
Regulatory Alternatives
Sidney Shapiro
In 1982, U.S. Supreme Court Justice Stephen Breyer, who was then a professor of law at the Harvard Law School, suggested a metaphor to describe regulatory reform. He proposed that regulatory failure should be understood as an issue of match and mismatch. According to Breyer, a regulatory failure occurs when government fails to match correctly the problem and the regulatory tool.
A mismatch can occur for either of two reasons. First, government can misdiagnose the problem that it is attempting to solve and therefore apply the wrong regulatory approach. For example, it is now widely recognized that government regulation of transportation markets was based on the erroneous perception that unrestrained competition would result in monopolization.
Second, even if the problem is correctly identified, regulatory tools vary in their effectiveness and cost. A partial mismatch occurs when government relies on a regulatory tool that is less effective or more expensive than another option would have been.
REGULATORY DIAGNOSIS AND MISMATCH
Critics of regulation argue that the failure to balance the costs and benefits of health and safety regulation has burdened economic development and wasted scarce resources. The Delaney Clause is the most prominent example of this criticism. It requires Food and Drug Administration (FDA) to ban the use of any food additive that causes cancer in laboratory animals, even if there is only a de minimis risk to humans.
A related problem is the government's attempt to remove what Breyer calls "the last 10 percent of a risk." Breyer notes that it is often so expensive to buy this extra margin of safety that the cost of doing so is likely to exceed the benefits to be gained. In such cases the country might be better off with more modest attempts at reducing health and safety risks: because of the high cost of excess risk reduction, critics point out that less money is available to
address more significant risks. For example, it would be far less expensive, and perhaps no less safe, to fence off some Superfund sites instead of cleaning them up to the point where someone can eat the dirt with little or no risk.
In summary, the reformers are critical of policy choices that cannot be reconciled with cost-benefit analysis. They object to spending more money to reduce a risk than the economic benefits that are gained from reducing it.
However, other policy analysts respond that citizens may use a non-economic yardstick when they decide what level of risk is appropriate. We need not presume or deny that consumer attitudes are "irrational" or "unscientific" or that individual citizens necessarily misperceive the actual probabilities of risk. This is a different point. Assume that consumers adequately understand the probabilities of risk, but that they object to quantifying or comparing the costs and benefits of those risks. Citizens may choose certain risk policies, for example, because they promote such values as individual autonomy or fairness rather than a balancing of risks and benefits.
As the philosopher Carl Cranor notes, citizens may decide that some things are more important than producing net community benefits. If risk policies are to serve both economic and non-economic goals, regulators face difficult questions concerning the trade-offs. Mark Sagoff, also a philosopher, has written concerning environmental law: "The role of the policy maker and of the legislature may be to balance what we believe in and stand for as a community with what we want and need as a functioning economy. The future of environmental policy rests on facilitating the balance of interests with morality and one morality with another morality." This is no easy job.
Thus, the choice of the appropriate regulatory goal is confounded by the definition of the regulatory problem. The definition of risk as an economic problem produces one diagnosis, whereas the definition of risk as a problem of other social values produces another.
MAXIMIZING EFFECTIVENESS AND EFFICIENCY
Even after the problem is defined, there remains the question of which regulatory option is the most appropriate. As Justice Breyer has indicated, a partial mismatch occurs when the government relies on a regulatory tool that is less effective and more expensive than an alternative.
According to many critics, a significant source of regulatory failure is the use of rigid, highly bureaucratic command-and-control regulations. In command and control regulation, government specifies the method of compliance that a regulated entity is to use. For example, the regulation of air, water and workplace conditions relies on the use of the "best available technology"; that is, all firms are required to act to the level of the best
available technology to reduce a particular risk. Another example is FDA requiring drug manufacturers to adopt a particular method to ensure the sterility of a product.
Critics have made three general criticisms of command-and-control regulation. The first is an efficiency-based criticism, that is, that command-and-control approaches are inefficient in terms of society's resources. They are also expensive because uniform standards ignore diversity among firms' abatement costs and because they may not permit the regulated entities to adopt new technologies that are equally effective and less costly.
A second criticism relates to innovation. Command-and-control regulations discourage research and innovation concerning new regulatory technology if they do not permit the adoption of such technology.
Finally, command-and-control regulations encourage strong political opposition because they are inefficient and discourage innovation. By comparison, the critics assert, less stringent—that is, more reasonable—regulation would be easier to adapt and adopt because there would be less opposition. Thus, critics of command-and-control contend that it is inefficient, that it discourages innovation, and that it encourages regulatory opposition.
These charges are true, no doubt, to some extent but they also overstate for several reasons the failure of the existing systems. First, the implementation of command-and-control schemes tends to take into account intraindustry and other differences that affect efficiency. Agencies typically use variances and other devices to account for these differences. Secondly, agencies more often tend to use efficient systems of command-and-control than inefficient systems. Finally, some research has indicated that regulatory entities tend to oppose less stringent regulatory initiatives with the same vigor as they oppose stricter proposals. The reason is simple: delay typically saves regulated entities money, and given enough time, the political environment could change in favor of the regulatory entity.
Nonetheless, reformers urge three kinds of reforms of command-and-control: more flexible regulations, market-based incentives, and voluntary approaches. It is important, first, to note that command-and-control regulations also come in three flavors. A command-and-control regulation can be a specification standard, a design standard, or a performance standard. A specification standard is one that specifies a particular technology to be used. For example, the U.S. Congress has directed that new hazardous waste landfills and surface impoundments install two or more plastic liners, and specifies the exact nature of the plastic to be used and the width to be used. That is a specification standard.
A design standard is one that specifies the use of a model technology that meets a legislatively articulated requirement, but design standards also permit individuals the freedom to achieve the same outcome by any other means. As
noted, air and water pollution goals are usually stated in terms of a best available technology. The Environmental Protection Agency (EPA), for example, chooses a pollution limitation based on the amount of abatement that can be achieved by the current best available technology. A regulated entity, however, may use any abatement method as long as it will reduce pollution to the level of the model best available technology.
A performance standard is stated in terms of some regulatory goal. It does not specify the method that must be used to achieve that goal. For example, the Occupational Safety and Health Administration (OSHA) has set a limit on the amount of accumulated grain dust in grain elevators to reduce the risk of explosion. An elevator may choose any method it wishes to reduce dust to the specified level, as long as it meets that regulatory goal.
PROPOSED ALTERNATIVES TO COMMAND-AND-CONTROL
Some of the suggested alternatives are based on market incentives. Regulations can be written to take advantage of market-based or financial incentives to direct behavior toward a regulatory goal. This approach includes the following options:
-
Taxes or charges. Under this strategy, the regulator imposes a tax or a charge on the behavior to be regulated. For example, the government might tax each pound of a particular pollutant at a flat or even an escalating rate. Other examples include bottle deposits, increased gasoline taxes, or even higher taxes on old cars that pollute more.
-
Liability provisions. This approach requires a regulated entity or person to pay damages attributed to the harm that the entity or person has caused. Although liability regimes have notorious problems, modified approaches, such as no-fault compensation, might be used. For example, the focus of malpractice litigation might be shifted from individual physicians to the health organizations under whose auspices they practice.
-
Information reporting. This approach requires regulated entities to. report certain kinds of information to the public, which in turn can create political and legal pressure via liability provisions to reduce risks. OSHA, for example, requires employers to distribute a Material Safety Data Sheet concerning each chemical or toxic substance to which a worker is exposed. Under the Clean Water Act, companies must file monthly Discharge Monitoring Reports that are public, and indicate the extent to which that firm is polluting.
-
Subsidies, grants, and tax breaks. These provide various forms of financial assistance from the government.
-
Technical assistance. These programs provide government advice and technical help to induce targeted entities to prevent or reduce pollution.
The final set of reforms that appear in the literature deal with voluntary approaches, of which two should be mentioned here:
-
So-called "challenge regulations." Under this approach the government identifies a goal and gives targeted entities time to select and implement effective means of achieving that goal. In back of this challenge lies the implicit threat that, "If you do not do something, we will regulate." For example, in the early 1980s OSHA adopted several enforcement programs that promised reduced inspections for voluntarily achieving below-average workplace injury rates. In the late 1980s, as another example, EPA established the 35-50 Program, in which companies that emitted 17 targeted toxic chemicals were challenged to reduce their emissions by 50 percent by 1995.
-
"Consensus standards." This approach has an agency using industry-generated standards as the basis for regulation. For example, the regulation of many toxic substances under OSHA's jurisdiction is based on permissible exposure limitations recommended by the American Council of Governmental Industrial Hygienists.
Serious consideration should be given to these regulatory alternatives. Yet, at the same time, it can be argued that the status quo has not performed as badly as many of its critics contend. Moreover, the alternatives that reformers have proposed are hardly perfect themselves. Critics have succeeded in exposing the maladies of current approaches to regulation for all to see, and from a distance, their alternative approaches look better by comparison. A closer look at the alternatives, however, reveals that they also pose implementation problems.
The correct public policy question, therefore, remains "Which method of regulation produces the fewest problems in reaching the intended regulatory goal?" As Neal Komisar has aptly concluded, "In a society of millions of persons with ever-changing technology, selecting the best means of preventing injury means carefully considering and comparing highly imperfect alternatives."
Specific Techniques
David Pritzker
The Administrative Conference of the United States (ACUS) is a federal agency, although a tiny one. It has been in existence for more than 25 years, for the purpose of studying administrative procedures and for advising the U.S. Congress, federal agencies, and the President on improving those procedures.1
ACUS STUDY AND RECOMMENDATIONS
ACUS has been studying various problems of administrative procedure. Some of these are quite narrowly focused and some are very broad based. In the late 1970s ACUS studied governmental use of voluntary consensus standards. That investigation disclosed a substantial private-sector engine that had been generating standards for many decades. There was a time, for example, when fire companies' hoses might not hook up to different sources of water because they were not all the same size.
A standards development industry thus arose. Through the 20th Century the industry has grown to encompass a variety of standards such as technical, safety, health, library, data and handling standards, among others. Many of these came into being by bringing together a committee of experts (or of purported experts), sometimes with a balance of manufacturers or producers and those who were buyers or users.
This is the model that ACUS had in mind in the early 1980s when it promulgated its recommendations to Congress and federal agencies on regulatory negotiation (sometimes called negotiated rule-making or ''reg-neg''). The model was, in appropriate circumstances, to bring together a committee of representatives who could speak not necessarily by authority of particular
interest groups but who could represent the points of view of the different interests affected.
THE ADMINISTRATIVE PROCEDURE ACT
Understanding of how this model evolved requires knowledge of some historical facts. In the 1940s Congress passed the Administrative Procedure Act, a recognition that Congress and the courts could not do everything that the people wanted the federal government to do. Numerous agencies were in existence by then, and many more were established in the succeeding decades, particularly in the areas of health, safety, and the environment. The Act passed by Congress in the 1940s established the ground rules for how these agencies were to work.
Agencies have a quasijudicial function (so-called adjudication), and they have a quasilegislative function (so-called rule-making). The Administrative Procedure Act requires the agencies, when they are considering the adoption of a rule, to let the public know through an announcement in the Federal Register that the agency has identified the problem, to give some indication (which might be quite brief or it might be quite extensive and detailed) as to what the agency is thinking about doing about the problem, and then to give the public an opportunity to submit its comments, generally in writing. This is called a Notice of Proposed Rule-making.
The Act does not specify how much time the agency must give the public to respond. In some instances, either through other legislation or through subsequent court decisions, agencies were required to give the public an opportunity for an oral hearing, particularly if the public asked for it. Essentially all the agency must do, however, is to say what it has in mind, give the public an opportunity to respond, and then publish a notice of a final rule-making containing the text of the rule and an explanation of how it responded to the comments that had been received.
Often, that is not the end of it, because those who do not like the regulations can go to the federal courthouse and try to do something about it. This is largely what has been tying the process up.
THE ALTERNATIVE: NEGOTIATION
To address this problem ACUS had in mind creating, in some instances, a committee that could try to "negotiate" a rule. This is not simply a process of turning regulation over to a committee that would largely comprise private citizens, and thus an abdication of agency responsibility. It is a procedure that
gives people with different viewpoints about the problem—and about what the solutions might be—an opportunity to talk with each other in a public session in which the agency participates. The group would have incentives to come to agreement—primarily, that if they did not agree, the agency would make the decision by itself.
The objective was to offer to people of goodwill an opportunity to recognize the legitimate needs of the others represented and to see if through a negotiating process, including with the agency, a consensual resolution in the public interest could be achieved.
RECENT DEVELOPMENTS AND THE FEDERAL ADVISORY COMMITTEE ACT (FACA)
A number of agencies have tried the regulatory negotiation process, with some success. In 1990, to encourage its further use and to answer some of the legal questions that had been raised, Congress passed the Negotiated Rule-Making Act of 1990. Increasing numbers of agencies have been trying it ever since. The National Performance Review under the Clinton Administration has called for greater use of this technique. Figure 1 puts the negotiation process in a broader context of ways to build consensus.
Several consultation and consensus building approaches is available to the regulatory agencies, a number of which are in use by the Food and Drug Administration (FDA). Figure 1 notes the significance of the Federal Advisory Committee Act (FACA). FACA was passed about 20 years ago for the purpose of regulating the establishment and behavior of advisory committees created by statute or by the federal agencies to get advice. The problem that FACA was trying to solve was a proliferation of uncontrolled advisory committees that might be acting behind closed doors. Essentially, FACA now guarantees that the proceedings of advisory committees are open to the public, and it regulates other aspects of a committee's work. A committee that is subject to FACA cannot operate confidentially, for example, and must observe certain requisites of form and process. For example, with certain exceptions for matters of national security, a FACA committee must hold open meetings.
What that means is that if a committee is dealing with the public's business, there may be some limitations on the ability of the members of the committee to meet privately outside of the publicly announced schedule. That has been a problem that we have had to deal with in the negotiated rule-making context, since these negotiating groups are usually FACA committees. When Congress considered the Negotiated Rule-Making Act, it was urged to make modifications to FACA to facilitate the ability of the committee members to negotiate, but Congress was not enthusiastic about doing so.
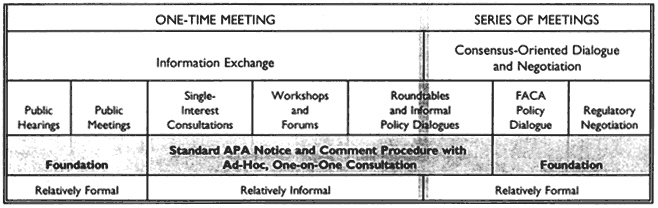
The objective of regulatory negotiation is usually to develop the actual text of the rule rather than only general principles or policies to guide the agency in its own drafting of a rule. The process is thus at the FACA-governed end of the scale.
Examples of techniques that are subject to the act are illustrated to the right of the shaded vertical line. In some cases roundtables or informal policy dialogues may in time become preferred sources of advice for the agency, and therefore fall under FACA.
Outside of FACA coverage are the relatively informal types of procedures, which can be forums, workshops, or consultations with single interests or more formal but one-time consultations, which can be either announced public meetings or public hearings that may have the formalities and additional procedure of cross-examination or other protection and that can perhaps be opportunities to respond to what the speakers have said at the hearings. Agencies can use a variety of techniques to develop consensus.
AUDITED SELF-REGULATION
ACUS is currently conducting a two part study of alternatives to direct regulation by federal agencies. The first half of the study led to a recommendation, adopted in June 1994, under the heading "use of audited self-regulation." In some industries and for some sectors of the economy it has proved to be workable and effective for an agency, under congressional authorization, to delegate to private, self-regulatory organizations the ability to implement and enforce laws or regulations, with the agency retaining the power of independent action and review. Among the best-known examples of this audited self-regulation are the stock and commodity exchanges. They in turn are monitored and audited by the Securities and Exchange Commission and the Commodity Futures Trading Commission.
These regulatory schemes have generally been fairly successful. They have also been applied in a number of other settings, including standards of medical care under government insurance programs, agricultural marketing agreements, certification of medical testing laboratories, and others. ACUS's work was directed at identifying the procedures that would ensure that they would work well, that is, both effectively and fairly. Effectiveness seemed to require finding a private group, the self-regulatory organization, with both the ability and the incentive to carry out the regulatory program. The agency also had to have the ability and the incentives to oversee the process and make it work. This included the need for substantive expertise on both sides. On the part of the agency, this includes knowledge of organizational behavior as well as the statutory authority to engage in this form of delegation.
As for fairness, the self-regulatory organization had to have sufficient procedural protection to constrain it to behave essentially as the agency would with respect to the regulated community and the public. Control would initially lie with the private organization, with periodic monitoring and right of appeal to the agency in case of improper behavior.
ACUS is now examining a further step: What safeguards are needed when there is no such self-regulatory private organization? There are different terms for this—self-implementation and sometimes self-enforcement—although the essence is the government's reliance on agents or employees of the regulated entities themselves to interpret and enforce the applicable laws and regulations, with the agency again being limited to monitoring the effort.
The private entities must have sufficient incentives to want to comply with the rules. Current research involves looking at what these incentives might be. For example, a regulated entity that has undergone a markedly successful inspection might be exempted from inspection again for a longer period of time instead of annually or biannually. Other self-regulatory schemes are built around reporting requirements. If the regulated firm reports its own violation, the potential penalties would be mitigated. The incentive systems are important ingredients in many such regulatory schemes.
Negotiated Rule-Making
Philip Harter
When considering alternative regulatory approaches it is important to ask not only whether it will work but also whether it is politically acceptable. The structure of negotiated rule-making (Reg-Neg) is a response to both sets of considerations.
"REG-NEG"—DOES IT WORK?
What is the experience with negotiated rule-making? Does this idea of empaneling a group of affected people and the agency to negotiate a rule actually work? The answer is yes, in numerous instances, of which several examples may be instructive.
Recently, the reformulation of gasoline to reduce toxic emissions was a negotiated rule. The Clean Air Act required a dramatic reduction. The Environmental Protection Agency (EPA), however, realized that, politically, the agency could not get a rule out through traditional rule-making. Thus, it empaneled representatives from the refiners, the auto industry, clean air advocacy groups, environmental groups, and the states. Over a 4-month period, the group did indeed develop the formula for reformulated gasoline.
To focus on how the process works, consider another example, the Federal Aviation Administration's (FAA's) rule involving pilot flight duty status time. FAA regulates how long pilots can fly, how long they must rest, how many hours a month they can fly, and the like. The original duty time rule, issued in 1947, was found in two paragraphs in the Code of Federal Regulations. In 1947 the pilots were former B-25 pilots fresh from World War II. They were 25 years old. They flew DC-3s nonstop all the way from New York to Philadelphia.
Over the years, things changed: DC-3s became 747s, the 25-year-olds became 65-year-olds, and the route became nonstop to Tokyo. The situation became very different, with many different demands placed on the pilot. FAA attempted to effect the necessary changes, but every time that it promulgated
a proposal, if it favored the pilots the airlines had the wherewithal to block it. If it favored the airlines, pilots had the power to block it. FAA was stopped dead in its tracks. Unable to issue a new rule, FAA began to issue interpretive bulletins. Over time, what had been a two-paragraph rule became a set of interpretations of more than 1,000 pages.
FAA, having heard about negotiated rule-making but dubious about the probability of success, created a negotiating committee and set to work. Its doubts were based on the failures of the industry and agency to come to terms in the past; the issue seemed too controversial. Then, an odd thing happened in that committee. They agreed. They were able to talk about the real practical needs in the cockpit and the needs of the pilot—topics that had not been open to discussion before. For example, the pilots coming in from Paris land at Kennedy Airport, but there is no good place to stay at Kennedy Airport. They have to go into the city, adding another half hour to flight duty time. Now, every time that you get in an airplane you are trusting your life to a negotiated rule.
Another example is the ongoing negotiation of rules for the construction of steel buildings. The Occupational Safety and Health Administration (OSHA) looked at the various statistics, from which it was clear that the construction of steel buildings was probably the most hazardous occupation outside of mining. The agency's first response was that everybody who is exposed to a fall of more than 6 feet should be tied off. Interestingly, both industry and the union showed that it would be hazardous to tie off the first people who tie the structure together. They really ought to have freedom up to 30 feet.
One of the people on the committee said, "It is really pretty interesting that those guys don't fall. All the accidents are either above 30 feet, where people are already violating the existing rule, or below 10 feet, which is where the safety precautions really kick in, or doing something else." Getting the people together at the table to talk about how to build a building, what the needs are, and where the real risk is changed the perspective of the regulatory agency fundamentally—something that can occur only through this kind of direct dialogue.
THE PUBLIC AT THE NEGOTIATION TABLE
The beginning of negotiated rule-making is a sophisticated analysis of the kinds of interests at stake (who is substantially affected by the rule) and then the selection of representatives of those interests. Creativity sometimes is needed to identify members of the general public. For example, in the reformulated gasoline rule, who could represent everybody who drives? No one is going to spend 6 months of their life negotiating a rule to save $200 a
year; yet, someone who can aggregate those interests and represent the common approach must be found.
The answer in this case turned out to be farmers. Gasoline is a major cost for their business, they are certainly well organized, and they are certainly aggressive in protecting that interest. Therefore, farmers were at the table to represent people interested in gasoline costs. The regulatory agency was also represented at the table, participating in the give and take of it and working toward a consensus on the recommended rule.
CONSENSUS
The goal of the process is consensus. For the purposes of a negotiated rule, "consensus" means that each interest represented at the table concurs—basically, unanimity. Each interest that participates has a veto.
Although critics of the process have expressed doubts about the workability of this process, it in fact has two major beneficial effects. First, if each interest has a veto, it is safe to come to the table. Recall the flight duty rule: each side had enough power to make sure that the agency could not issue rule that it did not like. Neither side however, had enough power to force it to issue a rule that it did like. A veto allows protection in the same way.
The second effect is that the group develops to some degree a "lifeboat mentality." Each party may feel about the other, "I do not like you very much and I hope I never see you again after we reach the island, but you know, we are not going to reach the island unless we row together." It converts the committee from a group of disparate interests into one with a common problem: How are we going to write a rule that reconciles all of these divergent interests within the statutory prescription?
It also has the effect of forcing people to look at the rule as a whole, as opposed to its individual parts. Negotiating rules is akin to buying a house. You may feel that a particular house is a great house but it has a bad yard, or it is a great house but you do not like the kitchen, or it is a great house but it needs another bathroom, or it is a great house but . . . Unless one is exquisitely unimaginative, there is always something wrong with a house.
Then there comes a time when some house must be bought. Resource constraints are realities. The perfect house is not available. For every house, there are going to be parts you do not like. The same goes for a rule. All of this comes with the definition of consensus.
DECISION-MAKING DYNAMICS
In traditional technical data-based rule-making, people fight over the facts. Each interest argues that the facts prove that they are right. Why do they do that? They do it because they do not have control of the decision. They do it to constrain the discretion of the decision-maker. The facts are pushed well beyond what the science and technology can bear, and the parties are on the fringe of what is unknowable. In negotiating a rule the question is, how much information does a group of knowledgeable people really need to make a responsible decision?
It is usually possible to get rid of the 10 or 20 percent fringe. For example, the OSHA limit on occupational exposure to benzene had been kicked around for 9 years. OSHA issued the benzene standard, it went to the U.S. Supreme Court, and the Supreme Court reversed it and sent it back. OSHA then tried to negotiate it.
When the people got together they talked about that history. It had been going on for about 9 years, but that group of people, all the prominent people for all the interests, had been in the same room together only once before—and that was when they heard the oral arguments in the Supreme Court, where they certainly did not talk to each other. In the negotiating process, everyone was together, talking to each other for the first time.
As to the central issue of exposure limits, it was very clear that a doseresponse curve for benzene would never be resolved. It is still being fought out in the technical journals. One of the parties then said, "We are never going to agree on the facts. The question is, what are we going to do about it? What is a responsible regulatory approach to it?" That turnaround is critical. It allows the parties to grapple with the real concerns of the driving policy on the basis of what can be agreed upon.
BENEFITS OF COLLABORATION
Negotiated rules tap very practical insight. Very practical people from the shop floor may be involved, and not just those from an agency who are necessarily distant from the action, working with derivative knowledge. The participants are people who are actually doing the work. They are then probed by others who are concerned about it.
Again, the reformulated gasoline process offers an example. The Clean Air Act prescribed one number for the evaporation limits. The petroleum industry, however, kept advocating for a very high number, that is, for highly volatile gasoline. Yet, it became clear that they could manufacture gasoline that was much less volatile. Through the probing and discussion among the
group, it emerged that what the oil companies were really concerned about was that they could not achieve an effective limit on a gallon-for-gallon basis. Feed stocks differ, there are different kinds of equipment in the refinery, and some old material remains in the pipelines. However, they certainly could make it on the average. Hence the solution: the rule provides a very low number and very reduced volatility on average.
Here was a creative approach that could not have been done before. It could not have been done through notice and comment, and it probably could not have been done in legislation because it took a long time to figure out what the real problem was and how to fix it.
POLITICAL CONSENSUS
There is a further benefit to the negotiated rule-making process. Representatives of the parties sit at the table, haggling it out, and at the end they find an agreement. Although they are building the technical rule, they are also building a political consensus. That means that people are not going to endrun the rule to the Congress or challenge it in the courts.
Of the 20 or 30 negotiated rules—and they tend to be the most controversial ones—there has so far been only one challenge to a rule in which an agreement was reached, and that is in a bizarre setting. Again, it was the reformulated gasoline rule. After the rule had been negotiated, one of the producers of ethanol persuaded President Bush to overturn EPA, which he did in the final throes of an election. There are some people who have used this example as a criticism of the process, but in fact it is not. What happened in fact was that the committee came back together and persuaded the new President to overturn the old President, with one change. The parties to the original negotiation are now suing EPA to put the rule back to the way it was originally. In effect, the process had built the political consensus.
REG-NEG OUTCOMES
The result of the negotiation process is anything but a least-common-denominator approach. Interestingly, a negotiated rule is usually significantly more stringent than the one that the agency would have issued on its own, and yet, it is cheaper to implement.
How can that be? The result is practical. The agency knows where the shoals are and what to avoid. Everyone learns what is wasteful. The averaging in the reformulated gasoline rule would have never been done if it were a rule made by the agency. If EPA had issued that rule on its own, the Reid vapor
pressure of reformulated gasoline would very likely have been somewhere halfway between what it is now and what was originally proposed, without any averaging. The air would have lost, and so would the refiners.
GETTING REG-NEG STARTED
What do we look for when we consider a Reg-Neg? Talk to the agency first, to get its description of the issues and the people who would be affected. Then talk to the people on that list. Ask everyone the same thing: what are the issues and who is affected? Frequently, the views of the private sector are very different from the agency's. Part of looking at the prospects of negotiated rule-making is issue definition.
For the process to work well there should be a limited number of interests that will be significantly affected, somewhere between 15 and 20. On the one hand that seems very small for a rule of major consequence. The burden, however, is usually carried by about that number of organizations. Caucuses will coalesce; it is not as small as it looks. On the other hand, 20 to 25 people can seem like a lot. It turns out that only five people talk anyhow, so it does boil down to a smaller number, but that larger number is still needed for representation.
The second thing to do is to identify appropriate individuals to represent those interests and to make sure that each issue, each major point, is discussed.
CHOOSING THE PROBLEM
The process works fairly well, although it does not work in all instances. There are conditions for likely success, and some degree of analysis is necessary to determine if the issues are of the sort for which a negotiated rule would be appropriate. This is the next question. Are the issues known, mature, and ripe for discussion? Nobody, let alone a committee, debates things in the abstract. Solving a problem that is not here yet is not a compelling circumstance. What this means is that the issue should be on the political agenda. The train is moving, something is going to happen. The decision-making process is beginning, and the outcome is on the horizon. One of the reasons for negotiating is to control that outcome.
OTHER REQUISITES
For the process to succeed, no party to it should have to compromise a
fundamental value. This does not mean an important value; it means a value that is more like an article of faith. For example, no one could negotiate an end to the abortion issue right now or negotiate which of several religions is better.
Another criterion is that the issue should involve diverse aspects or elements. The essence of a negotiation is trading off one issue against another. For example, in the reformulated gasoline rule-making the petroleum companies agreed to a very stringent rule in return for an averaging process.
The next question is whether the outcome is really in doubt, whether there is a countervailing power. People do not negotiate if they can achieve their goals themselves. They come together in committees to control the outcome and to make sure that what they are concerned about is discussed. The pilots could not get what they wanted out of FAA. Neither could the airlines, nor could FAA. Everybody had a different kind of access. Some might have access to the White House, some have access to the Congress, and some have access to the Washington Post. It makes sense to come together to negotiate a truce in this war of attrition. If all of these conditions are met, the parties will likely view it to be in their interest to negotiate.
Last, and critically important, is the fact that the agency must be willing to rely on the process and to participate in it. Some agencies have been exquisitely creative in sabotaging things they do not like. As to whether this is an impermissible delegation of governmental authority to private individuals, the answer is ''no.'' By the definition of consensus, everybody must agree—including the agency. As one senior agency official has said, "I am the one authorized by law to make the decision, but it does not invalidate my decision simply because everybody else agreed with me."
If the agency does not participate, the group will talk and talk until somebody gets tired and walks out, because the committee will not reach agreement. It is essential for the agency to be there.
CONSTITUENCY PRESSURES
As to the possibility that public pressure on the Congress is operating on the issues in question, the experience has been that once a committee is formed to address an issue, Congress usually does not intervene. If all of the constituents are sitting at the table saying that this is the forum in which to work it out, Congress tends to stay out of the loop. In several instances in which Congress was actively involved, the parties said, "We are finally solving the problem legitimately here and we will keep you posted." Several of the congressional representatives came to the first couple of meetings, and being confident that the issues were being addressed legitimately, they went away.
ACCOUNTABILITY
Before the agency engages in a negotiated rule, it publishes a notice in the Federal Register that it is about to engage in the process, usually with a list of the interests that would participate in it. That notice serves as an invitation to others to request participation. A few additional interests will often show up in this way, so save a few seats.
That initial public notice has two purposes. One is to make sure that anybody who is interested has an opportunity to participate. It also ensures that nobody can subsequently complain that they were not invited or that they could not participate. That is enormously important politically.
A critical part of the process is that all of the meetings be open to the public. Sometimes, if it is an area of considerable interest, there can be a lot of people, but the representatives get used to it after awhile and continue to talk candidly. This public openness makes an important contribution to accountability. People can see what is happening; people in the audience can present issues to be considered.
Reg-Neg Process
The following outline of the steps in a negotiated rule-making process was provided by David Pritzker:
-
Evaluation
-
Identify issues and deadlines.
-
Identify interested parties.
-
Compare with selection criteria.
-
Confirm management interest.
-
Select convener.
-
-
Convening, Phase I
-
Identify additional parties.
-
Discuss the negotiated rule-making process with parties.
-
Discuss issues with parties.
-
Determine willingness of parties to negotiate.
-
Report to agency.
-
Obtain agency management commitment.
-
Preliminary selection of 15 to 20 participants.
-
-
Convening, Phase II
-
Obtain parties' commitments to negotiate.
-
Publish Notice of Intent to Negotiate in Federal Register.
-
-
Process Federal Advisory Committee Act (FACA) charter.
-
Select a facilitator or mediator.
-
Respond to public comments on the Notice.
-
Adjust committee membership if necessary.
-
Arrange organizational meeting.
-
Arrange committee orientation and training.
-
Negotiations
-
Establish ground rules and protocols.
-
Define consensus.
-
Set meeting schedule.
-
Publish notices of meetings.
-
Review available information and issues.
-
Review draft rule or proposals.
-
Establish work groups or subcommittees.
-
Negotiate text or outline of proposed rule.
-
-
Rule-Making
-
Negotiations concluded.
-
If consensus is reached on language of rule:
-
Agency circulates draft for internal and external review.
-
Agency publishes consensus on draft rule.
-
-
If consensus is reached only on issues or outline:
-
Agency drafts proposed rule.
-
Agency circulates draft for internal and external review.
-
Agency publishes Notice of Proposed Rule-Making (NPRM).
-
-
If consensus is not reached:
-
Agency proceeds with rule-making with discussions as guide.
-
Agency drafts and publishes NPRM.
-
-
Draft rule is subject to public comment.
-
Committee is notified of public comments.
-
Agency revises rule if necessary.
-
Agency publishes final rule.
-
QUESTIONS ABOUT REG-NEG
Question: Who decides whether an issue deserves a guideline, a recommendation, or a regulation: the industry, the agency, or the public? Who is it that does the negotiating? Who represents the industry and the other interests? How is the venue established, and the shape of the table determined? What is the work product of the negotiation? How does an agreement get established? Does the requirement of unanimity reduce the likelihood of success?
David Pritzker: As to who decides who is going to be there, in a technical sense, under the provisions of the statutes, the agency ultimately decides. As a practical matter, however, the way that it has actually worked is that the agency or someone on behalf of the agency talks to the interested parties, and through these consultations, specific suggestions emerge as to who might best be representative of the different points of view. It has usually worked better if, in such consultations, the agency decides what organizations or what interest groups should be represented and then leaves it to the groups or the organization to determine exactly who will come to the table.
As to how appropriate issues are determined, like so many other things, it depends. To the extent that the agency knows the issues that it wants the group to address, the agency can say what it has in mind. If they have some general ideas but are not sure which ones the group could best work at, it can ask for their opinion, either before the process gets started or once the committee has been convened. Always bear in mind that this process is a flexible tool.
In part the choice of issues may depend on how well formed the agency's ideas are. If they are less well formed and they either want—or for political or other reasons are being pressed to have—that question decided by the other nonagency participants, that is okay, too. The process is broad enough to allow for the politics of the situation. Ideally, the entire group, including the agency, should come to a consensus on which issues will be negotiated.
As Phil Harter just mentioned, one of the criteria for Reg-Neg success is the outcome is genuinely in doubt. The issues are known and ripe for a decision. Those are questions for the agency and the other interested participants to explore. The process is broad enough to deal with various combinations. The notion of the convening process is that of an exploration that the agency starts to determine what is out there. Given all of these factors, the ultimate decision on whether to use reg-neg is simply whether it makes sense to bring together a committee to try to work out the issues. There is no black-and-white rule as to who decides what. Once the committee is together, the committee must be able to arrive at some consensus about its agenda and the nature of the consensus they are seeking on the rule itself. The flexibility should be fully stressed, for some of the regulatory agencies believe that the process has much less flexibility than is the case. In fact, an agency can mold this to its needs and its particular circumstances.
"Unanimity" and "veto" also need to be properly understood. Everyone at the table does have a theoretical veto. What that means in practice, however, can be different. First, the group could agree to proceed without the requirement of unanimity. Assume that unanimity is required and that the group has debated for many months but all members cannot agree. Suppose the count is 24 to 1. Does the group have consensus? No. What is the agency going to do?
If the basis for the negotiations is "if you don't have unanimous agreement you have nothing," then there is no consensus. What the agency does have, however, is the experience and the record of the negotiations. It knows what the issues are, it knows the feelings of the group as a whole, and it knows the feelings of the individual representatives of the constituencies. The agency is now free to do what it will, but in general, the agencies that have found themselves in those circumstances have come forth with rules based on the majority view. On a couple of occasions they have been sued, but for the most part the courts readily upheld the agency's rule. The standard traditionally used in court is whether the agency acted arbitrarily and capriciously. It is likely to be difficult to show that the agency was arbitrary when it has been through this negotiation process and has only one or two holdouts. The agency has the benefit, even if it does not have consensus, of having heard the debate. It has heard suggestions for solutions to the problems, and it has heard the responses. It knows what the real-world consequences are likely to be. That is what happens in practice.
Panel Discussion on Applications of Negotiated Rule-Making to Issues in Blood Banking
Kathryn C. Zoon: It has been helpful to have the opportunity to discuss these issues regarding FDA and its regulatory responsibilities. The mission of the Center for Biologics Evaluation and Research (CBER) is to protect and enhance the public health through the regulation of the products for which it is responsible, and to make those products available. Whatever regulatory course is taken and whatever changes may ultimately be made, that mission will not change.
With that preface, it is worth noting that the discussion of regulatory alternatives actually relates to a number of areas that CBER and other FDA components have discussed of late—new regulatory paradigms.
REGULATORY INITIATIVES AT FDA
I would like to share with you some of the initiatives currently being taken by CBER, and how they integrate with broader initiatives such as government streamlining/government reinvention, including the National Performance Review activities.
CBER has begun a strategic planning process. Some very important and fundamental issues continue to be in our minds. One is a reaffirmation of science-based decision making. If we lose sight of the science in regulation, the public health's interests are not well served.
We also need to examine how we are using our resources. With downsizing and refining how we do business, the expectations are not to do less in all cases. We need to be creative and still fulfill the mission, but to do it with less. That creates some challenges and controversies with respect to a number of areas.
We are also looking at partnerships. As we go into the next decade, the ability and the need to rely on partnerships—partnerships within the government, partnerships with industry, and partnerships with academia—in order to effect useful and important regulations to protect the public health, become very critical. We see this now in a number of our international harmonization efforts. We have been actively engaged in partnerships. Those partnerships will
continue to exist and will be enhanced. We need to have outside input to understand what the agency is asking people to do, how we do that, and how in the end we can serve the public in terms of having a blood supply in which they feel confident.
We must think globally. We do not live in an isolationist community where decisions made outside our country do not affect us. We must become more involved in those decisions and become partners in making those decisions. They affect the area of blood regulation as well as the other areas that come under our regulatory purview. This is true for FDA as a whole; CBER is not unique from that point of view.
FDA IN THE FUTURE
In our strategic planning process, as we look to the year 2004, we see a more closely managed center, one in which our processes are defined and managed to set up level playing fields, internal and external. It is important for our own staff as well as people in the outside communities to be able to work and interact and have input into the agency. That is very important in public decision-making.
We see that this transcends not only our regulatory processes but also our research processes. Focusing and enhancing their contributions to our regulatory mission will continue to evolve. It is important to have public confidence. We cannot stop trying to regain public confidence if it is not where it should be. That is a job that for all of us—people in the blood banking industry, and people in the government. If we need to take steps to do that, then that is part of our job.
For us to do our business more effectively and more efficiently, we also see the need for information management systems. Again, in times of limited resources the ability to deal with the issues will require an interactive database in which information can be drawn not only from our own agency but also from other sources inside and outside the federal government. We need to be able to adapt and manage that, and to share that information where appropriate.
INITIATIVES AT CBER
A number of specific initiatives are going on at CBER. One involves the re-write of the general biologics regulations, as well as the blood regulations. This is an excellent opportunity to look at our processes and the flexibility of our regulatory structure. Some of our rules and regulations that may have served their purposes decades ago are now outdated and need more flexibility.
Science continues to move at a rapid pace in the area of biologics. To keep up with that pace, one needs to be able to make adaptations relatively quickly and to not be confined by changes in rule-making, which tend to be very cumbersome. Over the years we have tried to use workshops, "points to consider" documents, and memoranda in a more flexible fashion. There are ups and downs to all of these things. Alternatives are never perfect, and we keep looking for ways to improve the ability to get input, to communicate and have decision-making based on scientific data as well as other factors that affect a particular area under discussion.
In the next 5 to 10 years the focus on performance-based management will be our future. Under the National Performance Review, some of our initial negotiations with industry in developing the user fee program for new prescription drugs provide an example of not quite negotiated rule-making for the agency, but this is probably as close as we have come to collaborative discussion of what we thought would help improve processes as well as performance. It has had a lot of benefit to the public, the industry, and the agency.
WILLINGNESS TO EXPLORE PILOTS
FDA has not been reluctant to do pilot projects to examine new regulatory paradigms. That is still the case, although in fairness, while issues have been raised about FDA regulation, I do not perceive the process and the results of the process to be broken. Our processes have served the public health in this country, but that does not mean that we cannot look at new ways of doing business and making it better. At the end of the analysis, are we willing to look at and evaluate how we do our business? I believe the answer to that is "yes."
In order to have a good and effective pilot project there are several things we need to do. One is to define clearly what the pilot should address. Two, the pilot should have substantial public health impact. Three, it would need to be viewed as something that is useful.
It is important not only to develop the pilot project, but also to do an evaluation of the pilot. Has it been effective? Then one must determine whether or not those changes are doing what you anticipated they would do: Are they making it better? Are they making it worse? Have they made no change at all? Although one can experiment, as with any experiment one needs to evaluate the information critically and decide what has been learned and gained. That way one can then apply the process to other areas and other issues with the expectation that it will have a positive impact. What I would like to see is some discussion of areas and targets that might be pilot experiments that we can bring back to our organization and discuss candidly.
The discussion brought up here is certainly the beginning of a dialogue. I look forward to continuing the dialogue. The issues raised about negotiated rule-making are intriguing, and require additional thought and discussions with this group as well as within CBER. Further discussion will occur, within FDA and between FDA and other organizations. I welcome those discussions.
Jay Epstein: Kathryn Zoon has described the broad initiatives. I would like to embellish a few points, with some particulars in the area of blood regulation.
INITIATIVES IN BLOOD REGULATION
The regulation review process is ongoing to identify sections of the regulations that require updating. The ongoing debate is whether we should create additional standards for specific products under the regulations or move toward more flexible models of regulation such as those described during this Forum. In that discussion a lot is at stake for the future of regulation.
A set of initiatives is related to report reduction. This was started under the pharmaceutical drug user fee negotiation, but it is clearly on the agency's agenda to extend it to the area of blood regulation. The idea is to determine the minimum necessary change requiring reporting and determine what requires prior approval versus what can be implemented with a subsequent report to the agency. This is an area particularly relevant to the implementation of computerized systems, when the blood banks do not want to be at the agency's doorstep for every minor tinkering or fix.
There are other initiatives, for example increasing exemptions from lot release. That is an issue which, in the area of blood and blood products, will mainly focus on diagnostic tests. We find ourselves, however, repeatedly dealing with the question of shortages because of the impact of FDA control procedures, and a lot of that discussion tends to focus on lot release and how we should use lot release generally versus how we should control particular problems. This is an area that needs a sound debate.
The computerized product license application review is another fruitful area, not only because of the possibility of expediting review procedures through the use of information management technology but also because of the goal of reducing at least part of the applications procedure to automated checklists. We have an initiative related particularly to the source plasma application in that domain.
Another example of creativity in the area of streamlining has to do with the delegation of responsibility for inspections. The agency has been moving toward more reliance on the field offices to play a primary role in inspections
other than the prelicense approval inspections. The idea is to create a more homogeneous vantage point for the field with respect to the entire agency in looking across the board at inspection-related issues and trying to concentrate expertise in an appropriate way.
Managed review under the Pharmaceutical Drug User Fee Act (PDUFA), has also had a tremendous impact on other areas of regulation. It has already had an impact on forming time frames and developing performance standards in the area of blood regulation, particularly affecting devices.
Within the strategic planning process a similar concept is an attempt to have management take control over research so that research can be coordinated in a fashion that would focus it more on the mission of FDA.
PARTICIPATORY DECISION-MAKING
An issue intriguing issue is the role of negotiation—or perhaps in a broader construct, a more public decision-making process. Without having had in mind the formal negotiated rule-making (Reg-Neg) construct, which is very exciting, we have been moving in that direction as a response to the forces that have been acting upon us. I would cite as examples the degree to which we have been seeking to perform our regulatory duties by improving guidance statements and educating industry.
Other examples would include the quality assurance guideline that was developed through a workshop process and a similar guidance document on the validation of computer systems in the blood bank. Perhaps a less well recognized example is the fact that we have been taking error and accident analysis, which are done in the agency, and going public with them as a form of industry assistance through memoranda. We did that in March 1991 and will do that again periodically. Another model is using workshops to develop license standards. We have done this now for irradiated red cells, and we have the intention to do it with leuko-filtered blood components.
Another change that has been made concerns the way that we are using the Blood Products Advisory Committee. We received a great deal of industry comment about opening up that committee process and trying to use it as a communications tool. For example, we have deliberately brought new products before the advisory committee for discussion before products are approved.
We have taken very seriously the charge that industry is in an impossible position when it learns of a deadline on the day that the memorandum is published and has pressure to implement a major change within 48 or 72 hours. Instead, we are trying to give industry the "heads up" that it needs by making known the state of progress toward approval of critical new products to the advisory committee.
Another change has to do with using the advisory committee to discuss decision making related to risk assessment and risk management. We have been doing this quite consistently in the last decade, since the start of the AIDS era, and have applied that model to dealing with many issues.
Another change has been the way that we are using liaison meetings with the industry. We now have regular liaison meetings with more groups. That is not seen by the agency as a wonderful thing, because it tends to result in a proliferation of the number of meetings, and, some discussion of the subject matter tends to be repetitive. That aside, however, we have a regular liaison on the Transfusion-Transmitted Diseases Committee, regular liaison meetings with American Association of Blood Banks, and liaison meetings together and separately with the Council of Community Blood Centers and the American Blood Resources Association. We are benefitting from those meetings, as opportunities for the agency to listen and get the word from the ''shop floor.
PUBLIC MEETINGS
The Center for Biologics, Evaluation and Research (CBER) has also been using public meetings as part of the conventional process of notice and comment rule-making. That effort has been proactive, in that we are trying much harder to identify the concerned interests and bring them to the table. We still must work through the mechanism of an open public hearing, but we are endeavoring to get the word out so that all of the concerns are heard at these meetings and we try not to make it a haphazard process in which the right people must read the notice in the Federal Register.
Another change has been the effort to broaden the representation on the Blood Products Advisory Committee. In the wake of the discovery of the human immunodeficiency virus (HIV) in the 1980s, there was an accusation that the advisory committee was too dominated by blood bank concerns and was not sufficiently responsive to consumer concerns. We have addressed that by revising the charter to add to the number of members permitted on the committee and have appointed special government employees, individuals with interest in particular sides of the issue who can function as consultants or guests of the committee. That has been the method, by which we have tried to increase the level of representation of the hemophilia community.
NATIONAL PERFORMANCE REVIEW
Under the National Performance Review we are expected to come up with performance standards for all of our activities. We have opened that
process up to the blood industry by requesting specifically that the industry form some cooperating forum and get back to the agency on its recommendations and expectations for the performance standards to which the agency will hold itself.
What we are talking about, for example, is the standard defining the length of a product licensing application (PLA) review or a 510k 2 review, the percentage of such reviews accomplished within the time frame based on the resource. It is the same model as PDUFA but without any added external resource beyond the federal tax dollar. We have extended that opportunity to industry and advocated it at some of the liaison meetings.
EXPERIMENTS IN REGULATION
So far I have been describing ways in which we are trying to make the present system work better. We could do a lot more but we have already performed some experiments. I want to note a few of them.
First, there is the manner in which we are regulating the banking of human tissue. We do that under the Communicable Disease Control Authority of the Public Health Services Act Part 361, not under the Product Approval Authority of Part 351. That is a bit of an experiment because it is an alternative to device regulation.
Second, there is the issue of the extent to which CBER will elect to review more of the in vitro diagnostic devices under premarket approval versus product licensing application. That is an experiment because it shifts the balance of the establishment review from the preapproval stage to the field.
Third, there is the question of how far we will pursue good practice (GMP) regulation of unlicensed products. We tend to see this as an alternative to product review when we do not have licensing standards. We are engaging in an experiment in which we are trying to find ways of approaching vendor software and approaching human stem cells as a product subject to GMP, but not yet subject to a license review. We still have the mind-set that these products will become licensable once we understand the standard, but it is an experiment to try to achieve standards of safety under the GMP approach without the license review.
The impact of federal legislation dealing with risk assessment and cost-benefit analysis is uncertain. We have not ventured very far into that domain
does cover risk benefit assessment, which we commonly discuss in our advisory committee proceedings, but except in the domain of rule-making, FDA is not supposed to engage in the discussion of cost per se. I think that is an important problem, and I think that much good discussion will come out of the proposed legislation, although at this point there is no official FDA response to it.
THE FORCES OF CHANGE
We are living in a complex world. There are international forces relating to harmonization, and there is the changing tide of congressional oversight, which, whether it is of liberal or conservative persuasion, is still very deterministic toward the activities of the agency.
We have the agenda of downsizing. We have the agenda of accountability under performance review. We have the conflicting agendas of using fewer resources, while performing regulatory activities and ensuring safety more effectively. I do not know how we will accomplish that balancing act, but examples of experimentation exist at FDA.
FDA has permitted the use of voluntary standards. For example, the question of whether to formally regulate the human tissue industry has been debated for almost 20 years. In the interim voluntary standards were accepted as the basis for self-regulation, and it was only in the wake of certain incidents such as HIV transmission by transplanted organs and tissues that a more formal regulatory approach has been taken.
The agency has a history of flexibility. What remains to be examined is the exact issues that are ripe for either negotiated rule-making or an alternative mode of regulation. That is where the thoughtfulness and effort need to be applied. It is not fundamentally an issue of rigidity of the agency. It is more a question of trying to define what our expectations are and to determine the specific areas where it is reasonable to try these novel approaches.
THE ROLE OF THE RULE-MAKING PROCESS
Question from the Audience: Two questions were put to Jay Epstein concerning the fact that FDA has not often used the "Code of Federal Regulations CFR process"—the formal notice-and-comment procedure for rule-making under the Administrative Procedure Act, but has, rather, relied more on issuing guidelines and recommendations than on promulgating rules. Thus, the following questions were asked. First, given the structure of Reg-Neg, could adopting it have the ironic consequence of causing a shift away from the
informal structure of rule-making and back to the formal structure? Second, given the parallel tracks in FDA's regulatory activities, including both CFR rule-making and the recommendations and "points to consider procedures" involving public comment through the Blood Products Advisory Committee, is there not still a need for some alternative process, one that involves more rather than less negotiation, with the end products being decisions that come out not necessarily as rules in the CFR?
Jay Epstein: CFR-type rule-making has not been and could never be abandoned. But the agency has learned just how hard it is to get rules out, and so there is a higher threshold for engaging in formal rule-making.
The question is, at how many levels can you change the system. Under the current legal structure, agencies must promulgate regulations as the basis for their authorities. They derive their authorities from their governing statutes and then promulgate regulations on the basis of authority and, in the case of FDA, their authority for enforcement. That must remain as the agency's fundamental legal framework. The issue that we have addressed is what is the necessary level of detail in regulations for us to regulate effectively. A lively debate on that question is occurring. One camp contends that we should be more specific with our regulations because that is the only way to resolve the ambiguities that crop up with enforcement. How do you rein in compliance? The answer is, by specifying things more precisely in the regulations, and eliminating the ambiguity. The other argument is that when there are regulations written in that they do not stand up against the test of time. They become outdated; they tend to be too particular; there are too many circumstances not captured in the regulations. What one really wants are general framework regulations that then permit the agency to periodically issue and update interpretations that are enforceable. There is a debate about that, too, about how enforceable interpretations are. I do not have the answer to these things, but I do not see abandoning regulations in CFR as an endpoint that we can reach. I do think, though, that trying to develop flexible ways to use more spare regulations is a realistic end point.
Toby Simon: Even in the current political climate, blood bankers have not been opposed to regulation by FDA. There is a general consensus that regulation is an important part of the profession and that it is essential in many ways. To deal with the public it is important for FDA to be there, reassuring the public about the safety of blood by virtue of its activities.
In addition to the new regulatory tools already discussed, another one worth mentioning is "error management," looked at from the point of view of both the transfusion service and the blood center. The Federal Aviation Administration (FAA) has used reporting of near misses anonymously to
encourage full reporting, and the reports are processed under FAA guidance to make sure that everybody knows of and can benefit from them.
Negotiated rule-making itself might allow us to move more rapidly in areas that we have been dealing with from a regulatory point of view, particularly in terms of inspection and enforcement. Likewise, self-audit with FDA review is an interesting concept. Today, GMP audit results are ordinarily not shared, theoretically to stimulate a complete investigation. One might take the opposite point of view and share the audit, having FDA determine whether the audit has been adequate and thereby improving the efficiencies of its inspection.
If asked to define the characteristics of an appropriate regulatory process for the issues facing us, I would suggest that one characteristic would be involvement. That is, there would be a significant input from different parties who have a potential impact—government, the private sector, patients potentially affected, and specialists in related areas (for example, infectious disease specialists and neurologists). We would have some way to bring that group together to formulate a consensus, and to have the flexibility to keep measuring what we do, to come back and look at it, and to change it again.
COMMENTS AND QUESTIONS
Question from the Audience: As a practical matter, there is an increased risk of civil liability when FDA issues a recommendation and a blood bank does not abide by it. Thus, in effect, the blood industry complies as fully with recommendations as with regulations. From FDA's point of view, do recommendations and guidelines have a potency or effect different from those of regulations themselves?
Jay Epstein: Because FDA does not make recommendations that are not within its authorities under the regulations or, in some fairly rare cases, direct interpretations of statutes, the agency would regard recommendations as enforceable to the degree that they are grounded in a regulation or statute. There are occasional challenges in court, and FDA does not always win.
Thomas Zuck: One difficulty with the present system, which a negotiated rule-making process might help to alleviate, is illustrated by the question of stem cells. The agency indicated that it was going to regulate stem cells and somatic cell lines. But then, whether it is by regulation or by guideline, there is a long period during which we do not know whether to file an investigational new drug (IND) application, whether to ship in interstate commerce, or whatever else we are supposed to do. In that kind of situation we have to wait
until somebody is cited for a violation or is brought to court. That is not a very good way to make rules. These alternatives offer a way to get on with it, to convene a process to decide.
Unidentified Participant: The present process is one in which FDA receives input and issues a decision. These negotiation alternatives allow all of us, and others who have an interest in the rule, to sit at the table together, communicate, and hammer out those guidelines and "points to consider."
James MacPherson: The present system is not serving us well, and we do not believe that it is serving the public well. Most of the current initiatives, about which I agree there has been progress, have largely been internal. It has so far been FDA creating policy entirely internally and then seeking comment. We should now be talking about a true partnership, not a partnership between FDA and the field office or between CBER and the field office, but a true partnership with all of the parties involved and in which there is no risk to anyone. The only risk is that we may fail again.
Pinya Cohen: Negotiated alternatives seem difficult but challenging and exciting; they could add substantial value to the regulatory process, particularly given the need for depoliticization. Politics and ideology have little to do with safety, yet the Commissioner's office has a need to respond to the U.S. Congress. There is really no need for a body like that to be active other than to monitor in a general sense what is going on. We can take the reins fully in our own hands if we hold ourselves to some new standards.
William Sherwood: We have heard in these Forums several looming issues. We have heard of the deterioration or the erosion of the public trust in our industry. We have heard that the public has a far more dire expectation or understanding of the safety of our blood supply compared with what we think it is. And we have discussed the regulatory environment that we are in and have looked at it as perhaps a problem. That question—dealing with the regulatory environment—is, at least from inside the industry, one of the most important.
Is the problem rule-making? As I look at the rules that have been made, I do not have much quibble. Rule-making has been slow: slow in coming and slow in eliminating rules that are there. Maybe that is a problem that can be corrected.
Are the issues more that of how the rules are applied? How they are interpreted by the agency? How they are laid out? Is there an unevenness or a lack of consistency in the way in which these rules are looked at, and is our industry kept off balance? These are issues and problems on which we can focus. Maybe now we have some alternatives that we can work with the
agency to develop.
Pinya Cohen: With all due respect to the inspections in the field, there is a growing sense that there is a lack of coordination or a question as to who reports to whom with regard to the inspectors in the field. In the application process, we have certainly had a dialogue with CBER. But once our applications are approved, one gets the impression that the inspectors in the field may feel free to challenge and reopen that dialogue. If these are the ground rules, we should know them in a clear way. We need to pull together and get back to a more centralized sense of where the science is coming from. A science-directed approach rather than a compliance-directed approach might be helpful.
Miriam Sparrow: We have heard today about countervailing power, and we have heard about rule-making and the legislative function of FDA. It is the enforcement function of FDA, however, that we also need to work with, specifically, the inspection process, which does have a role to play in improving blood safety. Part of where we should be headed, in terms of trying to work together, is to build not only an FDA that has credibility with the public at large and with the congressional constituency, but also an FDA that has the same credibility with the industry that is being regulated. That objective will help the industry itself to meet some of the concerns about public perception.
Henrik Bendixen: How have advances in blood safety come about? Have we done it all ourselves? It is fair to say that we have done a lot ourselves, through the introduction of new concepts, good research, the introduction of more technology, and the writing of guidelines, standards, and the like. All of this has contributed to increased safety in our field. At the same time, we have not been alone. The intervention of professional societies has been very important. Then, of course, there has been attention from the press and the voices of the consumer and special interest groups, and we cannot deny that even lawsuits may have moved us in the right direction.
The other participating change agents have been regulations, whether they are those of the federal government or the states. Advances in safety do not logically argue toward deregulation. Nonetheless, as we live in a modern industrial society, a most important thing is the word "predictability." You need to have rules of the game. You do not mind even tough rules of the game as long as you know what the rules are. If McDonald's stops selling hamburgers in the People's Republic of China it will be because they cannot predict from one day to the next who has to be persuaded or bribed. I have never minded regulation, even tough regulation, but it must be predictable.
Jay Epstein: I would like to say "message received" with respect to the issue of integrating the enforcement side into these discussions. I see that as an area that could be developed as some kind of model for a negotiated resolution. The points made about accountability are also quite perceptive. Ultimately we are a federal agency; we are accountable to our leaders, to the U.S. Department of Health and Human Services, and ultimately to the Congress, and that should never be forgotten. As an example, increased role of the field offices in decision-making and policy generation is a direct result of congressional oversight, which faulted the agency for not responding enough to the role of the field in that process.
We cannot have it both ways; the implication is that there is a need for a careful examination of the political environment and figuring out who the constituents are and how they interact. The system must be looked at as a whole, and the players must be recognized. We must not forget that the Congress as the voice of the people is the ultimate master of the agency.
General Discussion of Issues for Negotiated Rule-Making
Editor's Note: A portion of a later meeting of the Forum was set aside for discussion of the topics that could be considered as candidates for a pilot project in negotiated rule-making. David Pritzker offered a checklist of qualifications that a candidate issue should have, day Epstein suggested the problem of new tests, and Celso Blanco presented the particular case of Creutzfeldt-Jakob disease (CJD). Other members of the Forum proposed specific candidate issues, a list of which follows. Consistent with its charter, the Forum as a group took no position on recommending any particular issue or program.
CRITERIA FOR SELECTING ISSUES
David Pritzker: The following is an amalgam of several of the published checklists describing the features of problems for which Reg-Neg seems most promising:
-
Both parties must be dissatisfied with the current situation.
-
There must be a limited number of interests that are significantly affected.
-
The appropriate individuals who can represent those interests can be identified.
-
The issues must be known, mature, and ripe for discussion.
-
No party should be able to reach the result by themselves—that is, the outcome is genuinely in doubt and no interest should be able to dominate the proceedings.
-
No party can be required to compromise a fundamental value.
-
The problem must involve diverse issues, so there are things to trade off.
-
The agency must be willing to rely on the process and actively participate in it.
-
The parties see it as in their interest to use the process.
-
There should be a deadline for achieving a result.
CANDIDATES FOR REG-NEG
Jay Epstein: One issue that has been pressing and that may be appropriate is development of a framework for dealing with changes in the institution of tests. The effort is to come up with a set of principles that can be used to work through the problems that have faced blood safety. There has been a sense that we have dealt ad hoc or case by case with issues, not always to the benefit of the system as a whole, and that there is a need to focus on the decision-making process—not perhaps to fault or revise the process as a system but to put it on a better foundation. That has been seen as one of our pressing issues. The structure of the regulatory agency-industry relationship is certainly another, but that is a little bit more long term than an effort to develop guiding principles for risk decision making.
Celso Bianco: CJD is an interesting problem for these models. At the Blood Products Advisory Committee in December 1994, we presented a model by which certain decisions could be made. The idea was that each blood center would report those diseases to a central place, for example, to the Food and Drug Administration, which would file them and once a year convene a panel of experts, including patients and others, and ask the questions, "Would notification of recipients of these products be beneficial to them? Is there anything known that could help them?" That is one example of a small pilot project that could encompass not only CJD but other diseases that are ill-defined. There are diseases for which there are theoretical possibilities of transmission by transfusion but for which there is no documented evidence of actual transmission.
The following are additional "Reg-Neg" topics suggested by members of the Forum:
-
Progenitor cells. The technology is moving very fast.
-
p24 antigen testing for HIV. For detection of HIV during the "window period" after infection but prior to detectable antibody response.
-
Recommendations concerning "lookback." The cost and benefits; how the information is and is not used, whether lookback actually serves the recipient population.
-
Stem cells and gene therapy regulations.
-
Transplantation regulations.
-
Test implementation decision process: implementing new tests and eliminating old ones. It would be desirable to establish a decision-making process outside of the "heat of battle." Specification of steps, timing, and procedures is needed.
-
Prevention of Chagas disease transmission. This is linked to the issue of implementing new tests.
-
Organizational alternatives to current U.S. blood banking system: federation (e.g., Scotland) or national blood banking system.
-
Definition and role of ''donor incentives".
-
Donor deferral and reentry.
-
How should the safety of the blood supply be monitored?
-
Regulation of new classes of products such as hemoglobin solutions: safety and efficacy; adequacy of substrate supply.
-
Approval of significant blood-processing techniques: virus inactivation; blood filtration.
-
Recipient screening. Is this a practical solution, or a logistical nightmare?
-
Establishment inspections. Increase uniformity of the inspection process; develop internal audits.
-
Computer validation: computer models and quality systems; integration into the inspection process.












































