2
Clinical Trial
INTRODUCTION
This chapter focuses on the Clinical Trial (CT), which is the costliest, most complicated, and most controversial component of the Women' s Health Initiative (WHI). The CT is designed to test the benefits and risks of dietary modification (DM), hormone replacement therapy (HRT), and calcium and vitamin D supplements (CaD) on the health of postmenopausal women. The primary hypotheses of these three branches of the CT are: (1) whether a low fat dietary pattern reduces the risks of breast and colorectal cancers; (2) whether hormone replacement therapy reduces the risk of coronary heart disease (CHD); and (3) whether combined calcium and vitamin D supplementation reduces the risk of hip fractures.
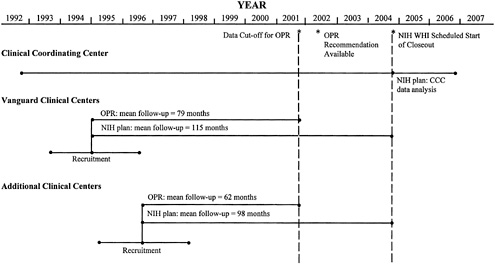
NIH has structured the CT as a 3 × 2 × 2 partial factorial design involving 63,000 women between the ages of 50 and 79 (in addition, 100,000 women will be enrolled in the observational study). NIH has funded a Clinical Coordinating Center and the first 16 of 45 expected Clinical Centers. These 16 centers, called Vanguard Clinical Centers, began a three-year recruitment period on September 1, 1993. The additional clinical centers, to be named in September 1994, would begin recruitment in January 1995. Clinic closeout is scheduled to begin September 2004, followed by two years of data analysis by the Clinical Coordinating Center.
The CT is the most thoroughly designed aspect of the WHI thus far. The committee's assessment of it was therefore based on more information than was available for the Observational Study ( Chapter 3 ) or the Community Prevention Study ( Chapter 4 ); that is reflected in the size and scope of this chapter. The chapter begins with trial-wide issues of rationale and study design. This is followed by a presentation of more detailed information on each branch. Cost details for the entire CT are presented. The chapter, concludes with a presentation of the committee's findings and suggestions, and major recommendations regarding the CT component of the WHI.
RATIONALE
General Issues
Cardiovascular disease, breast cancer, and osteoporotic fractures are among the leading causes of morbidity and mortality in postmenopausal women. As such, they are reasonable and defensible targets for a large prevention study. Coronary heart disease is the leading cause of death in U.S. women. The mortality and incidence rates of breast cancer are high; over an average 85-year lifespan, one in nine women develop breast cancer and approximately one in thirty die of it. Osteoporotic fractures, which are associated with aging, affect many more women than men; complications are life threatening and reduce both longevity and quality of life.
These diseases are not alone among the severe disablers of women, however. The CT does not directly address arthritis, dysmobility, poverty and isolation, depression, dementia, hearing, vision, and dental losses, and institutionalization. Neither does it address other compelling outcomes, such as dysfunction or pain, that are not linked to solitary etiologies. This should not imply that these issues are not troubling sources of morbidity, nor that they would be inappropriate targets for future prevention and treatment research. Similarly, that the focus of the CT is on postmenopausal women should not be mistaken for a disregard of the myriad unanswered questions about younger women, or about the effects of behavior and disease in earlier stages of life on morbidity and mortality in later stages. One study cannot answer all questions.
The primary hypotheses of the CT are as follows:
-
A low fat dietary pattern reduces the risk of breast cancer.
-
A low fat dietary pattern reduces the risk of colorectal cancer.
-
Hormone replacement therapy reduces the risk of coronary heart disease.
-
Calcium and vitamin D (combined) supplementation reduces the risk of hip fracture.
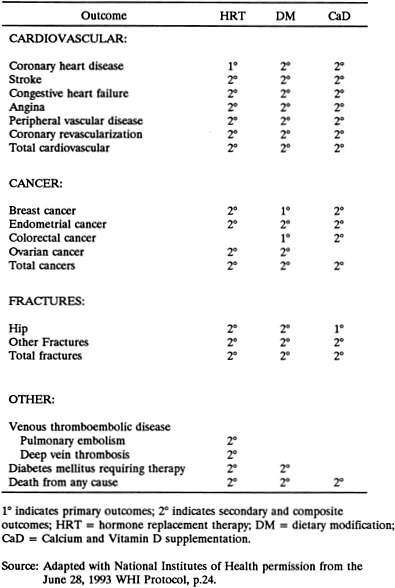
The numerous secondary hypotheses include: DM reduces risk of CHD; HRT increases risks of breast and endometrial cancers; HRT reduces risk of fractures; and CaD reduces risk of colorectal cancer. The CT outcomes are presented in Figure 2-1; the CT hypotheses are listed in Appendix F.
There are reasonably good rationales for some aspects of each of the three branches of the CT, although evidence for the central hypothesis for the DM branch–that a change to a low fat dietary pattern by women over the age of 50 will reduce the incidence of breast cancer over the following nine years—is the weakest and least consistent of the three. There are stronger rationales for expecting that there are effects of DM on colorectal cancer and various cardiovascular disease endpoints. Similarly, there is a strong rationale for the HRT branch, which will test not only the relationship between HRT and coronary heart disease,
but also quantify the secondary adverse and beneficial outcomes such as cancers of the breast and endometrium, fractures (especially hip fractures), quality of life, and total mortality. It is defensible to test the effect of CaD on risks of hip fracture and colorectal cancer within the context of a study that has been mounted for other purposes; these hypotheses would not stand alone as a rationale for an expensive trial. The DM branch drives the size of the study, the DM-breast cancer hypothesis drives the length of the study, and the DM and HRT branches generate most of the complexity of the study. Each outcome to be measured is hypothesized to be affected by more than one of the CT intervention branches. For example, DM and HRT may affect coronary heart disease; DM and HRT may affect breast cancer; and HRT and CaD may affect fractures.
Integration of the CT with Other Components of the WHI
The goal of the WHI CT is to test whether the interventions being used will reduce the morbidity and mortality associated with breast cancer, cardiovascular disease, and osteoporotic fractures. The WHI Observational Study (OS) is designed to follow women for an average of nine years. The goals of the OS are to: (1) improve risk prediction of coronary heart disease, breast cancer, colorectal cancer, fractures, and total mortality in postmenopausal women; (2) create a resource of data and biologic samples that can be used to identify new risk factors and/or biomarkers for disease; and (3) examine the impact of changes in individual characteristics on disease and total mortality. The OS can provide quantitative assessments of risk factor associations with major chronic diseases in women and it will enable the calculation of improved risk estimates for cardiovascular disease, cancers, bone fracture, and other disease endpoints in older women. Such information is expected to improve the quality of life of postmenopausal women by facilitating the identification and preventive treatment of high-risk women.
Many women must be screened to determine their eligibility for the CT, and this is costly. The marginal cost of following these women in the OS is small relative to the expense of mounting an independent OS. Thus, it is appropriate to conduct the OS in tandem with the CT.
The details of the Community Prevention Study (CPS) are unknown, so the committee cannot judge whether that component of the WHI will draw on the experience or results of the CT or OS. The CPS would fit well into the overall vision of improved women's health if its goals were to develop lifestyle change strategies in diet, exercise, smoking, and early disease detection that are accepted as national goals and for which major gaps in development exist, especially as pertain to women of low socioeconomic status (SES) and minority women. The CPS could also furnish an infrastructure of trained personnel to aid in carrying out interventions and policies that might flow from the CT and OS.
DESIGN AND METHODS
The committee's examination of the CT design concentrated on two fundamental questions:
-
Can the study design—if no operational difficulties occur—answer the questions it addresses?
-
If the study design is appropriate, what other threats are there to the successful completion of the study?
The committee focused on twelve issues central to these questions, which are discussed below. Seven of these issues involve conceptual problems that are built into the design of the CT. Even if all study operations were to proceed without incident, these conceptual issues threaten the validity of the findings:
-
factorial design
-
sample characteristics
-
proposed analytic techniques
-
ethics: consent and stopping rules
-
minority analysis plan
-
specificity of intervention and effect
-
outcome definition and measurement
In addition to these conceptual problems, any study—no matter how well designed—is subject to setbacks by operational problems. The CT is particularly vulnerable to such problems because of its size, complexity, and duration. The committee has identified five operational issues that could jeopardize the study's success:
-
recruitment and retention
-
adherence
-
secular trends
-
provision of health care services to participants
-
study management
In the ensuing discussion of these twelve issues, certain specific suggestions are made. More global recommendations will be discussed at the end of this chapter.
Factorial Design
NIH argued that conducting a partial factorial design would reduce the required number of women and attendant costs and allow assessment of interactions among intervention branches. The partial factorial design is presented in Figure 2-2 , which has been reprinted with NIH permission from the June 28, 1993 WHI Protocol, page 18. The committee feels
that the factorial design has serious weaknesses. The factorial design is criticized because the difficulty of maintaining adherence to one intervention, such as DM, is magnified greatly in a design that requires adherence to two interventions, DM and HRT. The 15.9 percent overlap between the DM and HRT interventions is insufficient to provide adequate statistical power to assess interactions between the interventions. Therefore, the complexity of the design is not compensated by an increase in statistical power.
NIH also argued that it will be more economical for the clinical centers to screen simultaneously for the two branches, DM and HRT, rather than to mount each one separately. As now planned, there are effectively two separate studies, HRT and DM, done within the same administrative structure. It is mostly the efficiency of shared administration which make this plan more economical. In essence, the integrated design has become primarily a matter of efficiency; it is not essential to hypothesis testing.
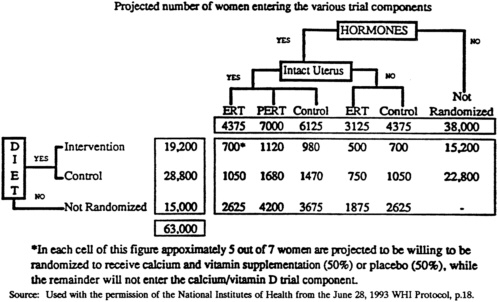
FIGURE 2-2 Women's Health Initiative Clinical Trial partial factorial design.
Sample Characteristics
Size
The CT is one of NIH's largest clinical trials: 63,000 women are expected to enroll. The large sample size is one of the primary reasons that the CT is expensive: the CT and OS are expected by NIH to cost approximately $586 million.
The sample size is driven by the choice of endpoints. The primary endpoints of the CT are incidence of breast cancer, colorectal cancer, CHD, and hip fractures. Because the incidence rate of each outcome differs, and because the study interventions have different hypothesized effects, setting sample size requirements for the overall CT is a complicated task. The cost of the trial, strongly linked as it is to sample size, would vary based on assumptions made. In reviewing the WHI Protocol, the committee was concerned about a number of the assumptions. For example, a continuing linear decline of CHD mortality is assumed; this should be examined in more detail, especially by age group.
For each of the main hypotheses in the CT, the sample size is also determined by the need to achieve a specified power to test the effect of the intervention at a given significance level. Take, for example, the DM intervention. To test whether the difference in breast cancer event rates in the intervention and control groups is an effect of DM, a significance level α = 0.025 and a one-sided test is used in the WHI Protocol. The power to test for effects, and hence the required sample size, depends on assumptions made by NIH about the following factors:
-
Age distribution. Women aged 50-54, 55-59, 60-69, and 70-79 are to be enrolled in the ratio 2:4:9:5, by design.
-
Loss to follow-up. For the breast cancer endpoint, loss to follow-up is assumed to be 3.0 percent per year due to deaths from other causes or disappearance.
-
Adherence. Based on the Women's Health Trial Vanguard Study (Henderson et al., 1990), it is assumed that the average percentage of calories from fat will drop from 39 percent at baseline to 20.9 percent at six months, will increase to 21.6 percent at one year, and to 22.6 percent at two years. It is then assumed to increase linearly to 26 percent at 10 years (June 28, 1993 WHI Protocol). For the control group, average percent calories from fat is assumed to decrease linearly from 38 percent at baseline to 34 percent at 10 years.
-
Magnitude and Lag of Dietary Effects. Based on international correlations between dietary fat disappearance data (rate of use or wastage in the population) and breast cancer incidence rates (Prentice et al., 1988), the WHI Protocol assumes that the risk ratio decreases linearly from RR = 1.0 at baseline to RR = 0.5 at 10 years for fully adherent women. When this effect is averaged over nine years and nonadherence is taken into account, it is projected that the DM effect is a 14 percent reduction in breast cancer incidence.
-
Incidence Rates. The protocol uses published age-specific incidence data from the SEER program for the years 1985-1989. The resulting percentage of cases, assuming 14 percent DM effect, are 2.92 percent and 2.52 percent for the control and intervention groups, respectively, after nine years.
With the above assumptions, the WHI Protocol states that the power of the test is 86 percent based on a sample of 48,000 women. * It should be noted that the power of the test, and hence the required sample size, can vary drastically depending on changes in the above assumptions. For example, if the intervention effect is only 11 or 12 percent, rather than the expected 14 percent, then the power of the test would drop to 63 or 75 percent for a sample of 48,000 women. In fact, the protocol shows that reasonable changes in just three of the underlying assumptions—follow-up, effect size, and number of enrollees—produce enormous variation in the power, which could be as low as 25 percent (six years follow-up, 11 percent intervention effect, and 42,000 participants) or as high as 89 percent (nine years follow-up, 14 percent intervention effect, and 54,000 participants). All are within reasonable ranges of assumptions.
To illustrate the effect of additional assumptions on sample size, the committee considered an example given by Lakatos (1988). In this example, when the lag time (the interval needed to achieve full intervention effect) increases from instantaneous (zero years) to one full year, the sample size needed to achieve 90 percent power at α= 0.05 (two-sided) can increase more than fourfold. Thus, the necessary sample size is very sensitive to the assumed timing of effect on the relative risk.
The proposed protocol assumes a linear halving of the risk over five years. The existing data neither support nor contradict this claim, but the biology of breast cancer would seem to make this an optimistic projection. If diet does have an effect on breast cancer, but the lag time for halving the risk is, for example, 20 years, then the currently proposed project has very little chance to detect an effect. The uncertainty of the lag effect is crucial to the reliability of the sample size estimates. Short lag times would enable results to be acquired more quickly and longer lag times would likely preclude a result in the trial as planned. Information gained in the first five years of the WHI may be critical in setting bounds on these estimates.
Recruitment
Because the sample size determines what recruitment efforts are required, it is necessary to assess recruitment assumptions. The June 28, 1993 WHI Protocol estimates that 33 percent of the 189,000 women who are expected to attend the first screening visit are expected to enter the CT. The CT will randomize 25,000 women to the HRT component (40 percent of whom are expected to agree to be in the DM component as well); 48,000 women to the DM component (21 percent of whom are expected to agree to be in the HRT component as well); and 45,000 women to the CaD component (71 percent of the total), all of whom will be participating in at least one of the other components.
|
* |
This is based on a modified version of a program designed by Lakatos (1988). |
Each Vanguard Clinical Center expects to enroll 336 women in the HRT arm; 846 in the DM arm; 224 in both the HRT and DM arms; and 2,220 women in the OS. As currently planned, each Clinical Center is expected to enroll 39 women per month. Therefore, for each month ahead of schedule a clinic becomes, there is a gain in power from the increase of three person-years (39 person-months) of follow-up. Similarly, for each month a clinic falls behind in recruitment, three person-years of follow-up are lost. Because of the recent delay in bringing on the 29 additional clinics, several months of follow-up are already lost to the study. This delay threatens the power and sample size computations, adding to the level of uncertainty.
Participant Characteristics
Postmenopausal women between the ages of 50 and 79 will be invited to join the CT. It is the goal of the WHI to have the study sample represent initial age categories in the following allocations:
-
50-54 years old—10 percent
-
55-59 years old—20 percent
-
60-69 years old—45 percent
-
70-79 years old—25 percent
The WHI is also striving for, but not requiring, a “representative” accrual of participants with regard to race/ethnicity and SES. This will further complicate recruitment, although it will strengthen the generalizability of the results. It is not clear to the committee how this goal would be enforced.
General inclusion criteria for the CT, according to the June 28, 1993 WHI Protocol, are postmenopausal status, with or without a uterus or ovaries; 50-79 years of age, inclusive, at first screening contact; likely to be residing in the study area for at least three years after randomization; and providing written informed consent.
Exclusion criteria include competing risks such as a medical condition associated with a survival rate of less than five years, invasive cancer of any type in the past ten years, or breast cancer (in situ or invasive) at any time; characteristics that could affect adherence or retention, such as alcohol or drug dependency, mental illness, dementia, or current active participation in another intervention trial; and unwillingness to give up current HRT or calcium supplementation. See Appendix A for more a detailed description of exclusion criteria.
Participants will not be categorized by risk for breast cancer, colorectal cancer, or coronary heart disease. This allows a more generalizable study, but the lack of risk restrictions requires a much larger sample size. The factorial design does not allow specific branches to focus on the most efficient samples, such as women at high risk of CHD for an HRT trial or women at high risk of breast cancer for a DM trial.
Proposed Analytic Techniques
NIH has proposed carefully designed and deliberated analytic techniques. A weighted logrank test will be used to test for the hypothesized effects in the CT (Lakatos, 1988). The logrank test is based on the time it takes until the event occurs. If the event does not occur within the observation period, the case is considered a censored observation. The null hypothesis (i.e., that the intervention made no difference) of the logrank test is that the distribution of time-to-events is the same in the intervention and the control groups.
Although a one-sided test with α = 0.025 is mathematically equivalent to a two-sided test at α = 0.05 (yielding equivalent sample size estimates), the difference has implications for conceptualizing and monitoring the results. The committee, as well as a number of the investigators, feels that a two-sided test should be used.
Statistical adjustments using relative risk regression methods will be used to consider the effects of including other covariables, the ability of intermediate variables to explain an intervention effect, the estimation of full adherence relative risk as a function of time since randomization, and a reliability substudy.
No multiple comparison adjustments are planned for primary endpoint analysis. Subsidiary outcome analyses will rely on multivariate response analyses when appropriate. While it is legitimate to forego formal multiple comparison adjustment, as long as that is clearly stated in the protocol, the practice stands in stark contrast to the proposed use of the Bonferroni adjustment, one of the most conservative adjustments, in the analyses to be presented to the Data and Safety Monitoring Board (DSMB). The Bonferroni adjustment to the significance level consists of dividing the alpha by the number of tests simultaneously performed and using the result as the level of significance for the test. It seems likely, and preferable, for the DSMB to receive uncorrected data.
Data Safety and Monitoring Board
As in many blinded NIH studies involving human participants, there will be a Data and Safety Monitoring Board (DSMB) with oversight responsibilities. To address the tasks of the DSMB, plans for interim analysis have been drafted. The committee was told that the Clinical Coordinating Center will present data on primary, subsidiary, and intermediate outcomes to the DSMB after Bonferroni adjustments for multiple comparisons are made. Each CT branch will be monitored for early stoppage based on summary measures of benefits and risks. Since the DSMB will have the responsibility of stopping a CT branch if adverse effects produce a risk to participants, these interim plans are not well enough formulated to be adequate. These plans are extremely complicated and are slated to be addressed by the DSMB. This onerous task has major implications. If certain monitoring plans are adopted, it might be decided to provide the participants with some study results. Alternatively, using the severe corrections for all the multiple comparisons, key results may
be obscured, delaying the release of important public health results. The DSMB will no doubt address these issues, but the lack of information on how these decisions will be made over the duration of the trial increases the uncertainty about the ability of the CT to achieve specific goals.
The committee suggests that the DSMB prespecify a number of outcomes and situations to monitor concerning stopping the trial.
Ethics: Consent and Stopping Rules
Any clinical trial must incorporate adequate protection for the well-being and self-determination of human participants. This study has such a broad population base and such high visibility that its procedures in this regard are likely to come under special scrutiny. A randomized study is ethically justifiable only when competent professionals cannot discern a reason why one arm of the study is clearly better or worse than others for the potential participants. Allowing a participant to join a randomized trial is ethically defensible only when the participant has enough information to evaluate whether all arms are reasonably equal in her own view or, alternatively, that the differences between arms are of a magnitude and seriousness that she is willing to accept in order to contribute to the common good. After much discussion, the committee decided that it is currently defensible to offer the randomizations to each of the CT branches.
Ensuring that each participant can knowingly accept randomization requires that she know the key information about the risks, benefits, and uncertainties involved, as individualized to her situation. Conventionally, this means that a certain minimum of information is given to the potential participant, who is then encouraged to ask any additional questions that may be of special relevance or interest. Obviously, the respondent to these inquiries must be knowledgeable in the subject area. This information and consent requirement can pose challenges to the achievement of a study's implementation goals.
If the WHI CT proceeds as currently designed, it will require substantial resources to meet the obligation to inform actual and potential participants adequately. This obligation will require much more information about the interventions at the outset, as discussed below, as well as a commitment to provide evolving scientific information over the course of the project.
Informed Consent
The committee found the proposed informed consent measures to be inadequate. The committee was provided with Appendix IV “Informed Consent Guidelines” in the WHI Protocol, approved by the DSMB on June 16, 1993, and feels that the consent forms give no understanding of the likelihood or magnitude of major risks and benefits. Certain women at substantial risk of particular problems would not necessarily learn of the currently known
effects of their choice. For example, women at high risk of osteoporosis and/or CHD would not learn from existing materials that HRT has been shown to slow or prevent the acceleration of bone loss (as opposed to fractures), and to reduce the risk of clinical CHD in high risk patients. Women at high risk of breast cancer would not learn that they might increase that risk by using HRT.
The committee's concerns about informed consent were raised with representatives from the Clinical Coordinating Center (CCC) and Vanguard Clinical Centers at its meeting in July 1993. These concerns, when presented to the investigators, were met with three types of responses: that the institutional requirements are higher, that the videotapes will provide the appropriate information, and that the counseling sessions will also provide that information.
Most of the Vanguard Clinical Centers reported that the NIH forms passed their Institutional Review Board without substantial modification. Thus, their institutions are not serving as gatekeepers to rectify the problems observed by the committee.
WHI investigators told the committee that the videotapes being prepared for use at all centers would obviate the concerns expressed. The committee subsequently received the scripts (dated July 12, 1993) for these videotapes. After reviewing the scripts, the committee determined these studywide materials to be unbalanced and inadequate to inform women about their choices. The deficiencies apply to literate and economically advantaged women and even more so to disadvantaged women. The videotape scripts simply do not address risks; on the contrary, their tone and presentation are entirely aimed at reassurance and inspiration, and they do not make clear that support would be available for women seeking more information or declining to be randomized. Thus, adequate informed consent would actually be dependent on individual counseling sessions.
Individual counseling, together with the recruitment material, could be a strong and flexible way to ensure truly informed consent. However, ensuring adequate consent for 63,000 women at 45 centers with tight budgets would require focused attention. The counselors would have to be knowledgeable individuals on the “front line,” armed with algorithms and guidelines, and, probably, printed and graphic material about known risks and benefits. These persons would need supervision, training, and monitoring. It is not clear that any of the above is included in the CCC plans or in the site budgets.
The investigators must set higher standards than currently exist in the all-study material, including introductory brochures, consent forms, and videotape information. These materials must provide sufficient information about potential benefits and risks to enable most women to make reasonable choices about whether to be randomized. This material should be available in Spanish and perhaps, also, in more conversational English. In addition, case-specific counseling about consent to randomization must be ensured, must be of high quality, and must be monitored. Interactive video programs might be an appealing and effective way to tailor education and decision-making assistance for prospective patients.
The committee strongly recommends that the consent process be outlined more carefully, be implemented well and monitored across all centers, and be evaluated and updated as needed. This is important for ensuring respect for the self-determination of the participants, ensuring the continuation of participants in the study, and maintaining a favorable public evaluation of the project.
Stopping Rules
The CT involves interventions with effects that may occur within a few years (e.g., protection of estrogen against CHD), after at least five years (e.g., protection of low fat diet against breast cancer) or perhaps many years later (e.g., increased risk of breast cancer in women who use estrogen). The inclusion of several interventions with several endpoints in a single trial makes the stopping rules difficult to formulate. Stopping rules are very important because, otherwise, randomized participants may not be informed of changed understanding of risks or benefits in a timely manner.
The very issues that are worthy of studying in a randomized fashion are those for which other kinds of information are likely to become available during the course of a randomized trial, particularly a prolonged trial. As substantial new information becomes available, the question of whether it remains reasonable to randomize new participants must be addressed, usually by a group of experts assembled for that purpose. Ordinarily, if they decide that the new information makes it unreasonable to randomize new participants, then those currently randomized participants receiving treatment under one branch or the other must also be informed and offered the opportunity to select their treatment. Once this is done, the trial has effectively terminated. This is obviously a serious step, and the evidence for taking it must ordinarily be quite persuasive.
In other cases, new information that is insufficient to change the justification for randomization may nevertheless be sufficient to change the decisions of individual participants as to whether they would accept randomization. There is an ethical obligation to continue to inform participants during a trial, by providing information that might change their personal decisions to continue receiving a blinded treatment. This situation arises less frequently, however, and its effects upon the trial are less certain. If additional information causes cross-over or disenrollment of a small or fairly representative set of participants, the effects may be small. If the information causes these changes in a large or highly biased set of participants, the study might effectively be terminated.
The emergence of new information that may require closing a branch of the CT is not unlikely over the next nine years. One branch is at special risk: the near-term effects of hormones on reducing cardiovascular risk factors and event rates may be confirmed early in this project. Other studies, such as the Postmenopausal Estrogen/Progestin Interventions (PEPI) trial and the Heart Estrogen-Progestin Replacement Study (HERS), might provide additional corroboration, sufficient to make it imperative to tell at least women at
moderate-to-high risk that estrogens are somewhat protective against CHD and to be able to give reliable estimates of the size of this effect.
The committee believes that such information must be shared with the participants in order for them to make their own decisions about the possible long-term risks of breast cancer as compared with the opportunities to have reduced risks of cardiovascular disease and possibly reduced risks of fractures. Sharing this information a few years into the project might curtail it prematurely. A reasonable response to this likely threat to the study would be to tell women prior to randomization of the current, still uncertain, estimates of the association between estrogen and CHD, and the long-term health risks. The number of women willing to be randomized might shrink, but the threats to completion of the study would be diminished, once women consenting to be randomized despite these risks were enrolled and randomized.
Data and Safety Monitoring Board
The “stopping rule” in the current protocol appears to be based on “all cause” mortality, supplemented by unspecified intervention-specific outcome rates. The committee is concerned that this complex and interlocking study provides even more than the usual substantial impetus for the DSMB to be reluctant to stop the trial or to provide the participants with additional information. Several suggestions were made by the committee, including the following:
-
The DSMB should use preexisting or external information to establish a prior probability that internal data then would have the role of confirming. This might mean accepting an earlier “stop” conclusion than would be justified by data arising solely from the CT.
-
The DSMB should perform prespecified subset analyses on participant groups especially likely to evidence harm or benefit.
-
The committee was told that the DSMB would only receive data if an intervention group in the study was significantly different statistically from control after Bonferroni correction for multiple comparisons. The DSMB should be able to do any analyses it feels are warranted and should examine uncorrected estimates of effect.
-
The DSMB should review the monitoring of the consent process, especially to confirm the propriety of proceeding in the face of an expected range of new findings with regard to estrogen and cardiovascular disease.
-
The DSMB should evaluate prespecified event rates for morbid and mortal potential outcomes, not only “all cause” mortality.
Minority Analysis Plan
A driving impetus of the WHI is to begin to ameliorate the effects of the historical exclusion of women from clinical trials. A parallel situation exists for minorities, men as well as women.
Public statements regarding the CT describe it as explicitly designed to include minority women. In its granting process, NIH issued distinct Requests for Applications for minority centers. The CT goals include an average 20 percent minority recruitment, with the goal of the minority centers set at 60 percent. To judge whether the recruitment and analysis plans for minority participants in the CT are adequate, one must consider what NIH's intent may be in focusing on minority participation. Motivations for increased minority recruitment in a research study include the following:
-
The enduring injustice of restricting participation to any one group whose future members will thereby be primary beneficiaries;
-
The unfortunate reality that it is only by research participation that some people have access to promising experimental treatments, and the associated inequity of excluding some groups from that opportunity; and
-
The possibility that minorities may have differing risks or likelihood of response to treatment, or a differing disease process (which could affect risks and/or treatment response).
The committee concluded that the CT as now designed would satisfy the first two considerations. The assumptions and implications of the final consideration will now be discussed.
Four of the Vanguard Clinical Centers are designated contractually as Minority Centers, representing African Americans, Native Americans, and Latinas. The data from these groups will not allow definitive conclusions, however, due to the heterogeneity among and within the three populations, and due to the small size of the minority sample. The committee notes that parallel difficulties beset analyzing heterogeneity within the white population as well. The anticipated power of the CT will be insufficient to compare individual minority groups to the majority population. The study will be able to observe trends, if they exist, but will probably not have adequate power for conclusive statistical tests. The committee feels that the inability to analyze subsamples should be made clear to groups that are proponents of the WHI precisely because it might be believed—in error—that the study will provide the opportunity to test such comparisons.
Although the intent of the CT is to generalize the data to the general population, it is not clear that there is a uniform effort to stratify the recruitment efforts by SES for the Minority Centers or at the other Vanguard Clinical Centers or additional centers. It is expected, but not necessarily correct, that many of the minority participants will be at the
low end of the socioeconomic spectrum. The committee felt that attempts should be made to include the entire range of SES, both for the majority and minority populations.
The available research instruments may well be systematically biased in assigning SES categories to minority populations. For example, some indices of SES incorporate education, income, and occupation; all ethnic groups are assumed to be equal with regard to these factors. However, a considerable difference in income often exists between ethnic minority and white individuals, even with the same level of education and the same occupation. (Potential distortion by gender is eliminated within the study, since the CT includes only women.) In measuring the SES of minority participants, NIH should consider such recognized difficulties. For example, scales have been adapted for use in minority populations that include only education and occupation, not income. If income is to be a variable included in scales or analyses, geographical differences must also be taken into account.
Specifying the Relationship of Intervention and Effect
While the committee understood the constraints that gave rise to the specific design disadvantages, it pointed out several worthy of note so that expectations do not exceed the capabilities of the study design. If an association were to be demonstrated between the DM and decreased risks of CHD or breast cancer, the scientific and lay audience would want to know whether it was the low fat component or other changes in the diet that decreased risk. The CT is not designed to acquire data from which to respond to such inquiries. Any estimates calculated by CT investigators using regression techniques would not be as useful as a straightforward test set up in a randomized design.
If the women randomized to CaD do indeed experience fewer osteoporotic fractures, CT investigators will not have definitive data with which to separate the effects of the two elements. Also, investigators hypothesize potential effects on breast cancer risk in different directions for the DM and HRT. Despite the partial factorial design of the CT, the amount of controlled overlap of intervention subgroups will not be adequate to test interactions with sufficient power. Finally, given sample size constraints, there is insufficient power to test the merits of ERT in comparison with PERT on primary and secondary endpoints. This last comparison is one with substantial clinical impact.
Outcome Definition and Measurement
NIH and Clinical Coordinating Center documents discuss in detail the clinical definitions of CT endpoints. The committee noted two additional endpoint detection problems. The first lies in the uncertain meaning of tiny malignancies detected by mammograms. If, as it seems to be the case, large numbers of these cases are nests of cells that appear to be malignant at pathology but which do not behave as malignant during the woman's lifespan,
then it would be important to be able to distinguish who would have experienced invasive cancer. However, based on current knowledge, there is no way to do this prospectively (or even retrospectively). There is no reason to think that the effect of the proposed diet is similar in tumors of both sorts: clinically malignant, or clinically benign but pathologically malignant. If the population of the two types cannot be separated and if the effect of intervention differs substantially, then results may well be misleading.
Second, despite colorectal cancer being of primary interest in the DM branch and of secondary interest in the CaD supplementation branch, there are no plans to detect it systematically. This is especially important with a condition that can progress undetected for a prolonged period. The committee acknowledges that there are no easy solutions, and encourages NIH and the WHI investigators to consider alternate ways for more complete and unbiased detection of colorectal cancer outcomes. Such detection might entail more prolonged follow-up.
In addition, regarding the definition and measurement of endpoints, the committee suggested that additional constructs that will be measured in the course of the CT be examined in connection with intervention-endpoint relationships. Pain, mobility, HRT-associated mood changes, or concern about a possibly unpalatable diet, all influence adherence, disease endpoints, and total morbidity and mortality, both independently and through the same pathways. Furthermore, the quality of life, as measured in part by these variables, may be as important to individual women as years-of-life-gained or lost.
Recruitment and Retention
General Issues
The recruitment plans for the WHI CT reflect the extensive experience of the investigators and their recognition of the challenges of recruiting for such a massive clinical trial. A national media campaign, which would serve as a catalyst for the local recruitment efforts, is planned. Production of a variety of studywide materials is well underway, as are local recruitment activities.
Although the investigators expect the national media campaign to begin later in 1993, it is suggested in the WHI Manual of Operations and Procedures that the campaign may be delayed until all 45 clinical centers are operational. The media campaign includes a variety of elements such as public service announcements, a celebrity spokeswoman, and media appearances by the investigators on national media such as “Good Morning America.” The investigators correctly note that the national campaign will spark the local campaigns, where the heart of recruitment activity will take place.
A clinic-specific recruitment plan was prepared by each clinical center; the plans have already commenced. Some Vanguard Clinical Centers have established a community
network that includes as many as 60 diverse groups drawn from civic, religious, government, and other nonprofit groups. The investigators clearly recognize that recruitment involves development of working relationships with these community groups and the media community, as well as use of a wide variety of strategies such as direct mail and print/broadcast media.
The investigators have decided to produce a set of studywide support materials for recruitment. These include a study logo, a brochure, and four videos—one for use in community presentation or general orientation to the study, and three for use as adjuncts to the on-site recruitment process. In addition, a slide presentation for use with professional groups and a sample press release have also been prepared.
Recruitment activity will be reported by the clinical centers on a monthly basis to the Clinical Coordinating Center, which will monitor and report studywide participant accrual. The investigators have organized a recruitment coordinators' group composed of the recruitment staff from the clinical centers. This group will regularly share information about recruitment experiences by conference call, and will report to the Recruitment and Retention Working Group, which includes representatives from the Clinical Coordinating Center, NIH, and six clinical centers.
Despite these efforts, however, the IOM committee has identified three remaining areas of concern that may have significant impact on the viability of the CT recruitment plan and the realistic costs associated with the successful completion of recruitment:
-
The “message” of the study is not adequately developed and may be misleading.
It cannot be assumed that the general importance and scope of the study will be adequate to convey a powerful appeal to the target group. Although some centers have developed an altruistic or family-oriented appeal for their recruitment campaigns, the study overall lacks a clear message and theme. Experience in past clinical trials suggests that a successful recruitment campaign involves presenting the study in a way with instant, easily recognizable appeal to potential participants. For example, the PEPI trial used a “Women Have Hearts, Too!” theme with the queen of hearts logo. Given the size, complexity, and length of the CT, the study 's message must be clearly developed in order for recruitment to be successful.
The committee recommends, however, that great care be taken in the articulation of a theme, since media coverage thus far has emphasized only one of the CT hypotheses: low fat dietary pattern-breast cancer. Since this is the weakest hypothesis, it should not be the central theme. An expert public relations/marketing consultant might help the investigators develop an appealing message for the study and spearhead a comprehensive national media campaign. Experience in other clinical trials currently underway with postmenopausal women suggests that this type of strategic planning in the early phase of the study is a wise investment of time and resources. Such an investment produces a message that stimulates
national and local attention by increasing the recognizability of the study and its appeal to large numbers of women. It is not clear, however, if the current budget has the flexibility to absorb the costs for such consulting services. One method of saving costs might be to explore collaborative association with other groups in similar efforts. For example, the American Dietetic Association has recently begun a national public relations campaign designed to improve the dietary habits of postmenopausal women.
-
The increased percentage of the total population in the over-70 cohort (25 percent of the study sample) will affect effort required.
This recent change in the protocol has implications for recruitment, since specialized approaches may be required to attract women of this age group to the study. The degree of experience with this age group varies considerably among the investigators, and only a few Vanguard Clinical Centers have developed approaches for this older group of women. The clinical centers should be encouraged to develop specific recruitment plans for the oldest cohort of women. This should involve sources of recruitment, transportation to the clinical center, and any other considerations that may be unique to this group. The clinical centers should also provide an estimate of the additional costs associated with this age-specific recruitment effort.
-
The recruitment plans do not specify if and how the clinical centers plan to adjust their recruitment plans over the long course of recruitment.
Given the very long recruitment period, a general plan for the entire course of the recruitment phase will not adequately address the well-recognized seasonal variations in the community's and the media's receptivity to recruitment efforts. Specialized recruitment efforts will be needed to maintain interest in the study after the study loses its initial news appeal with the media. Experience in past studies has shown that such efforts often require considerable financial support. Additional funds for the CT mid-course recruitment effort do not appear to be included in the current budget, and delays in recruitment decrease the power of the study.
Minority Recruitment Issues
The HRT branch raises many concerns with regard to the Minority Centers. There are few data available on the use and effects of HRT in the minority populations. For example, the effect size for the minority populations may differ from that of the majority population in the study. In addition, the dropout and adherence rates for the minority groups are established on majority group data. Based on the literature, both participation rates and adherence rates are likely to be lower in the minority population than in the majority population. Therefore the sample size for this sector may not be adequate.
The WHI CT Minority Centers have linked their recruitment efforts to the established networks within the local minority community. Religious and political leaders should be
involved in the recruitment effort, as well as local women. In these clinical centers, attention has also been paid to recruiting staff from the target community. There is strong concern that the Minority Centers will probably require considerably more personnel at the community level, which implies that those personnel will not be in-kind contributions from the institution.
The Women's Health Trial Minority Feasibility Study, sponsored by the National Cancer Institute and the National Heart, Lung, and Blood Institute, is currently underway to test the feasibility of the DM component in African American, Latina, and low-income women. This 29-month study, carried out at three clinical centers, is designed to note the effect of a changed diet on blood lipids, lipoproteins, and hormones, and to measure the influence of culture and economic status on the maintenance of a low fat diet. The recruitment goal is 2,250 women between the ages of 50 and 79. Recruitment began in August 1992, and as of June 30, 1993 the study had reached 44 percent enrollment. It is too early in the study to establish whether retention and dietary adherence have either succeeded or failed. Recruitment is approximately on target at one site, lagging at another, and drastically behind at the third due to a natural disaster.
Adherence
The problem of getting participants to adhere to treatment regimes in randomized trials is more or less difficult depending on the nature of the interventions, the size of the study population, and the duration of the trial. The WHI CT is particularly difficult because it involves three intervention branches (two of which involve major lifestyle changes or side effects), a large number of participants, and a very long duration. Elements of the three treatment branches in the CT have been tested separately—theDM in the two-year Women's Health Trial, the HRT in the three-year PEPI trial, and CaD in various smaller randomized trials.
The participants in the Women's Health Trial were primarily well-educated white women who, because of their high risk of breast cancer, were highly motivated to adhere to a low fat diet. This trial demonstrated that such women could reduce the fat in their diet to a level close to the target of 20 percent and remain on that diet with some small amount of recidivism for two years. It is uncertain how successfully low and moderate risk women of a wider range of SES and race/ethnicity, including quite elderly women, would fare on such a diet. The problem of adherence is exacerbated by the fact that changes in diet may affect the entire family, not just the woman herself, and they may involve costs and access issues (to fruits, etc.) that are difficult for poor or elderly women.
HRT has various side effects that can also impede adherence. For example, PERT often results in breast tenderness, breakthrough bleeding and acne. Moreover, long-term adverse effects are serious: the risk of endometrial cancer is increased in women who use ERT, and there is serious concern that HRT increases the risk of breast cancer. Adherence
can be seriously affected by news reports of adverse (or beneficial) effects of these drugs. The PEPI trial demonstrated that a population of women who were primarily white and well educated could, with intensive staff effort, adhere to HRT for up to three years.
CaD has few adverse effects and adherence is expected to be adequate. The CT intends to randomize women to at least two and up to three interventions. Maintaining good adherence to any single intervention over a period of a decade is a difficult task. As noted, one intervention —DM—involves lifestyle changes, and a second—HRT—has side effects, some of which are serious. The feasibility of achieving adherence over a period of a decade, among women of varying SES, ethnicity, and age, is of great concern as a threat to the study in terms of cost and study success.
Secular Trends
There has been a trend toward decreasing the fat content of the diet in the United States during the last decade. The figures used for planning the trial express an expectation of a change in dietary fat intake in control subjects participants from 38 to 34 percent of calories from fat over the duration of the trial. The assumptions made by NIH about secular trends in dietary fat may well underestimate the actual decline. If secular trends are greater than expected, the differential between intervention and control participants will decrease, especially if there is appreciable nonadherence in the DM intervention group, unless the intervention participants are similarly affected and decrease fat intake more than expected. If secular trends among the control participants bring less change than estimated, the ability to test the main hypothesis is enhanced.
It is very difficult to estimate these secular trends. The committee noted the considerable diffusion of low fat health messages to the population, due in part to dietary recommendations by the National Cancer Institute; National Heart, Lung and Blood Institute; the American Heart Association; other health organizations; and purveyors of low fat foods. In opposition to these messages are firmly ingrained food habits, advertising, and the availability of high-fat foods to most segments of the population. Therefore, any estimate of secular trends must be considered uncertain.
Secular trends also apply to HRT. For example, if the PEPI trial publishes favorable results, many women may elect to start HRT. This sort of change would seriously impair the ability of the CT to proceed as planned.
Provision of Health Care Services to Participants
Research funding typically does not cover routine medical care. However, the identification in research studies of health problems in participants without adequate health
care is a difficult problem, whether or not the study had a role in inducing the problem. The current protocol vaguely refers to a regular source of care.
If any study test suggests that a health problem needs further study, you will be sent back to your doctors or clinic, who will evaluate the need for further study. (“Consent form for the hormone replacement therapy part of the women's health initiative clinical trial,” June 28, 1993 WHI Protocol.)
This is not adequately responsible. At least for potentially serious health conditions, and especially for those conditions that might be linked to the study interventions, reliable referral for effective follow-up is essential. The clinical centers should continue to develop adequate links with reliable community providers and adequate follow-up to ensure that care is available. Once this is investigated, it may become essential for the study to pay for some kinds of follow-up for some poor or uninsured women.
Investigators in ongoing NIH projects involving HRT have indicated to the committee that research staff need to spend “considerable time” discussing side effects, associated apprehensions, and decisions with their participants, both in the clinic and on the telephone. Adequate staff time for these activities may not be included in the WHI contract budget. Participants who do not feel their concerns are being taken seriously may drop out, impairing the chances of the study's success.
Study Management
The management of a project of this size represents an unprecedented challenge. Compared to past clinical trials, the WHI CT involves a large number of centers, participants, and scientific questions. Successful management is essential to ensure that protocols are successfully executed and that the primary hypotheses are tested. This depends in part on rapid and effective communication among clinical centers and between clinical centers and NIH staff.
NIH has developed a detailed subcommittee organization to address the different components of the study, a structure that incorporates many Vanguard Clinical Center investigators and staff (see Appendix G ). The committee encourages NIH to enlist staff from additional clinical centers as they are identified. A graphic representation of the study management plan is reprinted with NIH permission, from the June 28, 1993, WHI Protocol, page 55 ( Figure 2-3 ).
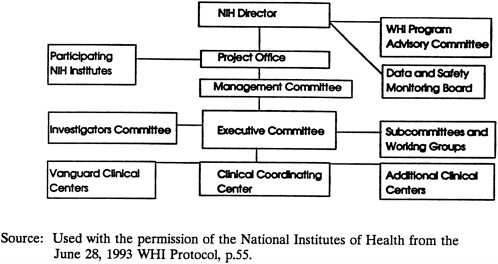
FIGURE 2-3 NIH Organization for the WHI.
DIETARY MODIFICATION BRANCH
Rationale
The DM branch of the WHI CT examines the health effects of reducing total dietary fat to 20 percent of daily calories by reducing saturated fats to less than 7 percent of calories, increasing complex carbohydrate and fiber-containing foods to five or more daily servings of vegetables and fruits, and six or more daily servings of grain products. Each participant will work toward a grams-of-fat-per-day goal based on her weight, height, and activity level. The intervention is considered a “low fat dietary pattern.” Endpoints of interest are breast cancer, colorectal cancer, and coronary heart disease (CHD).
Breast Cancer
The low fat dietary pattern-breast cancer hypothesis is based largely on international comparisons: countries with diets of apparently lower fat content have lower rates of breast cancer. Supporting this association are the results of migrant studies in which, for example, U.S. women whose parents emigrated from Japan, a country with low breast cancer incidence, begin to shift toward the higher American rate of breast cancer. However, in the migrant studies it is not clear whether dietary changes, or other changes, were responsible for the increases of breast cancer incidence. In the international comparisons, it cannot be inferred that those who eat the higher fat diets are the ones who develop breast cancer. In fact, many of these studies estimate the fat content of a country's diet by measuring the
amount of fat produced and sold; it is not known whether it is eaten by women, eaten by their husbands and sons, used as animal feed, or wasted. Some investigators have shown that much of the international variation in breast cancer can be explained by the variation in reproductive factors across populations. Other factors may also be involved.
In attempts to explain the results of the international and migrant studies, researchers have mounted case-control and cohort studies. Evidence from these types of studies, considered to be stronger in causal elucidations than correlational studies, have shown no associations, or at best weak associations, between diet and breast cancer. Results from prospective cohort studies range from slight protection against breast cancer to slightly increased risks associated with a low fat diet. A recent meta-analysis of data from 12 case-control studies (Howe et al., 1990) demonstrated a statistically significant weak association (RR = 1.4) between estimated fat consumption and postmenopausal breast cancer risk, but three large cohort studies have reported conflicting results. If a low fat diet in adulthood affects breast cancer risk, most epidemiologists agree that its effect is likely to be small. Thus, the low fat dietary pattern-breast cancer hypothesis is considered to be quite weak. In addition, other evidence indicates that nutrients other than fat may be important in the etiology of CHD and certain cancers. The anti-oxidant vitamins (particularly vitamins E, A, and possibly C) are of interest as protective factors against CHD and certain cancers.
There are other concerns with the low fat dietary pattern-breast cancer hypothesis; for example, factors early in life, such as diet during adolescence, may be more relevant to breast cancer risk. Alternatively, the number of years during which the woman ate a high fat diet may be more important in establishing breast cancer risk than intervention on dietary practices later in life.
A strong, consistent association of dietary fat with breast cancer has not been established. To plan the trial, it was necessary to make assumptions based on the strength of this putative association, expected lag between behavior change and change in risk, etc. The committee felt that existing information is not sufficiently certain so as to place the assumptions on firm ground.
Colorectal Cancer
While hypotheses regarding breast cancer risk reduction have focused primarily on dietary fat, hypotheses concerning colorectal cancer include dietary fiber as well. International correlational data between dietary factors and colon cancer are much weaker than the corresponding correlations for diet and breast cancer, although the case-control and limited cohort literature show a stronger strength of association than do comparable epidemiologic studies regarding breast cancer. For example, results from a large follow-up study of nurses indicate a positive association between dietary fat and colon cancer (Willett, 1992). A recent literature review by Potter et al. (1993) notes that a diet high in meat, protein, and fat is consistently associated with a higher risk of colon cancer. There is good evidence that increased consumption of fiber-rich foods reduces the risk of colorectal cancer.
In a meta-analysis of 13 case-control studies, Howe et al. (1992) found similar reductions in risk across gender and age (a marker for menopausal status).
Coronary Heart Disease
There is a strong rationale for a dietary study designed to examine the effects of a lipid lowering diet on various cardiovascular disease endpoints. It is known that a diet low in fat, saturated fat, and cholesterol lowers total cholesterol and low-density lipoprotein cholesterol (LDL-C) levels. It is also known that blood lipid levels are a strong risk factor for CHD outcomes.
While lowering LDL-C reduces the risk of CHD, elevated high-density lipoprotein cholesterol (HDL-C) levels have been shown to be the strongest protective risk factor for CHD. Since a low fat diet, in general, reduces HDL-C, it is important to learn how changes in diet affect HDL-C and cardiovascular outcomes.
In practice, many women, including many elderly women, receive low fat, high fiber diet prescriptions from health care professionals. A study the size and duration of the WHI could add to the understanding of the relationships among diet, its components, physiologic results (such as blood lipid levels), and cardiovascular event endpoints. There have been no major intervention studies conducted in women to address the issue of efficacy of primary prevention efforts.
The weight loss or maintenance of appropriate body weight associated with a change to a low fat dietary pattern could contribute to improved health as well. Decreased rates of obesity could relate to decreased risk of many chronic disorders, including hypertension and hyperlipidemia associated with coronary heart disease. However, significant weight loss in elderly women could possibly have negative effects. Certainly the WHI might provide extensive information about the feasibility of following a low fat dietary pattern and the barriers women encounter in achieving adherence, and the relationship between percent calories from fat and obesity and weight control.
Design and Methods
The research objectives of the DM intervention are to study whether a low fat dietary pattern reduces the incidence of breast cancer, colorectal cancer, and CHD. A total of 63,000 women will enter the CT, of whom 48,000 will be in the DM group and 25,000 will be in the HRT group. About 45,000 of these are expected to subsequently enter the CaD component. The average treatment period is nine years. Post-trial mortality and breast and endometrial cancer incidence surveillance is planned (but not funded) for an additional five years to protect against the possibility of missing any adverse effects that may require a longer period of time to develop.
The DM is well planned and intensive. Women will be assigned to a permanent group of 8 to 15 members led by a nutritionist. They will meet weekly for six weeks, biweekly for six weeks, and monthly for nine months. Individual counseling sessions will be scheduled early in the intervention, and women in the intervention arm will receive nutrition materials as well as self-monitoring tools. While the intervention during the first year is standardized among clinics, there is individual flexibility in actual dietary modification and the rate at which changes will be made. Various other activities are aimed at promoting social support among group members.
The intervention program is founded on theory-based research and past experiences of the investigators. It is expected to lead to the desired outcomes (i.e., the nutritional goals) of the intervention, and the committee was impressed that the investigators will use sophisticated approaches to maximize adherence. However, while the past experiences of the investigators have been successful, there are no data that demonstrate the effectiveness of such a low fat diet over a nine-year period in women of the age-range encompassed in the CT. Furthermore, while many of the investigators have had experience working with postmenopausal women, the literature does not reflect substantial experience with 70- to 79-year-old women following a low fat eating pattern for nine years.
The safety of a low fat eating pattern remains to be established, an issue that is of particular importance in elderly women, for whom many nutritional problems are prevalent. The ability of women with limited financial means to adhere to such a diet is a concern, as well. From discussions with WHI CT investigators, the committee learned that the safety of the diet is an issue of primary importance to the DSMB, and CT nutritionists plan to monitor all intervention women (and pay particular attention to elderly women) for possible adverse effects of a low fat dietary pattern. However, this attentiveness is not apparent in the written WHI Protocol, and the concern is not apparent in the consent process in which participants need to be informed of the need to maintain an adequate caloric intake.
Dietary assessment will be conducted using several different techniques. A four-day baseline food record will be analyzed using the University of Minnesota Nutrition Data System, which has one of the most comprehensive food product and nutrient data bases in the world. It has become the leading U.S. nutrient data base resource for scientific research. A food frequency questionnaire will be collected at selected annual visits on all CT participants. A subsample of these women will be asked to provide a four-day food record. In addition, a subsample will complete a 24-hour dietary recall every 12 months. A semi-quantitative food frequency questionnaire will be administered to women in both the CT and the OS at the first screening visit. The lack of sensitivity of that instrument may seriously underestimate fat intake and thereby hinder recruitment efforts.
Accurate dietary assessment methodologies are essential to the success of the DM. Major sources of error include data-collection methodologies (i.e., the sensitivity and validity of the instruments and methods available); data analysis (i.e., nutrient data base completeness and accuracy); and poor reporting of food intake due to participants ' inability
to remember total intake, estimate portion sizes accurately, include important descriptors about foods and food preparations techniques, and/or provide truthful information (which is limited for a variety of reasons).
Many potential problems inherent in the collection and analysis of dietary data can be avoided or minimized by having trained nutritionists responsible for data collection and the quality assurance. Moreover, choosing data collection instruments that have been tested widely and using a reputable nutrient data base will also minimize errors and help ensure that the data collected are accurate. The data collected in this trial are likely to be the best possible given the limitations of current, state-of-the-art dietary assessment methodologies.
There is considerable debate in the literature about the validity of these measures. The absence of a clear biological marker for dietary fat makes validity difficult to establish. The trial proposes the use of measures of dietary intake that are state-of-the-art at the present time. It is clear that women in feasibility trials are reporting impressive reductions in dietary fat. What is less clear is the degree to which these reports reflect actual intake. At the present time, there is no viable alternative to self-report techniques.
Weighing Benefits and Uncertainties of the Breast Cancer Arm
Some degree of uncertainty in the hypothesis and feasibility of the methods is inherent in any clinical trial; if one were certain about the outcome, one would not need to do a clinical trial. With uncertainty comes risk and potential benefits. Considerable benefit is gained when assumptions are met, a trial is successful, cause and effect have been addressed, and the public health implications are clear. Uncertainty arises from the possibility that assumptions are faulty and the study hypotheses cannot be tested.
The potential benefits of a study that would demonstrate an effect of diet on breast cancer are enormous. Breast cancer is a particularly frightening disease for women. The existence of a lifestyle change that could reduce risk would be of considerable benefit, both because of the risk reduction itself and the perceived control placed in the hands of potential victims. The primary threat to the DM branch of the CT is that the results will not clearly answer whether diet modification affects incidence of breast cancer. This arm of the study stands in jeopardy if key assumptions are incorrect, and the costs of such an outcome would be enormous:
-
A massive expenditure would bring little tangible benefit.
-
Funds would have been diverted from other studies that might ultimately have proven more beneficial to women's health.
-
Ambiguous results might result in the belief that dietary change is not important to breast cancer, when in fact a link may exist.
-
The negative effects of altered diet would be magnified in the absence of clear benefits.
-
The failure of this very visible study would erode support for further initiatives in the women's health arena, or in respect to diet effects on health.
Such threats are encountered in any clinical trial and are not unique to the WHI CT. It was the committee's finding, however, that elucidating the breast cancer outcome of the DM contains more than the usual hazard of a clinical trial because of uncertainties of fundamental assumptions. The link between dietary fat and breast cancer is weak and inconsistent. Whether intervention in the specified age group is the most advantageous time for change is in doubt. Whether women will adhere to such a strict diet for so many years is uncertain. Whether secular reductions in dietary fat will be modest is also uncertain.
Because of these uncertainties, there was some disagreement among committee members about whether a trial designed to test the diet and breast cancer hypothesis was justified. The committee agreed, however, that the DM branch of the CT has the potential to test the effect of DM (i.e., decreased fat and increased fruits, vegetables, and grains) on risk of cardiovascular disease and colorectal cancer, and was justifiable on those grounds. Therefore, despite disagreement among committee members over the strength of the scientific evidence supporting the low fat dietary pattern-breast cancer hypothesis, and the feasibility of testing that hypothesis, the committee agreed that the DM branch of the CT could proceed, with the recommendations specified.
The committee also noted that because public expectation is high that this trial will have definitive results regarding the diet and breast cancer hypothesis, NIH should act to limit those expectations.
HORMONE REPLACEMENT THERAPY BRANCH
Rationale
Numerous studies have examined the relationship between exogenous estrogen use and coronary heart disease (CHD), and have generally reported beneficial effects. It is not clear whether the apparent benefits of HRT (from observational data) are due to a process of self-selection by which healthier women are prescribed HRT, or by other selection biases in the inclusion of participants or in the reporting of results. These biases may both exaggerate the apparent benefit of HRT and underestimate the magnitude of adverse effects. This branch of the CT is designed to assess the benefits and risks of HRT on CHD, cancers of the breast and endometrium, fracture rates (especially hip fractures), quality of life, and total mortality. The HRT branch also will provide information on the factors (such as effects on plasma lipids, clotting factors, blood pressure, plasma insulin, and body fat distribution) that may influence the putative protective effect of estrogens on CHD.
Women use estrogens during the menopause primarily to decrease various unpleasant symptoms—such as hot flashes and vaginal dryness—related to estrogen deficit. Other positive
and negative effects have been observed, including fewer cardiovascular events and deaths, decreased bone loss, and increased endometrial cancer. Adding a progestin to balance the estrogen restores the endometrial cancer risk to its lower rate. In many women, however, the addition of a progestin causes unpleasant and sometimes serious symptoms.
It is hypothesized that if estrogen decreases bone loss, and bone loss is a risk factor for fractures, then HRT will result in fewer fractures. A critical question is whether HRT increases breast cancer risk. It is unknown whether adding a progestin to HRT increases the risk of breast cancer, attenuates or enhances the estrogen-induced decrease in the rate of bone loss, or attenuates the putative cardiovascular advantage conferred by estrogen.
Small randomized trials have shown that estrogen replacement affects HDL-C and LDL-C levels in directions that would be expected to reduce the risk for coronary heart disease (CHD). The largest proportion of deaths from a single cause in the age group on which the WHI is focused will be from CHD, and therefore the effect of HRT on CHD will greatly influence the effect on mortality from all causes.
HRT with both estrogen and progestin (PERT) has been in common use for a shorter period of time than HRT with estrogen alone (ERT) in the United States, and evidence about the long-term effects of PERT is less certain. Cyclical use of PERT prevents or greatly retards bone loss, although it is uncertain whether the beneficial effect of PERT is greater than that of ERT alone. Any effects of PERT on hip fracture and colorectal cancer risk have not been reported to date. Based on studies of effects of PERT on lipoprotein levels, the beneficial effect of combined therapy may be less than that of ERT alone, although that would be dependent to some extent on the particular progestin used. Whether to use ERT or PERT is an important question among many postmenopausal women and the clinicians who advise them. However, the CT is not designed to test ERT versus PERT.
The effects of initiating HRT at various ages after the menopause have not been well studied. The proposed trial would offer the opportunity to study risks and benefits associated with initiating HRT at older ages.
Design and Methods
In the HRT branch of the trial, 25,000 women will be stratified on the basis of the presence or absence of a uterus. Women with a uterus will be randomized to one of three arms: (1) conjugated equine estrogen (0.625 mg per day); (2) conjugated equine estrogen (0.625 mg per day) plus medroxyprogesterone (2.5 mg per day continuously); and (3) placebo. They will be randomized to the three groups in the ratio of 7:5:8. Women without a uterus will be randomized to one of two arms: (1) conjugated equine estrogen (0.625 mg per day); and (2) placebo. They will be randomized to the two groups in the ratio of 7:5. The percentage of women with a hysterectomy at baseline will be restricted to 30 percent. Power to compare the effects of HRT versus placebo on CHD incidence will be adequate,
while the power to detect differences in effects of women in one hormonal group compared to the other will be limited. A variety of exclusion criteria will be applied (see Appendix A).
The committee identified the following unresolved issues with regard to this design:
-
The study is likely to terminate early because of evidence demonstrating protection against CHD, thereby precluding the identification of later occurring outcomes.
-
Limiting study enrollment to moderate and high-risk women for CHD might maximize the likelihood of early detection of a possible protective effect. A disadvantage would be that the trial likely would need to be stopped even earlier (before the breast cancer association could be learned) because of the greater difference of protective effect on CHD between the treated and control groups.
-
The trial would be more informative if the effects of ERT could be compared to the effects of PERT. To do so, the study would need to either increase the sample size, use women at higher risk of the primary endpoint, or change the ratio of participants randomized to different regimens to increase the power of comparison.
-
Blinding to the study participants will only be partial because of symptoms associated with the various regimens.
-
Knowledge of reported symptoms and the relationship between such knowledge and adherence would be a useful result to come from this study.
-
Endometrial aspiration will be done annually on all women on ERT, 5 percent of women on PERT, and 5 percent of women on placebo. Endometrial biopsy will be done only at the request of the clinic consulting gynecologist. The committee is concerned that a 5 percent sample of women on PERT may not be adequate. Also, the committee is aware that some women (with estimates ranging to 40 percent) receiving an unopposed dose of 0.625 mg of conjugated equine estrogen will develop endometrial hyperplasia within one year. It has been suggested that a short course of medroxyprogesterone periodically would reduce this incidence.
-
Adherence will be measured by the relatively weak method of pill counts. Alternatives might be blood or urine checks at already scheduled six-month visits.
-
It may be more difficult than anticipated to enroll women in this branch of the CT, since many women (or their physicians) will already have decided whether they wish to be on one of these regimens, especially among women in their 70s at randomization.
-
Hormones may have different effects on risks for colon and rectal cancers.
-
Perimenopausal bone loss requires differential informing of these women and probably also subset analyses that focus upon this effect.
Threats to Completion of the HRT Branch
It is generally accepted that ERT increases the risk for endometrial cancer and decreases the risk for low bone mass. The few observational studies with adequate numbers of long-term users of ERT suggest that the risk for breast cancer is increased somewhat (30
to 80 percent) among long-term users. The effect of ERT on risk for colorectal cancer is uncertain.
The increased risk of endometrial and breast cancer among women on ERT is also an ethical concern, especially since women at high risk for these cancers will be randomly assigned to this treatment group. It is reassuring that they will be followed more closely. Nonetheless, it is important for these women to be fully informed of the risks of HRT during the informed consent process.
Since PERT has been in common use for a shorter period of time than ERT, evidence about the long-term effects of this regimen is less certain. In addition, the effects of starting these hormonal regimens at various ages after the menopause have not been well studied. At present women and their doctors are often making HRT decisions with conflicting information. The proposed trial would offer the opportunity to study risks and benefits associated with initiating HRT at different ages. Given the importance of HRT to a large number of women, work in this area should be a high priority.
Successful completion of this branch of the WHI CT will require a great deal of effort to ensure an acceptable level of adherence with the HRT regimen. In addition, it will be important to minimize cross-over between the treatment and control groups during the trial. There is concern about both of these major potential threats to the successful completion of the HRT branch. With respect to adherence, it is unclear whether women will tolerate the side effects of HRT. This is an especially important point for the older cohort, for which there are no adequate data. These women may be more sensitive to the side effects of HRT and less tolerant of them. A high dropout rate would compromise the integrity of this branch of the CT. Another important concern deals with probable “drop-ins” from the control group initiating HRT due to new scientific findings during the scheduled course of the WHI. For example, the results of the highly visible PEPI trial are forthcoming and likely will have a significant impact on medical practice. Depending on what the results of PEPI are, there could be an appreciable control group contamination, which would confound the findings of the CT.
It is also likely that some women in the intervention group will change their hormone replacement formulations, upon the advice of their own physicians, as a result of side effects or new information, and thus will cause a contamination of the intervention group. Should this happen with a sizeable number of women in the intervention group, the results of the HRT branch would be uninterpretable.
While the issues of dropouts and drop-ins are a potential threat to the successful completion of the HRT branch, it is important to appreciate that there are no data to predict the magnitude of this potential problem. The WHI will thus provide important information about adherence with HRT regimens in women aged 50-79. Finally, it is likely that the effect of ERT on CHD will be seen before the scheduled completion of the CT. The participants of the study should be informed of this finding and asked whether they wish
to continue their participation. Given this option, there is concern that this branch of the trial will be discontinued and the effects of HRT on cancer risk will be unable to be assessed.
Use of HRT in Elderly Women
The committee does not believe that the WHI Protocol addresses sufficiently the risks versus the benefits of initiation of HRT in the older study cohort (70-79 years). Therefore the committee presents a more extensive literature review in this section than in the other sections. Among the issues of interest with respect to the introduction of estrogens in an elderly population are several that are reasons to include the elderly in the HRT branch. The committee urges NIH to carefully examine those of the following issues that are amenable to analysis in the WHI trial:
-
Will estrogens still be of value in patients with a high degree of osteoporosis?
-
Will they afford protection against age-associated cardiovascular disease?
-
Are ERT and PERT tolerated by the elderly?
-
Will ERT and PERT increase the risk of thromboembolism in an age group already vulnerable to thromboembolism?
-
Will ERT and PERT increase the risk of breast cancer in this age group?
-
Will the benefits outweigh the potential risks?
-
What dose(s) of conjugated estrogens should be used in the elderly?
Osteoporosis
The effect of estrogens in the elderly has been studied by a number of investigators. Quigley et al. (1987) found that patients who begin estrogens in the early postmenopausal years or even in the seventh decade of life will continue to receive benefit with continued use. Those who have never used estrogens until the eighth decade may or may not benefit, depending, probably, upon how much estrogen-dependent bone they still have to lose. Overall, the mean percentage decrease in bone density per year was low in women over the age of 70, and the decrease was similar in estrogen users and nonusers. Adverse events were not discussed. The authors recommended that a double-blind placebo-controlled study be performed in women over the age of 70 to answer the question of whether initiation of ERT would be useful in this group.
In a placebo-controlled trial, Christiansen (1991) observed an increase in bone mineral content in women aged 70 and over treated with estradiol plus norethindrone acetate, versus a decrease in bone mineral content in the placebo group. The effect was greater in trabecular than in cortical bone. Adverse events were not discussed.
A dose of 0.625 mg of conjugated estrogens has been found to be effective in postmenopausal women for prevention or treatment of osteoporosis. Some investigators feel
that .3 mg together with calcium is sufficient for women over the age of 70, while others feel that as much as 1.25 mg is required. A dose of 1.25 mg in the elderly may not be advisable, however, because of possible adverse effects on clotting factor and symptomatic side effects.
Morbidity, Mortality, and Potential Risks of HRT
There do not appear to be any systematic studies on whether estrogen used for the first time after the age of 70 is associated with any change in cardiovascular risk or overall mortality. Data do, however, exist on “current” estrogen users and ever-users. For example, in a report on cognitive function in a cohort of 800 estrogen users and non-users aged 65 to 95 years (mean 77 years), Barrett-Connor and Kritz-Silverstein (1993) mention that women using ERT had an age-adjusted risk of death of 0.75 compared with nonusers. Most studies of postmenopausal estrogen users have found a decrease in cardiovascular events or all-cause mortality as compared with nonusers. These are summarized in review papers by Stampfer and Colditz (1991) and Wren (1992). However, in two studies, examination of cardiovascular events or all-cause mortality in elderly women showed no clear-cut advantage and, perhaps, an increased risk of cardiovascular events. Bush et al. (1983) reported a significant overall advantage in all-cause mortality in white female estrogen users aged 40 to approximately 79. This was also true in the 70-79 year age range (RR = 0.68). However, in the non-hysterectomized, non-oophorectomized subfraction of the 70- to 79-year-olds (which constituted 56 percent of this age range), the relative risk was higher (1.6) in estrogen users than in nonusers. It is possible that hysterectomized and oophorectomized women began estrogens at an earlier age than non-hysterectomized women, and perhaps the beneficial cardiovascular effects in the oophorectomized women outweighed any potential risks from the estrogen effects on blood clotting factors.
In an analysis of women 50-83 years of age in the Framingham study, Wilson (1985) reported no beneficial effect on all-cause mortality in estrogen users and an increased risk of cardiovascular events, including stroke and coronary heart disease, in estrogen users of all age groups. The risk was somewhat less in the age range of 70 to 83 but there were few subjects in that subgroup. As noted above, most other studies found reduced risk of CHD in estrogen users. The authors speculated that the difference between their results and those of others may have been due in part to the ascertainment in the Framingham study of unsuspected cases of myocardial infarction. (Thirty-five percent of the myocardial infarction cases were clinically unrecognized but were observed by changes in ECG from previous readings). In addition analysis of “interval use” of estrogens was used as compared with cross-sectional classification.
Women taking oral contraceptives are at increased risk of myocardial infarction, stroke, and venous thromboembolism, due probably in large part to the effect of estrogens on blood clotting factors and, perhaps, also to the decrease in HDL-C from the progestogen component and an increase in blood pressure from the estrogen component. The risk is greatest in those with underlying cardiovascular risk factors, especially smoking, and the risk increases with age.
In postmenopausal women with lower levels of estrogen, HRT should pose a lesser risk than in premenopausal women. There have been a number of papers on clotting factors in postmenopausal women receiving estrogens. Stangel et al. (1977) reported low antithrombin III activity in 57 percent of postmenopausal women receiving estrogen, as compared with 15 percent of those not using estrogen. The dose of estrogen (1.25 mg conjugated estrogens) was, however, high by current standards. In a review paper, Wren (1992) noted that although several groups reported an increase in various clotting factors with 1.25 mg conjugated estrogens, other studies reported no differences in various clotting factors between estrogen users (0.625 mg to 1.25 mg conjugated estrogens) and nonusers. This was postulated to be due to a spontaneous increase in antithrombin III and other anticlotting factors with increasing age, thus negating any possible adverse effect of estrogens. In a case-control study of women between ages 48 and 87 (mean 65) who experienced venous thrombosis, Devor et al. (1992) reported a similar incidence of current estrogen use in cases (5 percent) and in controls (6 percent). The study had the power to detect only a twofold or greater risk.
Some women appear to be very sensitive to the clotting effects of estrogen. In addition, some older women appear to have a surprisingly ample degree of estrogenic activity. It is possible that, in these women—particularly if they are obese, smokers, sedentary, diabetic, hypertensive, or hyperlipidemic—administrationof estrogens may sufficiently increase the risk of thrombotic events to counteract the salubrious effect of estrogens on HDL- and LDL-cholesterol and vascular endothelium. (The same caveat applies to younger postmenopausal women as well; the same dosage is typically dispensed by physicians and will be administered in the WHI regardless of age and body size.)
Finally, elderly women experience a higher incidence of unacceptable breast tenderness and breakthrough bleeding when estrogens are administered than do younger postmenopausal women. These side effects may cause women to drop out of the CT; clinical staff should be aware of these issues and respond to participants' concerns appropriately. It may be that elderly women require lower doses to produce a given estrogenic effect than do younger postmenopausal women.
Summary
Studies conclusively demonstrate that estrogen therapy has a positive effect on bone mineral density in younger postmenopausal women, and that this effect continues with continued use into the elderly age range. Data also strongly suggest a positive effect on cardiovascular disease in younger and older postmenopausal women. Some elderly women may experience a benefit on osteoporosis from introduction of estrogens if they have sufficient estrogen-dependent bone remaining. It is, however, unknown whether introduction of estrogens in the elderly will result in a positive or negative effect on cardiovascular events and mortality. Also, the relative magnitude and timing of these effects remain fairly uncertain. Thus, the CT will serve an especially important role in helping to elucidate the benefits and/or risks of HRT in women over the age of 70.
CALCIUM AND VITAMIN D SUPPLEMENT BRANCH
Rationale
There is some evidence that the use of calcium in the form of supplements reduces the risk of osteoporosis and resulting fractures, which are serious causes of morbidity for older women. Approximately one-third of cortical bone and one-half of trabecular bone is lost through osteoporosis in postmenopausal women. The rates of bone loss may reach three to five percent per year immediately following menopause, and one percent per year in older women. Although fractures are not a major overall cause of mortality, death from complications of hip fractures (such as thromboembolism, fat embolism, pneumonia, and surgical deaths) are high, and fractures account for much morbidity and dysmobility. The annual incidence of fractures is 0.5 percent of women aged 55-64, doubles to 1 percent of women ages 65-74, and more than doubles again to 2.3 percent in women aged 75-84. Hip fractures will be the primary endpoint for the CaD branch of the CT.
Most women do not have an adequate daily intake of calcium. Postmenopausal women require 1,500 mg per day, yet 75 to 80 percent of women have daily intakes below 800 mg per day (1984 NIH Consensus Conference, referenced in the June 28, 1993 WHI Protocol). The intestinal absorption of calcium declines with age, increasing the probability that calcium in the diet is insufficient to prevent bone loss.
Some investigators have found that the addition of vitamin D increases the effect of supplemental calcium on the prevention of bone loss. It is uncertain if this is because the absorption of calcium is enhanced, or if vitamin D exerts an independent effect on bone (Dawson-Hughes et al., 1991, referenced in the June 28, 1993 WHI Protocol). A subsidiary aim of the CaD branch will be to test the effect of supplementation on bone mineral density. Bone mineral density measurements will be made at only three Vanguard Clinical Centers (it is unclear how many of the additional clinical centers will measure bone mineral density). Changes in bone density over the course of the study will be examined in relation to each branch of the CT.
The CaD branch of the CT is not the primary motivator for the WHI, and it could not stand alone as the justification for the trial. However, it can be justified as part of the CT. In addition, it may provide valuable information on the interaction of CaD supplementation and the DM and HRT interventions. For example, estrogen is known to increase intestinal calcium absorption (as well as reduce renal calculi formation). Therefore, it may be possible to test the hypothesis that HRT and calcium together protect women from osteoporosis. In contrast, low fat diets are frequently low in calcium because of the reduction of dairy foods, and although a reduction of calcium has not been seen in the feasibility studies for the WHI, it will be useful to have a subsample of women in the DM who are also taking calcium.
Colorectal cancer may be related to intake of calcium, and will be a secondary endpoint for the CaD branch. The association between calcium, vitamin D, and colon cancer has also been studied in several correlation, case-cohort, and control studies. The evidence is mixed, with some studies suggesting inverse associations between calcium intake and colon cancer, and others showing no association. Fewer studies have focused on the role that vitamin D plays in colorectal cancer risk, but a strong association has not been identified. It is of course not possible to separate the effects of calcium and vitamin D when they are issued together.
Many women of all ages are currently taking calcium supplements. A clinical trial in which definitive results are provided is necessary for women and their physicians to make informed choices, particularly if responsive subgroups can be identified, such as women who are and are not on HRT.
Design and Methods
Women who are already randomized to the HRT and DM branches will be asked at their one-year anniversary if they are interested in joining the CaD supplementation branch. It is anticipated that 45,000 of the 63,000 women in the CT will be randomized in a 1:1 ratio to either (a) calcium carbonate containing 1,000 mg elemental calcium per day plus vitamin D3 400 International Units per day, with meals (dispensed as two tablets, each with 500 mg elemental calcium plus 200 IU vitamin D3), or (b) placebo dispensed as two tablets. Participants and clinic staff will not be told who is in which group.
Outcome information on hip fractures will be collected by annual mailed questionnaires and at all follow-up visits, and documented primarily by X-ray report and discharge summary. Outcome information for other fractures will be collected by annual mailed questionnaires and at all follow-up visits with self-report, and documented by a physician's diagnosis or hospital discharge summary. Outcome information on colorectal cancer will be collected by annual mailed questionnaires and at all follow-up visits, and will be documented by medical report.
The committee is concerned about the inability of this branch to separate the effects of calcium and the effects of vitamin D. Several documents sent to the committee by the NIH provide a fragmented picture of the process through which the decision was made to do only a two-way randomization (CaD versus placebo). The minutes of the Concept Review group for the CT/OS Component of the WHI reflect mixed views in the 10-person panel, and recommend that the NIH planning group “reconsider the question of a 3-group versus 2-group design and assess power for combined fractures and hip fractures.” At an advisory meeting on August 15, 1991, the participants strongly encouraged a three-way randomization (calcium, calcium and vitamin D, or placebo). This would permit assessment of whether vitamin D made an independent contribution, although the magnitude of the effect of vitamin D given alone would not be known in this trial.
In February 1992, NIH considered the statistical implications of a three-way randomization. * The authors assumed that the fracture rate is reduced by 30 percent when either calcium alone or vitamin D alone is used, and that adding vitamin D to calcium would increase the effect an additional five or ten percent. With the designed sample size there would be insufficient power to test this well. Therefore, the statisticians recommended not using a full factorial design for this component.
While acknowledging that the practical elements of power, sample size, and cost are necessarily limiting issues, the committee is concerned that the decision to forego testing a three-way randomization was based, apparently, on statistical expediency. There is no evidence provided that NIH considered increasing the sample size of this branch, which could be considered relatively inexpensive since there would be many CT participants not already in the CaD test.
Threats to Successful Completion of CaD Branch
As was discussed above with regard to DM endpoints, colorectal cancer will not be detected systematically in the study. Its detection will rely on information from follow-up visits.
Although compliance is expected to be adequate, it is also quite difficult to monitor. Motivation may be more limited. If a substantial number of pills are not actually taken, the opportunity to see an effect will rapidly diminish.
CLINICAL TRIAL COST
In addressing the issue of cost, the IOM committee considered two different cost components. The first deals with basic accounting: Did the applicants correctly estimate, for example, the cost of one full-time equivalent dietitian? Did NIH check that the applicants correctly multiplied that cost by the number of dietitians to be used? The routine contract-processing done by NIH performs this audit-type function, and the committee chose not to pursue this form of additional audit.
The second—and more appropriate to the committee's charge—cost issue involves whether the Center applicants and NIH assessed appropriately the nature and amount of human and material resources (staff level and distribution, equipment, etc.) necessary to perform adequately the tasks required by the contracts. To determine whether the choices of resources seem adequate, the committee engaged in three activities:
|
* |
L. Freedman and E. Lakatos, National Cancer Institute, February 11, 1992 memorandum. |
-
Staff and other expenses were compared across the funded Vanguard Clinical Centers, to see if the distribution and/or outliers might provide clues to consistency of funding and whether problems, if any, were localized or studywide;
-
Cost information from other NIH-funded clinical trials was compared, to see whether the relationship between estimated WHI CT costs and work scope was similar to that of other trials; and
-
Using personal professional experiences with similar interventions, communities, or endpoints, committee members individually judged whether staffing patterns, intervention components, and overall budget seemed sufficient.
Data Available to Committee Deliberations
The fact that this committee was formed after the funding of the Vanguard Clinical Centers should have been helpful to the budget evaluation, since, rather than mere plans for funding, negotiated contracts existed. However, the task of this committee was made difficult by the repeated failure of NIH to provide cost information in a usable format. For example, NIH considered the institution-specific data on indirect costs to be confidential and insisted that release of that information would affect future NIH negotiations with contract applicants. On the other hand, the NIH did provide average budgets from which to judge some of the issues related to long-term cost assessments.* The Clinical Coordinating Center and some of the Vanguard Clinical Centers were forthcoming, providing specific information about the budget issues. Of necessity, however, much of the analysis of budget was, however based on expert judgment, comparison to other historical and similar information, and the contractual liabilities to which NIH and the Vanguard Clinical Center host institutions had agreed.
|
* |
On July 28, 1993, the NIH Research Contracts Branch, DCG/OA/OD, issued an amendment to its contract solicitation for additional WHI clinical centers (NIH-WH-93-30 E/W dated July 2, 1993, “Clinical Centers for the Clinical Trial and Observational Study of the Women's Health Initiative—East/West”). The amendment included the cost information regarding the WHI that NIH had provided to the National Academy of Sciences (NAS). It also notified potential applicants that other materials that had been made available to NAS would be available in the RFP Reading Room in the Federal Building in Bethesda, Maryland. The NIH project officer assigned to the IOM study noted that this was done so that the IOM committee members who may be affiliated with institutions applying for contracts did not bring information to the contract application that put their institutions at an advantage. |
WHI Cost Relative to Other Large NIH Studies
From the time of its initial contract negotiations with NIH, IOM sought data on the scope and cost of other clinical trials, particularly multicenter, long-term trials. On August 3, 1993, the committee received from NIH a packet of extremely useful scope and cost information, from which it built the display in Appendix H.
In order to gain a quantitative comparison of the WHI relative to other similar, albeit smaller, efforts at NIH, the cost per participant per year was computed from the total costs, total duration, and sample size of 55 NIH studies. These studies were conducted from the 1970s to the 1990s. No adjustments or discounts for inflation were applied. Because some studies provided are clinical trials, some observational studies, some large and some small, some multicenter and some single center, the comparison of costs per participant per year for some studies may be problematic for the comparison with WHI CT costs.
The average cost of all studies for which total costs, duration, and sample size were provided was slightly over $2,300 per participant per year. For studies that were initiated in the 1990s, this figure exceeds $3,000. Information on the cost of the CT was estimated by the Clinical Coordinating Center to be $586 million, consisting of approximately $168 million for the Vanguard Clinical Centers, $142 million for the Clinical Coordinating Center, and the remainder for the 29 additional clinical centers about to be selected. Whereas completed NIH studies include all costs (start-up, trial, follow-up, and close-out), the CT calculation can only consist of estimated costs. To arrive at the estimated cost per participant per year the committee used the estimated average follow-up period of nine years (from WHI power computations) to reflect the person years of follow-up. This number was multiplied by the 63,000 participants to yield the total number of person-years of participation. The anticipated NIH contribution of $586 million was then divided by the resulting 567,000 person-years, yielding a cost per participant per year of $1,034. The costs associated with the OS are not excluded, which makes the estimate conservative in that including the OS participants further deflates the cost per participant per year. For example, if the cost of the OS is $15 million, and this is subtracted from the total cost, the cost per participant per year drops to $1,007.
Comparisons of the WHI costs with those of similar trials yield similar disparity. For example, the PEPI trial costs slightly in excess of $2,000 per participant per year, and the Women's Health Trial (WHT) Minority Feasibility pilot studies in excess of $3,000. Despite the size and complexity of the WHI, requiring a great deal of staff emphasis, the WHI funding per person per year is less than half that for other NIH studies of women's health, including specifically those that use similar drug regimens (e.g., PEPI) and approach (e.g., WHT Minority Feasibility Study).
Clinical Center Funding
The average 12-year budget for a Vanguard Clinical Center is approximately $10.5 million. Appendix I presents the budget as provided by NIH. While the additional clinical centers have not been selected or contracts negotiated, NIH officials told the committee that they expect to fund these centers at lower rates, even though they would be expected to cover the increases in sample size that are now required. *
The average indirect cost rate shown on NIH summary documents is lower than on-campus rates of most universities. The acceptance of lower indirect cost rates implies the institutions will be covering the costs for some services that would normally have been paid for by higher indirect cost rates. Some Vanguard Clinical Center representatives suggested that support for WHI activities provided by their institution is through accepting off-campus rates. None of the centers that responded to the committee stated that their institutions were actually providing the direct 0.23 full-time equivalent support shown in the average data from NIH (however, only a few Vanguard Clinical Centers provided information). Taking these factors into account, the cost per new clinical center could be $10 million.
The investigators in the Vanguard Clinical Centers have made numerous adjustments and attempts at efficiencies in the CT—such as proposing the use of trained volunteers and economies of scale in purchasing —in order to arrive at acceptable cost projections. The committee appreciates the enormous efforts that have already gone into the cost cutting and protocol modifications designed to produce cost efficiencies and savings. Nonetheless, the committee felt that the costs budgeted for the Vanguard and additional clinical centers are low for the extensive effort necessary.
NIH negotiated contract funding amounts with each Vanguard Clinical Center based on estimates of future costs, including an official inflation factor. What is unpredictable in the specific but to be expected in general is that, over the course of this 12-year trial, medical technologies, practice patterns, and pharmaceutical agents will continue to evolve. Past experience demonstrates that these advances are often accompanied by increased costs. The Vanguard Clinical Center representatives did not have a shared view of what would be expected of their projects, their institutions, or NIH, should increased costs impinge on the functioning or quality of the trial. This led the committee to explore NIH funding in relationship to the total cost of the WHI.
|
* |
The June 28, 1993 WHI Protocol notes an increase based on changes in age distribution, overlap of interventions, and other revised assumptions. |
Total Cost
Critics say that all three WHI components cannot be done for the announced costs of $625 million or, alternatively, that the announced costs are excessive. To determine whether there are sufficient resources to carry out the CT, OS, and CPS as designed, the committee also looked at non-NIH sources of funding committed to WHI activities. *
Institutional Support
The direct cost contributions of the contracting institutions have not generally been considered in the critics' costing of the study. The cost assessment must therefore address both the funded and hidden costs of the studies. Institutions have agreed to support the CT to varying degrees, and that support represents real costs to be considered in the committee's assessment. This is not unique to the WHI nor does it represent a departure for NIH in its research funding, but there are also tangible and intangible institutional benefits to giving specific contributed support in order to secure a clinical or coordinating center. It is the extent of these hidden costs, and the financial basis upon which they are to be covered, that is an issue.
The committee has two very different concerns about the apparent expectation of and reliance on institutional support. First, it would like to consider the effects such a policy has on which centers receive NIH research funding. If it is essential that an institution make significant extra-contract support available to a project in order to receive research funding, some institutions are necessarily excluded from the application process, possibly creating an unintended effect of skewing contracts away from very capable investigators who are located at less well-endowed, less established, or more financially wary research institutions. Second, but with more direct relevance to the WHI, the committee is concerned that when seeking additional centers, the NIH might not be able to identify 29 institutions with both qualified investigators and the ability to provide substantial institutional support. The Vanguard Clinical Centers are very likely to be more sophisticated and experienced centers, which may enable them to be more efficient and have more resources with which to support the efforts of the trial. Will the forty-third, forty-fourth, and forty-fifth centers chosen have the experience to carry out the tasks of high quality research with very limited resources? If not, the overall quality of the WHI is threatened. In that context, this issue falls under this committee's domain.
Changes in Scope of Work
A related issue is the extent to which the investigators understand these institutional commitments. There was a diversity of opinion among the Vanguard Clinical Center
|
* |
Staff effort at the NIH Office of the Director, NHLBI, NCI, and ORWH has been substantial and is not accounted for in the WHI $625 million budget. |
investigators as to what the contract actually requires financially in terms of anticipated or unanticipated changes in the sample size, scope of work, etc. This must be clarified. For example, if several years into this project a PI faces greater expenses than were budgeted, how will this be handled? Will NIH provide additional support, will the institution in which the clinical center is based provide necessary funding—at the expense of the PI's other projects or from general funds? Or will the quality of work on the WHI tasks suffer? Some PIs believe that by signing the contract, the institution agreed to pick up any necessary additional costs. Others see a more standard contract, in which NIH would allocate more money if the scope of work were to change. What if, though, the scope were to remain the same but the costs increase? Still other investigators assume that NIH would not allow its investment to founder and would provide additional funding if necessary.
Ancillary Studies
The committee recognizes that Vanguard Clinical Center investigators and others have plans to request funds—from NIH and other public and private sources—to carry out studies ancillary to the WHI. Although adding in the funding amounts of those studies would increase the total cost associated with the WHI, the committee believes ancillary studies represent anticipated and desired side benefits to such a large trial. NIH has already proposed a mechanism for review of ancillary studies.
Potential Causes of Budget Shortfalls
Despite elegant planning and budgeting, there are predictable threats to maintaining a study of this scope within its budget. These include difficulty in recruiting participants, unanticipated staff turnover, inadequate adherence to the protocol, and larger than estimated cross-overs. As any one of these occurs, the budget will affected; for each additional problem encountered, the budget will be further challenged.
Participant Recruitment
If participant recruitment were to lag, there is no money available for increased—and usually more costly—staff time, clinic hours, or promotional materials.
In response to questions by the committee regarding the ability to recruit minorities and older women and to monitor various demographic characteristics, such as SES and age level, during recruitment and enrollment, NIH pointed out repeatedly that the Vanguard Clinical Center investigators have signed contracts to produce specific recruitment results for the monies allocated. If a clinical center has insufficient funds in reserve to accomplish this, however, it will threaten the validity of the science in a number of ways, but most prominently in diminishing the power of the CT by reducing the person years of follow-up. If NIH plans to drop centers experiencing recruitment delays (the possibility for which has been adequately planned by randomizing within centers), those person-years attributable to
a clinical center will be lost, thus weakening the study. These person-years of follow-up can be regained, but only at additional cost by increasing enrollment at other centers and/or extending the study. Thus, if the funding is not adequate to recruit and to implement the interventions with appropriate intensity, the tests of the hypotheses will suffer.
If the informed consent interview (discussed above) were to include, as recommended by the committee, a fuller description of possible risks and benefits, along with estimated probabilities of their occurring for a given participant, fewer women may consent to be randomized. This could slow the recruitment rate or increase the efforts needed to compensate for the higher refusal rate. In either case, increments in costs would ensue.
Staffing
If there is unanticipated staff turnover, the study will incur additional costs. Especially in a 12-year study involving 45 centers, new staff must be recruited and trained, and this will raise related costs. Staff turnover also has the potential to delay recruitment and threaten adherence to the protocols, thus delaying the study with the accompanying financial and validity costs.
Adherence
Similar threats to the budget lie in attempts to achieve adequate adherence to the intervention regimens. If adherence to the DM or the medication schedule is weak, a clinical center could direct increased effort at education and incentives leading to increased adherence. Increased effort would translate into more staff time or more highly skilled staff, both at higher cost. If such additional resources were not available, any potential difference in outcomes between the intervention and control groups would be attenuated because of poor adherence to the intervention regimen and, therefore, the diminished difference in group exposures.
Cross-Over
Investigators anticipate some cross-over of study participants from intervention regimens to control and vice versa. The extent of that cross-over activity is difficult to estimate, especially in a clinical trial involving more than one intervention with potentially problematic side effects. Expected cross-over in the DM branch is exceptionally uncertain, because few studies have attempted dietary change over a 12-year duration. Percentages in excess of the small percentages planned of participants changing their dietary or medication patterns could affect necessary sample size needed to test various hypotheses. To overcome the effect of cross-overs, investigators would need to increase sample size at concomitant expense.
The committee recognizes that NIH has considered many of these threats as well as others. Memoranda from NIH statisticians, for example, note that the occurrence of these difficulties will be monitored and sample size and power calculations would be adjusted as
necessary. The committee remains concerned, however, that while such adjustments would be required, the adjustments alone could not ameliorate the effects on the study. Additional source of funding would be needed to maintain sufficient power for meaningful statistical comparisons.
Summary
The committee feels that all three WHI components cannot be done for the announced costs of $625 million. In terms of the total cost of the WHI, with the Clinical Coordinating Center at $142 million, if the Vanguard Clinical Centers are funded at $10.4 million each and if additional centers are funded at $8 to $9 million each, this will account for approximately $570 to 580 million of the $625 million that has been committed before consideration of the funding of the Community Prevention Study. If additional centers are funded at $10 million each, the total committed funding would be, however, $600 million, leaving only $25 million for the CPS.
Are the proposed Vanguard Clinical Center budgets adequate? There is apparently a good deal of variability in Vanguard Clinical Center budgets and in expected institutional commitments. Certainly many of the Vanguard Clinical Center representatives stated comfort with their ability to achieve the requirements of the study for the budget, especially relying on institutional contributions. Some were not as certain. The integrity (and cost estimates) of WHI depend on the collective whole, not just those who are confident.
The committee concluded that the planned expenses are not excessive in relation to the research tasks they are to cover. NIH's publicizing of the WHI as one mega-study may enhance its chances at recruitment and public health promotion publicity. However, this characterization masks the fact that WHI is several studies of lesser cost combined in a single package.
After extensive formal and informal conversations with NIH, PIs, and other Vanguard Clinical Center representatives, the committee gets the picture of a very tightly budgeted trial—if nothing goes wrong. Should things not all go according to plan and estimate, however, there is little room for correction. The committee feels that the majority of the Vanguard Clinical Centers could probably function with the formal and informal arrangements in place but this is probably not true for all of the additional clinical centers. This does not give the committee the confidence to state that the funding of the WHI, as now designed, is adequate.
Because of the many uncertainties, the committee is uneasy regarding whether the budgeted funds will be adequate to carry out the WHI even if nothing unanticipated goes awry. The committee believes that the WHI CT will face enormous difficulties along the lines discussed above. In addition, it is impossible to assess the firmness of the nebulous soft costs that many institutions have committed to over the 14 years, which will probably span
different institutional administrations. In sum, the committee believes that the project cannot be fully completed as planned within the current budget.
FINDINGS AND SUGGESTIONS
The committee feels that the Women's Health Initiative (WHI) had inadequate peer review from within NIH or from outside scientists. Although various elements of the WHI were reviewed at one time or another (e.g., the dietary modification trial was reviewed many times in earlier proposals, none of which were allowed to proceed), the committee's impression is that the complicated interlocking combination of the clinical trial and the observational study at the inter-Institute level was not reviewed as rigorously as the usual Institute-initiated project. It seems that this inter-Institute study fell outside the established review process.
-
The committee suggests that NIH reexamine and strengthen the mechanism through which it reviews future inter-Institute proposed projects.
The committee concentrated on two fundamental questions.
Can the design answer the questions it addresses, if no operational difficulties occur?
If the study design is appropriate, what threats are there to the successful completion of the study?
The committee identified seven issues involving conceptual problems that are built into the design. Even if all study operations were to proceed without incident, these design issues threaten the validity of the findings. Where appropriate, the committee has also suggested strategies to overcome the difficulties.
Factorial Design
NIH argued that conducting a partial factorial design would reduce the required number of women and attendant costs and allow assessment of interactions among intervention branches. The committee feels that the factorial design has major drawbacks. The overlap of 15.9 percent between the DM and HRT interventions is insufficient to provide adequate statistical power to assess interactions, and the difficulties of maintaining adherence to two or three interventions detracts from the attractiveness of a factorial design. In essence, the integrated design has become primarily a matter of economic efficiency; it is not essential to hypothesis testing.
Sample Characteristics
In determining sample size, the study design relies heavily on extremely uncertain assumptions regarding magnitude of effect and lag times. This concern is a factor in the recommendation described below regarding study duration.
Participants will not be categorized by risk for breast cancer, colorectal cancer, or coronary heart disease. This allows a more generalizable study, but the lack of risk restrictions requires a much larger sample size. The factorial design does not allow specific branches to focus on the most efficient samples, such as women at high risk of CHD for an HRT trial or women at high risk of breast cancer for a DM trial, according to NIH assumptions.
Proposed Analytic Techniques
Committee concerns center on choice of endpoints for trial closeout and the planned use of methods to adjust for multiple comparisons when considering interim decisions by the Data and Safety Monitoring Board (DSMB).
-
The committee believes that studywide material must inform potential participants of risks as well as benefits. The committee suggested that unadjusted data be made available to the DSMB. The committee felt that the Bonferroni statistical adjustment, for which current analysis plans call, might be too conservative and therefore might deprive many participants of an appropriately timed conclusion to the study.
-
The committee also suggested the use of two-sided tests of significance to maintain a scientifically-justified neutral stance regarding whether the interventions might yield beneficial or adverse effects.
Ethics
The informed consent measures do not provide an adequate understanding of the likelihood or magnitude of major risks and benefits. The obligation to inform potential and current research participants would require much more information at the outset, as well as a commitment to provide evolving information over the course of the project.
-
The committee suggested that the counselors at the clinical centers be knowledgeable and have access to algorithms, guidelines, and printed material about known risks and benefits. These counselors would need supervision, training, and monitoring. In addition, new information from this as well as other pertinent trials (as judged by the WHI coordinators and the DSMB) must be shared with the participants to allow them to make their own decisions about ongoing risks and benefits of the interventions.
The inclusion of several interventions with several endpoints in a single trial makes the stopping rules difficult to formulate.
-
Therefore, the committee suggested that the DSMB should (a) use preexisting or external information to establish a prior probability that internal data could confirm (this might mean accepting an earlier “stopping” conclusion than would be justified by data arising solely from the CT); (b) perform pre-specified subset analyses on participant groups that are especially likely to evidence harm or benefit; (c) ask to examine uncorrected estimates of effect and do any analyses it feels are warranted; (4) review the monitoring of the consent process; and (5) evaluate pre-specified event rates for potential morbidity and mortality outcomes.
Minority Analysis Plan
As currently designed, the study will have insufficient power to compare individual minority groups to the majority population. The study will be able to observe differences, if they exist, but will not be able to test them with adequate power.
-
The committee encourages NIH to make these limitations known to those who may be expecting definitive comparative findings among minority and majority groups.
Specificity of Intervention and Effect
The CT design does not distinguish which element of the low fat dietary pattern may be responsible for any observed outcome. Similarly, the design will not allow analyses to distinguish whether calcium or calcium plus vitamin D is responsible for any observed outcome. Because some endpoints can be affected by more than one of the study interventions, and because the factorial design is modified by participant decisions, the overlap and interactions will be difficult to analyze.
Outcome Definition and Measurement
Threats to accurate and unbiased endpoint detection include the obscure meaning of many mammography-detected tiny malignancies; the unstandardized method of detecting colorectal cancer; and the inadequate development of behavioral, psychological, and quality of life measures for use in the study.
-
The committee encourages NIH to include measures of constructs such as pain, mobility, and psychological status.
In addition to the conceptual problems described above, any study —nomatter how well designed—is subject to setbacks by operational problems. The WHI CT is particularly vulnerable to such problems because of its size, complexity, and duration. The committee has identified five operational issues that could jeopardize the success of the study:
Recruitment, Retention, and Adherence
The message of the study is not adequately developed and may be misleading.
-
The committee suggests that NIH and the clinical centers develop an overall message for the study that pays particular attention to long-term recruitment strategies for older and minority participants, and does not emphasize the WHI as a breast cancer prevention trial. In addition, investigators should set higher standards for studywide materials than currently appears to exist, including introductory brochures, consent forms, and videotape information. This information should be available in conversational language.
NIH has made overly optimistic assumptions about recruitment, retention, and adherence, especially in subgroups with which researchers have less clinical trial experience, such as older women, minority women, and the spectrum of socioeconomic status (SES) and in recruitment plans that cover many years.
-
Nevertheless, the committee encourages NIH to seek diversity within the sample and suggests that attempts should be made to include the entire SES range in this study.
The acceptability of the various branches of the CT to women is unclear at this stage, especially since the interventions are difficult and have potential side effects.
-
To maintain adequate statistical power, the CT must have funds available to boost recruitment efforts if, as the committee expects, recruitment rates are lower than anticipated.
Secular Trends
If secular trends toward a decreasing fat content in the U.S. diet continue, and if there is appreciable nonadherence in the DM treatment group, the difference between the treatment and control diets is likely to be too small to show a treatment effect.
Provision of Health Care Services to Participants
The current protocol includes a referral to a regular source of care. This is not adequately responsible.
-
The committee suggests that the clinical centers must continue to develop adequate links with reliable community providers to ensure that adequate follow-up care is available. It may become essential for the project to pay for some kinds of follow-up for some poor or uninsured women.
Research staff need to spend considerable time discussing side effects with participants, and dealing with associated apprehension, both in the clinic and on the telephone. To fail
to do so is to risk unethical behavior and increased study dropout. The current budget may not include adequate staff time for these activities.
Cost
The committee believes that the total costs of the CT will be greater than the $625 million provided by NIH. NIH and Vanguard Center representatives have indicated that the additional funds necessary for successful completion of the trial will be covered by the institutions at which the Vanguard Centers are based. This reliance on institutional support may be reasonable in the case of the Vanguard Centers, but the committee felt it is unlikely that an additional 29 institutions can be identified that have both the experience to carry out the tasks of high quality research and the ability to provide additional resources.
Potential sources of budget shortfall include lagging participant recruitment, which could require increased staff resources; staff turnover, which could require training and travel resources and might delay recruitment, threaten adherence, and, therefore, affect study validity; and cross-over of participants between study intervention regimens and control status.
The CT funding per person per year is less than half that for other recent NIH studies of women's health, including, specifically, those that use similar drug regimens and approaches.
There does not seem to be a budget adjustment plan for unanticipated changes in either the scope of work or medical technology during the course of the trial.
-
In addition to its concerns about initial funding levels, the committee was concerned about long-term funding and suggested that NIH clarify what the contract requires financially in terms of anticipated or unanticipated changes throughout the duration of the study.
RECOMMENDATIONS
Finally, the committee was charged to begin with the existing WHI design, consider threats to its successful completion—whether design, financial, or ethical—and to consider whether it would yield reliable results.
-
The committee recommends that the dietary modification-breast cancer hypothesis be considered a subsidiary rather than a primary hypothesis, shifting the emphasis to the effect of dietary modification on coronary heart disease outcomes, making those the primary hypotheses.
-
The committee recommends that the consent process be outlined more carefully, be conscientiously implemented and monitored across all centers, and be evaluated and updated as needed.
-
The committee recommends that the CT be scheduled to end in mid-2002, rather than close out the interventions by April 2005, and that the findings of an Objective Prescheduled Reassessment (OPR) be available by April 2002 (see Figure 2-4).
The OPR, managed through an internal or external review board, would consider whether continuation or modification of the CT could be justified. Recruitment for the CT began in September 1993, so the project would run unimpeded for more than eight years (unless the Data Safety and Monitoring Board moves to stop the trial sooner based on external or interim data). Data analysis would begin in October 2001 and conclude with a recommendation by April 2002. Between October 2001 and the decision to extend, modify, or terminate, the CT would continue in its active mode. Sufficient time would be provided for closeout or redesign and data analyses.
This recommendation addresses the primary concerns of the committee in the following ways:
-
Data from nearly six years mean follow-up time would be available for the OPR. According to NIH power calculations (see Appendix J), this timeframe would allow hypotheses regarding stronger, expected associations (HRT and coronary heart disease; and HRT and combined fractures) to be tested and findings disseminated in a timely manner. If the intervention effect is strong, this timeframe also allows the hypotheses regarding the weaker, expected associations (DM and CHD; CaD and hip fractures; and HRT and hip fractures) to be tested. This timeframe does not allow for adequate follow-up for the DM and breast cancer hypothesis, the DM and colorectal cancer hypothesis, or the HRT and breast cancer hypothesis. However, the committee feels that, as currently designed, the CT does not have a high probability of yielding statistically significant results for the DM and breast cancer hypothesis or the HRT and breast cancer hypothesis, even after more prolonged follow-up. The committee would therefore prefer to see the other hypotheses analyzed in an appropriate timeframe. While the DM and colorectal cancer hypothesis is reasonable, it alone does not justify continuing the CT.
-
This recommendation allows an assessment that would be informed by recruitment, retention, adherence, and incidence experience; if any of these estimates have not been or are not being met, the problem can be addressed. For example, if HRT is demonstrated to be favorable compared with control, the CT could reassign the control participants (with their permission) to ERT or PERT, thus increasing statistical power for that direct comparison, which as designed is not currently adequate. If there is evidence that the DM-breast cancer investigation should continue, justifications for that should be offered at the same time. If recruitment or adherence experience is so poor
-
that an adequate test of a hypothesis would not be possible in any reasonable time frame, the CT or a branch of it could terminate. If, on the other hand, recruitment or adherence problems are discretely identifiable, the study could be redesigned for the remaining duration to compensate for these problems.
-
Any clinically beneficial findings of the CT can be made available to participants. Clinical knowledge resulting from other studies can also be applied to participants in both intervention and control arms of the CT. Therefore, WHI investigators would not be pressured to deny benefits to women in the CT to keep intact its overlapping studies.