
Page 277
6
Future Strategies for MS Therapies
This chapter focuses on the development of therapies that can halt or slow the disease process in multiple sclerosis (MS). Therapies for the relief of specific symptoms are discussed in Chapter 3.
The plethora of potential therapeutic agents and the multiplicity of patterns and stages of disease to which each might be applied will demand tailoring of pivotal clinical trial designs to the specific clinical situation. Such tailoring will involve trial duration, selection of outcome measures, and as a consequence, sample size.
Modification of the course of MS presents opportunities for five types of interventions:
1. Primary prophylaxis in at-risk individuals. These trials will be aimed at preventing the appearance of overt disease in two populations of patients: (1) individuals who have presented clinically with an episode of monosymptomatic demyelinating disease and (2) individuals known to be genetically at risk.
2. Relapse prevention via immune modulation. In this category, one would place the recent and ongoing trials of the beta-interferons—those whose primary clinical targets were relapse rate, with secondary outcome measures of magnetic resonance imaging (MRI) progression. In these studies, the interferons, all to a similar degree, decreased relapse rates as well as the number of gadolinium-enhancing lesions. However, glatiramer acetate reduced clinical activity to a similar degree without as profound an effect on the MRI. Thus, different agents might produce similar clinical
Page 278
results, but with different imaging profiles because each targets a slightly different aspect of the underlying disease pathology.
3. Relapse limiting. Current practice is to treat acute relapses with intravenous methylprednisolone. However, no criteria have been adopted that permit uniform designation of the onset and completion of a relapse. The difficulty in this area arises from the fact that relapses can take numerous forms, from monosymptomatic optic neuritis to acute transverse myelitis, and so forth. Nonetheless, if treatments are to be tested for the ability to shorten individual relapses, then criteria for determining precise clinical onset and end of relapse episodes will be required.
4. Progression altering. To date, clinical measures of progression of disability have correlated poorly with imaging measures in MS clinical trials. The exception appears to be the estimation of brain atrophy. Studies evaluating the effects of agents on disease progression will have to incorporate measures of brain and spinal cord parenchymal volume as well as more sensitive and reproducible measures of neurological function than those currently in use.
5. Neuroprotective and restorative. Chapter 5 discusses the potential of and rationale for the use of various neuroprotective and potentially restorative therapies for MS. Many of the current putative neuroprotective or restorative agents in clinical or pre-clinical research are protein growth factors that must somehow be delivered to the central nervous system (CNS), either directly or across the normally restrictive blood-brain barrier. However, it should be noted that insulin-like growth factor-1 (IGF-1) appears to cross the blood-brain barrier in certain experimental autoimmune encephalomyelitis (EAE) models, so the inflammatory nature of the acute MS lesion might permit the use of such agents in the acute setting. Designs for trials of neuroprotective or glioprotective agents in MS will depend on the nature of the question being asked, as well as the specificities and properties of the agent being tested.
STRATEGIES FOR DISEASE MODIFICATION
Advances in understanding the molecular neurobiology of myelinated axons, as described in Chapter 2, have revealed much about the contribution of impaired impulse conduction to symptom production in MS. Nonetheless, there is much more to learn about the mechanisms underlying demyelination and axonal injury in MS. What are the precise molecular steps that lead from the initial immunologic assault to the death of oligodendrocytes and the degeneration of axons? Are there steps at which these cascades can be halted? It has recently been suggested that cytokines might play a role in injuring myelinated nerve fibers in MS. More information is needed about which cytokines are involved and which molecular
Page 279
pathways carry out their injurious actions. Cytokines and the toxic levels of nitric oxide (NO) that they produce can be manipulated in a variety of ways and could provide a target for therapeutic interventions in demyelinating disorders.
Immune-Based Therapy
Antigen-specific tolerance, a method of antigen-specific immunomodulation, relies on administering an MS-related antigen in a manner that induces tolerance, thereby reducing the immune response to that antigen. Myelin basic protein (MBP) acts as a classical encephalitogenic autoantigen in certain EAE models, which has raised hopes for the development of T-cell-based therapies in which MS patients could be vaccinated with targeted portions of the MBP molecule to induce tolerance and prevent further T-cell-mediated attack. However, as reviewed in the “Immunopathology” section of Chapter 2, the human response to MBP is more complex than that in these EAE models. Indeed, MBP does not act as a classic autoantigen in humans, and even among EAE models, it is highly variable. Generally, it appears that the T-cell response against myelin proteins also differs greatly between individual patients, suggesting that immune therapies might have to be individually tailored for different patients.
Another concern of this approach is that the immune response to antigen administration follows an unusual dose-response curve. Although low-dose administration of most drugs is generally the safest course when beginning clinical trials, low-dose administration of antigen can, in fact, sometimes induce unsafe immune responses, whereas high doses can induce tolerance.
Vaccination
Vaccination, of course, would be an attractive therapeutic avenue. In fact, numerous variants of vaccination have been proposed. These include vaccination with whole myelin-specific T cells,8,33 with T-cell receptor peptides,13,107,116 DNA-encoding autoantigens, or T-cell receptor (TCR) sequences.64,111 Some of these therapies were quite impressive in EAE models when immunization was induced against a known target autoantigen (for example, MBP, MOG, or PLP), but at least in the case of TCR vaccination, the therapy failed in human MS. This is already an active area of corporate research, which has thus far has not fulfilled our hopes. Although potentially of significant therapeutic value, the induction of tolerance remains poorly understood, making it difficult to test clinically.
Suppressor Cells
For the most part, T cells are considered to underlie the immune-mediated attack on myelin, but one class of T cells—suppressor cells—can suppress the activity of pathogenic T cells and might play a protective role in MS.57 Several
Page 280
types of cells, including CD4 minus and CD8 minus positive suppressor cells have been shown to suppress EAE in some animal models, but the role of suppressor T cells and their potential for therapeutic use in MS are far from clear. CD25+ T cells are closest to the classic suppressor cells, but they are enigmatic and not well characterized in terms of antigen specificity, function, and mechanism of action. Their existence has been postulated on the basis of transfer and depletion studies in vivo (Don Mason, Ethan Shevach), as in the NOD mouse model of insulin-dependent diabetes mellitus (IDDM), in neonatal thymectomy models of multiorgan infiltration (Sakaguchi), and also in T-cell receptor transgenic, “monoclonal” mice (Tonegawa; Lafaille). Evidence for a role of these cells in CNS-specific autoimmune diseases is indirect at best.
Immunodeviation
Approach potential involves inducing immunodeviation of myelin-specific T cells, that is, shifting the balance of production from Th1 to Th2 cells. This is a crowded field in MS research, as in other putative autoimmune diseases. Several private firms are investigating this strategy, and the committee feels this approach is already receiving adequate attention and is not lacking for encouragement. Further, in the pathogenesis of EAE and MS, there is no clear distinction between “good” Th2 and “bad” Th1 T cells. Both cell types might produce pathogenic inflammation in different situations.
Genetic Engineering
Another possible innovative therapy would be to use genetically engineered autoimmune T cells. Even though the precise pathological roles of T cells and their autoantigens are unresolved, this line of research has generated many approved or emerging therapies. These include vaccination strategies, which use either attenuated myelin-specific T cells97,33 or peptides representing myelin-specific T-cell receptors3 as vaccines to strengthen the body's own regulatory responses against pathogenic T cells (reviewed in 1998 by Zhang et al.).120 Also under development are “altered peptide” therapies that use peptide analogues of myelin protein segments to induce autoreactive T cells to produce protective, rather than pathogenic, cytokine mediators.96
Neuroprotection
Recent studies have elegantly demonstrated the loss of axons in chronic MS lesions. As discussed elsewhere in this report, the precise mechanisms leading to axonal degeneration in MS are at present unknown. Axons can degenerate as a result of the loss of trophic support of their myelin sheaths. Alternatively, they
Page 281
can be victimized as “innocent bystanders” in the surrounding inflammatory milieu. In addition to the structural loss of axons in MS lesions, axonal dysfunction and conduction block in demyelinated foci also contribute to neurological symptoms in MS patients. Axonal loss occurs early in disease and is possibly the major cause of irreversible neurological impairment.104 Neuroimaging studies suggest that axonal loss begins as early as the onset of disease.
Axonal damage leads to Wallerian degeneration and the loss of neuronal cell bodies. MRI and computed tomography (CT) scans of MS victims show evidence of widespread atrophy. It is unclear at present whether the loss of axons leads to neuronal cell death in MS. However, accumulated axonal loss and dysfunction, especially in the progressive phase of the disease, underlie the progressive neurodegeneration seen in MS. Thus, therapeutic strategies aimed at preventing neuronal damage might be a key to preventing permanent disability. There are a variety of approaches to anti-inflammatory strategies, remyelination, and the use of specific neuroprotective agents, and these are considered in turn below.
Anti-inflammatory Strategies
The most obvious “neuroprotective” strategy for the treatment of MS is prevention of the inflammatory lesion that leads to demyelination and oligodendrocyte loss. Anti-inflammatory approaches are discussed elsewhere in this volume. However, it should be noted that in the course of inflammation, cytokines such as tumor necrosis factor-α (TNF-α) and lymphotoxin are produced, and they can be damaging to neurons and glia. Furthermore, the reactive oxygen species, nitrous oxide and glutamate, produced by invading inflammatory cells can be toxic to both oligodendroglia and axons that pass through regions of active demyelination. Therapies aimed at preventing oxidative damage to neurons might, therefore, be effective in minimizing neurological impairment during acute MS attacks.
Remyelination
Trophic and other interactions between axons and ensheathing glial cells contribute to the structural and functional integrity of axons. Therefore, administration of agents that promote remyelination or ameliorate damage to myelin should also protect neurons. Such strategies might include administration of gliotrophic factors or transplantation of glial precursors or stem cells that repopulate demyelinated regions of the CNS.112
Attempts at myelination occur in and around MS lesions.54,85,92 Especially in recent lesions, an abundance of oligodendrocytes may be found, as well as axons with thin myelin coatings and shortened internodes. This presumably indicates de novo myelin formation as it does in remyelination of peripheral nerves. Lassmann
Page 282
and colleagues have categorized MS lesions into five types, based on the degree of oligodendrocyte destruction and the stage of apparent repair.54
Having noted that attempts at myelin repair are not uncommonly found in MS brains, it may be assumed that at some level, this phenomenon is responsible to greater or lesser degree for the recovery of function that occurs after relapses. Thus, the time course and the degree of recovery from individual bouts of demyelination give us some notion of the capacity of the CNS to repair myelin damage. The obvious questions then are why remyelination and recovery are not universal, what factors inhibit remyelination, and what therapeutic options might exist to promote remyelination in the future?
The cell type responsible for initiating the process of remyelination in MS plaques is at present unknown. Mature oligodendrocytes are incapable of mitosis and migration.44 However, it appears from animal models that there is a pool of progenitor cells available that can, under appropriate circumstances and in the absence of continued immune attack, regenerate myelinating oligodendrocytes.11,28 However, it would also appear that this pool of potential oligodendrocyte precursors is small and rapidly depleted.45
There appear to be two potential strategies for promoting myelin replacement in MS. While both are conceptually appealing, they have their potential drawbacks as well. One strategy might be to attempt to alter the cellular environment of the CNS so that it becomes more permissive, instead of inhibitory, to myelination by providing factors that promote the proliferation, migration, and maturation of oligodendrocytes. Oligodendrocyte precursors appear to originate from the O-2A precursor cell, so named because it gives rise to both oligodendrocytes and astrocytes.
Several protein growth factors are involved in the maturational sequence of oligodendrocytes. Current evidence suggests that initial proliferation of oligodendrocyte precursors is driven by basic fibroblast growth factor (bFGF). Platelet-derived growth factor (PDGF) is also involved in the growth of oligodendrocyte precursors, as well as in migration. The intracellular signaling pathways activated by these growth factors include at least two members of the mitogen-activated protein kinase (MAPK) family as well as pp70 S6 kinase.11 Activation of cyclic AMP5 or the PDGF antagonist, trapidil,85 blocks growth factor-induced proliferation of oligodendrocyte precursors. Withdrawal of PDGF and FGF results in differentiation of oligodendrocyte precursors into mature oligodendrocytes.5 Insulin-like growth factors 1 and 2 appear to be survival factors for mature oligodendrocytes; transforming growth factor-beta (TGFβ) is also involved in the differentiation of oligodendrocytes, as is the neurotrophic factor neurotrophin-3.
The complexity inherent in enhancing the gliogenic milieu of the CNS can be readily appreciated. The proliferation, migration, and differentiation of progenitor cells into mature, myelinating oligodendroglial cells require a precisely timed sequence of growth signals that, for the treatment of patients, must be delivered to multiple lesions disseminated in space and time, inherently differing
Page 283
in their states of demyelination and remyelination. Furthermore, the success of such a strategy depends on the availability of an endogenous pool of progenitor cells ready to be induced to divide, migrate, and mature into functional myelinating oligodendrocytes. Finally, the newly formed myelinating cells must be protected from further immune attack.
An alternative strategy might be to supply the diseased MS brain with cells that would develop into mature oligodendrocytes, perhaps in conjunction with a source of the requisite growth factors. Many of the same caveats apply to such transplantation strategies as attempts to promote regeneration of endogenous myelinating cells. In addition, the potential for both immune rejection and malignant transformation of transplanted cells must be overcome.
Specific Neuroprotective Agents
Damaged neurons in the CNS attempt to repair themselves. These attempts are usually not successful and, as indicated previously, can lead to maladaptive effects. An important component of restoration of function, especially in a disease such as MS in which axons are damaged, must include the possibility of axonal repair. Several conditions interfere with attempts at axonal regrowth after lesions develop. These include the presence of a glial scar (depending on the site of the lesion), the lack of neurotrophic factors that support growth, or the presence of inhibitory molecules that impede axonal growth. One of the more promising approaches to encourage axonal growth is the administration of growth-supporting molecules, particularly specific growth factors.
As noted above, axonal transection in areas of demyelination can lead to Wallerian degeneration and neuronal cell loss. Thus, agents that induce protective reactions in neurons or enhance axonal sprouting might preserve or promote recovery of neuronal function. Application of such strategies is not, however, without its challenges. Recent clinical trials of neurotrophic factors for the treatment of amyotrophic lateral sclerosis (ALS), which is a relatively simple, monophasic degenerative disorder of the motor system, have so far failed to produce positive results. Neurotrophic factors are large protein molecules. For therapeutic use, these complex molecules are synthesized in bacteria or cultured animal cells from which they can be purified and then administered by injection. To the best of our knowledge, systemically administered neurotrophic factors do not cross the blood-brain barrier. For the treatment of MS, it would seem that they must be administered directly into the CNS, either in soluble form via an implanted pump or using some novel cellular means of delivery such as transformed cells or direct gene therapy approaches. Small-molecule agonists and enhancers or inducers of neurotrophic activity are in development.
Specificity is the final challenge of using neurotrophic factors to treat degenerative disorders, including MS. Many neurotrophic factors act only on certain cell types within the CNS. For example, the receptor for nerve growth factor
Page 284
(NGF) appears localized to only basal forebrain cholinergic neurons and a few other cell types in the brain; receptors for the related trophic factor, brain derived neurotrophic factor (BDNF) are widespread throughout the cortex and subcortical structures. Neuronal damage in MS, however, affects axons belonging to multiple cell types, not all of which will necessarily respond to the same neurotrophic factors. In addition to having trophic actions on neurons, a number of neurotrophic factors, including neurotrophin-3 (NT-3), ciliary neurotrophic factor, and IGF-1, exert effects on glia. Many of the “neuroprotective” agents today are protein growth factors.
Neurotrophins. The neurotrophins are a family of related 22-kDa proteins that are crucial for the survival and differentiation of neurons during development and in response to injury. They include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin 4/5 (NT-4/5).59 Originally discovered because of their ability to promote neuronal survival in vitro, the neurotrophins have been found to participate in many aspects of neuronal function, including plasticity, neurotransmitter synthesis and release and regulation of cytoskeletal proteins, receptor proteins, and so forth. The specificity of action of the neurotrophins is dictated by the distribution of their receptors. Neurotrophins bind to two populations of cellular receptors. The so-called low-affinity neurotrophin receptor (p75LNGFR) belongs to the TNF family and binds all members of the family with varying affinities. In some systems, occupancy of this receptor by a cognate ligand can prevent apoptotic cell death. The “high-affinity” receptors for the neurotrophins belong to the Trk family of tyrosine kinase receptors. The three members of this family TrkA, TrkB, and TrkC bind NGF, BDNF, NT4/5, and NT-3, respectively. NT-3 is somewhat “promiscuous” because it can bind to and signal through TrkA as well as TrkC.
Neurotrophins can be produced by inflammatory cells. Both activated lymphocytes9 and glia49 possess neurotrophin receptors and can produce neurotrophins. Different cytokines stimulate different patterns of neurotrophin expression. Beta-interferon stimulates NGF production by astrocytes;14 activated lymphocytes and monocytes can produce BDNF.9,46 Thus, the neurotrophins appear to be expressed in the milieu of the MS lesion, perhaps as part of the reparative or restorative response.
Neurotrophic factors can protect and rescue neurons in a large number of experimental models. Of particular relevance to MS is the demonstrated ability of BDNF and NT-3, in particular, to promote regeneration of long tracts in the spinal cord.117
Neurotrophins are also involved in the development and maintenance of glia, including oligodendroglia. NT-3 stimulates the proliferation of oligodendrocyte progenitors in vitro, and both NT-3 and NGF enhance the survival of differentiated oligodendrocytes in culture.20 NGF receptors are found on mature human oligodendrocytes.51 Intracranial injection of NT-3 appears to cause an increase in
Page 285
the proliferation of oligodendrocyte precursors and an inhibition of proteases involved in cell death signaling.48
The role of the neurotrophins in demyelinating diseases is presently unclear. Levels of NGF are increased in the CSF of MS patients,55 in the optic nerve of MS patients,73 and in the brains of animals with EAE.72 However, it is not known whether the increase in NGF is due to its release from inflammatory cells or whether it is part of a protective response of the CNS. There is no information available concerning the regulation of BDNF or NT-3 in animal or human demyelinating disease. Exogenous NGF can prevent autoimmune demyelination in marmosets.110 Whether this is due to inhibition of the immune attack or elicitation of protective responses in oligodendrocytes remains to be determined.
BDNF has been studied in clinical trials in ALS, although the initial results suggested that the doses studied might have been too low.7 Subsequent studies are examining the safety and efficacy of BDNF administered either at systemic doses higher than those studied in the original trial or intrathecally. These studies are in progress at the time of this writing. Nerve growth factor and NT-3 have both been studied in peripheral neuropathy patients, but not in CNS disorders.
Neuropoietic Cytokines. Neuropoietic cytokines are the family of neural growth factors that act on both the nervous and the hematopoietic or immune systems, and which include ciliary neurotrophic factor (CNTF). CNTF is further a member of the interleukin-6 (IL-6) family of cytokines. It was originally isolated from ocular tissues as a protein factor that supports the survival of ciliary ganglion neurons in cell cultures and from the cytoplasm of myelinating Schwann cells of peripheral nerves. It has neuroprotective properties in a number of in vitro and in vivo systems, including models of motor neuron disease and spinal cord injury. CNTF appears to protect oligodendrocytes from TNF-induced cell death in vitro.21,65 CNTF has been studied in clinical trials of patients with ALS by both subcutaneous and intrathecal routes of administration.2,83 When administered subcutaneously, CNTF did not affect the course of disease in ALS patients, and the stability of the pharmaceutical formulation limited its use via the intrathecal route.
Other Growth Factors. Glial cell line-derived neurotrophic factor (GDNF) is a member of the TGF-β family of growth factors. GDNF was originally discovered based on its ability to support the survival of embryonic dopamine neurons in tissue culture. It was subsequently discovered to have effects on several classes of CNS neurons including corticospinal tract cells.43 GDNF has been studied in clinical trials using intracerebroventricular administration in patients with ALS and Parkinson's disease. These trials were stopped due to the severe side effects of the drug.
Insulin-like growth factor 1 (IGF-1) is a 7.65-kDa polypeptide that has multiple endocrine, metabolic, and neurotrophic actions.42 It is structurally and func-
Page 286
tionally related to insulin, and its effects are mediated through a tyrosine kinase receptor of the insulin receptor family. The activity and distribution of IGF-1 are tightly modulated by a group of IGF-specific binding proteins. IGF-1 participates in the development of neurons and glia, and its expression in the developing brain correlates spatially and temporally with the onset of myelination.56 Myelin content is increased in the brains of mice that overexpress the IGF-1 gene17 and decreased in IGF-1 knockout animals. The IGF-1 gene is induced in reactive astrocytes by experimental demyelination47 and in human MS lesions.31
In vitro, IGF-1 can protect oligodendrocytes from TNF-induced injury;119 cellular survival under appropriate conditions can be affected by the balance between IGF and TNF signaling due to cross-talk between postreceptor signal transduction cascades.108 IGF-1 also modulates the activity of immune-competent cells. Several recent studies have suggested that systemically administered IGF-1 can ameliorate EAE in animals.60
IGF-1 has been studied in clinical trials in ALS with inconclusive results.
“Gliotrophic” Factors. Oligodendroglial precursor cells respond to several growth factors in the course of their development. Signaling through some of these same pathways appears to be important for remyelination and proliferation of oligodendrocyte progenitor cells in the injured adult CNS as well. IGF-1 has already been mentioned, but it is only one of several growth factors that are important in oligodendroglial function. Some of these molecules can ameliorate the course of EAE. Fibroblast growth factor-2 (FGF-2) and thyroid hormone influence early pluripotential glial precursors to develop into oligodendrocytes. Platelet-derived growth factor controls the migration and proliferation of later-stage oligo precursors (see 1999 review by Rogister et al.87). Glial growth factor 2 (GGF-2; neuregulin), a member of the epidermal growth factor family, promotes survival and proliferation of pre-oligodendrocytes, but prevents differentiation to fully mature cells.16 GGF-2, however, has been reported to delay onset, decrease severity, and reduce relapse rate in a murine EAE model.67 Yet another neurotrophic factor, ciliary neurotrophic factor (CNTF), has been shown to protect oligodendrocytes from TNF-mediated cell death.65
Augmentation of Local Mechanisms
Neurotrophic factors might be produced in the MS lesion itself. For example, NGF is increased in inflammatory foci and in the CSF of MS patients. Interleukin-1 can stimulate astrocytes to synthesize NGF, which is probably the mechanism through which NGF is increased in inflammatory foci. Therapeutic enhancement of local neurotrophic factor production is a pathway worth exploring.
Page 287
Small Molecules
There are many small molecules that can protect neurons. These include glutamate antagonists, neuroimmunophilins, and nitric oxide and are discussed in turn below.
Riluzole is a putative neuroprotective agent that can modestly prolong survival in ALS74 patients. It is believed to work by blocking neuronal glutamate release and is currently being studied in other neurodegenerative disorders, such as Parkinson's disease and Alzheimer's disease.
Activated macrophages in MS lesions appear to release glutamic acid, as well as other potentially neurotoxic molecules. Neurons possess two types of glutamate receptors, both of which might participate in the neurotoxic effects of glutamate: NMDA and AMPA/kainate receptors. Oligodendrocytes also carry AMPA/kainate receptors. Recent studies have suggested that, like neurons, oligodendroglia might be susceptible to AMPA/kainate receptor-mediated glutamate excitotoxicity.69 Glutamate antagonists have been found to ameliorate myelin damage in an EAE model.32,113 AMPA antagonists are currently under study in stroke and neurodegenerative disease trials. Exploration of AMPA antagonism in MS is warranted as both a neuroprotective and a glioprotective strategy.
SR7657A is a small molecule that has been found to enhance and/or mimic the effects of neurotrophins in tissue culture. It is currently being studied in ALS. Thus far, studies of SR7657 on the course of ALS have failed to demonstrate a statistically significant effect of the drug, as measured by the trials' chosen end points.
Neuroimmunophilins are cyclosporin-related molecules that inhibit the signaling pathways that lead to cell death. They have been reported to protect brain dopamine neurons from injury and are currently being tested in early-stage clinical trials in Parkinson's disease.
Nitric oxide is a small, highly reactive molecule that is normally a gas at room temperature. It is synthesized in biological organisms where it exists as a highly lipid-soluble, dissolved nonelectrolyte solute. NO diffuses rapidly in tissues and enters into redux reactions via its unpaired electron. It is less reactive than other free radical species. Under appropriate conditions, NO is synthesized by one of a family of nitric oxide synthase (NOS) enzymes. It reacts with superoxide anion to produce peroxynitrite (ONOO−), a highly reactive substance that can interact with and modify proteins, lipids, and nucleic acids. NO is produced as a neurotransmitter in certain neurons and is also produced by endothelial cells and by active inflammatory cells.22 The inducible form of nitric oxide synthase (iNOS) has been localized in active MS lesions in human brain specimens.12 Selective inhibitors of both the neuronal (nNOS) and inducible (non-neuronal, iNOS) enzymes are becoming available18 and might be another class of neuroprotective and glioprotective agents that merit testing for their therapeutic value in MS.
Page 288
Interrupting the Secondary Injury Cascade in Axons
Axons, like neurons, do not always die immediately after an insult. Regeneration ensues after a latent period of hours to days and occurs as a result of a process of “secondary axonal injury.” Within gray matter regions of the brain and spinal cord, “excitotoxic” mechanisms trigger this secondary injury cascade. These excitotoxic mechanisms involve the abnormal, exaggerated activation of excitatory postsynaptic receptors, which allow the entry of damaging amounts of calcium into neurons, triggering a destructive cascade of damaging molecular events.
Glutamate is the most common excitatory neurotransmitter within the CNS and appears to play a key role in the excitotoxic process. In contrast to neuronal cell bodies, axons do not express glutamate receptors. Therefore, excitotoxic mechanisms would not be expected to play a major role in triggering the death of axons in white matter. Indeed, glutamate does not injure axons within white matter.86 Nonetheless, calcium-mediated axonal injury does occur in white matter.98 A wide variety of cellular insults will cause a sustained influx of sodium via “persistent” or non-inactivating sodium channels. This influx of sodium triggers calcium import via reverse mode operation of the Na+−Ca2+ exchanger.100 The result is a surge of damaging levels of calcium inside the axon. In in vitro experimental models, drugs that block the Na+−Ca2+ exchanger or sodium channels can protect axons so that they do not degenerate following injury.25,99,100 Neurotransmitters and neuromodulators such as γ-aminobutyric acid (GABA) and adenosine appear to have a modulatory effect on the axonal injury cascade within white matter, and indeed, GABA and adenosine have a neuroprotective effect in white matter where they can preserve axonal function after various insults.26 Calcium channels also appear to play a role in admitting injurious calcium into axons following some injuries.29 Blocking calcium channels, as well as sodium channels, thus should be considered in the search for neuroprotective strategies that will preserve axons in MS.
Delivering Neuroprotective Agents
The various possibilities for delivering neuroprotective agents include direct administration either systemically or directly into the CNS, or “indirectly” via gene therapy in which genes can be introduced that will increase the body's ability to produce more neuroprotective agents.
Systemic Delivery. One of the earliest events in the development of an MS lesion is the opening of the blood-brain barrier, which might thus provide a unique window of opportunity to deliver therapeutics to the CNS via systemic delivery. For example, systemically delivered IGF-1 has been found to be protective in EAE models.114 Studies in primates and in MS patients may be warranted.
Page 289
CNS delivery. Until the last decade or so, direct delivery of drugs into the CNS was too dangerous for human use, but with the emergence of ever-smaller implantable devices and genetically engineered cells, CNS drug delivery is increasingly possible.
Many MS patients have been treated with intrathecal baclofen for spasticity using an implanted pump. Recent clinical trials in ALS with BDNF82 and CNTF83 have demonstrated the feasibility of this method for delivering protein therapeutics to the CNS. Penetration to deep structures may be a problem. However, the side-effect profile of intrathecal BDNF, which includes insomnia and other behavioral effects, suggests that active concentrations of the drug are achieved in higher centers.82
Studies by Gage and others have demonstrated the potential of transplantation of genetically modified cells to deliver protein trophic factors to the CNS, with promising results. Such an approach has the advantage that multiple agents can be delivered simultaneously through one vehicle. However, like replacement transplantation approaches, the multifocal nature of the MS disease process could make clinical application of this approach problematic.
Gene Therapy Approaches. The term “gene therapy” refers to the insertion of specific genetic elements into either intact or diseased cells in the body with the aim of either restoring lost biochemical functions or introducing a molecular compensation for the consequences of a disease process. This may be accomplished by injecting either naked DNA or specially modified “vector” viruses containing the therapeutic genes either systemically or into a restricted location in the body. Gene therapy holds great promise for the treatment of focal or generalized CNS disorders. However, the plethora of protective factors that would have to be invoked for neuroprotective gene therapy of the CNS to be feasible will probably render this approach impractical, at least for the near future.
Summary of Neuroprotective Strategies
Axons in the MS brain exist in a threatening and destructive milieu, surrounded by toxic cytokines and free radicals, and stripped of their comforting myelin sheaths. Thus, the mechanisms by which axons are damaged in MS might be quite similar to those postulated in other neurodegenerative diseases such as Parkinson's disease. In addition, oligodendroglial cells are likely susceptible to many of the same cytotoxic mechanisms. Several potential therapeutic strategies have been described that could be investigated as neuroprotective approaches in MS.
The ideal neuroprotectant(s) would be able to protect, foster repair, and promote regeneration of all the different types of cells affected in MS and would further have a narrow enough range of function that there would be no undesir-
Page 290
able side effects. Unfortunately, this is unrealistic. Few of the agents currently under investigation are specific enough in their action on the cells they affect to avoid undesirable side effects.
In the future, application of neuroprotective or neurorestorative treatments is likely to involve combinations of agents that can be tailored to the stage of the disease. Examples of combination therapy might include the use of an anticytokine agent (for example, interferon or glatiramer acetate); an antiexcitotoxic agent, such as an AMPA antagonist (none of which have yet reached the clinic); a mitogenic or differentiating factor for oligodendrocyte precursors, such as IGF, NT-3, GGF-2, or a synthetic small-molecule agonist for one of these growth factors; and an agent that promotes repair or regeneration of central axons, such as BDNF or NT-3. Perhaps these drugs, or others like them, can be administered as “cocktails.” More likely, strategies of alternating agents that have different, yet complementary, activities will be adapted from those devised by oncologists.
Finally, the development of neuroprotective and glioprotective treatments will depend on advances in protein drug delivery and high-throughput drug screening. In addition, innovative clinical trial designs and novel outcome measures will be required to screen potential therapies in the clinic.
CNS Repair and Restoration
Strategic Questions About Remissions
The converse of the concept of relapse prevention, which has driven the most recent round of clinical trials in MS, is the concept of active induction of remission. Remissions, in which there is restoration of neurologic function, such as vision or the ability to walk, that had previously been lost, commonly occur in multiple sclerosis. One potential goal of MS treatment could thus be to try to take therapeutic advantage of the tendency of MS to naturally remit in its early stages in the majority of patients, with the ultimate goal of inducing sustained or even permanent improvement. This concept is not new. For many years, patients have been treated with high-dose intravenous steroids or adrenocorticotropic hormone (ACTH) with the goal of inducing immediate and sustained reversal of acute relapses. The challenges facing remission induction as a therapeutic approach in MS include the fact that some MS patients do not experience remissions and that the transition from relapsing-remitting disease to progressive disease represents an event in the disease course that is not fully understood.
Can we use what we know about the molecular basis for remissions in order to induce them? This question is under study in a small number of laboratories, but our understanding is just in its infancy. More fundamentally, however, the question of the basis for the transition from relapsing-remitting disease to progressive MS also remains unanswered. There are several possible explanations for why MS follows a relapsing-remitting course in some patients.
Page 291
Axons, as well as myelin and glial cells, are damaged in MS. It is currently believed that axons within the CNS do not regenerate, or regrow, after they degenerate consequent to injury. Thus, one scenario is that irreversible (non-remitting) deficits in MS arise from axonal injury, as opposed to injury to myelin, which either is fundamentally reversible or can be more readily repaired. If axonal injury is, in fact, a basis for the acquisition of irreversible deficits, treatments aimed at reversing disability will have to focus efforts on protecting axons.
A second scenario is that, for some reason, sodium channel plasticity (which is necessary for restoration of impulse conduction in demyelinated axons) might not occur in some demyelinated or degenerating axons. To the extent that molecular plasticity at the channel level permits regeneration of critical channels, deficits might be reversible, but when channel numbers or distributions cannot be reconstituted, conduction fails and permanent disability results. Molecular neuroscience can teach us more about the molecular mechanisms that are responsible for synthesis and deployment of sodium channels in myelinated and demyelinated axons and about ways to manipulate channel distribution along the demyelinated axon therapeutically.
A third scenario is that other mechanisms of plasticity, such as synaptic sprouting or recruitment of previously uninvolved parts of the brain and spinal cord, can compensate for demyelination and even axonal loss early in the disease course, but that these processes become overwhelmed as the burden of disease accumulates, so that they can no longer contribute to recovery. This “threshold” model would focus attention on these mechanisms of plasticity and on strategies for enhancing them so that they are more robust. However, there is a need for information about these mechanisms in MS and about how they can be enhanced or promoted.
Harnessing Endogenous Neuroplasticity Through Exercise
Several aspects of current thinking about the mechanisms that mediate neuroplasticity are potentially relevant to the restoration of function for people with MS. One interesting concept is the effect of training or exercise. In accordance with the theory that many plastic changes in the CNS are activity dependent, increases or decreases in input to the CNS can result in reorganization of sensory maps and changes in synaptic strength. In experimental and disease states, neurons can be induced to respond to stimuli that were previously not effective in eliciting a response. Similarly, neurons change the efficiency with which they respond to stimuli. Several groups have demonstrated that exercise, or an enriched environment, can improve motor outcomes after cortical lesions.81 An interesting therapy has been attempted in individuals with impaired limb use due to stroke, by restricting use of the normal limb. Taub and colleagues observed that in both primates and humans, the use of the impaired limb was improved after limiting activity on the normal side.101 In rats, this type of therapy results in
Page 292
adaptive changes in the motor representation in the spared cortical sites. This overall procedure must be approached with caution, however, since restricting use of the normal limb too quickly after a cortical lesion can also result in persistent motor deficits; this unfortunate result is thought to be due to increases in sensitivity to NMDA toxicity.80 Although restricting the use of a limb might not be appropriate for people with MS, these observations suggest that achieving a careful balance of excitation and inhibition might substantially enhance appropriate neural responses. It might be possible to reach this goal by appropriately timing a combination of exercise techniques that are known to induce changes in synaptic strength with pharmacotherapy known to alter excitation and inhibition.
Transplantation
Over several decades, the use of transplantation has emerged as a viable technique for repair of the nervous system. Immature fetal cells are required for transplanted cells to survive and integrate into the host nervous system. These cells have been used in many experimental animal models, as well as in human diseases, such as Parkinson's disease and Huntington's chorea. Although some of the results have been encouraging, the use of fetal cells in human diseases is probably not practical, in large part because of the restricted availability of fetal tissue.
Transplantation offers the opportunity to replace defective or destroyed tissue, cells, or organs with functional ones. A number of potential sources of neural tissue exist. Allotransplantation refers to transplantation from one donor of the same species to another of the same species. Xenotransplantation refers to transplantation of tissue from one species to another. Both are being investigated as potential therapeutic approaches to replace neural tissue destroyed by MS.
The possibility of harvesting animal cells for transplantation into humans is an active area of research. These xenografts could provide either specific replacement cells, such as cell populations that contain a specific neurotransmitter, or immortalized cell lines, such as fibroblasts genetically engineered to produce specific substances, including neurotrophic factors.39
Major limitations in transplantation include the requirement for immunosuppressive agents to prevent graft rejection, the toxicity associated with the use of these agents, and difficulty with control of rejection of xenografts even with immunosuppression. Strategies to control rejection, induce tolerance, and enhance xenograft survival have to be developed, and the mechanisms underlying xenoreactivity must be better defined.
Stem Cells
A mature animal originates from a single cell, which initially has the potential to produce a diverse array of tissues, organs, and cells. That original cell is
Page 293
thus pluripotent, meaning it can give rise to cells of many different types. Certain cells retain this feature throughout the lifetime of an organism and are known collectively as stem cells. They are currently being studied as a possible way of replacing cells that have been destroyed by disease.
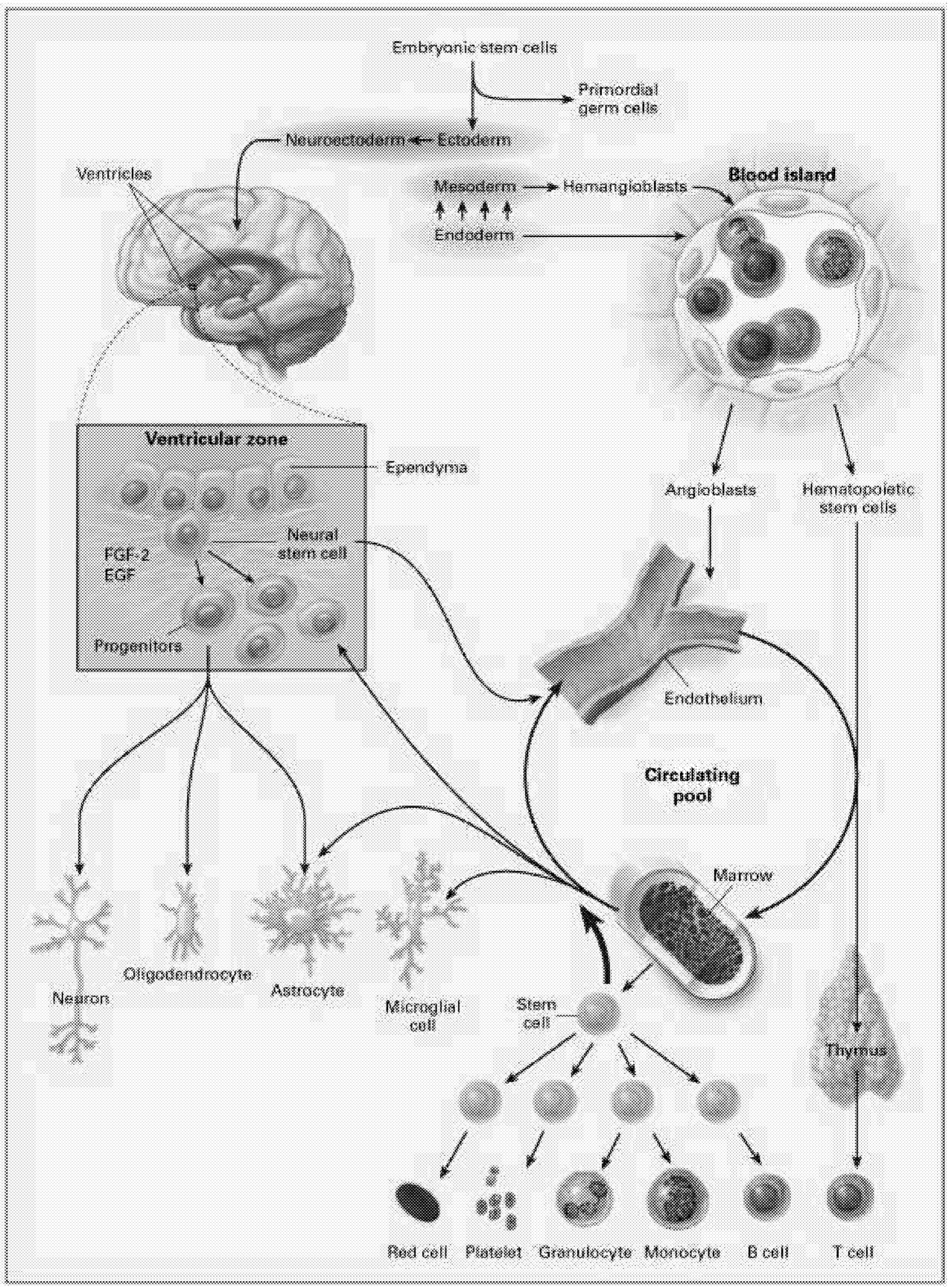
The most intensively studied stem cells are the hematopoietic stem cells (HSCs). These stem cells produce at least 11 different cell types, or lineages, including the immune system components (T cells, B cells, NK [natural killer] cells, granulocytes, dendritic cells), microglia of the brain and central nervous system, platelets, and red blood cells (Figure 6.1). Even for HSCs, the mechanisms underlying their regulation, survival, maintenance of pluripotency, and cell fate are still being worked out.
Recently, reports that stem cells from adults can differentiate into developmentally unrelated cell types have offered promise that this ability could be exploited to replace damaged or destroyed neural cells. Intrinsic and extrinsic signals regulate stem cell fate. Although some of these mechanisms have been identified, continuing studies will provide important information to bring this technology closer to clinical application. What a stem cell is, what regulates self-renewal and differentiation, the intrinsic controls of stem cell fate, the microenvironment and its role in regulation, and the control of plasticity are all important unresolved questions.
Neural Stem Cells. Another approach to neural repair exploits the observation that the CNS appears to contain multiple cell populations that are capable of self-replication and behave as endogenous stem cells in the adult brain.6 In certain regions of the brain, these stem cells have been identified and studied for several years, but they were thought to be restricted to repopulating a few specific sites such as the olfactory bulb and the hippocampus.71 More recently, immature progenitor cells with the potential to participate in neural repair have been identified throughout the forebrain. These are located in the subventricular zone, a region close to the ependymal cells that line the ventricles. Although these cells do not seem to engage independently in neural repair, it might be possible either to isolate and harvest a pluripotential population of stem cells or to induce endogenous self-repair by administering the appropriate growth factors or other substances that will induce these cells to differentiate and repopulate lesioned sites.
Until recently, the concept of a neural stem cell was not considered because the CNS has been regarded as a structure incapable of regeneration. However, a number of prototype neural stem cells have been identified in brain and spinal cord. Neural stem cells can also be derived from embryonic stem cells. Before the full potential of neural stem cells can be realized, we must better understand what controls their proliferation, what tissues they reside in, how the regulation of pluripotency occurs, and how differentiation to daughter cells occurs.
More recently, attention has focused on the potential of stem cells for replacement and repair in the CNS. Very early during embryonic development, true
Page 294
~ enlarge ~
Page 295
“stem” or primordial cells exist. These are progenitors for all cells in the body. As the organism develops, cells become progressively more committed to their fate as a particular type of cell. During development, different lines of stem cells develop, including those dedicated to the hematopoietic system that produces cells related to blood, bone marrow, and other systems. Other lines of stem cells develop into neurons and glial cells that populate the brain and spinal cord. There are intermediate levels of commitment in the lineage from primordial cells to cells committed to becoming a specific type of neuron or glial cell. Partially committed cells can be tapped at different levels of development to be used as stem cells to repair the nervous system. At this point, most studies investigating neural repair have used cells that are more or less committed to differentiate into either glia or neurons. Several reports have recently suggested, however, that with the right culture conditions, it might be possible to obtain a pluripotent stem cell that can differentiate into either neurally related cells or cells related to the hematopoietic system.10
Stem cells taken from embryonic brain are capable of extended self-renewal, but they lose this capacity as they mature. However, the capacity for self-renewal can be retained either by inserting immortalizing genes or by using specific growth factors in the cell culture media that encourage cells to exit the cell cycle and proliferate or return the cells to their immature state. Fetal cells maintained in this way can be transplanted into the CNS where they subsequently develop into both neurons and glial cells. Of particular relevance to MS is the transplantation of precursor stem cells into “shiverer” mice. These mice are missing a portion of the gene that encodes for myelin basic protein and, as a consequence, have poor myelination. When Yandava and colleagues transplanted neural stem cells into the brains of newborn shiverer mice, they migrated widely throughout the brain, and many of them differentiated into oligodendrocytes, producing improved myelination with reduced tremor in the mice.118 It remains to be shown, however, whether similar transplants in adult animals can produce equally beneficial effects.
Although it is well known that young brains recover from injury more easily and respond better to treatment than adult brains, there are examples of effective transplantation treatment in adult brains. For example, in a model of targeted apoptosis in a specific population of neocortical cells, Snyder and colleagues94 found that precursor cells were able to selectively repopulate and form appropriate connections in adult brains. They suggest that a number of factors in the microenvironment of both the host and the transplanted cells are required for this kind of transplant to succeed.
Cells from many different sources are being studied, including various animal models, progenitor cells from different regions of immature brain, and cell lines of multiple lineages, including human. Stem cells can also be transfected with viral vectors that allow the delivery of substances capable of inducing neural repair. Various growth factors have been successfully induced in neural stem
Page 296
cells to promote repair in many lesion models. Liu and colleagues grafted cells that were genetically modified to express the growth factor NT-3 into lesioned spinal cords.61 The transplant cells dispersed for substantial distances within the spinal cord, differentiated into both neurons and glia (probably as a result of environmental cues from the host), and encouraged axonal growth from host axons. Many other studies report positive findings after delivery of growth factors into lesioned spinal cord, including functional improvements in gait.19,63,84
Bone Marrow Transplantation. The bone marrow is the source of all stem cells responsible for the formation of the blood or hematopoietic system (red blood cells, platelets, immune cells, and white blood cells). Bone marrow transplantation (BMT) and the HSC are associated with a number of autoimmune diseases.1,52,76,79,103 Bone marrow from disease-prone donors transfers the disease process, and conversely, transplantation of bone marrow from disease-resistant donors into autoimmune recipients reverses the autoimmunity.37,58,76,77 BMT has therefore been suggested as a potential new strategy to interrupt some autoimmune diseases.
Animal Experiments Support a Role for HSC in Autoimmunity. Observations that autoimmunity could be transferred through different types of stem cells in mouse models led investigators to examine clinical correlates. In 1974, Morton and Siegel showed that autoimmunity in New Zealand Black (NZB) mice could be transferred by hematopoietic cells into irradiated disease-resistant recipients76 Conversely, BMT from disease-resistant donors prevented autoimmunity in NZB mice, an animal model for autoimmune hemolytic anemia.23,77 Subsequently, disease prevention was reported in two diabetes-prone animal models, the non-obese diabetic (NOD) mouse37 and the BioBreeding (BB) rat,78 that had been fully conditioned and transplanted with bone marrow from disease-resistant donors. Mixed chimerism achieved by partial conditioning was sufficient to reverse the autoimmunity in NOD mice and prevent the development of diabetes. Transfer of autoimmunity by BMT has also occurred in other animal models, such as the MRL/lpr (lupus/rheumatologic disorders) mouse, BXSB mouse, and HLAB27 transgenic rat.15,40,102 Unmodified marrow was administered in the early studies, implicating T cells within the donor inoculum as potential mediators of the effect. Splenocytes or lymph node lymphocytes did not transfer disease, pointing to a role for the HSC itself. When bone marrow from nude mice or bone marrow with reduced T-cell levels from normal allogeneic donors reversed autoimmunity in disease-prone recipients, data in support of a role for the HSC, rather than its progeny (T cells), emerged.15,35,37,38,52,78,93,102,115 Finally, transplantation of purified HSC from NOD mice into genetically identical disease-resistant recipients transferred the autoimmune state.36 Therefore, one can hypothesize that the immune dysfunction in autoimmune diseases such as Type 1 diabetes is due to an inherited defect of the pluripotent HSC that is also expressed in their progeny.
Page 297
van Bekkum recently suggested that autoimmune diseases should be classified as HSC defects.106
Bone marrow used for transplantation can be either autologous or allogeneic. In autologous transplants, the recipient serves as his or her own donor. In allogeneic transplants, previously extracted marrow from a different donor provides the stem cells for transplantation to a genetically different recipient. Both types of bone marrow transplantation have an impact upon autoimmune diseases. Further research is needed to understand the mechanisms by which BMT helps to reestablish self-tolerance in the context of autoimmune diseases. Better understanding of this process should significantly improve transplant outcomes. In addition, support of well-developed Phase I pilot studies in which optimal information is obtained about posttransplant outcomes and impact on disease progression should be supported to evaluate the impact of BMT on MS.
Human Studies Involving Allogeneic Bone Marrow Transplantation. Recent case reports have confirmed a strong association between the HSC and autoimmunity in humans. BMT has been shown to transfer autoimmunity to disease-free human recipients who underwent BMT to treat leukemia.34,53,109 Conversely, BMT from disease-resistant donors has reversed the autoimmune process.68 Seven patients with rheumatoid arthritis who received an allogeneic BMT from disease-free HLA (human leukocyte antigen) identical siblings experienced a dramatic reduction in the severity of the disease, with complete remission in six of the seven patients.4,41,66,88,89 Other autoimmune diseases including Crohn's colitis,62 psoriatic arthritis,24,62 MS, and Type 1 diabetes in the honeymoon period105 have gone into remission following allogeneic BMT for leukemia. Because life-threatening autoimmune diseases are not controlled by conventional immuno-suppressive therapy, BMT is currently being tested in pilot clinical trials as a therapeutic strategy in patients with life-threatening disease. This seminal observation has opened a door for a potentially new strategy to treat autoimmune diseases such as MS. However, a number of challenges have to be better understood and refined through research in this area.
Limitations of Bone Marrow Transplantation. The four major limitations of BMT are (1) the toxicity of the conditioning; (2) the requirement for close HLA matching; (3) graft-versus-host disease (GVHD); and (4) failure of engraftment. The toxicity associated with conditioning required in conventional BMT is one of the main limitations that has prevented the widespread application of BMT to nonmalignant diseases, including MS. In fully ablative BMT, the marrow of the recipient is first removed and then replaced by donor marrow, resulting in a fully allogeneic chimeric state. Fully ablative BMT carries a 10 percent mortality risk because of the conditioning process used to make space for the new marrow to become engrafted. If the recipient is only partially conditioned, the result is a mixed chimeric state. The morbidity and mortality associated with partial conditioning are significantly lowered. Strategies to allow engraftment with partial
Page 298
conditioning should be developed. A second major limitation of BMT is the requirement for clone matching. Methods to eliminate this need would make BMT more widely available. The need for clone genetic matching is due to the fact that GVHD is directly correlated to the closeness of genetic matching between donor and recipient. A third major limitation in BMT is GVHD itself. In GVHD, the donor immune system accompanies the HSC. The mature donor T and B cells attack the recipient, destroying the liver, skin, and gastrointestinal tract. Historically, strategies to remove GVHD-producing cells avoided GVHD but resulted in high rates of failures of engraftment. Methods to engineer a BMT that contains the essential cells but not GVHD-producing cells are needed. Research to understand the mechanism of failures, engraftment, GVHD, and graft take will allow this approach to more readily be applied clinically.
In summary, mixed chimerism might be a useful therapeutic approach for MS in two ways: (1) to induce tolerance to neural stem cell transplants and (2) to reverse the underlying autoimmune process. Strategies to apply BMT safely to patients with MS may allow a novel approach for therapeutic intervention. If the risk of the procedure is reduced to a minimum, one could envision intervening to interrupt the autoimmune process at a much earlier time, prior to the development of irreversible complications.
CHALLENGES IN MS CLINICAL TRIALS
The art of performing clinical trials in MS has been greatly stimulated by the availability of disease-modifying MS drugs—glatiramer acetate and several forms of interferon beta-1b. However, as the field has advanced, new and complex issues have arisen. This section highlights some of these areas, but does not attempt to address the entire universe of clinical therapeutic research in MS. Nor does it review the individual clinical trials of therapeutic agents (for a summary of some of these trials, see Appendix D).
The National Multiple Sclerosis Society has been highly effective in proactively addressing many of these issues through its Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis and its Clinical Outcomes Assessment Task Force and Task Force on Use of MRI in MS Clinical Trials. These efforts have resulted in (1) a set of guidelines for the conduct of clinical investigations and (2) development of the MS Functional Composite, a brief impairment measure for assessing patient outcomes in clinical trials. These documents have been published in respected clinical neurology journals (see Box 6.1).
The major challenges facing the researcher who plans clinical trials in any area of medicine are the needs to:
-
select promising new candidate therapies;
-
reject agents that are either potentially harmful or unlikely to prove efficacious, and do so at an early stage in the development process; and
Page 299
BOX 6.1Published Guidelines for the Conduct of MS Clinical TrialsWhitaker JN, McFarland HF, Rudge P, Reingold SC. Outcomes assessment in multiple sclerosis clinical trials: a critical analysis. Multiple Sclerosis 1995;1:37-47. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. Neurology 1996;46:907-911. Miller DH, Albert PS, Barkhof F, et al. Guidelines for the use of magnetic resonance techniques in monitoring the treatment of multiple sclerosis. Ann Neurol. 1996;39:6-16. Rudick R, Antel J, Confavereux C, et al. Clinical outcomes assessment in multiple sclerosis. Ann Neurol. 1996;40:469-479. Whitaker JN, et al. Expanded clinical trials of treatments for multiple sclerosis. Ann Neurol. 1996;34:755-756. Lublin FD, Reingold SC. Guidelines for clinical trials of new therapeutic agents in multiple sclerosis. Relations between study investigators, advisors and sponsors. Neurology 1997;48:572-574. Rudick, R, Antel J, Confavreux C, et al. Recommendations from the National Multiple Sclerosis Society Clinical Outcomes Assessment Task Force. Ann Neurol. 1997;42:379-382. Lublin FD, Reingold SC. Combination therapy for treatment of multiple sclerosis. Ann Neurol. 1998;44:7-9. Fischer JS, Rudick RA, Cutter GR, Reingold SC. The Multiple Sclerosis Functional Composite Measure (MSFC): an integrated approach to MS clinical outcome assessment. Multiple Sclerosis 1999;5:244-250. Goodkin DE, Reingold SC, Sibley W, et al. Guidelines for clinical trials of new therapeutic agents in multiple sclerosis: reporting extended results from Phase III clinical trials. Ann Neurol 1999; 46;132-134. |
-
convincingly establish the utility, safety, and efficacy of agents that proceed through the drug development process.
Drug development in MS faces major challenges in a number of these arenas. In addition to challenges that apply to all areas of medicine, MS presents its own challenges. It is a heterogeneous disorder of long duration and—relative to other disabling and life-threatening disorders such as rheumatoid arthritis, diabetes, and cardiovascular diseaseæ its prevalence is low. MS is a difficult disease to study, and the potential market for profit-dependent firms is small relative to the investment needed to produce an approved drug.
Specific challenges to the design of MS clinical trials include the general challenges presented by therapies that alter immune responses, therapies for which there is so much demand that they are applied even before solid clinical data are available; situations in which placebo controls are not appropriate, despite the
Page 300
analytic power they add to a clinical trial; and blinding designs that control for subject and observer biases about expected outcomes. The considerable natural variation in the manifestation of MS also complicates the design and interpretation of clinical trials. In some cases, seemingly reasonable decisions to exclude “atypical patients” from the data analysis or about which diagnostic criteria to use can generate spurious results.30
Need for a Standardized Testing Program
With the plethora of agents becoming available for testing in MS and four approved drugs already on the market, efficient systems are needed to facilitate drug development and comparisons between agents or combinations of treatments. A uniform currency of clinical assessment that incorporates several types of assessments, not all of which may be used in any given trial, is needed. An example is the Airlie House guideline for clinical trials in amyotrophic lateral sclerosis, which were formulated by a working group of the World Federation of Neurology.32 The authors of this document included academic physicians, pharmaceutical company representatives, and representatives of the Food and Drug Administration (FDA). The Airlie House document provides a menu of evaluations that form the core assessment for most modern ALS trials. The MS Society could contribute by continuing its efforts on behalf of MS clinical trials and convening a similar gathering to consolidate its efforts to this point and help develop standards for future clinical trials.
An ongoing challenge will be to determine what represents a continuum of the spectrum of disease as opposed to unique etiologic or immunopathogenic mechanisms. This is especially complicated because human genetic variation is so extensive.
Translating Research Results to Treatment Results
A critical underfunded and neglected area for MS is the application of basic research discoveries to the bedside in clinical feasibility (Phase I) studies (see Box 6.2). In the past, hospital centers and universities contributed funds to obtain proof of concept in early testing. In a time of health care reform, this option is no longer available. Funding for well-constructed, high-risk, potentially high-yield clinical trials for exploring feasibility and proof of concept in testing new technologies discovered at an academic center should be considered.
Need for Controlled Clinical Trials
Another area of concern focuses on those therapies that, although they might provide very real therapeutic benefit, are not being appropriately studied in controlled clinical trials. Allogeneic peripheral blood stem cell therapies currently
Page 301
BOX 6.2Clinical Trial Phases
|
fall under this category. In general, these treatments are not being developed by large pharmaceutical companies but are most often studied in small clinical trials under the auspices of independent researchers. These researchers have neither the funding nor the infrastructure for large, well-controlled, multicenter trials. The resulting publications describe only a handful of experimental patients and do not include control groups. Of danger to the field as a whole is that if controlled, adequately powered trials are not performed soon, the time for clinical trials may pass; clinicians will form an opinion of the treatment based on inadequate trials. Whether the treatment is truly beneficial or not will remain unknown.
The proper choice of control groups is an issue that affects any clinical trial but becomes especially difficult for MS clinical trials as patients gain access to more effective therapies. Asking individuals to forgo effective treatment is an unacceptable option. Yet if a treatment has a modest benefit, this benefit is far more difficult to detect when it is compared with another treatment of modest benefits than when compared to a placebo control. As a result, clinical trials comparing different treatments must be more elaborate; for example, they might test more subjects or for longer periods or collect more data, and will thus be more expensive.
Some modifications of placebo control design might provide viable alternatives for MS studies. So-called add-on trials test a new treatment on top of another beneficial treatment. For example, all patients in a clinical trial would receive beta-interferon. On top of this, half of these patients would also receive
Page 302
some novel therapy. As long as the therapies are thought to have significantly different mechanisms of action and toxicity, these trials are relatively straightforward.
Another option is the early-escape placebo-controlled trial, in which control patients are withdrawn from the trial at the first sign of any exacerbation. This technique, of course, relies on a clinically relevant and sensitive method for measuring impairment. An alternative is the randomized withdrawal approach, in which all patients start on a novel therapy and maintain this treatment until they become stabilized. At this point, researchers withdraw treatment randomly from some patients and closely monitor the clinical outcome of both groups. Again, the early-escape approach applies to any patients who worsen in the absence of the treatment.
Active controlled trials, in which two treatments are compared side-by-side, can establish the efficacy of a new treatment through two methods. The first involves demonstrating that a new treatment is more beneficial than an old one. In studies of MS, this approach works well, although as treatments become more and more beneficial, it will become more difficult to find a new treatment that is superior to the old ones even though it may be an effective form of therapy.
The second method involves demonstrating the efficacy of a new drug by showing that it is similar, but not superior, to a known effective treatment. When such similarity is shown, the statistical confidence interval around the estimated difference between the treatments includes the possibility that the new treatment is somewhat inferior. In order to conclude that the new treatment retains some efficacy, it is essential to determine that the amount of potential inferiority of the new treatment is less than the total efficacy of the active control treatment under the conditions studied. However, since trial conditions vary, patient populations are heterogeneous, and data regarding active control therapies in MS are limited, determination of the latter amount can be very difficult or impossible.
Controlling for Subjective Factors
Two critical elements of clinical trials are (1) the ability to blind both the participants and the observers with respect to treatment so that neither are influenced by their expectations of a treatment's effect (double blinding) and (2) the existence of unbiased, objective measurements of disease severity (objective outcomes measures). Both elements can prove difficult in MS studies. (For a further discussion of the latter factor, see “Outcome Measures in Clinical Trials” below.) Many therapies cause adverse events that can unblind subjects or investigators. In the case of transplantation-based treatments, such as stem cell and bone marrow therapies, blinding would require sham surgeries, which can be unethical or impractical. When a study cannot be blind, it may be possible to reduce bias by having the outcomes assessed by individuals who are blinded to the treatment. It is also particularly desirable in incompletely blinded trials that
Page 303
observers rely on objective end points to avoid subjective bias in interpreting the outcome of the trial.
Heterogeneous Patient Populations
The great variation in the clinical manifestations of MS presents additional issues in the design of a well-controlled clinical trial. Although MS is inevitably progressive, the rate and pattern of progression are highly variable from person to person. Responsive and unresponsive patient groups, starting with similar clinical or MRI deficits, have not yet been identified, although relapsing-remitting disease (characterized by inflammatory lesions) seems more amenable to immunotherapy than secondary progressive MS (where progression is less related to recurrent inflammation). However, currently used distinctions between relapsing-remitting MS and secondary progressive MS overlap considerably and are not necessarily the optimal distinctions for determining who is likely or unlikely to respond to a given therapy. Identification of disease factors that determine therapeutic response is very important, but difficult.
Some therapies are under development to target subsets of patients based on testing that is not standard in the clinical setting, for example, patients with a specific T-cell receptor subtype or antigen-specific T-cell activity. Several principles are critical in developing such targeted therapies, including the use of validated, standardized outcome measures in the trial, well-defined criteria for participating in a clinical trial and for defining patient subsets, storage of samples from patients for use in validating future tests as methodologies evolve, and finally, giving early consideration to developing a test that will be broadly available clinically should the need arise.
Response to Withdrawal of Therapy
For any therapy that modulates the immune response, time-course studies are needed. While most MS clinical trials follow patients on a course of treatment for one to three years, few follow these patients after they come off the treatment. For example, could one year of beta-interferon treatment be enough to “reset” the immune response of an individual so that he or she can then stop taking it? Might continued therapy increase long-term toxicity while yielding no additional benefit? These are important questions, yet there is little incentive for pharmaceutical companies to study them, and physicians who have seen their patients improve may not want to risk taking them off the therapy.
Multiple Agents
Since none of the treatments currently cure MS or halt disease progression entirely, it is important to test multiple agents in combination. Not only are such
Page 304
trials expensive, but they also will be lengthy and require large numbers of patients. Agents of different classes will have to be tested in sequence and in combination. Such trials are best done when the dose range and safety profile of each individual agent to be employed in the trial is known. The potential for adverse drug interactions should be carefully monitored. Separate end points might be required for each agent as appropriate to its individual pharmacological profile. Most importantly, standardized protocols and assessments will have to be devised and agreed upon, including Phase II studies that allow the abandonment of ineffective combinations to avoid unnecessary time, expense, and patient exposure in large, multicenter efficacy trials. At the time of this writing, there are no published reports comparing the efficacy of multiple agents given in combination to their efficacy when given alone. The results of several Phase I studies to test the safety of this approach have been presented at scientific meetings, but so far, none have been published in the peer-reviewed literature.
Assessment Instruments
The development and validation of efficient, accurate, sensitive and reproducible assessment instruments is critical to the success of clinical trials. As noted in Chapter 4, the MS Society has contributed to this area by promoting the development of the Multiple Sclerosis Functional Composite Measure (MSFC). The MSFC has not yet been prospectively validated in the setting of a multicenter clinical trial. Further development of outcome measures to supplement or replace the Expanded Disability Status Scale (EDSS; see Appendix D) are also needed. Finally, there is a variety of MRI protocols currently available, and it is important to determine which measures can give the most comprehensive assessment of the degree of CNS damage in MS.
Impairment. Impairment of neurological function can be measured directly by examination or testing of the patient. Examples of impairment assessments include the Scripps scale, the Functional Systems grading instrument,50 and so forth. The new MSFC is, in reality, an impairment measure, since it directly quantitates deficits of dexterity, ambulation, and cognitive test performance.
Disability. The assessment of disability is complex in a disease as varied as MS. The Kurtzke EDSS is the most widely used disability rating instrument, but it has been criticized for its apparent nonlinearity and insensitivity to change (see discussion in Chapter 4). As currently performed, it does not behave as a continuous outcome measure and does not equally weigh all areas of neurological dysfunction and disability in MS. There is consensus that the EDSS can be improved to yield a more comprehensive clinical disability measure for use in MS clinical trials.
Page 305
Attempts have been made to apply other, standardized outcome measures such as the Functional Impairment Measure (FIM) to the assessment of clinical outcomes in MS. Application of these scales often suffers from their general nature and lack of specificity for MS-related disabilities.
Clinical investigators in the fields of movement disorders and ALS have developed disease-specific rating instruments that incorporate quantitative or semiquantitative recording of aspects of neurological function together with questionnaire-based assessments of patients' level of function in activities of daily living. Multidimensional ordinal scales such as the Unified Parkinson Disease Rating Scale (UPDRDS), the Unified Huntington Disease Rating Scale (UHDRS) and the Amyotrophic Lateral Sclerosis Rating Scale (ALSRS) have been validated in multicenter studies and found to reflect the progression of disability as well as demonstrating good inter- and intrarater reliability. Protocols have also been developed for the standardized assessment of neural transplant patients. A comparable, comprehensive rating instrument and/or protocol does not yet exist for treatment trials in MS. Attempts to develop and validate comprehensive rating scales are under way. An intriguing pair of instruments is the Symptom Inventory and Performance Scales.91 However, none of these instruments has yet been tested and validated in the setting of a treatment trial.
Quality of Life. The application and validation of quality-of-life measures in MS is an ongoing field of investigation. Generic quality-of-life measures, which have the advantage of being able to permit comparisons of deficits and treatment outcomes across different disease states, often suffer from “floor” effects in the evaluation of aspects of physical functioning of neurological, including MS patients. Recently, an MS-specific quality-of-life measure (the MSQLI), has been developed and validated under the aegis of the Consortium of Multiple Sclerosis Centers and the National MS Society.27
Neuroimaging
The rapid advance in the development and sophistication of magnetic resonance imaging techniques is detailed elsewhere in this volume. Whereas some measures, for example gadolinium enhancement, are most sensitive to acute blood-brain barrier breakdown and inflammation, other measures such as assessments of brain volume or atrophy give a better picture of accumulated tissue loss. The former measure correlates best with acute and even subclinical disease activity; the latter, with long-term disability. Thus, the choice of imaging paradigm for a clinical trial should be selected based on the goals of the trial. A study of a new cytokine-modifying agent might call for assessments of gadolinium-enhancing lesions. A trial of a neuroprotective agent intended to slow or reverse loss of function in patients with progressing disease might instead rely on a measure of brain volume. Additional issues regarding the use of imaging techniques include the timing of imaging relative to treatment administration.
Page 306
Electrophysiology
If therapies to aid in remyelination of the CNS in MS are to be developed and tested, the efficiency of remyelination must be assessed. Recently it has been proposed that central conduction times might be useful in this regard,95 although the lack of positive studies makes validation of this measure in humans difficult at present.
Outcome Measures in Clinical Trials
The past decade has featured clinical trials that have led to the first approved therapies for MS. The initial definitive trials focused on relapsing-remitting forms of the disease and were combined with MRI studies. One primary end point was clinical relapse rate. Most studies have used relatively standardized definitions of a relapse, although in some trials, efficacy was more apparent where severe relapse rather than overall relapse rate was used as the outcome measure. The sensitivity of MRI to identify the rate of new lesion formation, which is the apparent counterpart to clinical relapse rate, probably makes it less imperative to develop better definitions of relapse.
A more critical issue would seem to be the clinical measures that can be used to assess neurologic disability. Most clinical trials are conducted over periods of one to three years, but the current standard disability scale, the EDSS, changes relatively little over that length of time. (See Chapter 4 for further discussion of problems with the EDSS.) Some, but not all, clinical trials of relapsing-remitting MS showed significant effects of interferons as measured by the EDSS disability scale, which raises the issue of whether the “successful” trials reflected better training of investigators in the use of these measures. An international task force convened by the National MS Society during 1994-1997 recommended use of the MSFC, a modified scale that incorporates measures of ambulation, upper limb function, and cognitive functions (discussed further in Chapter 4 and Box 4.6).90 Such measures are likely to be especially important in assessing therapies aimed at arresting the more progressive phases of disease or at improving functional capacity. MRI measures of tissue injury are being developed and might serve to support clinical observations regarding disease progression. Clinical studies aimed at functional improvement rely more on clinical measures. It seems unlikely that dramatically improved clinical scales can be developed given the rate of disease progression. Finally, even when the same clinical outcome measures have been used, it is not always statistically valid to compare different clinical trials.
Standardized measures are also needed to demonstrate the effects of clinical therapies on quality of life. There are numerous measures that can be utilized, as discussed in Chapter 4.
Page 307
MRI as a Surrogate Outcome Measure
The objective, sensitive, and quantitative changes measured using MRI make it an attractive tool for measuring the outcome of new therapies. This is in distinction to clinical outcome measures that are characteristically insensitive, often poor at reflecting disease activity, and inconsistently defined (for example, different studies define relapse differently). Indeed, the changes seen on MRI, used as a secondary outcome measure a pivotal trial of interferon beta-1b, had an important influence on the approval of this therapy in MS. In 1994, an international group of experts convened by the MS Society concluded that MRI could serve as a primary outcome measure in Phase II or exploratory clinical trials but that it should be used only as a secondary outcome measure in Phase III or pivotal trials.
The recent approval and widespread use of several partially effective therapies for MS have changed the options for testing new therapies and, as a result, interest in using MRI as an outcome measure has intensified. Large, long-term clinical studies conducted over several years raise ethical concerns about the use of placebo controls when approved therapies are already available. Further, the number of untreated patients available for studies will diminish. Also, trials comparing new treatment to active therapies will require larger sample sizes than placebo-controlled studies and will be very expensive. Overall, the MS research community is faced with serious difficulties in testing new and potentially effective therapies thus rendering the ability to conduct trials using small numbers of patients over a shorter duration than previously used in MS pivotal trials of considerable potential merit.
The following discussion of MRI as a surrogate outcome measure is based on a workshop of an international group of MS researchers that was convened jointly by the National Institute of Neurological Disease and Stroke (NINDS) and the National MS Society in November 1999.70
Role of Surrogate Outcome Measures
The issue of surrogacy is complex. In general terms, a surrogate is an outcome other than disability that can reliably predict clinical outcome. An example is the use of blood pressure as a surrogate for heart disease. The concept of surrogacy in MS research has often been misunderstood, and various concepts of surrogacy exist.
Validated Surrogates. For an outcome measure to be accepted as a “validated surrogate” measure of treatment efficacy, it must meet the following criteria: First, the surrogate must predict future clinical disease. Second, the effect of treatment on clinical disease must be explained by its effect on the surrogate; the treatment has to affect clinical outcome by working through the surrogate. Third,
Page 308
evidence must exist that treatments of various classes affect the surrogate in the same and predictable manner.
Demonstration of a relationship between a marker and a prognostic outcome in natural history studies is not sufficient to establish the marker as a validated surrogate. Finally, since therapies of different classes must be shown to have a concordant effect on the surrogate as well as on the clinical outcome, evidence from a single class of therapy is not sufficient since other therapies could affect the clinical and surrogate outcomes through different mechanisms.
These stringent criteria make it unlikely that single studies will be sufficient to assess a surrogate marker. Unless treatment differences in clinical outcomes are highly significant, single studies generally lack the precision to provide a robust validation. Interpretation is also difficult if the treatment has multiple mechanisms but the surrogate explores only one. Single studies are more likely to be helpful in excluding useless surrogates than in validating good ones. Metaanalysis of multiple, large studies allows the relationship between the surrogate marker, clinical outcome, and treatment effect to be explored in comparative trials in a more robust manner. Meta-analysis may define the proportion of treatment effect that is explained by the surrogate marker. Collaboration and data sharing between investigators will facilitate validation of surrogate outcome markers.
Unvalidated Surrogates. In contrast to the criteria for a validated surrogate, current FDA regulatory guidelines allow for the use, under some conditions, of an unvalidated surrogate. The condition placed on the use of an unvalidated surrogate is that the surrogate outcome must be considered reasonably likely to predict future clinical outcome or disease activity. As discussed below, studies to date that have examined the relationship between MRI measures of disease and clinical disease either in cross section or as future disease have been, with one notable exception, disappointing. Despite this, changes on MRI clearly reflect some of the underlying pathological process in MS, implying that MRI is a reasonable, albeit unvalidated, surrogate. However, serious mistakes have been made in clinical medicine using unvalidated surrogates. For example, it is now clear that CD4 counts do not necessarily predict mortality in AIDS. Thus, the use of an unvalidated surrogate must be viewed with caution by both investigators and regulatory authorities, who must balance the risks against the potential benefits of rapid identification of effective therapies for MS.
It is also important to note that the acceptance of unvalidated surrogates by regulatory authorities in countries other than the United States is less clearly defined. In the European Community, MRI findings in MS trials have, thus far, been given less consideration than in the United States. The use of a surrogate in a clinical trial must be consistent with the guidelines of the countries in which approval for treatment will be sought. Discussion of this issue at an international level is imperative since most pivotal clinical trials are now international studies.
Page 309
MRI as an Unvalidated Surrogate Outcome Measure
The use of MRI data as surrogate outcome measures in pivotal trials of new therapies in MS requires an appreciation both of what is being measured and of the potential errors and difficulties. At present, there is insufficient evidence to support any single MRI measure, or combination of measures, as a fully validated surrogate. However, there is evidence to support the concept that various MRI changes reflect changes in the underlying pathology of the disease, and consequently, it has been suggested that under some conditions, MRI changes can be appropriately used as a primary outcome measure in pivotal trials in MS. In other words, the changes in MRI measures can appropriately serve as an unvalidated surrogate for changes in pathology.
If MRI is used as a surrogate outcome, the following potential problems will have to be addressed, and trial designs will have to be devised to minimize the chance of coming to incorrect conclusions.
First, the possibility exists that clinical trials using MRI as an outcome would be of short duration and that an effect could be identified that was temporary and would not translate into persistent clinical effect. This criticism can be countered by incorporating an extension phase that could be used to provide confirmatory clinical data in those studies having a successful early MRI outcome—for example, to include formal Phase IV components to follow-up short-term studies using MRI outcome measures. However, in this setting, lack of comparative control data would be a limitation.
These questions also arise with respect to the nature of MRI as a surrogate outcome and the relationship of the effects of the interventions studied on the appearance of MRI scans and the clinical state of the patients. First, as mentioned above, the justification for using MRI as a surrogate will be based on the belief that MRI reflects the pathology of the disease. Thus, a treatment could produce a change in the pathological process but have little or no effect on clinical disease. Second, trials using surrogate outcome measures will often be of insufficient duration to identify adverse effects of treatment. Extension phases of short trials using MRI as the primary outcome will also be necessary to define the safety profile of a treatment. An additional aspect of this issue relates to defining the risk-benefit ratio of treatments that are established as effective through the use of surrogate MRI outcomes.
Third, a drug could act through mechanisms not easily identified by MRI, which would result in a false-negative study. To minimize this possibility, the decision to use MRI and the choice of imaging paradigm must be made on a case-by-case basis, with careful thought given to the proposed mechanism of action of the new treatment. The MRI protocol might have to be adapted to be compatible with the proposed mechanism of action, for example, serial magnetic resonance spectroscopy (MRS) to evaluate a neuroprotective therapy as opposed to the use of gadolinium enhancement to monitor an anti-inflammatory or anti-cytokine
Page 310
drug effect. It might not be suitable to test some new therapies using currently available MRI outcome measures as the primary assessment.
Fourth, it is possible that a treatment might affect MRI but not be mediated by an action on the underlying pathology reflected by the MRI measure chosen. For example, measures of brain atrophy are thought to reflect tissue loss and to be sensitive to loss of myelin as well as axons. Thus, atrophy is an attractive MRI measure to assess the ability of a therapy to reduce the rate of progression of MS. However, changes in measures of atrophy can also occur through other mechanisms. When acute inflammation subsides, there is some reduction in brain volume. Depending on the relative timing of changes in inflammation and MRI scans, a postinflammation change in brain volume could be misinterpreted as an increase in atrophy. Thus, attention must be given to the sensitivity and specificity of the MRI measure in relationship to the experimental therapy being used.
Finally, careful thought should be given to how MRI data are used as an outcome measure. No single MRI measure is sufficient for evaluating new therapies at every stage of the disease process. Instead, a combination of MRI measures that measure different aspects of lesion development should be used. Various MRI measures are listed in Table 6.1.
|
TABLE 6.1 MRI Outcomes for Clinical Trials of New Agents Aimed at Preventing Relapses or Slowing Progression: Recommendations of the November 1999 Workshop Convened by NINDS and the MS Society. |
|||||
|
Process Measured |
Imaging Parameter |
Clinically Isolated Syndrome |
Relapsing-Remitting |
Secondary Progressive |
Primary Progressive |
|
Inflammation |
T2 lesion load |
X |
X |
X |
X |
|
Gadolinium enhancing lesions |
X |
X |
X |
X |
|
|
Tissue loss |
T1 hypointense lesions (“black holes”) |
X |
X |
X |
X |
|
Atrophy-brain volume |
X |
X |
X |
X |
|
|
Spinal cord area |
X |
X |
|||
|
Dysfunctional tissue burden/biochemical abnormalities |
MR spectroscopy |
X |
X |
||
|
MTR histograms |
X |
X |
|||
|
Diffusion imaging |
X |
X |
|||
|
T2 decay analysis |
X |
X |
|||
|
T1 relaxation time |
X |
X |
|||
Page 311
However, many of these techniques are not well validated, and some are difficult to standardize across centers. Further, how various MRI measures would be used to provide a single outcome measure needs to be carefully thought through.
In summary, measures of disease activity provide a robust reflection of the underlying pathobiology of the disease process, and under some conditions, MRI could represent a primary, albeit unvalidated, surrogate outcome in pivotal trials of new therapies for MS. No MRI measure represents a validated surrogate outcome measure using the previously defined criteria; the likelihood that MRI reflects the underlying pathology of the disease implies that if the aim of a treatment is to prevent damage to the brain, then MRI represents an acceptable outcome measure.
Thus, continued follow-up of patients in studies of short duration using MRI as the primary outcome is essential. Further, the decision to use MRI as an outcome measure and the type of outcomes chosen should be made after considering the mechanism of action of the new therapy.
It is critical that the application of MRI as a surrogate outcome measure be done thoughtfully. Additional information on standardization of techniques is necessary. No single MRI measure represents an outcome measure of sufficient predictive value or validation to be used alone. Thus, for each new therapy, the most effective use of the various MRI measures should be carefully considered (see Table 6.1). In general, most investigators seem to feel that multiple MRI measures should be used. The next step in trial design will be to determine the most appropriate use of multiple MRI outcomes to assess the effect of a new therapy. Different MRI measures will be needed at different stages of disease, especially in the relapsing versus progressive phases.
Finally, assessment of MRI outcomes and clinical changes obtained in previous trials must be examined in greater detail, with an emphasis on the degree to which the effect of treatment on the clinical outcome is concordant with MRI outcomes.
Overall, the use of MRI as a surrogate outcome measure is an attractive possibility at a time when clinical trials using conventional clinical outcomes in placebo-controlled studies that require large numbers of patients and a long duration are becoming increasingly problematic. However, the use of MRI as a currently unvalidated surrogate must be approached with caution and with careful thought to the potential errors that could result as well as the possible benefits. The MS research community must next focus on issues of trial design and standardization of imaging techniques. In planning such studies, the ultimate need to validate the surrogate should not be forgotten.
Page 312
SHARED RESOURCES
Data Registries
International Multiple Sclerosis Trials Research and Resource Center
The approval in some countries of up to four treatments that favorably modify the course of multiple sclerosis has created a problem for future clinical trials. Because these agents are only modestly effective, there is a pressing need to develop new, more effective therapies. There is general agreement that double-blind placebo-controlled clinical trials for relapsing-remitting multiple sclerosis, of a size and duration such as have been necessary in the past to demonstrate effectiveness, are no longer ethically justified. Such trials can be justified in certain small clinical subgroups of patients, but their results cannot be assumed to apply to MS patients as a whole. A new approach is needed. In response to this need, the International Federation of Multiple Sclerosis Societies* proposes to set up an International Multiple Sclerosis Trials Research and Resource Center with the primary aim of answering two questions: (1) Is it possible to model the “expected” clinical behavior of multiple sclerosis using clinical, laboratory, and imaging markers of disease in order to determine that “observed behavior” in a treatment trial suggests treatment effectiveness or futility? (2) Does any single MRI measure, or combination of measures, of disease activity have sufficient predictability of future clinical course to be considered a surrogate marker of clinical disease activity?
The center is in an advanced stage of planning. It will be headed by a statistician. Data will be entered from the placebo arms of clinical trials carried out over the past decade, together with data from population-based epidemiological studies. Approval in principle to provide data from these sources has already been given. The center is intended to be an international resource. Access to the data will be open to investigators subject to the approval of hypothesis-driven proposals following peer review. The analyses will be carried out by the staff of the center.
A Model for MS Research: Transplant Registries
As noted elsewhere, clinical trials using cellular transplant protocols are often, by nature, small and institution based rather than being sponsored by corporations and, because of the attendant risks, are often unblinded and uncontrolled. Support, organization, and collation of data from such studies can be greatly facilitated through the use of clinical registries.
*In January 2001, the name was change to the Multiple Sclerosis International Federation.
Page 313
Two associated data registries, the International Bone Marrow Transplant Registry (IBMTR) and the Autologous Blood and Marrow Transplant Registry of North America (ABMTR) provide an exceptionally productive resource for researchers and clinicians. These registries represent a unique example of international cooperation that has greatly benefited the fields of transplantation and cancer treatment. They require considerable investment that is not easily matched, but nonetheless, they offer an insight to the MS community of what might be possible.
The IBMTR and the ABMTR are voluntary, nonprofit organizations of more than 400 institutions in 47 countries that submit data on consecutive blood and marrow transplant recipients to the IBMTR-ABMTR Statistical Center at the Medical College of Wisconsin in Milwaukee. The IBMTR was established in 1972 and the ABMTR in 1990. Together, they maintain databases of comprehensive clinical information for more than 60,000 transplant recipients. These are not donor registries that find donors for patients needing transplants. Rather, they collect information on transplant outcomes. The information gathered by the IBMTR and ABMTR is used to identify trends in transplant use and outcome, to guide physicians and patients in making treatment choices, and to conduct scientific studies of issues pertinent to improving transplant outcomes. Researchers planning clinical trials use the database to aid study design; they are an invaluable resource for estimating effect sizes, targeting appropriate study populations, designing efficient protocols, and determining accrual feasibility. Health care organizations and institutes use the database to better inform policy decisions.
The IBMTR-ABMTR database facilitates analysis of transplant outcomes by focusing on questions that are difficult to answer in randomized trials, including:
-
descriptions of transplant results in various disease states and patient groups;
-
analysis of prognostic factors;
-
comparison of transplant with nontransplant therapy;
-
identifying intercenter variability in diagnosis, practice, and outcome;
-
developing analytic approaches for evaluating transplant outcome; and
-
evaluating costs.
This sort of database is particularly valuable for research on complex and long-term diseases. As with MS treatment outcomes, assessing transplant outcome is complex. Outcomes are influenced by many patient- and disease-related factors such as age, disease stage, and prior treatment, as well as differences in treatment protocols. Ideally, most transplant issues would be addressed by large randomized clinical trials. However, many factors limit the application of randomized trials. Many diseases treated with transplantation are uncommon; single centers might treat only a few patients with a given disorder. This makes randomized trials difficult and also limits the ability to perform nonrandomized trials (Phase II) with sufficient statistical power to detect meaningful effects. Small
Page 314
trials, even when randomized, can give misleading results. Even when randomized trials are performed, enrolled patients often represent only a small proportion of the target population and may not be representative of the larger group. With the participation of many centers in a prospective, observational database, large numbers of subjects representative of the general population can accrue rapidly. This increases the power to detect meaningful changes in outcome and increases the precision with which outcomes can be measured.
It is important to emphasize that studies of observational data from multiple centers do not replace the need for carefully conducted, single-center, Phase I and II studies and single- or multicenter randomized studies. Rather, they offer a complementary approach for addressing issues and can help facilitate trials.
The database serves as an efficient source of information for evaluating new treatments, especially in the preliminary stages of investigation. It also provides information that is unlikely to be extensively pursued in industry-funded clinical trials—for example, long-term effects, continuation of therapy, and different responses to different therapies. In addition, a large, longitudinal database such as the IBMTR-ABMTR is uniquely suited to late and infrequent effects of transplantation—or any other long-term consequence such as accumulated disability in the case of MS.
Since the mid-1990s, HSC transplantation has been used to treat a variety of autoimmune diseases in about 300 patients. Some, but not all, have shown long-lasting improvement, and many issues require further clarification through coordinated clinical trials. The European Bone Marrow Transplant (EBMT) group is working with IBMTR-ABMTR to develop consensus and standardization for data collection related to HSC transplantation in patients with autoimmune disease, as well as guidelines for trials and patient selection.
There are 35 full-time staff dedicated to the registry, including physicians, statisticians, database managers, and a development office for fund raising. The activities of the IBMTR and ABMTR are supervised by more than a dozen volunteer committees that design and conduct studies, develop general policies for use of IBMTR and ABMTR data, review study proposals, and plan and conduct scientific workshops. The success of the registry programs is due, in large part, to the voluntary efforts of the hundreds of physicians, basic scientists, and clinical research associates who contribute their time and expertise.
The annual budget for the IBMTR is about $3 million, with about 60 percent of the funding coming from the National Institutes of Health (Melody Nugent, personal communication). The remaining support comes from corporations, foundation grants, and individual donors. The major expense of the registry is for reimbursement of fully participating centers, who are asked to submit their data in a 75-page form. Institutions can also participate on a limited basis, in which case they submit reports with only 20 items and are not reimbursed for their services. The registry is supported by the Medical College of Wisconsin, the Department of Defense, and the following institutes within the National Institutes
Page 315
of Health (NIH): National Cancer Institute; National Institute of Allergy and Infectious Disease; and National Heart, Lung and Blood Institute.
Comparable data registries for multiple sclerosis would be invaluable. Each of the challenges presented by transplantation is mirrored in those presented by MS. Most importantly, such registries provide a larger data set than would be possible through individual efforts. Although such registries are likely to receive generous federal and industry support, they should be led through collaborative public and nonprofit efforts. An independent, investigator-driven database will likely have more scientific credibility than for-profit ventures and thus ultimately be of greater value to investigators as well as pharmaceutical firms.
The IBMTR-ABMTR includes data on MS patients, but only in the context of transplantation. The most efficient approach for MS research might be to develop a database that would permit comparison with the IBMTR-ABMTR database.
Human Tissue in MS Research
The scientific value of human tissue is immeasurable. A small amount of diseased human tissue can reveal clues to a disease that no other research strategy can. For MS research, brain tissue is crucial, but this can be obtained only after death or by biopsy. However, brain biopsy for research purposes is generally not possible because of the risk of brain injury to the patient, which means that researchers must rely on postmortem tissue.
In terms of understanding disease mechanisms and developing therapeutic interventions, the most informative tissue would be that of MS patients in the earlier stages of diseases, but these patients are generally young and less likely to die than patients in the later stages of MS. Another complicating factor is that unless brain tissue is processed quickly and according to standard protocols, it degrades to the point where is useful only for relatively crude research techniques, and not for the newer genetic and molecular techniques.
Access to tissues from a wide array of MS patients either with different disease phenotypes or in different phases of the disease not only permits important research on the pathogenesis of the MS disease process, but enables research on the specific causes of neurologic disability, as well as disease heterogeneity. Tissue collection from patients is complex, and cooperative efforts are necessary to achieve sufficient numbers. Some tissues are readily available, such as peripheral blood samples and CSF. Others, such as brain tissue, are not.
Collections of serum and lymphocytes have been built into many natural history and clinical trial studies. Access to such samples has, for the most part, involved collaborations with those who collected the samples. At issue is whether such samples will be stored in optimal condition, which is particularly important with regard to serial samples needed to study the natural cause of disease.
Page 316
Lymphocyte samples are valuable with regard to both RNA- and DNA-based investigations. RNA would be the basis for evaluating single molecules as well as for use in the emerging DNA microarray technology. DNA is used for the molecular genetic analyses aimed at identifying disease susceptibility genes or genes that modulate disease course. The challenges of collecting, storing, and sharing such resources are dealt with in other sections.
CSF fluid is becoming more difficult to obtain because sampling for diagnostic purposes is done less routinely than in the past. Although private CSF banks continue to exist, most tend to have samples suitable for analysis of the fluid but not for any of the cell constituents that were present (RNA is needed to measure gene expression, but it is highly degradable under normal tissue storage conditions).
Brain and Tissue Banks
Many brain banks, wherein patients arrange to donate their brains after death, have been established for the advancement of research in a variety of neurological disorders, most commonly Alzheimer's disease. These brain banks have been instrumental in identifying the mechanisms of Alzheimer's disease and should prove equally useful in MS research. However, MS is less prevalent than Alzheimer's disease, and there are fewer available samples. Thus, it is important to be sure that any available tissue is collected using protocols that will optimize its scientific value—for example, by establishing standards for collection, quality control, and collaborations among multiple centers. Finally, brain banks require concerted outreach efforts since this need is not something of which the public is generally aware.
CNS tissue from cases of MS has been collected for many years at many institutions, forming the basis for the classical pathologic descriptions of the disease. As expected, the majority of tissues were collected from long-standing cases. Most of the early disease stage samples represent the most aggressive form of the disease. Samples were not usually preserved in a manner that is ideal for optimal immunohistochemical or molecular analyses. A number of brain banks have been established over the years. Limitations of these collections include the paucity of clinical information available on the cases and the lack of recent modern neuroimaging investigation.
The NIH currently funds 14 brain banks, several of which also receive funding from other sources. Ten of these banks are for research into Alzheimer's disease, and none of them collect tissue from MS patients. The MS Society has funded two brain banks for many years. One is at the Veteran's Administration in California and includes tissue collected from patients with a variety of neurological diseases in addition to MS. The other is in Colorado and collects only tissue from MS patients. According to user surveys conducted by the MS Society, the needs of researchers have been adequately met by these brain banks. There are
Page 317
also MS brain banks in Britain and in the Netherlands. The latter provides services unmatched by any other MS brain bank (Box 6.3). The amount of brain tissue at the British brain bank has been too limited to supply researchers outside
BOX 6.3Netherlands Brain BankThe Netherlands Brain Bank was organized in 1985 and, in 1990, established its program related to MS, whose main purpose is to obtain rapid autopsies from which tissues can be used for studies in neuropathology, immunocytochemistry, molecular biology, and tissue culture. The tissues are properly prepared for special studies, and the program has the flexibility to initiate new procedures. Sufficient amounts of all tissues are retained for potential follow-up for future studies. Thus far, 110 MS autopsies have been performed, averaging 10 to 12 cases per year. The program also emphases obtaining “control” autopsies, of which about 30 or more are performed annually. Only about 10 percent of the rapid autopsy cases performed are for MS; the control cases (20 to 30 percent) are also part of the program. The brain bank also obtains cases of various other neurological diseases, with an emphasis on Alzheimer's disease (30-40 cases per year). Overall, the brain bank team performs 100 to 120 rapid autopsies per year. Of note, the protocols involved in an MS autopsy include MRI pathology studies and lesion dissection, which result in such cases taking three to four times longer (six to seven hours) than many others. The tissues must be analyzed as completely as possible, ideally including assessment of the extent of tissue injury, activity of the disease, and extent of remyelination. Indeed, a key strength of this program is the ability to perform MRI scans in the middle of the night. The MRI-pathology correlation effort of the brain bank is internationally recognized. This effort includes postmortem in situ imaging, as well as imaging of tissue sections. The approach of using MRI-defined lesions to direct the site of neuropathology examination, as well as more traditional MRI correlation with pathologically obvious lesions, is considered very promising. Because it is part of the larger brain bank, the MS brain bank can share the highly expert staff (secretaries, pathologists, technicians, and undertaker) who are available full-time. Without this type of infrastructure, the rapid response to MS cases could not take place. There is a well-established program in which patients agree to tissue donation. Donations are organized through direct efforts of the brain bank with the public. The entire staff is attuned to the ethical issues and specific restrictions on tissue access opportunities that exist in the Netherlands. There is increasing demand on the bank for tissue, and an impressive number of research papers have been published using its materials. A 1999 review of the program indicated that it should be able to deal with any concern within the Netherlands regarding access to this national resource. Established users of the bank pay an annual subscription fee of 3000 df ($1,100 U.S.). Commercial companies are charged at higher rates. There is also a “pilot” program for which no charges are made. The brain bank is funded approximately equally by the Dutch MS Society, the hospital in which it operates, and research grants. |
Page 318
Britain, and tissue distribution from the Netherlands brain bank has been largely restricted to European researchers. Finally, some researchers maintain their own brain banks and do not publicly distribute the tissue. Private collections are necessary for researchers who need to tailor tissue collection procedures for specific research techniques.
The issue of access to cases of MS early in the disease course remains an unresolved challenge. Surgical samples are available mainly from cases that represent diagnostic difficulties and cannot be expected to be representative of all early MS cases. These patients can be followed and their subsequent disease course established. They provide an excellent opportunity to correlate neuroimaging data with tissue analyses. The hope is to establish imaging criteria that will correlate with the suspected different pathologic forms of MS. Serial samples are not likely to be available from these cases. Additional surgical samples may become available if other invasive therapies such as deep brain stimulators come into use to treat some of the disabling symptoms of MS such as tremor.
Combined with the increased demand, the limitations in obtaining MS tissues for research studies suggest a need for international cooperative efforts. Such efforts would include ensuring that all those involved in tissue collection aim to meet the ideals described above with regard to tissue handling. Collected tissue should be preserved to allow as many types of analyses as possible. Specifically, tissue collection and storage protocols that allow for histologic, immunocytochemical, and molecular analyses should be established and consistently followed. Tissues that are to be provided to investigators for biochemical and molecular analyses of disease activity must be carefully evaluated pathologically. Standard criteria for rating disease activity would improve comparisons between different studies. It is also important to have facilities that can respond rapidly when cases of MS become available for postmortem analyses, including the ability to perform immediate pre-autopsy MR scanning and scanning of tissues immediately after collection. Education of MS patients about the value of such programs is also important. As discussed under communication in Chapter 4, it is important that patients and their families participate in the development of informational materials. The protocols of the Amsterdam brain bank offer an instructive model for brain banks in other countries (Box 6.3).
REFERENCES
1. , , . 1978. Expression of autoimmunity by NZB/NZW marrow. Clin Immunol Immunopathol.; 10: 247-50.
2. . 1996. The Amyotrophic Lateral Sclerosis Functional Rating Scale. Assessment of activities of daily living in patients with amyotrophic lateral sclerosis. The ALS CNTF treatment study (ACTS) phase I-II Study Group. Arch Neurol.; 53: 141-1.
3. , , . 1996. Immunotherapy for multiple sclerosis: from theory to practice. Nat Med.; 2: 1074-5.
Page 319
4. , , , . 1977. Bone marrow transplantation in patients with gold-induced marrow aplasia. Arthritis Rheum.; 20: 1043-8.
5. , , , , . 2000. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol Cell Neurosci.; 15: 314-29.
6. . 1999. A new role for glia: generation of neurons! Cell.; 97: 667-70.
7. . 1999. A controlled trial of recombinant methionyl human BDNF in ALS: The BDNF Study Group (Phase III). Neurology.; 52: 1427-33.
8. , , . 1981. Vaccination against autoimmune encephalomyelitis with T-lymphocyte line cells reactive against myelin basic protein. Nature.; 292: 60-1.
9. , 1999. Cutting edge: clonally restricted production of the neurotrophins brain-derived neurotrophic factor and neurotrophin-3 mRNA by human immune cells and Th1/ Th2-polarized expression of their receptors. J Immunol.; 162: 6303-6.
10. , , , , . 1999. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo. Science.; 283: 534-7.
11. , . 1999. The origin of remyelinating cells in the central nervous system. J Neuroimmunol.; 98: 69-76.
12. , , , . 1994. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol.; 36: 778-86.
13. , , , . 1998. Immunity to T cell receptor peptides in multiple sclerosis. III. Preferential immunogenicity of complementarity-determining region 2 peptides from disease-associated T cell receptor BV genes. J Immunol.; 161: 1034-44.
14. , , . 1997. Interferon-beta is a potent promoter of nerve growth factor production by astrocytes. J Neurochem.; 69: 939-46.
15. , , , . 1993. Transfer of the inflammatory disease of HLA-B27 transgenic rats by bone marrow engraftment. J Exp Med.; 178: 1607-16.
16. , , , , , . 1996. GGF/neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron.; 17: 229-43.
17. , , , . 1993. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron.; 10: 729-40.
18. , , . 1999. Nitric oxide synthases: targets for therapeutic strategies in neurological diseases. Cell Mol Life Sci.; 55: 1029-35.
19. , , , . 1997. Gait analysis of adult paraplegic rats after spinal cord repair. Exp Neurol.; 148: 544-57.
20. , , , , . 1996. Nerve growth factor and neurotrophin-3 differentially regulate the proliferation and survival of developing rat brain oligodendrocytes. J Neurosci.; 16: 6433-42.
21. , , . 1996. Ciliary neurotrophic factor selectively protects human oligodendrocytes from tumor necrosis factor-mediated injury. J Neurosci Res.; 43: 289-98.
22. , . 1998. Nitric oxide in neurodegeneration. Prog Brain Res.; 118: 215-229.
23. , . 1977. Evidence for a B lymphocyte defect underlying the anti-X anti-erythrocyte autoantibody response of NZB mice. J Immunol.; 118: 1858-63.
24. , , , . 1990. Clearance of severe psoriasis after allogenic bone marrow transplantation. BMJ.; 300: 908.
25. , , , . 1993. Pharmacological protection of CNS white matter during anoxia: actions of phenytoin, carbamazepine and diazepam. J Pharmacol Exp Ther.; 266: 1549-55.
Page 320
26. , , . 1995. Endogenous GABA attenuates CNS white matter dysfunction following anoxia. J Neurosci.; 15: 699-708.
27. , , , , , . 1999. Recent developments in the assessment of quality of life in multiple sclerosis (MS). Multiple Sclerosis.; 5: 251-9.
28. , . 1997. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron.; 19: 197-203.
29. , , . 1995. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci.; 15: 6445-52.
30. . 1999. Perils and pitfalls in the interpretation of clinical trials: a reflection on the recent experience in multiple sclerosis. Neuroepidemiology.; 18: 53-63.
31. , , . 1999. Insulin-like growth factors and binding proteins in multiple sclerosis plaques. Neuropathol Appl Neurobiol.; 25: 215-25.
32. , , . 2000. The age-range of risk of developing multiple sclerosis: evidence from a migrant population in Australia. Brain.; 123: 968-74.
33. , , , . 2000. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci.; 20: 6421-30.
34. , , , . 1991. Concordant Graves' disease after bone marrow transplantation: implications for pathogenesis. J Clin Endocrinol Metab.; 72: 837-840.
35. , , , . 1985. Rationale for bone marrow transplantation in the treatment of autoimmune diseases. Proc Natl Acad Sci U S A.; 82: 2483-7.
36. , , , . 1990. Organ-specific and systemic autoimmune diseases originate from defects in hematopoietic stem cells. Proc Natl Acad Sci U S A.; 87: 8341-4.
37. , , , . 1985. Prevention of type I diabetes in nonobese diabetic mice by allogenic bone marrow transplantation. Proc Natl Acad Sci U S A.; 82: 7743-7.
38. , , , . 1989. Long-term observations of autoimmune-prone mice treated for autoimmune disease by allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A.; 86: 3306-10.
39. , . 1997. Benefits and risks of hosting animal cells in the human brain. Nat Med.; 3: 964-9.
40. , , , . 1994. Requirement of donor-derived stromal cells in the bone marrow for successful allogeneic bone marrow transplantation. Complete prevention of recurrence of autoimmune diseases in MRL/MP-Ipr/Ipr mice by transplantation of bone marrow plus bones (stromal cells) from the same donor. J Immunol.; 152: 3119-27.
41. , , . 1986. Prolonged remission of severe refractory rheumatoid arthritis following allogeneic bone marrow transplantation for drug-induced aplastic anaemia. Bone Marrow Transplant.; 1: 237-239.
42. , . 1995. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev.; 16: 3-34.
43. , . 1998. CNTF and GDNF, but not NT-4, support corticospinal motor neuron growth via direct mechanisms. Neuroreport.; 9: 3749-54.
44. , . 1997. Identification of post-mitotic oligodendrocytes incapable of remyelination within the demyelinated adult spinal cord. J Neuropathol Exp Neurol.; 56: 1191-201.
45. , . 1999. The role of oligodendrocytes and oligodendrocyte progenitors in CNS remyelination. Adv Exp Med Biol.; 468.
46. , , , . 1999. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med.; 189: 865-70.
Page 321
47. , , , . 1992. Insulin-like growth factor I gene expression is induced in astrocytes during experimental demyelination. Proc Natl Acad Sci U S A.; 89: 1894-8.
48. , , , . 1998. NT-3-mediated TrkC receptor activation promotes proliferation and cell survival of rodent progenitor oligodendrocyte cells in vitro and in vivo. J Neurosci Res.; 54: 754-65.
49. , , . 1993. CNS glial cells express neurotrophin receptors whose levels are regulated by NGF. Brain Res Mol Brain Res.; 17: 163-8.
50. . 1983. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology.; 33: 1444-52.
51. , , , , , . 1998. p75 neurotrophin receptor expression on adult human oligodendrocytes: signaling without cell death in response to NGF. J Neurosci.; 18: 1297-304.
52. , . 1989. Reciprocal allogeneic bone marrow transplantation between NOD mice and diabetes-nonsusceptible mice associated with transfer and prevention of autoimmune diabetes. Diabetes.; 38: 894-901.
53. , , , . 1993. Transfer of insulin-dependent diabetes between HLA-identical siblings by bone marrow transplantation. Lancet.; 341: 1243-1244.
54. , , , . 1997. Remyelination in multiple sclerosis. Mult Scler.; 3: 133-6.
55. , , , . 1992. Multiple sclerosis patients express increased levels of beta-nerve growth factor in cerebrospinal fluid. Neurosci Lett.; 147: 9-12.
56. , , . 1992. Coordinate expression of insulin-like growth factor system components by neurons and neuroglia during retinal and cerebellar development. J Neurosci.; 12: 4737-44.
57. , . 1999. Suppressor cells in demyelinating disease: a new paradigm for the new millennium. J Neuroimmunol.; 100: 53-7.
58. , , , , , . 1996. Mixed allogeneic chimerism induced by a sublethal approach prevents autoimmune diabetes and reverses insulitis in nonobese diabetic (NOD) mice. J Immunol.; 156: 380-8.
59. . 1996. Role of neurotrophins and trk receptors in the development and maintenance of sensory neurons: an overview. Philos Trans R Soc Lond B Biol Sci.; 351: 365-73.
60. , , . 1995. Insulin-like growth factor I treatment reduces clinical def cits and lesion severity in acute demyelinating experimental autoimmune encephalomyelitis. Mult Scler.; 1: 2-9.
61. , , , . 1999. Intraspinal delivery of neurotrophin-3 using neural stem cells genetically modified by recombinant retrovirus. Exp Neurol.; 158: 9-26.
62. , . Resolution of immune-mediated diseases following allogeneic bone marrow transplantation for leukemia. Bone Marrow Transplantation. 1992; 9.
63. , , , . 1999. Transplants of fibroblasts genetically modified to e press BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. J Neurosci.; 19: 4370-87.
64. , , , . 1998. Vaccination with DNA encoding an immun dominant myelin basic protein peptide targeted to Fc of immunoglobulin G suppresses experimental autoimmune encephalomyelitis. J Exp Med.; 187: 1543-8.
65. , , , . 1993. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science.; 259: 689-92.
66. , , , . 1993. Apparent cure of rheumatoid arthritis by bone marrow transplantation. J Rheumatol.; 20: 137-140.
Page 322
67. , , , . 1999. Neuregulin in neuron/glial interactions in the central nervous system. GGF2 diminishes autoimmune demyelination, promotes oligode drocyte progenitor expansion, and enhances remyelination. Adv Exp Med Biol.; 468: 283-295.
68. . 1995. Immune ablation followed by stem cell rescue: a new radical approach to the treatment of severe autoimmune diseases. Forum.; 5: 24.
69. , , , , . 1998. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med.; 4: 291-7.
70. , , , . 2001. The role of MRI as a surrogate outcome measure in MS. Ann Neurol.; in press.
71. . 1999. Brain stem cells change their identity. Nat Med.; 5: 261-2.
72. , , . 1995. Elevated levels of nerve growth factor in the thalamus and spinal cord of rats affected by experimental allergic encephalomyelitis. Arch Ital Biol.; 133: 131-42.
73. , , , . 1999. Altered nerve growth factor level in the optic nerve of patients affected by multiple sclerosis. Mult Scler.; 5: 389-94.
74. , , . 2000. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev.; CD001447.
75. . 1999. “Turning brain into blood”—clinical applications of stem-cell research in neurobiology and hematology. N Engl J Med.; 341: 605-7.
76. , . 1974. Transplantation of autoimmune potential. I. Development of anti-nuclear antibodies in H-2 histocompatible recipients of bone marrow from New Zealand Black mice. Proc Natl Acad Sci U S A.; 71: 2162-5.
77. , . 1979. Transplantation of autoimmune potential. IV. Reversal of the NZB autoimmune syndrome by bone marrow transplantation. Transplantation.; 27: 133-4.
78. , , , , , . Prevention of diabetes in rats by bone marrow transplantation. Annals of Surgery. 1981; 194: 328.
79. , , , . 1985. Abnormal stem cells in autoimmune-prone mice are responsible for premature thymic involution. Thymus.; 7: 151-60.
80. , , , . 1997. Hippocampal N-methyl-D-aspartate and kainate binding in response to entorhinal cortex aspiration or 192 IgG-saporin lesions of the basal forebrain. Neuroscience.; 77: 649-59.
81. . 1999. Recovery after damage to motor cortical areas. Curr Opin Neurobiol.; 9: 740-7.
82. , , , . 1999. Epi-arachnoidal drug deposit: a rare complication of intrathecal drug therapy. J Pain Symptom Manage.; 18: 229-32.
83. , , , . 1997. Intrathecal ciliary neurotrophic factor delivery for treatment of amyotrophic lateral sclerosis (phase I trial). Neurosurgery.; 40: 94-9; discussion 99-100.
84. , , , , , . 2000. Basic fibroblast growth factor (bFGF) enhances functional recovery following severe spinal cord injury to the rat. Exp Neurol.; 164: 280-91.
85. . 1996. Multiple sclerosis: prospects for remyelination. Mult Scler.; 2: 195-7.
86. , , . 1990. Anoxic injury of CNS white matter: protective effect of ketamine. Neurology.; 40: 1399-403.
87. , , . 1999. From neural stem cells to myelinating oligodendrocytes. Mol Cell Neurosci.; 14: 287-300.
88. , . 1993. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today.; 14: 426-430.
89. , , , . 1987. Remission of rheumatoid arthritis with the successful treatment of acute myelogenous leukemia with cytosine arabinoside, daunorubicin, and m-AMSA. Arthritis Rheum.; 30: 1187-1190.
Page 323
90. , , , . 1997. Recommendations from the National Multiple Sclerosis Society Clinical Outcomes Assessment Task Force. Ann Neurol.; 42: 379-82.
91. , , . 1999. Reliability and validity of two self-report measures of impairment and disability for MS. North American Research Consortium on Multiple Sclerosis Outcomes Study Group. Neurology.; 52: 63-70.
92. , . 1997. Remyelination in demyelinating disease. Baillieres Clin Neurol.; 6: 525-48.
93. , , , . 1988. NOD marrow stem cells adoptively transfer diabetes to resistant (NOD x NON)F1 mice. Diabetes.; 37: 252-5.
94. , . 1995-1996. Multipotent neural progenitor or stem-like cells may be uniquely suited for therapy for some neurodegenerative conditions. Clin Neurosci.; 3: 310-6.
95. , , , , . 2000. Placebo controlled pilot trial to study the remyelinating potential of intravenous immunoglobulins in multiple sclerosis. J Neurol Neurosurg Psychiatry.; 68: 89-92.
96. . 2000. Despite epitope spreading in the pathogenesis of autoimmune disease, highly restricted approaches to immune therapy may still succeed [with a hedge on this bet]. J Autoimmun.; 14: 278-82.
97. , , . 1997. Autoimmune pathogenesis of multiple sclerosis: role of autoreactive T lymphocytes and new immunotherapeutic strategies. Crit Rev Immunol.; 17: 33-75.
98. , , , . 1990. Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc Natl Acad Sci U S A.; 87: 4212-6.
99. , , , . 1993. Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci U S A.; 90: 6976-80.
100. , , . 1992. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci.; 12: 430-9.
101. , , . 1999. Constraint-Induced Movement Therapy: a new family of techniques with broad application to physical rehabilitation—a clinical review. J Rehabil Res Dev.; 36: 237-51.
102. , , , . 1985. Association of lpr gene with graft-vs. host disease-like syndrome. J Exp Med.; 162: 1-18.
103. , . 1985. Murine models of systemic lupus erythematosus. Adv Immunol.; 37: 269-390.
104. , , , . 1999. Neurodegeneration in multiple sclerosis: relationship to neurological disability. The Neuroscientist.; 5: 48-57.
105. . 1997. Hematopoietic stem cell transplantation in rheumatic diseases other than systemic sclerosis and systemic lupus erythematosus. J Rheumatol Suppl.; 48.
106. . 1993. BMT in experimental autoimmune diseases. Bone Marrow Transplant.; 11: 183-7.
107. , , , . 1996. Treatment of multiple sclerosis with T-cell receptor peptides: results of a double-blind pilot trial. Nat Med.; 2: 1109-15.
108. , , , , , . 1999. A new mechanism of neurodegeneration: a proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc Natl Acad Sci U S A.; 96: 9879-84.
109. , , , . 1993. Autoimmune polyendocrine failure—type 1 (insulin-dependent) diabetes mellitus and hypothyroidism—after allogeneic bone marrow transplantation in a patient with lymphoblastic leukaemia. Diabetologia.; 36: 541-546.
110. , , , . 2000. Human nerve growth factor protects common marmosets against autoimmune encephalomyelitis by switching the balance of T helper cell type 1 and 2 cytokines within the central nervous system. J Exp Med.; 191: 1799-806.
Page 324
111. , , , . 1996. Suppressive vaccination with DNA encoding a variable region gene of the T-cell receptor prevents autoimmune encephalomyelitis and activates Th2 immunity. Nat Med.; 2: 899-905.
112. , , , . 2000. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci U S A.; 97: 6820-6825.
113. , . 2000. Methodological problems in evaluating efficacy of a treatment in multiple sclerosis. Pathol Biol (Paris).; 48: 104-13.
114. . 1997. Growth factors and myelin regeneration in multiple sclerosis. Mult Scler.; 3: 113-20.
115. , , , , . 1988. Expression of genetically dete mined diabetes and insulitis in the nonobese diabetic (NOD) mouse at the level of bone marrow-derived cells. Transfer of diabetes and insulitis to nondiabetic (NOD X B10) F1 mice with bone marrow cells from NOD mice. J Exp Med.; 167: 1801-10.
116. , , , . 1997. Results of a phase I clinical trial of a T-cell receptor peptide vaccine in patients with multiple sclerosis. I. Analysis of T-cell receptor utilization in CSF cell populations. J Neuroimmunol.; 76: 15-28.
117. , , , , . 1995. A combination of BDNF and NT-3 promotes supraspinal axonal regeneration into Schwann cell grafts in adult rat thoracic spinal cord. Exp Neurol.; 134: 261-72.
118. , , . 1999. “Global” cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated shiverer mouse brain. Proc Natl Acad Sci U S A.; 96: 7029-34.
119. , . 1999. Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endocrinology.; 140: 3063-72.
120. , , , , . T-cell vaccination for the treatment of multiple sclerosis. In Zhang J, ed. Immunotherapy in Neuroimmunologic Diseases. London, England: Martin Dunitz; 1998.
















































