
B
International Research Oversight and Regulations
The governance of research and clinical trials using human genome editing is expected to draw on the foundation of international and national regulations, policies, and guidance that apply to other areas of clinical research and development, including other types of genetic technologies, stem cells, reproductive medicine, and research involving human embryos. This appendix provides further information on some of these systems. It is not meant to be comprehensive, but rather to provide perspectives on how issues are addressed in countries other than the United States.
PUBLIC CONSULTATION CAN FORM PART OF A GOVERNANCE STRATEGY
There are numerous examples around the world of the use of public consultation on a wide range of biomedical and environmental policies (see Box B-1 for two examples described in greater detail). In the United States, the National Environmental Policy Act is unusual among environmental laws, because rather than directly regulating action, it simply provides that when the government makes a major decision, it must be subjected to a higher than usual degree of public scrutiny. By incorporating public comment, such public scrutiny creates political pressure that can drive decisions in one way or another, and it allows for some interplay between government expertise/authority and public consultation. Canada, when it looked at assisted reproduction across many different forms, formed a royal commission on new reproductive technologies that traveled the country from east to west, holding public hearings on the topic. In the European Union
(EU), genetically engineered foods are of special interest, and an EU directive requires public access to information whenever a product potentially affects biodiversity or other environmental elements. Public consultation is considered an alternative to a directive centralized form of governance, in which the public can, through its own decentralized processes, exert pressure on government or on industry and alter the direction or the speed of biotechnology innovation (Charo, 2016b).
VOLUNTARY REGULATION THROUGH GUIDELINES IS ANOTHER COMPONENT OF GOVERNANCE
Beyond consultation are voluntary self-regulation and nonbinding agreements. These are self-imposed rules that are seriously constraining with respect to donation of tissues, recruitment of donors, and experimentation that raises concerns, such as the use of chimeras. Examples include the guidelines adopted by the International Society for Stem Cell Research,
which have been amended to cover all forms of embryo research, from basic science to clinical trials with stem cells (ISSCR, 2016). Guidance also comes in the form of persuasive, albeit unenforceable, international instruments, such as those issued by the Council for International Organizations of Medical Sciences (CIOMS) for global standards for human subjects research (Gallagher et al., 2000).
At the far end of the spectrum, of course, there is regulation and legislation. Specifically with respect to gene therapy and germline manipulation, there are a number of international instruments of varying degrees of enforceability. For example, the Council of Europe’s Oviedo Convention says that predictive genetic tests should be used only for medical purposes. It specifically calls for a prohibition on the use of genetic engineering of the germline or changing the makeup of the following generations. It builds on earlier European conventions, but like many international instruments, it is not ratified by every member country and, even when ratified, does not
necessarily get implemented with domestic legislation. It has great normative value, but its enforcement-level value is uneven.
REGULATORY APPROACHES VARY BY COUNTRY
According to one recent review of gene-transfer trial information from regulatory and other sources, as of June 2012 more than 1,800 trials have been approved, initiated, or completed in 31 countries (Ginn et al., 2013; IOM, 2014). By mid-2016 that number had grown to more than 2,400, with trials primarily located in the Americas and Europe but nonetheless ongoing on every populated continent, and the number of studies generally growing every year.1 The 2013 review reported that 65.1 percent of the trials were based in the Americas, 28.3 percent in Europe, and 3.4 percent in Asia. Data from 2015 and 2016 show a similar pattern. Because more than half of all trials (63.7 percent, or 1,174) are associated with U.S. investigators or institutions (Ginn et al., 2013), U.S. regulations that govern research funded by the National Institutes of Health (NIH) or subject to the NIH rules due to a federal-wide assurance will have some effect on how the work proceeds outside the United States. The U.S. Food and Drug Administration (FDA) rules will also apply for products for which FDA approval is sought so that sale can proceed in the United States, regardless of funding source or whether the trial site is in the United States or abroad.
Countries approach the structure of their regulatory pathways in different ways. Japan has a regulatory pathway that tries to identify prospectively those things that are going to be high, medium, or low risk, and regulate them accordingly. The United States follows a similar process in its regulation of medical devices. But for drug regulation, the United States treats everything ab initio as equally dangerous and runs every proposed drug through the same rules for testing safety and efficacy. By contrast, in Japan there is an initial determination about the level of risk that is likely to be present for each proposed drug and the degree of stringency that the regulatory process must apply as a result. Japan has also added a conditional approval pathway specifically for regenerative medicine and gene therapy products, but it is too new for evaluation (Charo, 2016b).
Singapore also has a risk-based approach similar to Japan’s, and for cell therapy it uses variables that include whether the manipulation is substantial or minimal, whether or not the intended use is homologous or nonhomologous, and whether or not this is going to be combined with some drug, device, or other biologic.
___________________
1 Gene Therapy Clinical Trials Worldwide, provided by the Journal of Gene Medicine. http://www.abedia.com/wiley/years.php (accessed January 30, 2017).
Brazil provides an example of regulation and governance by accretion. It has approved laws related specifically to genetically engineered foods and stem cell research and cell therapy, but they are layered on top of earlier, more general rules, including constitutional prohibitions on the sale of any kind of human tissue and 1996 laws on the patenting of human biological materials, creating a situation of confusion. The result has been a degree of paralysis while the interplay among the laws is being managed.
More generally, discourse in Latin America on human somatic cell genome editing has been informed by concerns about genetically modified plants and animals, biopiracy, biosecurity, and use of stem cells for clinical care. Mexico addresses genetic engineering in the context of GMOs and biosecurity in its general health law and in its research regulations.2 Brazil addresses gene editing in its Biosafety Law, implicitly permitting at least some somatic gene-editing research in humans, although its primary focus is clearly on GMOs.3 Similarly, Ecuador’s Constitution has provisions addressing genomic heritage in the setting of GMOs and biopiracy, and in its guarantee of personal integrity it prohibits the use of genetic material for scientific research in violation of human rights.4
A few jurisdictions in Latin America have explicitly addressed somatic genome editing, typically imposing restrictions aimed at prohibiting uses that might be perceived as “enhancement” rather than treatment or prevention of disease and injury. Chile states that gene editing “in somatic cells will be authorized only for the treatment of diseases or to prevent their occurrence” in a far-reaching law that also addresses intellectual property, discrimination, and protection of genetic identity, as well as prohibiting “eugenic practices” (with an exception for genetic counseling).5 In Panama and Mexico City, use of genetic manipulation except for the elimination or treatment of a serious defect or disease is punishable by a prison sentence of 2 to 6 years.6 Colombia’s penal code similarly permits genetic modification
___________________
2 Ley General de Salud, Titulo Decimo Segundo. Capitulo XII, Artículo 282. And Regulamento de la Ley General de Salud en Materia de Investigacíon para la Salud, Titulo Cuarto, Capitulo II, Articulos 85-88 (recombinant DNA research).
3 Public Law No. 11.105, Chapter 1, Article 6, as translated by WIPO.
4 Constitución de la Republica del Ecuador 2008, Titulo II, Articulo 66, 1.3(d). Interestingly, Ecuador promulgated an extensive set of regulations governing the use of biological samples and genomic data, specifically citing, inter alia, the biopiracy of DNA from an indigenous population in that country. Ministero de Salud Pública (MSP), Reglamento para uso del material genético humano en Ecuador. Ministereo de Salud Pública, Dirección Nacional de Noamrtización y Programa Nacional de Genética, 2013.
5 Public Law No. 20.120 On the Scientific Investigation of the Human Genome, Its Genoma, & Prohibition of Human Cloning, Articles 1, 3, 4, 7, 8, 12, and 13, 2006 (English translation).
6 Ley Penal en General. Capítulo II, Artículo 145. (2010); Código Penal para el Distrito Federal. Capítulo II, Artículo 154 (also bars employment and other benefits this period).
for treatment, diagnosis, and research to alleviate suffering or improve human health, while imposing a prison sentence of 1 to 5 years for other uses.7
In the European Union, the European Medicines Agency (EMA) has the responsibility to evaluate and supervise human and veterinary medicines to protect public and animal health (EMA, 2013). In 2007, the EMA established the Committee for Advanced Therapies as the unit responsible for assessing the quality, safety, and efficacy of medicines made from genes and cells—medicines that are termed “advanced therapy medicinal products.” This committee provides a centralized procedure for the assessment and approval of medicines for marketing in the European Union. The process is mandatory for biologics, including gene and cell therapy products, and a number of other product categories, including medicines for the treatment of HIV/AIDS and cancer (Cichutek, 2008).
The EMA, however, does not have authority to review and approve protocols for clinical research, including gene-transfer research (Pignatti, 2013). That authority resides with national regulatory agencies, but every EU state has adopted the EU Directive on Clinical Trials (Kong, 2004). It requires member states to adopt a system for the review of clinical research consistent with internationally recognized standards for good clinical practice for the ethical and scientifically valid design, conduct, and report of trials (Kong, 2004). The FDA, which participated in the international process for developing these standards, also recognizes these standards and publishes them as guidance documents (FDA, 2012).8
As with Europe, China has a regulatory framework for the development and use of human medical products. Although it is not yet amended to address genome editing specifically, frameworks governing gene and cell therapies have been implemented and the State Food and Drug Administration (the predecessor of the current China Food and Drug Administration [CFDA]) approved a gene therapy product for marketing. In addition, regulatory guidelines for human embryo research and in vitro fertilization (IVF) practices have been published by Chinese agencies (China Ministry of Health, 2001, 2003). Within the current regulatory framework, human somatic cell genome editing may be considered a third category therapeutic technology rather than a drug. If so, it would be regulated by CFDA, and procedures would include evaluations for safety and efficacy through preclinical testing and clinical trials, similar to processes used by the FDA and EMA. In addition to CFDA, the Health and Family Planning Commission
___________________
7 Código Penal Colombiano, Capítulo Octavo, Artículo 132 (2015).
8 Information on the national regulatory frameworks likely to apply to somatic and germline human genome editing in a number of European countries is described in a background document produced for a 2016 workshop on human genome editing in the EU. http://acmedsci.ac.uk/file-download/41517-573f212e2b52a.pdf (accessed January 30, 2017).
(HFPC), which regulates IVF clinics, is likely to be involved in oversight of human genome editing. Consultations would likely occur with agencies such as the Ministry of Science and Technology, Chinese Academy of Sciences, Chinese Academy of Medical Sciences, and Chinese Academy of Engineering to enable their positions to be incorporated into regulations.
HERITABLE GENETIC MODIFICATIONS RAISE ADDITIONAL ISSUES
A number of special regulatory and governance issues may arise around use of human embryos and the potential for genetic changes to be made to the human germline. Discussions of such issues have been informed by debates about topics such as embryonic stem cells, cloning assisted reproductive technologies, and the beginning of life.
As noted above, the Convention for the protection of Human Rights and Dignity of the Human Being with regard to the Application of Biology and Medicine: Convention on Human Rights and Biomedicine (“Oviedo Convention”) develops principles concerning a number of medical topics that raise bioethics issues. For countries that have signed and ratified the treaty, the genetic constitution of the individual is to be protected against unlawful interventions seeking to modify the germline. A number of countries have also enacted national laws, regulations, or guidelines that restrict human germline modifications (see Figure B-1).
INTERNATIONAL COOPERATION ON THE GOVERNANCE OF HUMAN GENOME EDITING IS DESIRABLE ALTHOUGH FORMAL REGULATORY HARMONIZATION SEEMS INFEASIBLE
Given the global nature of scientific and medical advances and the diversity and complexity of approaches to regulating human genetic technologies, there have been calls for international collaboration, cooperation, and even harmonization of regulations governing human genome editing. Arguments can be made both for and against an international convergence of national regulation of genomic technologies (Breggin et al., 2009; Marchant et al., 2012). A compelling argument for creating uniform or consistent regulations of gene editing is to avoid “regulatory havens” that circumvent restrictions if providers or consumers travel to jurisdictions with more lenient or non-existent regulations in order to undertake the restricted procedures (Charo, 2016a). The potential for lucrative medical tourism may create a “race to the bottom” that encourages laxer standards in nations seeking revenues from medical tourism (Abbott et al., 2010). Consistent standards may also promote equal health protection for citizens of all nations and provide consistent requirements for companies and sci-
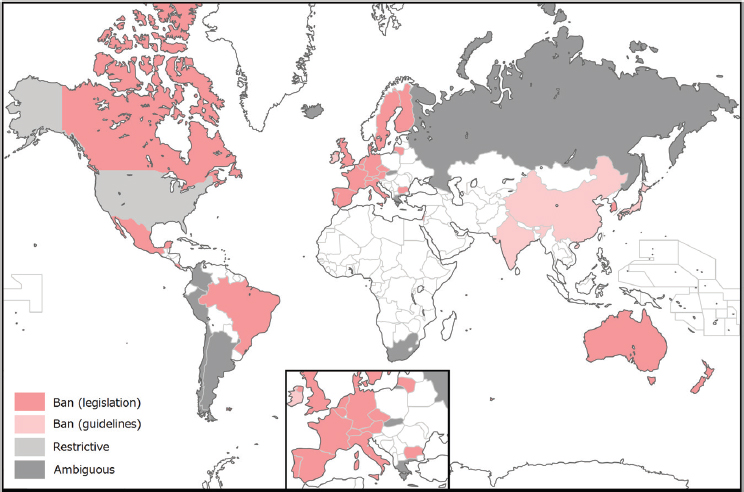
SOURCE: Araki and Ishii, 2014.
entists in the field, reducing transaction costs and increasing economies of scale (Vogel, 1998). Harmonized standards also provide economies of scale for regulators, reduce administrative costs in adopting and administering national laws, and increase opportunities to share regulatory resources and workload. Finally, the process of harmonization can promote the exchange of good practices and build regulatory capacity (OECD, 2013).
On the other hand, nations have different historical, economic, social, and cultural systems and values, which may translate into different approaches to the regulation of a powerful technology such as human genome editing. Uniform national regulations may also subject every nation to the lowest common regulatory denominator, depending on how harmonization is achieved. As a practical matter, reaching consensus among 100 or more nations on regulatory requirements for any technology is a laborious and resource-intensive undertaking that in the end may not be successful. Diversity in regulatory approaches also provides a natural experiment to evaluate
TABLE B-1 Three General Approaches to International Regulatory Convergence
| Convergence Process | Definition | Example |
|---|---|---|
| Transnational Regulatory Dialogue and Networking | Informal process of communication and policy learning between regulators | International Dialogue on Responsible Nanotechnology |
| International Coordination/Cooperation | Nonbinding international instruments such as guidelines, principles, and standards | ISSCR Guidelines for Embryonic Stem Cell Research |
| Treaty-Based Harmonization | Formal negotiation of binding treaties | United Nations Convention on Cloning (failed) |
SOURCE: Adapted from Breggin et al. (2009). We are grateful to Chatham House, the Royal Institute of International Affairs, for permission to reproduce a figure from the work titled: Securing the Promise of Nanotechnologies: Towards Transatlantic Regulatory Cooperation, authored by Linda Breggin, Robert Falkner, Nico Jaspers, John Pendergrass, and Read Porter, 2009.
the effects of different regulatory frameworks, providing a “laboratory of nations” that “fosters innovation and rapid learning about the impact of striking different balances between innovation and precaution” (Evans, 2015). However, realizing this benefit requires procedures that facilitate the exchange of information and promote learning.
As summarized in Table B-1, there is a continuum of approaches to aligning national regulatory requirements (Breggin et al., 2009). Harmonization usually involves the enforcement of identical or equivalent regulatory requirements under the national laws of participating countries. Harmonization is usually accomplished through an international treaty or other formal and binding legal instrument, implemented through the amendment of national laws to conform to treaty requirements (OECD, 2013). International treaties and other formal agreements are difficult and time consuming to negotiate, and often present difficult enforcement issues.9 Given these obstacles, there has been a trend away from treaties in the international governance of technologies and products in favor of mechanisms of international cooperation and coordination (Falkner, 2013; Susskind, 2008).
Informal mechanisms of international coordination and cooperation do not create legal requirements to implement specific provisions, but
___________________
9 An example of the challenges associated with negotiating and enforcing treaties was the unsuccessful attempt through the United Nations system in the early 2000s to create a binding international treaty banning human cloning (Cameron and Henderson, 2008).
rather provide general agreement between governments in the form of nonbinding guidelines, recommendations, consensus documents, statements of principles, or voluntary standards. Such normative guidelines may be agreed upon in free-standing negotiations, but are often negotiated within an appropriate international organization (Abbott, 2014). International cooperation and coordination approaches can also be achieved through nongovernmental organizations, such as scientific societies, for example through the International Society for Stem Cell Research (ISSCR) Guidelines for Embryonic Stem Cell Research (Daley et al., 2007).
The least formal mechanism of international convergence is policy diffusion through transnational regulatory dialogue and networking. This approach usually does not involve the creation of instruments that set forth specific substantive or procedural recommendations for nations to follow. Rather, it provides a forum for regulators from different nations to share information, approaches, challenges, and ideas. An example is the International Dialogue on Responsible Nanotechnology, in which experts from 25 national governments convened in a series of meetings every 2 years to report on their regulatory activities and challenges (Meridian Institute, 2004). Regulators who have been involved in international coordination activities state that such person-to-person contacts and communications provide one of the most effective mechanisms for promoting international cooperation and understanding (Saner and Marchant, 2015).
A convergence of regulatory approaches for human genome editing would have some beneficial effects as described above, but countries have already adopted diverse laws relevant to human genome editing and a formal or complete harmonization does not seem feasible—and may not even be entirely desirable—at this time. Moreover, national responses to human genome editing reflect unique historical, cultural, economic, and social factors. Notwithstanding these important differences that prevent uniform international standards, there are important benefits for providing for robust communication and coordination between regulators in different countries, and potential opportunities for identifying common ground on specific substantive or technical aspects as well as opportunities to produce learning benefits (e.g., Zhai et al., 2016).
REFERENCES
Abbott, K. W. A. 2014. International organisations and international regulatory co-operation: Exploring the links. In International regulatory co-operation and international organisations: The Cases of the OECD and the IMO. Paris: OECD Publishing. Pp. 17-44.
Abbott, K. W. A., D. J. Sylvester, and G. E. Marchant. 2010. Transnational regulation: Reality or romanticism? In International handbook on regulating nanotechnologies, edited by G. Hodge, D. Bowman, and A. Maynard. Cheltenham, UK: Edward Elgar Publishing. Pp. 525-544.
Araki, M., and T. Ishii. 2014. International regulatory landscape and integration of corrective genome editing into in vitro fertilization. Reproductive Biology and Endocrinology 12:108. http://www.rbej.com/content/12/1/108 (accessed January 25, 2017).
Breggin, L., R. Falkner, N. Jaspers, J. Pendergrass, and R. Porter. 2009. Securing the promise of nanotechnologies: Towards transatlantic regulatory cooperation. London, UK: Chatham House. https://www.chathamhouse.org/sites/files/chathamhouse/public/Research/Energy,%20Environment%20and%20Development/r0909_nanotechnologies.pdf (accessed November 7, 2016).
Cameron, N. M. de S., and A.V. Henderson. 2008. Brave new world at the General Assembly: The United Nations Declaration on Human Cloning. Minnesota Journal of Law, Science and Technology 9(1):145-238.
Charo, R. A. 2016a. On the road (to a cure?): Stem-cell tourism and lessons for gene editing. New England Journal of Medicine 374(10):901-903.
Charo, R. A. 2016b. The legal and regulatory context for human gene editing. Issues in Science and Technology 32(3). http://issues.org/32-3/the-legal-and-regulatory-context-for-human-gene-editing (accessed November 7, 2016).
China Ministry of Health (People’s Republic of China Ministry of Health). 2001. Guidelines on human assisted reproductive technologies [in Chinese]. http://go.nature.com/1ztc8qb (accessed November 7, 2016).
China Ministry of Health (People’s Republic of China Ministry of Health). 2003. Guidelines on human embryonic stem cell research. http://www.cncbd.org.cn/News/Detail/3376 (accessed November 7, 2016).
Cichutek, K. 2008. Gene and cell therapy in Germany and the EU. Journal fur Verbraucherschutz und Lebensmittelsicherheit 3(Suppl. 1):73-76.
Daley, G. Q., L. Ahrlund-Richter, J. M. Auerbach, N. Benvenisty, R. A. Charo, G. Chen, H. K. Deng, L. S. Goldstein, K. L. Hudson, I. Hyun, S. C. Junn, J. Love, E. H. Lee, A. McLaren, C. L. Mummery, N. Nakatsuji, C. Racowsky, H. Rooke, J. Rossant, H. R. Scholer, J. H. Solbakk, P. Taylor, A. O. Trounson, I. L. Weissman, I. Wilmut, J. Wu, and L. Zoloth. 2007. The ISSCR guidelines for human embryonic stem cell research. Science 315:603-604.
EMA (European Medicines Agency). 2013. Legal foundation. http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/general/general_content_000127.jsp&mid=WC0b01ac0580029320 (accessed January 25, 2017).
Evans, B. J. 2015. Panel: Governance at the institutional and national levels: National regulatory frameworks. Presentation at International Summit on Gene Editing, Washington, DC, December 2.
Falkner, R. 2013. The crisis of environmental multilateralism: A liberal response. In The green book: New Directions for liberals in government, edited by D. Brack, P. Burall, N. Stockley, and M. Tuffrey. London, UK: Biteback Publishing. Pp. 347-358.
FDA (U.S. Food and Drug Administration). 2012. Vaccine, blood, and biologics: SOPP 8101.1: Scheduling and conduct of regulatory review meetings with sponsors and applicants. Rockville, MD: FDA. http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/ProceduresSOPPs/ucm079448.htm (accessed January 25, 2017).
Gallagher, J., S. Gorovitz, and R. J. Levine. 2000. Biomedical research ethics: Updating international guidelines: A consultation. Geneva, Switzerland: Council for International Organizations of Medical Sciences (CIOMS). http://www.cioms.ch/index.php/publications/available-publications/540/view_bl/61/bioethics-and-health-policy/3/biomedical-research-ethics-updating-international-guidelines-a-consultation?tab=getmybooksTab&is_show_data=1 (accessed November 7, 2016).
Ginn, S. L., I. E. Alexander, M. L. Edelstein, M. R. Abedi, and J. Wixon. 2013. Gene therapy clinical trials worldwide to 2012—An update. Journal of Gene Medicine 15(2):65-77.
IOM (Institute of Medicine). 2014. Oversight and review of clinical gene transfer protocols: Assessing the role of the Recombinant DNA Advisory Committee. Washington, DC: The National Academies Press.
ISSCR (International Society for Stem Cell Research). 2016. Guidelines for stem cell research and clinical translation. http://www.isscr.org/docs/default-source/guidelines/isscr-guidelines-for-stem-cell-research-and-clinical-translation.pdf?sfvrsn=2 (accessed November 7, 2016).
Kong, W. M. 2004. The regulation of gene therapy research in competent adult patients, today and tomorrow: Implications of EU directive 2001/20/EC. Medical Law Review 12(2):164-180.
Marchant, G. E., K. W. Abbott, D. J. Sylvester, and L. M. Gaudet. 2012. Transnational new governance and the international coordination of nanotechnology oversight. In The nanotechnology challenge: Creating law and legal institutions for uncertain risks, edited by D. A. Dana. Cambridge, UK: Cambridge University Press. Pp. 179-202.
Meridian Institute. 2004. International dialogue on responsible research and development of nanotechnology. Washington, DC: Meridian Institute. http://www.temas.ch/nano/nano_homepage.nsf/vwRes/SafetyAlexandria/$FILE/Final_Report_Responsible_Nanotech_RD_040812.pdf (accessed November 7, 2016).
OECD (Organisation for Economic Co-operation and Development). 2013. International regulatory co-operation: Addressing global challenges. Paris: OECD Publishing.
Pignatti, F. 2013. Harmonizing across regions. Presentation at Implementing a National Cancer Clinical Trials System for the 21st Century, Washington, DC, February 12.
Saner, M. A., and G. E. Marchant. 2015. Proactive international regulatory cooperation for governance of emerging technologies. Jurimetrics 55(2):147-178.
Susskind, L. 2008. Strengthening the global environmental treaty system. Issues in Science and Technology 25(1):60-68.
Vogel, D. 1998. Globalization of pharmaceutical regulation. Governance 11(1):1-22.
Zhai, X., V. Ng, and R. Lie. 2016. No ethical divide between China and the West in human embryo research. Developing World Bioethics 16(2):116-120.












