
4
Chemical Utilization of CO2 into Chemicals and Fuels
INTRODUCTION
Scientists have been aware of the potential economic and environmental benefits of using CO2 as a feedstock for the synthesis of commodity chemicals and fuels for decades. It is a generally inexpensive waste product, which contributes significantly to global warming. Nevertheless, despite the large amount of fundamental research that has been performed regarding the conversion of CO2 into more valuable products there are relatively few examples of industrially viable processes. The challenges associated with the conversion of CO2 are primarily related to both its kinetic and thermodynamic stability. CO2 cannot be converted into commodity chemicals or fuels without significant inputs of energy and contains strong bonds that are not particularly reactive. As a consequence, many of the available transformations of CO2 require stoichiometric amounts of energy-intensive reagents. This can often generate significant amounts of waste and can result in large greenhouse gas footprints. The grand challenge for converting CO2 waste streams into useful products is to develop processes that require minimal amounts of nonrenewable energy, are economically competitive, and provide substantial reductions in greenhouse gas emissions compared to existing technology. In this chapter we provide an assessment of the current state of research into chemical pathways for the conversion of CO2 into commodity chemicals and fuels. Areas of promise are identified and technological limitations are highlighted, focusing on the technical features of the CO2 conversion step. Issues related to enabling technology are covered in Chapter 7, and economic considerations of conversion processes are covered in Chapter 9. Additionally, niche processes which could only be practiced on a small scale and likely could not be generalized to a larger application, for example, the late-stage carboxylation of organic molecules for pharmaceutical synthesis, are not covered, even though there is considerable research activity in this area.
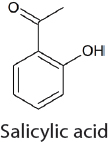
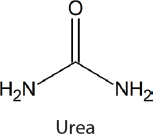
Currently only a limited number of commercial processes that involve the conversion of CO2 into value-added chemicals exist (Table 4-1) (Patricio et al., 2017; Quadrelli et al., 2011; Topham et al., 2014). Two notable examples are the synthesis of salicylic acid (89.8 kilotons produced worldwide in 2015), which was developed in the 19th century (Lindsey and Jeskey, 1957), and the synthesis of urea (164 million tons produced worldwide in 2013), which was developed in 1922 (Artz et al., 2018). CO2 is combined with ammonia for urea synthesis. However, it should be noted that the CO2 used in urea synthesis typically is produced from methane steam reforming, which also produces the H2 required for ammonia synthesis. The use of CO2 from a waste stream for sustainable urea synthesis would require using water electrolysis to make H2 or an alternative ammonia synthesis.
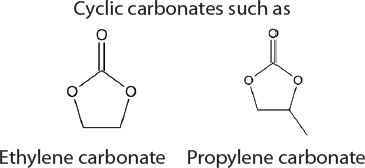
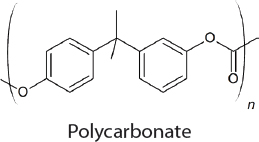
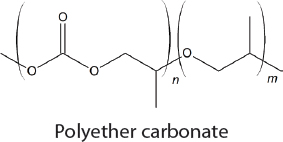
In the 1950s, methods for the commercial synthesis of cyclic carbonates from CO2 were developed (Sakakura et al., 2007). Specifically, treatment of CO2 with ethylene or propylene oxide in the presence of a basic catalyst generates ethylene carbonate or propylene carbonate, respectively. Similarly, styrene oxide, cyclohexene oxide, and 1,3-propylene oxide can also be used as substrates with CO2 but the cyclic carbonates that are produced are made on a significantly smaller scale. Overall, approximately 80,000 tons of cyclic carbonates were produced worldwide from CO2 in 2010 (Alper and Yuksel-Orhan, 2017). CO2 has also been used as a feedstock for the synthesis of aromatic and aliphatic polycarbonates. The Asahi-Kasei process, which generates 600,000 tons of polycarbonate per year, uses CO2, ethylene oxide, and bisphenol A as feedstocks (Fukuoka et al., 2007). Ethylene glycol, a commodity chemical, is produced as a stoichiometric by-product. Recently, Covestro developed a plant for the copolymerization of CO2 with propylene oxide to generate polymeric polyols (polyether carbonates), branded as cardyon® (Langanke et al., 2014). These polyols are used to make polyurethanes, which are found in foam mattresses. The CO2 is obtained from nearby ammonia production. Approximately 5,000 tons per year of polymeric polyols are produced in this facility. Novomer developed a related process, purchased by Saudi Aramco and branded Converge®, for the generation of polyols from CO2 and propylene oxide, which is being performed on a scale similar to the Covestro system, using the facilities of Centauri Technologies in Texas. In the case of both the Converge® and cardyon® processes, the scale at which the CO2-derived polyols are produced is approximately 60 times smaller than conventional polyol plants. Whether this technology will eventually replace existing plants and can be performed on a larger scale remains unclear.
The limited examples described above are established technologies (with the exception of polyether carbonate production, which is still undergoing development related to the scale of the process) that are generating products which are available commercially. They will not be discussed further. Instead in the following sections emerging technologies for the conversion of CO2 into commodity chemicals and fuels are evaluated. Initially, specific products are discussed, before a section on research challenges that extend across products. At the conclusion of the chapter short- and long-term research needs and opportunities are described.
TABLE 4-1 Major commodity chemicals that are currently synthesized from CO2 on an industrial scale globally.
| Chemical | Scale per yeara | |
|---|---|---|
 |
30 kilotons | |
 |
112,000 kilotons | |
 |
40 kilotons | |
 |
600 kilotons | |
 |
10 kilotons | |
a Global amount produced using a process involving CO2.
SOURCE: Alper and Yuksel-Orhan, 2017.
EMERGING TECHNOLOGIES FOR CO2 CONVERSION INTO COMMODITY CHEMICALS AND FUELS BASED ON PRODUCT
Methanol Production
Methanol (CH3OH), produced globally on a scale of approximately 70 million tons in 2015, typically is synthesized from syngas (H2 + CO) obtained directly from fossil fuels (Álvarez et al., 2017; Ganesh, 2014; Pérez-Fortes et al., 2016b; Saeidi et al., 2014). A small amount of CO2 (up to 30 percent) is generally added to the feed to improve performance (Jadhav et al., 2014). This is successful in part because the mechanism of methanol production involves the initial conversion of CO and H2O to CO2 and H2 via the water gas-shift reaction (Eq 1). In fact, the development of methods to increase the amount of CO2 in the syngas feed without causing a large decrease in methanol yield represents an opportunity to utilize waste CO2 that is produced during syngas production. Although this strategy is only viable if excess H2 is available, it could improve current technology and increase plant efficiency.
| CO + H2O ⇌ H2 + CO2 | (Eq 1) |
The direct hydrogenation of CO2 to methanol could provide a more sustainable synthetic route if coupled with low-carbon methods for the production of H2 (refer to Chapter 7) (Álvarez et al., 2017; Ganesh, 2014; Pérez-Fortes et al., 2016b; Saeidi et al., 2014). Furthermore, the development of a practical method for the synthesis of methanol from CO2 could also facilitate a transition toward a methanol economy, in which methanol is used either directly as a fuel or as a source of H2 (Olah, 2005).
Researchers have developed several catalysts and reactors for direct hydrogenation of CO2 to methanol, but high rates and high methanol selectivity have only been possible using high pressures (>300 bar) (Álvarez et al., 2017; Klankermayer et al., 2016; Wang et al., 2015). The cost of this technology presently is not competitive with the cost of methanol synthesis from syngas. Nevertheless, due to special circumstances related to location, presently two large pilot plants for direct methanol production from CO2 are in operation. The Mitsui Chemical Company in Japan produces around 100 tons of methanol per year from CO2 (Table 4-2) (Quadrelli et al., 2011). The CO2 is a waste product generated in an adjacent petrochemical facility, while the H2 is either also a waste product from the petrochemical industry or generated through the electrolysis of water using photovoltaic devices. The close links to the petrochemical industry make the process economically viable. Second, Carbon Recycling International, located in Iceland, produces approximately 4,000 tons of methanol from CO2 each year (Klankermayer et al., 2016). The plant uses hydro and geothermal energy to produce H2 and uses CO2 captured from the flue gas of a geothermal power plant, which is located next to the CO2-to-methanol facility. The process is economically feasible because of the availability of
TABLE 4-2 Major fine and commodity chemicals that are currently synthesized from CO2 on a pilot plant scale
| Chemical | Company (Location) | Scale per yeara |
|---|---|---|
| Methanol |
|
4,000 tons 100 tons |
| Methane |
|
1,000 tons |
| Carbon monoxide (via SOEC)b |
|
12 N m3/h |
| Fuel (via CO2-based Fischer-Tropsch) |
|
3 tons 200 L |
 |
|
500 tons 1,000 tons |
 |
|
2.4 tons |
aAmount of product that is produced per year. b SOEC = solid oxide electrolyzer cell.
NOTE: A pilot plant is defined as a precommercial system, which produces a chemical or fuel on a smaller scale than a full plant and is used for learning purposes.
low-cost electricity, required to generate the H2, in Iceland, and because the composition of the flue gas is 85-90 percent CO2. This substantially lowers the cost of CO2 compared with more traditional flue gas streams which contain lower amounts of CO2 (see Chapter 2).
Improved catalysts are critically needed if the direct hydrogenation of CO2 to methanol is to replace methanol production from syngas. At this stage, significant amounts of research into the direct hydrogenation of CO2 to methanol have focused on using heterogeneous copper-based catalysts that are closely related to those used for CO conversion to methanol (Ganesh, 2014). In recent years there have also been a number of reports of catalysts for CO2 hydrogenation to methanol which use metals other than copper and show promising activity (Martin et al., 2016; Studt et al., 2014). Two general challenges for catalyst development
are product inhibition by water (the by-product of CO2 hydrogenation) and poor selectivity because of the competing reverse water gas-shift reaction between CO2 and H2 to generate CO and H2O. Once more efficient catalysts are developed, further attention can be given to factors such as stability, cost, sustainability, and scale-up potential. Additionally, although ultimately a large-scale catalyst for direct methanol hydrogenation will almost certainly be heterogeneous, research into homogeneous catalysts, which is occurring in the academic community, may prove valuable for guiding the development of heterogeneous systems and for niche applications where a small amount of methanol is generated, for example as fuel to power a portable device (Wang et al., 2015).
Finally, research is currently ongoing into the electrochemical reduction of CO2 to methanol in which protons and electrons are used as the H2 source. To date, however, most work reports the formation of methanol as a by-product, at selectivities less than 15 percent (Costentin et al., 2013). Several intermediates formed along the six-electron reduction pathway can release from the catalytic surface to form other products. Recently, it was reported that a molybdenum-bismuth bimetallic chalcogenide electrocatalyst could generate methanol with a Faradaic efficiency (see Glossary in Appendix A) exceeding 70 percent, although this catalyst requires an acetonitrile/ionic liquid electrolyte solution (Sun et al., 2016b). Further exploratory and mechanistic research will be required to identify even more selective (and stable) catalysts that do not require organic electrolytes before electrocatalytic methanol production from CO2 can be considered for larger-scale application. Alternatively, methanol could be synthesized indirectly, via the initial electroreduction of CO2 to CO (see below), followed by the conversion of CO to methanol using the processes highlighted above.
Dimethyl Ether Production
Dimethyl ether, which is closely related to methanol (dehydration of methanol gives dimethyl ether), is a platform chemical widely used as an alternative to liquified petroleum gas as a clean fuel (Quadrelli et al., 2011). Currently, dimethyl ether is synthesized either via methanol dehydration or from syngas. For similar reasons to those described for methanol, the direct conversion of CO2 to dimethyl ether is attractive. The Korea Gas Corporation has developed an indirect strategy for dimethyl ether synthesis from CO2 that is performed on a scale of 100 tons per day. It involves the tri-reforming of methane, CO2, and H2O into syngas (see Chapter 6). The syngas is then converted into dimethyl ether. Although this route is scalable it is not economically viable and does not involve the direct conversion of a CO2-only feedstock into dimethyl ether. Most likely research into CO2 hydrogenation to methanol will also provide insight into the formation of dimethyl ether and, if a practical catalyst for direct methanol formation is obtained, a system for dimethyl ether may follow shortly thereafter.
Formic Acid Production
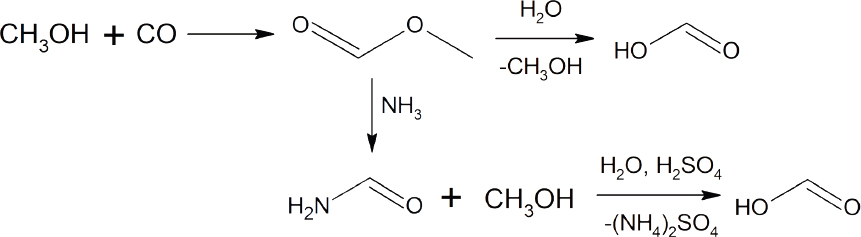
Formic acid is used as a preservative, insecticide, or reducing agent primarily in the food, textile, and pharmaceutical industries (Klankermayer et al., 2016). In 2013, the global production of formic acid was 620 kilotons. The most common method for the synthesis of formic acid is a two-step process involving the initial reaction of CO with methanol to form methyl formate, followed by the conversion of methyl formate to formic acid either through reaction with ammonia to generate formamide and subsequent acidification with H2SO4, or through direct hydrolysis with water (see Scheme 4-1). This direct hydrolysis requires high pressure and a large excess of water, followed by a rapid reduction of the pressure and cooling to generate the formic acid. The conversion of CO2 to formic acid using either H2 or protons and electrons represents an atom-economical approach to a valuable commodity chemical, which could potentially become even more important if formic acid is used as a H2 vector (Pérez-Fortes et al., 2016a; Sordakis et al., 2018).
The thermal hydrogenation of CO2 to formic acid is thermodynamically unfavorable when starting from gas-phase reactants but becomes slightly exergonic when performed under aqueous conditions (Álvarez et al., 2017; Sordakis et al., 2018). Nevertheless, in academic research, a variety of strategies are typically used to drive the reaction, including using a base to deprotonate the formic acid, generating an ester by reacting formic acid with methanol (or a higher-order alcohol) in situ, or removing formic acid as it is formed. On a laboratory scale, a vast number of homogeneous and heterogeneous catalysts for CO2 hydrogenation to formic acid have been developed (Álvarez et al., 2017; Sordakis et al., 2018). In some cases these catalysts give high turnover numbers and frequencies but typically only in the presence of a base, which reduces atom economy and increases cost. Challenges for future research into the development of systems for the hydrogenation of CO2 to formic acid include (i) the discovery of catalyst systems which give high turnover numbers in the absence of base or with recycling of the base, (ii) the need for cheaper ligands to stabilize homogeneous catalysts, and (iii) the
refinement of strategies to separate formic acid from the reaction media, which will be a crucial part of successful catalyst commercialization.
Electrochemical and photochemical systems for the conversion of CO2 and protons and electrons to formic acid have also been developed (Costentin et al., 2013; White et al., 2015). A number of photocatalysts (most typically Si and TiO2 semiconductor-based materials) have been reported for reduction of CO2 to a variety of C1 products, including formic acid, but their rates tend to be very low (White et al., 2015). At this point these catalysts and associated photoreactor designs do not show promise for efficient and economic CO2 reduction at scale. The electrocatalytic reduction of CO2 to formate has been reported at Faradaic efficiencies (FEs) in the range 80-95 percent using tin-based catalysts (Kumar et al., 2017; Zhang et al., 2014) at moderate to high overpotential, both in standard three-electrode electrochemical cells and in liquid electrolyte-based electrolysis flow cells, the latter at a rate of 200 mA/cm2 (Whipple et al., 2010). Palladium nanoparticle electrodes reduce CO2 to formate with >90 percent FE at <200 mV overpotential, but the palladium requires periodic regeneration because of poisoning by trace CO, which is produced as a by-product (Jiang et al., 2018; Min and Kanan, 2015; Stalder et al., 1984). Palladium has also been used recently as a cathode in a photoelectrochemical device for formate synthesis. The Joint Center for Artificial Photosynthesis has demonstrated 10 percent efficient light-to-formate energy conversion using a Pd/C cathode wired to a tandem III-V GaAs/InGaP photoanode coated with a protective conductive TiO2 layer (Zhou et al., 2016).
More recently Dioxide Materials reported the reduction of CO2 to formic acid at a Faradaic efficiency of 94 percent at a rate of 200 mA/cm2 using a three-compartment electrolyzer with a cation-anion exchange membrane that exhibited stable performance for 500 hours (Yang et al., 2017). Technoeconomic and life-cycle analyses suggest that electroreduction of CO2 to formic acid has promise and gradually is moving toward scale-up and commercialization (Pérez-Fortes et al., 2016a). Remaining challenges for electrochemical conversion of CO2 to formic acid include (1) achieving sufficient stability and durability for both catalysts and electrodes and (2) the refinement of energy-efficient strategies to separate formic acid from the product stream (typically a 5-20 wt% aqueous solution).
Methane Production
Methane is widely used as a fuel and to make syngas (3,500 billion cubic meters of natural gas were consumed in 2014) (EIA, 2017). Industrially, it is predominantly obtained directly from natural gas and is rarely synthesized. Nevertheless, the Sabatier reaction, which hydrogenates CO2 to methane using a nickel catalyst, has been known for more than a hundred years (Eq 2) (Su et al., 2016).
| CO2 + 4 H2 → CH4 + 2 H2O | (Eq 2) |
Small-scale pilot plants based on related heterogeneous catalysts that typically operate at around 300°C were tested in Norway using CO2 from flue gas and low-carbon H2, and in Japan and the United States using H2 from water electrolysis (Quadrelli et al., 2011). This occurred before the widespread production of methane in natural gas from shale resources. Currently, Germany has several small-scale plants, including a pilot plant run by Audi (Table 6-2), developed due to Germany’s dependence on renewable energy, which requires systems for energy storage. However, research is also continuing on the design of improved catalysts (Su et al., 2016). Specific challenges that need to be addressed include the development of catalysts that operate at lower temperatures (<200°C) where the reaction is more favorable and the sintering and oxidation-induced deactivation in nickel-based catalysts can be prevented. Although ruthenium catalysts have shown advantages compared to nickel systems for solving these problems, they have additional cost. At this stage, the hydrogenation of CO2 to methane is not practical on a large scale and is unlikely to be so in the near future given the low price and abundant availability of methane from natural gas. Additionally, there will be a significantly greater economic value in converting CO2 to many other chemicals compared with methane.
Electrochemical reduction of CO2 to methane is also a widely studied process with reported Faradaic efficiencies in the range of 80 to 94 percent using N-doped carbon or copper-on-carbon catalysts in standard three-electrode or H cells (Manthiram et al., 2014; Qiu et al., 2017; Sun et al., 2016a). Partial current densities for methane formation as high as 38 mA/cm2 have been reported for a Cu catalyst electrodeposited on a carbon gas diffusion electrode (Qiu et al., 2017). As for the aforementioned thermochemical CO2-to-methane processes, at this time the electrocatalytic conversion of CO2 to methane, despite continued progress in the development of more selective catalysts, probably will not be pursued on a large scale given the global availability of low-cost methane derived from natural gas.
Carbon Monoxide Production
Carbon monoxide (CO) is an important feedstock in the synthesis of many chemicals and fuels (Keim, 1989). For example, hydrogenation of CO can generate methanol, or through the Fischer-Tropsch process higher-order hydrocarbons can be produced (Eq 3).
| (2n + 1) H2 + n CO → CnH2n+2 + n H2O | (Eq 3) |
Typically, CO is obtained through the partial oxidation of coal or hydrocarbons at very high temperatures (>800°C). The CO generated from these reactions is not pure, with H2 being the main contaminant. For many applications a mixture of CO and H2 is required, but for some applications gas separation is subsequently needed to isolate pure CO.
One method to convert CO2 to CO is the reverse water gas-shift reaction (Eq 1), in which CO2 and H2 are converted into CO and H2O (Wang et al., 2011). The reverse water gas-shift reaction is an endothermic reaction and consequently high temperatures (~500°C) are typically utilized to favor the formation of CO. Even then a large excess of either CO2 or H2 or the constant removal of products are often used as strategies to increase conversion. A range of heterogeneous catalysts have been used for the reverse water gas-shift reaction, including copper-, iron-, or ceria-based systems, but in general they have poor thermal stability, and methane is commonly formed as an undesired side product (Wang et al., 2011). Fluidized bed reactors give greater conversion to CO than fixed bed reactors and this is one approach to improve conversion. Nevertheless, given the thermodynamic limitations of the reverse water gas-shift reaction and the fact that other potential routes to generate CO from CO2 are at a significantly more advanced state (see below), it is unlikely that research on the reverse water gas-shift reaction for generating large amounts of CO will advance beyond the research stage. However, there may be benefits associated with combining the reverse water gas-shift reaction and Fischer-Tropsch chemistry to generate fuels as described in a subsequent section.
Currently, there are no catalysts (either homogeneous or heterogeneous) for the direct thermochemical reduction of CO2 to CO and O2. In contrast, the direct electrochemical splitting of CO2 to CO and O2 may provide an alternative to the conventional fossil fuel–based synthetic route (Qiao et al., 2014; White et al., 2015). This reaction can be performed either at high temperature using a solid oxide electrolysis cell (SOEC) or at low temperature using a solution-phase or gas diffusion electrolysis cell. SOECs for CO2 splitting have recently been brought to market, while low-temperature systems are still in a research phase. Below we summarize the status and remaining challenges for developing both of these technologies.
SOECs are monolithic devices composed of two electrodes separated by a solid oxide-conducting (O2–) electrolyte (Ebbesen et al., 2014; Zhang et al., 2017). A commonly used combination consists of an yttria-stabilized zirconia (YSZ) solid oxide electrolyte, a composite Ni-YSZ cathode, and a perovskite-type anode such as strontium-doped lanthanum manganate. For CO2 electrolysis, CO2 is supplied to the cathode side and the anode side is swept with air or another gas. The cathode reduces CO2 to CO and O2–, which migrates through the oxide-conducting electrolyte to the anode, where it is oxidized to O2. High temperatures (700-900°C) are required to attain sufficient oxide conductivity in the electrolyte. Under operating conditions, the temperature is maintained by the Joule heating from the internal resistance in the cell itself, which imposes a small energetic penalty but avoids the need to actively heat the system.
SOECs operate at high current densities (0.2 up to 2 A cm-2), achieve high energy efficiency (typically >95 percent), and can produce CO product streams of high purity (>99 percent). Researchers have demonstrated relatively stable performance for CO2 electrolysis at current densities up to 0.5 A cm2, with small cell voltage increases observed over several hundred hours of continuous operation (Ebbesen and Mogensen, 2009; Ebbesen et al., 2014).
Degradation is faster at higher current densities. Haldor Topsoe1 currently sells a commercial SOEC system for onsite CO production that provides CO at 99.0 to 99.999 percent purity and requires 6-8 kWh of power per normal cubic meter (Nm3) CO produced. The first commercial system with 12 Nm3 h-1 capacity began operating at Gas Innovations in Texas in 2016 (Mittal et al., 2017).
There is room for substantial improvement in SOECs to increase deployment of this technology. Major challenges include developing new electrodes with enhanced stability at higher current densities, finding electrolytes that provide high oxide conductivity at lower temperatures, and increasing the tolerance to impurities.
In the past 20 years a large number of molecular and heterogeneous systems for low-temperature CO2 electroreduction to CO have been developed (Costentin et al., 2013; Ganesh, 2016; Lim et al., 2014; Spurgeon and Kumar, 2018). Faradaic efficiencies in the range 95-98 percent can be achieved routinely in standard three-electrode or H-type cells using a variety of neutral or acidic electrolytes, typically using silver cathode catalysts, resulting in rates of 2-10 mA/cm2 (Mistry et al., 2017; Neubauer et al., 2016). Interestingly, there are also a number of reports that use non–precious metal catalysts, such as N-doped carbon or carbon nanofibers, achieving performance levels that are similar to those obtained with silver catalysts (Kumar et al., 2013).
Use of silver catalysts or supported gold catalysts in electrolyzers in which the electrodes are separated by a flowing liquid electrolyte, either alkaline or neutral, significantly increases the CO production rate to 150-450 mA/cm2, while still maintaining Faradaic efficiencies of 60-98 percent (Verma et al., 2016, 2018). Although there will always be a trade-off between maximizing rate and energetic efficiency (see Glossary in Appendix A), the overpotentials for CO reduction using silver or gold catalysts are fairly low, which means that rates of 150 mA/cm2 can be achieved at energetic efficiencies exceeding 50 percent. In all these cases the oxygen evolution reaction takes place at the anode, typically using an IrO2 catalyst. Based on these prior efforts that typically employ electrodes with a geometric area of 1-2 cm2, Siemens performed experiments on a larger scale, using first 10 cm2 and then 100 cm2 gas diffusion electrodes in electrolyzer configurations with a flowing liquid electrolyte (Jeanty et al., 2018). These cells were successfully operated for 200 hours using a neutral electrolyte at a rate of 150 mA/cm2, with a Faradaic efficiency for CO formation of 60 percent. Subsequently, Siemens collaborated with Evonik to connect their 10 cm2 CO2-to-CO electrolyzer (running at 300 mA/cm2 for over 1,200 hours) with a fermentation process in which the formed CO is combined with unreacted CO2 to form butanol and hexanol, at close to 100 percent Faradaic efficiency (Haas et al., 2018). Detailed economic analysis of this hybrid system highlights the promise for sustainable production of first CO and then other chemicals at scale using this approach.
___________________
1 See https://www.topsoe.com/processes/carbon-monoxide/site-carbon-monoxide (accessed October 10, 2018).
In addition to low-temperature and high-temperature electrochemical cells described in the chapter, intermediate-temperature carbon dioxide electrolysis, with cell temperatures ranging from 200oC to 500oC, is a possible new direction that could provide access to products that are not made efficiently at either temperature extreme currently.
While these electrolyzer systems exhibit encouraging performance levels for the reduction of CO2 to CO, the use of gas diffusion electrodes, especially in combination with an alkaline electrolyte, can cause problems. Typically, CO2 flow rates that greatly exceed the rate of CO2 reduction are used to maximize the current density and Faradaic efficiency, which results in product streams that are diluted with a large excess of CO2 (e.g., 3:1 CO2:CO in the Siemens example above). In addition, some of the CO2 is lost by diffusion through the gas diffusion electrodes into the electrolyte, where it can react with OH– to form carbonates, which can precipitate on the electrode or migrate to the anode and release CO2 into the O2 stream. The rate of these undesired processes (often completely ignored in CO2 electrolysis work) depends on the specific gas diffusion electrode used, the nature of the catalyst layer, as well as the flow rates and pressures of the CO2 feed and the electrolyte. Achieving high CO2 conversion with minimal loss of CO2 into the electrolyte will be critical for advancing CO2 electrolysis technology.
Dioxide Materials has reported a different electrolyzer configuration: an anion-conductive membrane-based electrolyzer with a silver cathode catalyst that sustains CO production rates in the range 100-200 mA/cm2 over more than 1,000 hours (Kutz et al., 2017). The use of a membrane as the electrolyte between the electrodes significantly reduces some of the aforementioned issues (CO2 loss and electrolyte degradation) associated with the use of liquid electrolytes. In recent collaborative work Dioxide Materials and 3M explored the electrochemical generation of syngas from water and CO2, either (1) by operating a flowing liquid-electrolyte CO2 electrolyzer cell for CO production and a polymer electrolyte-based water electrolyzer for H2 production in parallel, or (2) by performing co-electrolysis of CO2 and H2O in a single anion-exchange membrane-based electrolyzer (Liu et al., 2016). Both systems produced CO and H2 at industrially relevant rates (for example, CO2 to CO at 100 mA/cm2). The various technoeconomic analyses reported to date suggest that the electroreduction of CO2 to CO has promise to become economically feasible.
Remaining challenges for electrochemical conversion of CO2 to CO include (1) achieving sufficient stability and durability for both catalysts and electrodes, (2) the need to develop strategies that minimize loss of CO2 to the electrolyte, and (3) the need to further reduce the overall energy requirement under operating conditions at practical CO production rates.
Ethylene and Ethanol Production
Ethylene is a high-volume commodity chemical that is used to produce a large number of other chemicals and is an important monomer in a range of different polymers. In 2016,
over 150 million tons of ethylene were produced, almost exclusively from fossil fuel–derived precursors. Although the conversion of CO2 to ethylene requires a large energy input, the prospect of using CO2 as a carbon source for a large fraction of commodity chemical production has motivated many research efforts. One potential strategy is to hydrogenate CO2 to ethylene, using a low-carbon source of H2. Recent studies have described iron catalysts with promising selectivity for CO2 hydrogenation to ethylene and other light olefins (up to 65 percent) (Satthawong et al., 2015; Wang et al., 2013). However, these systems operate at low CO2 conversion and also produce lower-value chemicals such as CO and methane, along with alkanes. At this stage a significant amount of catalyst improvement is required to develop systems which generate ethylene in higher yields. A more realistic short-term goal is the development of systems for the production of a mixture of hydrocarbons (Fischer-Tropsch chemistry) from CO2 (see below).
Another route to ethylene production from CO2 is electroreduction using copper catalysts. This reaction requires a significantly larger overpotential than CO2 reduction to CO using gold or silver catalysts. Extensive investigations of different copper nanostructures and operating conditions have led to systems that produce ethylene, but achieving selectivities exceeding 40 percent has been difficult. Strategies such as alloying silver with copper, in which the silver enhances formation of the needed CO intermediate (Hoang et al., 2018), and precise engineering of the copper catalyst layer inside a sandwich-type gas diffusion electrode have increased Faradaic efficiencies for ethylene to 60-70 percent at rates of 160 to 250 mA/cm2 (Dinh et al., 2018). Often a sizeable amount of ethanol (Faradaic efficiencies of 10 to 30 percent) is co-produced with the ethylene. Co-production of ethylene and ethanol—a valuable commodity chemical itself (with an annual global production of approximately 80 million tons in 2016)—may bring economic feasibility for ethylene production closer as a result of the additional income generated from the produced ethanol. Additionally, given that ethylene is a gas and ethanol is a liquid, separation of the products should be relatively straightforward, although extracting ethanol from the electrolyte may be challenging. In related chemistry, a promising class of boron- and nitrogen-co-doped nanodiamond catalysts was reported, which can achieve Faradaic efficiencies for ethanol production as high as 93 percent (Yanming et al., 2017). It is unclear whether this catalyst can be integrated on a gas diffusion electrode in a liquid electrolyte or membrane-based electrolyzer in order to achieve practical production rates.
Despite the progress in the electroreduction of CO2 to ethylene (and ethanol), a large number of challenges remain. The few studies that have explored the stability of the various copper catalysts suggest that catalyst degradation and remodeling is a serious issue. Indeed, sustained operation of an ethylene-producing electrolyzer for more than 2 hours has not been demonstrated. Furthermore, as described above, loss of CO2 to the electrolyte has the potential to be a major drawback. Despite these challenges, the prospect of converting CO2 into a valuable and versatile chemical such as ethylene using low-carbon electricity is a powerful
reason to continue to develop better and more durable catalysts, electrodes, and electrochemical cell designs.
Dimethylcarbonate and Diphenylcarbonate Production
Dimethylcarbonate is used to synthesize polycarbonates, as a mild methylating agent in organic chemistry, and as a solvent (Ma et al., 2009). It is currently produced on a scale of 90 kilotons per year. Potential applications for dimethylcarbonate also exist in the fuel industry where it could be blended with gasoline as an oxygenate. Historically, dimethylcarbonate was synthesized from phosgene and methanol, but now it is prepared either via transesterification of ethylene carbonate or propylene carbonate and methanol or using CO, methanol, and O2 as feedstocks. There has been considerable research into the development of catalysts for the conversion of CO2 and two equivalents of methanol into dimethyl carbonate and water (Tamboli et al., 2017). Both homogeneous catalysts based on tin and heterogeneous catalysts based on ceria-zirconia oxides are capable of promoting the reaction. However, unfortunately, the thermodynamics for dimethylcarbonate formation from CO2 are limiting and under standard reaction conditions (160-180°C, 90-300 atm CO2), yields range between 1 and 5 percent. Although more advanced catalysts would be beneficial the crucial challenge for development of practical systems for dimethylcarbonate production is to discover cost-effective strategies for the removal of water to drive the equilibrium and increase the yields of dimethylcarbonate. An alternative strategy to make dimethylcarbonate from CO2 involves using urea as a CO2 carrier (Shukla and Srivastava, 2017). In this approach, the reaction of urea and methanol generates dimethylcarbonate and ammonia. The ammonia can then in principle be recycled back into urea through a reaction with CO2. A number of catalysts are known for the conversion of urea and methanol into dimethylcarbonate and ammonia, and yields of up to 50 percent have been reported. However, improvements in selectivity are required, as well as the developments of methods to remove ammonia as it is formed, as this will assist in driving product formation.
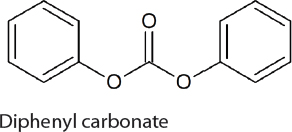
Diphenylcarbonate is used as a feedstock for the synthesis of aromatic polycarbonates and is produced on a 250 kiloton scale each year (Gong et al., 2007). It is prepared either using toxic phosgene and phenol or via the transesterification of dimethylcarbonate. Therefore, if a CO2-based route for the synthesis of dimethylcarbonate is developed it will likely also lead to an improved method from the production of diphenylcarbonate. This is supported by the recent development of an indirect method to produce diphenylcarbonate from CO2 and phenol by Asahi Kasei. The first step in the process is the reaction of CO2 with n-butanol to generate di-n-butylcarbonate. Subsequently, the di-n-butylcarbonate is treated with phenol to generate diphenylcarbonate and n-butanol, which is recycled. To date, a 1,000 tons/yr facility has been operational for more than 1,000 hours. The direct synthesis of diphenylcarbonate from phenol and CO2 is also possible but suffers from similar problems to the reaction of
methanol with CO2. In 2011, Shell opened a 500 ton per annum demonstration plant for the synthesis of diphenylcarbonate from phenol and CO2 using propylene oxide as a water scavenger. In this reaction propylene glycol is also generated. Nevertheless, this method for the synthesis of diphenylcarbonate has yet to be used on a larger scale and is limited by the market for propylene glycol.
Polymer Production
Plastics such as polycarbonates and polyurethanes have a wide range of applications ranging from insulating electronic components to cushioning materials (Qin et al., 2015). Petroleum is the key feedstock for the synthesis of most plastics. Replacing petroleum feedstocks with CO2 could be desirable for both economic and environmental reasons and could also lead to biodegradable polymers with novel properties. CO2 can be used as either a direct or indirect feedstock for the synthesis of polymers. In the direct approach CO2 is used as a monomer unit, which is directly incorporated into the polymer. In the indirect approach CO2 is first converted into a different monomer, for example, methanol, organic carbonates, carbon monoxide, ethylene, dimethylcarbonate, or urea, which is then polymerized. Other sections in this chapter evaluate the feasibility of CO2 conversion into chemicals such as methanol, so this section will focus on the direct use of CO2 in the synthesis of polymers.
Polycarbonates—plastics which contain carbonate groups—are typically synthesized through the reaction of phosgene with 1,2-diols (Lu and Darensbourg, 2012; Poland and Darensbourg, 2017; Qin et al., 2015). The copolymerization of CO2 with epoxides to produce polycarbonates is an alternative synthetic route. Numerous heterogeneous and homogeneous transition-metal catalysts have been developed which selectively form polycarbonates as opposed to the cyclic carbonates described earlier (see the section “Commercial Technologies for the Chemical Utilization of Methane” in Chapter 6) from a range of comonomers including ethylene oxide, propylene oxide, cyclohexene oxide, vinyl oxide, and styrene oxide, among others (Lu and Darensbourg, 2012; Poland and Darensbourg, 2017; Qin et al., 2015). Typically homogeneous catalysts are preferred because they give higher selectivity. Additionally, by changing the nature of the catalyst, polymers can be produced which are either alternating and contain only carbonate groups (one molecule of CO2 followed by one molecule of epoxide) or statistical (often referred to as polyether carbonates) and contain ether linkages that are formed when two ring-opened epoxides are adjacent to each other.
In general, alternating copolymers have low glass transition points, meaning that they will only be used in niche applications such as binders in ceramics and adhesives (Qin et al., 2015). Nevertheless, Empower Materials currently sells poly(ethylenecarbonate) made from ethylene oxide and CO2, and Econic manufactures polycarbonates that contain up to 50 percent CO2 by weight (Quadrelli et al., 2011). A problem which companies such as Novamer are trying to address is the tendency of the polycarbonate to decompose to the cyclic carbonate,
especially when electron-deficient epoxides are used. Both this problem and the tendency of polycarbonates with high CO2 content to react with water can in principle be solved through the judicious use of additives but this still needs to be studied further. At this stage research is also still required to develop catalysts that are highly active with a wide range of epoxides, which may lead to alternating polymers with higher glass transition temperatures. Additionally, further development of strategies to incorporate another monomer like cyclohexene oxide into the copolymer to improve the properties of the polymer should be pursued. The reaction of CO2 with epoxides is highly exothermic so finding catalysts that are thermally stable is also a key issue.
In contrast to the limited applications for alternating copolymers, polyether carbonates derived from epoxides and CO2 are useful for a much wider range of applications (Qin et al., 2015). In particular, they can be used as a component in polyurethanes, and, apart from the commercial application described in the Introduction to this chapter, Novomer and Covestro are both selling polyether carbonates for use in polyurethane synthesis. Nevertheless, despite the relative maturity of this field there is still room for growth. Further understanding of how catalyst structure effects polymer properties would be useful, as this could lead to the development of polymers with tailor-made structures.
Recently research has begun on using epoxide starting materials which are derived from renewable feedstocks such as cyclohexadiene oxide, limonene oxide, and α-pinene oxide, and catalysts have been developed for the copolymerization of CO2 with these substrates (Poland and Darensbourg, 2017). This is a promising area and further research to develop more efficient catalysts should be performed. Additionally, research into the copolymerization of CO2 with aziridines to produce polycarbamates and oxetanes is also only at an early stage and should also be encouraged as it may lead to polymers with different properties than those currently available.
Carboxylic Acid Production
Carboxylic acids comprise a broad class of commodity chemicals that are used as solvents, reagents, and monomers for polymer production, among other applications. With the exception of acetic acid, which is made by methanol carbonylation (Sunley and Watson, 2000), commodity chemical–scale carboxylic acids are typically synthesized by aerobic oxidation of hydrocarbons. Although these processes have been optimized over many years, they generally require highly corrosive conditions that necessitate expensive reactors. Conceptually, inserting CO2 into a C–H bond is an attractive alternative synthesis of carboxylic acids that would eliminate the need for a difficult oxidation step. However, CO2 insertion is thermodynamically disfavored by ~10-18 kcal mol-1 depending on the type of C–H bond (it is both endothermic and entropically disfavored). As a result, catalytic reactions either need to be performed in the presence of base (to drive the reaction forward by carboxylate formation) or
need to be combined with a very effective separation process to remove the carboxylic acid in situ. The vast majority of research focusing on the preparation of carboxylic acids from CO2 insertion has focused on the former. Another major challenge is the kinetic difficulty of activating C–H bonds in the absence of a strong oxidant. Researchers have typically resorted to the use of highly reactive, resource-intensive reagents (e.g., strong organic bases, Lewis acids, and zero-valent metals) to overcome this barrier, which renders these methods impractical. Even though research into the production of carboxylic acids from CO2 is still in an early stage, the large number of potential applications of carboxylic acids motivates further efforts. Below, an assessment is provided about the current state of research toward a number of carboxylic acid targets.
Acrylic and Methacrylic Acid Production
Acrylic acid (5.8 million tons were produced in 2014; Limbach, 2015) and methacrylic acid (3 million tons of methyl methacrylate, the methyl ester of methacrylic acid, were produced in 2007) are large commodity-scale chemicals used in the synthesis of polymers and water superabsorbers (Limbach, 2015). Acrylic acid can also be converted into acrylonitrile, which is a feedstock for carbon fiber synthesis. The current production of acrylic acid and methacrylic acid from petrochemical starting materials leaves open opportunities to use CO2 as a more sustainable feedstock. In principle, acrylic acid and methacrylic acid could be formed directly from CO2 insertion into a C–H bond of ethylene or propylene, respectively (Limbach, 2015). As noted above, the thermodynamics of these processes are unfavorable, which has led to a stepwise approach in which the carboxylate is formed first and subsequently protonated in a second step. Research into the development of catalysts for the synthesis of acrylate (the deprotonated form of acrylic acid) has primarily focused on homogeneous systems and is still at a very preliminary stage. State-of-the-art methods utilize Pd and Ni catalysts, an organic base (amine or alkoxide), and a stoichiometric amount of a Zn0 promoter to convert ethylene and CO2 into acrylate with low turnover frequencies (<0.1 h–1) and turnover numbers less than 1,000 (Hendriksen et al., 2014; Limbach, 2015; Stieber et al., 2015). A viable CO2 route to acrylic acid will require much more development to improve the catalyst performance, eliminate the requirement for Zn0, and engineer a process which can operate without a base or with efficient base recycling.
Furan-2,5-Dicarboxylic Acid Production
Furan-2,5-dicarboxylic acid (FDCA) is a monomer that has attracted strong commercial interest for polyester synthesis (Sousa et al., 2016). In particular, polyethylene furandicarboxylate, the condensation polymer of FDCA and ethylene glycol, has superior gas barrier properties to polyethylene terephthalate, which is produced on a scale of 60 million tons per year (Pang et al., 2016). One potential method for FDCA synthesis involves
edible fructose as a feedstock and requires difficult oxidation and purification steps. The carboxylation of furoic acid is potentially an advantageous route because furoic acid is produced from furfural, which is made industrially from inedible biomass. Recently, researchers have reported that simple alkali carbonate salts (M2CO3; M+ = alkali cation) can promote CO2 insertion into C–H bonds to form carboxylates under solvent-free conditions at elevated temperatures (200°C to 350°C) (Banerjee et al., 2016). This chemistry was used to convert furoic acid and CO2 into FDCA in high yield on a 1 mol scale (Dick et al., 2017). The absence of transition-metal catalysts allows for the use of relatively low-purity CO2 (industrial grade). Nevertheless, using carbonate-promoted carboxylation for commodity chemical synthesis will require substantial development to increase the reaction rates and integrate the process with efficient methods to regenerate carbonate.
Benzoic Acid Production
Benzoic acid is a relatively small-scale commodity chemical (600,000 tons per year) used as an intermediate in phenol synthesis and in the production of preservatives (alkali benzoates), plasticizers (benzoate esters), and solvents. Currently, benzoic acid is synthesized industrially through the aerobic oxidation of toluene. The preparation of benzoic acid by inserting CO2 into a C–H bond of benzene has been investigated as an alternative strategy in a few early stage research efforts. These methods have relied on very energy-intensive stoichiometric reagents to activate benzene, such as Al2(CH3)3(OCH2CH3)3 or a combination of AlCl3 and Al0 (Olah et al., 2002; Suga et al., 2014). While these examples have provided insight into chemical strategies for activating aryl C–H bonds, the CO2 footprint associated with the production of strong Lewis acids far outweighs the CO2 consumed in the benzoic acid synthesis. Further research is needed to uncover strategies for avoiding stoichiometric reagents altogether.
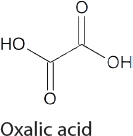
Oxalate and Oxalic Acid Production
Oxalates and oxalic acid, while not commodity chemicals, are still produced at a scale of 120,000 tons per year (Qiao et al., 2014). Their synthesis normally involves the reactions of alcohols with CO and O2 to form diesters of oxalic acid, which can be hydrolyzed with acid to give oxalic acid. This is a relatively cumbersome process. If efficient and economical methods to convert CO2 into oxalic acid are developed, the amount of oxalic acid produced may increase dramatically, as oxalic acid represents a convenient feedstock for the synthesis of a range of chemicals. For example, there are commercial processes for the conversion of dimethyl oxalate into monoethylene glycol. Currently, dimethyl oxalate is made from CO and H2, but the esterification of oxalic acid made from CO2 may represent a more sustainable process. Liquid Light (now owned by Avantium) developed a process for the synthesis of ethylene glycol through the initial electrochemical conversion of CO2 to oxalic
acid (Table 4-2), which is then converted into ethylene glycol using standard chemistry (Twardowski et al., 2016).
Researchers continue to work on more efficient methods to synthesize oxalates and oxalic acid from CO2. In early research (prior to 2000), Pb and other catalysts were used to generate oxalic acid in electrochemical cells with Faradaic efficiencies in the 70-98 percent range, but this was only done at low rates in a standard electrochemical cell (Rudolph et al., 2000; Shoichiro et al., 1987). More recently, both mononuclear and binuclear copper complexes have shown promise for electrochemical reduction of CO2 to oxalic acid (Angamuthu et al., 2010; Pokharel et al., 2014). Nevertheless, further mechanistic understanding of this process is required to lower the overpotential and to improve selectivity.
Fuel (Hydrocarbon) Production
Given that the majority of CO2 that is released in the atmosphere is from the combustion of fossil fuels, the development of methods to synthesize fuels from CO2 could result in a closed carbon cycle, where increases in concentration of CO2 in the atmosphere will be minimal. This can only be achieved if the electricity or H2 that is used to reduce CO2 is generated from carbon-free sources, and if the carbon waste gases created by the combustion of the fuel are recaptured and reutilized. In principle, methane and methanol could be used as fuels and as discussed earlier systems for the conversion of CO2 into these molecules are actively being pursued. Therefore, in this section only the state of technology for the conversion of CO2 into hydrocarbon fuels with more than two carbons will be described.
The Fischer-Tropsch process is used to convert CO and H2 into liquid fuels and has been commercialized on a large scale (Artz et al., 2018). In addition to large-scale units for Fischer-Tropsch, smaller units, which may be better suited for use with waste carbon streams, are also being developed. In an earlier section of this chapter methods for the conversion of CO2 to CO were described. One approach for producing fuels from CO2 could involve initially electrochemically synthesizing CO from CO2 and then in a second thermal step combining the CO with sustainably produced H2 to produce fuels via the conventional Fischer-Tropsch process. Alternatively, a significant amount of research is currently being performed to develop systems that can perform Fischer-Tropsch chemistry starting from CO2 in a single reactor using a single catalyst. In this chemistry the first step is generally the reverse water gas-shift reaction to generate CO from CO2 (Eq 1). This CO then reacts with H2 to form liquid fuels through a mechanism based on the conventional Fischer-Tropsch reaction (Eq 3). Both Sunfire and INERATEC have developed small-scale demonstration plants for CO2-based Fischer-Tropsch chemistry (Table 6-2). However, these rely on water electrolysis to supply H2, which is not economically competitive on a large scale. One of the challenges associated with the Fischer-Tropsch process using CO2 is that there is only a small concentration of CO present during the reaction. This limits chain growth and consequently the product distribu-
tion is normally rich in light hydrocarbons, which are not suitable as liquid fuels. To date, iron-based catalysts, which are active for both the reverse water gas-shift reaction and Fischer-Tropsch chemistry, have been the most extensively explored (Wei et al., 2017). Various transition metal–based promotors have been added to iron-based catalysts and give improved product distributions, but further research to understand the mechanism by which these operate is required. Similarly, supporting catalysts on materials such as SiO2 gives improved performance, but there is relatively little understanding of these effects. Further research into the development of new catalysts and the optimal design of reactors is likely to result in even better systems, given the relatively early stage of research in this field.
The electroreduction or photoreduction of CO2 to chemical fuels has also attracted significant research interest (Ganesh, 2016; Lim et al., 2014; White et al., 2015). Although most commonly CO2 is reduced to C1 feedstocks such as CO, formic acid, methanol, or methane, there are a number of systems that can form products containing new carbon-carbon bonds. In particular copper catalysts are effective for forming products containing C–C bonds such as ethylene (see above), ethane, and higher-order hydrocarbons (Ren et al., 2015a). Apart from selectively forming C2 or higher products over C1 products the other major challenge in electrochemical CO2 reduction to fuels is inhibiting the hydrogen evolution reaction which produces H2 as a by-product. At this stage there is also a lack of mechanistic information about the elementary processes which facilitate C–C bond formation and further research on this topic would likely be beneficial. There is still significant room to design tailor-made catalysts for electrochemical CO2 reduction and even though this problem is extremely challenging it is likely that further advances can be made.
Carbon Nanotube Production
The controlled synthesis of carbon nanotubes is a relatively recent discovery, which has already led to the commercial application of nanotubes in sports gear, such as bicycles and skis, boat hulls, and water filters (De Volder et al., 2013). In fact, as a result of their unusual thermal conductivity, mechanical, and electrical properties, there are many potential applications for carbon nanotubes. However, the preparation of carbon nanotubes is relatively expensive, which limits potential uses, and the current market size is small. Carbon nanotubes are typically produced using CO from fossil feedstocks as the carbon source. Recently, it has been demonstrated that carbon nanotubes can be produced electrochemically from CO2 (Licht et al., 2016; Ren et al., 2015b). The process, which to date has only been performed on a laboratory scale, involves molten carbonate electrolysis in the presence of CO2. Specifically, molten Li2CO3 can be reduced to generate carbon nanotubes and Li2O. The Li2O then reacts with CO2 to regenerate Li2CO3, meaning that CO2 is the ultimate carbon source for the nanotubes. Although carbon nanotubes are a valuable item, at this stage their range of applications is limited compared to conventional carbon fibers. The carbon nanotubes that have been pro-
duced from CO2 to date do not have suitable properties to act as replacements for the types of carbon fibers which are used extensively in the aerospace and automotive industry (Rahaman et al., 2007). However, if methods can be developed to produce more widely useful carbon nanotubes or carbon fibers from CO2, this has the potential to be a disruptive technology.
INTERSECTING RESEARCH CHALLENGES FOR CO2 CONVERSION
The section above describes the status of technology and research related to CO2 conversion separated by product type and identifies some of the challenges that need to be addressed to develop improved systems. A summary of the key technological barriers for different products is provided in Table 4-3. Although in some cases the problems are specific to a certain product, some challenges are common to a number of different products. In this section, some of these common challenges associated with developing improved systems for CO2 conversion are discussed.
Challenges in Catalyst Development
As described above, many catalysts for the conversion of CO2 into valuable chemicals have been developed. Although in some cases there are reasons specific to a particular product for why these catalysts are not commercially viable, in general, catalysts for CO2 conversion lack the required durability and stability. Even in cases where turnover frequencies are high, catalyst decomposition is a problem. This challenge is perhaps even more severe than realized, as most systems tested on a small scale use purified CO2. If these catalysts were utilized with nonpurified CO2 they could decompose even more rapidly. Purifying CO2 increases the cost of a process, and the development of systems that are compatible with CO2 which contains the type of impurities present in flue gas would make commercialization easier. Similarly, the utilization of CO2 from gaseous waste streams would be aided by improving the interface between CO2 capture and conversion. There are relatively few catalysts for CO2 conversion which have been utilized in conjunction with systems for CO2 capture and this could play a key role in developing efficient approaches to utilize CO2 from waste streams.
Challenges for Low-Temperature Electrochemical CO2 + H2O Conversion
Electrochemical CO2 conversion is appealing because it could be powered directly by renewable electricity sources. As noted in Chapter 7, increased deployment of low-carbon electricity generation from solar and wind has, at certain locations and times, led electricity prices to decrease to zero or negative values, indicating a possible future with greater incentive for utilization of renewable electricity. Most of the activity in this area has focused on electrochemical systems in which CO2 reduction to a C1 or C2 chemical intermediate is coupled to
TABLE 4-3 Key barriers for commercialization of the products from Figure 4-1.
| Product | Key Barriers | |
|---|---|---|
| Methanol |
|
|
| Dimethyl ether |
|
|
| Formic acid |
|
|
| Methane |
|
|
| Carbon monoxide |
|
|
| Ethylene and ethanol |
|
|
| Dimethylcarbonate and diphenylcarbonate |
|
|
| Polymers |
|
|
| Acrylic and methacrylic acid |
|
|
| Furan-2,5-dicarboxylic acid |
|
|
| Benzoic acid |
|
|
| Oxalate and oxalic acid |
|
|
| Hydrocarbon fuel |
|
|
| Carbon nanotubes |
|
|
water oxidation to O2 (oxygen evolution reaction). The overall process is effectively the combustion reaction in reverse, with electric power providing the energy input. Although significant progress has been made in this area over the past 10 years, multiple interrelated technical challenges remain—challenges that must be addressed to allow for practical devices.

NOTE: Fundamental research is defined as observation and reporting of fundamental principles of a scientific or engineering process, and formulation of a technology concept. The three proof-of-concept stages are defined as progressively larger-scale reactions to produce product. These include bench-scale processes where critical functions are proved and components or systems are validated in a laboratory environment and at a laboratory scale. Pilot plant scale is defined as a system validated in a relevant environment and at an engineering scale. Demonstration plant scale is defined as a full-scale system demonstrated in a relevant environment. Commercial-limited is defined as an actual system operating at a stage where product is being sold in the market in limited areas with specific advantageous geographical, regulatory, or other factors. Commercial-broad is defined as an actual system operating at a stage where product is sold in the market with opportunities not limited to specifically advantaged locations (see Box 9-1).
Enhancing Conversion per Pass and Avoiding Carbonate Formation
The development of an effective cathode for CO2 reduction is a major obstacle that has captured much of the attention in this area. An ideal cathode will reduce CO2 to a single desired product at a high rate with minimal overpotential and maintain its activity for long periods of continuous operation. Some systems capable of these transformations are described above. High synthesis rates require effective mass transport of CO2 to the catalyst material while maintaining high ionic conductivity. These requirements have been met by using gas diffusion electrodes, which are used in commercial technologies such as fuel cells and
electrolyzers. As summarized in preceding sections of this chapter, electrolysis cells employing gas diffusion electrodes have been used successfully for electroreduction of CO2 to products such as formic acid, CO, and ethylene at potentially commercially relevant rates exceeding 100 mA cm–2. Most of these flow electrolysis systems, however, are operated such that (1) only a modest percentage of CO2 (1-20 percent) is converted to the desired product, that is, a low single-pass conversion, and (2) some of the CO2 is captured by OH– in the electrolyte to form carbonate. A low single-pass conversion implies that more energy will be needed to separate gaseous products from the product stream, and to recycle the unreacted CO2 back into the feed. Second, formation of carbonates represents a net loss of CO2 and causes electrolyte degradation and electrode performance degradation (carbonate precipitation). Given the energy required to obtain CO2 of sufficient purity to begin with, low CO2 utilization per pass and loss of CO2 in the electrolyte imposes additional energy penalties that are typically not accounted for in system analyses. In fact most experimental work reported does not explicitly acknowledge, let alone quantify, these parasitic losses. Achieving practical electrochemical systems for conversion of CO2 to valuable intermediates such as CO and ethylene requires the development of systems that achieve high single-pass conversion of CO2 to the desired product with minimal loss of CO2 into the electrolyte.
Energy Requirements of the Anode
Another key factor to make the overall electrolysis process (more) economically feasible is to enhance energy efficiency and minimize overall energy requirements in order. Some gains can still be made by reducing the overpotential of the various cathode catalysts. However, those potential gains are small compared to the energy requirements of an overall electrolysis process that typically couples the CO2 electroreduction at the cathode to the highly energy intense oxygen evolution reaction (OER) at the anode. An analysis of the energy requirements based on only Gibbs free energies shows that about 90 percent of the energy needed to drive the CO2 electrolysis process is required for OER at the anode, to form O2, for which there is very limited commercial value at scale. Indeed efforts have started to identify alternative anode reactions that involve the oxidation of readily available chemicals. For example, glycerol (a by-product of biodiesel production), glucose, and even methane, for which the thermodynamic potentials are 0.8-1.1 V lower than the minimum potential needed for the OER, have potential in this regard. In fact, it has been demonstrated that glycerol oxidation can lead to a 37-50 percent reduction in the energy requirement for the overall CO2 electrolysis process, and with the cost of electricity often the prime cost-determining factor (Jouny et al., 2018), this significantly changes the likelihood of achieving economic viability. Of course, no chemical is as abundantly available as water for the OER; however, vast amounts of (waste) chemicals like glycerol are available in various places around the world, for example, in Brazil’s
biofuel production. In those locations CO2 electrolysis to CO, ethylene, and/or ethanol has the potential to become a viable technology when coupled with glycerol oxidation.
A RESEARCH AGENDA FOR CHEMICAL UTILIZATION OF CARBON DIOXIDE
Stages of development and key barriers for various chemical utilization approaches are shown in Figure 4-1 and Table 4-3.
Priority Research Areas
In previous sections of this chapter specific challenges related to converting CO2 to certain products, general challenges related to CO2 conversion regardless of the product, and the technological barriers to commercialization have been described. Based on the findings of these sections, a general list of scientific research needs and opportunities related to the conversion of CO2 to commodity chemicals and fuels is provided below.
Chemical Catalysis
Both in the short and long term there is a need for improved catalysts for CO2 conversion. Depending on the application, this could include homogeneous, heterogeneous, and electro- and photochemical systems. Where possible, an emphasis placed on developing catalysts that use sustainable raw materials would be beneficial. Additionally, catalyst stability and durability need to be improved for many CO2 conversion technologies.
Avoiding Stoichiometric Additives
Many current processes for CO2 conversion, especially in academic settings, use stoichiometric amounts of additives (such as base). Although these additives are sometimes required for thermodynamic reasons, they limit atom economy, generate waste products, and increase the net carbon footprint of the process. Research focusing on developing systems which can use catalytic quantities of additives instead of stoichiometric amounts or removing additives altogether through the use of reactors that allow reactions to be performed at low conversion should be encouraged.
Integrating Catalysis and Reactor Design
When using waste CO2 as a feedstock, catalysts need to be compatible with the impurities present in the waste stream and able to operate with the concentration of CO2 in the waste stream, or the waste gas needs to be concentrated and/or purified. Research into interfacing systems for CO2 conversion with capture technology is required. This includes both chemical and reactor design considerations. Alternatively, the development of catalysts that can react directly with captured CO2 (for example, CO2 which has been captured using an amine) should be explored further.
Pathways to New Products
Research exploring CO2 conversion into commodity chemicals has traditionally focused on a relatively small number of molecules, as outlined in this chapter. Most of these tend to be C1 products. Recent efforts to make molecules such as ethylene or materials such as carbon nanotubes from CO2 represent departures from the norm. The identification of other nontraditional targets and subsequent catalyst development could have transformative impacts.
Coupling Oxidation and Reduction Reactions
The focus of most CO2 electroreduction efforts has been on improving the cathode in its efficiency and activity in converting CO2 to a desired product. Typically the overall process requires stoichiometric amounts of water for the oxygen evolution reaction on the anode, with that reaction requiring more than 85 percent of the total electrical energy input. Though O2 will in some cases be a valuable co-product, at scale, the amount of produced oxygen is likely to vastly exceed need, and thus the produced oxygen would be released. Converting some other feed into a valuable product at the anode, which at the same time would reduce the anode’s energy requirement, would significantly improve the overall economic feasibility and life-cycle assessment of CO2 electrolysis.
System Engineering and Reactor Design
Improving the efficiency of CO2 conversion chemistry would be aided by the integration of catalysts with the most efficient reactor technology. For example, the integration of CO2 conversion catalysts with reactors that allow for the efficient removal of products that must be formed at low conversion for thermodynamically limited reactions would be beneficial. Similarly, separation challenges can be mitigated by developing reactor designs that improve conversion per pass. In electrochemical CO2 conversion, developing improved membrane-based electrolyzers instead of liquid electrolyte-based electrolyzers should be a goal. Furthermore, integrating catalyst into durable electrode configurations, while still maintaining catalyst activity, will need further attention. To achieve these important objectives chemists and engineers should be encouraged to collaborate to jointly solve problems and develop new processes.
FINDINGS, CONCLUSION, AND RECOMMENDATIONS
Finding 4-1 At present, there are very few industrial processes that convert CO2 into fuels or chemicals.
Finding 4-2 Most of the research and development of CO2 utilization processes has targeted C1 compounds (methane, CO, methanol, and formic acid) with a few prominent exceptions such as organic carbonates and polymers. Processes that
produce compounds with C–C bonds have received less attention and are at an earlier stage of development.
Finding 4-3 Catalyst performance is a limiting factor in many CO2 conversion processes. There is a substantial need for improved heterogeneous, homogeneous, and (photo)electrochemical catalysts. Major challenges include minimizing the energy input required for CO2 conversion and improving catalyst selectivity, stability, and tolerance to common impurities in waste CO2 streams.
Finding 4-4 Research on catalysts for CO2 conversion is often conducted using reactors and reaction conditions (e.g., single-pass conversion) that are not suitable for the economic large-scale synthesis of chemicals or fuels at reasonable rates. The factors that affect catalyst performance in simplified laboratory reactors may differ from those that affect performance in a reactor engineered for larger-scale and higher rates.
Finding 4-5 Many CO2 conversion processes that have been investigated require stoichiometric additives that cannot readily be regenerated. This requirement likely precludes commodity chemical or fuel synthesis because such additives can have an economic value or energy demand that exceeds that of the chemical or fuel product.
Finding 4-6 In electrochemical CO2 conversion, water oxidation to O2 is typically used as the anodic reaction to supply the protons and electrons for CO2 reduction. Although water is by far the most scalable substrate, processes that use alternative oxidation substrates (e.g., those derived from agricultural waste or industrial byproducts) could allow for CO2 conversion with lower energy demand and generate a more valuable co-product than O2.
Conclusion 4-1 The grand challenge for converting CO2 waste streams into useful products is to develop processes that require minimal amounts of nonrenewable energy, are economically competitive, and provide substantial reductions in greenhouse gas emissions compared to existing technology.
Recommendation 4-1 Researchers should continue research efforts to improve existing catalysts or discover entirely new catalysts to advance many CO2 conversion processes to industrial viability.
Recommendation 4-2 Researchers should focus on CO2 conversion processes that avoid stoichiometric additives or use additives that are easily regenerated for developing processes that could be used for commodity chemical or fuel production.
Recommendation 4-3 Researchers should integrate catalysis research with reactor design at an early stage to accelerate the development of CO2 conversion processes by rapidly identifying the factors that affect catalyst performance on larger scales at synthetically relevant rates.
Recommendation 4-4 Researchers should increase attention to CO2 conversion processes that produce nontraditional targets, especially those with C–C bonds, to have transformative impacts.
Recommendation 4-5 Researchers should explore processes that combine CO2 reduction with the oxidation of substrates from other waste streams (e.g., agricultural or biomass waste or industrial by-products) to open new pathways that would reduce the cost of CO2 conversion and create multiple high-value products.
Recommendation 4-6 Researchers should develop reactor technologies that are tailored to the demands of carbon dioxide conversion processes, including systems that integrate capture with conversion.
REFERENCES
Alper, E., and O. Yuksel-Orhan. 2017. CO2 utilization: Developments in conversion processes. Petroleum 3(1):109-126. doi: 10.1016/j.petlm.2016.11.00351T.
Álvarez, A., A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon, and F. Kapteijn. 2017. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chemical Reviews 117(14):9804-9838. doi: 10.1021/acs.chemrev.6b00816.
Angamuthu, R., P. Byers, M. Lutz, A. L. Spek, and E. Bouwman. 2010. Electrocatalytic CO2 conversion to oxalate by a copper complex. Science 327(5963):313-315. doi: 10.1126/science.1177981.
Artz, J., T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow, and W. Leitner. 2018. Sustainable conversion of carbon dioxide: An integrated review of catalysis and life cycle assessment. Chemical Reviews 118:434-504. doi: 10.1021/acs.chemrev.7b00435.
Banerjee, A., G. R. Dick, T. Yoshino, and M. W. Kanan. 2016. Carbon dioxide utilization via carbonate-promoted C–H carboxylation. Nature 531:215. doi: 10.1038/nature17185.
Costentin, C., M. Robert, and J.-M. Saveant. 2013. Catalysis of the electrochemical reduction of carbon dioxide. Chemical Society Reviews 42(6):2423-2436. doi: 10.1039/C2CS35360A.
De Volder, M. F. L., S. H. Tawfick, R. H. Baughman, and A. J. Hart. 2013. Carbon nanotubes: Present and future commercial applications. Science 339(6119):535-539. doi: 10.1126/science.1222453.
Dick, G. R., A. D. Frankhouser, A. Banerjee, and M. W. Kanan. 2017. A scalable carboxylation route to furan-2,5-dicarboxylic acid. Green Chemistry 19(13):2966-2972. doi: 10.1039/C7GC01059A.
Dinh, C.-T., T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton, and E. H. Sargent. 2018. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360(6390):783-787. doi: 10.1126/science.aas9100.
Ebbesen, S. D., and M. Mogensen. 2009. Electrolysis of carbon dioxide in solid oxide electrolysis cells. Journal of Power Sources 193(1):349-358. doi: 10.1016/j.jpowsour.2009.02.093.
Ebbesen, S. D., S. H. Jensen, A. Hauch, and M. B. Mogensen. 2014. High temperature electrolysis in alkaline cells, solid proton conducting cells, and solid oxide cells. Chemical Reviews 114(21):10697-10734. doi: 10.1021/cr5000865.
EIA (U.S. Energy Information Administration). 2017. International Energy Outlook 2017. September 14, 2017. Available at www.eia.gov/ieo.
Fukuoka, S., M. Tojo, H. Hachiya, M. Aminaka, and K. Hasegawa. 2007. Green and sustainable chemistry in practice: Development and industrialization of a novel process for polycarbonate production from CO2 without using phosgene. Polymer Journal 39:91. doi: 10.1295/polymj.PJ2006140.
Ganesh, I. 2014. Conversion of carbon dioxide into methanol—a potential liquid fuel: Fundamental challenges and opportunities (a review). Renewable and Sustainable Energy Reviews 31(Supplement C):221-257. doi: 10.1016/j.rser.2013.11.045.
Ganesh, I. 2016. Electrochemical conversion of carbon dioxide into renewable fuel chemicals—The role of nanomaterials and the commercialization. Renewable and Sustainable Energy Reviews 59(Supplement C):1269-1297. doi: 10.1016/j.rser.2016.01.026.
Gong, J., X. Ma, and S. Wang. 2007. Phosgene-free approaches to catalytic synthesis of diphenyl carbonate and its intermediates. Applied Catalysis A: General 316(1):1-21. doi: 10.1016/j.apcata.2006.09.006.
Haas, T., R. Krause, R. Weber, M. Demler, and G. Schmid. 2018. Technical photosynthesis involving CO2 electrolysis and fermentation. Nature Catalysis 1(1):32-39. doi: 10.1038/s41929-017-0005-1.
Hendriksen, C., E. A. Pidko, G. Yang, B. Schäffner, and D. Vogt. 2014. Catalytic formation of acrylate from carbon dioxide and ethene. Chemistry—A European Journal 20(38):12037-12040. doi: 10.1002/chem.201404082.
Hoang, T. T. H., S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis, and A. A. Gewirth. 2018. Nanoporous copper–silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to ethylene and ethanol. Journal of the American Chemical Society 140(17):5791-5797. doi: 10.1021/jacs.8b01868.
Jadhav, S. G., P. D. Vaidya, B. M. Bhanage, and J. B. Joshi. 2014. Catalytic carbon dioxide hydrogenation to methanol: A review of recent studies. Chemical Engineering Research and Design 92(11):2557-2567. doi: 10.1016/j.cherd.2014.03.005.
Jeanty, P., C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen, and M. Fleischer. 2018. Upscaling and continuous operation of electrochemical CO2 to CO conversion in aqueous solutions on silver gas diffusion electrodes. Journal of CO2 Utilization 24:454-462. doi: 10.1016/j.jcou.2018.01.011.
Jiang, B., X.-G. Zhang, K. Jiang, D.-Y. Wu, and W.-B. Cai. 2018. Boosting formate production in electrocatalytic CO2 reduction over wide potential window on Pd surfaces. Journal of the American Chemical Society 140(8):2880-2889. doi: 10.1021/jacs.7b12506.
Jouny, M., W. Luc, and F. Jiao. 2018. General techno-economic analysis of CO2 electrolysis systems. Industrial & Engineering Chemistry Research 57(6):2165-2177. doi: 10.1021/acs.iecr.7b03514.
Keim, W. 1989. Carbon monoxide: Feestock for chemicals, present and future. Journal of Organometallic Chemistry 372(1):15-23.
Klankermayer, J., S. Wesselbaum, K. Beydoun, and W. Leitner. 2016. Selective catalytic synthesis using the combination of carbon dioxide and hydrogen: Catalytic chess at the interface of energy and chemistry. Angewandte Chemie International Edition 55(26):7296-7343. doi: 10.1002/anie.201507458.
Kumar, B., M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin, and A. Salehi-Khojin. 2013. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nature Communications 4:2819. doi: 10.1038/ncomms3819.
Kumar, B., V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara, and J. M. Spurgeon. 2017. Reduced SnO2 porous nanowires with a high density of grain boundaries as catalysts for efficient electrochemical CO2 into HCOOH conversion. Angewandte Chemie International Edition 56(13):3645-3649. doi: 10.1002/anie.201612194.
Kutz, R. B., Q. Chen, H. Yang, S. D. Sajjad, Z. Liu, and M. I. Richard. 2017. Sustainion imidazolium functionalized polymers for carbon dioxide electrolysis. Energy Technology 5(6):929-936. doi: 10.1002/ente.201600636.
Langanke, J., A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner, and C. Gurtler. 2014. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chemistry 16(4):1865-1870. doi: 10.1039/C3GC41788C.
Licht, S., A. Douglas, J. Ren, R. Carter, M. Lefler, and C. L. Pint. 2016. Carbon nanotubes produced from ambient carbon dioxide for environmentally sustainable lithium-ion and sodium-ion battery anodes. ACS Central Science 2(3):162-168. doi: 10.1021/acscentsci.5b00400.
Lim, R. J., M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang, and K. H. Lim. 2014. A review on the electrochemical reduction of CO2 in fuel cells, metal electrodes and molecular catalysts. Catalysis Today 233(Sup-plement C):169-180. doi: 10.1016/j.cattod.2013.11.037.
Limbach, M. 2015. Chapter Four: Acrylates from alkenes and CO2, the stuff that dreams are made of. In Advances in Organometallic Chemistry, edited by Pedro J. Pérez. New York: Academic Press, pp. 175-202.
Lindsey, A. S., and H. Jeskey. 1957. The Kolbe-Schmitt reaction. Chemical Reviews 57(4):583-620.
Liu, Z., R. I. Masel, Q. Chen, R. Kutz, H. Yang, K. Lewinski, M. Kaplun, S. Luopa, and D. R. Lutz. 2016. Electrochemical generation of syngas from water and carbon dioxide at industrially important rates. Journal of CO2 Utilization 15:50-56. doi: 10.1016/j.jcou.2016.04.011.
Lu, X.-B., and D. J. Darensbourg. 2012. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chemical Society Reviews 41(4):1462-1484. doi: 10.1039/C1CS15142H.
Ma, J., N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei, and Y. Sun. 2009. A short review of catalysis for CO2 conversion. Catalysis Today 148(3):221-231. doi: 10.1016/j.cattod.2009.08.015.
Manthiram, K., B. J. Beberwyck, and A. P. Alivisatos. 2014. Enhanced electrochemical methanation of carbon dioxide with a dispersible nanoscale copper catalyst. Journal of the American Chemical Society 136(38):13319-13325. doi: 10.1021/ja5065284.
Martin, O., A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré, and J. Pérez-Ramírez. 2016. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angewandte Chemie International Edition 55(21):6261-6265. doi: 10.1002/anie.201600943.
Min, X., and M. W. Kanan. 2015. Pd-catalyzed electrohydrogenation of carbon dioxide to formate: High mass activity at low overpotential and identification of the deactivation pathway. Journal of the American Chemical Society 137(14):4701-4708. doi: 10.1021/ja511890h.
Mistry, H., Y.-W. Choi, A. Bagger, F. Scholten, C. S. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon, K. Kisslinger, E. A. Stach, J. C. Yang, J. Rossmeisl, and B. Roldan Cuenya. 2017. Enhanced carbon dioxide electroreduction to carbon monoxide over defect-rich plasma-activated silver catalysts. Angewandte Chemie International Edition 56(38):11394-11398. doi: 10.1002/anie.201704613.
Mittal, C., C. Madsbjerg, and P. Blennow. 2017. Small-scale CO from CO2 using electrolysis. Chemical Engineering World 52(3):44-46.
Neubauer, S., R. K. Krause, B. Schmid, D. M. Guldi, and G. Schmid. 2016. Overpotentials and Faraday efficiencies in CO2 electrocatalysis–the impact of 1-ethyl-3-methylimidazolium trifluoromethanesulfonate. Advanced Energy Materials 6(9):1502231. doi: 10.1002/aenm.201502231.
Olah, G. A. 2005. Beyond oil and gas: The methanol economy. Angewandte Chemie International Edition 44(18):2636-2639. doi: 10.1002/anie.200462121.
Olah, G. A., B. Török, J. P. Joschek, I. Bucsi, P. M. Esteves, G. Rasul, and G. K. Surya Prakash. 2002. Efficient chemoselective carboxylation of aromatics to arylcarboxylic acids with a superelectrophilically activated carbon dioxide−Al2Cl6/Al system. Journal of the American Chemical Society 124(38):11379-11391. doi: 10.1021/ja020787o.
Pang, J., M. Zheng, R. Sun, A. Wang, X. Wang, and T. Zhang. 2016. Synthesis of ethylene glycol and terephthalic acid from biomass for producing PET. Green Chemistry 18(2):342-359. doi: 10.1039/C5GC01771H.
Patricio, J., A. Angelis-Dimakis, A. Castillo-Castillo, Y. Kalmykova, and L. Rosado. 2017. Method to identify opportunities for CCU at regional level—Matching sources and receivers. Journal of CO2 Utilization 22(Supplement C):330-345. doi: 10.1016/j.jcou.2017.10.009.
Pérez-Fortes, M., J. C. Schöneberger, A. Boulamanti, G. Harrison, and E. Tzimas. 2016a. Formic acid synthesis using CO2 as raw material: Techno-economic and environmental evaluation and market potential. International Journal of Hydrogen Energy 41(37):16444-16462. doi: 10.1016/j.ijhydene.2016.05.199.
Pérez-Fortes, M., J. C. Schöneberger, A. Boulamanti, and E. Tzimas. 2016b. Methanol synthesis using captured CO2 as raw material: Techno-economic and environmental assessment. Applied Energy 161(Supplement C):718-732. doi: 10.1016/j.apenergy.2015.07.067.
Pokharel, U. R., F. R. Fronczek, and A. W. Maverick. 2014. Reduction of carbon dioxide to oxalate by a binuclear copper complex. Nature Communications 5:5883. doi: 10.1038/ncomms6883. Supplementary information available at https://www.nature.com/articles/ncomms6883#supplementary-information.
Poland, S. J., and D. J. Darensbourg. 2017. A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes. Green Chemistry 19(21):4990-5011. doi: 10.1039/C7GC02560B.
Qiao, J., Y. Liu, F. Hong, and J. Zhang. 2014. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chemical Society Reviews 43(2):631-675. doi: 10.1039/C3CS60323G.
Qin, Y., X. Sheng, S. Liu, G. Ren, X. Wang, and F. Wang. 2015. Recent advances in carbon dioxide based copolymers. Journal of CO2 Utilization 11(Supplement C):3-9. doi: 10.1016/j.jcou.2014.10.003.
Qiu, Y.-L., H.-X. Zhong, T.-T. Zhang, W.-B. Xu, X.-F. Li, and H.-M. Zhang. 2017. Copper electrode fabricated via pulse electrodeposition: Toward high methane selectivity and activity for CO2 electroreduction. ACS Catalysis 7(9):6302-6310. doi: 10.1021/acscatal.7b00571.
Quadrelli, E. A., G. Centi, J.-L. Duplan, and S. Perathoner. 2011. Carbon dioxide recycling: Emerging large-scale technologies with industrial potential. ChemSusChem 4(9):1194-1215. doi: 10.1002/cssc.201100473.
Rahaman, M. S. A., A. F. Ismail, and A. Mustafa. 2007. A review of heat treatment on polyacrylonitrile fiber. Polymer Degradation and Stability 92(8):1421-1432. doi: 10.1016/j.polymdegradstab.2007.03.023.
Ren, D., Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi, and B. S. Yeo. 2015a. Selective electrochemical reduction of carbon dioxide to ethylene and ethanol on copper(I) oxide catalysts. ACS Catalysis 5(5):2814-2821. doi: 10.1021/cs502128q.
Ren, J., F.-F. Li, J. Lau, L. González-Urbina, and S. Licht. 2015b. One-pot synthesis of carbon nanofibers from CO2. Nano Letters 15(9):6142-6148. doi: 10.1021/acs.nanolett.5b02427.
Rudolph, M., S. Dautz, and E.-G. Jäger. 2000. Macrocyclic [N4P2-P] coordinated nickel complexes as catalysts for the formation of oxalate by electrochemical reduction of carbon dioxide. Journal of the American Chemical Society 122(44):10821-10830. doi: 10.1021/ja001254n.
Saeidi, S., N. A. S. Amin, and M. R. Rahimpour. 2014. Hydrogenation of CO2 to value-added products—a review and potential future developments. Journal of CO2 Utilization 5:66-81.
Sakakura, T., J.-C. Choi, and H. Yasuda. 2007. Transformation of carbon dioxide. Chemical Reviews 107(6):2365-2387. doi: 10.1021/cr068357u.
Satthawong, R., N. Koizumi, C. Song, and P. Prasassarakich. 2015. Light olefin synthesis from CO2 hydrogenation over K-promoted Fe–Co bimetallic catalysts. Catalysis Today 251:34-40. doi: 10.1016/j.cattod.2015.01.011.
Shoichiro, I., T. Takehiko, and I. Kaname. 1987. Selective formation of formic acid, oxalic acid, and carbon monoxide by electrochemical reduction of carbon dioxide. Bulletin of the Chemical Society of Japan 60(7):2517-2522. doi: 10.1246/bcsj.60.2517.
Shukla, K., and V. C. Srivastava. 2017. Synthesis of organic carbonates from alcoholysis of urea: A review. Catalysis Reviews 59(1):1-43. doi: 10.1080/01614940.2016.1263088.
Sordakis, K., C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller, and G. Laurenczy. 2018. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chemical Reviews 118(2):372-433. doi: 10.1021/acs.chemrev.7b00182.
Sousa, A. F., A. C. Fonseca, A. C. Serra, C. S. R. Freire, A. J. D. Silvestre, and J. F. J. Coelho. 2016. New unsaturated copolyesters based on 2,5-furandicarboxylic acid and their crosslinked derivatives. Polymer Chemistry 7(5):1049-1058. doi: 10.1039/C5PY01702E.
Spurgeon, J. M., and B. Kumar. 2018. A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products. Energy & Environmental Science. doi: 10.1039/C8EE00097B.
Stalder, C. J., S. Chao, and M. S. Wrighton. 1984. Electrochemical reduction of aqueous bicarbonate to formate with high current efficiency near the thermodynamic potential at chemically derivatized electrodes. Journal of the American Chemical Society 106(12):3673-3675. doi: 10.1021/ja00324a046.
Stieber, S. C. E., N. Huguet, T. Kageyama, I. Jevtovikj, P. Ariyananda, A. Gordillo, S. A. Schunk, F. Rominger, P. Hofmann, and M. Limbach. 2015. Acrylate formation from CO2 and ethylene: Catalysis with palladium and mechanistic insight. Chemical Communications 51(54):10907-10909. doi: 10.1039/C5CC01932J.
Studt, F., I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff, and J. K. Nørskov. 2014. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nature Chemistry 6:320. doi: 10.1038/nchem.1873. Supplemental information available at https://www.nature.com/articles/nchem.1873#supplementary-information.
Su, X., J. Xu, B. Liang, H. Duan, B. Hou, and Y. Huang. 2016. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. Journal of Energy Chemistry 25(4):553-565. doi: 10.1016/j.jechem.2016.03.009.
Suga, T., H. Mizuno, J. Takaya, and N. Iwasawa. 2014. Direct carboxylation of simple arenes with CO2 through a rhodium-catalyzed C-H bond activation. Chemical Communications 50(92):14360-14363. doi: 10.1039/C4CC06188H.
Sun, X., X. Kang, Q. Zhu, J. Ma, G. Yang, Z. Liu, and B. Han. 2016a. Very highly efficient reduction of CO2 to CH4 using metal-free N-doped carbon electrodes. Chemical Science 7(4):2883-2887. doi: 10.1039/C5SC04158A.
Sun, X., Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang, and B. Han. 2016b. Molybdenum-bismuth bimetallic chalcogenide nanosheets for highly efficient electrocatalytic reduction of carbon dioxide to methanol. Angewandte Chemie International Edition 55(23):6771-6775. doi: 10.1002/anie.201603034.
Sunley, G. J., and D. J. Watson. 2000. High productivity methanol carbonylation catalysis using iridium: The Cativa™ process for the manufacture of acetic acid. Catalysis Today 58(4):293-307.
Tamboli, A. H., A. A. Chaugule, and H. Kim. 2017. Catalytic developments in the direct dimethyl carbonate synthesis from carbon dioxide and methanol. Chemical Engineering Journal 323:530-544. doi: 10.1016/j. cej.2017.04.112.
Topham, S., A. Bazzanella, S. Schiebahn, S. Luhr, L. Zhao, A. Otto, and D. Stolten. 2014. Carbon dioxide. In Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi: 10.1002/14356007.a05_165. pub2.
Twardowski, Z., E. B. Cole, J. J. Kaczur, K. Teamey, K. A. Keets, R. Parajuli, A. Bauer, N. Sivasankar, G. Leonard, and T. J. Kramer. 2016. Method and system for production of oxalic acid and oxalic acid reduction products. Google Patents. EP2935654A4.
Verma, S., X. Lu, S. Ma, R. I. Masel, and P. J. A. Kenis. 2016. The effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodes. Physical Chemistry Chemical Physics 18(10):7075-7084. doi: 10.1039/C5CP05665A.
Verma, S., Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima, and P. J. A. Kenis. 2018. Insights into the low overpotential electroreduction of CO2 to CO on a supported gold catalyst in an alkaline flow electrolyzer. ACS Energy Letters 3(1):193-198. doi: 10.1021/acsenergylett.7b01096.
Wang, J., Z. You, Q. Zhang, W. Deng, and Y. Wang. 2013. Synthesis of lower olefins by hydrogenation of carbon dioxide over supported iron catalysts. Catalysis Today 215:186-193. doi: 10.1016/j.cattod.2013.03.031.
Wang, W., S. Wang, X. Ma, and J. Gong. 2011. Recent advances in catalytic hydrogenation of carbon dioxide. Chemical Society Reviews 40(7):3703-3727. doi: 10.1039/C1CS15008A.
Wang, W.-H., Y. Himeda, J. T. Muckerman, G. F. Manbeck, and E. Fujita. 2015. CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chemical Reviews 115(23):12936-12973. doi: 10.1021/acs.chemrev.5b00197.
Wei, J., Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu, and J. Sun. 2017. Directly converting CO2 into a gasoline fuel. Nature Communications 8:15174. doi: 10.1038/ncomms15174. Supplementary information available at https://www.nature.com/articles/ncomms15174#supplementary-information.
Whipple, D. T., E. C. Finke, and P. J. A. Kenis. 2010. Microfluidic reactor for the electrochemical reduction of carbon dioxide: The effect of pH. Electrochemical and Solid-State Letters 13(9):B109-B111. doi: 10.1149/1.3456590.
White, J. L., M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev, and A. B. Bocarsly. 2015. Light-driven heterogeneous reduction of carbon dioxide: Photocatalysts and photoelectrodes. Chemical Reviews 115(23):12888-12935. doi: 10.1021/acs. chemrev.5b00370.
Yang, H., J. J. Kaczur, S. D. Sajjad, and R. I. Masel. 2017. CO2 conversion to formic acid in a three compartment cell with Sustainion™ membranes. ECS Transactions 77(11):1425-1431. doi: 10.1149/07711.1425ecst.
Yanming, L., Z. Yujing, C. Kai, Q. Xie, F. Xinfei, S. Yan, C. Shuo, Z. Huimin, Z. Yaobin, Y. Hongtao, and M. R. Hoffmann. 2017. Selective electrochemical reduction of carbon dioxide to ethanol on a boron- and nitrogen-co-doped nanodiamond. Angewandte Chemie International Edition 56(49):15607-15611. doi:10.1002/anie.201706311.
Zhang, L., S. Hu, X. Zhu, and W. Yang. 2017. Electrochemical reduction of CO2 in solid oxide electrolysis cells. Journal of Energy Chemistry 26(4):593-601. doi: 10.1016/j.jechem.2017.04.004.
Zhang, S., P. Kang, and T. J. Meyer. 2014. Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. Journal of the American Chemical Society 136(5):1734-1737. doi: 10.1021/ja4113885.
Zhou, X., R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis, and C. Xiang. 2016. Solar-driven reduction of 1 atm of CO2 to formate at 10% energy-conversion efficiency by use of a TiO2-protected III–V tandem photoanode in conjunction with a bipolar membrane and a Pd/C cathode. ACS Energy Letters 1(4):764-770. doi: 10.1021/acsenergylett.6b00317.
This page intentionally left blank.


































