from the formulation is determined by sampling and quantitative analysis of the receiver fluid at different time points. The in vitro release rate is the slope of the amount of drug released from the formulation to the square root of time (Higuchi, 1961, 1962).
In Vitro Permeation Testing
Furthermore, the performance of topical formulations is evaluated by subjecting them to in vitro skin permeation testing studies using a suitable
SOURCES: PermeGear, 2018; © 2014 United States Pharmacopeial (USP) Convention.
skin model. In vitro permeation testing is performed using Franz diffusion cells across an appropriate skin model. The skin is sandwiched between donor and receiver compartments with the epidermis facing upward. The receiver fluid would be a buffer system that would serve as a perfect sink. The receiver compartment fluid is withdrawn at different time intervals and analyzed for the amount of drug permeated across the skin (Friend, 1992). Generally, two types of dosing protocols are reported in the literature. Finite dosing would mean the application of a clinically relevant dose applied on the skin surface using a suitable application technique. The objective of applying a finite dose is to mimic the in vivo phenomenon of postapplication metamorphosis of the product (Kasting and Miller, 2006). In an infinite dose study, an excess dose is applied on the skin, and the permeation study does not exactly reflect the in vivo situation.
There are several skin models used in dermal and transdermal delivery research. Excised skin from rodents and other animals have been the most common. However, the permeation studies across rodent skin can lead to an overestimate of the drug’s permeability. This is generally attributed to differences in stratum corneum thickness and composition of the intercellular lipid domain (Bond and Barry 1988a,b; Bronaugh et al., 1982; Wester and Noonan, 1980). Nevertheless, such skin models would serve as great and inexpensive tools during product development and optimization (Scott et al., 1986). Porcine ear skin has been suggested as a valid model for human skin in vivo (Sekkat et al., 2002). Sekkat and coworkers performed a systematic biophysical evaluation study and observed that the barrier function of the pig ear skin model closely resembled the human skin in vivo. However, additional evidence is needed to demonstrate that porcine ear skin has comparable permeability characteristics to drug molecules as that of human skin.
Excised human skin is most commonly obtained from cadavers or from patients undergoing plastic surgery. Skin from the abdominal region, breast, thigh, or back is most convenient, due to the availability of adequate area for performing multiple replicates (Rougier et al., 1987). The use of dermatomed human cadaver skin for bioequivalence testing of topical products is increasing in the pharmaceutical industry as regulatory agencies are accepting the study data (Abd et al., 2019).
Variability in skin permeability due to age, sex, region, processing, storage, and diffusion study protocols are common issues seen in the case and of all these skin models. An adequate number of replicates should be implemented in the study design to generate reliable data that helps draw valid conclusions.
DRUGS USED IN COMPOUNDED PAIN CREAMS
Compounded topical creams typically use a combination of two or more medications to achieve multiple complementary effects (Keppel Hesselink and Kopsky, 2017). Common APIs used in compounded topical creams are listed in Table C-1. The key physicochemical characteristics influencing the dermal penetration of drugs are also tabulated. Apparently, all are small molecules and meet the size criteria to be a candidate for topical administration. However, for most of the molecules, not all of the other physicochemical characteristics seem to favor permeation. Either the log P is out of the 1–3 range or the molecule has a high melting point or an unfavorable dissociation constant (pKa).
TABLE C-1
Physicochemical Properties of APIs Commonly Used in Pain Medications
| Drug | Mol. Wt (g/mol) | Log P (Oct-water) | Melting Point (°C) | pKa |
|---|---|---|---|---|
| Amitriptyline | 277.40 | 4.92 | 197 | 9.4 |
| Baclofen* | 213.66 | 1.3 | 207 | 9.62 and 3.67 |
| Bupivacaine | 288.43 | 3.41 | 107 | 8.2 |
| Cannabidiol | 314.46 | — | 66 | — |
| Carbamazepine | 236.27 | 2.45 | 191 | 13.9 |
| Clonidine | 230.09 | 1.59 | 130 | 8.12 |
| Cyclobenzaprine* | 275.39 | 5.2 | 218 | 8.47 |
| Dexamethasone | 392.46 | 1.83 | 262 | 12.42 |
| Doxepin | 273.38 | –0.548 | 184 | 8.96 |
| Gabapentin | 171.24 | –1.1 | 166 | 3.68 and 10.70 |
| Ketamine | 237.73 | 3.12 | 92.5 | 7.5 |
| Lidocaine | 234.34 | 2.44 | 68.5 | 8.01 |
| Meloxicam | 351.40 | 3.43 | 254 | 4.08 |
| Memantine | 179.31 | 3.28 | 258 | 10.27 |
| Naproxen | 230.26 | 3.18 | 153 | 4.15 |
| Nifedipine | 346.30 | 2.20 | 173 | 3.93 |
| Orphenadrine | 269.39 | 3.77 | 156 | 8.91 |
| Pentoxifylline | 278.31 | 0.38 | 105 | — |
| Topiramate | 339.36 | –0.5 | 125 | 8.6 |
| Tramadol | 263.38 | 1.34 | 181 | 9.4 |
* Zwitter ionic in nature.
SOURCES: Sources for the data in this table include DrugBank, 2020; Expert Committee on Drug Dependence, 2017; Plumley et al., 2009; PubChem, 2020.
Owing to greater lipid partitioning ability, generally the un-ionized moiety is more permeable across the lipid pathways over the ionized counterpart. Depending on the pKa, the drugs undergo dissociation to different extents (Chantasart et al., 2015). For example, Watkinson and coworkers reported a significant order of magnitude decrease in the permeability coefficient of ibuprofen (pKa = 4.41) across the skin when the pH was increased from 4 to 7 owing to the significant increase in dissociation of ibuprofen into its ion (Watkinson et al., 1993). The skin surface is acidic in nature (pH 4–5). Skin is not only capable of resisting any change in pH upon exposure to extreme pH formulation, but it also has the ability to drive the formulation to come into homeostasis with the skin’s pH (Levin and Maibach et al., 2007). Therefore, the percent of the un-ionized drug present at skin pH is critical in determining the absorption. Although the ionized form of drug generally gets absorbed by polar pathways in the skin, it is relatively many-fold smaller than the amount that could get absorbed via nonpolar pathways.
In the case of formulations with a combination of drugs of different pKa, it is challenging to adjust the pH to favor the permeation of all of the drugs. For example, in a topical pain product containing amitriptyline (pKa 9.4) and ketamine (pKa 7.5) if the formulation pH happened to be 9, most of the ketamine (96.93 percent) would exist in ionized form. But at the same pH only 28.47 percent of amitriptyline would exist in ionized form. If meloxicam or nifedipine are in combination with the above drugs, they would exist almost completely in an ionized state at a pH of 9, which could hamper their penetration significantly.
When oppositely charged medications are combined in the same formulation, they tend to form ion pairs. Ion pairing is generally known to favor the absorption of drugs owing to the relatively higher partitioning ability of neutral ion pairs compared to the charged moieties of the API. Valenta and coworkers have used an ion-pairing mechanism to enhance the delivery of a local anesthetic, lignocaine (Valenta et al., 2000). The advantage of an ion-pairing mechanism is jeopardized if the size of the complex exceeded the acceptability limit of the skin. Particularly, when APIs have multiple ionizable groups, they will be combined; the chances of forming larger ion pair complexes are even higher. The ion pairing could happen between excipient and the API as well. Jain and Panchagnula reported an enhanced permeation of amitriptyline owing to ion pairing with oleate (Jain and Panchagnula, 2003).
PERMEATION STUDIES OF PAIN MOLECULES
Some studies in the literature have specifically discussed the permeation of APIs from topical pain products. In one of the studies by Wang and Black
(2013), the investigators studied the permeation of a few pain medications from cream formulations across excised trunk skin from human cadavers. The objective was to compare a custom-made cream base against respective reference cream products. The amount of drug present in the respective reference cream products and the amount of drug permeated in the creams are given in Table C-2. The amount of pain medication permeated was less than 1 percent of the applied dose (except ketamine, which was a little more than 1 percent). The custom-made cream did not enhance the permeation of pain medications significantly compared to the reference creams (Wang and Black, 2013).
In another study, Bassani and Banov investigated the absorption of ketamine, gabapentin, clonidine, and baclofen across the human cadaver trunk skin (see Table C-3). All of the drugs permeated poorly except ketamine hydrochloride, which achieved a 35 percent permeation across the skin (n = 3) (Bassani and Banov, 2016).
Sznitowska and other researchers investigated the effect of absorption promotes on percutaneous permeation of baclofen, a zwitterion. Baclofen
TABLE C-2
The Amount of Drug Absorbed Across the Human Cadaver Skin from Topical Pain Creams (n = 6 ± SD)
| Active Pharmaceutical Ingredient | % w/w API in the Cream Formulation | % Absorbed (dose 50 mg of formulation across 1.77 cm2 in 48 h) |
|---|---|---|
| Bupivacaine hydrochloride | 1 | 0.28 ± 0.11 |
| Diclofenac sodium | 3 | 0.84 ± 0.23 |
| Gabapentin | 6 | 0.38 ± 0.43 |
| Ketamine hydrochloride | 10 | 1.03 ± 0.32 |
| Orphenadrine citrate | 5 | 0.13 ± 0.05 |
SOURCE: Wang and Black, 2013.
TABLE C-3
The Amount of Drug Absorbed Across the Human Cadaver Skin from Topical Creams (n = 3 ± SD)
| Active Pharmaceutical Ingredient | Percent Drug Absorbed Across the Cadaver Skin | |
|---|---|---|
| Lipoderm | Lipoderm ActiveMax | |
| Ketamine | 35.48 ± 9.03 | 45.52 ± 2.42 |
| Clonidine | 3.955 ± 2.60 | 4.38 ± 0.95 |
| Gabapentin | 0.41 ± 0.34 | 0.19 ± 0.08 |
| Baclofen | 0.27 ± 0.27 | 0.10 ± 0.08 |
SOURCE: Bassani and Banov, 2016.
is one of the commonly used drugs in neuropathic pain management via topical administration alongside other pain management drugs. The drug was found to be poorly permeable across the full-thickness cadaver skin. All of the highly efficient lipophilic and hydrophilic permeation enhancers such as oleic acid-propylene glycol, oleic acid, azone, ethanol, sodium lauryl sulfate, propylene glycol, and dimethyl sulfoxide failed to enhance its permeation significantly for up to 30 hours (Sznitowska et al., 1996).
It is evident from the above reports that APIs used in the compounded pain creams are poorly permeable and require appropriate enhancement strategies to improve their delivery into and across the skin. In the above studies, the absorption did not differ significantly with the use of different types of bases. A rational selection of base composition might be one of the factors needed to be considered in optimizing dermal delivery of pain medications. For example, Lehman and Raney (2012) studied the permeation of ketoprofen across excised human skin at finite dose conditions, from different formulation bases. The performance of Pentravan base (Vanishing o/w base) was compared with the PLO gel. The ketoprofen permeation from Pentravan base (~13.12 percent) was almost 4-fold higher than the PLO gel (~3.63 percent). This study demonstrated the importance of utilizing the appropriate formulation base for the development of a pain product.
The bioavailability of drugs from topical products also depends on the area of application. Any accidental or intentional application of medicated creams over a larger skin surface area can be associated with a potential risk of toxicity. Pomerleau and coworkers reported a case of severely elevated levels of clonidine in a 23-year-old patient who had rubbed a specially compounded medicinal cream over his entire body (Pomerleau et al., 2014). The formulation contained clonidine 0.2 percent w/w, gabapentin 6 percent w/w, imipramine 3 percent w/w, ketamine 10 percent w/w, lidocaine 2 percent w/w, and mefenamic acid 1 percent w/w. The patient suffered severe hypertension, bradycardia, and altered mental status.
SUMMARY
The physiochemical characteristics of the API are critical in determining the suitability of a drug for dermal delivery. Only a few of the drugs currently used come close to meeting all of the criteria to penetrate the skin. Moreover, the use of multiple drugs increases the potential for drug–drug and drug–excipient interactions influencing the dermal absorption of actives. The skin barrier can be modulated using formulation approaches, compositional approaches, and chemical permeation enhancers to improve the delivery of drugs. There is a need to determine appropriate methods and approaches to evaluate the topical cream products. In vitro release testing and in vitro permeation testing are great tools to compare the compounded
products with benchmark reference products. However, the fact that there are no exact reference products or FDA-approved products available for the comparison of compounded products complicates the challenge significantly. Preclinical and clinical studies are necessary to determine the safety and efficacy of compounded products.
REFERENCES
Abd, E., S. A. Yousef, M. N. Pastore, K. Telaprolu, Y. H. Mohammed, S. Namjoshi, J. E. Grice, and M. S. Roberts. 2016. Skin models for the testing of transdermal drugs. Clinical Pharmacology: Advances and Applications 8:163–176.
Barry, B. W. 2001. Novel mechanisms and devices to enable successful transdermal drug delivery. European Journal of Pharmaceutical Sciences 14(2):101–114.
Bassani, A. S., and D. Banov. 2016. Evaluation of the percutaneous absorption of ketamine HCl, gabapentin, clonidine HCl, and baclofen, in compounded transdermal pain formulations, using the Franz finite dose model. Pain Medicine 17(2):230–238.
Binder, L., J. Mazál, R. Petz, V. Klang, and C. Valenta. 2019. The role of viscosity on skin penetration from cellulose ether based hydrogels. Skin Research and Technology 25(5):725–734.
Bond, J., and B. Barry. 1988a. Damaging effect of acetone on the permeability barrier of hairless mouse skin compared with that of human skin. International Journal of Pharmaceutics 41(1–2):91–93.
Bond, J. R., and B. W. Barry. 1988b. Hairless mouse skin is limited as a model for assessing the effects of penetration enhancers in human skin. Journal of Investigative Dermatology 90(6):810–813.
Bronaugh, R. L., R. F. Stewart, and E. R. Congdon. 1982. Methods for in vitro percutaneous absorption studies II. Animal models for human skin. Toxicology and Applied Pharmacology 62(3):481–488.
Campbell, C. S., L. R. Contreras-Rojas, M. B. Delgado-Charro, and R. H. Guy. 2012. Objective assessment of nanoparticle disposition in mammalian skin after topical exposure. Journal of Controlled Release 162(1):201–207.
Chantasart, D., S. Chootanasoontorn, J. Suksiriworapong, and S. K. Li. 2015. Investigation of pH influence on skin permeation behavior of weak acids using nonsteroidal antiinflammatory drugs. Journal of Pharmaceutical Sciences, 104(10):3459–3470.
Chattaraj, S., and I. Kanfer. 1996. “The insertion cell”: A novel approach to monitor drug release from semi-solid dosage forms. International Journal of Pharmaceutics 133(1–2):59–63.
DrugBank. 2020. DrugBank database. https://www.drugbank.ca (accessed April 6, 2020).
Elias, P. M. 2005. Stratum corneum defensive functions: An integrated view. Journal of Investigative Dermatology 125(2):183–200.
Expert Committee on Drug Dependence. 2017. Cannabidiol (CBD). Geneva, Switzerland: World Health Organization Technical Report Series. https://www.who.int/medicines/access/controlled-substances/5.2_CBD.pdf (accessed April 6, 2020).
FDA (U.S. Food and Drug Administration). 1997. SUPAC-SS nonsterile semisolid dosage forms, scale-up and post approval changes: Chemistry, manufacturing, and controls. In vitro release testing and in vivo bioequivalence documentation. Boca Raton, FL: Center for Drug Evaluation and Research (CDER). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/supac-ss-nonsterile-semisolid-dosage-forms-scale-and-post-approval-changes-chemistry-manufacturing (accessed March 11, 2020).
Friedman, D. I., J. S. Schwarz, and M. Weisspapir. 1995. Submicron emulsion vehicle for enhanced transdermal delivery of steroidal and nonsteroidal antiinflammatory drugs. Journal of Pharmaceutical Sciences 84(3):324–329.
Friend, D. R. 1992. In vitro skin permeation techniques. Journal of Controlled Release 18(3):235–248.
Green, P. G., and J. Hadgraft. 1987. Facilitated transfer of cationic drugs across a lipoidal membrane by oleic acid and lauric acid. International Journal of Pharmaceutics 37(3):251–255.
Haque, T., and M. M. U. Talukder. 2018. Chemical enhancer: A simplistic way to modulate barrier function of the stratum corneum. Advanced Pharmaceutical Bulletin 8(2):169–179.
Hatanaka, T., T. Kamon, C. Uozumi, S. Morigaki, T. Aiba, K. Katayama, and T. Koizumi. 1996. Influence of pH on skin permeation of amino acids. Journal of Pharmacy and Pharmacology 48(7):675–679.
Higuchi, T. 1961. Rate of release of medicaments from ointment bases containing drugs in suspension. Journal of Pharmaceutical Sciences 50(10):874–875.
Higuchi, W. I. 1962. Analysis of data on the medicament release from ointments. Journal of Pharmaceutical Sciences 51(8):802–804.
Izquierdo, P., J. Wiechers, E. Escribano, M. Garcia-Celma, T. F. Tadros, J. Esquena, J. C. Dederen, and C. Solans. 2007. A study on the influence of emulsion droplet size on the skin penetration of tetracaine. Skin Pharmacology and Physiology 20(5):263–270.
Jain, A., and R. Panchagnula. 2003. Transdermal drug delivery of tricyclic antidepressants: Effect of fatty acids. Methods and Findings in Experimental and Clinical Pharmacology 25(6):413–422.
Kasting, G. B., and M. A. Miller. 2006. Kinetics of finite dose absorption through skin 2: Volatile compounds. Journal of Pharmaceutical Sciences 95(2):268–280.
Keppel Hesselink, J., and D. Kopsky. 2017. Topical compounded analgesic treatment in neuropathic pain: 8 years of experience. Journal of Pain Management & Medicine 3(2):128.
Ki, H.-M., and Choi, H.-K. 2007. The effect of meloxicam/ethanolamine salt formation on percutaneous absorption of meloxicam. Archives of Pharmacal Research 30(2):215–221.
Ktistis, G., and I. Niopas. 1998. A study on the in-vitro percutaneous absorption of propranolol from disperse systems. Journal of Pharmacy and Pharmacology 50(4):413–418.
Lauer, A. C., C. Ramachandran, L. M. Lieb, S. Niemiec, and N. D. Weiner. 1996. Targeted delivery to the pilosebaceous unit via liposomes. Advanced Drug Delivery Reviews 18(3):311–324.
Lehman, P. A., and S. G. Raney. 2012. In vitro percutaneous absorption of ketoprofen and testosterone: Comparison of pluronic lecithin organogel vs. pentravan cream. International Journal of Pharmaceutical Compounding 16(3):248–252.
Levin, J., and H. Maibach. 2008. Human skin buffering capacity: An overview. Skin Research and Technology 14(2):121–126.
Martin, C. J., and A. Axelrod. 1957. The proteolytic enzyme system of skin I. Extraction and activation. Journal of Biological Chemistry 224(1):309–321.
Meidan, V. M., M. C. Bonner, and B. B. Michniak. 2005. Transfollicular drug delivery—Is it a reality? International Journal of Pharmaceutics 306(1–2):1–14.
Mohammadi-Samani, S., G. Yousefi, F. Mohammadi, and F. Ahmadi. 2014. Meloxicam transdermal delivery: Effect of eutectic point on the rate and extent of skin permeation. Iranian Journal of Basic Medical Sciences 17(2):112–118.
Moser, K., K. Kriwet, Y. N. Kalia, and R. H. Guy. 2001a. Enhanced skin permeation of a lipophilic drug using supersaturated formulations. Journal of Controlled Release 73(2–3):245–253.
Moser, K., K. Kriwe, A. Nai, Y. N. Kalia, and R. H. Guy. 2001b. Passive skin penetration enhancement and its quantification in vitro. European Journal of Pharmaceutics and Biopharmaceutics 52(2):103–112.
Naik, A., Y. N. Kalia, and R. H. Guy. 2000. Transdermal drug delivery: Overcoming the skin’s barrier function. Pharmaceutical Science & Technology Today 3(9):318–326.
Nallagundla, S., S. Patnala, and I. Kanfer. 2014. Comparison of in vitro release rates of acyclovir from cream formulations using vertical diffusion cells. AAPS PharmSciTech 15(4):994–999.
Osborne, D. W., and J. Musakhanian. 2018. Skin penetration and permeation properties of Transcutol—Neat or diluted mixtures. AAPS PharmSciTech 19(8):3512–3533.
Otto, A., J. Du Plessis, and J. Wiechers. 2009. Formulation effects of topical emulsions on transdermal and dermal delivery. International Journal of Cosmetic Science 31(1):1–19.
Padula, C., S. Nicoli, S. Pescina, and P. Santi. 2018. The influence of formulation and excipients on propranolol skin permeation and retention. BioMed Research International 1281673:7.
Park, Y.-D., J.-M. Yang, and Z.-R. Lü. 2011. Skin diseases-related enzymes: Mechanisms and clinical applications. Enzyme Research 464507:2.
PermeGear. 2018. General Catalog, Franz Cell stands image, p. 29. https://permegear.com/wp-content/uploads/2018/08/PermeGear-Catalog.pdf (accessed May 27, 2020).
Plumley, C., E. M. Gorman, N. El-Gendy, C. R. Bybee, E. J. Munson, and C. Berkland. 2009. Nifedipine nanoparticle agglomeration as a dry powder aerosol formulation strategy. International Journal of Pharmaceutics 369(1–2):136–143.
Pomerleau, A. C., C. E. Gooden, C. R. Frantz, and B. W. Morgan. 2014. Dermal exposure to a compounded pain cream resulting in severely elevated clonitinde concentration. Journal of Medical Toxicology 10(1):61–64.
PubChem. 2020. PubChem: Explore chemistry search engine. https://pubchem.ncbi.nlm.nih.gov (accessed April 6, 2020).
Rougier, A., C. Lotte, and H. I. Maibach. 1987. In vivo percutaneous penetration of some organic compounds related to anatomic site in humans: Predictive assessment by the stripping method. Journal of Pharmaceutical Sciences 76(6):451–454.
Schmid-Wendtner, M.-H., and H. C. Korting. 2006. The pH of the skin surface and its impact on the barrier function. Skin Pharmacology and Physiology 19(6):296–302.
Schwarz, J. S., M. R. Weisspapir, and D. I. Friedman. 1995. Enhanced transdermal delivery of diazepam by submicron emulsion (SME) creams. Pharmaceutical Research 12(5):687–692.
Scott, R., M. Walker, and P. Dugard. 1986. A comparison of the in vitro permeability properties of human and some laboratory animal skins. International Journal of Cosmetic Science 8(4):189–194.
Sekkat, N., Y. N. Kalia, and R. H. Guy. 2002. Biophysical study of porcine ear skin in vitro and its comparison to human skin in vivo. Journal of Pharmaceutical Sciences 91(11):2376–2381.
Shaker, D. S., R. A. H. Ishak, A. Ghoneim, and M. A. Elhuoni. 2019. Nanoemulsion: A review on mechanisms for the transdermal delivery of hydrophobic and hydrophilic drugs. Scientia Pharmaceutica 87(3):17.
Sotoodian, B., and H. I. Maibach. 2012. Noninvasive test methods for epidermal barrier function. Clinics in Dermatology 30(3):301–310.
Sznitowska, M., S. Janicki, and T. Gos. 1996. The effect of sorption promoters on percutaneous permeation of a model zwitterion baclofen. International Journal of Pharmaceutics 137(1):125–132.
Tiffner, K. I., I. Kanfer, T. Augustin, R. Raml, S. G. Raney, and F. Sinner. 2018. A comprehensive approach to qualify and validate the essential parameters of an in vitro release test (IVRT) method for acyclovir cream, 5%. International Journal of Pharmaceutics 535(1–2):217–227.
USP (United States Pharmacopeial) Convention. 2019. <1724> Semisolid Drug Products—Performance Tests, United States Pharmacopoeia 42—National Formulary 37, pp. 8311–8322.
Valenta, C., U. Siman, M. Kratzel, and J. Hadgraft. 2000. The dermal delivery of lignocaine: Influence of ion pairing. International Journal of Pharmaceutics 197(1–2):77–85.
Wang, X., and L. Black. 2013. Ex vivo percutaneous absorption of ketamine, bupivacaine, diclofenac, gabapentin, orphenadrine, and pentoxifylline: Comparison of versatile cream vs. reference cream. International Journal of Pharmaceutical Compounding 17(6):520–525.
Watkinson, A., K. Brain, and K. Walters. 1993. The penetration of ibuprofen through human skin in vitro: Vehicle, enhancer, and pH effects. Prediction of Percutaneous Penetration 3:335–341.
Wester, R. C., and P. K. Noonan. 1980. Relevance of animal models for percutaneous absorption. International Journal of Pharmaceutics 7(2):99–110.
Wiedersberg, S., and R. H. Guy. 2014. Transdermal drug delivery: 30+ years of war and still fighting! Journal of Controlled Release 190(28):150–156.
Williams, A. C., and B. W. Barry. 2012. Penetration enhancers. Advanced Drug Delivery Reviews 64(Suppl):128–137.
This page intentionally left blank.






















