
17
Genomes, Phylogeny, and Evolutionary Systems Biology
MÓNICA MEDINA*
With the completion of the human genome and the growing number of diverse genomes being sequenced, a new age of evolutionary research is currently taking shape. The myriad of technological breakthroughs in biology that are leading to the unification of broad scientific fields such as molecular biology, biochemistry, physics, mathematics, and computer science are now known as systems biology. Here, I present an overview, with an emphasis on eukaryotes, of how the postgenomics era is adopting comparative approaches that go beyond comparisons among model organisms to shape the nascent field of evolutionary systems biology.
Systems biology is in the eye of the beholder.
Leroy Hood
Only in the last decade have we had access to nearly complete genomes of a diversity of organisms allowing for large-scale comparative analysis. The access to this immense amount of data
|
* |
Department of Evolutionary Genomics, Department of Energy Joint Genome Institute, 2800 Mitchell Drive, Walnut Creek, CA 94598. Present address: School of Natural Sciences, University of California, P.O. Box 2039, Merced, CA 95344. E-mail: mmedina@ucmerced.edu. |
is providing profound insight into the tree of life at all levels of divergence (Fig. 17.1A). It is thus not surprising that understanding phylogenetic relationships is a prevalent research goal among not only evolutionary biologists but also all scientists interested in the organization and function of the genome. New genome sequences and analysis methods are helping improve our understanding of phylogeny, and at the same time improved phylogenies and phylogenetic theory are generating a better understanding of genome evolution. Currently however, the level of genome sequencing for different branches of the tree of life is far from equivalent. Prokaryotic genome projects are abundant, mainly due to their small genome sizes, with >200 genomes already published and at least 500 currently in progress (www.genomesonline.org). In contrast, <300 eukaryotic genomes are either finished or in progress (www.genomesonline.org). Nevertheless, these data are starting to have a major impact on our understanding of eukaryotic evolution.
These new genomic data have informed our understanding of phylogenetic relationships, and the emerging consensus topologies are adding new insight to the small subunit ribosomal RNA phylogenies. For example, the topology of the ribosomal eukaryotic tree has been recently redrawn with the use of genomic signatures that place the root of all eukaryotic life between two newly uncovered major clades, Unikonts and Bikonts (Fig. 17.1A). Unikonts, which contain the heterotrophic groups Opisthokonta and the Amebozoa, share a derived three-gene fusion of enzymeencoding genes in the pyrimidine synthesis pathway (Stechmann and Cavalier-Smith, 2003), whereas Bikonts, which contain the remaining eukaryotic clades, share another derived gene fusion between dihydro folate reductase and thymidine synthase (Stechmann and Cavalier-Smith, 2002). All photosynthetic groups of primary and secondary plastid symbiotic origins are now thought to be within the Bikonts. Although the animal, fungal, and plant lineages are the most widely represented in terms of genome initiatives (Fig. 17.1B–D), it is significant that multiple protistan genome projects have also been initiated by the interest of diverse scientific communities, including parasitologists (Gardner et al., 2002), plant pathologists (Waugh et al., 2000), oceanographers (Armbrust et al., 2004), and evolutionary biologists (www.biology.uiowa.edu/workshop).
As more whole-genome projects are being completed, postgenomic biology is also providing insight into the function of biological systems by the use of new high-throughput bioanalytical methods, information technology, and computational modeling. This new revolution in biology has become known as systems biology (Hood, 2003). In addition to shifting approaches to biological research from reductionist strategies to pathway- and system-level strategies (Hartwell et al., 1999), another paradigm
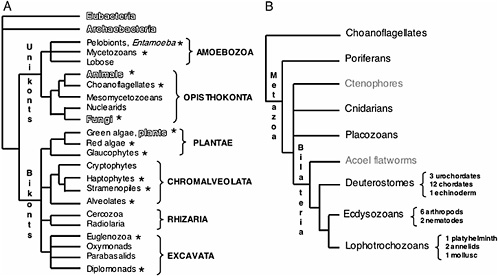
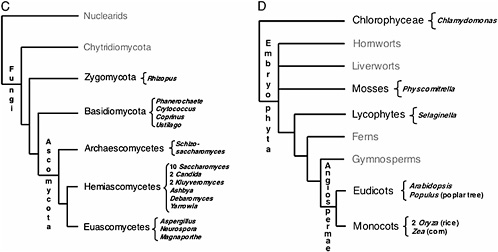
FIGURE 17.1 Current consensus eukaryotic tree. (A) The large subclades within Unikonts and Bikonts are recovered by a combination of multiple gene phylogenies, EST data, and genomic level characters (Stechmann and Cavalier-Smith, 2003; Simpson and Roger, 2004; Bhattacharya et al., 2004). Six major eukaryotic groups are now recognized although resolution within them is still lacking. The placement of the root is based on two gene fusion events (Stechmann and Cavalier-Smith, 2002, 2003). Lineages where whole-genome projects are in progress are marked with asterisks. Lineages being studied by large postgenomic initiatives are shadowed. (B) Metazoan consensus phylogeny of major branches (Adoutte et al., 1999; Medina et al., 2001; Ruiz-Trillo et al., 2002) and a conservative estimate of finished and ongoing genome projects (highlighted in black). (C) Fungal consensus phylogeny (Berbee and Taylor, 1993; Hedges, 2002) and estimate of ongoing genome projects (www.broad.mit.edu/annotation/fungi/fgi) (highlighted in black). (D) Consensus phylogeny of green plants (Hedges, 2002; Pryer et al., 2002) and estimate of ongoing genome projects (highlighted in black).
is rapidly emerging, namely the use of phylogenetically based inference in systems biology. Before the genomic revolution, research questions were typically addressed within a single model organism, with only occasional comparative studies when similar information was available for another organism. These comparisons were made between distantly related taxa, and the evolutionary implications were rarely mentioned or taken into account. The increasing importance of comparative analysis is evident in the growing proportion of new prokaryotic genome projects that have been chosen primarily because of their phylogenetic relationship to model organisms, such as Escherichia coli and Bacillus subtilis and their corresponding related taxa. This same trend is occurring for eukaryotes. Some prominent examples are the multiple Saccharomyces genome projects and those of other ascomycote fungi, the several Plasmodium projects and other genome initiatives for apicomplexan taxa, the numerous Caenorhabditis and other nematode genome projects, the multiple Drosophila and arthropod genome projects, and the large number of primate and mammalian genome projects.
GENOMES AND PHYLOGENY OF HIGHER EUKARYOTES
Metazoa
The sampling of the metazoan tree, and in particular of the chordate branch, was undertaken primarily due to the usefulness of the genomes in understanding human biology. However, this larger genomic dataset is already providing a powerful tool for comparative analysis and more accurate evolutionary inference. Deeper divergences in the Metazoan tree have become the target of major scrutiny due to the interest in comparative developmental genetics (Fig. 17.1B). Based on molecular phylogenies, the bilaterian phyla have been rearranged into three large clades, deuterostomes, lophotrochozoans, and ecdysozoans, these last two being sister taxa inside the protostome clade. At present, there is still debate regarding the placement of nematodes in the tree (i.e., the Ecdysozoa vs. Coelomata hypotheses) because analysis of genomic data currently challenges the placement of Caenorhabditis elegans as an ecdysozoan (Dopazo et al., 2004; Wolf et al., 2004).
In addition to the traditional developmental model organisms, genomes from unrepresented protostome (Annelida, Platyhelmintha, and Mollusca) and basal phyla are now being sequenced (Porifera, Placozoa, and Cnidaria) (www.jgi.doe.gov/sequencing/cspseqplans.html). Finally, another node in the tree of life that has gained recent interest is that of the choanoflagellates, a unicellular sister group to metazoans (King, 2004).
Ribosomal phylogenies suggest that choanoflagellates are the most likely unicellular lineage to have shared common ancestry with the multicellular animals (Medina et al., 2003), but there are a few other unicellular protists that also fall out in this part of the tree in other gene phylogenies (Ruiz-Trillo et al., 2004). A choanoflagellate genome project is now in progress, and multiple EST initiatives for unicellular opisthokont protists are also in place.
In summary, postgenomic research on the metazoans is advancing rapidly because of the large number of model organisms, e.g., C. elegans (nematode), Drosophila melanogaster (fruitfly), Danio rerio (zebrafish), Mus musculus (mouse), Rattus norvegicus (rat), and Homo sapiens (human). On the other hand, sequencing metazoan genomes is a major technical challenge, because of higher level of complexity associated with multicellularity and tissue compartmentalization. These challenges are giving a leading role to the yeast and other unicellular systems described in the next section.
Fungi
The initial driving force behind the choice of genome projects in fungi was the prime status of yeast (Saccharomyces cerevisiae) as a model organism. Additionally, the relatively small genome size in other fungi has facilitated the explosion of numerous large scale sequencing projects (www.broad.mit.edu/annotation/fungi/fgi). Consensus phylogenies of fungi place the Chitridiomycota as the most basal lineage, followed by the Zygomycota, with Ascomycota and Basidiomycota as sister crown clades (Berbee and Taylor, 1993; Hedges, 2002). Ribosomal phylogenies suggest that the Nuclearid amoeba are the likely unicellular sister group to Fungi (Amaral Zettler et al., 2001; Medina et al., 2003).
After the completion of S. cerevisiae, subsequent fungal genome projects were chosen within the Ascomycota (Fig. 17.1C) mainly based on phylogenetic proximity (within the Hemiascomycetes) (Dietrich et al., 2004; Dujon et al., 2004; Kellis et al., 2004), although now more distantly related taxa including additional model organisms such as Neurospora crassa and Aspergillus nidulans have also been sequenced. Basidiomycete genomes have been sequenced (Martinez et al., 2004) or are in progress as well (www.broad.mit.edu/annotation/fungi/fgi). The combination of both S. cerevisiae as the best characterized unicellular eukaryote and the thorough comparative genomics allowed by the numerous fungal genome projects have made this branch of the eukaryotic tree an ideal target for validation and improvement of postgenomic approaches.
Plantae
Most of the species diversity of plants is represented in the crown group, the angiosperms, which encompasses the Monocots and the Eudicots. Consensus phylogenies place paraphyletic gymnosperms basal to angiosperms and ferns as a sister group to this clade (Pryer et al., 2002) (Fig. 17.1D). The placement of some of the basal groups in the Embryophyta (hornworts, liverworts, and mosses) is still unresolved, although lycophytes are now considered the sister group to the clade containing ferns, gymnosperms, and angiosperms (Hedges, 2002; Pryer et al., 2002) (Fig. 17.1D). Finally, multiple sources of evidence point to the green algae as the unicellular sister group to plants (reviewed in Archibald and Keeling, 2002) (Fig. 17.1D).
Genome projects for green plants have been hampered by the larger genome sizes of most members of this group. Nonetheless, the first draft Plantae genome published was from Arabidopsis thaliana, a flowering plant model organism (Arabidopsis Genome Initiative, 2000). Genome drafts of two different rice strains (Oryza sativa) have been recently published (Goff et al., 2002; Yu et al., 2002). This effort is now complemented by the completion of a unicellular alga (Chlamydomonas reinhardtii), the poplar tree (Populus trichocarpa), and partial genome data from corn (Zea mays), whereas two basal lineages, the moss Physcomitrella patens and the lycophyte Selaginella moellendorffii, will be sequenced this year (www.jgi.doe.gov/sequencing/cspseqplans.html). Thus, although more sparse than for metazoans and fungi, the Plantae branch of the eukaryotic tree is rapidly expanding in terms of genomic data. Agricultural interests will likely drive future choice of Plantae genomes to some degree, but decisions will also be influenced by phylogenetic implications as reflected in the recent choice of the P. patens and S. moellendorffii for genome sequencing.
EVOLUTIONARY SYSTEMS BIOLOGY
With whole-genome data allowing reconstruction of more robust phylogenies for the major eukaryotic groups, new biological questions can now be addressed. Genomic and postgenomic data offer a new “global” view of the function of living systems across the tree of life. These new data suggest that biological systems (e.g., a cell) are composed of discrete “modules” of interacting components with different functions, and in turn these modules form biological networks that carry out the myriad functions of living systems (Hartwell et al., 1999). Multiple metabolic and regulatory networks are now being characterized in diverse organisms for which reasonably annotated genomes are available. Metabolites, be-
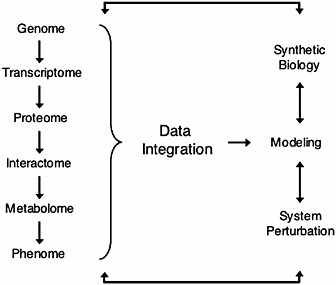
FIGURE 17.2 Overview of systems biology. Hierarchical information from the genome (DNA) to the phenome (phenotype) is integrated to predict mathematical models. These models can then be tested by “synthetic biology” (de novo design of biological modules) and/or by system perturbations that generate a cycle of hypothesis-driven science (Ge et al., 2003; Ideker et al., 2001; Kitano, 2002).
ing the end products of cellular regulatory networks, are one of the most directly accessible windows into the cell’s dynamic phenotype (Fiehn, 2002).
Systems biology is a rapidly expanding field that integrates widely diverse areas of science such as physics, engineering, computer science, mathematics, and biology, toward the goal of elucidating the hierarchy of metabolic and regulatory systems in the cell, and ultimately leading to a predictive understanding of the cellular response to perturbations (Ideker et al., 2001; Kitano, 2002) (Fig. 17.2). As the theoretical and experimental tools of systems biology rapidly advance, multiple fields are embracing systems biology approaches as a mainstream method of research. Because postgenomics research is taking place throughout the tree of life, comparative approaches are a way to combine data from many organisms to understand the evolution and function of biological systems from the gene to the organismal level. Therefore, systems biology can build on decades of theoretical work in evolutionary biology, and at the same time evolutionary biology can use systems approaches to go in new uncharted directions. For instance, although comparative genomics has benefited from a long tradition of theoretical work by molecular evolutionists (Wolfe and Li, 2003), new datasets being provided by systems biology are allow-
ing theoreticians new ways to study evolutionary processes (Barabasi and Oltvai, 2004).
Comparative studies can give insight into even the highest-level principles of life. For example, revolutionary findings in network theory have in part come from genomic data from a wide range of organisms, leading scientists to propose laws that seem to govern biological networks (Jeong et al., 2000; Ravasz et al., 2002). Different types of cellular networks (e.g., protein interaction and metabolic networks) seem to share properties with other complex abiotic networks such as their “scale-free” nature and “small world” organization. In scale-free networks, a few nodes (hubs) have the largest number of connections to other nodes, whereas most of the nodes have just a few connections. This property is reflected in a power-law distribution. In practical terms, this relationship means that, in a protein interaction network, most proteins interact with a couple of others whereas a few proteins (hubs) interact with a large number, and that, in a metabolic network, a few molecules (hubs) participate in most reactions whereas the rest participate in one or two. The “small world” concept refers to the property of such spoke-and-hub networks that there is a small path length between nodes, just as in modern air travel where only a few flights connect any two cities in the world. This property means that a path of just a few interactions or reactions will connect almost any pair of molecules in the cell (Barabasi and Oltvai, 2004).
Additional levels in the hierarchy of biological networks and the interactions between them are now being characterized that will allow for integration of data and new theoretical predictions (Ge et al., 2003). Processes widely studied by evolutionary biologists such as selection, gene duplication, and neutral evolution are being examined in the context of network models as opposed to at the level of individual genes or molecules (Hahn et al., 2004; van Noort et al., 2004; Wagner, 2003a,b; Wuchty, 2004).
EVOLUTION OF BIOLOGICAL NETWORKS
Transcriptional Networks
High-throughput global gene expression approaches such as EST sequencing and microarrays are now common practice for functional assessment of the genome. The extensive microarray gene expression datasets available for model and non-model organisms are starting to be incorporated into a comparative approach to study transcriptome evolution at multiple levels of divergence. At lower levels of divergence, studies in organisms including fish (Olesiak et al., 2002), fruitfly (Meiklejohn et al., 2003; Ranz et al., 2003), and yeast (Townsend et al., 2003) have now
shown that extensive variation exists in the transcriptome in natural populations and that this variation is likely to be an important factor in organismal evolution. Transcriptome comparisons across several primate and mouse species, however, suggest that the majority of gene expression differences within and between species evolve in a selectively neutral or nearly neutral fashion (Khaitovich et al., 2004). At intermediate levels of divergence, less information is available at present due to lack of genomic data. Although analytically challenging, the use of gene expression profiling by heterologous hybridization to a single species cDNA microarray is starting to be explored, potentially opening the door to comparative analyses of taxa as divergent as 200 mega-annum (Ma) (Renn et al., 2004). This application would be of great significance for the comparative study of non-model organisms that are only distantly related to an already sequenced species. At deep levels of divergence, coexpression of large aggregates of functionally related genes seems to be conserved across evolution. Two recent comparisons of the transcriptomes of several of the model organisms [S. cerevisiae, D. melanogaster, C. elegans, and H. sapiens in one case (Stuart et al., 2003), and these four plus A. thaliana and E. coli in the second case (Bergmann et al., 2004)] support the hypothesis that coexpression networks can be split into multiple components enriched for genes involved in similar functional processes. Some of these identified components can be unique to a certain clade, such as the signaling pathway and neuronal function components present only in metazoans in the four-species comparison (Stuart et al., 2003). These cross-species comparisons promise to provide more information about coexpression network evolution as the transcriptomes of additional diverse lineages becomes available (Zhou and Gibson, 2004).
Central to postgenomic analysis is the accuracy of genome annotation. The degree of accuracy in which genomes are annotated is affected by the quality of sequence assembly, gene prediction, and functional annotation by both bioinformatics and experimental data. This relationship is particularly critical in genome projects of non-model organisms where little genetic work has been performed in the past.All these factors, combined with the lack of network information outside the model organisms, point to the tradeoff between a comprehensive systems analysis of a particular network within a well-studied organism, versus the historical perspective introduced by evolutionary conservation or divergence of systems through time in phylogenetic comparisons. Therefore, although only partial inference is possible at present, studies have already shown that the comparative approach to coexpression not only is giving insight into the universal rules that govern biological systems but also has practical implications by helping improve functional annotations of both model and non-model organisms (Bergmann et al., 2004; Stuart et al., 2003). Be-
cause comparative analyses of coexpression data from several model organisms have shown high levels of conservation between such divergent taxa as prokaryotes (E. coli and B. subtilis) (Snel et al., 2004), opisthokont eukaryotes (Stuart et al., 2003), and even prokaryotes and eukaryotes (Bergmann et al., 2004), some efforts are now targeting the coupled evolution of regulatory networks and the transcriptome.
Regulatory Networks
The characterization of the transcriptome is only a fraction of the information needed to understand global cellular processes because gene expression is driven by the spatio-temporal localization of regulatory networks and details of specific protein–DNA and protein–protein interactions. Genomewide efforts to characterize transcriptional regulatory networks have already been fruitful in model organisms like yeast (Lee et al., 2002) and E. coli (Shen-Orr et al., 2002). In multicellular organisms, fractions of the regulatory networks are being characterized for sea urchins (Davidson, 2001), Drosophila (Berman et al., 2004), and mammals (ENCODE Project Consortium, 2004).
Transcription factors are regulatory proteins that influence the expression of specific genes. They work by binding to cis-regulatory elements (short and often degenerate sequence motifs frequently located upstream of genes) where they interact with the transcription apparatus to either enhance or repress gene expression. Even though identifying cisregulatory elements in new genomes is an inherently difficult task due to their short sequence length and as yet unknown syntax, comparative approaches have been helpful. By aligning orthologous regions flanking a gene from multiple species, conserved noncoding sequence motifs can be distinguished. These evolutionary conserved motifs are then hypothesized to be potential functional elements. This method, called phylogenetic footprinting (Tagle et al., 1988), has successfully been used to identify a limited number of regulatory regions in vertebrates (Dermitzakis et al., 2003; Gumucio et al., 1992) and plants (Hong et al., 2003; Kaplinsky et al., 2002). More sophisticated comparative approaches are starting to combine computational prediction and laboratory validation of regulatory networks. Coexpression data and known cisregulatory elements from S. cerevisiae were used in a multispecies comparison of 13 published ascomycete genomes, finding multiple cases of regulatory conservation but also some cases of regulatory diversification (Gasch et al., 2004). It has become apparent, however, that sequence conservation alone will not help identify all cis-regulatory elements by phylogenetic footprinting, and additional data and experimental approaches have to be integrated (Richards et al., 2005).
Gene expression can be regulated not only at transcriptional initiation but also at other levels, such as during mRNA editing, transport, or translation, and characterizing these interactions and their evolution is one of the many future challenges of systems biology (Wei et al., 2004). For example, comparative work on populations of yeast and fruitfly has recently shown that protein–protein interactions are negatively associated with evolutionary variation in gene expression (Lemos et al., 2004). A comparative analysis of the E. coli and yeast regulatory networks has demonstrated that gene duplication has a key role in network evolution both in eukaryotes and prokaryotes (Teichmann and Babu, 2004). Finally, introducing concepts of network dynamics has revealed new topological changes in the regulatory network in yeast (Luscombe et al., 2004), an approach that, incorporated into a comparative framework, will eventually provide answers to the evolution of morphological divergence in multicellular taxa (Howard and Davidson, 2004).
Protein Networks
The proteome for several of the model organisms is now characterized, and this global scale information has been used to predict protein–protein interaction networks (interactomes) for D. melanogaster (Giot et al., 2003), C. elegans (Li et al., 2004), and S. cerevisiae (Uetz et al., 2000). Assuming some degree of evolutionary conservation, these data can also be used to transfer interactome annotations to genomes that have not been characterized experimentally. Comparisons across multiple species have shown conserved protein interactions that allow for initial drafts of protein–protein interaction maps of human (Lehner and Fraser, 2004) and A. thaliana (Yu et al., 2004). When formulating evolutionary hypotheses, however, attention to the phylogenetic relationships is necessary. For example, some of the conclusions from the analysis of the C. elegans interactome (Luscombe et al., 2004) are weakened by the incorrect assumption that plants (A. thaliana) and animals (C. elegans, D. melanogaster) are more closely related to each other than to yeast. Current phylogenies show that multicellularity has occurred independently in metazoans, fungi, and plants (Fig. 17.1A), and that unicellularity in yeasts is a derived rather than ancestral state (Fig. 17.1C).
Metabolic Networks and “Ome” Data Integration
The metabolome is made up of all of the low-molecular weight molecules (metabolites) present in a cell at a particular time point, and their levels can be regarded as the functional response of biological systems to genetic or environmental stimuli (Fiehn, 2002). Challenges faced in the
global study of metabolites, such as their dynamic behavior and chemistry, are being addressed by emerging technologies such as liquid and gas chromatography mass spectrometry and NMR (Stitt and Fernie, 2003). Plant biologists have led in the application of these advances (Oksman-Caldentey et al., 2004), and soon there will likely be large datasets for multiple plant and other eukaryotic species. Although high-throughput metabolome projects are just now being initiated, comparative analysis of 43 known metabolic networks has already shown that they seem to follow a power-law distribution (Barabasi and Oltvai, 2004; Jeong et al., 2000).
The integration of data from the different levels of cellular networks (transcriptome, regulome, interactome, and metabolome) is the next obvious step to identify patterns of network interactions in individual species and in multispecies comparisons (Castrillo and Oliver, 2004; Ge et al., 2003; Papin et al., 2004). This integrative approach has already been fruitful in model organisms such as C. elegans (Walhout et al., 2002) and S. cerevisiae (Ge et al., 2001).
It is clear that producing a large scale comparative systems biology analysis will have to involve the work of many research groups and that many challenges will need to be overcome. For example, rigorous standards will need to be established to facilitate the comparison of results from high-throughput “omic” analyses before we can make conclusive evolutionary inferences (Levesque and Benfey, 2004). A pioneer example is the ENCODE initiative, which aims to identify all functional elements in the human genome by using coordinated computational and experimental efforts in a multispecies framework (ENCODE Project Consortium, 2004).
Although we can already find global patterns of network evolution, in the future we should be able to look at trends and patterns in the evolution of biological systems within phylogenies. For instance, we should be able to look at how many of the biological network similarities are due to homoplasy as opposed to phylogenetic constraints due to common ancestry. Thus, by using the theoretical framework developed for the comparative method, phylogenetic information can allow only for improvement of evolutionary inference at the systems level. Finally, to bring evolutionary systems biology to the highest level of biological organization, ecosystem-level factors have to be taken into consideration. To this end, the use of highthroughput approaches for the study of interactions among organisms and between organisms and their natural environments is engaging the interest of ecologists (Benfey, 2004; Thomas and Klaper, 2004).
HISTORICAL PERSPECTIVE
Darwin’s theory of natural selection and, later on, the integrative nature of the modern synthesis consolidated the study of evolution as a solid discipline to address fundamental questions in biology. The scientific advances that allowed for the discovery of the structure of DNA and the development of molecular biology eventually led to large-scale wholegenome initiatives. This unfolding was a revolutionary moment in the scientific mentality of 20th century researchers, because it generated the integrative approaches of systems biology, which will most likely become the standard of 21st century biology. Organismal biologists have been thinking along these lines for the past few decades, advocating integrative and multidisciplinary approaches to evolutionary questions (Wake, 2003). Thus, bridging knowledge between evolutionary theory and systems biology will be only a natural process. Together, these approaches offer the promise to solve two of the ultimate questions in biology: the function of biological systems and an understanding of the evolution of life’s diversity.
ACKNOWLEDGMENTS
I thank Francisco Ayala, Jody Hey, and Walter Fitch for the invitation to participate in the Mayr colloquium. Pilar Francino and Paramvir Dehal provided thoughtful insight for both the seminar and the manuscript. I also thank Mike Colvin, Benoît Dayrat, Jodi Schwarz, Rick Baker, Kevin Helfenbein, and an anonymous reviewer for helpful comments on previous versions of the manuscript. Benoît Dayrat and Peter Brokstein helped with figure design. I thank Caturro Mejía for introducing me to Mayr’s writings many years ago. This work was performed under the auspices of the U.S. Department of Energy, Office of Biological and Environmental Research, University of California, Lawrence Berkeley National Laboratory under contract DE-AC03-76SF00098. I also acknowledge support by National Science Foundation Grant OCE 0313708.
REFERENCES
Adoutte, A., Balavoine, G., Lartillot, N. & de Rosa, R. (1999) Animal evolution. The end of intermediate taxa? Trends Genet. 15, 104–108.
Amaral Zettler, L. A., Nerad, T. A., O’Kelly, C. J. & Sogin, M. L. (2001) The nucleariid amoebae: More protists at the animal-fungal boundary. J. Eukaryotic Microbiol. 48, 293–297.
Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815.
Archibald, J. M. & Keeling, P. J. (2002) Recycled plastids: a “green movement” in eukaryotic evolution. Trends Genet. 18, 577–584.
Armbrust, E. V., Berges, J. A., Bowler, C., Green, B. R., Martinez, D., Putnam, N. H., Zhou, S., Allen, A. E., Apt, K. E., Bechner, M., et al. (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306, 79–86.
Barabasi, A. L. & Oltvai, Z. N. (2004) Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113.
Benfey, P. N. (2004) Development and ecology in the time of systems biology. Dev. Cell 7, 329–330.
Berbee, M. L. & Taylor, J. W. (1993) Dating the evolutionary radiations of the true fungi. Can. J. Bot. 71, 1114–1127.
Bergmann, S., Ihmels, J. & Barkai, N. (2004) Similarities and differences in genome-wide expression data of six organisms. PLoS Biol. 2, E9.
Berman, B. P., Pfeiffer, B. D., Laverty, T. R., Salzberg, S. L., Rubin, G. M., Eisen, M. B. & Celniker, S. E. (2004) Computational identification of developmental enhancers: conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 5, R61.
Bhattacharya, D., Yoon, H. S. & Hackett, J. D. (2004) Photosynthetic eukaryotes unite: endosymbiosis connects the dots. BioEssays 26, 50–60.
Castrillo, J. I. & Oliver, S. G. (2004) Yeast as a touchstone in post-genomic research: strategies for integrative analysis in functional genomics. J. Biochem. Mol. Biol. 37, 93–106.
Davidson, E. H. (2001) Genomic Regulatory Systems: Development and Evolution (Academic, San Diego).
Dermitzakis, E. T., Reymond, A., Scamuffa, N., Ucla, C., Kirkness, E., Rossier, C. & Antonarakis, S. E. (2003) Evolutionary discrimination of mammalian conserved nongenic sequences (CNGs). Science 302, 1033–1035.
Dietrich, F. S., Voegeli, S., Brachat, S., Lerch, A., Gates, K., Steiner, S., Mohr, C., Pohlmann, R., Luedi, P., Choi, S., et al. (2004) The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304, 304–307.
Dopazo, H., Santoyo, J. & Dopazo, J. (2004) Phylogenomics and the number of characters required for obtaining an accurate phylogeny of eukaryote model species. Bioinformatics 20, Suppl. 1, I116–I121.
Dujon, B., Sherman, D., Fischer, G., Durrens, P., Casaregola, S., Lafontaine, I., De Montigny, J., Marck, C., Neuveglise, C., Talla, E., et al. (2004) Genome evolution in yeasts. Nature 430, 35–44.
ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306, 636–640.
Fiehn, O. (2002) Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 48, 155–171.
Gardner, M. J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R. W., Carlton, J. M., Pain, A., Nelson, K. E., Bowman, S., et al. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511.
Gasch, A. P., Moses, A. M., Chiang, D. Y., Fraser, H. B., Berardini, M. & Eisen, M. B. (2004) Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biol. 2, e398.
Ge, H., Liu, Z., Church, G. M. & Vidal, M. (2001) Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat. Genet. 29, 482–486.
Ge, H., Walhout, A. J. & Vidal, M. (2003) Integrating “omic” information: a bridge between genomics and systems biology. Trends Genet. 19, 551–560.
Giot, L., Bader, J. S., Brouwer, C., Chaudhuri, A., Kuang, B., Li, Y., Hao, Y. L., Ooi, C. E., Godwin, B., Vitols, E., et al. (2003) A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736.
Goff, S. A., Ricke, D., Lan, T. H., Presting, G., Wang, R., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100.
Gumucio, D. L., Heilstedt-Williamson, H., Gray, T. A., Tarle, S. A., Shelton, D. A., Tagle, D. A., Slightom, J. L., Goodman, M. & Collins, F. S. (1992) Phylogenetic footprinting reveals a nuclear protein which binds to silencer sequences in the human gamma and epsilon globin genes. Mol. Cell. Biol. 12, 4919–4929.
Hahn, M. W., Conant, G. C. & Wagner, A. (2004) Molecular evolution in large genetic networks: Does connectivity equal constraint? J. Mol. Evol. 58, 203–211.
Hartwell, L. H., Hopfield, J. J., Leibler, S. & Murray, A. W. (1999) From molecular to modular cell biology. Nature 402, C47–C52.
Hedges, S. B. (2002) The origin and evolution of model organisms. Nat. Rev. Genet. 3, 838–849.
Hong, R. L., Hamaguchi, L., Busch, M. A. & Weigel, D. (2003) Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell 15, 1296–1309.
Hood, L. (2003) Systems biology: Integrating technology, biology, and computation. Mech. Ageing Dev. 124, 9–16.
Howard, M. L. & Davidson, E. H. (2004) cis-Regulatory control circuits in development. Dev. Biol. 271, 109–118.
Ideker, T., Galitski, T. & Hood, L. (2001) A new approach to decoding life: systems biology. Annu. Rev. Genomics Hum. Genet. 2, 343–372.
Jeong, H., Tombor, B., Albert, R., Oltvai, Z. N. & Barabasi, A. L. (2000) The large-scale organization of metabolic networks. Nature 407, 651–654.
Kaplinsky, N. J., Braun, D. M., Penterman, J., Goff, S. A. & Freeling, M. (2002) Utility and distribution of conserved noncoding sequences in the grasses. Proc. Natl. Acad. Sci. USA 99, 6147–6151.
Kellis, M., Birren, B. W. & Lander, E. S. (2004) Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428, 617–624.
Khaitovich, P., Weiss, G., Lachmann, M., Hellmann, I., Enard, W., Muetzel, B., Wirkner, U., Ansorge, W. & Paabo, S. (2004) A neutral model of transcriptome evolution. PLoS Biol. 2, E132.
King, N. (2004) The unicellular ancestry of animal development. Dev. Cell 7, 313–325.
Kitano, H. (2002) Systems biology: A brief overview. Science 295, 1662–1664.
Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., Hannett, N. M., Harbison, C. T., Thompson, C. M., Simon, I., et al. (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804.
Lehner, B. & Fraser, A. G. (2004) A first-draft human protein-interaction map. Genome Biol. 5, R63.
Lemos, B., Meiklejohn, C. D. & Hartl, D. L. (2004) Regulatory evolution across the protein interaction network. Nat. Genet. 36, 1059–1060.
Levesque, M. P. & Benfey, P. N. (2004) Systems biology. Curr. Biol. 14, R179–80.
Li, S., Armstrong, C. M., Bertin, N., Ge, H., Milstein, S., Boxem, M., Vidalain, P. O., Han, J. D., Chesneau, A., Hao, T., et al. (2004) A map of the interactome network of the metazoan C. elegans. Science 303, 540–543.
Luscombe, N. M., Babu, M. M., Yu, H., Snyder, M., Teichmann, S. A. & Gerstein, M. (2004) Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431, 308–312.
Martinez, D., Larrondo, L. F., Putnam, N., Gelpke, M. D., Huang, K., Chapman, J., Helfenbein, K. G., Ramaiya, P., Detter, J. C., Larimer, F., et al. (2004) Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 22, 695–700.
Medina, M., Collins, A. G., Silberman, J. D. & Sogin, M. L. (2001) Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA. Proc. Natl. Acad. Sci. USA 98, 9707–9712.
Medina, M., Collins, A. G., Taylor, J. W., Valentine, J. W., Lipps, J. H., Amaral Zettler, L. A. & Sogin, M. L. (2003) Phylogeny of Opisthokonta and the evolution of multicellularity and complexity in Fungi and Metazoa. Int. J. Astrobiol. 2, 203–211.
Meiklejohn, C. D., Parsch, J., Ranz, J. M. & Hartl, D. L. (2003) Rapid evolution of male-biased gene expression in Drosophila. Proc. Natl. Acad. Sci. USA 100, 9894–9899.
Oksman-Caldentey, K. M., Inze, D. & Oresic, M. (2004) Connecting genes to metabolites by a systems biology approach. Proc. Natl. Acad. Sci. USA 101, 9949–9950.
Olesiak, M. J., Churcill, G. A. & Crawford, D. L. (2002) Variation in gene expression within and among natural populations. Nat. Genet. 32, 261–266.
Papin, J. A., Reed, J. L. & Palsson, B. O. (2004) Hierarchical thinking in network biology: the unbiased modularization of biochemical networks. Trends Biochem. Sci. 29, 641–647.
Pryer, K. M., Schneider, H., Zimmer, E. A. & Ann Banks, J. (2002) Deciding among green plants for whole genome studies. Trends Plant Sci. 7, 550–554.
Ranz, J. M., Castillo-Davis, C. I., Meiklejohn, C. D. & Hartl, D. L. (2003) Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300, 1742–1745.
Ravasz, E., Somera, A. L., Mongru, D. A., Oltvai, Z. N. & Barabasi, A. L. (2002) Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555.
Renn, S. C., Aubin-Horth, N. & Hofmann, H. A. (2004) Biologically meaningful expression profiling across species using heterologous hybridization to a cDNA microarray. BMC Genomics 5, 42.
Richards, S., Liu, Y., Bettencourt, B. R., Hradecky, P., Letovsky, S., Nielsen, R., Thornton, K., Hubisz, M. J., Chen, R., Meisel, R. P., et al. (2005) Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 15, 1–18.
Ruiz-Trillo, I., Inagaki, Y., Davis, L. A., Sperstad, S., Landfald, B. & Roger, A. J. (2004) Capsaspora owczarzaki is an independent opisthokont lineage. Curr. Biol. 14, R946–R947.
Ruiz-Trillo, I., Paps, J., Loukota, M., Ribera, C., Jondelius, U., Baguña, J. & Ruitort, M. (2002) A phylogenetic analysis of myosin heavy chain type II sequences corroborates that Acoela and Nemertodermatida are basal bilaterians. Proc. Natl. Acad. Sci. USA 99, 11246–11251.
Shen-Orr, S. S., Milo, R., Mangan, S. & Alon, U. (2002) Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 31, 64–68.
Simpson, A. G. & Roger, A. J. (2004) The real “kingdoms” of eukaryotes. Curr. Biol. 14, R693–R696.
Snel, B., van Noort, V. & Huynen, M. A. (2004) Gene co-regulation is highly conserved in the evolution of eukaryotes and prokaryotes. Nucleic Acids Res. 32, 4725–4731.
Stechmann, A. & Cavalier-Smith, T. (2002) Rooting the eukaryote tree by using a derived gene fusion. Science 297, 89–91.
Stechmann, A. & Cavalier-Smith, T. (2003) The root of the eukaryote tree pinpointed. Curr. Biol. 13, R665–R666.
Stitt, M. & Fernie, A. R. (2003) From measurements of metabolites to metabolomics: an “on the fly” perspective illustrated by recent studies of carbon-nitrogen interactions . Curr. Opin. Biotechnol. 14, 136–144.
Stuart, J. M., Segal, E., Koller, D. & Kim, S. K. (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255.
Tagle, D. A., Koop, B. F., Goodman, M., Slightom, J. L., Hess, D. L. & Jones, R. T. (1988) Embryonic epsilon and gamma globin genes of a prosimian primate (Galago crassicaudatus). Nucleotide and amino acid sequences, developmental regulation and phylogenetic footprints. J. Mol. Biol. 203, 439–455.
Teichmann, S. A. & Babu, M. M. (2004) Gene regulatory network growth by duplication. Nat. Genet. 36, 492–496.
Thomas, M. A. & Klaper, R. (2004) Genomics for the ecological toolbox. Trends Ecol. Evol. 19, 439–445.
Townsend, J. P., Cavalieri, D. & Hartl, D. L. (2003) Population genetic variation in genomewide gene expression. Mol. Biol. Evol. 20, 955–963.
Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627.
van Noort, V., Snel, B. & Huynen, M. A. (2004) The yeast coexpression network has a smallworld, scale-free architecture and can be explained by a simple model. EMBO Rep. 5, 280–284.
Wagner, A. (2003b) How the global structure of protein interaction networks evolves. Proc. R. Soc. London Ser. B Biol. Sci. 270, 457–466.
Wagner, A. (September 30, 2003a) Does selection mold molecular networks? Sci. STKE, 10.1126/stke.2003.202.pe41.
Wake, M. L. (2003) What is “Integrative Biology”? Integr. Comp. Biol. 43, 239–241.
Walhout, A. J., Reboul, J., Shtanko, O., Bertin, N., Vaglio, P., Ge, H., Lee, H., Doucette-Stamm, L., Gunsalus, K. C., Schetter, A. J., et al. (2002) Integrating interactome, phenome, and transcriptome mapping data for the C. elegans germline. Curr. Biol. 12, 1952–1958.
Waugh, M., Hraber, P., Weller, J., Wu, Y., Chen, G., Inman, J., Kiphart, D. & Sobral, B. (2000) The Phytophthora genome initiative database: informatics and analysis for distributed pathogenomic research. Nucleic Acids Res. 28, 87–90.
Wei, G. H., Liu, D. P. & Liang, C. C. (2004) Charting gene regulatory networks: strategies, challenges and perspectives. Biochem. J. 381, 1–12.
Wolf, Y. I., Rogozin, I. B. & Koonin, E. V. (2004) Coelomata and not Ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Res. 14, 29–36.
Wolfe, K. H. & Li, W. H. (2003) Molecular evolution meets the genomics revolution. Nat. Genet. 33, Suppl., 255–265.
Wuchty, S. (2004) Evolution and topology in the yeast protein interaction network. Genome Res. 14, 1310–1314.
Yu, J., Hu, S., Wang, J., Wong, G. K., Li, S., Liu, B., Deng, Y., Dai, L., Zhou, Y., Zhang, X., et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296, 79–92.
Yu, H., Luscombe, N. M., Lu, H. X., Zhu, X., Xia, Y., Han, J. D., Bertin, N., Chung, S., Vidal, M. & Gerstein, M. (2004) Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 14, 1107–1118.
Zhou, X. J. & Gibson, G. (2004) Cross-species comparison of genome-wide expression patterns. Genome Biol. 5, 232.



















