
MARY ELLEN JONES
December 25, 1922–August 23, 1996

BY THOMAS W. TRAUT
FOR ALMOST 50 YEARS Mary Ellen Jones was actively engaged in research related to amino acid metabolism and pyrimidine nucleotide metabolism. In collaboration with Leonard Spector she was a codiscoverer of carbamoyl phosphate, a compound essential for the biosynthesis of arginine and urea, and also for the biosynthesis of pyrimidine nucleotides. The discovery of carbamoyl phosphate was truly significant, as it rapidly influenced research in many other laboratories. An indicator of its importance is that within a few years it became commercially available. By the early 1970s she was among the first to define the new area of multifunctional proteins with her studies of dihydroorotate synthase (also called CAD) and UMP synthase. Jones continued to demonstrate talent for devising new analytical procedures, for cleverly designing experimental approaches, and for insightful analyses of the emerging information.
Jones was the first woman scientist to hold an endowed chair at the University of North Carolina and the first woman to become a department chair at the medical school. Jones was widely recognized for her scientific accomplishments and for her leadership roles. Among her awards were the Wilbur Lucius Cross Medal from Yale University (1982),
the North Carolina American Chemical Society Distinguished Chemist (1986), the Thomas Jefferson Award from the University of North Carolina (1990), and the Award in Science awarded by the state of North Carolina (1991). She was elected to the Institute of Medicine (1981), the National Academy of Sciences (1984), the American Association of Arts and Sciences (1991), and the American Philosophical Society (1994). She was very active in the affairs of the Biochemistry Division of the American Chemical Society. She was frequently elected to preside over national organizations: president of the Association of Medical School Departments of Biochemistry in 1985, president of the American Society for Biochemistry and Molecular Biology in 1986, and president of the American Association of University Professors in 1988. Her final honor was to have an 11-story research center dedicated with her name at the University of North Carolina.
Mary Ellen Jones was born on December 25, 1922, in La Grange, Illinois, one of four children of Elmer and Laura Klein Jones. She received an undergraduate degree at the University of Chicago in 1944, having majored in biochemistry. Lacking financial support, she delayed her graduate school career for four years while working full time and saving for her future. She entered Yale University in 1948 and soon began to see enzymology as most intriguing. She completed her dissertation studies in 1951, under the direction of Joseph Fruton.
Her earliest scientific training began at Armour and Company, where she worked half time during her undergraduate school years. After graduation from the University of Chicago, she remained at Armour until 1948. Initially she worked as a bacteriologist in quality control before becoming a research chemist.
At Armour she met Paul Munson, who then was director
of the research laboratory. With Munson she would do her first research on androsterone and monopalmitin, leading to two publications in the Journal of Biological Chemistry. Jones and Munson were married in 1948, and had two children: Ethan V.Munson (born 1956), currently an associate professor of computer science at the University of Wisconsin-Milwaukee, and Catherine Munson (born 1960), currently a psychiatrist in Charlotte, North Carolina. Jones would later reminisce about how she and Munson had agreed to maintain a dual-career family. To make this possible they would dedicate one salary to obtain help for household work and childcare, enabling Jones to be a parent while continuing as a productive scientist.
When Paul Munson became an assistant professor of pharmacology at Yale, Jones was able to begin graduate studies at that university in 1948. She did her dissertation research under the supervision of Joseph Fruton, completing her studies in only three years while characterizing the catalytic properties of cathepsin C.Fruton was a very helpful mentor and instilled in her the paradigms for good experimental design.
When Munson moved to Boston in 1951, Jones was able to find a postdoctoral position at the Massachusetts General Hospital, with Fritz Lipmann with whom she worked until 1957. By 1957 Jones had two papers published while she was a technician, two more from her graduate studies at Yale, plus another nine papers from her work in the Lipmann laboratory. This productivity enabled her to become an assistant professor in the newly established graduate Department of Biochemistry at Brandeis University in 1957, where she became an associate professor by 1960. When Paul Munson was offered the chair of pharmacology at the University of North Carolina at Chapel Hill in 1966,
Jones relocated to that campus in the Department of Biochemistry, where she became professor in 1968.
The move to North Carolina was not easy. By 1965 she had developed a very active laboratory at Brandeis and had become well established in her field. The biochemistry department in Chapel Hill had not been seeking a new faculty member because of a lack of space. Jones therefore found herself located in the basement of a building in the zoology department, a space that was not designed for laboratory research and was also isolated from her new home department. Reminiscing about this time in later years, Jones would note a few unhappy elements of her new situation, but she would normally follow this with a chuckle while emphasizing how she and her new laboratory associates found ways to triumph over their circumstances.
After divorce from Paul Munson a few years later, Jones moved to Los Angeles in 1971, where she worked at the University of Southern California School of Medicine for seven years. In 1978 she returned to the Department of Biochemistry at the University of North Carolina as chair. She led the department until 1989, and she continued active research until the spring of 1995. Unfortunately, her retirement—planned almost two years earlier—came only a few months after she was diagnosed with cancer of the esophagus. She was barely able to initiate her retirement plans for moving to New Mexico, occupying a newly built house and resuming her love for painting. After less than two months she abandoned her new retirement to return to Boston, where she spent her last year as a recipient of vigorous chemotherapy. She died in Waltham, Massachusetts, on August 23, 1996.
Mary Ellen Jones was an energetic and almost tireless worker, highly flexible, and yet very focused. These qualities are important for maintaining an active, independent
scientific laboratory, especially while being a department chair who continued very much to be concerned with the current well-being and future development of her department. Given her special circumstances as a developing scientist, these personal traits were essential. She was an ardent supporter of all her students and postdocs, and she continued to be a lifelong advocate for women in science and for minorities in general.
THE EARLY YEARS WITH LIPMANN
Jones’s productive three years as a graduate student at Yale had prepared her for enzymology, and she always maintained that each enzyme “develops a character uniquely its own.” She had been absolutely delighted to start in the Lipmann laboratory, as she had seen Lipmann’s recent work on the importance of high-energy phosphate bonds as truly exciting. This assessment of Fritz Lipmann would be shared two years later by the committee awarding him the Nobel Prize for medicine and physiology in 1953. In the Lipmann laboratory she would study how coenzyme A became activated and would become familiar with the use of ATP to activate biosynthetic molecules. Her initial triumph in this period was the unexpected demonstration that pyrophosphate, rather than orthophosphate, was the direct product in the action of acetyl-CoA synthetase (1953). This was a novel demonstration of ATP being involved in a reaction other than the well-known phospho-transfer reaction. Jones and Lipmann correctly hypothesized that the reaction mechanism involved an enzyme-adenylate intermediate. When some years later DNA polymerase was first being characterized by Arthur Kornberg and his colleagues, they would observe a similar reaction with a pyrophosphoryl cleavage of the nucleotide precursors, thereby helping to establish the general nature of the mechanism described by Jones and Lipmann.
Lipmann proved to be a good mentor, and Jones often mentioned how he coached his postdocs and students to look at all possibilities and to ever be flexible.
Jones’s major scientific direction was determined in 1955, when she and Leonard Spector deduced that carbamoyl phosphate should be the most likely agent involved in the synthesis of citrulline from ornithine (see Figure 1). She sometimes spoke of those early exciting days when she and Spector discussed and explored the most plausible structure for the mysterious compound that had to be an intermediary in joining with ornithine to form citrulline. The difficulty then was to actually synthesize carbamoyl phosphate so as to test its possible function. Jones has recounted how Spector was at an early concert of Debussy’s Afternoon of a Fawn. While the music was compelling, and his date was very attractive, his mind returned to the synthesis of carbamoyl phosphate. A simple strategy for using cyanate with lithium phosphate occurred to him and he dashed back to the laboratory right after the concert. Before mid-night he would have crystals of carbamoyl phosphate.
Then Jones demonstrated that liver cell extracts converted it to citrulline (1955). The cellular biosynthesis of carbamoyl phosphate and its subsequent utilization were rapidly pursued by a number of laboratories. The central position of this compound is depicted in Figure 1, as are the many enzymes involved in the formation or utilization of this metabolite that were subsequently investigated by Jones. Jones and Lipmann then demonstrated that carbamate was first formed from bicarbonate and required ATP (1960). By including the enzyme carbonic anhydrase, they were able to demonstrate that bicarbonate was the immediate substrate for the carboxy phosphate step in the synthesis of carbamoyl phosphate.
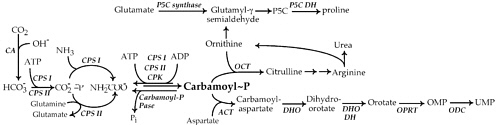
FIGURE 1 Metabolic pathways studied by Mary Ellen Jones. All the enzymes identified were studied by her group: ACT, aspartate carbamoyltransferase; carbamoyl-P pase, carbamoyl phosphate phosphatase; CA, carbonic anhydrase; CPK, carboxyphosphate kinase; CPS, carbamoyl phosphate synthetase; DHO, dihydroorotase; DHO DH, dihydroorotate dehydrogenase; OCT, ornithine carbamoyltransferase; ODC, OMP decarboxylase; OPRT, orotate phosphoribosyltransferase; P5C synthase, pyrroline-5-carboxylate synthase; P5C DH, pyrroline-5-carboxylate dehydrogenase.
As depicted in Figure 1, carbamoyl phosphate is directly involved in joining with aspartate for the synthesis of the pyrimidine base orotate, or in linking with ornithine to form citrulline. This latter reaction was the original focus of research during her years with Fritz Lipmann. Because ornithine is also utilized in the synthesis of proline, defining this pathway also became a challenge for Jones. Her first studies on pyrimidine biosynthesis began shortly thereafter.
Today we know that at least three enzymes can synthesize carbamoyl phosphate. In 1955 it was completely unclear as to whether the synthesis began with the formation of carboxyphosphate and whether this came from bicarbonate or from a carboxyl group on an amino acid, because it was already established that N-acetyl-glutamate was a necessary factor in the synthesis of citrulline. Furthermore, with bacterial extracts it appeared that either ammonia or glutamine could serve as a nitrogen donor. Jones appreciated that the utilization of ammonia in bacterial cell extracts might not be
physiologically relevant. Earlier studies with liver extracts had clearly demonstrated that incorporation of ammonia into urea was an important function, and this pathway presumably required the same carbamoyl phosphate synthetase. For almost 10 years there would be uncertainty and confusion about why two ATP molecules were needed for the synthesis of carbamoyl phosphate, what the true nitrogen source was, and whether there was more than one carbamoyl phosphate-synthesizing enzyme in any specific cell or tissue. By 1967 this was largely settled, and Mary Ellen Jones had been pivotal in clarifying the most important details in Figure 1.
In her subsequent career she would pursue both the amino acid metabolism shown in Figure 1 and all the enzymes in the pyrimidine pathway. She would thus become an authority in these two areas of metabolism, publishing a major review on amino acids (1965) and then on pyrimidine biosynthesis (1980). Instead of looking at her career chronologically, it will be more understandable to explore her separate research areas.
A YOUNG INDEPENDENT INVESTIGATOR STUDYING PYRIMIDINE BIOSYNTHESIS IN EUKARYOTES
Having established the existence and function of carbamoyl phosphate while in the Lipmann laboratory (1955), Jones and Spector (now at Brandeis) established that carbon dioxide or bicarbonate was the direct source for the initial activation step leading to carboxy-phosphate (1960). The focus in these early years was completely on the synthesis of citrulline and arginine, and the assay being used always measured the incorporation of bicarbonate plus ammonia as the cosubstrate. The product being measured was citrulline, detected by the only method then available, a colorimetric assay that was convenient but not very sensitive. To
assure that formation of citrulline was not limited by the lack of a cellular ornithine carbamoyl transferase (OCT), this enzyme, purified from S. faecalis, was always added to the assay.
The Jones group then observed that, of 18 rat tissues, only the liver contained abundant carbamoyl phosphate synthetase activity, while it was barely detectable in kidney and intestine (1961). Equally important was their finding that this activity was localized in the mitochondrial fraction. This result caused some concern as to how most cells were able to synthesize pyrimidines, since carbamoyl phosphate was clearly implicated in that pathway, and since all cells were expected to synthesize nucleotides. In reviewing this subject (1963), Jones for the first time speculated about the existence of two carbamoyl phosphate synthetases: one enzyme in liver using ammonia and a second enzyme dependent on glutamine.
The significant breakthrough came with the next four papers reporting work done with Sally Hager. The incorporation of 14C-bicarbonate by whole cells into possible products was a much more sensitive assay. For the first time they could infer a glutamine-dependent carbamoyl phosphate synthetase activity in mouse tumor cells by readily measuring 14C-bicarbonate incorporation into carbon 2 of uracil (1965). This experimental design was excellent, as it combined a tissue source likely to have higher enzyme levels (tumors consume nucleotides more steadily) with a more sensitive enzyme activity assay. Her laboratory had already established that carbamoyl phosphate is not detectable in fresh blood from rabbits, thereby excluding the liver as the unique source for this compound to be used by other tissues. Therefore Jones now proposed the likelihood that there were two separate carbamoyl phosphate synthetase isozymes, one requiring ammonia for the synthesis of arginine or
urea and a second enzyme requiring glutamine for the synthesis of orotate. It simply remained for her group to prove this.
In the following year Jones and Hager reported the first isolation of the glutamine- dependent carbamoyl phosphate synthetase from cytoplasmic extracts of Ehrlich ascites cells and from rat liver (1966). The critical factor impeding earlier efforts was now apparent with their demonstration that the enzyme was extremely unstable. With customary resourcefulness, they had developed an improved purification strategy in which they used a substrate, ATP, to stabilize the enzyme, and this now allowed them to characterize the enzyme. Thereby, they now could explain the fact that this enzyme had never been detected in mammalian cells previously. Their paper was the first to actually demonstrate the existence of carbamoyl phosphate synthetase II. This important achievement was followed with a detailed presentation of the new isolation procedure devised to maintain the enzyme in a more stable form (1967,1). The addition of ATP to the homogenization buffer had a dramatic effect on maintaining the enzyme activity during several purification steps. They showed that the enzyme has a high affinity for glutamine as the nitrogen donor, but shows a modest and unphysiological activity with ammonia. This paper helped to clarify some of the earlier confusion between the two separate sources for nitrogen.
The subsequent paper in the same year again showed a keen sense for experimental design. Jones and Hager used fetal rat liver as a source for the enzymes, since it was clearly evident that liver should have the ammonia-dependent CPS, but could well have the glutamine-dependent CPS in addition. They therefore used their new isotope assay to measure activity with either ammonia or with glutamine as the nitrogen source. In addition, they carefully fractionated the
fetal liver extracts by centrifugation to prepare a mitochondrial fraction and a cytoplasmic supernatant fraction (1967,2). The results produced one of those occasions where the light goes on and all becomes clear. Fetal liver had two carbamoyl phosphate synthetase activities, now designated as CPS I and CPS II. CPS I is only in the mitochondrion and preferentially utilizes ammonia in the formation of citrulline by the enzyme ornithine carbamoyltransferase (OCT). CPS II is in the cytoplasm and only uses glutamine for the formation of carbamoyl-aspartate by the enzyme aspartate carbamoyl transferase (ACT).
These results truly helped to resolve the existing confusion in this field. It was becoming evident that bacteria had but a single CPS enzyme, whose product—carbamoyl phosphate—was used for either arginine or for pyrimidine synthesis. In contrast, rats (and presumably other higher eukaryotes) had evolved two CPS enzymes and had physically separated the synthesis of urea (only in mitochondria, and mostly in liver) and the synthesis of orotate or UMP (only in the cytoplasm, and presumably in all cells). The significance of this was that mammals could now have separate control of the initial metabolic step for either of the two pathways by having the respective enzymes in separate subcellular compartments. The work in these last three papers, largely completed at Brandeis, must have been a real boost for Jones, as they were published just as she was reestablishing her laboratory in Chapel Hill.
ASPARTATE CARBAMOYLTRANSFERASES IN BACTERIA
By the mid-1960s the Jones group had devised a procedure to synthesize 14C-carbamoyl phosphate. This increased the sensitivity by twenty-fold for measuring the activity of aspartate carbamoyltransferase with carbamoyl phosphate as the varied substrate. Initially working with the E. coli
ACT, an enzyme already well characterized by several laboratories, Jones and her colleagues were the first to demonstrate positive cooperativity by the enzyme for this substrate (1968). Expanding these studies to eight bacterial species, they showed that the sizes of the ACT enzymes, measured by column chromatography, suggested that there were three major classes for this enzyme in the different types of bacteria (1969). Using the same methodology for cell extracts from the eight bacterial species, they now characterized the three types of ACT. Class A enzymes from P. aeruginosa and P. fluorescens were the largest and were subject to inhibition by pyrimidine nucleotides or ATP. They did not appear to have a separate regulatory sub-unit. Class B enzymes from E. coli or C. freundii were of intermediate size. They were sensitive to the same regulatory nucleotides, but these appeared to act at a separate regulatory sub-unit. Class C enzymes from C. freundii or S. faecalis were the smallest enzymes. These showed no regulatory features.
Her postdoc Mary Sue Coleman showed that aspartate carbamoyltransferase from C. freundii included both classes B and C enzymes (1971). The larger species could dissociate to form the smaller species, presumably by loss of a regulatory sub-unit, as could be done with the same enzyme from E. coli. The combined efforts of her associates Lansing Prescott, T.-Y. Chang, and Linda Adair led to purification of ACT from several bacterial species and characterization of ACTs in each of the three classes above.
MULTIFUNCTIONAL PROTEINS
By the early 1970s Jones had become quite interested in the possibility that in mammals the fusion of genes for enzymes that are consecutive in a pathway could produce much larger proteins containing two or more catalytic activities. Such work in her laboratory was initiated by Tom Shoaf,
who demonstrated that Ehrlich ascites cells probably had two such “enzyme complexes,” as they were initially called (1973). Shoaf and Jones showed that the enzyme activities for CPS II, ACT, and DHO (see Figure 1) always appeared to be joined, as were the activities for OPRT and ODC.
Although UMP synthase had not yet been purified, my own studies with Jones established that the two activities had to be joined with experiments showing that both activities underwent changes in sub-unit association in tandem, as the enzyme was progressively converted from the monomer to the dimer form (1979). This conversion to the active dimer form could be titrated with various nucleotides, or analogs. Comparing the effectiveness of such ligands in these molecular-size experiments to their Ki in kinetic studies led to the awareness that such effector ligands could produce a conformational response at two binding sites on the UMP synthase protein.
Postdoc Richard Christopherson then worked with the dihyroorotate synthase multifunctional protein (1980,1,2). In some very well-designed experiments, they achieved the first really detailed examination of the mechanism of the dihydroorotase (DHO) reaction. The possibility of channeling had always appeared intriguing, and Christopherson, by doing appropriate kinetic studies with two isotopes for the initial bicarbonate or an exogenous carbamoyl phosphate, was able to quantitate partial channeling of carbamoyl phosphate between the domain where it was formed (CPS II) and the domain where it was utilized (ACT).
The purification of UMP synthase from human tissue by her student Laura Livingstone was extended with her postdoc B.D.Han to produce an expression system for the human protein (1995). This was also an achievement, since years of work by Jones and colleagues on UMP synthase had established that this protein is not very stable. Because one of
her initial triumphs came from the search for an optimum buffer system to stabilize carbamoyl phosphate synthetase in the 1960s, this last paper by Jones and her colleagues now defined the conditions for maintaining both catalytic domains of UMP synthase at their optimum.
A genetic disease, orotic aciduria, results from mutations in either catalytic domain of UMP synthase, leading to loss of enzyme activity. A standard expectation is that such mutations are most likely to be harmful if they affect the catalytic site of the enzyme. Jones explored this with her student Mary Perry, who used fibroblasts from a human patient to demonstrate that the mutant enzyme was actually highly unstable to heat denaturation or to proteolysis. A clever strategy involved culturing cells in the presence of azauridine, a nucleoside easily absorbed by cells, and then converted to azaUMP, a very strong inhibitor of OMP decarboxylase. It was anticipated that the nucleotide would bind to the enzyme in the cells and thereby stabilize it during purification experiments. Perry was successful in isolating the mutant protein and established that the mutant enzyme had the same apparent molecular weight and immunoactivity. The defect in the mutant enzyme was therefore one that affected the structural integrity of this bifunctional protein.
PROLINE SYNTHESIS
Studies in the early years with Lipmann had focused on amino acid metabolism. In later years, Jones would continue the effort to demonstrate that mammals could synthesize the amino acid proline, with ornithine as the likely precursor. The scheme that Jones deduced for this synthesis involved the initial formation of glutamyl-semialdehyde, either from ornithine or from glutamate in tissues where ornithine is not abundant. The intermediate glutamylsemialdehyde spontaneously converts to pyrroline-5-carboxy-
late (P5C), which can then be reduced to proline. Wakabayashi and Jones published the first demonstration of the enzyme P5C synthase in mammals (1983). Because bacteria were known to have this pathway, they used germ-free rats to demonstrate the activity in the animal tissue. Later, her student Curtis Small accomplished the difficult purification of P5C dehydrogenase (1990). This required new assay procedures to increase sensitivity and permitted them to define the kinetics for this enzyme.
MECHANISM OF OMP DECARBOXYLASE
The mechanism for this reaction is not immediately evident, and at least four schemes had been proposed over the years. Now that Jones had a source of pure protein from yeast, and the cDNA coding for it, she began to pursue this question. Jones and her student Jeff Smiley and postdoc Juliette Bell, working in collaboration with Marion O’Leary, made a strong case for a mechanism where cleavage of the scissile C-C bond was the rate-determining step (1990). Using kinetic isotope effects with 13C-OMP as a function of pH, they now proposed that a proton from the enzyme was needed in the transition state to stabilize a nitrogen ylide, thereby facilitating the elimination of the CO2. These studies were extended as Smiley performed systematic mutagenesis at a key residue, lysine 93. All mutants had no detectable activity, even though binding studies showed that the affinity for OMP was largely unchanged (1992). This implied the importance of lysine 93 directly in catalysis. One mutant, Lys93Cys could be “rescued.” This protein was covalently modified at the new cysteine residue with bromoethylamine to yield an enzyme with a cysteine-ethylamine at residue 93, to mimic the normal lysine. This modified mutant protein had recovered much of the enzymatic activity, confirming the most likely role for lysine 93.
Although the above studies were done as Mary Ellen Jones was in her late sixties, she continued to show a spirit for pursuing new directions and new technologies. Jeff Smiley has spoken of how open she was to his using molecular biology for mutation studies—a procedure completely new to her laboratory. She was equally quick to explore using the kinetic isotope effect as an approach to evaluating the mechanism of the enzyme.
The last scientific effort of Mary Ellen Jones was a review paper that we wrote together (1996). The plan for this review had been initiated late in 1994: I would cover the enzymes uridine kinase and ß-alanine synthase, while she would focus on UMP synthase. Though her illness became an important factor by early 1995, she still had enough strength and determination to supervise her laboratory for those last few months, while also attempting to put her normal effort into this final manuscript. In our frequent interactions to discuss the progress of this review article a decline in her normal energy, resulting from chemotherapy, became apparent. Although I quickly volunteered to absolve her of all responsibility for the completion of this manuscript, she did not relinquish her commitment. It was still early enough in the progress of her cancer that she projected an outward optimism to continue for many years her enjoyment of science, the arts, and all the colleagues to whom she felt close. By all these people she will always be remembered with great affection.
SELECTED BIBLIOGRAPHY
1953 With S.Black, R.M.Flynn, and F.Lipmann. Acetyl-coenzyme A synthesis through a pyrophosphoryl split of ATP. Biochim. Biophys. Acta 12:141–49.
1955 With L.Spector and F.Lipmann. Carbamyl phosphate, the carbamyl donor in enzymatic citrulline synthesis. J. Am. Chem. Soc. 77:819–20.
1960 With L.Spector. The pathway of carbonate in the biosynthesis of carbamyl phosphate. J. Biol. Chem. 235:2897–901.
With F.Lipmann. Chemical and enzymatic synthesis of carbamyl phosphate. Proc. Natl. Acad. Sci. U. S. A. 46:1194–205.
1961 With A.D.Anderson, C.Anderson, and D.Hodes. Citrulline synthesis in rat tissues. Arch. Biochem. Biophys. 95:499–507.
1963 Carbamyl phosphate. Science 140:1373–79.
1965 Amino acid metabolism. Annu. Rev. Biochem. 34:381–418.
1966 With S.E.Hager. Source of carbamyl phosphate for pyrimidine biosynthesis in mouse Ehrlich ascites cells and rat liver. Science 154:422.
1967 With S.E.Hager. Initial steps in pyrimidine synthesis in Ehrlich ascites carcinoma in vitro. II. The synthesis of carbamyl phosphate by a soluble, glutamine-dependent carbamyl phosphate synthetase. J. Biol. Chem. 242:5667–73.
With S.E.Hager. A glutamine-dependent enzyme for the synthesis of carbamyl phosphate for pyrimidine biosynthesis in fetal rat liver. J. Biol. Chem. 242:5674–80.
1968 With M.R.Bethell, K.E.Smith, and J.S.White. Carbamyl phosphate: An allosteric substrate for aspartate transcarbamylase of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 60:1442–49.
1971 With M.S.Coleman. Aspartate transcarbamylases of Citrobacter freundii. Biochemistry 10:3390–96
1973 With W.T.Shoaf. Uridylic acid synthesis in Ehrlich ascites carcinoma. Properties, subcellular distribution, and nature of enzyme complexes of the six biosynthetic enzymes. Biochemistry 12:4039– 51 .
1979 With T.W.Traut. Interconversion of different molecular weight forms of the orotate phosphoribosyltransferase:orotidine-5'-phosphate decarboxylase enzyme complex from mouse Ehrlich ascites cells. J. Biol. Chem. 254:1143–50.
1980 Pyrimidine nucleotide biosynthesis in animals: Genes, enzymes, and regulation of UMP biosynthesis. Annu. Rev. Biochem. 49:253–79.
With R.I.Christopherson. The effects of pH and inhibitors upon the catalytic activity of the dihydroorotase of multienzymatic protein pyr1–3 from mouse Ehrlich ascites carcinoma. J. Biol. Chem. 255:3358–70.
With R.I.Christopherson. The overall synthesis of L-5,6-dihydroorotate by multienzymatic protein pyr1–3 from hamster cells. Kinetic studies, substrate channeling, and the effects of inhibitors. J. Biol. Chem. 255:11381–95.
1983 With Y.Wakabayashi. Pyrroline-5-carboxylate synthesis from glutamate by rat intestinal mucosa. J. Biol. Chem. 258:3865–72.
1989 With M.E.Perry. Orotic aciduria fibroblasts express a labile form of UMP synthase. J. Biol. Chem. 264:15522–28.
1990 With W.C.Small. Pyrroline-5-carboxylate dehydrogenase of the mitochondrial matrix of rat liver. Purification, physical and kinetic characteristics. J. Biol. Chem. 265:18668–72.
1991 With J.A.Smiley, P.Paneth, M.H.O’Leary, and J.B.Bell. Investigation of the enzymatic mechanism of yeast orotidine-5'-monophosphate decarboxylase using 13C kinetic isotope effects. Biochemistry 30:6216–23.
1992 With J.A.Smiley. A unique catalytic and inhibitor-binding role for Lys93 of yeast orotidylate decarboxylase. Biochemistry 31:12162– 68.
1995 With B.D.Han, L.R.Livingstone, D.A.Pasek, and M.J.Yablonski. Human uridine monophosphate synthase: Baculovirus expression, immunoaffinity column purification and characterization of the acetylated amino terminus. Biochemistry 34:10835–43.
1996 With T.W.Traut. Uracil metabolism—UMP synthesis from orotic acid or uridine and conversion of uracil to beta-alanine: Enzymes and cDNAs. Prog. Nucleic Acid Res. Mol. Biol. 53:1–78.




















