
1
Introduction
A quality health care system has three critical components: quality-assured practitioners, quality-assured facilities, and quality-assured health care products1 (i.e., vaccines, pharmaceuticals, and medical devices). All three of these components must be present; if even one is absent, the system cannot provide the care patients deserve, expect, and require. It is the role of regulators to ensure that quality health care products reach the market. Moreover, although affordability and accessibility are tangential to the scope of most regulatory authorities, both are essential for ensuring a well-functioning public health system, in which quality-assured essential medicines reach those for whom they are intended.
Patients and those who work in health care expect the products they use to be what they purport to be and to work as described in the products’ packaging. The fundamental issue is trust: just as patients need to be able to have confidence in the expertise of health care workers and the quality of their health care facilities, they and health workers need to be able to have confidence that the products they take and prescribe prevent or treat illness, alleviate suffering, and improve health.
A robust regulatory system that provides effective oversight of health care products throughout their lifecycle, from the laboratory to the patient, is the linchpin of product assurance. The highest priority for regulatory authorities responsible for medicines is ensuring the availability of medicines that are of high and consistent quality, safe, and effective. To this end,
___________________
1 For quality-assured pharmaceutical health care products, this component encompasses assuring a favorable benefit/risk profile.
regulators must have access to scientifically robust, reliable data to inform their decision making. Fulfilling this mission in today’s world of increasingly complex medicines that are produced in increasingly complex global manufacturing and supply chains presents significant new challenges, and meeting these challenges requires 21st-century best collaboration practices among regulatory authorities.
STUDY CONTEXT
While regulatory authorities are responsible for a wide array of health care products, this report focuses only on medicines. It does not include any discussion of medical devices, illicit drugs, naturopathic substances, or veterinary medicines, although the committee recognizes that such products are often part of the overall mandate of regulatory authorities. Moreover, this report is not designed to focus on medicines per se but to look at the regulatory authorities overseeing their quality, safety, and efficacy, and how those authorities in different jurisdictions can and do cooperate and collaborate to fulfill their responsibilities in protecting public health most effectively. These responsibilities typically involve ongoing assessment of the safety, efficacy, and manufacturing quality of all medicines, from their first introduction into human clinical trials to their widespread use following marketing authorization. The public expects not only that medicines on the market will be safe to use, work as labeled, be affordable, and be rapidly available to patients, but also that regulatory systems will make the best, most efficient use of their time and financial resources.
In the United States, the U.S. Food and Drug Administration (FDA) works hard to meet its obligations while navigating the increasing challenges and complexities associated with regulating products in a global market. Congress recognized these challenges in establishing FDA’s mission by including—in addition to its public health protection mandate—a requirement that it “participate through appropriate processes with representatives of other countries to reduce the burden of regulation, harmonize regulatory requirements, and achieve appropriate reciprocal arrangements.”2 According to FDA’s Center for Biologics Evaluation and Research (CBER), the agency supports this mission element through exchange mechanisms with international counterparts and organizations (FDA, 2018b). Given that medicines in the 21st century are global commodities, information exchanges have helped CBER fulfill its mandate of ensuring safe and effective medicines.
Medicines research, development, production, and distribution are decidedly global in nature and often occur in a multitude of jurisdictions
___________________
2 Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 393.
during the lifecycle of a single product. FDA has estimated that nearly 40 percent of finished medicines/medicinal products and 80 percent of active pharmaceutical ingredients consumed by Americans are made abroad (GAO, 2016). Accordingly, regulatory authorities have had to bring a more global perspective to their work. In this global context, it has become clear that higher public health purposes and better resource utilization are best served through harmonization of technical standards and coordination of processes among regulatory authorities around the world. By sharing work and information and supporting one another through collaborative activities, regulatory authorities can bring drugs to market more quickly, avoid drug shortages, lower drug prices, communicate the risks of medicines, and become better prepared to address emerging threats. They also can optimize the use of their limited resources.
Regulatory authorities around the world have access to varying levels of resources (human, financial), but the totality of these resources is finite. Reliance allows regulatory authorities to leverage resources and expertise more efficiently. All of these relationships start with gaining knowledge of potential partners’ regulatory systems and cultures, thereby building trust and confidence in their work. These relationships then evolve over time. As shown in Figure 1-1, when a regulator gains confidence in the equivalency of a counterpart’s regulatory oversight, the relationship between those regulators has the potential to grow into recognition and reliance arrangements whereby one regulatory authority is willing to rely on the outputs of another. Such arrangements may start with a pilot, which often provides a safe space in which both parties can learn from each other, and in which trust is verified and built. In some instances, the trust gained during a pilot phase is enough to motivate partners to enter into more formal arrangements, the highest level of which is a mutual recognition agreement, generally agreed upon between two countries under the auspices of a trade agreement.
Many such arrangements have been implemented between well- and moderately well-resourced regulatory authorities, although lower-resourced authorities also engage in a variety of arrangements that rely on the work of other trusted authorities. A number of these collaborations (catalogued in Appendixes B and C) have the potential to enable more effective use of resources and help agencies meet the expectations of their people and their governments. By directing efforts away from duplicative inspections of low-risk manufacturers, regulatory authorities can channel more of their resources toward areas that pose greater public health challenges, including those that may stem from medicines produced and manufactured in regions with weaker regulatory oversight. In this era of globalization, moreover, strengthening and leveraging the capacities of well-resourced regulatory authorities is essential for maximal protection of global public health, as is
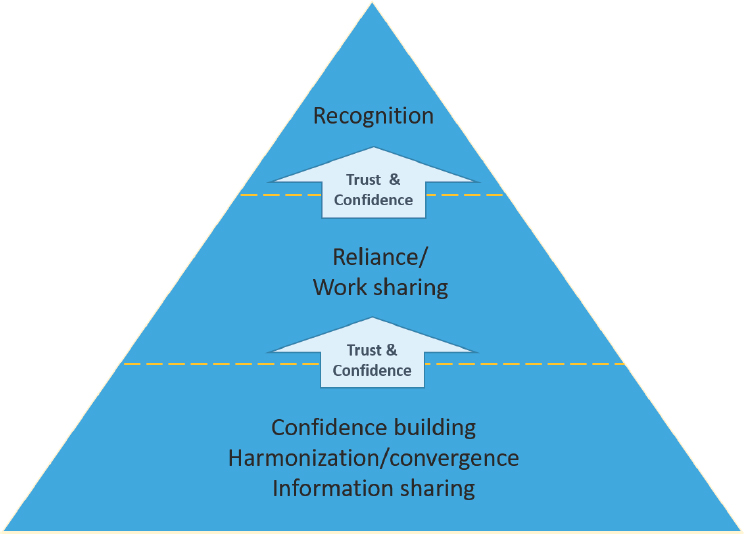
SOURCES: Adapted from a figure created by Dr. Petra Doerr, Swissmedic, and presented by Emer Cooke, World Health Organization (WHO, 2019i).
improving regulatory oversight and decision making in low-income nations. Figure 1-1 shows how trust and confidence are necessary for any form of cooperation among and between regulatory authorities, from reliance and work-sharing activities to recognition agreements (mutual and others). The arrows indicate that while confidence and trust build toward more formal arrangements, there is a certain amount of fluidity in the dotted lines for regulatory authorities to draw upon such activities as information sharing to build trust while demonstrating equivalence.
CHARGE TO THE COMMITTEE
In this context, in September 2018 FDA’s Office of International Programs (now the Office of Global Policy and Strategy) charged the National Academies of Sciences, Engineering, and Medicine with convening an ad hoc committee to conduct a landscaping and analysis of mutual recognition and reliance arrangements for pharmaceutical products. The work was to include engaging relevant stakeholders in discussions that could contribute to and facilitate potential future action with respect to collaboration and
cooperation among and between regulatory authorities. These discussions would include descriptions of how specific types of arrangements are or could be used, as well as the associated risks, benefits, challenges, and opportunities. The committee’s report was to include a landscape of the various arrangements employed by regulatory authorities and to describe various strategies FDA and others might consider for making these arrangements even more helpful in meeting the expectations of their people and governments with respect to protecting public health and promoting quality-assured medicines (see Box 1-1 for the committee’s full Statement of Task).
The committee formed to conduct this study and prepare this Consensus Study Report comprised 12 members with a broad range of expertise
in areas relevant to the committee’s charge, including national and international medicines regulation, law, global health policy, public health, economics, risk management, and pharmaceutical policy and manufacturing. Biographical sketches of the committee members are presented in Appendix E.
STUDY SCOPE AND APPROACH
The committee’s Statement of Task drew the boundaries of the scope of this study. Because recognition and reliance arrangements have implications for virtually all countries and regulatory authorities around the globe, this report describes how different arrangements, particularly those involving regulatory authorities with higher levels of resources, could benefit the work of all regulators and provide a more universal level of quality, safety, and efficacy for medicines globally. The report touches upon but does not delve deeply into optimization efforts, including harmonization of technical standards or convergence in other appropriate areas. Nor does the report focus heavily on recognition and reliance arrangements among low-income nations. A limited discussion in these two areas is not intended to minimize the critically important harmonization work within medicines regulation, but reflects the time limitations of this study. Additionally, as noted earlier, the scope of this report is limited to medicines, encompassing both chemically and biologically based medicines and vaccines.
To address the issues raised in its Statement of Task, the committee used multiple sources in compiling the content for this report. Those sources included literature and online searches, as well as written supplemental details from regulators and information-gathering sessions involving representatives of regulatory authorities, industry, and patients. See Appendix D for a full description of the methodology used in carrying out this study.
ORGANIZATION OF THE REPORT
Following this introductory chapter, this report includes five chapters designed to respond to the Statement of Task for this study. Chapter 2 provides key background information that encompasses the issues characterizing the environment for medicines regulation, definitions of recognition and reliance, and the essential features of recognition and reliance arrangements. Chapter 3 describes how these arrangements can be viewed by policy makers and presents an overview of the regulatory functions of oversight over good manufacturing practice, batch certification, good laboratory practice, and good clinical practice. This is followed in Chapter 4 by a landscape of recognition and reliance arrangements focused on the associated challenges, benefits, and opportunities identified by stakeholder
representatives who informed the committee’s work, as well as the other sources noted above. Chapter 5 presents the committee’s distillation of the challenges and opportunities identified in Chapter 4 as the basis for a conclusion and six recommendations, including a strategy that engages all the key stakeholders in taking action to address the key messages of this report: the importance of encouraging greater regulatory cooperation and of removing impediments that could prevent recognition and reliance arrangements from progressing. Chapter 6 closes with ideas to explore in the future, in particular, the important area of regulatory cooperation on ensuring the safety of medicines after they are available to consumers in the market.
This page intentionally left blank.








