
This paper was presented at a colloquium entitled “Genetics and the Origin of Species,” organized by Francisco J.Ayala (Co-chair) and Walter M.Fitch (Co-chair), held January 30–February 1, 1997, at the National Academy of Sciences Beckman Center in Irvine, CA.
Evolution by the birth-and-death process in multigene families of the vertebrate immune system
MASATOSHI NEI*, XUN GU, AND TATYANA SITNIKOVA
Institute of Molecular Evolutionary Genetics and Department of Biology. The Pennsylvania State University, 328 Mueller Laboratory, University Park, PA 16802
ABSTRACT Concerted evolution is often invoked to explain the diversity and evolution of the multigene families of major histocompatibility complex (MHC) genes and immunoglobulin (Ig) genes. However, this hypothesis has been controversial because the member genes of these families from the same species are not necessarily more closely related to one another than to the genes from different species. To resolve this controversy, we conducted phylogenetic analyses of several multigene families of the MHC and Ig systems. The results show that the evolutionary pattern of these families is quite different from that of concerted evolution but is in agreement with the birth-and-death model of evolution in which new genes are created by repeated gene duplication and some duplicate genes are maintained in the genome for a long time but others are deleted or become nonfunctional by deleterious mutations. We found little evidence that interlocus gene conversion plays an important role in the evolution of MHC and Ig multigene families.
Multigene families whose member genes have the same function are generally believed to undergo concerted evolution that homogenizes the DNA sequences of the member genes (1–3). A good example of this type of gene family is the cluster of ribosomal RNA genes, where all the member genes (several hundred genes) have very similar DNA sequences within species even in nontranscribed spacer regions. For example, the member genes of this cluster in humans are more similar to one another than to most of the genes in chimpanzees (4). This high degree of sequence homogeneity within species is believed to have been achieved by frequent inter locus recombination or gene conversion (Fig. 1A).
Concerted evolution was also invoked to explain the diversity and evolution of the multigene families of major histocompatibility complex (MHC) genes (5–7) and immunoglobulin (Ig) genes (8–11). In this case the genetic diversity (polymorphism) for a locus or a set of loci is assumed to be generated by introduction of new variants from different loci through interlocus recombination or gene conversion. However, we have questioned these claims of concerted evolution on grounds that the member genes of MHC and Ig gene families from the same species are not necessarily more closely related to one another than to the genes from different species (12–16). For example, Ota and Nei (14) showed that the Ig heavy chain variable region (VH) genes in humans can be divided into three major groups, and these major groups are shared by mice, amphibians, and teleost fishes, yet gene duplication apparently occurs quite frequently and many duplicate genes subsequently become nonfunctional. Therefore, they concluded that the evolution of this gene family is characterized by the “birth-and-death model of evolution” (Fig. 1B). However, some authors (10, 17–20) still maintain that
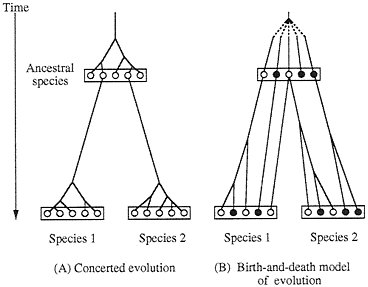
FIG. 1. Two different models of evolution of multigene families. ○, Functional gene; ●, pseudogene.
interlocus gene conversion or recombination plays an important role in the generation and maintenance of genetic diversity of MHC and Ig gene families.
To resolve this controversy, it is important to conduct a comprehensive study of the evolutionary patterns of immune syste, genes. Fortunately, the amount of DNA sequence data for immune system genes is rapidly increasing, mainly because of the genome project that is under way in various organisms. We have therefore compiled DNA sequence data for immune system genes from GenBank and other sources and studied the general evolutionary pattern of these genes. In vertebrates there are three major gene families that play an important role in identifying and removing invading foreign antigens or parasites (virus, bacteria, and eukaryotic parasites). They are the MHC, T cell receptor (TCR), and Ig gene families. The function of MHC molecules is to distinguish between self- and nonself- antigens and present foreign peptides to TCRs of T lymphocytes (T cells), whereas the role of TCRs is to interact with peptide-bound MHC molecules and stimulate T cells to kill virus-infected cells or to give signals for B lymphocytes (B cells) to produce immunoglobulins. Immunoglobulins are primarily responsible for humoral immunity, removing foreign antigens or parasites circulating in the bloodstream. The gene families encoding MHC, TCR, and Ig molecules are all evolutionarily related and are composed of several
© 1997 by The National Academy of Sciences 0027–8424/97/947799–8$2.00/0 PNAS is available online at http://www.pnas.org.
|
|
Abbreviations: MHC, major histocompatibility complex; Ig, immunoglobulin; TCR, T ceil receptor; β2m, β2-microglobulin; Myr, million years. |
|
* |
To whom reprint requests should be addressed. e-mail: nxm2@psu.edu. |
separate gene clusters, each of which spans more than one megabase (Mb) of DNA in the human genome.
The purpose of this paper is to present major findings of our recent study on this subject. The immune system is one of the most complicated genetic systems in vertebrates, and detailed results will be published elsewhere. In this paper, we will be concerned primarily with the evolution of MHC and VH gene families.
EVOLUTION OF MHC GENES
MHC molecules in vertebrates can be divided into two groups; class I and class II molecules. The class I MHC molecule consists of an a chain and a ß2-microglobulin (ß2m). The a chain is encoded by a class I MHC gene, whereas ß2m is produced by a gene that lies outside the MHC. The class I a chain has three extracellular domains (a1, a2, a3), a transmembrane portion, and a cytoplasmic tail. The a3 domain associates noncovalently with ß2m. The class II MHC molecule consists of noncovalently associated a and ß chains, which are encoded by class II a-chain (A) and ß-chain (B) loci, respectively. Each chain is composed of two extracellular domains (designated as a1 and a2 in the a chain and ß1 and ß2 in the ß chain), a transmembrane portion, and a cytoplasmic tail.
In humans class I and class II genes are located on chromosome 6 and form two separate clusters (Fig. 2). The class I MHC consists of three highly expressed and highly polymorphic loci, A, B, and C (classical class I or class Ia loci), and 25–50 nonclassical (class Ib) loci, including pseudogenes. Nondefective class Ib genes are usually monomorphic and expressed in limited tissues, and their function is not well understood. So the definition of class Ib genes is somewhat vague (23). However, the recently discovered class Ib gene HLA-HH seems to have some important function, because mutants of this gene apparently cause the genetic disease hemochromatosis (21). The human class II gene cluster contains six major gene regions: DP, DN, DM, DO, DQ, and DR. Each of the DP, DM, DQ, and DR regions consists of at least one a-chain and at least one ß-chain functional gene. The a-chain and ß-chain genes in the regions DP, DQ, etc., are designated as DPA1, DPA2, DPB1, DPB2, DQA1, etc. The class II gene cluster also includes many poorly expressed genes and pseudogenes.
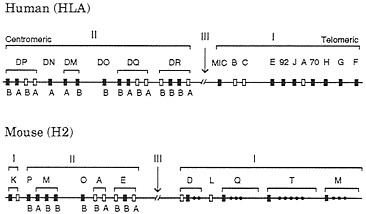
FIG. 2. Simplified genomic organizations of the human and mouse MHC genes. Only relatively well characterized genes are presented here, and there are many other genes or pseudogenes in both organisms. The recently discovered class Ib gene HLA-HH (21) is located about 4 Mb away from gene F on the telomeric side. (The original authors used the gene symbol HLA-H, but we changed it to HLA-HH to avoid the confusion with an already established Ib locus with the same name.) The number of genes also varies with haplotype in both class I and class II regions. Open boxes refer to polymorphic or classical loci, whereas closed boxes stand for monomorphic or nonclassical loci. Class III genes or other genes unrelated to class I and class II genes are not shown. Class II genes A and B refer to class II a- and ß-chain genes, respectively. The MHCs in humans and mice are often called HLA and H2, respectively. The gene maps in this figure are based on information from Trowsdale (22) and other sources.
The mouse MHC genes have also been studied extensively. The mouse class Ia genes are not orthologous with the human class Ia genes (24–26), and therefore different gene symbols are used for them (Fig. 2). Actually, most different orders of mammals seem to have nonorthologous class Ia genes. The number of class Ia genes in mammals is usually 1–3, but there are often a large number of class Ib genes. By contrast, class II genes from different orders of mammals usually have orthologous relationships, but the genes from birds and amphibians are not orthologous with the mammalian genes, with a few possible exceptions (27).
Polymorphism. The hallmark of MHC genes is the extremely high degree of polymorphism within loci, the extent of polymorphism being the highest among all vertebrate genetic loci (28). The mechanism of maintenance of this polymorphism has been debated for the last 30 years, and it still remains controversial (15, 19, 29). The hypotheses proposed to explain the polymorphism include those of maternal-fetal incompatibility, mating preference, overdominant selection, frequency-dependent selection due to minority advantage, and interlocus gene conversion. This problem has been discussed by Hughes and Nei several times (15, 16, 30, 31), and we are not going to repeat the discussion here. However, we would like to mention that in our view the simplest explanation is heterozygote advantage or overdominant selection. In this hypothesis, heterozygotes for a locus have selective advantage over homozygotes, because they can cope with two different types of antigens, whereas the latter can deal with only one type of foreign antigen. Since there are several different functional MHC loci, heterozygotes for all these loci should have substantial selective advantage over homozygotes. Evidence supporting overdominant selection is also increasing (19, 31).
In recent years a number of authors (19, 32) presented evidence that new alleles can be created by interallelic recombination at the B locus. However, interallelic recombination is powerless in producing new alleles unless there are abundant polymorphic alleles in the population, and the MHC polymorphism seems to be maintained primarily by point mutation and overdominant selection (15). Another interesting discovery in recent years is the relatively high degree of polymorphism at a class Ib locus, MICA (33). The function of this gene is unknown, but the average heterozygosity per nucleotide site (nucleotide diversity) for the three extracellular domains (exons 2, 3, and 4) is 0.011. Although this is lower than that for class Ia loci (0.04–0.08), it is considerably higher than that (0.0002–0.007) for other nuclear genes (15). The reason for this high degree of polymorphism is unclear, but it is possibly caused by a hitchhiking effect of overdominant selection operating at the B locus, which is closely linked with this locus.
In the past, population geneticists have been primarily interested in the extent of polymorphism within loci. In the MHC, however, there is a substantial amount of polymorphism due to gene duplication, insertion, or deletion. For example, in the class II DRB region of humans there are at least five different haplotypes, and the number of genes per haplotype varies from 2 to 5 (22). Furthermore, there seem to be at least 9 distinct gene copies in this region, and only one gene (DRB1) is shared by all haplotypes. The mouse population is also known to have many different haplotypes, and the type and number of class I genes vary considerably with haplotype. For example, class Ia locus D is missing in most haplotypes, and the number of class Ib genes varies considerably with haplotype (34).
Intralocus vs. Interlocus Variation Within Species. One way of studying the significance of interlocus recombination or gene conversion is to compare the interlocus and intralocus genetic variation within species. This can be done by constructing a phylogenetic tree for alleles from different loci. If there is any kind of interlocus genetic exchange, one would expect that alleles from each polymorphic locus do not necessarily form a monophyletic cluster in the phylogenetic tree. Fig. 3 shows the phylogenetic tree for different alleles from human MHC (HLA) class
I loci. The numbers of allelic sequences available for class Ia loci A, B, and C are now about 70, 150, and 40, respectively (19), but in this paper we used no more than 8 representative sequences (exons 2, 3, and 4 encoding domains α1, α2, and α3, respectively) from each locus, excluding partial sequences. It is clear that all alleles from the same locus form a single cluster and every allelic cluster is statistically supported. Essentially the same results were obtained when we used all available complete sequences (56 A alleles, 123 B alleles, and 34 C alleles) in the analysis. This indicates that despite the high level of polymorphism of classical loci interlocus variation is much greater than intralocus variation and there is no clear indication of interlocus genetic exchange. This conclusion is in agreement with that of Lawlor et al. (36).
As mentioned earlier, nonclassical and pseudogene loci are generally monomorphic, and when polymorphic alleles exist, their nucleotide differences are usually very small (Fig. 3). The genes at these loci (genes E, F, G, J, 70, and 92 in Fig. 3) also tend to evolve faster than class Ia genes (A, B, and C) apparently because of less stringent functional constraint. Exceptions are the alleles of locus H, which are believed to be pseudogenes because they have deleterious mutations (37). The nucleotide diversity among these alleles is quite high (0.021), and the alleles have not evolved very fast. These features are unusual for pseudogenes or unimportant genes, but the reason is unclear at this moment. It is possible that this gene became a pseudogene relatively recently or that it has a new but unknown function, since a frameshift mutation that
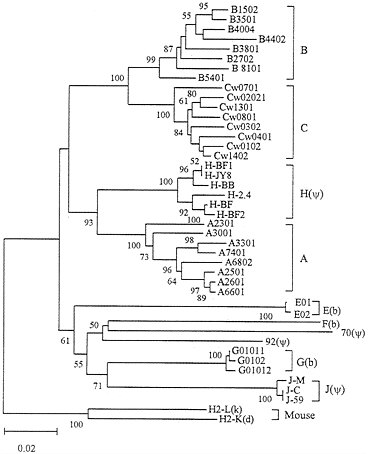
FIG. 3. Phylogenetic tree of human MHC (HLA) class I genes and alleles. The phylogenetic trees presented in this paper are neighbor-joining trees obtained by the computer software MEGA (35). The tree in this figure was constructed by using Jukes-Cantor distances for the nucleotide sequences (822 bp) of exons 2, 3, and 4, which encode class I MHC domains α1, α2, and α3, respectively. Genes E, F, and G, designated by (b), are functional class Ib loci, whereas genes H, J, 92, and 70, designated by (ψ), are class Ib pseudogenes. Class Ib genes MICA and HH were not included because they are distantly related (see Fig. 4). Two mouse class Ia genes were used as outgroups. The numbers for interior branches refer to the bootstrap values for 500 replications. The bootstrap values less than 50% are not given. The scale at the bottom is in units of nucleotide substitutions per site.
occurred in this gene generates a stop codon near the end of the gene (37). It should also be noted that the class I gene region contains many other defective genes lacking some exons or parts of exons (38). These are clearly dead genes.
Until recently, class Ib genes were thought to be relatively unimportant and possibly in the process of becoming pseudogenes (13, 25). However, some of them are apparently functional, as mentioned earlier, and Figs. 3 and 4 show that most class Ib loci in humans diverged earlier than class Ia loci and thus survived longer than class Ia loci. Therefore, it is possible that nondefective Ib genes play some important roles that are not identified at the present time.
As mentioned earlier, class II genes are composed of class II A and class II B genes (loci). The A and B genes seem to have diverged about 500 million years (Myr) ago, nearly at the same time when class I and class II genes evolved (27). We have therefore constructed phylogenetic trees for human class II A and B genes separately. Fig. 5 shows the tree for class II B genes from three polymorphic loci (DPB1, DQB1, and DRB1), two poorly expressed loci (DOB and DQB2), and one pseudogene (DPB2). The alleles from each of the three polymorphic loci again form a single cluster with a bootstrap value of 99–100%, whereas poorly expressed genes have survived for an unexpectedly long time. This pattern of evolution is very similar to that of class I loci, though the evolutionary time involved in gene turnover is much longer in class II loci than in class I loci. Our phylogenetic analysis of class II A genes also showed a similar evolutionary pattern (data not shown).
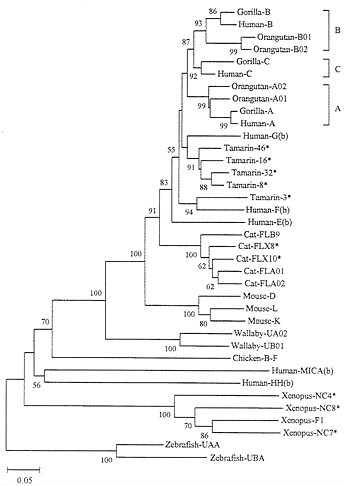
FIG. 4. Phylogenetic tree of MHC class I genes from various vertebrate species. This tree was constructed by using p-distances (39) for the amino acid sequences (274 residues) for domains α1, α2, and α3. Class Ib genes are denoted by (b) except for Xenopus genes NC4, NC7, and NC8. Other genes are primarily class Ia genes. Asterisks indicate cDNA data, where the identification of loci or alleles is ambiguous. In this paper we used common species or genus names to designate their MHC genes to make the paper understandable for nonspecialists. The wallaby is a marsupial species; Xenopus is Xenopus laevis.
DNA sequences for polymorphic alleles are also available for mice. Lawlor et al. (36) conducted a phylogenetic analysis of class I gene data, but there were some problems in their locus identification. Hughes (40) reanalyzed the data, rectifying the relationships between alleles and loci. This analysis indicated that interlocus genetic exchange is rare in mice as well, though it cannot be ruled out. In the case of class II loci, we conducted our own phylogenetic analysis, which showed that the alleles from the same locus always makes a monophyletic cluster with a bootstrap value of 99% or higher (data not shown). Therefore, the pattern of genetic differentiation of alleles and loci appears to be similar to that of humans. In rats there seem to be two class Ia loci (RT1.A and RT1.Eu), but there are many Ib loci and some of them are apparently expressed (41). Rada et al. (7) reported a possible case of interlocus genetic exchange among class I loci, but Hughes’ (40) reanalysis of the data did not support their claim.
At any rate, our phylogenetic analyses have shown that polymorphic alleles from the same locus almost always produce a monophyletic cluster. This finding does not rule out the possibility of gene conversion involving short gene segments (20) but indicates that the contribution of interlocus gene conversion or recombination to the entire genetic variation of MHC loci is small. This is in contrast to the apparent occurrence of interallelic gene conversion within loci, but our conclusion is not unreasonable because the likelihood of gene conversion is known to decline as the extent of sequence divergence increases (42).
Long-Term Evolution. To understand the evolutionary dynamics of MHC genes, we need to compare the genes from various organisms. It is already known that the class Ia loci from humans and mice are not orthologous; they were derived from different duplicate genes that originated before separation of the two species (13, 25). Fig. 4 shows a phylogenetic tree for class I genes (generally one allele from each locus whenever the locus is identifiable) from various organisms. As expected from previous studies (13, 25), distantly related organisms (e.g., humans, cats, mice, wallabies, and Xenopus laevis) have different sets of class Ia genes, indicating that the genes have differentiated by duplication
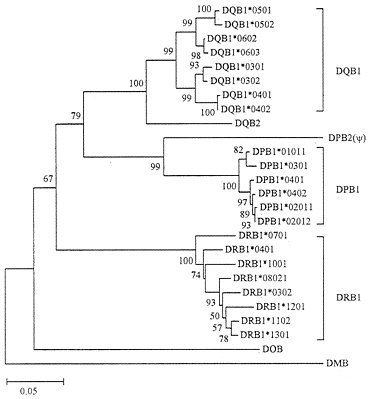
FIG. 5. Phylogenetic tree of human MHC (HLA) class II B genes and alleles. This tree was constructed by using Jukes-Cantor distances for the nucleotide sequences (564 bp) of exons 2 and 3 that encode the class II MHC ß-chain domains ß1 and ß2, respectively. DMB is known to be distantly related to other B genes (Fig. 6).
and deletion or dysfunctioning of the original genes. Even the class Ia loci from the New World monkey Saguinus oedipus (tamarin) are different from those of humans, though some nonclassical loci [e.g., tamarin 3 (So-3) and human F in Fig. 4] are orthologous and shared by the two species (43). However, closely related species such as humans, chimpanzees, and gorillas share the same loci A, B, and C. Gibbons and orangutans, which diverged from the human lineage about 17 and 13 Myr ago, respectively, share the loci A and B with humans but not C (43). Interestingly, orangutans appear to have gained a new duplicate A gene (A01) and a new B gene (B02) (43).
If interlocus genetic exchange occurs frequently, one would expect that some genes (loci) from a species are more similar to one another than to those from different species. For example, one would expect that the genes from loci A, B, and C of humans are more similar to one another than to those of gorillas. In practice, this is not the case, and the genetic relationships of genes from loci A, B, and C arc the same for humans and gorillas (Fig. 4). This indicates that for relatively closely related organisms different loci maintain their genetic identity despite the apparent occurrence of repeated gene duplication and that no or few interlocus genetic exchanges have occurred among different loci. However, the fact that different sets of class Ia genes exist in different orders of mammals indicates that some Ia genes are deleted or become nonfunctional after gene duplications. For this reason, Nei and Hughes (16) proposed that MHC genes are subject to the birth-and-death model of evolution.
We also note that some Ib genes have survived in the genome for a long time. For example, the human MICA and HH genes apparently diverged from other human class I genes before the human and chicken lineages separated. This suggests that these genes have survived in the human lineage at least for about 300 Myr. In fact, Southern blot analysis has suggested that the MICA gene exists in most mammalian orders (33). Human Ib genes E, F, and G seem to have survived longer than Ia genes A, B, and C, as was mentioned earlier. The Ib genes (NC4, NC7, and NC8) in Xenopus also apparently survived for a long time. This suggests that even class Ib genes are not subject to frequent interlocus genetic exchange.
The evolutionary pattern of class II genes is more or less the same as that of class I genes, though the time scale of gene turnover involved is much longer. Fig. 6 shows the tree for class II B genes, which indicates that distantly related organisms (zebrafish, Xenopus, chicken, and mammals) again have different sets of class II B genes. These genes therefore clearly experienced birth-and-death evolution. However, humans and rodents share the orthologous loci DP, DQ, DR, and DO. Similarly, loci DQ and DR are shared by humans, dogs, sheep, and cattle. The DP gene in the mouse is a pseudogene and is not shown in Fig. 6. Sharing of the DQ and DO genes by humans and rodents is understandable, because these two genes apparently diverged about 175 Myr ago (27) and humans and rodents apparently diverged about 100 Myr ago (44). The tree in Fig. 6 suggests that cattle and sheep gained a new locus, DIB/DYB, around the time of mammalian radiation. The phylogenetic position of marsupial (wallaby) genes DAB and DBB is unclear because of low bootstrap values for the interior branches surrounding this lineage, but they are apparently unique to marsupials (45).
The sequence data for class II A genes are still scanty, but available data indicate that the evolution of these genes is also characterized by birth-and-death evolution (46).
EVOLUTION OF IMMUNOGLOBULIN GENES
Immunoglobulin or antibody molecules consist of two heavy chains and two light chains. Both the heavy and light chains are composed of the variable region (V) and constant region (C). The variable region is responsible for antigen binding and the constant region for effector function. The heavy-chain variable region is encoded by the variable-segment (VH), diversity-segment (DH), and joining-segment (JH) genes, but here we consider only VH
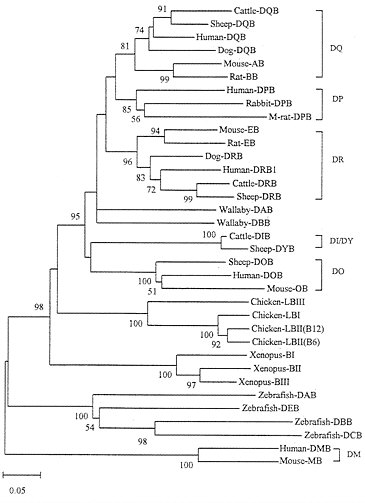
FIG. 6. Phylogenetic tree of MHC class II B genes from various vertebrate species. The tree was constructed by using p-distances for the amino acid sequences (188 residues) of domains β1 and β2. One allele from each locus was used as long as the locus was identifiable. In chicken and Xenopus the class II B loci have not been clearly identified, so that some sequences used may represent different alleles from the same locus. The human and mouse DM B genes, which are known to be distantly related to other class II B genes (31) were used as outgroups. The orthologous genes such as DQ, DP, etc., in different species are bracketed. M-rat, mole rat.
genes because DH and JH genes are very short and are not useful for our purpose. There are two types of light-chain gene families: κ-chain and λ-chain gene families. Each of these gene families consists of the variable-segment (Vκ or Vλ) and joining-segment (Jκ or Jλ) gene families, but Jκ and Jλ genes are again unimportant for our purpose. In higher vertebrates, the heavy-chain, κ-chain, and λ-chain gene families are located on different chromosomes and form separate clusters. The human genome contains about genes vary with haplotype. The numbers of VH, Vκ, and Vλ genes also vary with species, and some organisms (e.g., chicken) have no Vκ genes and others (e.g., mouse) have only a few Vλ genes. In cartilaginous fishes such as sharks and skates the genomic organization of Ig genes is different from that of higher vertebrates, which may be described as (Vn-Dn-Jn-Cn). In lower vertebrates, the basic unit of repeat of genes is (V-D-D-J-C). This unit of linked genes is repeated several hundred times in the genome, and the repeats are scattered on different chromosomes. Therefore, the Ig genes in these organisms may be represented by (V-D-D-J-C)n. In this paper we will be concerned primarily with the evolution of VH genes.
Diversity Within Species. The diversity of immunoglobulins is primarily caused by multiple copies of VH or VL genes. It has been shown that the rate of nonsynonymous nucleotide substitution is significantly higher than that of synonymous substitution at the complementarity-determining regions of VH and VL genes (47). This observation once suggested the possibility that VH and VL genes might also be subject to overdominant selection, but recent studies have shown that the extent of VH or VL gene polymorphism within loci is much lower than that of MHC genes (48–50). Therefore, it seems that a higher rate of nonsynonymous substitution than that of synonymous substitution in VH and VL genes is caused by directional selection rather than overdominant or balancing selection.
A DNA region of 1,100 kb containing the entire human VH repertoire has now been sequenced (51, 52). It contains about 50 functional genes and about 40 pseudogenes. In addition, polymorphic alleles for a substantial number of VH loci have been sequenced. Fig. 7 shows a phylogenetic tree for 40 functional genes and 6 additional polymorphic alleles (3 alleles for 3 loci). (Unexpressed nondefective genes with open reading frames were not used.) It is clear that the sequence divergence of polymorphic alleles is very small compared with that of MHC loci. By contrast, the extent of sequence variation among different VH loci is much higher than that of class I or class II MHC genes. The human VH genes have been classified into seven different families according to sequence similarity (51, 52). Each of these seven families of genes forms a monophyletic cluster, though there are three single-sequence families (families 5, 6, and 7). Ota and Nei (14) and Schroeder et al. (55) noted that the VH genes in mammalian species can be classified into three groups—A, B, and C—or three clans—I, II, and III. The genes belonging to each of these groups
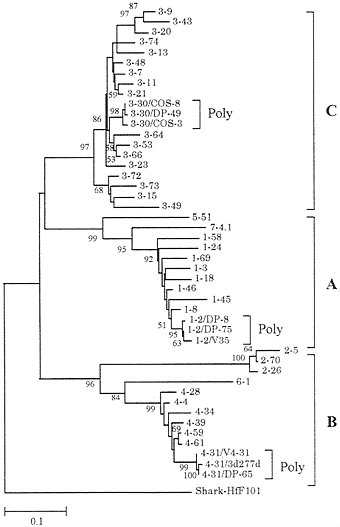
FIG. 7. Phylogenetic tree of 40 functional VH genes (loci) and 6 additional polymorphic alleles from humans. This tree was constructed by using Jukes-Cantor distances for the nucleotide sequences (228 bp) of framework regions 1, 2, and 3 (14). Loci 1–2, 3–30, and 4–31 are represented by three alleles. Gene and allelic notations are the same as those of Honjo and Matsuda (53) and the V BASE database (54), from which the sequence data were obtained. The human VH genes are classified into seven families, and the first number of each gene symbol designates the family number, whereas the second number refers to the order of the chromosomal location from the DH gene region.
in humans again form a monophyletic cluster with a high bootstrap value. As will be mentioned later, these groups of genes are-shared by amphibians and fishes. Therefore, the three groups of genes have persisted in the human lineage for more than 400 Myr. This evolutionary time is much longer than that for the gene groups in MHC class II A or B clusters.
Fig. 7 indicates that the present VH genes in the human genome were generated by repeated gene duplication. One may therefore wonder whether the VH gene repertoire was produced by repeated tandem gene duplication or by the combination of duplication, deletion, and translocation events. If tandem duplication is the major factor, one would expect that closely related genes in the phylogenetic tree are also located closely in the physical map of VH genes on the chromosome. In practice, this is not the case, and the genealogical relationships of the genes are almost independent of the chromosomal locations (Fig. 7). The same conclusion applies to the Vκ gene cluster (50) and somewhat weakly to the Vλ gene cluster as well (56). These results suggest that the VH, Vκ, and Vλ gene families in humans were formed by the combination of events of gene duplication, deletion, and translocation.
Haplotype Polymorphism. If duplication, deletion, and translocation are important factors of the evolutionary change of Ig genes, one would expect that haplotype polymorphism due to these events is observed in extant species. This is indeed the case, and the number of copies of VH or VL genes is known to vary with haplotype. For example, some human haplotypes have only one VH3–30 gene, but others have a group of five duplicate genes in the vicinity of this locus (53). A more dramatic example of gene duplication is observed in the human Vκ gene cluster (50). Human populations have two haplotypes, say H1 and H2. Haplotype H1 has 40 Vκ genes including pseudogenes, whereas H2 has 76 Vκ genes. Sequencing of these genes has shown that H2 was produced by a block duplication of 36 Vκ genes from haplotype H1, and the sequence similarity between the two sets of duplicate genes is very high. This duplication apparently occurred after humans and chimpanzees diverged about 5 Myr ago, because chimpanzees, gorillas, and orangutans do not have this duplication. The frequency of haplotype H1 in human populations is very low and seems to be about 4% (50).
Long-Term Evolution. The pattern of long-term evolution of VH and VL genes has been studied by a number of authors (12, 14, 57, 58). These studies are primarily based on sequence data from humans and mice and some limited data from other organisms. The general conclusion obtained from these studies is that the vertebrate genome contains a diverse array of VH and VL genes and they have been formed during hundreds of millions of years. However, recent studies of VH and VL sequences from chicken (59), rabbits (60), sheep (61), etc. suggest that this general conclusion does not apply to all organisms.
Fig. 8 shows the phylogenetic tree for representative VH genes from 15 different vertebrate species. According to this tree, the VH genes can be grouped into five major clusters, A, B, C, D, and E (14). The human and mouse genomes contain only group A, B, and C genes (14, 55), but since these three groups of genes are shared by Xenopus, teleost fishes, etc. as well as by other mammals, they must have diverged about 400 Myr ago. This indicates that VH gene diversity in these organisms was generated by gene duplication and diversification during a long evolutionary time.
The tree in Fig. 8, however, shows that all VH genes in some mammalian species are closely related and all sequences from a species belong to only one VH group (B or C). They are cattle, sheep, pigs, and rabbits, which are all domesticated animals. Particularly interesting are the cattle and sheep gene clusters which belong to group B. Since cattle and sheep belong to the same suborder (Ruminantia) and apparently diverged about 20 Myr ago, the divergence of the two clusters occurred quite rapidly. Another interesting observation is that pigs, which belong to the same order (Artiodactyla) as cattle, also have one cluster of closely related VH genes but this cluster belongs to group C genes rather than group B genes. At any rate, these organisms have a limited VH gene repertoire compared with that of humans and mice, but they have no problem in survival.
Another organism which has an unusual set of VH genes is chicken. In this organism the gene cluster has only one functional gene (VH1) and about 100 VH pseudogenes, and VH diversity is generated through somatic gene conversion of the functional gene by pseudogenes (59). Interestingly, all the VH1 and pseudogenes are closely related to one another and belong to group C (62). Furthermore, the Vλ gene cluster in chicken also shows essentially the same genetic property (63). A somewhat similar genetic system is observed with the rabbit VH genes (60). In this organism only one VH gene is usually expressed, and other VH genes or pseudogenes are used for somatic conversion of this expressible gene. At the present time, it is unclear how these types of genetic systems have evolved.
DISCUSSION
When the major aspects of genomic structure of MHC and Ig genes were first clarified in humans and mice, the evolutionary schemes of the two genetic systems appeared quite different. The diversity of MHC molecules is generated primarily by intralocus polymorphism, whereas the immunoglobulin diversity is generated by interlocus genetic variation barring somatic mutation or somatic gene conversion (64). However, as the genes from other organisms are studied, a number of common features have emerged between the two systems despite the difference in genomic organization and function. In both genetic systems the major force of evolution is the combination of events of gene duplication, deletion, and translocation as well as point mutation
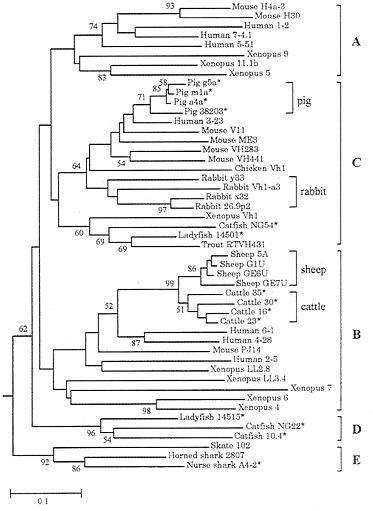
FIG. 8. Phylogenetic tree of 49 VH genes from various vertebrate species. This tree was constructed by using p-distances for the amino acid sequences of framework regions 1, 2, and 3. Most sequence data are from Ota and Nei (14), and additional sequences for cattle, sheep, pigs, and rabbits are from GenBank. The root of the tree was determined by using light chain genes as was done in ref. 14.
and selection. Dysfunctioning of genes due to deleterious mutation is also quite common in both systems. Furthermore, some organisms seem to have more homogeneous functional genes than other organisms. In the following, we discuss the general patterns of evolution of MHC and Ig genes in relation to concerted and birth-and-death evolution.
Concerted Evolution. Concerted evolution was originally proposed to explain a high degree of sequence similarity among member genes of a multigene family (1). The first mechanism considered for concerted evolution is interlocus recombination, which generates new duplicate genes and deletes some extant duplicate genes. If this process continues, duplicate genes in a multigene family tend to have similar nucleotide sequences even in the presence of mutation. Later, intergenic gene conversion was introduced as an additional mechanism for homogenizing the member genes of a multigene family (2, 65).
Concerted evolution was also invoked to explain the diversity and evolution of Ig and MHC genes, as mentioned earlier. In this second version of concerted evolution, interlocus gene conversion or recombination is regarded as a mechanism of increasing genetic diversity (polymorphism) for a locus or a set of loci by introducing new variants from different loci. Therefore, all loci are assumed to be polymorphic, and the polymorphism at different loci evolves in unison as a result of concerted evolution (5, 6, 11).
We have seen that in both MHC and Ig gene families repeated gene duplication occurs quite often and thus generates a set of closely related genes. At first sight, this appears to be consistent with concerted evolution that homogenizes the member genes. In practice, however, this gene duplication does not necessarily lead to deletion of preexisting heterogeneous genes and therefore does not contribute to homogenization of all member genes. Rather, duplicate genes gradually diverge by mutation and selection, though some of them become nonfunctional by deleterious mutations or are deleted from the genome. In fact, both MHC and Ig gene families contain various member genes that have diverged during tens of millions or hundreds of millions of years. Even the human MHC class Ia loci A and B, which were generated by a recent gene duplication, seem to have a history of about 30 Myr (25). Mammalian group A, B, and C VH genes have a history of about 400 Myr (14), and the extent of nucleotide differences among them is very high. We have seen that the VH genes in cattle, sheep, pigs, and chickens diverged relatively recently, but even these genes seem to have a history of 20–40 Myr. This conclusion is essentially the same as that of Gojobori and Nei (12) and does not lend support to the first version of concerted evolution.
Our results also contradict the second version of concerted evolution, in which intralocus polymorphism is generated by interlocus gene conversion or recombination. First, VH genes are not very polymorphic as was previously assumed (11), and thus there is no need to invoke interlocus genetic exchange to promote gene diversity in this gene family. Second, classical MHC loci are certainly highly polymorphic, but the phylogenetic trees do not show any significant intermingling of alleles from different polymorphic loci, suggesting that genetic exchange between loci does not contribute significantly to the extent of polymorphism. Some authors (18) claimed the importance of interlocus genetic exchange by finding that a few alleles from a locus were clustered with the alleles from other loci in MHC gene families. However, they have not done any statistical test of allelic clusters, and it seems that this intermingling of alleles from different loci was caused by stochastic or sampling errors. Of course, there are a few clear-cut cases of exon exchanges between different MHC loci (13, 66), but their contribution to MHC gene diversity seems to be minor (67).
Birth-and-Death Model of Evolution. In this model of evolution, duplicate genes are produced by various mechanisms, including tandem and block gene duplication, and some of the duplicate genes diverge functionally but others become pseudogenes owing to deleterious mutations or are deleted from the genome (Fig. 1B). The end result of this mode of evolution is a multigene family with a mixture of divergent groups of genes and highly homologous genes within groups plus a substantial number of pseudogenes. The model of birth-and-death evolution was formally presented by Nei and Hughes (16), but essentially the same process was envisaged at the genome level by Nei (68), who, on theoretical grounds, predicted that the mammalian genome contains a large number of duplicate genes and nonfunctional genes. Nei and his colleagues (12–16, 27) studied this problem statistically when DNA sequences for VH and MHC genes became available and obtained evidence supporting the model. The birth-and-death model of evolution is similar to Klein et al.’s (69) accordion model of MHC evolution, in which the number of MHC genes is assumed to be expanded or contracted, depending on the need to protect the host from ever-changing groups of parasites. The former model, however, allows some MHC genes (e.g., Ib genes) to stay in the genome for a long time or even to acquire slightly modified functions.
The results presented in this paper are consistent with what are expected from the birth-and-death model of evolution. We have seen that both the MHC and VH gene clusters in a species have experienced repeated gene duplication and many duplicate genes have become nonfunctional or deleted About 40% of VH genes and 50% of Vκ and Vλ genes in humans are known to be pseudogenes. The exact number of pseudogenes in the MHC cluster in humans is still unknown, but it seems to be quite large. In the class I gene region at least 16 pseudogenes or gene fragments have been identified (38). The mouse genome also contains many MHC pseudogenes (22).
In the early stage of study of evolution of VH genes it appeared that the number of VH genes per genome is roughly the same for different species and that the effects of the birth and death processes in the VH gene cluster are balanced (12). Recent data, however, show that the number of VH genes varies considerably with species and is probably less than 20 in sheep, cattle, and pigs (61, 70, 71). Therefore, many VH genes have apparently been deleted in these species. This suggests that the number of VH genes need not be very large for the survival of an individual and that in these organisms VH diversity may be enhanced by somatic mutation, somatic gene conversion, and deletions/insertions that are generated when the VH, DH, and JH genes are recombined somatically. The number of MHC genes per genome is also known to vary extensively with organism (22). Therefore, gene duplication and deletion seem to be quite common in both Ig and MHC genes.
In retrospect, it is quite reasonable that MHC and Ig genes are subject to birth-and-death evolution, whereas ribosomal RNA (rRNA) genes follow concerted evolution. The latter gene family is used to produce a large quantity of the same gene product (rRNA) and thus natural selection would favor a highly homogeneous group of member genes. Concerted evolution is an efficient way to achieve this homogeneity. By contrast, the function of MHC and Ig genes is to defend the host from various forms of invading parasites, and a great amount of diversity is required for them. Therefore, the evolutionary force needed is for diversification of genes, and this can be achieved by gene duplication, mutation, and diversifying selection.
Interlocus genetic variation may increase by independent mutation alone during a long evolutionary time, but in the case of MHC and Ig genes there is some evidence that different member genes are often adapted to cope with different types of parasites. For example, alleles from human HLA class I loci A, B, and C have different antigen (peptide) specificity (19). The long-term survival of mammalian group A, B, and C VH genes also suggests that these genes are adapted to different groups of antigens. Indeed, recent studies have shown that the human VH family 3 is adapted to cope with a special group of bacterial antigens (72). In this respect chickens are special in having very closely related
VH or Vλ sequences, but the paucity of VH and Vλ diversity is compensated by a high rate of interlocus somatic gene conversion, as mentioned earlier. Some authors (10, 73) suggested that these closely related sequences are subject to germ-line as well as somatic gene conversion. This may well be the case, but chicken and rabbit Ig genes are clearly exceptional and do not seem to follow the general pattern of Ig gene evolution in vertebrates.
The genome of higher organisms contains a large number of multigene families, some of which have evolved to produce a large quantity of the same gene product and others of which have evolved to produce a diverse array of proteins. If our conclusion is correct, the former group of gene families would be subject to concerted evolution and the latter to birth-and-death evolution. We hope that a detailed study will be conducted on this subject for many multigene families and the general pattern of evolution of multigene families will be clarified in the near future.
We thank Kei Takahashi for his help in the preparation of the manuscript and Austin Hughes, Jan Klein, Sudhir Kumar, and George Zhang for their comments on an earlier version of this manuscript. This work was supported by National. Institutes of Health Grant GM20293 and National Science Foundation Grant DEB-9520832 to M.N.
1. Smith, G.P. (1973) Cold Spring Harbor Symp. Quant. Biol. 38, 507–513.
2. Zimmer, E.A., Martin, S.L., Beverley, S.M., Kan, Y.W. & Wilson, A.C. (1980) Proc. Natl. Acad. Sci. USA 77, 2158–2162.
3. Irwin, D.M. & Wilson, A.C. (1990) J. Biol. Chem. 265, 4944–4952.
4. Arnheim, N. (1983) in Evolution of Genes and Proteins, eds. Nei, M. & Koehn, R.K. (Sinauer, Sunderland, MA), pp, 38–61.
5. Weiss, E.H., Mellor, A.L., Golden, L., Fahrner, K., Simpson, E., Hurst, J. & Flavell, R.A. (1983) Nature (London) 301, 671–674.
6. Ohta, T. (1991) Proc. Natl. Acad. Sci. USA 88, 6716–6720.
7. Rada, C., Lorenzi, R., Powis, S.J., Bogaerde, J.V.D., Parham, P. & Howard, J. (1990) Proc. Natl. Acad. Sci. USA 87, 2167–2171.
8. Hood, L., Campbell, J.H. & Elgin, S.C.R. (1975) Annu. Rev. Genet. 9, 305–353.
9. Bentley, D.L. & Rabbitts, T.H. (1983) Cell 32, 181–189.
10. McCormack, W.T., Hurley, E.A. & Thompson, C.B. (1993) Mol. Cell. Biol. 13, 821–830.
11. Ohta, T. (1983) Theor. Pop. Biol. 23, 216–240.
12. Gojobori, T. & Nei, M. (1984) Mol. Biol. Evol. 1, 195–212.
13. Hushes, A.L. & Nei, M. (1989) Mol. Biol. Evol. 6, 559–579.
14. Ota, T. & Nei, M. (1994) Mol. Biol. Evol. 11, 469–482.
15. Nei, M. & Hughes, A.L. (1991) in Evolution at the Molecular Level, eds. Selander, R., Clark, A. & Whittam, T. (Sinauer, Sunderland, MA), pp. 222–247.
16. Nei, M. & Hughes, A.L. (1992) in 11th Histocompatibility Workshop and Conference , eds. Tsuji, K., Aizawa, M. & Sasazuki, T. (Oxford Univ. Press, Oxford), Vol. 2. pp. 27–38.
17. Huber, C., Selläble, K.F., Huber, E., Klein, R., Meindl, A., Thiebe, R., Lamm, R. & Zachau, H.G. (1993) Eur. J. Immunol. 23, 2868– 2875.
18. Brunsberg, U., Edfors-Lija, I., Andersson, L. & Gustafsson, K. (1996) Immunogenetics 44, 1–8.
19. Parham, P. & Ohta, T. (1996) Science 272, 67–74.
20. Yun, T.J., Melvold, R.W. & Pease, L.R. (1997) Proc. Natl. Acad. Sci. USA 94, 1384–1389.
21. Feder, J.N., Gnirke, A., Thomas, W., Tsuchihashi, Z., Ruddy, D.A., et al. (1996) Nat. Genet. 13, 399–408.
22. Trowsdale, J. (1995) Immunogenetics 41, 1–17.
23. Klein, J. & O’hUigin, C. (1994) Proc. Natl. Acad. Sci. USA 91, 6251–6252.
24. Rogers, J.H. (1985) EMBO J. 4, 749–753.
25. Klein, J. & Figueroa, F. (1986) Crit. Rev. Immunol. 6, 295–386.
26. Hughes, A.L. & Nei, M. (1989) Genetics 122, 681–686.
27. Hughes, A.L. & Nei, M. (1990) Mol. Biol. Evol. 7, 491–514.
28. Klein, J. (1986) Natural History of the Major Histocompatibility Complex (Wiley, New York).
29. Hedrick, P.W. & Kim, T.J. (1997) in Evolutionary Genetics from Molecules to Morphology, eds. Singh, R.S. & Krimbas, C.K. (Cambridge Univ. Press, New York), in press.
30. Hughes, A.L. & Nei, M. (1988) Nature (London) 335, 167–170.
31. Hughes, A., Hughes, M.K., Howell, C.Y. & Nei, M. (1994) Phil. Trans. R. Soc. Lond. B 345, 359–367.
32. Watkins, D.I., McAdams, S.N., Liu, X., Strang, C.R., Milford, E.L., Levine, C.G., Garber, T.L., Dogon, A.L., Lord, C.I., Ghim, S.H., Troup, G.M., Hughes, A.L. & Letvin, N.L. (1992) Nature (London) 357, 329–333.
33. Fodil, N., Laloux, L., Wanner, V., Pellet, P., Hauptmann, G., Mizuki, N., Inoko, H., Spies, T., Theordorou, I. & Bahram, S. (1996) Immunogenetics 44, 351–357.
34. Weiss, E.H., Golden, L., Fahrner, K., Mellor, A.L., Devlin, J.J., Bullman, H., Tiddens, H., Bud, H. & Flavell, R.A. (1984) Nature (London) 310, 650–655.
35. Kumar, S., Tamura, K. & Nei, M. (1993) MEGA: Molecular Evolutionary Genetic Analysis (Pennsylvania State Univ., University Park).
36. Lawlor, D.A., Zemmour, J., Ennis, P.D. & Parham, P. (1990) Annu. Rev. Immunol. 8, 23–63.
37. Zemmour, J., Koller, B.H., Ennis, P.D., Geraghty, D.E., Lawlor, D.A., Orr, H.T. & Parham, P. (1990) J. Immunol. 144, 3619–3629.
38. Geraghty, D.E., Koller, B.H., Pei, J. & Hansen, J.A. (1992) J. Immunol. 149, 1947–1957.
39. Nei, M. (1996) Annu. Rev. Genet. 30, 371–403.
40. Hughes, A.L. (1991) Immunogenetics 33, 367–373.
41. Salgar, S.K., Kunz, H.W. & Gill, T.J. (1995) Immunogenetics 42, 244–253.
42. Liskay, R.M., Letsou, A. & Stachelek, J.L. (1987) Genetics 115, 161–167.
43. Chen, Z.W., McAdam, S.N., Hughes, A.L., Dogon, A.L., Letvin, N.L. & Watkins, D.I. (1992) J. Immunol. 148, 2547–2554.
44. Hedges, S.B., Parker, P.H., Sibley, C.G. & Kumar, S. (1996) Nature (London) 381, 226–229.
45. Schneider, S., Vincek, V., Tichy, H., Figueroa, F. & Klein, J. (1991) Mol. Biol. Evol. 8, 753–766.
46. Slade, R.W. & Mayer, W.E. (1995) Mol. Biol. Evol. 12, 441–450.
47. Tanaka, T. & Nei, M. (1989) Mol. Biol. Evol. 6, 447–459.
48. Li, H. & Hood, L. (1995) Genomics 26, 199–206.
49. Sasso, E.H., Buckner, J.H. & Suzuki, L.A. (1995) J. Clin. Invest. 96, 1591–1600.
50. Zachau, H.G. (1995) in Immunoglobulin Genes. eds. Honjo, T. & Alt. F.W. (Academic, San Diego), pp. 173–191.
51. Cook, G.P., Tomlinson, I.M., Walter, G., Riethman, H., Carter, N.P., Buluwela, L., Winter, G. & Rabbitts, T.H. (1994) Nat. Genet. 7, 162–168.
52. Matsuda, F., Shin, E.K., Nagaoka, H., Matsumura, R., Haino, M., Fukita, Y., Taka-ishi, S., Imai, T., Riley, J.H., Anand, R., Soeda, E. & Honjo, T. (1993) Nat. Genet. 3, 88–94.
53. Honjo, T. & Matsuda, F. (1995) in Immunoglobulin Genes, eds. Honjo, R. & Alt, F.W. (Academic, San Diego), pp. 145–171.
54. Tomlinson, I.M., Williams, S.C., Ignatovich, O., Corbertt, S.J. & Winter, G. (1996) V BASE Sequence Directory (MRC Centre for Protein Engineering, Cambridge, U.K.).
55. Schroeder, H.W.J., Hillson, J.L. & Perlmutter, R.M. (1990) Int. Immunol. 2, 41–50.
56. Williams, S.C., Frippiat, J.-P., Tomlinson, I.M., Ignatovich, O., Lefranc, M.-P. & Winter, G. (1996) J. Mol. Biol. 264, 220–232.
57. Andersson, E. & Matsunaga, T. (1995) Res. Immunol. 147, 233–240.
58. Rast, J.P., Anderson, M.K., Ota, T., Litman, R.T., Margittai, M., Shamblott, M.J. & Litman, G.W. (1994) Immunogenetics 40, 83–99.
59. Reynaud, C.-A., Dahan, A., Anquez, V. & Weill, J.-C. (1989) Cell 59, 171–183.
60. Knight, K.L. & Tunyaplin, C. (1995) in Immunoglobulin Genes, eds. Honjo, R. & Alt, F.W. (Academic, San Diego), pp. 289–314.
61. Dufour, V., Malinge, S. & Francois, N. (1996) J. Immunol. 156, 2163–2170.
62. Ota, T. & Nei, M. (1995) Mol. Biol. Evol. 12, 94–102.
63. McCormack, W.T., Tjoelker, L.W. & Thompson, C.B. (1991) Annu. Rev. Immunol. 9, 219–241.
64. Tonegawa, S. (1983) Nature (London) 302, 575–581.
65. Slightom, J.L., Blechl, A.E. & Smithies, O. (1980) Cell 21, 627–638.
66. Holmes, N. & Parham, P. (1985) EMBO J. 4, 2849–2854.
67. Hughes, A.L. (1995) Mol. Biol. Evol. 12, 247–258.
68. Nei, M. (1969) Nature (London) 221, 40–42.
69. Klein, J., Ono, H., Klein, D. & O’hUigin, C. (1993) Prog. Immunol. 8, 137–143.
70. Sinclair, M.C. & Aitken, R. (1995) Gene 167, 285–289.
71. Sun, J., Kacskovics, I., Brown, W.R. & Butler, J.E. (1994) J. Immunol. 153, 5618–5627.
72. Silverman, G.J. (1995) Ann. N.Y. Acad. Sci. 764, 342–355.
73. Roux, K.H., Dhanarajan, P., Gottschalk, V., McCormack, W.T. & Renshaw, R.W. (1991) J. Immunol. 146, 2027–2036.








