
3
What Policy Makers Need to Know About Today’s Regulatory Environment
In understanding the current regulatory environment, it is important to recognize the growing complexity of medicines research and development, production, and manufacturing. In many instances, manufacturing of medicines has become a global endeavor crossing multiple national borders before a product is distributed to numerous local markets around the world. Not only have supply chains grown in complexity, but medicines themselves have also become more complex, both biologically and chemically. Figure 3-1 shows the range of medicines, from the simplest products with well-defined chemical structures, to highly complicated gene or cell therapies using extremely advanced drug research, development, and manufacturing methodologies. Of note is that as medicines go from simple to complex, regulatory oversight will follow a similar path. However, not all regulatory authorities have the resources to train or hire those with the requisite skillset to assess more complex products for approval or to monitor their safety post-approval (Luigetti et al., 2016). Cooperation and collaboration in their many forms represent tools regulatory authorities can use to overcome resource constraints, avoid duplication of efforts, and leverage expertise across borders for improved decision making within a local jurisdiction or region. All of these tools fit under the umbrella of recognition and reliance arrangements.
OVERVIEW OF GOOD PRACTICES
It is industry’s responsibility to comply with rules and scientific and technical standards established for products, and it is the regulator’s respon-
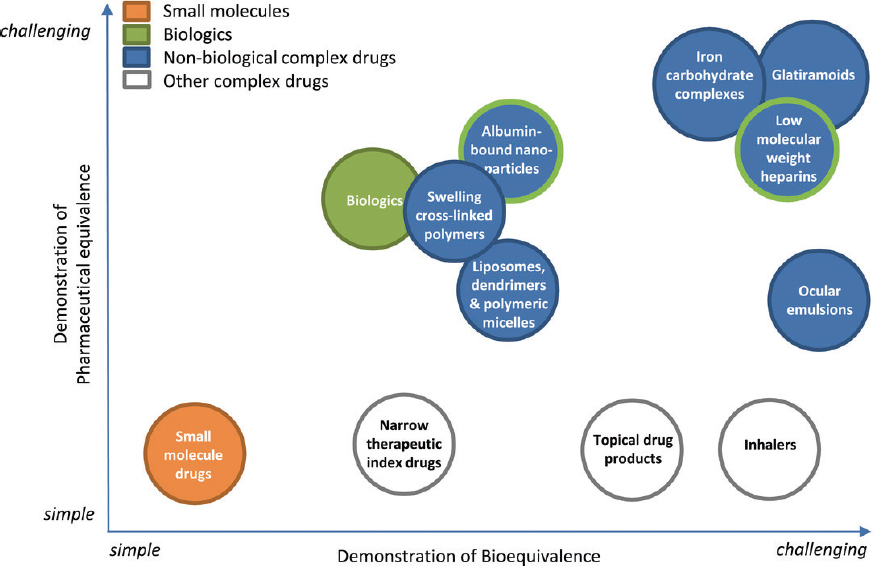
NOTES: Drug products are positioned on the basis of the challenge to assess the pharmaceutical equivalence (PE) and bioequivalence (BE) of two drug products (i.e., the reference product and its generic version). The percentage of new medicines categorized as “complex” is increasing. From a global perspective, the resources and expertise needed to assess the expanding number of complex medicines are limited. Complete execution of recognition and reliance arrangements will be essential if regulatory authorities plan to assess the broad spectrum of medicines being manufactured in a timely and reliable way.
SOURCE: Hussaarts et al., 2017.
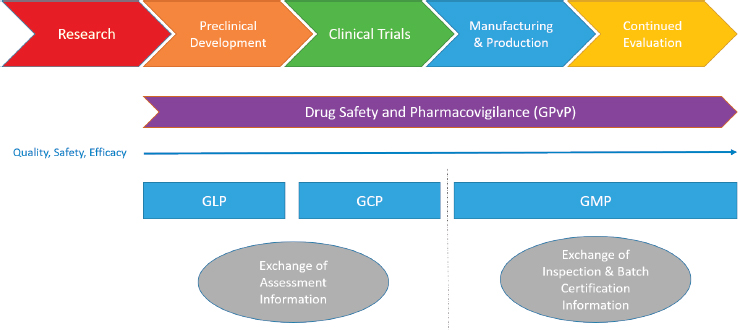
sibility to oversee compliance with these rules and standards (Rönninger et al., 2012) and to work with the larger community to develop new standards as technology and scientific knowledge improve. For medicines, there are regulations describing compliance activities and good practices across the lifecycle of a medicine from preclinical research to post-marketing surveillance. Guidance is provided by regulatory authorities at each stage (as shown in Figure 3-2), but it is up to industry to interpret that guidance and apply it to their situation. Good laboratory practice (GLP) sets guidelines for ensuring that laboratory data are of high quality and reliable in the preclinical phase, good clinical practice (GCP) guides aspects of studies involving human subjects, good manufacturing practice (GMP) provides guidance for the manufacturing, production and distribution of medicines, and good pharmacovigilance practice provides guidance for the appropriate oversight of products once they have been released onto the general market. Assuring an overall positive benefit/risk profile to the most appropriate level possible is essential throughout the entire medicine product lifecycle.
In this report, regulatory work products—administrative documents produced by a regulator as a result of a regulatory authority’s evaluation—are differentiated from medical products that include but are not limited to medicines. These work products might include an inspection report or certificate of compliance for GMP, official batch release, or an assessment report regarding the evaluation of the clinical and statistical elements of the product clinical dataset used in the application requesting approval to market a new medicine.
Good Manufacturing Practice
All current mutual recognition agreements (MRAs) regarding the regulation of medicines include provisions for relying on a partner for GMP inspections. Each MRA, with the exception of that between the United States and the European Union (EU), is based primarily on the exchange of GMP certificates (see Table 3-1), with the provision of inspection reports upon request (the U.S. Food and Drug Administration [FDA] does not issue GMP certificates in practice). Inspections covered under each of the MRAs are intended to confirm that the products are manufactured in compliance with current agreed-upon GMP. Of note is that none of the current MRAs include provisions for GMPs for advanced therapy medicinal products, although this may be an area of future interest between the United States and the EU.
Before countries sign MRAs, their regulatory authorities spend a significant amount of time evaluating each other’s systems and procedures to attain a level of confidence in each other’s output. In addition, one condition of an MRA or any reliance agreement is the conclusion that all
| Instrument Used for Information Exchange Between Manufacturer and Importer and Between Mutual Recognition Agreement (MRA) Partners | ||||
|---|---|---|---|---|
| MRA | Batch Testing | Good Manufacturing Practice (GMP) Certificate | Rapid Alert | Inspection Report |
| EU-AUS | Yes | Yes | Yes | No |
| EU-NZ | Yes | Yes | Yes | No |
| EU-CH | Yes | Yes | Yes | No |
| EU-CAN | Yes | Yes | Yes | No |
| EU-JAPAN | Yes | Yes | Yes | No |
| EU-ISRAEL | Yes | Yes | Yes | No |
| EU-US | Yes for EU | No | Yes | Yes |
| Australia-New Zealand | Yes | Yes | Yes* | No |
| Australia-Canada | Yes | Yes | Yes | No |
| Australia-Singapore | Yes | Yes | Yes | No |
| Canada-Switzerland | Yes | Yes | Yes | No |
NOTES: See Appendix B for a fuller description. Aus = Australia; CAN = Canada; CH = Switzerland; EU = European Union; NZ = New Zealand; US = United States.
*Rapid alert system not designated in MRA, but executed through Pharmaceutical Inspection Co-operation Scheme (PIC/S) membership.
SOURCES: Adapted from Saint-Raymond, 2019; EU-US MRA.
authorities involved are “capable” of evaluating GMP and taking appropriate actions when a facility is out of compliance. These evaluation activities foster trust and confidence. While MRAs are established on both a foundation of trust and years of cross-evaluation, each regulatory authority is ultimately responsible for the health of its population. As a result, under specified conditions, MRAs contain clauses stating that each party has the right to conduct an inspection in any facility even if a partner regulatory authority has already done so. At the time MRAs are established, procedures for communicating noncompliance findings to partner authorities are outlined. Each regulatory authority retains sovereignty and is responsible for interpreting inspection results. It is a regulatory authority’s privilege and responsibility to ask questions and take enforcement action if there is reason to believe that a manufacturer’s practices have compromised the quality of a product or are likely to do so, thereby compromising the health of that regulator’s population.
FDA defines enforcement as “action taken by an authority to protect the public from products of suspect quality, safety, and effectiveness or to assure that products are manufactured in compliance with appropriate laws, regulations, standards, and commitments made as part of the approval to market a product.”1 Common enforcement actions are a request for voluntary compliance, suspension of product distribution, detention at the border of the importing country, recall or withdrawal of the batch(es), and requests for additional information or inspections.2 Similarly, Health Canada has MRAs with the EU, Switzerland, and the European Economic Area (EEA). Health Canada’s enforcement actions in response to GMP noncompliance are similar to those of FDA and include, but are not limited to, written correspondence and requests for plans for corrective measures and/or additional assessments, public advisories, removal, and seizure and/or destruction of existing products.
Rather than providing specific details regarding the use of enforcement actions within the agreement, most MRAs make reference to the importance of honoring each party’s established regulatory commitments to quality, safety, and public health. Each regulatory authority can take enforcement actions it deems appropriate. Under MRAs, partner regulatory authorities are not required to take the same enforcement actions in response to an inspection report; however, given the MRA precondition of regulatory authority equivalence, authorities in an MRA are expected to take enforcement actions that are congruent with and imply similar reactions to noncompliant practices. At the same time, it should be understood that action taken to suspend manufacturing activities at a site in
___________________
1 21 CFR § 26.1.
2 21 CFR § 26.21.
an MRA partner’s own territory will directly impact supply to other territories, including MRA partners, without any direct intervention by the other authority. The MRA between Australia and Singapore highlights the independent authority of each party to “determine the level of protection it considers necessary with regard to health, safety, and the environment”; consequently, neither party is bound by contract to take the same enforcement actions as the other. Regardless of the actions either party takes, both have agreed to notify the other regulatory authority of any enforcement actions within 15 days.
Batch Certificate
While all MRAs are designed to minimize duplicative inspections of manufacturing sites of similar interest between the signatories, virtually all existing MRAs go further by waiving batch testing in the importing country (see Box 3-1). A batch certificate can be issued confirming that each individual batch (fully finished or partially manufactured) has been manufactured and checked in compliance with the appropriate laws, marketing authorization requirements, and GMP. The certificate is normally issued by the exporting manufacturer and may, if necessary, be validated or even issued by the competent regulatory authority of the exporting country (WHO, 2019f).
Some MRAs provide for acknowledging testing done by official laboratories or batch disposition decisions for certain types of products (e.g., blood products, some vaccines). At present, however, the scope of the EU-US MRA does not include regulatory activities associated with these product types, although it contains a provision for waiving batch testing for those medicines covered under the MRA. Having an MRA or other agreement that facilitates waiving batch testing in the importing country or mutually recognizes official batch testing reduces costs and product wastage and allows importers and regulatory authorities to redirect resources toward areas of greater risk instead of performing redundant regulatory laboratory testing (Garbe et al., 2015).
Good Laboratory Practice
GLP in the pharmaceutical industry is often guided by multiple resources, including a handbook from the World Health Organization (WHO, 2009), rules established by FDA for nonclinical laboratory studies,3 and principles set forth by the Organisation for Economic Co-operation and Development (OECD). These combined resources help set the boundaries for nonclinical
___________________
3 21 CFR Part 58.
testing conducted prior to the approval of a medicinal product through strict guidelines for generating quality-assured and reliable test data while using an environmentally safe and animal numbers reduction approach (OECD, 2019). Regulators are allowed to rely on certifications and inspections provided for GLP under some MRAs (e.g., EU-Japan; Switzerland-Canada, Switzerland-EU; Switzerland-EEA, which includes Norway, Iceland, and Liechtenstein [see Appendix B]). According to OECD, significant cost savings to industry and governments are possible through mutual acceptance of such laboratory data. Part of OECD’s estimated €309 million total savings each year (from all sectors) includes a reduction in the number of animals used for testing purposes (OECD, 2019).
In addition to GLP covered under MRAs, the United States and the Netherlands entered into a Memorandum of Understanding (MOU) under their confidentiality arrangement that reciprocally recognizes both countries’ GLP programs (FDA, 1988). This MOU was signed only after comparable standards of GLP had been established by the respective national authorities. As part of the agreement, both parties are required to regularly
communicate and share information and data unrelated to trade secrets or confidential commercial or financial disclosures.
The International Laboratory Accreditation Cooperation (ILAC) recognition arrangement links accrediting bodies around the world through a peer review system that includes a review of the impartiality and consistency of those performing inspections (ILAC, 2019). While ILAC signatories are not regulators, the program does call to light the importance of the International Organization for Standardization/International Electrotechnical Commission 17020:2012 requirements for establishing the competence of bodies performing inspections as a foundation for sharing in the area of GLP (ISO, 2012).
Good Clinical Practice
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use has published a GCP guideline (ICH E6 R1) for clinical trials involving human subjects (ICH, 2016). This guideline, agreed upon initially in 1996, establishes ethical and scientific standards for ensuring the safety and privacy of clinical trial participants and the credibility of data on trial results. The most recent addendum to the publication provides a “unified standard for the EU, Japan, the US, Canada, and Switzerland to facilitate the mutual acceptance of data from clinical trials by the regulatory authorities in these jurisdictions” (ICH, 2016, p. 1).
The European Medicines Agency (EMA) and FDA Good Clinical Practice Initiative that commenced after an 18-month pilot test between 2009 and 2011 involved sharing information on clinical trial site inspections, as well as collaborating on inspections (EMA, 2011). As with similar efforts, as discussed in Chapter 2, confidence and trust are built between partner regulatory agencies during the pilot phases of such initiatives. Thus, while GCP is not part of the recently functional EU-US MRA for pharmaceuticals, it is possible that the associated regulatory authorities could move beyond trust building and knowledge sharing and agree to expand the operational scope of the MRA to include recognition of each other’s GCP inspections of clinical trials. Doing so could help repurpose both agencies’ finite human and financial inspection resources.
Confidentiality Arrangements
Confidentiality arrangements are often what allow regulatory bodies to enter into cooperative arrangements such as the EU-US initiative. Confidentiality arrangements allow parties to exchange confidential information, although they are not a prerequisite for building trust and knowledge between regulators. For example, the GCP Inspectors Working Group
meets four times per year at EMA. The group includes representatives from the GCP inspectorates of the EEA Member States, as well as observers from candidate countries and Switzerland (EMA, 2013). In addition to the Working Group, WHO is invited to participate in these meetings. The Working Group’s regular interactions provide a forum for strengthening relationships among the peer groups from the regulatory authorities.
Because of the large number of redactions common with FDA inspection and assessment reports, a “super confidentiality commitment” (Super-CC) was established between the United States and the EU designed to minimize redactions, thereby making the reports more usable. The authority for FDA to enter into such super confidentiality arrangements under certain conditions is a specific provision added to U.S. law in the past few years.
PUBLIC HEALTH MANDATE
Part of the responsibility of national regulators in protecting the public’s health when relying on the work of other regulators is to interpret the information they receive and make a decision based on the context of their nation’s health care system, their population, and the public health situation in their country. In the approval space, for example, regulators working in countries with intense sunlight and high rates of skin cancers (such as Australia and New Zealand) might come to different decisions regarding the approval of certain dermatological medicines than those working in more temperate climates. This observation underscores another key message: that in the end, it is a national decision as to whether to approve a medicine.
In the beginning of a recognition or reliance relationship, as trust, confidence, and knowledge are being established between two regulatory bodies, it may be said that regulators must “trust but verify.” As the relationship strengthens and greater trust is built, the regulators may perceive less of a need to verify each other’s work. This decreased need to verify is where regulators can realize further cost savings while remaining confident that public health remains protected. This is critical because in the end, if something goes wrong, it is the responsibility of regulators to explain the decisions they made regarding the status of a product in their market.
Drug Shortages
Drug shortages are a major public health concern for high-, medium-, and low-income countries alike (Iyengar et al., 2016). As a result, patients risk being un- or undertreated for acute or chronic diseases, and the lack of available medicines creates opportunities for substandard and falsified medicines to make their way to consumers. These problems are especially
acute for those living in low- and middle-income countries, although those living in higher-income nations are not exempt from the risk. Regulatory authorities, such as FDA, EMA, Australia’s Therapeutic Goods Administration, and Health Canada, each have defined approaches and have set up task forces that focus on the problem of drug shortages, collecting data and seeking ways to address the problem. Regulatory authorities’ strategies for responding to this public health need include expedited inspections and/or reviews of existing manufacturing sites or new sites (FDA, 2019c). Another strategy is the use of reliance mechanisms. In the case of Mexico, for example, unilateral recognition of the medicines approvals of trusted authorities helped alleviate that country’s drug shortages (see Chapter 2), and reliance has also been proposed as a mechanism for helping higher-income nations deal with their drug shortage challenges (Vinther, 2016). Additionally, WHO encourages individual authorities, particularly those in low- and middle-income countries, to rely on products registered on its prequalified medicinal products list to address drug shortages in specific high-priority areas (WHO, 2018a).
Public Health Emergencies
Numerous public health emergencies require quick access to medicinal products. These include, but are not limited to, pandemics, natural disasters, military emergencies, disease outbreaks, and emerging diseases. As is the case with the drug shortages discussed above, government agencies must act in such circumstances to protect the health of their population, and regulatory authorities must weigh the benefits and risks of relaxing more stringent policies and practices.
The Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 empowers FDA to relax regulations so as to expedite access to “medical countermeasures” (e.g., antivirals, antidotes, vaccines, blood products, biological therapeutics) (FDA, 2017a). During public health emergencies, FDA can waive current GMP requirements and make available products it anticipates will be approved in the future, or approve existing drugs for alternative use (FDA, 2017a).
In 2017, Hurricane Maria created an example of a complex public health emergency. The water and debris created a breeding ground for bacteria. Puerto Rican residents were in desperate need of medicines, as they had been injured and exposed to disease-causing bacteria and toxins. Access to already prescribed medicines was hindered by the physical destruction of homes and businesses. In addition to being a public health emergency, the hurricane created shortages of medicines (FDA, 2018d), as 8 percent of all American drug expenditures are on products manufactured in Puerto Rico (FDA, 2017b). In such situations, if regulatory authorities are able to share
information via MRAs or other reliance arrangements, they can review assessments of alternative manufacturing sites producing the pharmaceutical products they need.
In a global example, in 2016 WHO declared the Zika virus a public health emergency of international concern (WHO, 2016d). There were no approved tests, vaccines, or treatments for Zika. Researchers and health officials were in constant communication with each other about how to address the problem (WHO, 2016c). As of this writing, Zika was no longer a WHO public health emergency; however, WHO has been encouraging reliance as a way to expedite market access to medicines used to treat and/or prevent priority diseases, given that traditional and/or duplicative inspections by each authority in need of pandemic treatments would result in loss of life (WHO, 2017b).
IMPEDIMENTS TO REGULATORY AUTHORITY RELIANCE
There are numerous impediments to the sharing of information between regulators. Examples of such impediments vary, and might include unusable overly redacted inspection and assessment reports, lack of common formats or standards or conformity, insufficient human and financial resources, conflicts of interest, and insufficient authority granted to regulators entering into informal and formal recognition or reliance arrangements. For regulatory authorities to build further upon their current recognition and reliance activities, impediments to entering into and using informal and formal recognition and reliance arrangements need to be removed. Some impediments originate from within the regulatory authority itself, while others are external to the agency and are influenced by policy makers, industry, or consumer/patient advocacy groups. Each of these stakeholder groups has a role to play in supporting efforts to enhance cooperation with, between, and among regulatory authorities with the aim of improving public health.
Impediments Between Regulatory Authorities
The premise behind sharing a report among regulatory authorities is that an industry sponsor has business interests in all the jurisdictions involved. Reports should be shared only with regulatory authorities representing jurisdictions in which a medicine is being considered for research or market placement. This being the case, it is essential for a regulatory authority with information about a medicine to share that information with a partner regulatory agency identified by an industry sponsor as representing a targeted market. It is equally essential that an industry partner expressing a legitimate intent to conduct business in a jurisdiction also express unambiguous support for that jurisdiction’s regulatory authority’s
gaining full access to assessment reports from partner regulatory authorities. As of this writing, heavily redacted reports represent a significant barrier to the optimal implementation of recognition and reliance arrangements. Unredacted reports (i.e., full reports without parts having been expunged, with the exception of personal privacy information) shared with receiving authorities could be used to inform their decision making. Doing so would uphold the personal privacy protection laws in most jurisdictions and maintain and protect the social contract made with clinical trial participants while facilitating information sharing and optimal execution of recognition and reliance arrangements.
Impediments Between Regulatory Authorities and Industry
The suggestion to ease current redaction practices may contravene current interpretations of trade secret and confidentiality laws in some jurisdictions. However, these laws and precedents were established at a time when most manufacturing was local and were intended to protect a company’s legitimate marketing rights in that original jurisdiction, where the regulator had full information as required by law. As discussed in Chapter 1, the current reality is that medicines are no longer primarily local products, but rather global commodities with development, manufacturing, and distribution in many countries; consequently, an examination is needed of whether current trade secret and confidentiality laws, as they apply to information shared between impacted regulatory authorities, are still fit-for-purpose.
Perceived Threats to Sovereignty
It must be emphasized that an agency relying on other countries’ work products is neither “outsourcing” that agency’s decision making nor giving up sovereignty, accountability, or responsibilities. Agencies retain the ability to make, and the responsibility for making, their own sovereign regulatory decisions. Reliance on materials received through informal information-sharing mechanisms and formal MRAs is, rather, a matter of establishing confidence in the other agency’s processes and trust in the information one receives. Such trust usually involves both agencies having a shared public health interest that is fundamental to their decision making. In addition, part of establishing trust is the development of systems for evaluating and validating the quality of the work of other agencies so that when one agency relies on the work of another, it can explain clearly to government officials and its people how it developed the confidence in the other agency that enabled partial or total reliance on information from that agency. Agencies must have the technical capacity, based in robust science and public health, to conduct independent assessments and produce
the reports that are shared. With such an arrangement comes the ability to make informed regulatory decisions without having to repeat the assessments of the trusted party. Clearly, avoiding the generation of redundant data for regulatory decision making is one of the main foci of these reliance arrangements. Thus, the receiving regulatory authority exercises sovereignty, interpreting information provided by other regulatory authorities in conjunction with its own information and within the context of the local health care system and public health state of play.
Limited Resources
There are significant costs to developing, adopting, implementing, and maintaining MRAs, particularly in terms of time and human resources (Correia de Brito et al., 2016). For countries establishing GMP MRAs with the EU, trust and confidence in all of the EU Member States must be established. In the case of the EU-US MRA, the agreement mandates that additional resources be used for auditing each EU Member State authority every 5 to 6 years because the actual inspection activities are performed by the individual Member State inspectorates rather than by a central agency inspectorate as in the United States.4 Often these maintenance costs are not anticipated when the parties first agree to establish an MRA.
Given the increasingly complex therapeutic innovations being tested and brought to market and the growing complexity of medicines supply chains, regulatory authorities will have to consider how relying on others could better ensure the adequate supply of safe and efficacious medicines—the mission of every regulatory authority being to help ensure that patients have access to quality medicines in a well-functioning health care system. In the end, the future of medicines regulation will be found in functional regulatory networks of agencies with increasing specialization and reliance on each other’s work, in parallel with decreasing duplicative efforts.
CHALLENGES TO RELIANCE BETWEEN REGULATORY AUTHORITIES IN A GLOBALIZED WORLD
Medicines Production in and Exportation from China and India
Active Pharmaceutical Ingredients
WHO (2017a) has recognized China as an emerging leader in the supply of active pharmaceutical ingredients (APIs). Over the past 30 years,
___________________
4 United States-European Union Amended Sectoral Annex for Pharmaceutical Good Manufacturing Practices (GMP) Appendix 4, Section 3.
China has seen growth in its pharmaceutical manufacturing with respect to the number of facilities and types of manufacturing, and, until recently, without a commensurate development of its medicines regulatory framework (WHO, 2017a). China is now the leading producer and exporter of APIs by volume, manufacturing more than 2,000 APIs (WHO, 2017a). While efforts are under way through cooperative agreements and other regulatory strengthening activities (WHO, 2017a) to help China implement international GMP, it appears that the manufacturing of falsified or substandard medicines and fraudulent clinical and manufacturing data remain challenges in many less resourced manufacturing sites (EMA, 2019d; Rees, 2019a; UNODC, 2019). The international regulatory capacity-building activities being undertaken are helping China better ensure the production of APIs that consistently meet international quality standards. However, the recent drug recall of valsartan highlights the continuing risks associated with the importation of medicines and APIs from countries with less stringent and less consistent regulatory controls (Byrd et al., 2019; EMA, 2019c; WHO, 2017a).
Generics
India, like China, is a major supplier of generic medicines to the world. According to the India Brand Equity Foundation (IBEF, 2019), the Indian pharmaceutical sector accounts for 71 percent of the market share of generic drugs and supplies more than 50 percent of the world’s vaccines. While the generics industry remains robust in India, the government has signaled a desire to expand its market while also making India a major hub for low-cost drug research and development (Indian Department of Pharmaceuticals, 2018). Yet, FDA and other regulators document repeated problems with quality manufacturing at many of the Indian sites that wish to export products to their countries (Rees, 2019b).
A Changing World
China has aspirations to shift the primary focus of its manufacturing from APIs for export to the more lucrative export of finished products and to become a world leader in research and development. However, it does not appear this will happen in the immediate future (Ni et al., 2017; WHO, 2017a). India, Brazil, and other middle-income countries also have aspirations to increase their abilities to develop and produce new medicines for both local and global consumption. These aspirations for expanding markets speak to the larger issue that the pharmaceutical landscape is constantly evolving and changing. India and China may be the world’s major suppliers of APIs and generics in today’s market, but as production costs in
China and India rise, new countries may enter into those market segments. Regulators need to be prepared for such shifts in the landscape to ensure the quality, safety, and efficacy of medicines regardless of the location of their production.
In addition, the pharmaceutical industry is continually advancing. The entire drug research and development enterprise is globalizing rapidly. Regulators cannot remain static in their acquisition of knowledge and their incorporation of the talent needed to understand new technological advances in precision medicine and drug development. It will be impossible to predict the exact skills needed to ensure the quality, safety, and efficacy of complex and innovative medicinal products such as complex biologics and gene and cell therapies. However, the establishment of a strong system of reliance can build a network of trusted experts from different agencies that can be drawn upon for their valuable insight when a product development or assessment challenge arises that has not previously been encountered or addressed.
The Need for Mutual Recognition of Third-Country Inspections
Given the large number of manufacturing sites in China and India that are involved in producing drug components and/or final products for the United States and Europe, it has not been possible for either FDA or EMA to inspect all of these sites as they would like to do in order to assure the quality of products being exported to their people. However, recent updates to the EU-US MRA on pharmaceutical GMP do make provisions for mutual recognition of inspections conducted by the parties to the MRA in third countries.5 Although these provisions regarding recognition of third-country inspections have not yet been implemented, the expectation is that this MRA, which became operational in July 2019, will allow greater sharing between regulatory bodies in the United States and Europe. This increased sharing will avoid duplication of efforts, thereby allowing those
___________________
5 Art. 1 “The provisions of this Annex apply to pharmaceutical inspections of manufacturing facilities carried out in the territory of a Party during the marketing of products (hereafter referred to as ‘post-approval inspections’) and, to the extent provided for in Article 11, before products are marketed (hereafter referred to as ‘pre-approval inspections’), as well as, to the extent provided for in Article 8.3, to pharmaceutical inspections of manufacturing facilities carried out outside the territory of either Party.” [emphasis added]
Art. 8.3 “A Party may accept official GMPs documents issued by a recognized authority of the other Party for manufacturing facilities located outside the territory of the issuing authority.” [emphasis added] (Brussels, 1.3.2017 C(2017) 1323 final ANNEX to the Commission Decision on determining the Union position for a Decision of the Joint Committee set up under Article 14 of the Agreement on Mutual Recognition between the European Community and the United States of America, in order to amend the Sectoral Annex on Pharmaceutical Good Manufacturing Practices [GMPs].)
regulators to redistribute their inspectional resources to higher-risk facilities both within their own borders and in third countries.
Predating the 2019 implementation of the EU-US MRA was the International API Inspection Program, which had grown from a pilot to a full program by 2011. The program created a framework for greater international collaboration and information sharing on GMP inspections of API manufacturers worldwide (EMA, 2018a). In 2016, it included regulatory agencies from Australia, Canada, Denmark, France, Germany, Ireland, Italy, Japan, the United Kingdom, and the United States.6 The work focused primarily on sharing plans for, outcomes of, and reports stemming from the GMP inspections. Most of the information shared was through monthly teleconferences and electronic exchanges. A spreadsheet (the Master List) of all API sites of interest to all those participating in the program formed the foundation for a mapping exercise carried out by the International Coalition of Medicines Regulatory Authorities showing the API manufacturing sites of common interest. This information is currently stored in the secure European database for GMP and good distribution practice inspections and certification platform, EudraGMDP, where participating countries take responsibility for updating their own information. Box 3-2 provides a summary of an evaluation of the program, and Appendix C presents a chart, adapted from the work of the International Coalition of Medicines Regulatory Authorities, showing the objective, scope, membership, and work products of global initiatives (EMA, 2016).
___________________
6 The European Directorate for the Quality of Medicines and HealthCare from the Council of Europe and WHO were also considered members; Germany’s engagement ended in 2016.
This page intentionally left blank.


















