
5
Marmoset Genomics and Genetic Diversity
Maintaining genetic variation in captive animal colonies helps to ensure survival of the colony and enables useful models of disease. Optimizing genome assembly and annotation is the first step in this process. Speakers discussed contributions to enhance understanding of the marmoset genome, measures to ensure the continuation of genetic diversity, and ways this knowledge can be utilized for clinical research applications.
CHARACTERIZING THE MARMOSET GENOME
Jeffrey Rogers and Ricardo del Rosario presented some of the latest developments in marmoset genome assembly, annotation, and identification. Rogers is an associate professor at the Human Genome Sequencing Center at the Baylor College of Medicine and core scientist at the WNPRC. del Rosario is a computational biologist at the Broad Institute.
Creating a Complete Genome Assembly
Ideally, using any animal as a model organism requires a complete DNA sequence, with little to no missing information. To date, different marmoset genome assemblies have had varying levels of success analyzing the genome (e.g., Sato et al. 2015), which was published in 2014 by the Marmoset Genome Sequencing and Analysis Consortium. del Rosario developed a new genome assembly that is more contiguous than previously
available. In doing so, he made strides toward creating a complete marmoset reference assembly for genomic analysis (ASM275486v11).
In genome assemblies, sequences are aligned to each other, and regions that overlap, known as contigs, are identified. The overlapping sequences end at repetitive or hard to sequence parts of the genome. Scaffolding, which involves pairing contigs with chromosomes, produces gaps in transcription. del Rosario’s new assembly maps external sets of contigs to the assembly to fill in some of the gaps.
Researchers use a number of measurements to evaluate the gaps in genome assemblies. One such reference for assembly completion is Contig N50. This calculation quantifies the length of continuous DNA sequence where half of the reference assembly is in pieces larger than or equal to Contig N50. Thus, a larger Contig N50 value translates to a more continuous reference assembly with fewer gaps. Using this evaluation and others for comparison, del Rosario’s new assembly has fewer gaps than with previous methods: 9 percent to the previous 34 percent. This assembly brings researchers closer to achieving a continuous Callithrix jacchus genome.
Genome Annotation
Once the genome assembly is complete, researchers need to identify the protein coding genes, microRNAs, and non-coding RNAs to enable functional analysis of the genome. Identifying genes that are expressed and then mapping RNA sequencing back to the reference assembly provides the locations of these coding genes. This process cannot be done in one iteration; additional RNA sequencing and replicating annotations are necessary steps to improve accuracy.
As part of the Nonhuman Primate Reference Transcriptome Resource project, researchers sequenced RNA samples from 10 different types of tissues. del Rosario used a marmoset sample from this project to create the new assembly. More than 19,000 protein coding genes and nearly 9,000 non-coding genes have been thus far annotated.
Identifying Functionally Significant Variation
Genomic sequencing is needed to identify functional variation in study colonies so that animals can be selected for specific experiments based on this information (e.g., knowing the genes involved in genetic or physiological pathways of interest). This reduces the amount of variation in a sample of animals and facilitates interpretation of research results when genetics
___________________
1 ASM275486v1, available at: https://www.ncbi.nlm.nih.gov/assembly/GCA_002754865.1.
may play a role. Furthermore, identifying functional variation informs potential gene editing experiments and subsequent breeding strategies.
del Rosario sequenced marmoset genomes using DNA from cultured fibroblasts to identify genetic variation. From 19 marmoset genomes (out of a group of 80 animals), he identified 15 million single nucleotide polymorphisms (SNPs). After frequency filtering and collapsing using linkage disequilibrium to ensure a high level of confidence in each, 167,000 SNPs remained. These reliable SNPs provide a starting point for creating a reference genotype chip. Acquiring genome sequences from other colonies of marmosets will be necessary to further develop the chip design.
Additionally, analyzing hetero- and homozygosity of SNP locations imparts useful information about genetic diversity in marmosets. Heterozygosity counts nucleotides within an individual that differ between the chromosomes inherited from the parents. By contrast, runs of homozygosity are contiguous lengths of inherited homozygous genotypes, often encountered in inbred animals. Current colonies of marmosets have greater heterozygosity than humans, a fact that researchers hope to maintain with careful breeding strategies to ensure maximal genetic variation. Obtaining better information about genetic structure would be critical to identify not only the genetic composition of members of colonies (intra) but differences between colonies (inter) as well as differences between captive and wild colonies.
Genetic Variation and Disease Models
Rhesus monkeys have greater genetic variation per individual than humans and extensive genetic variation between individuals (Xue et al. 2016). Rogers used examples from his research on the functionally significant genetic variation in these animals to show the phenotypic relevance of some of those findings to human diseases. For instance, a macaque model of colon cancer developed at the MD Anderson Keeling Center for Comparative Medicine and Research revealed clinical characteristics and pathologies similar to those seen in human patients affected by Lynch syndrome. Lynch syndrome, a hereditary form of colon cancer, is found in humans with mutations in DNA-mismatch-repair genes. These prevent the repair of DNA sequences and subsequently increase risk of cancer. Because this mutation is also observed in macaques, researchers are able to use macaques as a genetically valid model for studying this disease (Dray et al. 2018). Through collaborations with colleagues at the Baylor College of Medicine and the California National Primate Research Center, Rogers has been studying naturally occurring, spontaneous retinal diseases in macaques. After the team identified the genetic variation and damaging mutations at the root of retinal disease they further studied two juvenile macaques exhibiting partial
visual impairment. An ophthalmic exam confirmed cone dystrophy in the animals and whole genome sequencing revealed that both animals were homozygous for a missense mutation (Moshiri et al. 2019).
Applying these research strategies to marmosets would likely reveal similar levels of functionally significant genetic variation. This is important for a number of reasons:
- Discovery of novel “damaging” mutations can lead directly to new naturally occurring models of human genetic disease.
- Knowledge of functional variation among marmosets can facilitate better selection of animals for specific experiments.
- Information about functional variation among marmosets would allow more thorough interpretation of research results.
- Functional variation can be the subject of investigation on its own merit and lead to selection of animals for gene editing or new genetic models of disease.
GENETIC DIVERSITY IN MARMOSETS
Maintaining diversity in captive marmoset colonies requires careful attention to breeding. Diversity is influenced by genetic but also by environmental actors, such as the gut microbiome. Kenton Kerns discussed the development of protocols for tracking species survival in tamarins in his role as assistant curator of the small mammal house at the Smithsonian National Zoo. Joanna Malukiewicz, a postdoctoral research associate at the Biodesign Institute at Arizona State University and the Federal University of Viçosa in Brazil, discussed the effect of diet on the gut microbiome. Yasuhiro Go presented potential applications of sequencing specific genes in a marmoset model for study of neuropsychiatric diseases. Go is an associate professor affiliated with the Exploratory Research Center on Life and Living Systems (ExCELLS), the Department of Behavioral Development at the National Institute for Physiological Sciences (NIPS), and the National Institutes of Natural Sciences (NINS), Japan.
Methods for Maintaining Genetic Diversity
Using a tool known as the studbook,2 developed by the Association of Zoos & Aquariums (AZA), the Smithsonian National Zoo tracks select species’ births, deaths, and reproductive and medical histories to implement population survival, breeding, and transfer plans. Small, limited populations like those found in zoos, are rife with complications: risk of
___________________
population extinction, reduced ability to adapt to change, more common expression of negative traits, depressed immunity, and decreased reproductive success. Creating an environment where animals can live as they would in the wild is one of the crucial elements of conservation and welfare. Realistic and naturalistic social groups allow animals to breed while ensuring adequate genetic diversity is generated, without introducing new animals from the wild.
Each Species Survival Plan (SSP) is geared toward retaining 90 percent of a population’s genetic diversity over 100 years or 10 generations (whichever period is shorter) to ensure long-term population stability. This is a collaborative effort that extends across the AZA, which includes more than 200 accredited zoos in the United States. To track the progress of various species through their SSPs, participating zoos implemented a traffic light indicator. Populations with a green classification are at the desired level of 90 percent diversity; yellow signifies the population is close to the goal but needs more animals to attain the desired diversity; red indicates the lowest level of population diversity.
If there were an SSP for the 2,000 marmosets currently in the United States, each animal would receive a unique ID and its relevant information would be tracked in the studbook, including date of birth, date of death, transfers, and medical and reproductive updates. When genetic information is not available, as is the case in older marmoset colonies (see more details in Chapters 2 and 6), pedigree is used to calculate a kinship value describing an animal’s level of relatedness to the entire population plus to itself. This value is tied to the existing population and thus changes each time an animal is born or dies. Low kinship values are desirable as they represent diversity. A high kinship value may reflect either a failure to follow recommended guidelines or the fact that the original population lacked genetic diversity. Based on these values animals are assigned various designations: hold, breed, or transfer.
Kerns noted that this structure works well for the golden-headed lion tamarin (Leontopithecus chrysomelas) populations he oversees, as well as 500 other populations across many species in the network of zoos. An SSP can be restructured for optimal fit to a given population; thus, SSPs can help sustain small animal populations while maintaining and tracking their genetic diversity.
Genetic Variation and the Gut Microbiome
Protecting population growth alongside genetic diversity is particularly important for animals that experience the same risk of losing diversity in the wild. The Callithrix genus has six species of marmosets, half of which are endangered (see Chapter 6). Using whole genome low coverage
sequencing, Malukiewicz explored the natural and anthropogenic hybridization of Callithrix marmosets (Malukiewicz 2019).
The genomic sequence of these species demonstrated a high level of similarity within the C. jacchus marmosets sampled from several captive facilities in Northeast and Southeast Brazil, perhaps due to incomplete but variable levels of reproductive isolation (Coimbra-Filho et al. 1993). These preliminary data further suggest that C. aurita retain a high level of genetic diversity despite being endangered. Comparison between the genome sequence of C. jacchus and other Callithrix species highlights the lower genetic diversity specific to the Brazilian C. jacchus. This may be in part because C. jacchus is a relatively young species (Malukiewicz et al. 2017; Perelman et al. 2011).
Malukiewicz also collected anal swabs from the animals in this sample to study their gut microbiome. She classified them into one of three levels of captivity: wild (i.e., caught in the wild), semi-captive (i.e., caught in the wild and moved to captivity), and captive (i.e., always kept in captivity). Gut microbiomes provide information on an animal’s physiology, nutrition, and environment. As research into the microbiome of NHPs is increasing (McKenna et al. 2008), there are indications that captivity can have a significant impact on the composition of the gut microbiota, which tends to converge toward the modern human microbiome (Clayton et al. 2016).
A number of NHPs, including Callithrix marmosets, are obligate exudivores (Cabana et al. 2018), subsisting on indigestible oligosaccharides of tree gums or hardened saps as a large part of their diet. The effects of host taxonomy, hybridization, and habitat on the gut microbiome of gum eaters, including marmosets, remain largely unstudied. The abundance of bacterial species present in the gut microbiome samples Malukiewicz collected was related to captivity. Interestingly, in lieu of the Prevotella bacteria common to other captive primates, these captive hosts had Enterobacteriaceae. Helicobacteraceae and Campylobacteraceae were common to both semi-captive and wild hosts, although each group had other unique bacteria present as well. In line with their nature as gum eaters, Malukiewicz further identified carbohydrate metabolism as the most important metabolic pathway.
Genetic Diversity for Marmoset Models of Disease
Marmosets could play an integral role in developing a primate disease model for human neuropsychiatric diseases like autism, attention deficit hyperactivity disorder (ADHD), schizophrenia, and bipolar disorder. Go used genotype- and phenotype-driven methodologies to sequence neuropsychiatric-related genes in a spontaneous marmoset mutant and draw connections with diseases.
Beginning with the genotype-driven approach, Go sequenced genetic
data from more than 2,000 marmosets and macaques. He identified about 500 neuropsychiatric-related genes in each of these species. Of those identified, 53 and 142 genes, respectively, showed rare loss-of-function mutations. Some of these mutations can be connected to human diseases.
The phenotype-driven method examines candidate genes that are naturally occurring and expressed as behavioral or other kinds of symptoms. For instance, some of the marmosets in Go’s sample exhibited abnormal eye movement, which is a typical intermediate phenotype for Rett syndrome, a genetic mutation that affects brain development in girls. This phenotype was connected to loss-of-function mutations on the Methyl-CpG-Binding Protein 2 (MECP2) genes, thereby pointing to the link between the MECP2 gene mutation and Rett syndrome.
Continuing these genotype and phenotype-driven explorations, brain transcriptome analysis of diseased marmosets will shed light on the molecular basis of neuropsychiatric disease. However, monitoring the genetic variation in marmoset colonies is necessary to ensure success in devising effective marmoset models of disease. To this end, Go analyzed marmoset blood samples from marmoset centers in Japan. When compared with humans and macaques, the heterozygosity of Japanese marmosets was about equivalent to humans, but it was far less than the macaques. Furthermore, there was variation in heterozygosity among the samples from each center, but in comparing U.S. and Japanese marmoset populations, the genetic diversity was clustered between geographic samples (see Figure 5-1).
In his summation, Go called attention to the pros and cons of different methods for DNA sampling in marmosets: blood sampling provides significant amounts of high quality DNA but carries chimerism; nail sampling provides low amounts of low quality DNA but is not affected by chimerism.
Challenges and Incentives in Maintaining Genetic Diversity
Maintaining genetic diversity in research colonies is complicated by the limited information on historic pedigrees of animals imported into the United States in the 1960s and 1970s; a long-standing absence of record-keeping of marmoset pedigrees. Many of the current research colonies are derived from initial stocks of a few dozen animals, posing problems for genetic diversity. Genomic analysis of existing marmoset colonies lags behind that of macaques, though it is possible to generate a great deal of information on the genetic diversity of marmoset colonies with available technology.
SNPs are a useful and cost-effective tool for managing a colony’s genetic diversity through pedigree testing and genetic analysis. Although this approach does not offer the level of detail possible with whole genome sequencing, it can provide information about the degree of relatedness
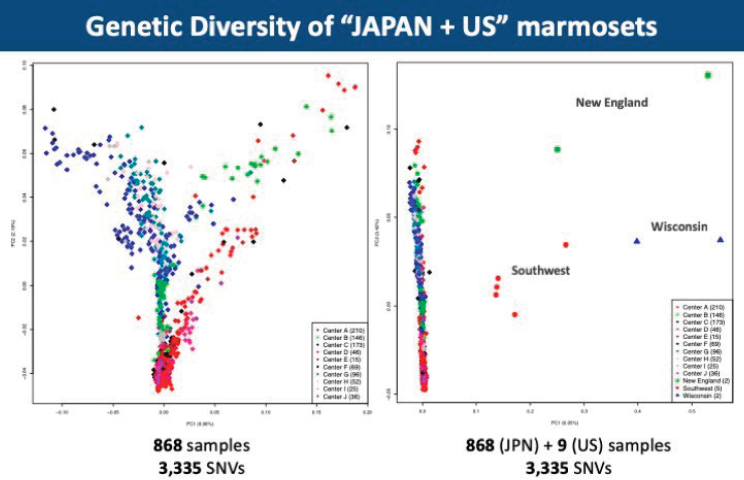
NOTE: JPN = Japan; SNV = single nucleotide variant; US = United States.
SOURCE: Yashuhiro Go.
between animals and also help researchers identify certain kinds of functionally significant variation.
The cost and time required for whole genome sequencing is rapidly decreasing. A marmoset’s genome could be sequenced in about 4 days and analyzed in about 1 week. The availability of new low-cost, portable genome sequencers is making sequencing increasingly feasible, as well. In addition to greater detail, whole genome sequencing offers the benefit of being able to identify novel variants, whereas SNP-based chips can only identify the variants that are built into the chip.
What characterizes wildtype animals? Even within a species, animals living in different environments, for example in Brazil’s Caatinga area versus the Atlantic forest region, show phenotypic differences and likely genetic ones as well. Such nuances are relevant when setting goals for maintaining genetic diversity in research colonies.
It is also important to recognize the inadvertent genetic selection that has likely gone into current marmoset colonies. The captive populations used in research today are the descendants of previous generations of
marmosets who were able to survive in captive environments under care and management conditions that have changed over time. It is possible that the process of maintaining generations of marmosets in captivity eliminated the more susceptible animals, so today’s research populations are hardier than wild populations.
Looking forward, the research community faces a fundamental question: Is the goal to establish research colonies that reflect the genetic diversity of wild populations, or to establish colonies that are well suited for captivity and research activities? Focusing on maintaining colonies that do well in laboratory conditions may inadvertently weed out animals with disease susceptibility that could offer important insights relevant to human diseases.
Researchers also face conflicting incentives affecting the maintenance of genetic diversity in research colonies. Generally speaking, most accept the notion that higher genetic diversity is beneficial and that it is valuable to implement breeding plans that increase diversity. But a laboratory’s specific research goals can interfere; for example, if a certain individual or group of marmosets consistently produces offspring with characteristics that are useful for the research questions being addressed, it is tempting to disproportionately favor those marmosets for breeding. Also, while introducing animals from other colonies can help increase genetic diversity in a research colony, it also imposes logistical complexities and increases the chance of disease transmission.
REFERENCES
Cabana F, Dierenfeld ES, Wirdateti GS, et al. 2018. Exploiting a readily available but hard to digest resource: A review of exudativorous mammals identified thus far and how they cope in captivity. Integr Zool 13:94–111.
Clayton JB, Vangay P, Huang H, et al. 2016. Captivity humanizes the primate microbiome. Proc Natl Acad Sci USA 113(37):10376–10381.
Coimbra-Filho AF, Pissinatti A, Rylands AB. 1993. Experimental multiple hybridism and natural hybrids among Callithrix species from eastern Brazil. Pp. 95–120 in Marmosets and Tamarins: Systematics, Ecology, and Behaviour, edited by AB Rylands. New York: Oxford University Press.
Dray BK, Raveendran M, Harris RA, et al. 2018. Mismatch repair gene mutations lead to lynch syndrome colorectal cancer in rhesus macaques. Genes Cancer 9(3–4):142–152. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6086002.
Malukiewicz J. 2019. A review of experimental, natural, and anthropogenic hybridization in Callithrix marmosets. Int J Primatol 40(1):72–98.
Malukiewicz J, Hepp CM, Guschanski K, et al. 2017. Phylogeny of the jacchus group of Callithrix marmosets based on complete mitochondrial genomes. Am J Phys Anthropol 62(1):157–169. doi: 10.1002/ajpa.23105.
McKenna P, Hoffmann C, Minkah N, et al. 2008. The Macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathogens 4(2):e20. Available at: https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.0040020.
Moshiri A, Chen R, Kim S, et al. 2019. A nonhuman primate model of inherited retinal disease. J Clin Invest 129(2):863–874. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6355306.
Perelman P, Johnson WE, Roos C, et al. 2011. A molecular phylogeny of living primates. PLoS Genet 7(3):e1001342. Available at: https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1001342.
Sato K, Kuroki Y, Kumita W, et al. 2015. Resequencing of the common marmoset genome improves genome assemblies and gene-coding sequence analysis. Scientific Reports 5:16894. Available at: https://www.nature.com/articles/srep16894.
The Marmoset Genome Sequencing and Analysis Consortium. 2014. The common marmoset genome provides insight into primate biology and evolution. Nature Genet 46:850–857.
Xue C, Raveendran M, Harris RA, et al. 2016. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res 26(12):1651–1662. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/27934697.










