
In his opening keynote, Adrian Gee, a professor of cell and gene therapy at Baylor College of Medicine, described the issues facing the field of regenerative medicine and set the stage for the ensuing panel presentation and discussions. There is increasing interest in the field from both researchers and the public, Gee said, and, accordingly, the global market for regenerative medicine therapies is growing rapidly. By some estimates, the market is projected to reach $53.7 billion by 2021 (Kelly Scientific, 2017). As patient demand for new forms of treatment continues to grow, Gee cautioned, the regenerative medicine industry must work closely with regulators to ensure that new therapies are safe and effective.
During the first session following Gee’s keynote presentation, a panel discussed the challenges and opportunities associated with bringing new discoveries from the laboratory to manufacturing and with navigating the process of scaling the production of new regenerative therapies. The three speakers—Bruce Levine, the Barbara and Edward Netter professor in cancer gene therapy at the University of Pennsylvania Perelman School of Medicine; Laura Niklason, a professor of anesthesiology and biomedical engineering at Yale University; and Robert Preti, the president and chief executive officer at PCT Cell Therapy Services and chairman of the Alliance for Regenerative Medicine—also described potential opportunities and models to enable the scaling of regenerative medicine therapies for production using the currently available infrastructure and addressed probable future needs as the regenerative medicine field evolves and grows. An open discussion moderated by Krishanu Saha, an assistant professor at the University of Wisconsin–Madison, followed the three presentations.
KEY CHALLENGES FACING THE REGENERATIVE MEDICINE INDUSTRY AS SAFE AND EFFECTIVE THERAPIES ARE DEVELOPED FOR PATIENTS
In the United States, FDA is responsible for regulating this field, and the agency is working closely with researchers and manufacturers to foster the responsible development of regenerative and cellular therapies, said Gee. FDA’s approach, he added, has been to hold meetings with stakeholders, publish guidance documents about how it will develop and implement regulations, and designate regenerative medicine as an advanced therapy.1
In setting the stage for the day’s panel discussions, Gee raised some of the issues that stakeholders face as they work to develop and gain approval for cellular and regenerative therapies. These issues and needs for the field
___________________
1 More information about FDA’s approach to regulating the regenerative medicine field can be found here: https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ucm537670.htm (accessed July 27, 2017).
are listed in Box 2-1. The ideal regenerative medicine product, Gee said, would have a starting material that is easy to collect or generate from induced pluripotent stem cells (iPSCs) or embryonic stem cells. The manufacturing method for this ideal product would be automated and would use simple closed systems, and there would be rapid and predictive testing methods to determine whether the product has met appropriate release standards. If possible, the ideal product would be made available any time a patient needed it, so that the product would either have a long shelf life under simple storage conditions or just-in-time shipping methods could be employed. Finally, the ideal regenerative medicine product would be easy to
distribute and administer, and each step in its production would be designed to ensure high quality at low cost.
The challenges involved in creating such a product are significant, Gee acknowledged. The collection process is often complex and involves using various ancillary agents such as anticoagulants and suspended media, some of which have not received regulatory approval for use in manufacturing. The source cells themselves may also vary, depending on the technical ability of the person collecting the starting material and the nature of the donor. If the starting material comes from the intended patient, those autologous cells are likely to be affected by the disease state of the patient and any prior therapies the patient has received, and they may be more difficult to collect. Cell therapy products that use allogeneic cells obtained from healthy donors are more likely to be uniform, Gee said, because fewer screened donors will provide the starting material for most of the product and their target cell populations are less likely to contain variations because of disease, but not all therapies will be amenable to using allogeneic cells (Malik, 2012).
While the ideal manufacturing process would use closed systems at every stage, some products require source cells that are not available in large quantities, and there do not yet exist closed systems for culturing and increasing the number of these cells, Gee said. A closed system is defined as “a process system with equipment designed and operated such that the product is not exposed to the room environment. Materials may be introduced to a closed system via mechanisms that avoid exposure of the product to the room environment (e.g., delivery through sterile ports and filtration), but the addition must be done in such a way to avoid exposure of the product to the room environment (e.g., by 0.2-µm filtration)” (Palberg et al., 2017). There is a need for approved manufacturing equipment that can conduct the in-process monitoring required during the production of cells for these products, Gee said. Such devices should be easy to use for scale-up (increasing the capacity of a single manufacturing process) or scale-out (increasing the number of processes performed in parallel), he added, noting that scale-up and scale-out are a significant challenge for many centers because they can be costly and time-intensive. Many centers also encounter difficulties when implementing manufacturing analytics to meet GMP operations and assess complex variables relating to quality control. It would be ideal for manufacturing devices to have integrated software capable of monitoring quality control variables as a product is being manufactured instead of having to rely on the relatively few available software packages that focus on quality systems variables, Gee said. Another issue, he said, is that there are not enough available people who have been trained to oversee these kinds of manufacturing processes, and there is an associated lack of training and certification programs for regenerative medicine product manufacturing.
To illustrate one approach to addressing the above challenges, Gee
discussed Baylor School of Medicine’s experience producing virus-specific T cells. In the early 1990s, researchers at Baylor developed procedures for producing Epstein-Barr virus-specific T cells that eventually went into clinical trials and produced promising results (Heslop et al., 2010; Louis et al., 2010). By 2013 the specificity of the cells had been expanded to include cytomegalovirus and adenovirus, and since then the Baylor team has expanded the specificity again to include activity toward BK virus and human herpesvirus 6. Since the early 1990s manufacturing time has been reduced from 3 months to 10 days, which makes the cells more easily available for any suitable patient in a therapeutically relevant time-frame, Gee said, and the manufacturing process has moved from an open system to a closed system. Most importantly, he said, the safety and efficacy of these cells has remained the same as those first put through clinical trials. Studies have shown that these cells can be used across human leukocyte antigen (HLA) compatibility barriers without the risk of triggering graft-versus-host disease, suggesting that they will be able to gain an “off-the-shelf” designation. “I think it shows that even in one particular field, advances can be made moving toward the goal of the ideal product,” Gee said.
With regard to testing and release protocols for regenerative medicine products, Gee said, there are a few rapid testing assays, such as that for endotoxin, but there is still a need for a rapid assay to test for sterility. There is also a need for potency assays that correlate with clinical efficacies, which are required to conduct registration-enabling clinical trials. The cost of testing is also a barrier, Gee said, noting that some testing procedures such as those used to evaluate cell products produced using viral vectors, can have significant costs. “We need to develop and get regulatory approval for new release assays,” he said, “and we desperately need standardization of these assays so that we can compare them between centers.” To do that, he said, will require standardization of the appropriate controls.
The storage of regenerative medicine products is another issue facing the field. For example, the cryopreservation of cellular therapy products can affect their potency. “We need to consider whether storing products at ultra-low temperatures is suitable or whether other forms of storage may be preferable,” Gee said. It may be possible, for instance, to develop techniques to keep cells viable and potent without cryopreserving them. Another opportunity for further development is improving formulation and packaging methods to eliminate the need to manipulate the product upon receipt at the treatment center, Gee said. This would allow hospitals to use the product with minimal on-site manipulation and would reduce reliance on laboratories and technicians to prepare the product for clinical application.
Concerning the distribution and transport of these products, both from the source collection site to the manufacturing center and then from the
manufacturing center to the therapeutic centers, standardized labels are needed to make the production and delivery of a cellular therapy more consistent, accurate, and safe for donors and patients, Gee said. The Information Standard for Blood and Transplant (ISBT) 128, a global standard for identifying and labeling medical products of human origin, is the most generally agreed upon system, Gee noted.2 He also said that improved dry shipping methods (i.e., those methods that do not involve using free liquid nitrogen), perhaps using warmer temperatures, may be needed and that the field should consider developing just-in-time shipping approaches that deliver fresh cells for administration to the patient immediately upon arrival.
Current regulations are based on pharmaceutical standards, but a question exists as to whether those standards are appropriate for cell- and tissue-based products, particularly as they continue to evolve, Gee said. A number of bodies have developed professional standards for collecting source cells, manufacturing cellular products, and administering these cell therapies, raising the issue of what the proper interface should be between these standards and the regulatory environment prescribed by FDA.
The final challenge, Gee said, is how patients and insurers will pay for these therapies, given that they are likely to be expensive. “Will the average patient have the ability to pay for these?” he asked. “Or will the insurance companies be willing to cover the costs?” He also raised the issue of developing new methods for compensation, beyond standard licenses and intellectual property (IP) agreements. Specifically, Gee wondered if it would be feasible to return a portion of the profits from the sale of cellular therapy products back to the original centers where they were developed since many products arise from publicly funded research. Additionally, many products are developed at academic manufacturing centers or hospitals to treat a small number of patients, making them unattractive for large-scale commercialization and the challenge, Gee said, is that “most hospitals do not want to be in the business of manufacturing cell products.”
“Cellular regenerative medicine therapies are likely to revolutionize the practice of medicine in the future,” Gee said, but ultimately, researchers and manufacturers of regenerative medicine therapies must adapt their production processes and environments to best suit the needs of patients.
___________________
2 More information about the ISBT 128 can be found here: https://www.iccbba.org (accessed August 23, 2017).
TRANSITIONING ENGINEERED T CELLS FROM DISCOVERY TO MANUFACTURING AND REGULATORY APPROVAL
Interaction between the fields of human immunodeficiency virus (HIV) and oncology research led to the submission of the first biologics license application (BLA) for a cellular gene therapy to FDA, Levine said. In an effort to better understand T cell growth and senescence, Levine created artificial dendritic cells that allowed for the delivery of growth signals to T cells through bead-linked antibodies. This accelerated the development of a chimeric antigen receptor (CAR) which was delivered to T cells using a lentivirus. The lentivirus genome could be edited to encode a genetic sequence for the expression of a specific CAR when transcribed and translated in a T cell. The lentivirus was used as a “Trojan horse”—that is, the virus would enter a T cell and deliver its double stranded RNA, which would then be reverse transcribed and integrated into the T cell DNA, permanently incorporating the sequence into the cell’s genome (a process called transduction) and allowing the cell to produce both its original antigen receptor and the CAR. In his presentation, Levine reviewed the technology and methodology behind the first CAR clinical trials that used a murine retrovirus to edit the T cell genome. The resulting CAR T cells were used to treat patients with HIV (Mitsuyasu et al., 2000; Walker et al., 2000). Levine described how the technology used in those trials led to the University of Pennsylvania group’s work to develop CAR T cells with potent and long-lasting antitumor effects in patients with advanced chronic lymphoid leukemia (Kalos et al., 2011; Porter et al., 2011) and the more aggressive acute lymphocytic leukemia (ALL) in children who had failed previous therapies (Grupp et al., 2013; Maude et al., 2014). In initial trials studying both conditions, Levine said he and his team found that their CAR T cell treatments demonstrated significant anti-cancer effects in patients, and in their trial on ALL he and his colleagues found that 93 percent of patients had a complete response rate at 1 month post treatment.
The technology for producing CAR T cells has since been transferred to Novartis, which will take this technology through pivotal clinical trials and develop it commercially, something that Levine and his colleagues at the University of Pennsylvania are not equipped to do, he said. The research team at Novartis refined the production technology to further enhance the control and consistency of the process by closing some process steps and automating some manual processes. The Novartis team also developed new analytical methods to demonstrate that the resulting product was the desired one; that it met purity, identity, and potency requirements; and that it was free of adventitious agents.
Following comparability studies, FDA accepted that the Novartis product was comparable to the one that Levine and his colleagues used in
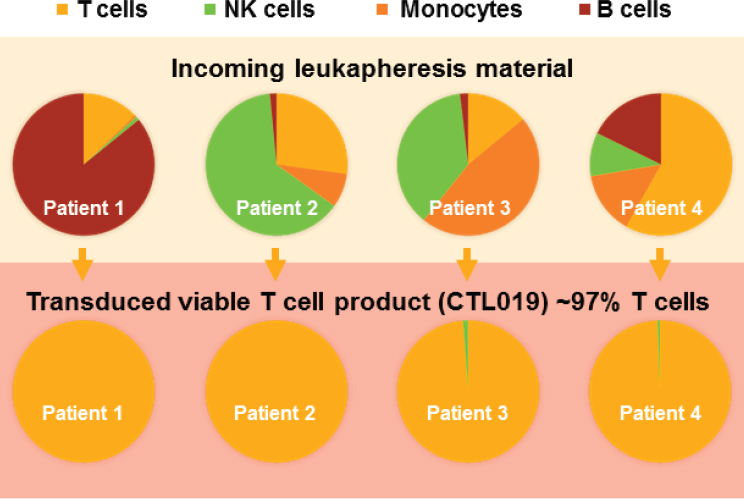
NOTE: CTL = cytotoxic T lymphocyte; NK = natural killer.
SOURCES: Bruce Levine, National Academies of Sciences, Engineering, and Medicine workshop presentation, June 26, 2017. Originally from Novartis Cell and Gene Therapy Analytical Development, 2016.
conducting its initial clinical trial and permitted the company to open its own investigational new drug application and conduct global clinical trials. Still, Levine said, there is the issue of individual patient variability. “We have some patients that come in with very low T cell counts,” he explained. The solution, he said, was to integrate a conditional manufacturing pathway based on a patient’s phenotype and incoming material (see Figure 2-1). The result has been a consistent transduced T cell product regardless of the makeup of the incoming patient material. A clinical trial using this approach in pediatric patients with relapsed and refractory B cell ALL, which was conducted at 25 sites in 11 countries on 4 continents, found a 6-month overall survival rate of 89 percent (Buechner et al., 2017). FDA accepted the resulting BLA on March 29, 2017, and granted priority review.3 Novartis is also conducting a Phase II clinical trial with
___________________
3 On August 30, 2017, FDA approved the first gene therapy available in the United States—Kymriah (tisagenlecleucel)—for certain applications in pediatric and young adult patients with acute lymphoblastic leukemia. See https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574058.htm (accessed September 14, 2017).
this product in adults with diffuse large B cell lymphoma, and preliminary results show that 79 percent of patients were relapse-free at 6 months after treatment (Schuster et al., 2017).
Addressing the lessons learned from efforts to develop materials for the first human clinical trials, Levine said that patient-derived materials such as cells from healthy humans, are not the same as cells from patients with advanced disease. To accommodate for those differences, researchers need to work early with reagents, other materials, and equipment that are clinical grade. The variability of human cell materials from patients currently requires human judgment and intervention, he said, which creates the opportunity for deviations and the need for more training of research personnel. “There are things we can automate,” Levine said, “but many things that we cannot yet automate.” Another lesson he mentioned was that studying a few patients thoroughly can “radically accelerate” clinical development. His final lesson was that an academic scientist can and should understand the industry terms, methodologies, and nuances associated with the commercialization of a new therapy.
Levine concluded his comments with a list of critical issues to consider when developing products for commercialization and increasing patient access to new cell therapies:
- Ensuring that there is a consistent supply chain for complex reagents and materials, such as serum-free media, and that there are alternatives available with comparable growth and potency properties.
- Near-term out-scaling and the mid- to long-term automation of manufacturing processes.
- Developing rapid and modified-release tests to assess product quality.
- Increasing the consistency and comparability of regenerative medicine products and managing complex manufacturing processes.
- Reducing the cost of goods, labor, and services.
- Recruiting, training, and retaining skilled technologists and engineers.
- Addressing ethical questions of patient access and moving from an investigational clinical trial for a potential new therapy with strong positive results to a larger clinical trial, given the complexities of scaling the production of these therapies and allocating enrollment in larger clinical trials.
LEARNING FROM PAST EXPERIENCES IN VASCULAR ENGINEERING
When Laura Niklason surveyed ClinicalTrials.gov, a registry and results database of publicly and privately supported clinical studies of human participants conducted around the world, last year, she found roughly 30 ongoing trials involving implanted engineered tissues. These trials, she said, mainly involved connective tissues—skin, cartilage, blood vessels, and others—rather than solid organs such as heart, kidney, or liver. The function of each of these connective tissues relies, in part, on cellular components, but the extracellular matrix confers many of their functional attributes, she explained.
In the late 1990s Niklason and her collaborators developed methods to collect autologous vascular smooth muscle cells, culture them in a bioreactor for approximately 8 weeks, and generate autologous blood vessels that functioned like normal blood vessels when transplanted into the host that provided the original cells. However, transitioning the application of this technology from healthy young adults to older individuals with vascular disease proved to be difficult. As a result, her group modified its approach to vascular regeneration, opting instead for an allogeneic approach that uses vascular smooth muscle cells harvested from the aortas of organ donors to generate a cultured blood vessel. Niklason’s team expands and banks these donor cells and, after testing them for function, seeds them onto a degradable scaffold in a single-use, tubular bioreactor where they are cultured for approximately 8 weeks (Dahl et al., 2011; Niklason et al., 1999). Once the cells grow into an artery-like structure in the bioreactor, the resulting construct is decellularized, producing a bioengineered graft with the mechanical characteristics of the remaining extracellular matrix. “The final product is an engineered, human vascular matrix that seems to remodel after implantation into patients,” Niklason said.
Except for the initial step when they first open a vial of cells, the process that Niklason and her colleagues have developed is functionally closed, she said. The engineered matrices are 6 millimeters in diameter and 42 centimeters long, and they are transplanted subcutaneously into kidney dialysis patients who need a conduit between an artery and a vein so they can undergo hemodialysis therapy. The grafts are allowed to heal for 1 month, and the transplanted vessels then undergo puncture with needles three times per week during the patient’s routine dialysis treatments.
Niklason and her team have conducted initial Phase I and Phase II trials involving 60 patients across 6 centers in the United States and Poland. The patients were followed for an average of 36 months, at which point Niklason and her colleagues found that the implanted vessels retained their structural and functional integrity, which Niklason said was better than
comparable synthetic materials had performed in clinical use (Lawson et al., 2016). She added that some of the first patients in the trial are still using their grafts for dialysis 4.5 years after implantation. FDA has since approved a Phase III trial that will compare the performance of Niklason’s grafts against expanded polytetrafluorethylene and Propaten vascular grafts in 350 patients, which she said is the largest clinical trial ever conducted in vascular access for hemodialysis. The first patient was enrolled in May 2016, and preliminary results should be available in late 2018.
Moving from the Lab to Commercialization
Humacyte, the company Niklason founded to commercialize this technology, has approximately 100 employees who have developed equipment and processes capable of producing 10 vessels at a time. While sufficient for a Phase III trial, commercial production will require tens of thousands of vessels, so the company is now working to develop the capacity to produce between 100 and 200 vessels in a batch using many small containers. While this approach has engineering challenges, it is preferable to growing 100 vessels in one large container, Niklason said, because it allows for greater control over the cell biology of the vessels.
Speaking of the lessons she has learned in moving this technology from her academic laboratory to a commercial enterprise and working with regulators and manufacturing experts, she said she would encourage academics who want to follow a similar path to first demonstrate the efficacy of their products. “I have seen many academic investigators become enthused about a mediocre outcome in a small number of animals, which leads them to get distracted and focus on trying to develop their product based on . . . fairly weak pre-clinical data,” she said, noting that she strives to obtain the most robust pre-clinical data possible before moving forward with product development. In her case, that meant conducting experiments in non-human primates matched to each other as closely as possible in terms of their immunological characteristics—an approach that cost $100,000 for each animal that she and her collaborators studied. That investment paid off, however, because the results from this non-human primate model were predictive of the subsequent outcomes in humans.
After demonstrating reproducible and quantifiable results, the next important step, Niklason said, is to attend to the biology and science in the early phases of development. “Cost reduction and the regulatory pathway will follow,” she said, emphasizing the importance of getting the science correct so that any following efforts will have a strong evidence base. Understanding potency and the important biological effects of a given cell or tissue therapy is the most critical aspect of being able to generate reproducible results and reach production scale, she said. Knowing what
characteristics make a product potent, she added, can ensure that potency is retained as the production scales up.
At the same time, Niklason said, although many academic scientists tend to characterize a product with a complex set of biological markers, in many cases such a complex set of markers will not be tied closely to the product’s important biological effect and desired potency. “You can actually muddy the waters by telling FDA that your cells need to express 55 different markers to be your cell target of choice,” she said. “Focusing on the biological effect and potency are probably more important than a large battery of markers.”
One stumbling block Niklason has seen when transitioning a technology from academia to Phase I trials is a failure to focus on reproducibility. After confirming that an effect is reproducible, she said, it is important to switch both mindset and personnel from “academic” to “industry,” which can necessitate hiring additional people with training that is relevant to the unique regulatory, engineering, and process needs of scaling up and testing a new product. “Identifying a robust process that is not dependent on a single technician is absolutely critical,” Niklason said. Concerning potency, Humacyte and FDA have had thoughtful discussions about what should constitute the potency of an implanted connective tissue. Her initial response to FDA’s question about potency was to describe how strong the implant is and how well it holds a needle and thread, but FDA wanted something different. Potency, she said, is turning out to be a combination of mechanical properties, biochemical composition, cell remnants, proteomic characterization, dimensions, and cell interactions.
Concluding her presentation, Niklason emphasized to the academic investigators attending the workshop that the key to developing a reproducible production process is to understand the vital aspects of a cell system, rather than cataloguing all potential identifying cellular markers, and to maintain control over the process parameters that affect those aspects. Her final piece of advice was to collaborate early and often with FDA. “On these types of projects, [FDA] can be incredibly helpful with regard to thinking about your cell source, your safety endpoints, and potency,” she said. “Conversations with FDA are helpful, but they are not binding. They are guidance and guidelines.” She also commented that the usefulness of FDA’s guidance depends on the quality of the questions they receive.
GETTING TO THE FACTORY OF THE FUTURE
PCT Cell Therapy Services, Robert Preti said, was started 18 years ago to help companies and academics develop cellular therapies. In May 2017 the company was acquired by Hitachi Chemical Company. Initially, the company’s focus was on delivering clinical manufacturing expertise,
which included helping with technology transfer, delivering GMP-grade manufacturing, and establishing global logistics and storage services around the world. Preti and his team soon realized they needed to integrate manufacturing and technology development, particularly engineering, analytical, and process development.
The field of regenerative medicine is challenged by the complexity of developing, analyzing, and producing new therapies, Preti said, and companies that are developing regenerative therapies are looking for methods to improve consistency. The search for faster, better, and cheaper production is likely to be frustrating, but this is a problem that needs to be solved if the industry is to move forward. The field is at an inflection point where mass production and treating patients with these transformative therapies is a reality, Preti said.
The goal for product developers and manufacturers such as Preti’s company is to achieve commercially viable manufacturing strategies, but the complexity of manufacturing regenerative therapies has proven to be a significant barrier to this goal. This complexity can create inefficiencies, Preti said, because every product and service solution must be a custom one. These inefficiencies need to be reduced, he said, which raises the question, “How can we, as a community, begin to establish consistency in these products and services to drive toward the creation of platform solutions and enable a reduced cost of goods?” Establishing platform solutions, he said, is what helped the biologics industry make substantial progress in manufacturing. The field of regenerative medicine would, he suggested, benefit from the expertise of engineers as researchers and manufacturers are working to adopt new strategies and infrastructure to facilitate the production of consistent products while recognizing that it is not yet possible to fully standardize manufacturing platforms. Toward that end, Preti’s company has taken a unit operations approach, which involves breaking down the manufacturing process into small pieces or clearly defined activities. Each activity that is examined is intended to accomplish a specific outcome based on the personnel, procedures, equipment, analytical methods, and materials in a given environment. Preti also reiterated Niklason’s statement that when developing a new regenerative medicine product, it is important to have a deep understanding of the key characteristics that influence potency, but it is not important to have the same understanding of every biomarker or aspect of a product.
Current challenges in the cell therapy industry can be categorized into four different areas, Preti said: quality, scalability, the sustainability and robustness of the supply chain, and the costs of goods. Within these areas, he highlighted three main challenges: the current state of manufacturing, idle capacity (and the effect it has on manufacturing business models), and scalability. Regarding the current state of manufacturing, Preti said
that the challenge is that manufacturing processes today are inadequate to sustainably produce large-scale and high-quality goods at a cost consistent with market expectations. He acknowledged the inadequate manufacturing processes that can lead to delays in getting viable therapies to patients. As a potential solution, Preti said, the field would benefit from sponsors developing strategic commercial manufacturing plans based on objective process capability analyses. These analyses break the manufacturing process into steps and use quality-by-design methodologies to improve the process one step at a time, reducing the risk of changing the product’s properties and performance. Developing the right business model, he said in closing, depends on the product and the company, and it may change over time. Some products may be amenable to in-house manufacturing, while others might require contract manufacturing or a combination of the two or, at some future date, production at the point-of-care. “Each product is different, and that model will fall out of a carefully devised strategic manufacturing plan,” Preti said. The ultimate goal, he added, is a scalability paradigm that results in the “factory of the future” for this industry.
DISCUSSION
Addressing Technical and Scientific Hurdles
The panel of speakers discussed the scientific and technical challenges of finding donors of cells and tissues that could be used for research and product development. There is variability in both the quality of donor cells and the ability to increase the number of donor cells in vitro prior to the inoculation step, said Laura Niklason, describing the challenges she and her team encountered in finding donors for the development of Humacyte’s vascular grafts. In the long term, she said, her goal is to use a finite number of “super donors” whose HLA phenotypes can yield grafts that may be tolerated by many recipients. She believes the company can reach that goal in a couple of years and that doing so will improve reproducibility even further.
Academic researchers sometimes encounter difficulties in obtaining cells or tissues from patients with disease or who may have the phenotype of the “super donors” that Niklason plans to use. In his experience, Levine said, it is easier in academia than in industry to obtain raw materials from patients with disease. For example, his group has protocols in place to collect leukapheresis products from healthy donors as well as from those with cancer and HIV. His group also receives tissue samples from resected tumors and lymph nodes.
The workshop participants discussed how researchers and manufacturers can be sure they have selected the correct type of cell to fill the right niche in vivo, given that cells can express different markers in different
environments. What matters more, Niklason said, is how the cell functions, not, say, what specific molecule is in the lipid bilayer, so one approach to product development is to “devise assays around [in vivo] function and capture that rather than relying on specific [cell] markers.” Levine added that while it is important to learn everything possible about a product, in the commercial world it is essential to set aside the desire for complete cell characterization and instead define what characteristics must be present and quantifiable in order to release a product for a clinical trial or for a particular development stage in the manufacturing process.
Specificity of cell identity and function is an extremely important part in analyzing a product to ensure that it is performing as it is supposed to and not causing any toxicities or off-target effects, Preti said. He pointed to GlaxoSmithKline’s achievement in determining the pharmacokinetics and pharmacodynamics of Strimvelis, a stem cell gene therapy product for a rare inherited immunodeficiency syndrome called severe combined immunodeficiency due to adenosine deaminase deficiency. “For a long time,” he said, “we said we could not [characterize the mechanism of action] in cellular therapy, but I have seen evidence that it is possible.”
Those in the regenerative medicine manufacturing industry, Preti said, need to begin to focus more heavily on critical process parameters when considering the types of assays that are needed for inline testing to monitor how cells and tissues are growing during the manufacturing process. There is not enough known about critical process parameters, he said. One approach to inline testing, he said, is to break the manufacturing process into discrete unit operations, create a technology roadmap that addresses those different unit operations, and then determine if better monitoring methods are needed for each of those operations. Levine added that simple inline testing, such as measuring glucose or lactate levels, provides information on the health of the cells and their potential for expansion, but it does not take the place of all of the testing required before the release of the final product.
Each of the panelists described the single biggest hurdle they faced in developing their therapies, specifying whether the challenge was technical, scientific, regulatory, intellectual property, legal, or ethical in nature. The biggest difficulties encountered were technical and remain technical, Preti said, adding that FDA has been extremely helpful in overcoming those challenges. Niklason agreed with Preti that aside from raising money, technical challenges were the biggest obstacles, and she added that identifying key inputs and process parameters for the 8-week protocol to grow vascular structures was particularly challenging. Intellectual property was not an issue, she said, because there is so much know-how available to help in that regard. Levine agreed that technical issues, particularly those relating to scaling up or scaling out production, have presented the biggest challenges.
Training a Workforce for Regenerative Medicine Manufacturing
According to a workshop participant, the bottleneck in training occurs with equipping students with manufacturing skills prior to the doctoral level. It is important for the people who will work in production facilities to be able to identify innovative solutions to problems they encounter during the manufacturing process. The issue is not just about training manufacturing operators and quality control technicians, Preti replied, but about retention and promotion. In his experience, he said, he found that hiring bright young people with advanced degrees can be challenging because they are excited to work in the clean room for about 1 year, and then they want a promotion or to move into development. At some level, he said, the manufacturing environment is not a thinking environment. “You need capable people who can do things over and over and over again, very consistently, and be good at it,” he said. Given how difficult it is to find such people, moving toward scalable manufacturing processes cannot include increasing the need for people that require training, he added.
Organizations such as NIIMBL (the National Institute for Innovation in Manufacturing Biopharmaceuticals)4 and BioFabUSA5 are interested in and play a role in training and education, as do professional societies, Levine commented. An additional challenge, he said, will be developing standards for training technicians or engineers who want to work in this field. The bulk of manufacturing personnel for other industries come from 2-year and technical colleges, said Krishnendu Roy of the Georgia Institute of Technology, adding that the National Cell Manufacturing Consortium,6 NIIMBL, and BioFabUSA are all interested in partnering with technical and 2-year colleges nationwide to train the manufacturing workforce this industry needs. That would be a good start in addressing these training issues, Preti agreed, one that in the long term would meet a growing industry’s needs.
There are training courses for those who work in blood processing and banking operations, and a workshop participant suggested that those programs could serve as a foundation for training programs for the regenerative medicine industry. There are a few potential candidates from blood banks and from medical technology schools, both of which train people in procedures and documentation, which are the underpinnings of GMP, Levine said. He added, however, that he is really looking for people with
___________________
4 More information on NIIMBL can be found here: http://www.niimbl.us (accessed August 28, 2017).
5 More information on BioFabUSA can be found here: https://www.armiusa.org (accessed August 28, 2017).
6 Resources from the National Cell Manufacturing Consortium can be found here: http://www.cellmanufacturingusa.org (accessed August 28, 2017).
cell culture experience, and those individuals are not coming from blood banking or medical technology schools. Relying on technicians from the blood banking industry could be a good short- to mid-term solution, especially since blood bank staff and medical technology school graduates would have the right mentality for the manufacturing environment, Preti said. He said, though, that he is concerned that the physical environment of a regenerative medicine manufacturing facility is restrictive and uncomfortable and that, as a result, the degree of burnout will be high, regardless of the workers’ training. He added that this is why he believes the industry cannot develop a model that will require finding thousands of individuals to fill manufacturing positions.
Convening Stakeholders for Collaboration and Standards Setting
The workshop participants discussed the challenge of balancing competition and the development of shared standards across the regenerative medicine industry. One workshop participant asked the panelists how they handle the transition from open academic science to proprietary commercial development and how they view the ethical considerations in making that transition. There is no defining line where one goes from the public sphere and publishing results to the commercial, proprietary space, Niklason said. She noted that disclosure and publication are important for policing science. “Once you stop disclosing what you are doing, except to FDA, then that policing function goes to the leadership of the company,” she said.
In addition to disclosure, reproducibility is another important consideration, Levine said. In his area of research on CAR T cells, he said, there are now many companies with products in advanced clinical trials, and the clinical results from these different trials appear to be validating the superior performance of this category of product versus conventional therapies.
Manufacturing standards are available, including clear standards for GMP and good tissue practices from FDA,7 Preti said, and the Alliance for Regenerative Medicine8 has standards covering different types of therapies. The Foundation for the Accreditation of Cellular Therapy has standards that draw on FDA regulations and guidelines and guidelines for quality in blood banking operations, Levine added. The University of Pennsylvania, for example, has an accredited facility. Standards are important, and a public–private partnership has established a standards coordinating body
___________________
7 Information on drug applications and Current Good Manufacturing Practice (CGMP) regulations can be found on FDA’s website here: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/Manufacturing/ucm090016.htm (accessed August 29, 2017).
8 The Alliance for Regenerative Medicine launched the Standards Coordinating Body for regenerative medicines in 2017. The Standards Coordinating Body website can be found here: https://www.standardscoordinatingbody.org (accessed August 29, 2017).
for the industry. However, he said, past attempts to develop common analytics and assays have failed, and he said that he did not see industry sharing assays and analytics.
Collaborations will be important in addressing technical challenges as the regenerative medicine industry moves forward, Levine said, and academic institutions and companies can only do so much in isolation. The National Institute of Standards and Technology (NIST) and FDA can play a critical role, he said, particularly where the regulatory realm and analytical standards are involved. The National Academies’ Forum on Regenerative Medicine has a unique and important role to play in bringing all of the necessary parties together to talk about how to address the challenges this industry faces as it moves forward, Levine said.


















