
4
Mercury
ATMOSPHERIC MERCURY PRIMER
The element mercury (Hg) is a unique metal that is liquid at ambient conditions, and easily volatilized to the atmosphere, where it is distributed on a global scale. This environmental contaminant emitted from any source has the potential for long-range transport, interaction with and assimilation by terrestrial and aquatic surfaces, and a ubiquitous presence in all environmental media. The dominant form of Hg in the atmosphere is elemental (Hg (0)) and concentrations are typically in the range of 1 to 2 ng m−3. Other forms, including divalent Hg (II) and monovalent Hg (I) gaseous compounds and particulate bound Hg (Hgp), are usually less than 5 percent of the total atmospheric burden (Schroeder and Munthe, 1998; Valente et al., 2007). These small concentrations make measurement of atmospheric Hg challenging. Gaseous divalent forms referred to here as reactive gaseous mercury (RGM) are thought to include compounds such as HgCl2, Hg (OH)2, HgBr2, and HgO (Lin and Pehkonen, 1999). RGM may be a primary or secondary air pollutant. In cloud water, Hg can be dissolved as Hg (0) or Hg (II) (Lin and Pehkonen, 1999).
The major forms that concern human exposure are Hg (0), and compunds containing methyl mercury (methyl Hg). These have different health effects and exposure pathways with Hg (0) being most harmful when inhaled and methyl Hg when ingested. Hg (0) affects the nervous system, kidneys, and lungs (Clarkson, 2002); methyl Hg is a neurotoxin, teratogen, and linked with cardiovascular problems (Clarkson, 2002; Mozaffarian and Rimm, 2006). Methyl Hg is produced in ecosystems from inorganic
mercury derived from the atmosphere or terrestrial landscape (Munthe et al., 2007). Although this form has been measured in rain, the concentrations are extremely low, and the atmosphere is not thought to be a direct source to ecosystems (Sakata and Marumoto, 2005; Hammerschmidt et al., 2007).
Hg is released to the atmosphere by both natural and anthropogenic sources. Natural sources emit almost exclusively Hg (0) (Bagnato et al., 2007; Gustin et al., 2008) ; anthropogenic sources emit varying combinations of Hg (0), RGM, and Hgp (Pacyna et al., 2003b, 2006a). Atmospheric RGM may also be generated by reactions of Hg (0) with oxidants such as O3, OH, and reactive halogens such as Cl, Cl2, Br, and BrO (Lin et al., 2006; Hynes et al., 2008; Ariya et al., 2009). These reactions, which may occur in the surface boundary layer, the free troposphere, and stratosphere, and are poorly understood.
To evaluate the potential for long-range transport of Hg once emitted from a natural or anthropogenic source, the atmospheric lifetime (mean time in the air before being removed) of the different forms needs to be considered. Gaseous Hg (0) has an estimated atmospheric lifetime of months to more than a year (Lindberg et al., 2007), thus a molecule of Hg in a fish may have its origin from a source far removed. Episodic events of elevated air Hg concentrations recorded at the Mt. Bachelor Observatory in central Oregon (Jaffe et al., 2005a, Figure 1.3; Weiss-Penzias et al., 2006) and in aircraft measurements (Friedli et al., 2003; Swartzendruber et al., 2008) have been linked to air masses passing over Asia. The form of Hg transported in these events is predominantly Hg (0). Elevated air Hg concentrations have also been measured in plumes associated with fires and industrial sources (Edgerton et al., 2006; Ebinghaus, 2008; Finley et al., 2009). In contrast RGM and Hgp are water soluble and have high deposition velocities, resulting in efficient removal from the atmosphere by dry and wet deposition (Schroeder and Munthe, 1998).
Field and laboratory data have shown that Hg (0) is recycled between the air and terrestrial and aquatic surfaces, and this can occur over the course of a day (Gustin et al., 2008). Limited field studies using stable isotopes and laboratory experiments have indicated that 5 to 40 percent of Hg added to an ecosystem as HgCl2 is released in the short term (Hintelmann and Evans, 1997; Hintelmann et al., 2001, 2002; Lindberg et al., 2002; Amyot et al., 2004; Ericksen et al., 2005; Xin et al., 2007). This recycling of Hg between the air and surfaces results in long-term availability of Hg to ecosystems and complicates source attribution.
Human and Ecosystem Health The major human and ecosystem health threat associated with long-range transport of Hg arises from the fact that Hg in the air is deposited to watersheds where it may enter aquatic systems
and be converted to methyl Hg (Munthe et al., 2007). Humans are exposed to this neurotoxin primarily by fish consumption (~ 95 percent of the Hg in fish muscle tissue is methyl Hg [Bloom, 1992]). Because Hg is globally distributed, fish in remote regions may be impacted by any source. In the United States, 48 states have Hg consumption advisories, (EPA, 2006b) and many are associated with water bodies located in areas with no apparent land-based Hg contamination or anthropogenic Hg source. Consumption advisories are set by the states; some states apply the 0.3 μg g−1 (0.3 ppm) EPA limit while others apply the 1 ppm FDA limit.
Since methyl Hg is bioconcentrated in organisms and biomagnified in aquatic food webs, large fish and those with high trophic stature (i.e., bass, walleye, and perch) tend to have higher concentrations. Thus, marine and freshwater advisories often target specific fish species and are size based. Of the marine fish, shark, tuna, swordfish, and tilefish have been shown to have elevated concentrations of Hg (Burger and Gochfeld, 2004; Chen et al., 2008). High blood levels of Hg have been reported for those eating large quantities of marine fish high in the food web (Hightower and Moore, 2003).
An NRC committee (NRC, 2000) concluded that the risk of adverse effects from current methyl Hg exposures to the majority of the U.S. population is relatively low. However, since the developing human nervous system is sensitive to methyl Hg, young children and children of women who consume fish during pregnancy are at risk (IPCS-WHO, 1990; NRC, 2000; Clarkson et al., 2003). The NRC committee recommended, as have others (Sakamoto et al., 2005; Mozaffarian and Rimm, 2006), that since fish are an important food resource benefiting human health, the long-term goal should be reduction of methyl Hg concentrations in fish rather than replacement of fish in the diet by other foods.
Humans are not the only organism at risk due to Hg exposures. Scheuhammer et al. (2007) concluded that dietary methyl Hg exposures at environmentally relevant concentrations may cause behavioral, neurochemical, hormonal, and reproductive effects in wild animals. Similar results were reported for Hg-exposed fish in national parks of the western United States (Landers et al., 2008). Other research has suggested that marine predators such as sharks, seabirds, seals and walruses, may also be at risk (Kemper et al., 1994; Braune et al., 2006; García-Hernández et al., 2007).
Policy and Regulatory Context Current U.S. regulations for Hg are limited to the listing as a hazardous air pollutant in the 1990 Clean Air Act amendments. In March 2005, the EPA tried to remove electricity-generating utilities from the listing, instead allowing for development of a cap-and-trade program called the Clean Air Mercury Rule (CAMR). This Rule was
vacated by the DC Circuit Court in February 2008 and the future of this legislation is unclear (Milford and Pienciak, 2009).
Discussions are ongoing within United Nations Environmental Program (UNEP), the European Union, and U.S.-Mexico-Canada about how to deal with this transboundary pollutant. Because Hg is a global pollutant, reducing Hg deposition to terrestrial and aquatic ecosystems ultimately requires an international protocol. At its session in February 2007 the UNEP Governing Council concluded that efforts to reduce risks from mercury were not sufficient to address the global challenges posed by mercury; and they concluded that further long-term international action is required (http://www.chem.unep.ch/).
ATMOSPHERIC MERCURY CONCENTRATIONS
Atmospheric Hg concentrations measured in remote locations are relatively consistent over space and time with values of 1.5 ± 0.2 ng/m3 reported for the Northern Hemisphere and ~ 1.2 ng/m3 for the Southern Hemisphere (Slemr et al., 2003; Lindberg et al., 2007). Fairly constant concentrations have been reported for the past 10 to 15 years, based on data collected during ship cruises and at Mace Head, Ireland. The ubiquitous nature of Hg in the atmosphere results in this reservoir being described as a “global pool” that is a mixture of Hg emitted from all sources (Lindberg et al., 2007). Higher atmospheric Hg concentrations are reported for areas directly affected by anthropogenic and natural sources (Ebinghaus, 2008 and references therein). Reductions in air Hg concentrations on a regional scale have been reported for areas where anthropogenic emissions have been reduced (Kellerhals et al., 2003; Wängberg et al., 2007). However there are limited long-term air concentration datasets and significant gaps in the spatial and temporal coverage (Keeler et al., 2009). Since Hg is a global pollutant, understanding Hg is critical for assessing potential sources to the United States.
RGM and Hgp concentrations in air are typically in the tens of pg m−3. Long-term trend data are not available for these forms, because automated methods to measure these forms have only been available during the past ten or so years, and regular monitoring has been done at only a few locations. In remote continental areas RGM concentrations often peak during the day and decline to zero at night (see references listed in Ebinghaus, 2008). RGM and Hgp are emitted directly by specific point sources. RGM may also be produced by oxidation of Hg (0) to RGM. While the exact nature of this oxidation is not understood, there is evidence that this process occurs in the Arctic boundary layer (Dommergue et al., 2008; Steffen et al., 2008), marine boundary layer (Laurier et al., 2003; Hedgecock and Pirrone, 2004; Sprovieri et al., 2008), surface boundary layer (Weiss-Penzias
et al., 2003; Liu et al., 2007; Peterson and Gustin, 2008), free troposphere (Swartzendruber et al., 2006; Sillman et al., 2007), and possibly in the stratosphere (Talbot et al., 2007; Slemr et al., 2009). A few studies have reported elevated RGM at high altitudes using mountain top and aircraft sampling platforms (Landis et al., 2005; Swartzendruber et al., 2006; Sillman et al., 2007). Subsidence of the air from the free troposphere and convective mixing may result in delivery of Hg, and specifically RGM, from the global pool to the surface. Recent field and modeling work has suggested that this is important in Nevada (Selin et al., 2007; Weiss-Penzias et al., 2009) and in the southeastern United States (Guentzel et al., 2001; Selin et al., 2008). The process of oxidation of Hg (0) in the free troposphere and subsequent transport to the surface could be an important means by which Hg is deposited from the global pool.
SOURCES AND SINKS OF ATMOSPHERIC MERCURY
A recent UNEP initiative (Pirrone and Mason, 2009a) estimated current annual emissions of Hg to the atmosphere as ~ 8000 Mg/yr, with approximately one-third directly derived from anthropogenic sources. Natural sources are thought to contribute an additional one-third of the total surface-to-air flux each year. The remainder of the annual Hg input to the atmosphere is that part previously emitted from natural and anthropogenic sources designated as “legacy Hg” but still being cycled between surface reservoirs and the atmosphere. Because Hg is not degraded, once emitted from a source it may become a component of the legacy pool until some process (geophysical, geochemical, biological) results in long-term removal and sequestration. Figure 4.1 illustrates the important fluxes and reservoirs associated with the Hg biogeochemical cycle. The atmosphere itself is a small reservoir.
The major anthropogenic sources of atmospheric Hg are coal combustion, waste incineration, chlor-alkali plants, and metal processing (Table 4.1). However emission inventories are evolving with new sources being identified and estimates for specific sources and developing countries having a high degree of uncertainty. The recent UNEP report and resulting book (Pirrone and Mason, 2009a,b) provide a summary of the current estimates of contributions from anthropogenic sources in developed and developing countries. Inventories for latter are incomplete and have uncertainties on the order of 35 to 50 percent (Lindberg et al., 2007). Based on the UNEP report, China and India are estimated to contribute 42 and 20 percent, respectively, of current global anthropogenic emissions, while North America contributes 9 percent (Table 4.1). As developing countries grow economically their fossil fuel combustion is projected to increase. Wu et al. (2006) estimated an increase in China’s Hg emissions of 3 percent
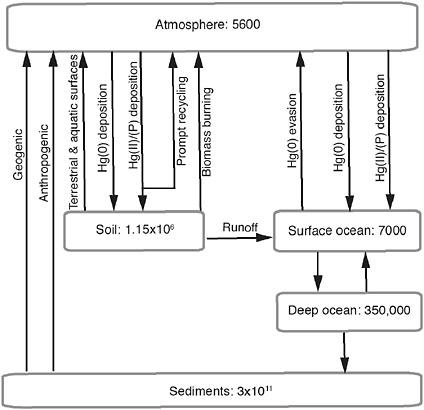
FIGURE 4.1 Major reservoirs and fluxes in the global Hg biogeochemical cycle, adapted from Selin et al. (2008). See text for discussion of values applied, as well as Pirrone and Mason (2009a,b) for additional information. Mercury reservoirs are shown in closed boxes in units of Mg.
per year from 1995 to 2003, primarily due to increasing coal combustion. Streets et al. (2009) suggested that the change of global anthropogenic Hg emissions may range anywhere from −4 to +96 percent by 2050, depending on the future of coal-fired utility releases. Estimated individual source contributions also have significant uncertainty on the order of ±25 percent for stationary fossil fuel combustion, ±30 percent for non ferrous metal production and industrial sources, and ±50 percent for gold production (Pacyna et al., 2006a; Lindberg et al., 2007).
Geogenic emissions of Hg to the atmosphere occur from areas with ongoing volcanic and geothermal activity and from substrates with Hg concentrations enriched by past geologic activity above the estimated natural background of < 100 ppb (Wedepohl, 1995). Estimated emissions of Hg from naturally enriched substrates range from 500 to 1500 Mg/yr (Gustin
TABLE 4.1 Global Anthropogenic Emissions by Countries and Regions
et al., 2008). The range in estimated releases from volcanic systems is large (1 to ~ 700 Mg/yr, Gustin and Lindberg, 2005). The only global estimate for geothermal emissions indicates a source of 60 Mg/yr (Varekamp and Buseck, 1984). Both volcanic and geothermal emissions will vary temporally depending on activity.
Wildfires (biomass burning) contribute Hg to the air, with estimates of global emissions ranging from 200 to 1000 Mg/yr (Brunke et al., 2001; Friedli et al., 2001; Weiss-Penzias et al., 2007). While most of the release from wildfires is in the form of Hg (0), particulate-bound Hg may be a component of the Hg released (Wiedinmyer and Friedli, 2007; Finley et al., 2009). Mercury derived from this source is largely legacy Hg (Wiedinmyer and Friedli, 2007; Gustin et al., 2008). Some component of the Hg emitted during fires may be derived from soils; the importance is unclear and most likely related to fire intensity (Engle et al., 2006; Biswas et al., 2007).
Hg emitted from terrestrial and aquatic surfaces (not enriched by geologic processes) and the oceans is natural and anthropogenic in origin. Most of the Hg emitted from the former can be accounted for by inputs from the atmosphere (Gustin et al., 2008; Hartman et al., 2009). All forms of atmospheric Hg may be deposited to soils; the potential for long-term sequestration after deposition is not clear. Recent work has suggested that terrestrial ecosystems are a net sink for atmospheric Hg (Hartman et al., 2009). The overall role of the oceans as a sink or source for atmospheric Hg, and the relative contributions of natural and legacy Hg are debated. Earlier work suggested that atmospheric inputs to the ocean were roughly equal to ocean emissions (Mason et al., 1994), but more recent global mass balances have suggested the oceans are a net sink of 500 to 2500 Mg/yr (Lamborg et al., 2002b; Mason and Sheu, 2002; Sunderland and Mason, 2007). A recent model-based study suggested that the majority of the oceanic emissions are legacy Hg (Strode et al., 2007).
Ocean sediments, plant foliage, and polar regions are sinks for atmospheric Hg. The latter are considered to be a small sink where Hg (0) is oxidized to RGM by atmospheric bromine compounds (< 100 Mg/yr) (Dastoor et al., 2008; Outridge et al., 2008). After formation, RGM is deposited to snow, with a portion remaining in the ecosystem and the rest emitted back to the atmosphere (Poulain et al., 2004; Kirk et al., 2006). Plant foliage is a significant sink for atmospheric Hg (0) (Gustin and Lindberg, 2005) and plant litter is a significant reservoir (Grigal, 2003). Ultimate removal of Hg from the atmosphere-ocean-land system occurs through settling of particulate Hg to the deep ocean where it is incorporated into sediments (Strode et al., 2007; Sunderland and Mason, 2007). Model studies suggest that the time scale for this removal is thousands of years (Selin et al., 2008).
ATMOSPHERIC MERCURY DEPOSITION
The potential for Hg introduction to aquatic food webs by way of wet and dry deposition is a primary concern. Wet deposition measurements of Hg are routinely made in the United States as part of the National Mercury Deposition Network (MDN) (http://nadp.sws.uiuc.edu/mdn/). Observations have been made for up to twelve years at some sites. It should be noted that most MDN sites were chosen to sample regional background air, and in most cases are removed from local sources. Spatial patterns in wet deposition measured as part of this network are similar from year to year, and recent work has suggested slight declines in deposition at specific locations in the eastern United States (Butler et al., 2007; Prestbo and Gay, 2009). Based on the maps available from the MDN network, the highest wet deposition rates occur in Florida and along the Gulf Coast (up to 16 μg/m2 yr); and the eastern states have higher wet deposition inputs (7 to 10 μg/m2 yr) than those in the west (1.2 to 5 μg/m2 yr). Several studies have reported that wet deposition is enhanced directly around point sources (Dvonch et al., 1999; Munthe et al., 2001; Keeler et al., 2006; Wängberg et al., 2007). White et al. (2009) reported that 42 percent of the mercury in summer-time wet deposition at the Steubenville, Ohio, MDN site, an area with a high density of coal-fired utilities, could be linked to local point source emissions.
There is no standard method for measuring Hg dry deposition, and this process is poorly understood. Spatial and temporal variability in dry deposition is likely to be large since it varies inversely with precipitation. In a few field studies dry deposition was shown to be of similar magnitude as wet deposition (Lamborg et al., 2002a; Caldwell et al., 2006; Lyman et al., 2007). Model studies suggest that for the United States as a whole, dry deposition is greater than wet deposition with significant spatial variability (Selin et al., 2008). A recent study that applied surrogate surfaces for measuring dry deposition found wet:dry inputs in Nevada to be 1:4.5, while in the southeastern United States this was 6:1 (Lyman et al., 2009).
Indirect methods that have been applied to estimate net Hg atmospheric deposition include the use of sediment cores (Swain et al., 1992; Lucotte et al., 1995; Landers et al., 1998, 2008; Bindler et al., 2001; Lamborg et al., 2002a; Yang et al., 2002; Fitzgerald et al., 2005), ice cores (Schuster et al., 2002), and peat bog profiles (Biester et al., 2007 and references therein). It is thought that sediment cores are the more reliable indicator of historic deposition (Biester et al., 2007). Data indicate that Hg deposition has increased significantly over the past 150 years in both remote and industrialized areas. In remote regions increases in deposition of two to five times above preindustrial rates are reported (Lucotte et al., 1995; Engstrom and Swain, 1997; Fitzgerald et al., 1998; Lamborg et al., 2002a). The measured
increase occurs concurrently with industrialization, suggesting that anthropogenic sources are responsible (Biester et al., 2007). Increases in deposition observed in remote regions indicate that Hg in the global atmosphere pool is an important component.
Although sediment cores clearly show an increase in deposition since the industrial revolution, interpretation of the sediment core data may be more complex than originally believed. Mercury in some cores has been found to be correlated with phytoplankton (Outridge et al., 2005) and organic carbon (Landers et al., 2008). Since Hg is readily bound to carbon, changes in watershed characteristics and climate that influence the available carbon may affect the ability of the sediments to permanently sequester Hg.
RELATIONSHIP BETWEEN MERCURY DEPOSITION AND ECOSYSTEM IMPACTS
For both freshwater and marine ecosystems the relationship between Hg deposition and Hg concentrations in fish is not straightforward because the properties of individual ecosystems will affect the efficiency with which Hg is converted into methyl Hg. It is believed that increased Hg deposition to aquatic ecosystems leads to increases in methyl Hg in fish, but there is limited quantitative data to test this hypothesis (Munthe et al., 2007).
One study that investigated the potential for fish uptake of atmospherically deposited Hg (the METAALICUS project) (Harris et al., 2007) demonstrated that increased Hg concentrations in aquatic organisms were clearly linked with the direct loading of a specific stable isotope of Hg in precipitation to the lake surface. However, < 1 percent of the stable isotope added to the land and vegetation surrounding the lake was found in the lake ecosystem after three years.
Other studies have suggested a direct link between atmospheric inputs and biota Hg concentrations. For example, Hammerschmidt and Fitzgerald (2006) found that Hg concentrations in fish were correlated with Hg in wet deposition across 25 states. Atkeson et al. (2005) found that reduced Hg concentrations in largemouth bass and egret nestlings in the Everglades region of Florida were correlated with reductions in point source emissions of RGM. In Sweden regional gradients in atmospheric deposition were linked to concentration gradients in biota (Munthe et al., 2004). Other studies however, have not found any clear relationship (Evers and Clair, 2005). Recent work by (Sunderland et al., 2009) in the eastern North Pacific Ocean found total Hg concentrations were higher than previously measured and suggested that oceanic circulation, and increasing atmospheric deposition due to sources in Asia, are linked to this increase. They also suggested this increase could impact Hg concentrations in pelagic marine fish.
SOURCE ATTRIBUTION FOR Hg DEPOSITED TO THE UNITED STATES
Estimating the source contributions of Hg deposited to the United States requires the use of regional and global models. Global models are best applied to assess impacts of long-range transport because boundary conditions for regional models will affect results (Bullock and Jaeglé, 2008), although one approach that allows the use of more detailed regional models is to apply global models to set the boundary conditions (Seigneur et al., 2004; Bullock and Jaeglé, 2008). As with all models the accuracy of the underlying meteorological data will affect results. Uncertainties that influence Hg modeling results include inadequate understanding of Hg atmospheric chemistry, magnitude of sources and sinks, limited data on speciation of anthropogenic emissions and on dry deposition, and the significance of Hg recycling between the air and terrestrial and marine surfaces (Seigneur et al., 2004; Lin et al., 2006; Jaeglé et al., 2008; Pirrone, 2008).
To understand the impact of long-range transport of Hg on deposition to the United States we first need to understand the impacts associated with domestic sources. Figure 4.2 shows the percentage of domestic anthropogenic source contribution to U.S. deposition, based on the GEOS-CHEM model (Selin et al., 2007). This figure shows that in the eastern United
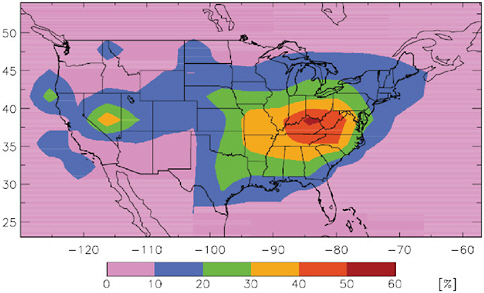
FIGURE 4.2 Percent contribution of anthropogenic North American emissions to total (wet + dry) deposition in the United States
SOURCE: Selin et al., 2007.
States, where most anthropogenic point sources of reactive and particle-bound Hg are located, domestic sources may contribute up to 60 percent to total deposition. For the western United States, the estimated contribution of domestic anthropogenic sources to deposition is much smaller. For both regions the remaining deposition (based on model results) is derived from regional and global natural emissions, current foreign anthropogenic emissions, and legacy Hg in the active pool.
Other models give similar results, although the details of these models (i.e., emission estimates, atmospheric reactions, dry deposition rates) vary. For example, Seigneur et al. (2004) applied a global chemical transport model (CTM) and a nested continental CTM, and found that across North America, depending on location, 10 to 80 percent of deposition was due to domestic anthropogenic emissions, with an area average of 25-32 percent; and Asian anthropogenic emissions contributed approximately 20 percent. The spatial variation for the latter was significant (5 to 36 percent), being higher in the west and declining across the continent. In this model, the deposition associated with natural sources also exhibited a large range (6 to 59 percent), with this source being more important in the western United States.
Travnikov (2005) used a hemispheric model (MSCE-Hg-Hem) to assess the long-range transport and deposition of Hg to North America. Results indicated that Asia contributed 24 percent, North America contributed 33 percent, and Europe contributed 14 percent (percentages include both natural and anthropogenic contributions). In this assessment North America was found to contribute 5 percent to the deposition in Europe and 4 percent to the deposition in Asia. More recently, Strode et al. (2008) applied the GEOS-Chem model and found that Asian and North American sources each contributed ~ 25 percent to deposition in the United States (including all sources), or 14 and 16 percent, respectively, if only current anthropogenic sources were considered.
Selin et al. (2008) applied the GEOS-Chem model in more detail and found that 20 percent percent of North American deposition was generated internally from anthropogenic sources, while 30 percent was derived from external anthropogenic sources. The remaining 30 percent was from natural sources and 20 percent from legacy Hg. Model simulations explain 50 to 65 percent of the variance of the MDN wet deposition data over the United States (Seigneur et al., 2004; Bullock and Jaeglé, 2008; Selin et al., 2008).
As part of the recent UNEP initiative (Pirrone and Mason, 2009a), four global models were used to assess the impact of a 20 percent anthropogenic emission reduction in four source regions: South Asia, East Asia, Europe, and North America. In all regions, the greatest effects of the emissions reductions occurred within the region itself, largely because of the significant contribution of RGM and Hgp was associated with point sources.
Reducing East Asian anthropogenic emissions by 20 percent resulted in a ~ 3 percent reduction of deposition to North America. In the longer term, a 20 percent reduction would have a greater impact because all emissions add to the global pool of Hg that is continually recycled among environmental reservoirs.
KEY FINDINGS AND RECOMMENDATIONS
Question: What is the role of long-range transport on Hg deposition to the United States?
Finding. Once emitted from any source, Hg has the potential to be transformed to different chemical forms, transported through the atmosphere, and deposited long distances from the point of origin. Hence, long-range transport is an important process that clearly affects U.S. exposures. Continued emissions will increase the amount of Hg in the global pool available for long-range transport and recycling between reservoirs.
Finding. Mercury deposition to the contiguous United States has increased since the beginning of the industrial revolution, as anthropogenic releases have increased. Recent modeling studies suggest a range of 10 to 80 percent of the Hg deposited to the United States is from domestic anthropogenic sources (depending on location) with an average of ~ 30 percent for the country as a whole. The rest is derived from natural sources, foreign anthropogenic emissions, and the active legacy pool.
Finding. Key limitations for understanding long-range Hg transport and its exchange among different reservoirs include our knowledge of atmospheric chemical processing, dry deposition, and the potential for Hg deposited by wet and dry processes to be emitted back to the atmosphere. Evaluation of model results is limited by the availability of key observational data.
Recommendation. Pursue further research focused on understanding Hg atmospheric chemistry and kinetics, the dry deposition of Hg species, the global spatial (horizontal and vertical) and temporal variability in air concentrations and deposition, and potential for recycling of current and legacy Hg. Develop methods that allow for the measurement of concentrations and deposition of Hg that are easy to apply and do not require elaborate field stations. We also recommend efforts to improve emission inventories for natural
and anthropogenic sources, including the magnitude and speciation of emissions in sectors where there is significant uncertainty. All such studies will be useful for improving and verifying model results. Model sensitivity analyses would be useful for further defining research needs.
Question: What are the potential implications of deposition of Hg from the global pool with respect to human health, ecosystems, and air quality management goals?
Finding. Hg exposure for humans occurs primarily through fish consumption and is a legitimate health concern for certain human populations (i.e., pregnant women and small children who consume fish and those that consume large quantities of fish with high Hg concentrations). Methyl Hg exposure may also adversely impact wildlife. A component of Hg in fish is derived from the global atmospheric Hg pool and therefore long-range transport is an important contributor to Hg found in U.S. ecosystems
Finding. Hg will continue to be deposited to the United States in the future, with the magnitude of these inputs depending on foreign and domestic emission controls. Point source controls can reduce local deposition of Hg, especially the more reactive forms; however, because Hg is globally distributed and recycled between surfaces and the atmosphere after being released from a source, major reduction in Hg deposition cannot occur without cooperation on a global scale.
Recommendation. Because the endpoint of concern for human exposure to Hg is fish consumption, research that focuses on development of a better understanding of the linkage between atmospheric Hg deposition and the means by which Hg enters aquatic food webs is needed.
Question: What factors might influence future Hg emissions and the potential for long-range transport?
Finding. It is expected that coal will continue to be an important and growing source of energy for much of the world, especially in developing countries. The implications for Hg emission and deposition will depend upon the rate of emissions growth, the specific type of coal used, and the pollution control technologies employed. Hg emissions from other sectors (such as mining, incineration, and
industrial operations) may also increase, as economic growth leads to greater extraction and processing of natural resources. Pollution control technologies that remove Hg (II) or Hgp may have some local benefit; however, without control of Hg (0) emissions, the globally available pool will continue to increase.
Finding. Climate change could affect the global cycling of Hg in a number of ways, for instance
-
warming temperatures could increase the release of Hg to the air from soils and oceans;
-
increased frequency of forest fires would increase the emissions of legacy Hg;
-
enhanced plant growth (resulting from elevated atmospheric CO2 levels) could increase Hg uptake from the air;
-
more rapid plant decomposition may result in release of the Hg stored in leaf litter;
-
changes in atmospheric circulation patterns could affect the dynamics of Hg transport and deposition;
-
changes in atmospheric oxidant concentrations (related to climate change or other causes) could affect the patterns of Hg deposition.
Predicting the impact of climate change on the global Hg cycle is challenging given the uncertainties associated with impacts on climate, weather, and atmospheric dynamics, and the complexity of air-surface-plant Hg exchange.
Recommendation. The United States should actively engage in international cooperation for reducing Hg emissions worldwide, including efforts to advance and globally disseminate technologies that reduce Hg emissions associated with energy use and other forms of industrial activity.
Recommendation. Continue research designed to understand the Hg biogeochemical cycle and potential implications of climate change on the global Hg cycle. This is especially important given the large pool of Hg in terrestrial and oceanic compartments and the possibility of increased mobilization of this Hg.
















