
A
The Basic Science of Genome Editing
This appendix provides technical and historical context for a number of issues related to the basic science of gene therapy and gene editing. Although an effort has been made to maximize the accessibility of this material, a simpler summary of this material can be found in Chapters 3 and 4. This appendix includes detailed material on the following topics:
- breakage and repair of DNA
- precursors of the clustered regularly interspersed short palindromic repeats (CRISPR) system/CRISPR-associated endonuclease (Cas9) gene editing—meganucleases, zinc fingers, and transcription activator-like effector nucleases (TALENs)
- development of CRISPR/Cas9
- the accuracy of gene editing
- enhancing the specificity of CRISPR/Cas9
- quality control and quality assurance for gene editing
- use of dead Cas9 (dCas9) to regulate transcription or to make epigenetic modifications
- gene targeting in transgenic animals
- gene editing in embryos
- alternative routes to heritable germline editing
- editing the mitochondrial genome
GENE THERAPY AND GENOME EDITING
The potential for gene therapy to address human disease has been evident for some years, and much progress has been made in its applications (Cox et al., 2015; Naldini, 2015). Gene therapy refers to the replacement of faulty genes, or the addition of new genes as a means to cure disease or improve the ability to fight disease. Genome editing is one aspect of gene therapy. Established approaches to gene therapy have been based on the results of extensive prior laboratory research on individual cells and on nonhuman organisms, establishing the means to add, delete, or modify genes in living organisms. Key advances include the development of techniques for generating molecular tools for cutting the DNA of genomes in specific places to allow targeted alterations in the DNA sequence. Over recent years, several such methods have been introduced and used effectively in clinical applications.
Within the past 5 years, a completely novel system has been developed based on fundamental research on bacterial systems of immunity to viral infections. The first such system to be developed for use in genome editing of human cells, known as CRISPR/Cas9, is based on RNA-guided targeting and is much simpler, faster, and cheaper than earlier methods. The ease of design, together with the remarkable specificity and efficiency of the CRISPR/Cas9 system has revolutionized the field of genome editing and reignited interest in the potential for editing of the human genome. The development of the CRISPR/Cas9 system as a programmable genome-editing tool was built on a firm foundation of earlier research.
BREAKAGE AND REPAIR OF GENOMIC DNA
Genomes and their constituent genes are made of double-stranded DNA; this DNA can be broken accidentally (e.g., by radiation) or purposefully, using proteins called endonucleases (often called nucleases) that can generate double-strand breaks (DSBs) in DNA.
Cells have mechanisms to repair DSBs in DNA, and these mechanisms can be used to generate alterations in the DNA sequence. Groundbreaking work in bacteria, yeast, and mammalian systems shows that DSBs dramatically stimulate the rate of DNA repair by nonhomologous end joining (NHEJ), in which the broken ends are reattached (see Figure A-1). Such NHEJ repair often results in the deletion or insertion of DNA sequences of varying length, which can disrupt gene function (Rouet et al., 1994).
However, if a homologous stretch of DNA is introduced into the cell as a donor template, homology-directed repair (HDR) can lead to more accurate repair or, if specific alterations are included in the homologous stretch, it can introduce specific precise changes into the recipient genomic
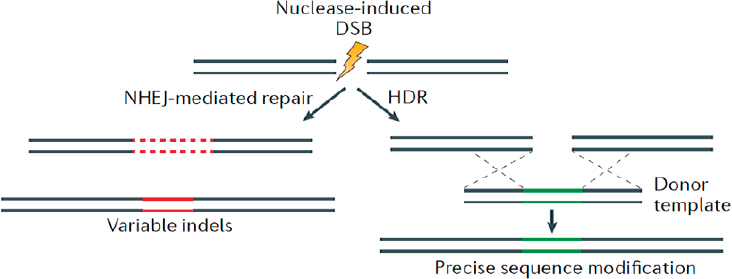
NOTE: DSB = double-strand break; HDR = homology-directed repair; NHEJ = nonhomologous end joining.
SOURCE: Modified from Sander and Joung, 2014.
DNA (see Figure A-1). These cellular DNA repair mechanisms have been used to develop several methods that allow genes or the genome to be edited in a very precise manner.
PRECURSORS OF CRISPR/CAS9 GENE EDITING
Three distinct strategies based on nuclease systems for generating targeted cleavages in DNA preceded the development of CRISPR/Cas9—meganucleases, zinc finger nucleases (ZFNs), and TALENs (see Figure A-2). All three have already enabled major advances in establishing the feasibility of using such targeted nucleases both to eliminate disease-causing genes as well as to repair damaged or mutated genes, ushering in a new era in biology and medicine.
Zinc Finger Nucleases
Zinc fingers are segments of protein that have evolved to recognize and bind to specific DNA sequences. Knowledge gained from natural zinc fingers led to the development of ZFNs as designer DNA-cutting enzymes (see Figure A-2), and their use in genome engineering represents pioneering, even heroic, protein engineering. Two major advances in protein engineering enabled the development of ZFNs: (1) the engineering of zinc finger proteins with designed DNA-binding specificity pioneered by Berg (Desjarlais and Berg, 1992), Pabo (Rebar and Pabo, 1994), and Wells (Jamieson et al., 1994); and (2) the generation of fusions between such designer zinc fingers
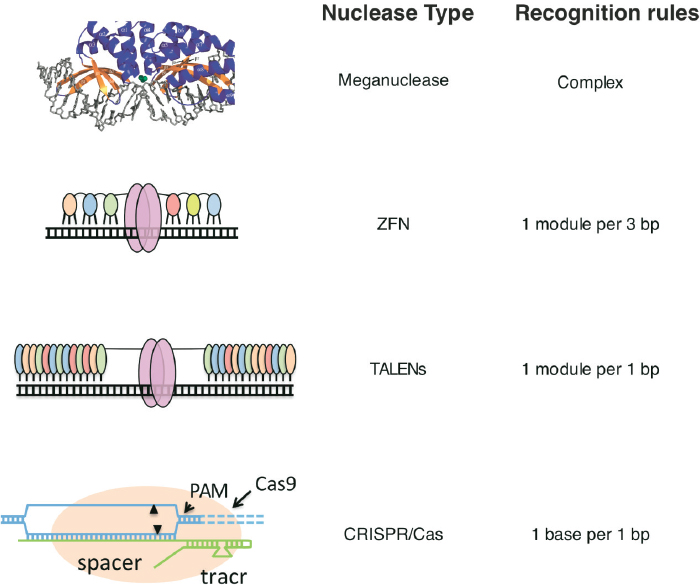
SOURCES: Carroll, 2014. (Modified with permission from the Annual Review of Biochemistry, Volume 83 © 2014 by Annual Reviews, http://www.annualreviews.org); Chevalier et al., 2002.
and a DNA-cleaving protein, FokI nuclease (Kim et al., 1996), yielding “artificial restriction enzymes” that could be used to promote site-specific genome engineering by creating DSBs at defined locations in the genome (Bibikova et al., 2001, 2002, 2003).
Using these findings, Sangamo Therapeutics developed ZFNs from laboratory tools into therapeutic agents that could be used to disable
disease-causing genes (by NHEJ) or to correct errors in existing genes (by HDR). The latter goal is particularly challenging, as the efficiency of HDR is usually much lower than the imprecise and mutagenic repair by NHEJ. By overcoming some of these difficult challenges, Sangamo now has several ongoing human clinical trials, the most advanced of which is in the treatment of HIV/AIDS where deletion of the CCR5 HIV coreceptor has the potential to enable the elimination of HIV following bone-marrow transplantation. Other applications of ZFNs have also been, or are being, tested in clinical trials (NCT02695160 [hemophilia B] and NCT02702115 [in vivo editing MPS1]).
Transcription Activator-Like Effector Nucleases
Like ZFNs, TALENs are composed of a DNA-binding protein that recognizes particular DNA sequences fused to a nuclease effector domain to achieve the cleavage (Joung and Sander, 2013) (see Figure A-2). TALEs are secreted bacterial proteins with DNA-binding domains that contain a series of conserved blocks of sequence 32-34 residues long, each with two divergent amino acids. These divergent amino acids are largely responsible for determining the DNA-binding specificity for a single DNA base pair, which allows engineering of specific DNA-binding domains by choosing combinations of repeat segments containing appropriate amino acids. The biotech firm Cellectis reported the successful conduct of the first-ever TALEN-based gene-editing clinical trial in the United Kingdom on a girl with incurable acute lymphoblastic leukemia (ALL) (Qasim et al, 2017). Although the applications of ZFNs and TALENs are largely overlapping, TALENS have the advantage of relative ease of design because of the robust recognition code.
Meganucleases
Meganucleases are nucleases with very long DNA-binding recognition sites, up to 40 nucleotides (Silva et al., 2011) (see Figure A-2). As a result of their length, it is exceedingly unlikely that natural sites would be present by chance even in complex human genomes. The challenge with meganucleases is the difficulty in designing new nucleases to target a sequence of interest at will. Some success has been achieved by designed changes to DNA-binding sites and combining meganucleases with TALE DNA-binding elements. However, meganucleases are unlikely to be much used in human genome editing given the relative simplicity of the alternative methods.
DEVELOPMENT OF CRISPR/CAS9
CRISPR as a Bacterial Adaptive Immunity System
The discovery that CRISPR systems provide adaptive immunity to bacteria represents a major conceptual advance in its own right. This discovery was also critical to the development of CRISPR/Cas9 genome engineering. A brief synopsis of the key findings is provided below (for a more complete review, see Doudna and Charpentier, 2014).
CRISPR loci were first identified based on analyses of bacterial genomes. From these studies, it was inferred that the spacer (i.e., nonrepetitive) regions of CRISPR loci were derived from the genomic DNA of bacteriophages (viruses that infect bacteria) leading to the hypothesis that CRISPR provided a defense mechanism against foreign genetic elements (Makarova et al., 2006; Mojica et al., 2005; Pourcel et al., 2005). The key experimental breakthrough came from research showing that CRISPR allowed bacteria to acquire resistance to bacteriophages by integrating segments of the bacteriophage genome into the CRISPR loci, demonstrating that CRISPR was a new form of adaptive immunity (Barrangou et al., 2007). In 2010, type II CRISPR/Cas systems were shown to mediate cleavage of invading bacteriophage DNA (Garneau et al., 2010). And in 2011, an associated RNA, tracrRNA, was identified (Deltcheva et al., 2011), and the CRISPR-associated gene, Cas9, was shown to be the only protein-coding gene in the type II Cas locus required for the defense function (Sapranauskas et al., 2011).
Development of Cas9 as a Programmable Endonuclease
The critical advance in the development of the CRISPR/Cas9-based genome-editing method came in 2012 from the laboratories of Doudna and Charpentier. They established that the CRISPR-associated protein Cas9, in complex with two small RNAs, CRISPR RNA (crRNA) transcribed from the CRISPR locus, and a trans-activating crRNA (tracrRNA), yields a site-specific endonuclease in which the site of cleavage is defined by base pairing of crRNA to the target DNA. This laid the groundwork for establishing that Cas9 is an endonuclease that can be programmed with a single “guide RNA” (gRNA, a chimera of crRNA transcribed from the CRISPR locus and the tracrRNA) to cleave at specific DNA sites (Jinek et al., 2012) (see Figure A-3a). Concurrent with the Jinek et al. (2012) manuscript, Siksnys and coworkers (Gasiunas et al., 2012) demonstrated that purified complexes containing Cas9 and a crRNA could mediate cleavage of double-stranded DNA in vitro at sites complementary to the crRNA. While this was clearly
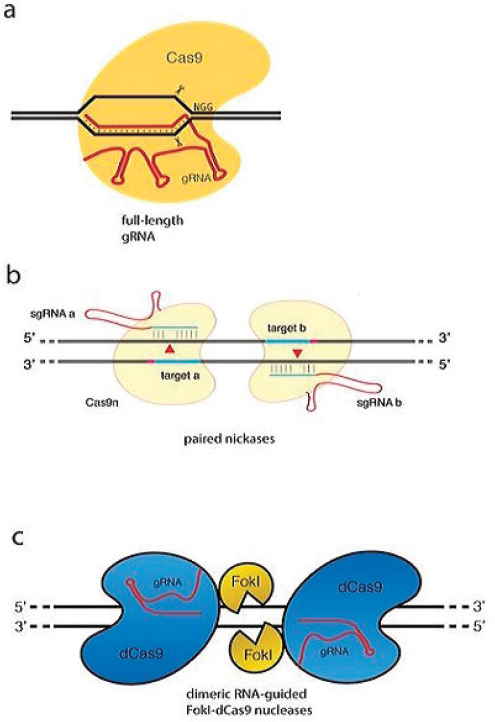
SOURCE: Modified from Tsai and Joung, 2016.
an important advance, a key missing element was the identification of the requirement for the tracrRNA to form an active endonuclease.
The Jinek et al. (2012) paper made the key breakthrough of the use of a single chimeric guide RNA (gRNA) that can fulfill the role of both the crRNA and tracrRNA (see Figure A-3a). Thus, mechanistically, the CRISPR/-
Cas9 system is superior to both ZFNs and TALENs in terms of ease of use; all that is required to generate a site-specific nuclease is design and synthesis of a single gRNA to target the Cas9 nuclease to the desired site of editing. Although not all predicted guide RNAs work, their synthesis is easy and multiple possible candidates can be synthesized easily and cheaply.
In Vivo Application of the Cas9 Programmable Nuclease
Within months of the publication of the Jinek et al. (2012) and Siksnys laboratory manuscripts (Gasiunas et al., 2012; Sapranauskas et al., 2011), there were six independent reports using the Cas9–guide RNA system to mediate programmable genome editing in vivo. These included four papers reporting Cas9 editing in mammalian cells (Cho et al., 2013; Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013); one on zebrafish (Hwang et al., 2013); and one on bacteria (Jiang et al., 2013). Additionally, there was a seventh paper (Qi et al., 2013) showing that dCas9 could be used to inhibit transcription. Since that time there has been an explosion in the application and refinement of Cas9-mediated cleavage, as well as the discovery of novel CRISPR systems that can be adapted for genome editing. These include the discovery of a novel RNA-guided endonuclease, Cpf1 (Zetsche et al., 2015), and more recent work has shown that further CRISPR-targetable nucleases with the potential for differing capabilities are being discovered.
ACCURACY OF GENE EDITING
The potential impact of unintended changes to DNA is a key challenge for safe use of genome editing as a therapeutic strategy. Unintended changes to the genome could be caused by cleaving DNA at sites other than those that are being deliberately targeted.
The Challenge of Off-Target Toxicity
The site at which Cas9 cleaves DNA is determined by the complementarity of the DNA target with the RNA guide (typically 20 base pairs) adjacent to a protospacer adjacent motif (PAM) sequence (e.g., NGG for Streptococcus pyogenes, the most commonly used species of Cas9). In principle, this 22-base sequence would give enough diversity such that the cut site should be unique within even a 3 billion base-pair human genome. In practice, however, some base mismatches are tolerated leading to significant potential for off-target cutting. This has motivated efforts both to monitor the sites of off-target cutting as well as to enhance the specificity of the targeted nucleases. Initial efforts to define off-target cutting of Cas9 focused
on specific searches for cutting at near-cognate sites. More recently these have been complemented by less biased genome-wide efforts. These can be divided into two broad classes: cell-based and cell-free (in vitro).
Genome-Wide Cell-Based Assays
Ostensibly, whole-genome sequencing (WGS), when conducted at a single-cell level, would seem to provide a definitive assessment of the accuracy of Cas9 genome editing. However, the depth of sequencing that would be required to certify the absence of off-target cutting is currently difficult to achieve for populations of cells. It should be possible, however, to estimate the sensitivity of the system for detecting off-target editing. Failure to detect editing with the assay would then indicate that the off-target editing rate was below the detection level.
Integrase-defective lentiviral vector (IDLV) capture (see Figure A-4a) is a genome-wide approach used to evaluate the specificity of genome-editing nucleases that was initially applied to engineered ZFNs and then later applied to TALENs and CRISPR/Cas9 (Gabriel et al., 2011; Wang et al., 2015). This method is based on the capture by NHEJ of IDLVs, which have linear double-stranded DNA genomes, into sites of nuclease-induced DSBs. Although the IDLV capture method directly identifies DSBs that occur in living cells, it is relatively insensitive and has a high background. To overcome these limitations, genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq) (see Figure A-4b) was developed (Tsai et al., 2015). GUIDE-seq exploits the efficient integration of a blunt, end-protected, double-stranded oligodeoxynucleotide (dsODN) tag, followed by tag-specific amplification and high-throughput sequencing. GUIDE-seq can detect off-target sites that are mutagenized by Cas9-sgRNAs with low frequencies (<0.1 percent) in a cell population, even with only a few million sequencing reads.
High-throughput genome-wide translocation sequencing (HTGTS) (see Figure A-4c) is another genome-wide method that identifies Cas9 off-target cleavage in live cells (Chiarle et al., 2011). HTGTS is based on the detection of translocations between a nuclease-induced “bait” DSB and off-target “prey” DSBs. A limitation of HTGTS is that nuclease-induced translocations represent rare events and thus require large numbers of input genomes for detection. A strategy for detecting genome-wide nuclease-induced DSBs in fixed cells, termed “BLESS” for breaks labeling, enrichment on streptavidin, and next-generation sequencing, captures a snapshot of transient DSBs that are present at a moment in time in a population of cells by direct in situ ligation of biotinylated hairpin adaptors in fixed and permeabilized cell nuclei (see Figure A-4d).
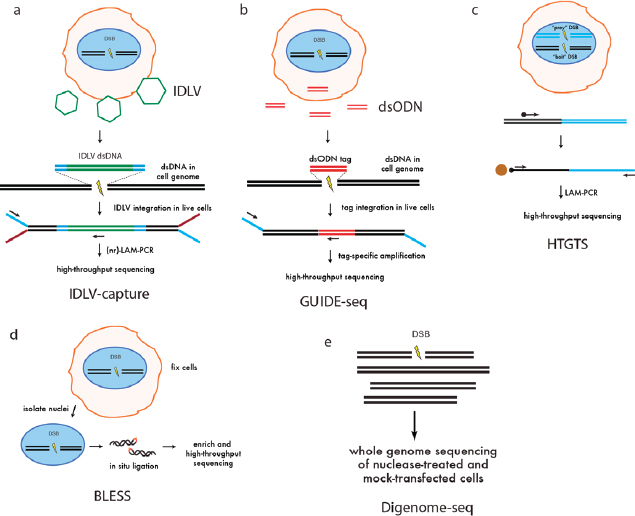
(a) Integrase-defective lentiviral vector (IDLV) capture. IDLVs (green) are integrated with a selectable marker into sites of nuclease-induced double-stranded breaks (DSBs) in living cells. Integration sites are recovered by linear amplification-mediated PCR (LAM-PCR), followed by high-throughput sequencing. (b) Genome-wide unbiased identification of DSBs enabled by sequencing (GUIDE-seq). An end-protected, short, double-stranded oligodeoxynucleotide (dsODN) is efficiently integrated into sites of nuclease-induced DSBs in living cells. This short sequence is used for tag-specific amplification followed by high-throughput sequencing to identify off-target cleavage sites. (c) High-throughput genome-wide translocation sequencing (HTGTS). Nuclease is expressed in a cell to generate a “prey” and “bait” DSB. Using a biotinylated primer designed against the targeted bait DSB junction, translocations between prey and bait are recovered by LAM-PCR and streptavidin-based enrichment for high-throughput sequencing. Off-target cleavage sites (prey) are identified by analysis of these translocation junctions. (d) Breaks labeling, enrichment on streptavidin, and next-generation sequencing (BLESS). Nuclease-treated cells are fixed, intact nuclei are isolated and permeabilized, and then sequencing adapters are ligated in situ to transient nuclease-induced DSBs. Adapter-ligated fragments are enriched and amplified for high-throughput sequencing. (e) Digested genome sequencing (Digenome-seq). Genomic DNA is isolated from cells and treated with Cas9 nuclease in vitro. Sequencing adapters are ligated and high-throughput sequencing is performed at standard whole-genome sequencing coverage. Absence of continuous sequence reads identifies cleavage sites.
SOURCE: Modified from Tsai and Joung, 2016.
Genome-Wide in In Vitro Assays
Digenome-seq (see Figure A-4e) is an in vitro method for detection of nuclease-induced DSBs in genomic DNA using whole-genome sequencing of Cas9-cleaved genomic DNA. Genomic DNA is isolated, and treated with high concentrations of Cas9–gRNA in vitro to maximize off-target cleavage and cleavage sites are identified by DNA sequencing. Because this assay is performed in vitro on purified DNA, it is not limited by cell-based factors such as chromatin context, epigenetic factors, subnuclear localization, or fitness effects. Digenome-seq thus may detect potential additional off-target cleavage at sites that would otherwise be obscured in cell-based methods. Thus, it could yield an overestimate of off-target events in vivo.
ENHANCING SPECIFICITY OF CRISPR/CAS9
Given its ease of use, flexibility, and versatility, the CRISPR/Cas9 system is rapidly becoming the tool of choice for gene editing. However, concerns about the potential risk of unwanted off-target effects have dominated many recent discussions. Most experiments that have detected significant off-targets have been performed in cancer cells (Fu et al., 2013; Hsu et al., 2013), which may have altered DNA repair pathways that could lead to elevated off-target events. In contrast, experiments in whole organisms such as mice (Yang et al., 2013), primates (Niu et al., 2014), zebrafish (Auer et al., 2014), or Caenorhabditis elegans (Dickinson et al., 2013) reported off-target frequencies that were low or not detectable, consistent with the high specificity of the CRISPR/Cas9-mediated gene targeting. It is possible that, in nontransformed cells, off-target cleavages are efficiently counter-selected by the endogenous DNA-damage response. Human pluripotent stem cells (hPSCs) are primary cells with genetically intact quality control mechanisms, and it seems possible that off-target events will accumulate less frequently in hPSCs or in normal somatic cells than has been observed in cancer cells. Nevertheless, it will be important to determine whether there are specific cell-types and conditions that predispose for the accumulation of off-target events. To address concerns about off-target events, diverse approaches to minimize mistargeting are being developed. Based on progress already made, it is anticipated that the risk of off-target events may be dramatically reduced, if not eliminated, in the near future for many genome-editing approaches. Below are three approaches and related progress.
Modification of Cas9 Structure
Protein engineering approaches can be deployed to develop better Cas9 variants that may be more accurate and efficient. Based on studies of
CRISPR/Cas9 structure, two recent papers report engineered substitutions such that the resulting Cas9 protein has a significantly reduced off-target rate (Kleinstiver et al., 2016; Slaymaker et al., 2016). The two studies focused on different DNA-binding domains of Cas9 but took the common strategy of reducing the relative affinity for nonspecific DNA binding. Remarkably this enhanced the specificity of Cas9 cutting without obviously impairing its overall efficiency. While these attempts are encouraging, other strategies will undoubtedly be forthcoming and should further improve the process, based on knowledge of CRISPR/Cas9 structure (Haurwitz et al., 2012; Jinek et al., 2014; Jore et al., 2011; Staals et al., 2013; Wiedenheft et al., 2009, 2011).
Engineered Combinations of Cas9 with Modified Cleavage Sites
This approach involves the use of two targeted DNA cuts, ensuring better fidelity than a single target cut. The basic rationale is that Cas9 protein has two active DNA-cleaving sites, involving aspartic acid, D10, and histidine, H840, each responsible for cutting a single strand of DNA thus generating the DSB (Jinek et al., 2014). Based on this feature, there are two ways of inactivating Cas9: single inactivation and double inactivation (Guilinger et al., 2014; Ran et al., 2013; Tsai et al., 2014). In one approach, Cas9 with a single inactivation, also named Cas9 nickase (Cas9n), in which, only one of the active site residues (D10 or H840) is replaced with alanine (A), yields a Cas9 protein capable of cutting one strand of double-stranded DNA. Consequently, providing two guide RNAs that direct cutting on opposite strands in close proximity mediated by a dimer of Cas9n single cutters leads to an effective DSB and stimulation of both NHEJ and HDR to yield a DSB (see Figure A-3b). Remarkably these nicks can be effective even as far apart as ~100bp. In a second approach, both cleavage sites of Cas9 are inactivated, yielding nuclease-deficient dCas9, which is then fused to the FokI cleavage domain. As with ZFNs and TALENs, two Cas9 monomers provide recognition specificity for FokI dimerization and DNA cleavage (see Figure A-3c). In addition, these two strategies require appropriate length spacers between the targets: if the spacers are too short, there will be two competing Cas9 proteins, and if the spacers are too long, it is more difficult to execute effective cutting. Because of these stringent requirements, the off-target rate is greatly reduced, albeit with increased difficulty in target selection. These strategies have been shown to significantly reduce off-target rates (Guilinger et al., 2014; Ran et al., 2013; Tsai et al., 2014).
Cas9 Base-Editor:
Genome Editing Without Double-Stranded DNA Breaks
In an effort to increase the efficiency and precision of making point mutations in genomic DNA, the Liu group recently developed the so called “base editor” variant of Cas9 (see Figure A-5), which consists of a highly engineered fusion protein that recruits a cytosine deaminase domain, which converts cytosine to uracil, thereby effecting an irreversible CàT substitution (or GàA by targeting the complementary strand) without double-stranded cleavage of the DNA backbone (Komor et al., 2016).
Because cytosine deaminase acts only on single-stranded DNA, the C to U conversion activity is targeted to a small window of ~5 nucleotides near the 5´ end of the guide RNA-specified protospacer sequence on the displaced DNA strand. By avoiding DSBs, exogenous DNA templates, and stochastic DNA-repair processes, base editing introduces point mutations in unmodified mammalian cells with an efficiency as high as 75 percent and with a ratio of point mutation correction:indels exceeding 20:1. Moving forward it will be important to develop alternate base-editors capable
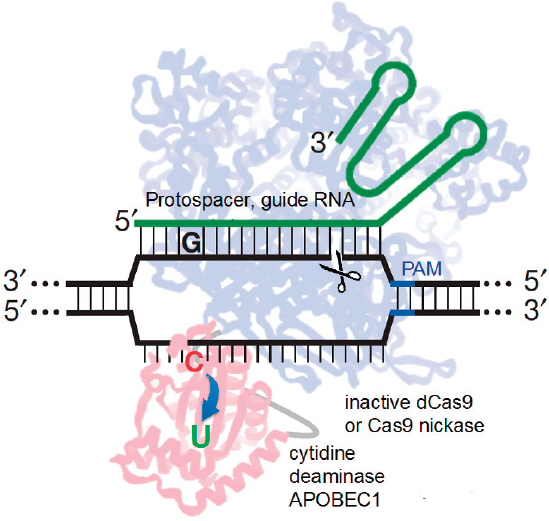
SOURCE: Modified from Komor et al., 2016.
of mediating a wider range of genetic changes ideally ultimately allowing conversion of any base to any other base within a user-defined window. Additionally, it will be important to robustly evaluate the propensity of the cytosine deaminase domain to introduce mutations outside of the DNA region targeted by the Cas9 guide RNA.
An alternative strategy for improving HDR, termed “CORRECT,” (consecutive re-guide or re-Cas steps to erase CRISPR/Cas-blocked targets), exploits the observation that HDR accuracy is increased dramatically by incorporating silent CRISPR/Cas-blocking mutations along with the desired functional mutations (Paquet et al., 2016). This prevents recleavage and potential NHEJ repair that might disrupt successful HDR products. The authors show that by controlling the location of the introduced point mutation relative to the Cas9-mediated DSB they can alter the efficiency of mutagenesis, generating either heterozygous or homozygous alterations in human-induced pluripotent stem cells (hiPSCs).
QUALITY CONTROL AND QUALITY ASSURANCE FOR GENE EDITING
The specificity of genome editing is even more important in clinical applications than in laboratory research. As a medical product or medical practice, genome editing must be safe, efficacious, and cost-effective. Development of a regulatory framework for genome editing will need to address various issues associated with these requirements. As discussed above, although technical advances will likely make off-target events a manageable issue, this consideration will remain a concern that must be addressed by quality control (QC) and quality assurance (QA) procedures. In somatic genome editing, it may be relatively easy to set up assays or procedures to address off-target changes but it is probable that such rates will vary among cell types, necessitating measurement of off-target events in each cell type targeted. Although the specifics may vary from case to case, the general principle of monitoring to ensure safety and efficacy should be implemented. In contrast, it would be quite difficult to monitor embryos if they were to undergo editing. Functional equivalence assays need to be developed and agreed upon to serve as QC measures. Alternatives may be considered, such as editing performed on sperm progenitor cells.
USE OF dCAS9 TO REGULATE TRANSCRIPTION OR TO MAKE EPIGENETIC MODIFICATIONS
An alternate strategy for genome editing involves the use of catalytically dead variants of Cas9. This yields a programmable DNA-binding protein that is incapable of generating either single- or double-stranded breaks
and thus does not typically lead to any changes in the DNA sequence of the genome. However, by fusing different effector domains to dCas9, it can be used either to turn on (CRISPRa) (Gilbert et al., 2014; Konermann et al., 2015; Perez-Pinera et al., 2013) or turn off (CRISPRi) transcription (Gilbert et al., 2013; Qi et al., 2013) or to make locus-specific changes to the epigenetic marks (modifications of the chromatin that regulate gene expression). It is likely that most if not all such epigenetic changes would fail to be passed to subsequent generations thus alleviating some of the concerns surrounding germline-editing approaches. By the same token, the transient nature of these changes limits their utility for correcting diseases caused by genetic mutations. Possible uses for such transient germline engineering, however, include the ability to expand germ cells, or the in vitro generation of desired stem cells or terminally differentiated cells. Additionally, the transient nature of the changes could expand the number of genes that could be safely targeted. For example, transient down-regulation of the HIV CCR5 coreceptor could protect against vertical passage of HIV, and this strategy could be expanded to other viral receptors. Additionally, it is possible to imagine that transient alterations to gene expression could lead to permanent developmental changes in an embryo, which could ameliorate the effects of disease-causing inherited mutations. Because no permanent, heritable changes are made to the individual, the use of dCas9 to alter gene expression does alleviate some ethical concerns. Nonetheless, at present the potential uses of dCas9 on embryos seem rather limited compared to approaches involving germline editing, and the more immediate therapeutic applications of dCas9 likely involve somatic alteration in gene expression.
GENOME TARGETING IN TRANSGENIC ANIMALS
Gene mutations can lead to abnormal development and to disease. Over the past several decades, a major advance in studying the consequence of mutations has been the development of the ability to experimentally introduce designed, targeted mutations into genes of organisms such as mice, fruit flies, and zebrafish, thus providing an important tool to understand the molecular-genetic basis of embryonic development and of disease. These methods can also be used to correct defective genes. Before describing the application of current precise genome-editing methods, we will summarize the major steps that were initially developed to genetically modify animals.
Random Insertion of Foreign DNA
The genetic manipulation of animals has been the basis for much of the research aimed at understanding embryonic development and human diseases. One powerful technology is based on manipulating mouse em-
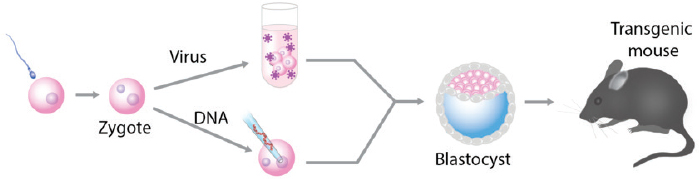
bryos in vitro and transferring the embryos to a foster mother to produce genetically altered animals (see Figure A-6).
Using these techniques, initially SV40 DNA (Jaenisch and Mintz, 1974) and later retroviruses (Jaenisch, 1976) were introduced into early mouse embryos leading to the generation of the first transgenic mice that transmitted the foreign DNA to the next generation according to Mendelian expectations. The most widely used method to generate genetically modified animals was microinjection of DNA into fertilized mouse or Drosophila eggs leading to the production of a large number of transgenic mice or flies that carried foreign DNA in their germline (Brinster et al., 1981; Costantini and Lacy, 1981; Gordon and Ruddle, 1981; Rubin and Spradling, 1982).
The random integration of foreign DNA into the genome of an animal can cause disruption of an endogenous gene leading to its inactivation. In this “insertional mutagenesis” approach the integrated DNA is used as a molecular tag for the isolation and identification of the mutated gene. The collagen I gene was the first endogenous gene inactivated by retroviral insertional mutagenesis resulting in mutant mice whose phenotype resembled brittle bone disease (Schnieke et al., 1983), a major disease of the skeletal system caused by mutations in a collagen gene. Similarly, injection of DNA into the zygote pronucleus produced mutant mice by insertional mutagenesis (Mahon et al., 1988). In addition to insertional mutagenesis, the integration of a gene into the genome can result in transcriptional activation of nearby genes, which has been widely used to study the consequences of ectopic transgene expression (Hammer et al., 1984).
While integration into the genome leading to insertional mutagenesis or ectopic transgene expression is efficient in generating transgenic animals, the approach suffers from unpredictability because the insertion of DNA
into the genome is random and does not allow the targeting of predetermined genes or predictable transgene expression.
Gene Targeting in Embryonic Stem Cells
Embryonic stem (ES) cells, initially isolated from mouse blastocysts, are able to differentiate into all cell types of the body (Evans and Kaufman, 1981; Martin, 1981). Of great interest was that ES cells, when injected into a mouse carrier blastocyst, could integrate into the developing embryo and contribute to all somatic tissues and generate “chimeric mice.” Of particular importance was the fact that the cells were able to contribute to the germline, thus allowing the derivation of animals from the cultured cells. Thus, the approach allowed the generation of mice carrying the alteration of an endogenous gene as a result of in vitro manipulation of the ES cells.
The first mouse strain derived from an ES cell carrying a mutation in a predetermined gene was a strain with inactivation of the HPRT gene, which is mutated in human patients with the severe mental disorder Lesch-Nyhan syndrome. The isolation of HPRT mutant ES cells was straightforward using a culture medium (HAT) that kills normal cells and selects for cells carrying an inactivated HPRT gene (Kuehn et al., 1987). This selective approach, while successful for HPRT, cannot be used for editing other genes.
Homologous Recombination
The discovery of homologous recombination represented a major breakthrough as it allowed the editing of any gene (Doetschman et al., 1987; Thomas and Capecchi, 1987). Targeting of genes requires the generation of a targeting vector containing DNA segments homologous to sequences of the endogenous gene flanking the desired modification (see Figure A-1). The vector is transfected into ES cells, and correctly targeted clones are selected (see Figure A-7a). Cells carrying the desired modification are injected into mouse blastocysts to generate chimeric mice (see Figure A-7b), which are bred with normal mice to obtain offspring carrying the mutant allele (see Figure A-7c). Homologous recombination in combination with ES cells has allowed scientists to efficiently create mice transmitting specific gene mutations to the next generation. Following the initial generation of mice carrying targeted mutations of the β2-microglubulin and the c-Abl gene (Schwartzberg et al., 1989; Zijlstra et al., 1989), homologous recombination in ES cells has become a widely used tool for the study of mammalian development and the generation of animal models of human genetic diseases (Solter, 2006). Because chimera–competent ES cells were only available in the murine system, gene editing by homologous recombination was restricted to mice and could not readily be used in other species.
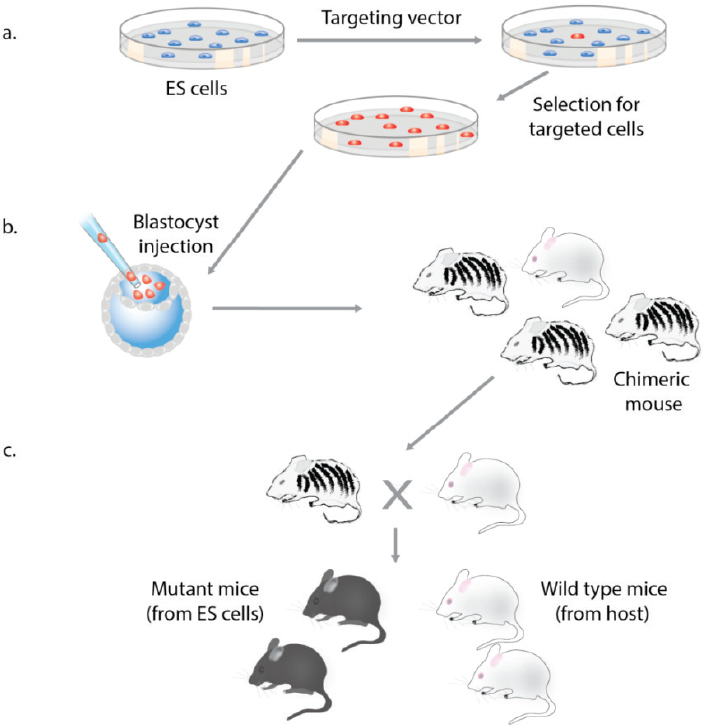
(a) In the first step a targeting vector with homology to the target gene is introduced into ES cells and correctly targeted clones are selected in culture. (b) The targeted ES cells are injected into an albino host blastocyst, which is transplanted into a foster mother to produce chimeric mice where the donor ES cells contribute to the tissues of the animal (as seen by coat color contribution derived from the ES cells). (c) Germline transmission of the ES cell clone is verified by mating of the chimeric mouse with the albino host strain. Pigmented offspring are derived from the donor ES cells.
Nuclear Cloning and the Generation of Mutant Animals
The transfer of a somatic nucleus into an enucleated egg resets the epigenetic state of the nucleus to an embryonic state and allows the generation of animals such as Dolly, the first cloned mammal (Wakayama et al., 1998; Wilmut et al., 1997). The production of animals from somatic cells by nuclear cloning allowed the generation of mutant animals in species where no ES cells were available. The first successful application of nuclear cloning in combination with homologous recombination to produce gene-altered farm animals used sheep fibroblasts. The human 1-antitrypsin gene was inserted into the 3’ UTR of the COL1A1 gene, a convenient “safe harbor” locus giving predictable expression of the transgene. Transgenic sheep were derived from the targeted fibroblasts by nuclear-cloning, generating animals that expressed the therapeutically important human 1-antitrypsin protein (McCreath et al., 2000).
The strategies to produce animals carrying engineered gene alterations as summarized above relied on the manipulation of ES cells or on nuclear cloning, both of which are labor-intensive and require special skills. This changed dramatically when the new genome-editing methods based on ZFNs, TALENs, and CRISPR/Cas9 became available (Doudna and Charpentier, 2014). These approaches, described above, revolutionized the ability of researchers to edit genes in any species and to produce genetically altered animals with a fraction of the effort and time and much less sophistication and experimental skills needed than were required for the generation of gene-edited animals by strategies based on ES cells or nuclear transfer.
GENOME EDITING IN EMBRYOS
Homologous recombination in conventional gene targeting is an inefficient process and requires the selection of correctly targeted cell clones in cell culture. In a second step the targeted ES cell clone is injected into a host blastocyst to create a chimeric animal, which, in a third step, is mated to produce the desired mutant animal, a process that may take as much as 1 or 2 years (compare Figure A-7). In contrast, gene targeting by TALEN or by CRISPR/Cas9 is so efficient that no selection for correct targeting is required (Sakuma and Woltjen, 2014), making it possible to derive genetically modified animals in one step by direct genetic manipulation of the fertilized egg (see Figure A-8).
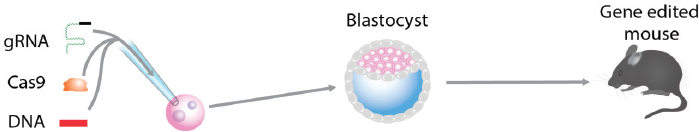
CRISPR/Cas9-Mediated Gene Editing in the Zygote
The injection of guide RNAs together with Cas9 RNA into the fertilized egg (zygote) was used to generate mice carrying mutations in several genes. The efficiency of Cas9-mediated DNA DSBs was high and resulted in 80 percent of pups carrying mutations in both alleles of two different genes (Wang et al., 2013). When the guide and Cas9 RNAs were co-injected with an oligonucleotide carrying a point mutation, the mutation was introduced into two target genes in 60 percent of the pups. In addition, the generation of conditional mutants requiring the insertion of two LoxP sites into the same allele was shown to be effective in the zygote. Thus, NHEJ-mediated mutation as well as insertion of DNA at the DSB site is extremely efficient, allowing the generation of mice carrying complex mutations within 3 weeks (the gestation time of the mouse) instead of 1 to 2 years when using ES cell–mediated gene targeting. The CRISPR/Cas9 gene-editing method was shown to work efficiently not only in mice but also in other species including rats (Li et al., 2013), zebrafish (Hwang et al., 2013), C. elegans (Friedland et al., 2013), and Drosophila (Zeng et al., 2015). Importantly, the approach allowed the generation of primates carrying mutations in specific genes (Niu et al., 2014). Most recently, two reports appeared that used genome editing in preimplantation human embryos that were defective and therefore could not be used to generate pregnancy (Kang et al., 2016; Liang et al., 2015).
The evidence summarized above indicates that animals carrying defined mutations in multiple genes can be generated in one step by manipulation of the fertilized egg. However, if intended for gene therapy (correction of mutant genes), several significant complications need to be considered. These include frequent mosaicism of manipulated embryos, the mutation of both
alleles when the goal is to correct one mutant allele, and the impossibility of genotyping the one-cell embryo.
Mosaicism
The cleavage of the target gene and the insertion of DNA at the double-strand breakpoint may occur at a later stage than the zygote—such as the two-cell stage. The consequence of integration at a stage later than the one-cell zygote stage is that half (or less, depending on the time of DNA insertion) of the embryo’s cells will carry the altered gene whereas the others will not. Animals with genetic alterations in only a subset of the cells are designated as “mosaics.” The available evidence indicates that the incidence of mosaicism may be as high as 50 percent or higher (Wang et al., 2013). The high incidence of mosaicism has an important practical consequence: Preimplantation genetic diagnosis (PGD)—the biopsy of one or a few cells of the manipulated embryo—cannot be used to ascertain whether gene targeting resulted in the desired mutation because the biopsied cells may not reflect the genotype of the other cells of the embryo.
Mutation of the Wild-Type Allele by Cas9-Mediated Cleavage
Cleavage by Cas9 is significantly more efficient than insertion of a donor DNA at the cleavage site by homologous recombination. This poses a complication if the goal of gene editing in embryos is the correction of a mutant allele. To correct a given mutation, a guide RNA and a DNA target construct are injected into the embryo. While the DNA will integrate into the mutant allele at the DSB and correct the mutation, the other allele will often be cleaved, creating a new mutation by NHEJ. Given present technology, this poses a possibly serious problem for gene therapy as the mutant allele is corrected, but a new mutant allele is created. The inhibition of repair by end joining using a small molecule may help to mitigate this problem as this has been shown to favor the insertion of DNA by HDR over that by NHEJ (Maruyama et al., 2015). However, the unwanted mutation of the normal allele currently remains a complication of CRISPR/Cas9-mediated gene correction.
Genotyping of the One-Cell Embryo
Genome editing in embryos with the goal being to correct a mutant allele faces another problem: how to distinguish a wild type from a mutant embryo. If one parent carries a dominant mutant gene, 50 percent of the embryos will be affected and 50 percent will be wild type, and if both parents carry a recessive mutation, 75 percent of the embryos will be normal
and 25 percent will be affected. Because it is not possible to use any current molecular test to distinguish mutant from normal embryos at the zygote stage, any gene-editing attempt will target (and modify) a large fraction of normal embryos. It is unlikely that a technological advance could resolve this dilemma in the foreseeable future.
GENE DRIVE: A MECHANISM TO SPREAD GENETIC ALTERATIONS THROUGH SEXUALLY REPRODUCING POPULATIONS
It has been proposed that naturally occurring “homing endonucleases” could cause mutant alleles to spread rapidly through sexually reproducing populations by a process designated as “gene drive” (Burt, 2003, 2014). The spreading of such a mutant allele through a population does not require a selective advantage for the carrier of the mutant gene but would rather propagate like a “selfish gene” (Esvelt et al., 2014; Oye et al., 2014).
Recently, a vector encoding both Cas9 and a guide RNA was introduced into a Drosophila genomic locus that governs cuticle color, creating a knock-out mutation. Cas9-mediated cleavage during development of the germline stimulated the copying of the insertion into the wild-type locus, such that all female gametes carried the insertion (see Figure A-9). Importantly, when these eggs were fertilized the mutation in the resulting heterozygous animals converted the wild-type allele into a mutant allele during meiosis by Cas9-mediated target gene cleavage, followed by HDR resulting in a homozygous mutation. Thus, the vector carrying the Cas9 gene and a guide RNA, when integrated into one allele, led to the conversion of the other allele during meiosis with an efficiency of 98 percent, causing the rapid spread of the mutant allele through the population (Gantz and Bier, 2015). This autocatalytic process was dubbed as a “mutagenic chain reaction.”
If a vector carrying sequences correcting a given mutation in addition to the Cas9 and the guide RNA was inserted into one allele, these sequences could serve as a template during meiosis and convert the other allele by homologous recombination resulting in two repaired alleles (see Figure A-9a). Similarly, if a vector carrying sequences coding for another gene in addition to the Cas9 and the guide RNA were integrated into one allele, Cas9-mediated cleavage of the wild-type allele during meiosis would transfer the exogenous “cargo” gene into the other allele, leading to homozygous transgenic animals (Esvelt et al., 2014). Thus, gene-drive mechanisms can effectively propagate and spread mutant alleles or newly inserted genes through animal populations (see Figure A-9b).
So far gene-drive constructs have been demonstrated to spread through insect populations and have been proposed for use in mosquito popula-
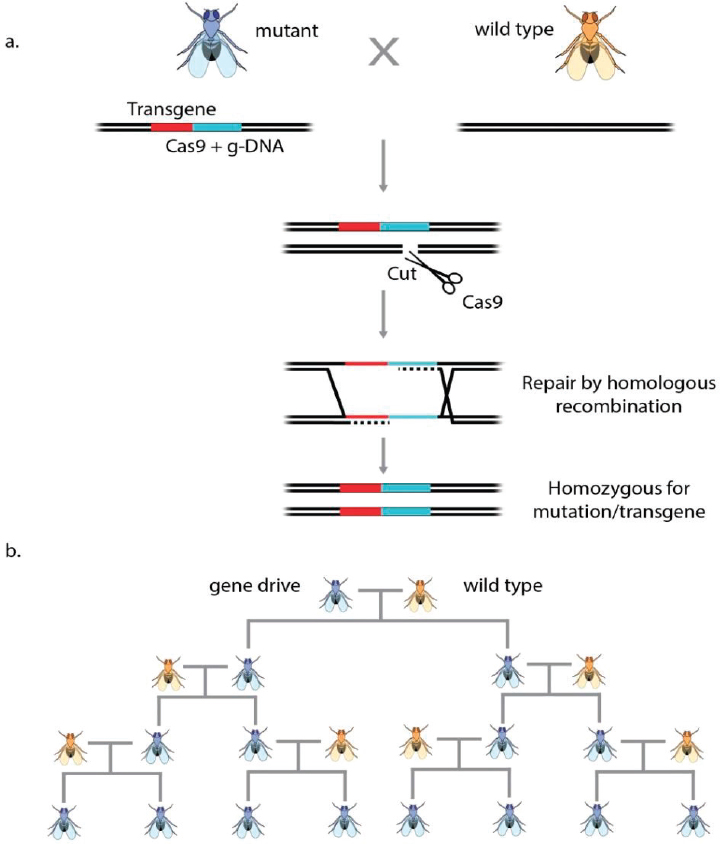
SOURCE: Information from Esvelt et al., 2014.
tion control. Gene drives have not as yet been shown to behave similarly in mammalian species. However, given the efficiency of genome editing in mammalian embryos it is possible, if not likely, that the underlying gene-drive mechanisms would also be effective in mammals and could in principle create gene modifications that could spread through the population. However, given the generation time and breeding patterns of humans, any such gene-drive application in the human species would require an inordinate numbers of years and seems inconceivable.
ALTERNATIVE ROUTES TO HERITABLE GERMLINE EDITING
The efficiency and precision of CRISPR/Cas9 gene-editing approaches have raised the potential that precise genome editing might be possible in cells that could contribute to the germline of the human species. For a genetic alteration to be passed on to the next generation, it has to be made in (1) progenitor cells that can give rise to the gametes (eggs and sperm), (2) the eggs and sperm themselves, or (3) in the fertilized zygote or early embryo, when all cells can still contribute to the future germline.
As discussed in the preceding section, methods for germline editing have been most highly developed in the mouse and applied to a number of other mammalian species, particularly in relation to agricultural needs or the generation of preclinical disease models for human genetic diseases. Until the recent advent of advanced genome-editing tools, germline gene alterations in mice were primarily achieved via nontargeted introduction of transgenic DNA into the genome of the zygote or by targeted mutagenesis in ES cells. The latter approach involved the homologous recombination of targeting vectors into the host genome, the selection of the correctly targeted clones, and the generation of germline-transmitting chimeric animals (see details in the above section on germline genetic alteration). While this approach has proven to be extraordinarily powerful in producing knockout mice, conditional mutations, reporter lines, and a variety of human disease models, it is still relatively inefficient and has not been readily applicable to direct targeted genome alterations in zygotes. Thus, the idea of potential targeted gene alterations or corrections in human embryos was not under consideration.
However, more recently, the efficiency of CRISPR/Cas in targeting nuclease-enhanced editing to specific sites in the genome has raised new vistas including possible human germline editing. Soon after the demonstration that CRISPR/Cas could very efficiently target specific sites in the genome of mammalian cells, it was shown that this approach could be applied directly to the mouse zygote without the need for the intermediate ES cell step (see above section and Figure A-8). Thus, it became possible to consider genome editing directly in human embryos. The only publications to date on
testing CRISPR on human zygotes demonstrated that targeted mutations could be generated, but also demonstrated that the resulting mutations were often complex and that only some embryos or embryonic cells carried the targeted event (Kang et al., 2016; Liang et al., 2015). As explained above, these intrinsic issues of CRISPR editing make the concept of using zygote genome editing to correct human genetic disease very challenging.
Editing the embryo genome, however, is not the only potential way to achieve modification of the genome in the germline. Approaches that directly modify the genome of the gametes—the eggs and sperm—before fertilization would overcome problems of mosaicism and would potentially allow preselection of the appropriately targeted gamete before in vitro fertilization.
Gamete Gene Editing: Current Status
There are a number of potential routes to gamete gene editing, some of which are already in use in the mouse and some of which remain to be fully developed.
Direct Introduction of Editing Factors into Oocytes
In the mouse it has been shown that maternally inherited Cas9 nuclease provides a very efficient means of generating targeted alterations in the resulting zygotes (Sakurai et al., 2016), presumably because of the immediate availability of the enzyme. While such an approach is, of course, not applicable to humans, it does suggest that preloading ovulated oocytes with the editing factors prior to in vitro fertilization might be a means of avoiding mosaic editing and enhancing efficiency. Whether this could actually promote gene targeting in the oocyte genome rather than after fertilization remains to be reported. Preselection of mutated or corrected oocytes would still be a challenge, but the reduction in mosaicism would allow PGD to be contemplated for identifying correctly targeted embryos.
Gene Editing in Sperm In Vitro
Sperm-mediated gene transfer is a fairly well established, although inefficient, route to transgenesis in a number of species, from fish to pigs (Lavitrano et al., 2013). Thus, it should be possible to introduce the components of genome-editing systems into sperm and have them carried into the zygote to promote genome editing there. More interesting is the possibility of direct genome editing in the sperm nuclei. Given that sperm are nondividing cells, currently only NHEJ-mediated gene editing would be possible, although the repair mechanism is presumably different from that in somatic
cells (Ahmed et al., 2010). Homologous recombination-mediated gene correction or alteration has to date only been possible in dividing cells. However, there are indications that this block can be overcome (Orthwein et al., 2015). Izpisua Belmonte’s group has recently developed an NHEJ-based gene knock-in method—homology-independent targeted gene integration (HITI). HITI allows the direct knock-in of DNA sequences at specific genomic loci in post-mitotic cells, for example, neurons (Suzuki et al., 2016b). HITI may open up new avenues to sperm (and even oocyte) gene editing. Insertion of a transient fluorescent reporter to identify sperm carrying the editing factors could help enrich for potentially gene-edited sperm. Final confirmation of the appropriately edited embryos after fertilization in vitro or intracytoplasmic sperm injection (ICSI) would require PGD.
Gene Editing in Germline Stem Cells
There is considerable biological and clinical interest in generating gametes from stem cell lines that can be propagated indefinitely in vitro. Spermatogonial stem cells (SSCs) have been isolated from mouse testes and have the capacity to regenerate fertilization-competent sperm when retransplanted to the germ cell–depleted adult testis (Kanatsu-Shinohara and Shinohara, 2013). Gene editing in SSCs would allow for the preselection of clonal lines with appropriate targeted mutations and the potential to prescreen for off-target effects or other unwanted genomic or epigenomic alterations before generating gametes. Proof of principle for such an approach has been published (Wu et al., 2015), in which the authors corrected a gene mutation that causes cataracts in mice by CRISPR/Cas9 editing in SSCs. SSCs were transferred back to the testis and round spermatids collected for ICSI. Offspring were correctly edited at 100 percent efficiency.
Translating this work into humans has many challenges. While SSC-like cells have been isolated from human testes (Wu et al., 2015), stable, self-renewing cell lines have not yet been achieved. If this challenge is overcome, there still remains the challenge of generating ICSI-competent gametes from the SSCs. In the mouse, this is achieved by transfer into the germ cell–depleted testis—not an easy solution in humans. Alternate approaches include generating a “reconstituted testis” with mixed SSCs and supporting cells of the testis and transplanting this under the testis capsule. This approach would also be ethically challenging in humans. The possible use of interspecies reconstitutions and transplants into immune-deficient mice would bring its own scientific and ethical challenges. The best solution would be to promote differentiation of the SSCs to mature haploid gametes in a fully defined culture system in vitro—a challenge not yet achieved in any system.
While the possibility of applying similar approaches to the female
germline is attractive, the evidence for the existence of oogonial stem cells is controversial (Johnson et al., 2004). Most evidence suggests that there is a limited resource of oocytes in the adult mammalian ovary (Eggan et al., 2006) and no evidence for any endogenous stem cells.
Gene Editing in Pluripotent Stem Cells Followed
by Germ Cell Differentiation
Pluripotent embryonic stem cells or induced pluripotent stem cells can be generated from both males and females, are readily amenable to CRISPR editing, and can be differentiated down the pathway toward meiotically competent germ cells. In the mouse, the most reliable reports of germ cell generation from ES cells have come from mimicking the known pathways that induce primordial germ cells from the pluripotent epiblast in the early embryo. Using this approach, Saitou’s lab has generated primordial germ cell–like cells (PGC-LCs) from both male and female ES cells. When PGC-LCs were reconstituted with support cells from the testis or ovary respectively and transplanted back to the testis or ovary environment, investigators were able to recover spermatids or oocytes that could be used to generate viable offspring when combined with normal eggs and sperm (Hayashi et al., 2011, 2012). Recent advances have further extended this approach, either by coculture of the PGC-LCs with testis cells in culture to generate spermatid-like cells in vitro (Zhou et al., 2016), or by derivation of SSCs in cocultures of pluripotent embryonic stem cells with somatic testis cells and subsequent maturation to spermatids in adult testes (Ishikura et al., 2016). In both cases, spermatids were derived that were capable of fertilizing oocytes after ICSI and generating viable offspring. There are some concerns about whether epigenetic reprogramming would be complete in this culture system, but the overall results are quite remarkable. Another interesting development is from the Izpisua Belmonte, Okuda, and Matsui groups, who have shown that knockdown or knockout of Max in mouse embryonic stem cells (ESCs) strongly activates expression of germ cell–related genes and results in profound cytological changes to resemble cells undergoing meiotic division (Maeda et al., 2013; Suzuki et al., 2016a). Whether functional haploid cells can be generated using this approach remains to be seen.
These results in murine systems raise expectations that human haploid gametes could be generated from human pluripotent cells, with implications for understanding gametogenesis and causes of infertility and potentially offering new avenues for reproduction in infertile couples. It also would open up genetic modification of the stem cells to repair known genetic causes of infertility or to repair dominant gene mutations. However, to date, human gametes have not been generated successfully from pluripotent stem cells,
although two recent papers report the generation of early PGC-LCs from human ES cells (Irie et al., 2015; Sasaki et al., 2015). Those studies revealed similarities and differences from the mouse germ cell differentiation pathway. This suggests that more knowledge of how germ cells actually develop in the human, or perhaps the nonhuman primate, embryo versus the mouse embryo is needed to move this research forward.
Gene Editing in Haploid ES Cells
Most animals are diploids, and natural haploid cells are typically limited to mature germ cells. Recently both androgenetic (male) and parthenogenetic (female) haploid ES cells (haESCs) have been derived in mice and rats (Leeb and Wutz, 2011; Li et al., 2012, 2014; Yang et al., 2012). haESCs contain only one copy of allelic genes of diploid cells and are amenable to genetic modification with traditional gene-targeting approaches and with new nuclease-based genome-editing strategies (Li et al., 2012, 2014). More interestingly, androgenetic haESCs, which contain a Y rather than an X chromosome, can produce viable and fertile offspring after intracytoplasmic injection into mature oocytes (Li et al., 2012, 2014). Haploid parthenogenetic mouse haESCs were also shown to be able to produce fertile mice when injected into oocytes in place of the maternal genome (Wan et al., 2013). Both strategies are possible to be used for introduction of genetic modifications to progeny. Most recently parthenogenetic human haESCs have also been successfully generated (Sagi et al., 2016). Human androgenetic haESCs have not yet been reported.
There are several limitations of haESCs. First, the haploid phenotype has been found to be unstable in culture. haESCs undergo spontaneous auto-diploidization and need several rounds of haploid purification by flow-activated cell sorting before becoming stable in culture. Also, there is a lack of androgenetic haESCs containing the Y chromosome (Li et al., 2012). This is due to the poor developmental potential of androgenetic embryos with YY chromosomes (Latham et al., 2000; Tarkowki, 1977). Therefore only female animals can currently be created. With further breeding, males can then be obtained. Another major drawback is that the efficiency for androgenetic haESCs to fertilize an egg is very low (less than 5 percent in mice and less than 2 percent in rats).
In summary, although the generation of human “artificial” gametes from stem cell lines is not currently achievable, work in the mouse suggests that this will likely be possible. Reconstituting the testis or ovary environment in vitro may be achieved by deriving both germ cells and supporting cells, such as Sertoli cells, and granulosa cells from in vitro differentiation of human ES cells. Further understanding of the endogenous signaling pathways that promote germ cell development and meiotic maturation will
aid in the future derivation of human gametes in vitro. Such cells will be immediately useful for understanding gametogenesis and dissecting fertility problems, but safety concerns will need to be overcome before they could be used for human reproduction, with or without genome editing. The germline is generally considered to be somewhat protected from genetic damage, unlike that of somatic cells, and also undergoes extensive epigenetic remodeling before completion of gametogenesis. Both aspects would need to be replicated in the artificial gametes generated in vitro.
EDITING THE MITOCHONDRIAL GENOME
Mitochondrial diseases are a group of maladies caused by the dysfunction of mitochondria due to mutations in mitochondrial DNA (mtDNA). Mitochondrial diseases are associated with the degeneration of tissues and organs that have high energetic demands—including muscle, heart, and brain—that lead, among other pathologies, to myopathies, cardiomyopathies, neuropathies, encephalopathies, lactic acidosis, stroke-like syndrome, and blindness (Taylor and Turnbull, 2005). The percentage of mtDNA molecules that is mutated generally determines whether or not a patient is symptomatic. Currently, there are no cures for mitochondrial diseases, and for patients healthy enough to have children, genetic counseling and PGD represent the best options for preventing disease transmission. However, due to the non-Mendelian inheritance of mtDNA and the potentially different heteroplasmy levels among different blastomeres, PGD can only reduce, not eliminate, the risk of transmitting the disease. Recently developed mitochondrial replacement techniques involve a series of complex technical manipulations of nuclear genome between patient and donor oocytes that results in the generation of embryos carrying genetic material from three different origins (Paull et al., 2012; Tachibana et al., 2012). For these reasons, mitochondrial replacement techniques have raised biological, medical, and ethical concerns (Hayden, 2013; Reinhardt et al., 2013). Mitochondrial replacement techniques have low rates of success, and studies in lower organisms have reported potential issues arising from incompatibility between nuclear and mtDNA upon mitochondrial replacement (Reinhardt et al., 2013).
A novel alternative therapeutic approach was recently developed to eliminate the mutated mtDNA in the germline. Using mitochondria-targeted endonuclease, the targeted mtDNA in the mouse germline was successfully prevented from transmission to the next generation (Reddy et al., 2015). Due to the limited number of mtDNA mutations that can be targeted by restriction endonucleases, efforts have been made to target most of the mtDNA mutations using mitochondria-targeted transcription activator-like effector nucleases (mito-TALENs) and ZFNs (Bacman et al.,
2013; Gammage et al., 2014). The mito-TALENs were able to specifically eliminate the targeted mtDNA in the mouse germline (Reddy et al., 2015). Importantly, the technique of injection of nucleases (e.g., mito-TALENs) into oocytes or early embryos involves a simple microinjection of mRNA that encodes the nucleases. Moreover, the use of mitochondrial localization signal (e.g., Cox8 and ATP5b) restricts the translocation to mitochondria alone. A caveat of this technology is that elimination of high levels of mutated mtDNA in oocytes will lead to the generation of embryos with a low number of normal mtDNA that, if failing to replicate after implantation, could lead to pregnancy loss. PGD could be used for the selection and transfer of embryos containing higher levels of normal mtDNA. Importantly, unlike nuclear editing, mtDNA editing is not aimed to correct the mutations, but to eliminate mutated DNA, which is possible due to the presence of multiple copies of mtDNA in the oocytes. Moreover, due to the very low activity of repair mechanisms in mitochondria, the frequency of re-ligation of target mtDNA and introduction of new mutations would be very rare. In addition, similar mitochondrial editing tools in the future could also be used to eliminate mutated mtDNA in gametes derived from stem cells. Finally, a combination of mitochondrial gene-editing tools with mitochondrial replacement techniques may represent an alternative option, in the future, to prevent the germline transmission of mutations in the mtDNA responsible not only for mitochondrial specific diseases but also for situations where alterations in mitochondrial function contribute to pathologies such as cancer, diabetes, and aging-associated diseases.
THE CHALLENGE OF DELIVERY
In addition to the technical advances being made to genome-editing systems, an important challenge for in vivo use is effective delivery. Table A-1 highlights a number of strategies being explored for the delivery of genome-editing components, including a discussion of their advantages and disadvantages.
TABLE A-1 General Approaches to Delivering Genome-Editing Components
| Method | Delivered Component | Explanation | Advantages | Disadvantages | Preferred Applications |
|---|---|---|---|---|---|
| Nonviral | |||||
| Transfection | Nuclease(s) as plasmid DNA, RNA, or protein | All components are assembled with a glyco/lipo-polymer vehicle that favors cell entry; the complex is applied to cells ex vivo or injected into a tissue or blood in vivo. | Relatively simple; can deliver all editing machinery together; transient expression limiting cytotoxicity and immunogenicity of editing machinery. | Cellular uptake and nuclear access can be rate-limiting; vehicle can cause cytotoxicity and inflammation in vivo; for in vivo delivery RNA needs to be modified for improved stability; cannot be targeted to specific tissues; formulation often proprietary. | Cell lines and some primary cells ex vivo |
| guideRNA as plasmid DNA or oligonucleotide is mixed with the nuclease RNA (can be complexed with nuclease as Ribonucleoprotein, RNP) | |||||
| Template as plasmid DNA or oligonucleotide (can be pre-complexed with RNP) (in some protocols nuclease(s) are delivered as mRNA, protein, or RNP by a nonviral method, most often electroporation, and template DNA is delivered separately by a viral vector, before or after delivery of the nuclease) | |||||
| Method | Delivered Component | Explanation | Advantages | Disadvantages | Preferred Applications |
|---|---|---|---|---|---|
| Nanoparticle | The complex, as above, is coupled to a nanoparticle (i.e., by gold, dextran) to enhance cellular uptake and delivery and biodistribution in vivo. | Cellular uptake can be enhanced; can be targeted to specific tissues; transient expression limiting cytotoxicity and potential immunogenicity of editing machinery. | Few | Tissues or systemic | |
| Electroporation | A brief electric pulse is passed across a population of cells in a solution that contains the editing nuclease reagents with or without template. | Very effective method of delivery to a wide variety of cell types ex vivo; transient expression limiting cytotoxicity and potential immunogenicity of editing machinery. | Can cause cytotoxicity (increasingly from protein to mRNA to DNA; alleviated by using modified nucleic acids) but better tolerated than transfection; challenging to get delivery in vivo. | Cell lines and primary cells ex vivo | |
| Squeeze Poration | A population of cells is passed through a small channel that is smaller than the diameter of the cells, creating small pores in the membrane to allow the nuclease and donor reagents to enter the cell. | New strategy that has not been widely validated. | Would not work for in vivo delivery. |
| Microinjection | Nuclease(s) as plasmid DNA, RNA, or protein; gRNA as plasmid DNA or RNA. The gRNA and nuclease can be pre-complexed as RNP. Template can be introduced as a plasmid, isolated DNA fragment, or oligonucleotide | The components can be introduced in a simple saline buffer, into the cytoplasm, or into the nucleus of cells or pronucleus of zygotes. | Efficiencies up to 100% | Technically more challenging than other methods, relatively few cells can be injected. | Zygotes and early embryos |
| Method | Delivered Component | Explanation | Advantages | Disadvantages | Preferred Applications |
|---|---|---|---|---|---|
| Viral | |||||
| Lentiviral Vector (LV) | Nuclease(s) and guideRNA as gene expression cassettes delivered by the vector genome (can be driven by separate and tissue specific promoters) | Replication-defective virus that can package ~8 kilobases of nucleic acid with an ability to enter almost all cell types; vector integrates quasi-randomly into cell genome, allowing for stable expression and transmission to the cell progeny; a modified version made with mutant integrase-defective lentiviral vector (IDLV), fails to integrate and is rapidly lost in proliferating cells. | Currently the most common tool for ex vivo gene transfer, also being explored for in vivo use; expression is stable for LV and transient for IDLV and can be made tissue-specific, regulated, or conditional; transduction of human cells well tolerated; IDLV provides for transient nuclease expression, thus limiting cytotoxicity and immunogenicity, and is well suited for template delivery. | Stable expression of nuclease by LV potentially leading to cytotoxicity and immunogenicity; potential risk of insertional mutagenesis by LV; vector manufacturing more complex and expensive than non-viral platform. | Cell lines and primary cells ex vivo |
| Template DNA (can be used for the delivery of template alone, in combination with electroporation of nuclease RNA, protein or RNP, or for the combined delivery of template and guideRNA and/or nuclease, on the same or separate vectors) | |||||
| Adeno-Associated Virus (AAV) Vector | Replication-defective virus that can package ~4.7 kilobases of DNA with a broad number of variants (both natural and engineered serotypes) that can target different cell types. | Currently the most common tool for gene transfer in vivo; remains mostly episomal in the target cell nucleus; efficient delivery and robust expression in wide variety of cell types and tissues; transduction of human cells well tolerated; transient expression in dividing cells; well suitable for nuclease and template delivery. | Limited capacity In non-proliferating cells and tissues, the vector can persist for years, leading to long-term expression of nuclease and potential cytotoxicity and immunogenicity; some people have pre-existing immunity to AAV, which can inhibit in vivo gene transfer or clear transduced cells. | Primary cells ex vivo and tissues or systemic in vivo | |
| Adenoviral Vector | Replication-defective virus that can package >20 kilobases of DNA able to transduce a wide variety of cell types both ex vivo and in vivo. | Able to package large fragments of DNA; can be used for in vivo and ex vivo delivery; transient expression in dividing cells and tissues. | Has shown severe acute toxicity in some clinical trials; many people have pre-existing immunity; no longer commonly used gene therapy vector. | Primary cells ex vivo |
NOTE: IDLV = integrase-defective lentiviral vector; RNP = ribonuclear protein complex; sgRNA = single guide RNA.
REFERENCES
Ahmed, E. A., P. de Boer, M. E. P. Philippens, H. B. Kal, and D. G. de Rooij. 2010. Parp1-XRCC1 and the repair of DNA double strand breaks in mouse round spermatids. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 683(1-2):84-90.
Auer, T. O., K. Duroure, A. De Cian, J. P. Concordet, and F. Del Bene. 2014. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Research 24(1):142-153.
Bacman, S. R., S. L. Williams, M. Pinto, S. Peralta, and C. T. Moraes. 2013. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nature Medicine 19(9):1111-1113.
Barrangou, R., C. Fremaux, H. Deveau, M. Richards, P. Boyaval, S. Moineau, D. A. Romero, and P. Horvath. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819):1709-1712.
Bibikova, M., D. Carroll, D. Segal, J. K. Trautman, J. Smith, Y. G. Kim, and S. Chandrasegaran. 2001. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Molecular and Cellular Biology 21(1):289-297.
Bibikova, M., M. Golic, M., K. G. Golic, and D. Carroll. 2002. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161(3):1169-1175.
Bibikova, M., K. Beumer, J. K. Trautman, and D. Carroll. 2003. Enhancing gene targeting with designed zinc finger nucleases. Science 300(5620):764.
Brinster, R. L., H. Y. Chen, M. Trumbauer, A. W. Senear, R. Warren, and R. D. Palmiter. 1981. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell 27(1 Pt. 2):223-231.
Burt, A. 2003. Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proceedings of The Royal Society B: Biological Sciences 270(1518):921-928.
Burt, A. 2014. Heritable strategies for controlling insect vectors of disease. Philosophical Transactions of the Royal Society B: Biological Sciences 369(1645):20130432.
Carroll, D. 2014. Genome engineering with targetable nucleases. Annual Review of Biochemistry 83:409-439.
Chevalier, B. S., T. Kortemme, M. S. Chadsey, D. Baker, R. J. Monnat, and B. L. Stoddard. 2002. Design, activity, and structure of a highly specific artificial endonuclease. Molecular Cell 10(4): 895-905.
Chiarle, R., Y. Zhang, R. L. Frock, S. M. Lewis, B. Molinie, Y. J. Ho, D. R. Myers, V. W. Choi, M. Compagno, D. J. Malkin, D. Neuberg, S. Monti, C. C. Giallourakis, M. Gostissa, and F. W. Alt. 2011. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147(1):107-119.
Cho, S. W., S. Kim, J. M. Kim, and J. S. Kim. 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnology 31(3):230-232.
Cong, L., F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini, and F. Zhang. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819-823.
Costantini, F., and E. Lacy. 1981. Introduction of a rabbit beta-globin gene into the mouse germ line. Nature 294(5836):92-94.
Cox, D. B. T., R. J. Platt, and F. Zhang. 2015. Therapeutic genome editing: Prospects and challenges. Nature Medicine 21(2):121-131.
Deltcheva, E., K. Chylinski, C. M. Sharma, K. Gonzales, Y. Chao, Z. A. Pirzada, M. R. Eckert, J. Vogel, and E. Charpentier. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340):602-607.
Desjarlais, J. R., and J. M. Berg. 1992. Toward rules relating zinc finger protein sequences and DNA binding site preferences. Proceedings of the National Academy of Sciences of the United States of America 89(16):7345-7349.
Dickinson, D. J., J. D. Ward, D. J. Reiner, and B. Goldstein. 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods 10(10):1028-1034.
Doetschman, T., R. G. Gregg, N. Maeda, M. L. Hooper, D. W. Melton, S. Thompson, and O. Smithies. 1987. Targeted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330:576-578.
Doudna, J. A., and E. Charpentier. 2014. Genome editing: The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213):1258096.
Eggan, K., S. Jurga, R. Gosden, I. M. Min, and A. J. Wagers. 2006. Ovulated oocytes in adult mice derive from non-circulating germ cells. Nature 441(7097):1109-1114.
Esvelt, K. M., A. L. Smidler, F. Catteruccia, and G. M. Church. 2014. Concerning RNA-guided gene drives for the alteration of wild populations. eLife e03401.
Evans, M. J., and M. H. Kaufman. 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature 292(5819):154-156.
Friedland, A. E., Y. B. Tzur, K. M. Esvelt, M. P. Colaiacovo, G. M. Church, and J. Calarco. 2013. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods 10(8):741-743.
Fu, Y., J. A. Foden, C. Khayter, M. L. Maeder, D. Reyon, J. K. Joung, and J. D. Sander. 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology 31(9):822-826.
Gabriel, R., A. Lombardo, A. Arenas, J. C. Miller, P. Genovese, C. Kaeppel, A. Nowrouzi, C. C. Bartholomae, J. Wang, G. Friedman, M. C. Holmes, P. D. Gregory, H. Glimm, M. Schmidt, L. Naldini, and C. von Kalle. 2011. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature Biotechnology 29(9):816-823.
Gammage, P. A., J. Rorbach, A. I. Vincent, E. J. Rebar, and M. Minczuk. 2014. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Molecular Medicine 6(4):458-466.
Gantz, V., and E. Bier. 2015. The mutagenic chain reaction: A method for converting heterozygous to homozygous mutations. Science 348(6233):442-444.
Garneau, J. E., M. E. Dupuis, M. Villion, D. A. Romero, R. Barrangou, P. Boyaval, C. Fremaux, P. Horvath, A. H. Magadan, and S. Moineau. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468(7320):67-71.
Gasiunas, G., R. Barrangou, P. Horvath, and V. Siksnys. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America 109(39):E2579-E2586.
Gilbert, L. A., M. H. Larson, L. Morsut, Z. Liu, G. A Brar, S. E. Torres, N. Stern-Ginossar, O. Brandman, E. H. Whitehead, J. A. Doudna, W. A. Lim, J. S. Weissman, and L. S. Qi. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2):442-451.
Gilbert, L. A., M. A. Horlbeck, B. Adamson, J. E. Villalta, Y. Chen, E. H. Whitehead, C. Guimaraes, B. Panning, H. L. Ploegh, M. C. Bassik, L.S. Qi, M. Kampmann, and J. S. Weissman. 2014. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159(3):647-661.
Gordon, J. W., and F. H. Ruddle. 1981. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 214(4526):1244-1246.
Guilinger, J. P., D. B. Thompson, and D. R. Liu. 2014. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nature Biotechnology 32(6):577-582.
Hammer, R. E., R. D. Palmiter, and R. L. Brinster. 1984. Partial correction of murine hereditary growth disorder by germ-line incorporation of a new gene. Nature 311(5981):65-67.
Haurwitz, R. E., S. H. Sternberg, and J. A. Doudna. 2012. Csy4 relies on an unusual catalytic dyad to position and cleave CRISPR RNA. The EMBO Journal 31(12):2824-2832.
Hayashi, K., H. Ohta, K. Kurimoto, S. Aramaki, and M. Saitou. 2011. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146(4):519-532.
Hayashi, K., S. Ogushi, K. Kurimoto, S. Shimamoto, H. Ohta, and M. Saitou. 2012. Offspring from oocytes derived from in vitro primordial germ cell-like cells in mice. Science 338(6109):971-975.
Hayden, E. C. 2013. Regulators weigh benefits of “three-parent” fertilization. Nature 502(7471):284-285.
Hsu, P. D., D. A. Scott, J. A. Weinstein, F. A. Ran, S. Konermann, V. Agarwala, Y. Li, E. J. Fine, X. Wu, O. Shalem, T. J. Cradick, L. A. Marraffini, G. Bao, and F. Zhang. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology 31(9):827-832.
Hwang, W. Y., Y. Fu, D. Reyon, M. L. Maeder, S. Q. Tsai, J. D. Sander, R. T. Peterson, J. R. Yeh, and J. K. Joung. 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology 31(3):227-229.
Irie, N., L. Weinberger, W. W. Tang, T. Kobayashi, S. Viukov, Y. S. Manor, S. Dietmann, J. H. Hanna, and M. A. Surani. 2015. SOX17 is a critical specifier of human primordial germ cell fate. Cell 160(1-2):253-268.
Ishikura, Y., Y. Yabuta, H. Ohta, K. Hayashi, T. Nakamura, I. Okamoto, T. Yamamoto, K. Kurimoto, K. Shirane, H. Sasaki, and M. Saitou. 2016. In Vitro Derivation and Propagation of Spermatogonial Stem Cell Activity from Mouse Pluripotent Stem Cells. Cell Reports 17(10):2789-2804.
Jaenisch, R. 1976. Germ line integration and Mendelian transmission of the exogenous Moloney leukemia virus. Proceedings of the National Academy of Sciences of the United States of America 73(4):1260-1264.
Jaenisch, R., and B. Mintz. 1974. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proceedings of the National Academy of Sciences of the United States of America 71(4):1250-1254.
Jamieson, A. C., S. H. Kim, and J. A. Wells. 1994. In vitro selection of zinc fingers with altered DNA-binding specificity. Biochemistry 33(19):5689-5695.
Jiang, W., D. Bikard, D. Cox, F. Zhang, and L. A. Marraffini. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology 31(3):233-239.
Jinek, M., K. Chylinski, I. Fonfara, M. Hauer, J.A. Doudna, and E. Charpentier. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816-821.
Jinek, M., A. East, A. Cheng, S. Lin, E. Ma, and J. Doudna. 2013. RNA-programmed genome editing in human cells. eLife 2:e00471.
Jinek, M., F. Jiang, D. W. Taylor, S. H. Sternberg, E. Kaya, E. Ma, C. Anders, M. Hauer, K. Zhou, S. Lin, M. Kaplan, A. T. Iavarone, E. Charpentier, E. Nogales, and J. A. Doudna. 2014. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343(6176):1247997.
Johnson, J., J. Canning, T. Kaneko, J. K. Pru, and J. L. Tilly. 2004. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 428(6979):145-150.
Jore, M. M., M. Lundgren, E. van Duijn, J. B. Bultema, E. R. Westra, S. P. Waghmare, B. Wiedenheft, Ü. Pul, R. Wurm, R. Wagner, M. R. Beijer, A. Barendregt, K. Zhou, A. P. L. Snijders, M. J. Dickman, J. A. Doudna, E. J. Boekema, A. J. R. Heck, J. van der Oost, and S. J. J. Brouns. 2011. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nature Structural & Molecular Biology 18(5):529-536.
Joung, J. K., and J. D. Sander. 2013. TALENs: A widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology 14(1):49-55.
Kanatsu-Shinohara, M., and T. Shinohara. 2013. Spermatogonial stem cell self-renewal and development. Annual Reviews Cell and Developmental Biology 29:163-187.
Kang, X., W. He, Y. Huang, Q. Yu, Y. Chen, X. Gao, X. Sun, and Y. Fan. 2016. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. Journal of Assisted Reproduction and Genetics 33(5):581-588.
Kim, Y. G., J. Cha, and S. Chandrasegaran. 1996. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences of the United States of America 93(3):1156-1160.
Kleinstiver, B. P., V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng, and J. K. Joung. 2016. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529(7587):490-495.
Komor, A. C., Y. B. Kim, M. S. Packer, J. A. Zuris, and D. R. Liu. 2016. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533(7603):420-424.
Konermann, S., M. D. Brigham, A. E. Trevino, J. Joung, O. O. Abudayyeh, C. Barcena, P. D. Hsu, N. Habib, J. S. Gootenberg, H. Nishimasu, O. Nureki, and F. Zhang. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536):583-588.
Kuehn, M., A. Bradley, E. Robertson, and M. Evans. 1987. A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 326(6110):295-298.
Latham, K. E., B. Patel, F. D. Bautista, and S. M. Hawes. 2000. Effects of X chromosome number and parental origin on X-linked gene expression in preimplantation mouse embryos. Biology of Reproduction 63(1):64-73.
Lavitrano, M., R. Giovannoni, and M. G. Cerrito. 2013. Methods for sperm-mediated gene transfer. In Spermatogenesis: Methods and protocols, Vol. 927, edited by D. Carrell and K. I. Aston. New York: Humana Press. Pp. 519-529.
Leeb, M., and A. Wutz. 2011. Derivation of haploid embryonic stem cells from mouse embryos. Nature 479(7371):131-134.
Li, W., L. Shuai, H. Wan, M. Dong, M. Wang, L. Sang, C. Feng, G.Z. Luo, T. Li, X. Li, L. Wang, Q. Y. Zheng, C. Sheng, H. J. Wu, Z. Liu, L. Liu, L. Wang, X. J. Wang, X. Y. Zhao, and Q. Zhou. 2012. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature 490(7420):407-411.
Li, W., F. Teng, T. Li, and Q. Zhou. 2013. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nature Biotechnology 31(8):684-686.
Li, W., X. Li, T. Li, M. G. Jiang, H. Wan, G. Z. Luo, C. Feng, X. Cui, F. Teng, Y. Yuan, Q. Zhou, Q. Gu, L. Shuai, J. Sha, Y. Xiao, L. Wang, Z. Liu, X. J. Wang, X. Y. Zhao, and Q. Zhou. 2014. Genetic modification and screening in rat using haploid embryonic stem cells. Cell Stem Cell 14(3):404-414.
Liang, P., Y. Xu, X. Zhang, C. Ding, R. Huang, Z. Zhang, J. Lv, X. Xie, Y. Chen, Y. Li, Y. Sun, Y. Bai, Z. Songyang, W. Ma, C. Zhou, and J. Huang. 2015. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & Cell 6(5):363-372.
Maeda, I., D. Okamura, Y. Tokitake, M. Ikeda, H. Kawaguchi, N. Mise, K. Abe, T. Noce, A. Okuda, and Y. Matsui. 2013. Max is a repressor of germ cell-related gene expression in mouse embryonic stem cells. Nature Communications 4:1754.
Mahon, K. A., P. A. Overbeek, and H. Westphal. 1988. Prenatal lethality in a transgenic mouse line is the result of a chromosomal translocation. Proceedings of the National Academy of Sciences of the United States of America 85(4):1165-1168.
Makarova, K. S., N. V Grishin, S. A Shabalina, Y. I. Wolf, and E. V. Koonin. 2006. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology Direct 1(1):7.
Mali, P., L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville, and G. M. Church. 2013. RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
Martin, G. R. 1981. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Sciences of the United States of America 78(12):7634-7638.
Maruyama, T., S. K. Dougan, M. C. Truttmann, A. M. Bilate, J. R. Ingram, and H. L. Ploegh. 2015. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature Biotechnology 33(5):538-542.
McCreath, K. J., J. Howcroft, K. H. Campbell, A. Colman, A. E. Schnieke, and A. J. Kind. 2000. Production of gene-targeted sheep by nuclear transfer from cultured somatic cells. Nature 405(6790):1066-1069.
Mojica, F. J. M., C. Diez-Villasenor, J. Garcia-Martinez, and E. Soria. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of Molecular Evolution 60(2):174-182.
Naldini, L. 2015. Gene therapy returns to centre stage. Nature 526(7573):351-360.
Niu, Y., B. Shen, Y. Cui, Y. Chen, J. Wang, L. Wang, Y. Kang, X. Zhao, W. Si, W. Li, A. P. Xiang, J. Zhou, X. Guo, Y. Bi, C. Si, B. Hu, G. Dong, H. Wang, Z. Zhou, T. Li, T. Tan, X. Pu, F. Wang, S. Ji, Q. Zhou, X. Huang, W. Ji, and J. Sha. 2014. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156(4):836-843.
Orthwein, A., S. M. Noordermeer, M. D. Wilson, S. Landry, R. I. Enchev, A. Sherker, M. Munro, J. Pinder, J. Salsman, G. Dellaire, B. Xia, M. Peter, and D. Durocher. 2015. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528(7582):422-426.
Oye, K. A., K. Esvelt, E. Appleton, F. Catteruccia, G. Church, T. Kuiken, S. B. Lightfoot, J. McNamara, A. Smidler, and J. P. Collins. 2014. Biotechnology: Regulating gene drives. Science 345(6197):626-628.
Paquet, D., D. Kwart, A. Chen, A. Sproul, S. Jacob, S. Teo, K. M. Olsen, A. Gregg, S. Noggle, and M. Tessier-Lavigne. 2016. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533(7601):125-129.
Paull, D., V. Emmanuele, K.A. Weiss, N. Treff, L. Stewart, H. Hua, M. Zimmer, D. J. Kahler, R. S. Goland, S. A. Noggle, R. Prosser, M. Hirano, M. V. Sauer, and D. Egli. 2012. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493(7434):632-637.
Perez-Pinera, P., D. D. Kocak, C. M. Vockley, A. F. Adler, A. M. Kabadi, L. R. Polstein, P. T. Thakore, K. A. Glass, D. G. Ousterout, K. W. Leong, F. Guilak, G. E. Crawford, T. E. Reddy, and C. A. Gersbach. 2013. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nature Methods 10(10):973-976.
Pourcel, C., G. Salvignol, and G. Vergnaud. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151(Pt. 3):653-663.
Qasim, W., H. Zhan, S. Samarasinghe, S. Adams, P. Amrolia, S. Stafford, K. Butler, C. Rivat, G. Wright, and K. Somana. 2017. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science Translational Medicine 9(374):eaaj2013
Qi, L. S., M. H. Larson, L. A. Gilbert, J. A. Doudna, J. S. Weissman, A. P. Arkin, and W. A. Lim. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):1173-1183.
Ran, F. A., P. D. Hsu, C. Y. Lin, J. S. Gootenberg, S. Konermann, A. E. Trevino, D. A. Scott, A. Inoue, S. Matoba, Y. Zhang, and F. Zhang. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380-1389.
Rebar, E. J., and C. O. Pabo. 1994. Zinc finger phage: Affinity selection of fingers with new DNA-binding specificities. Science 263(5147):671-673.
Reddy, P., A. Ocampo, K. Suzuki, J. Luo, S. R. Bacman, S. L. Williams, A. Sugawara, D. Okamura, Y. Tsunekawa, J. Wu, D. Lam, X. Xiong, N. Montserrat, C. R. Esteban, G. H. Liu, I. Sancho-Martinez, D. Manau, S. Civico, F. Cardellach, M. del Mar O’Callaghan, J. Campistol, H. Zhao, J. M. Campistol, C. T. Moraes, and J. C. I. Belmonte. 2015. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161(3):459-469.
Reinhardt, K., D. K. Dowling, and E. H. Morrow. 2013. Medicine: Mitochondrial replacement, evolution, and the clinic. Science 341(6152):1345-1346.
Rouet, P., F. Smih, and M. Jasin. 1994. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Molecular and Cellular Biology 14(12):8096-8106.
Rubin, G. M., and A. C. Spradling. 1982. Genetic transformation of Drosophila with transposable element vectors. Science 218(4570):348-353.
Sagi, I., G. Chia, T. Golan-Lev, M. Peretz, U. Weissbein, L. Sui, M. V. Sauer, O. Yanuka, D. Egli, and N. Benvenisty. 2016. Derivation and differentiation of haploid human embryonic stem cells. Nature 532(7597):107-111.
Sakuma, T., and K. Woltjen. 2014. Nuclease-mediated genome editing: At the front-line of functional genomics technology. Development, Growth & Differentiation 56(1):2-13.
Sakurai, T., A. Kamiyoshi, H. Kawate, C. Mori, S. Watanabe, M. Tanaka, R. Uetake, M. Sato, and T. Shindo. 2016. A non-inheritable maternal Cas9-based multiple-gene editing system in mice. Scientific Reports 6:20011.
Sapranauskas, R., G. Gasiunas, C. Fremaux, R. Barrangou, P. Horvath, and V. Siksnys. 2011. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Research 39(21):9275-9282.
Sander, J. D., and J. K. Joung. 2014. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology 32(4):347-350.
Sasaki, K., S. Yokobayashi, T. Nakamura, I. Okamoto, Y. Yabuta, K. Kurimoto, H. Ohta, Y. Moritoki, C. Iwatani, H. Tsuchiya, S. Nakamura, K. Sekiguchi, T. Sakuma, T. Yamamoto, T. Mori, K. Woltjen, M. Nakagawa, T. Yamamoto, K. Takahashi, S. Yamanaka, and M. Saitou. 2015. Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell 17(2):178-194.
Schnieke, A., K. Harbers, and R. Jaenisch. 1983. Embryonic lethal mutation in mice induced by retrovirus insertion into the a1(I) collagen gene. Nature 304(5924):315-320.
Schwartzberg, P., S. Goff, and E. Robertson. 1989. Germ-line transmission of a c-Abl mutation produced by targeted gene disruption in ES cells. Science 246(4931):799-803.
Silva, G., L. Poirot, R. Galetto, J. Smith, G. Montoya, P. Duchateau, and F. Paques. 2011. Meganucleases and other tools for targeted genome engineering: Perspectives and challenges for gene therapy. Current Gene Therapy 11(1):11-27.
Slaymaker, I. M., L. Gao, B. Zetsche, D. A. Scott, W. X. Yan, and F. Zhang. 2016. Rationally engineered Cas9 nucleases with improved specificity. Science 351(6268):84-88.
Solter, D. 2006. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell research. Nature Reviews Genetics 7:319-327.
Staals, R. H., J. Y. Agari, S. Maki-Yonekura, Y. Zhu, D. W. Taylor, E. van Duijn, A. Barendregt, M. Vlot, J. J. Koehorst, K. Sakamoto, A. Masuda, N. Dohmae, P. J. Schaap, J. A. Doudna, A. J. R. Heck, K. Yonekura, J. van der Oost, and A. Shinkai. 2013. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of Thermus thermophilus. Molecular Cell 52(1):135-145.
Suzuki, A., M. Hirasaki, T. Hishida, J. Wu, D. Okamura, A. Ueda, M. Nishimoto, Y. Nakachi, Y. Mizuno, Y. Okazaki, Y. Matsui, J. C. I. Belmonte, and A. Okuda. 2016a. Loss of MAX results in meiotic entry in mouse embryonic and germline stem cells. Nature Communications 7:11056.
Suzuki, K., Y. Tsunekawa, R. Hernandez-Benitez, J. Wu, J. Zhu, E. J. Kim, F. Hatanaka, M. Yamamoto, T. Araoka, Z. Li, M. Kurita, T Hishida, M. Li, E. Aizawa, S. Guo, S. Chen, A. Goebl, R. D. Soligalla, J. Qu, T. Jiang, X. Fu, M. Jafari, C. R. Esteban, W. T. Berggren, J. Lajara, E. Nuñez-Delicado, P. Guillen, J. M. Campistol, F. Matsuzaki, G. H. Liu, P. Magistretti, K. Zhang, E. M. Callaway, K. Zhang, and J. C. Belmonte. 2016b. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540:144-149.
Tachibana, M., P. Amato, M. Sparman, J. Woodward, D. M. Sanchis, H. Ma, N. M. Gutierrez, R. Tippner-Hedges, E. Kang, H. S. Lee, C. Ramsey, K. Masterson, D. Battaglia, D. Lee, D. Wu, J. Jensen, P. Patton, S. Gokhale, R. Stouffer, and S. Mitalipov. 2012. Towards germline gene therapy of inherited mitochondrial diseases. Nature 493(7434):627-631.
Tarkowki, A. K. 1977. In vitro development of haploid mouse embryos produced by bisection of one-cell fertilized eggs. Journal of Embryology and Experimental Morphology 38:187-202.
Taylor, R. W., and D. M. Turnbull. 2005. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics 6(5):389-402.
Thomas, K., and M. Capecchi. 1987. Site directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51(3):503-512.
Tsai, S. Q., and J. K. Joung. 2016. Defining and improving the genome-wide specificities of CRISPR-Cas9 nuclease. Nature Reviews Genetics 17(5):300-312.
Tsai, S. Q., N. Wyvekens, C. Khayter, J.A. Foden, V. Thapar, D. Reyon, M. J. Goodwin, M. J. Aryee, and J. K. Joung. 2014. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature Biotechnology 32(6):569-576.
Tsai, S. Q., Z. Zheng, N. T. Nguyen, M. Liebers, V. V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A. J. Iafrate, L. P. Le, M. J. Aryee, and J. K. Joung. 2015. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology 33(2):187-197.
Wakayama, T., A. C. Perry, M. Zuccotti, K. R. Johnson, and R. Yanagimachi. 1998. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394(6691):369-374.
Wan, H., Z. He, M. Dong, T. Gu, G. Z. Luo, F. Teng, B. Xia, W. Li, C. Feng, X. Li, T. Li, L. Shuai, R. Fu, L. Wang, X. J. Wang, X. Y. Zhao, and Q. Zhou. 2013. Parthenogenetic haploid embryonic stem cells produce fertile mice. Cell Research 23(11):1330-1333.
Wang, H., H. Yang, C. S. Shivalila, M. M. Dawlaty, A. W. Cheng, F. Zhang, and R. Jaenisch. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4):910-918.
Wang, X., Y. Wang, X. Wu, J. Wang, Y. Wang, Z. Qiu, T. Chang, H. Huang, R. J. Lin, and J. K. Yee. 2015. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nature Biotechnology 33(2):175-178.
Wiedenheft, B., K. Zhou, M. Jinek, S. M. Coyle, W. Ma, and J. A. Doudna. 2009. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure 17(6):904-912.
Wiedenheft, B., G. C. Lander, K. Zhou, M. M. Jore, S. J. J. Brouns, J. van der Oost, J. A. Doudna, and E. Nogales. 2011. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477(7365):486-489.
Wilmut, I., A. E. Schnieke, J. McWhir, A. J. Kind, and K. H. Campbell. 1997. Viable offspring derived from fetal and adult mammalian cells. Nature 385(6619):810-813.
Wu, Y., H. Zhou, X. Fan, Y. Zhang, M. Zhang, Y. Wang, Z. Xie, M. Bai, Q. Yin, D. Liang, W. Tang, J. Liao, C. Zhou, W. Liu, P. Zhu, H. Guo, H. Pan, C. Wu, H. Shi, L.Wu, F. Tang, and J. Li. 2015. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Research 25(1):67-79.
Yang, H., L. Shi, B. A. Wang, D. Liang, C. Zhong, W. Liu, Y. Nie, J. Liu, J. Zhao, X. Gao, D. Li, G. L. Xu, and J. Li. 2012. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell 149(3):605-617.
Yang, H., H. Wang, C. S. Shivalila, A. W. Cheng, L. Shi, and R. Jaenisch. 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154(6):1370-1379.
Zeng, H., S. Wen, W. Xu, Z. He, G. Zhai, Y. Liu, Z. Deng, and Y. Sun. 2015. Highly efficient editing of the actinorhodin polyketide chain length factor gene in Streptomyces coelicolor M145 using CRISPR/Cas9-CodA(sm) combined system. Applied Microbiology and Biotechnology 99(24):10575-10585.
Zetsche, B., J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. Van der Oost, A. Regev, E. V. Koonin, and F. Zhang. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163(3):759-771.
Zhou, Q., M. Wang, Y. Yuan, X. Wang, R. Fu, H. Wan, M. Xie, M. Liu, X. Guo, Y. Zheng, G. Feng, Q. Shi, X.Y. Zhao, J. Sha, and Q. Zhou. 2016. Complete meiosis from embryonic stem cell-derived germ cells in vitro. Cell Stem Cell 18(3):330-340.
Zijlstra, J., E. Li, F. Sajjadi, S. Subramani, and R. Jaenisch. 1989. Germ line transmission of a disrupted b2-microglobulin gene produced by homologous recombination in embryonic stem cells. Nature 342(6248):435-438.
This page intentionally left blank.












































