
6
Selected Central Nervous System Tumors
Pediatric central nervous system (CNS) tumors are a heterogeneous group of tumors that can form anywhere in the brain and spinal cord of children. The seven most common CNS tumors that are diagnosed and treated in children are glial, ependymal, pineal, embryonal, CNS germ cell, choroid plexus, and craniopharyngioma tumors. The focus of this chapter is on the diagnosis, prognosis, treatment, and outcomes of these pediatric tumors. The chapter begins with general information about CNS tumors, which is followed by a review of the diagnostic and prognostic information, standard treatments, and probable outcomes for each tumor. The chapter ends with a summary of the committee’s findings and conclusions. The epidemiology of pediatric CNS tumors is discussed in detail in Chapter 2.
OVERVIEW
This section is organized into the following four subsections that describe CNS tumors in general terms: the tumor classification scheme, common diagnostic methods, overall survival (OS) rates, and basic elements of treatment. The main sources of information on the CNS tumors discussed in this chapter include the National Cancer Institute’s Physician Data Query® (see Appendix C), the Children’s Oncology Group, and other published literature on CNS tumors.
Central Nervous System Tumor Classification
The 2016 World Health Organization (WHO) classification of CNS tumors is the standard scheme currently used by oncologists and pathologists (Louis et al., 2016). The classification scheme described in this edition of the WHO classification introduced for the first time tumor classification based on genetic and other molecular features in addition to the classic scheme based largely on histology. This is a rapidly evolving area of investigation that will most likely bring further modifications in the future. The classification of pediatric CNS tumors, therefore, is based on microscopic examination of the size, shape, structure, and staining characteristics of tumor cells/tissue, as well as the identification of genetic and molecular abnormalities found in the tumor (i.e., biologic markers). This integrated approach to tumor classification allows for a more precise clinical diagnosis of CNS tumors, which can enable more specific and more effective therapeutic approaches for children diagnosed with these tumors (Kristensen et al., 2019).
The CNS tumor classification scheme uses a grading system to describe certain features of the tumor that are linked to specific outcomes. WHO divides brain and spinal cord tumors into four grades (Gupta and Dwivedi, 2017). Tumor grade, determined by a pathologist, reflects the amount of cell abnormality and is an indicator of how quickly a tumor is likely to grow into nearby areas and destroy healthy tissue. In general, the lower the grade, the better the prognosis. The four grades are as follows:
- Grade I: These tumors typically grow slowly and do not grow into nearby tissue. They are associated with long-term survival and can often be cured with surgery alone.
- Grade II: These tumors also tend to grow slowly, but they can grow into nearby brain tissue and can reoccur as higher-grade tumors.
- Grade III: These tumors can grow into nearby brain tissue and are more likely than grade I or II tumors to need other treatments in addition to surgery. They often reoccur as grade IV.
- Grade IV: These are the fastest-growing tumors and generally require the most aggressive treatment.
Diagnostic Evaluation
Diagnostic evaluation of a CNS tumor involves a number of scans, tests, and procedures (PDQ Pediatric Editorial Board, 2020c). The assessment usually begins with imaging tests, most commonly magnetic resonance imaging (MRI) of the brain, spinal cord, or both, depending on the type of tumor suspected. The basis for a definitive diagnosis is a biopsy procedure to remove a sample of the tissue for microscopic evaluation. Biopsies of CNS tumors
may be performed with a needle or with surgery. With some diagnoses, a lumbar puncture to collect cerebrospinal fluid (CSF) is performed to evaluate for floating tumor cells as part of the grading procedure. Depending on various factors, the aim of the surgery is to remove either the entire area of abnormal cells (resection) or just part of the abnormal area. In some cases, molecular testing is performed on a tumor sample to identify specific genes, proteins, and other factors, such as tumor markers, unique to the tumor.
For many types of tumors in other parts of the body, a staging system is used to describe where a tumor is located and whether it has spread. For CNS tumors, however, there is no universally accepted or recommended staging system because, unlike other cancers, tumors that start in the brain or spinal cord rarely spread to distant organs outside of the CNS (PDQ Pediatric Editorial Board, 2020c). In general, pediatric CNS tumors are classified as either localized, meaning confined to one area of the brain or spine, or metastatic, denoting spread to other, distant parts of the brain and spine. Typically, patients with metastatic disease have a worse prognosis. The WHO grading system described above is used to determine how cancerous the tumor is, how quickly it will grow, and the likelihood it will spread within the CNS.
Figure 6-1 illustrates the elements of brain anatomy and provides a useful reference for subsequent discussion of the location of specific CNS tumors.
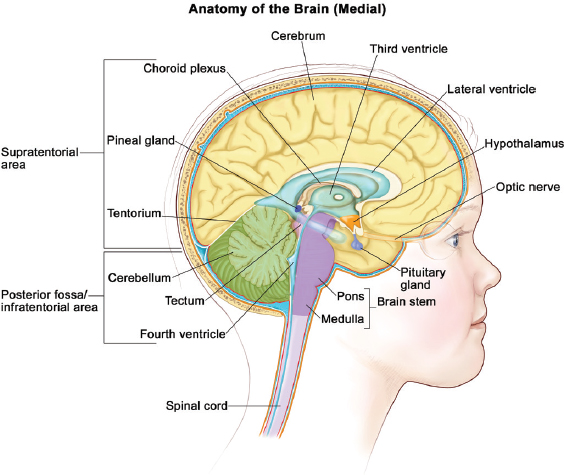
SOURCE: © 2013 Terese Winslow, U.S. Govt. has certain rights.
Overall Survival Rates
More than 70 percent of children diagnosed with brain tumors will survive for more than 5 years after diagnosis; however, survival rates are wide-ranging depending on tumor type and stage (PDQ Pediatric Editorial Board, 2020b). Although the survival rate is promising, moreover, it does not reflect the amount of impairment the child may experience as a result of the tumor or its treatment. In later sections of this chapter describing specific CNS tumors, the committee reports a 5-year OS rate for each tumor type and identifies the sequelae related to both the effects of the tumor and its treatment. The 5-year OS rate indicates the percentage of children who are living 5 years after diagnosis, but does not specify whether survivors are still undergoing treatment at 5 years or whether they have remained disease free. Note also that the survival time for an individual person may be higher or lower than the OS rate, depending on various tumor and patient characteristics.
Treatment Approaches
Treatment strategies for pediatric CNS tumors vary depending on the tumor type and its location, whether the tumor is newly diagnosed or recurrent, its grade and molecular features, and specific patient characteristics (such as age). The goal of treatment for patients with a CNS tumor is cure, but this goal is not always attainable, and within the realm of CNS tumors, stable disease is often considered a successful outcome. The standard therapies for CNS tumors are surgery, radiation therapy, and chemotherapy (discussed briefly below) (PDQ Pediatric Editorial Board, 2020c), sometimes in combination. In addition, new types of treatment are being tested in clinical trials. Chapter 3 presents a detailed discussion of the various treatment modalities for childhood cancers in general, including new treatments that are emerging, such as MEK and BRAF inhibitors for specific low-grade gliomas (LGGs).
On the basis of the research evidence and the committee’s clinical experience, the committee estimates that the duration of treatment for pediatric CNS tumors and a child’s recovery from acute treatment effects can range on average from less than 1 month to 2 years, depending on the type of tumor and treatment. Sequelae related to the effects of both the tumor and its treatment are common in CNS tumors (see the next section). Overall, survivors of pediatric cancer require close monitoring because the side effects of cancer therapy may persist or develop months or years after treatment. The functional limitations associated with the different treatment modalities are discussed in Chapter 4.
Surgery
The most common surgery for brain tumors is craniotomy, in which the skull is opened to remove the tumor. A patient’s prognosis is usually better when all of the tumor can be surgically removed. Although some brain tumors can be removed with little or no harm to the brain, many grow in areas that make them difficult or impossible to remove without damaging important parts of the brain. Many children with CNS tumors will require a ventriculoperitoneal shunt or third ventriculostomy for uncontrolled hydrocephalus. These are considered common and standard procedures in this population.
Radiation
Radiation therapy may be able to stop or slow the growth of childhood brain tumors that cannot be removed with surgery. Debilitating effects on the child’s growth and neurologic development have frequently been observed after radiation therapy, especially in younger children, and secondary tumors have increasingly been diagnosed in long-term survivors (PDQ Pediatric Editorial Board, 2020b). As discussed in the next section, however, there are some tumors for which radiation therapy is the best available treatment option, either alone or in combination with surgery and chemotherapy.
Chemotherapy
Chemotherapy, either alone or often in conjunction with radiation and surgery, is frequently a treatment for tumors that are growing quickly. Detail on when chemotherapy is indicated for particular CNS tumor types is included in the next section.
In sum, treatment of pediatric cancers is complex and requires a multidisciplinary approach. Optimal therapeutic management of children with primary brain or spinal cord tumors requires the coordinated efforts of a team of pediatric specialists who have expertise in the care of patients with these diseases (PDQ Pediatric Editorial Board, 2020c), including the following specialists: pediatrician, pediatric oncologist, neuro-oncologist, neurologist, neurosurgeon, radiation oncologist, neuroradiologist, neuropathologist, endocrinologist, ophthalmologist, audiologist, nurse specialist, psychologist, neuropsychologist, rehabilitation specialist, social worker, and others.
DIAGNOSIS, PROGNOSIS, TREATMENT, AND OUTCOMES BY TUMOR TYPE
This section provides information about the diagnosis, prognosis, treatment, and outcomes of the seven common pediatric CNS tumors addressed
in this chapter (glial, ependymal, pineal, embryonal, CNS germ cell, choroid plexus, and craniopharyngioma tumors). For each tumor, detail is provided on the tumor classification (type and subtypes); diagnostic information (methods, staging, and tumor location); survival prognosis; the standard therapies and duration of treatment; and long-term impairments specific to that tumor, which are a consequence of the tumor or its treatment.
Annex Tables 6-1 and 6-2 summarize the diagnostic, prognostic, and treatment information for each type of tumor discussed below.
Glial Tumors
The terms glioma and astrocytoma, commonly used interchangeably to describe the same type of CNS tumor, denote the most common types of pediatric CNS tumors. In general, these tumors are divided into two main categories: high-grade gliomas (HGGs) and LGGs (PDQ Pediatric Editorial Board, 2020a). The two are discussed separately below as they are treated differently and have distinct survival outcomes.
High-Grade Gliomas
This general category includes diffuse midline glioma (H3K27M+), anaplastic astrocytoma (WHO grade III), glioblastoma multiforme (WHO grade IV), oligodendroglioma (WHO grade III), oligoastrocytoma (WHO grade III), anaplastic pleomorphic xanthoastrocytoma (WHO grade III), gliosarcoma (WHO grade IV), and HGG not otherwise specified (NOS) (WHO grade III/IV) (Louis et al., 2016).
Pediatric HGGs behave very aggressively, and these children have a very poor prognosis even with use of a variety of therapies, including chemotherapy and radiotherapy, described below. Despite the histologic similarities to adult HGGs, pediatric HGGs are unique and distinct from their adult counterparts. They are classified by WHO as grade III and IV tumors. The etiology of these tumors is unknown, although there are predisposing risk factors in a very small group of patients, including previous exposure to CNS radiation (for treatment of leukemia, for example) and genetic cancer predisposition syndromes, such as Li-Fraumeni syndrome (Pollack et al., 2019). However, these predisposing risk factors are extremely rare, and most cases of pediatric HGG arise in children and adolescents for unknown reasons. Given their extremely poor survival outcomes, all HGGs are considered high risk: the vast majority of children will not survive the disease, and the published 5-year survival rates are less than 20 percent (Braunstein et al., 2017). Although rare, metastatic spinal disease and primary spinal HGG can occur, and for this reason, many practitioners will perform both baseline brain and spine MRI. HGGs in the midline with a unique
molecular abnormality called an H3K27M mutation have recently been designated by WHO as diffuse midline gliomas (Louis et al., 2016). Within the HGG category, these diffuse midline gliomas have a worse prognosis. One specific tumor in this category is a diffuse intrinsic pontine glioma (DIPG), which is usually fatal despite treatment (Vitanza and Monje, 2019).
The main treatment for HGG is a maximum surgical resection whenever possible; however, complete surgical resection is not safe or feasible for many patients with HGGs, such as those whose tumors are in midline locations, those with metastatic disease, and those with diffuse brain involvement. In these cases, a biopsy is often performed to confirm the diagnosis. In some patients, a DIPG diagnosis may be made by MRI alone with a classic appearance on imaging; however, biopsies are increasingly being performed on these lesions as a standard practice. In most children with HGG, the standard treatment approach is enrollment in a clinical trial pairing new agents or treatment modalities with radiation therapy (Fangusaro and Warren, 2013), focal radiation therapy being one of the only proven standard therapies that may provide some delay in progression of HGG. There are some published clinical trials involving the use of adjuvant classic chemotherapy (temozolomide and lomustine [CCNU], for example) in addition to radiation. Some practitioners may use these agents outside of a clinical trial as well, particularly in patients without diffuse midline gliomas (Cohen et al., 2011; Jakacki et al., 2016). In patients with metastatic disease, more extensive radiation (such as craniospinal radiation) may be utilized. In very young children with HGG (typically infancy through 3 years old), there may be a role for use of other standard chemotherapy as well. It appears that the biology of these HGGs in very young children is unique, and survival may be slightly improved for these children relative to their older counterparts.
Most impairments caused by HGG are due to the tumor location and treatment interventions. Patients may develop paralysis, seizures, swallowing difficulties, endocrine dysfunction, and complications from surgery and radiation therapy (Ullrich, 2009). Most children will not survive their disease, and the vast majority will die within 9 to 12 months from diagnosis despite the best available accepted therapy (Pollack et al., 2019).
Low-Grade Gliomas
This general category includes pilocytic astrocytoma, diffuse/fibrillary astrocytoma (WHO grade II), pleomorphic xanthoastrocytoma (WHO grade II), neuronal and mixed neuronal-glial tumors (WHO grade I and II), and LGG NOS (WHO grade I and II) (Louis et al., 2016).
LGGs are glial tumors classified by WHO as grade I and II. These are by far the most common pediatric CNS tumors. The etiology of these
tumors is largely unknown. However, children with the genetic disorder neurofibromatosis type 1 (NF1) are predisposed to developing LGG, most commonly in the optic tract and brainstem; approximately 20–30 percent of patients with NF1 will develop LGG (Packer et al., 2020). Although the vast majority of patients with LGG will survive their disease long-term, many will suffer long-term impairments and functional deficits, as described below. Among children with LGG, those with WHO grade II tumors (versus grade I), tumors in midline locations (such as hypothalamic and brainstem tumors), and metastatic disease to other parts of the brain and spine are often more difficult to treat and have worse long-term survival (Bandopadhayay et al., 2014). Although rare, metastatic spinal disease and primary spinal LGG can occur, and for this reason, many practitioners will perform both baseline brain and spine MRI.
The classic treatment for LGG is to resect as much of the tumor as is safe and feasible. For many children with tumors not in midline or other delicate locations (such as the optic tract or brainstem), a gross total resection of the tumor may be curative. After surgery, those patients with residual tumor or progression despite surgery are often treated with classic chemotherapy. The following chemotherapies are commonly utilized for children with LGG: a combination of carboplatin and vincristine; a combination of thioguanine, procarbazine, CCNU, and vincristine; carboplatin alone; vinblastine alone; and a combination of bevacizumab and irinotecan (de Blank et al., 2019).
Recently, a wealth of information about the molecular drivers of pediatric LGG has been discovered. These data have revealed that the mitogen-activated protein kinase (MAPK) pathway drives most LGG, and there commonly are abnormalities in the BRAF oncogene in most LGG (Jones et al., 2018). New oral medicines can target these abnormalities, and ongoing trials are comparing these new agents with classic chemotherapy. Some children with LGG may be enrolled in these trials and/or treated with these new oral biologically targeted agents both in and outside of clinical trials (Jones et al., 2018). The exact duration of treatment with these new targeted agents is as yet unknown; many children may be on these treatments indefinitely. Radiation is an effective treatment strategy in LGG; however, given the long-term risks of endocrine and neurocognitive dysfunction as well as the risk of secondary malignancy associated with this therapy, it is often used only in children with LGG for whom all other treatment options have been exhausted or who have a more clinically aggressive LGG (de Blank et al., 2019).
Although most children with LGG will survive their disease, they frequently are left with numerous long-term impairments and functional deficits, often due to the tumor location and treatment interventions. Patients may develop paralysis; motor dysfunction; seizures; swallowing difficulties;
vision dysfunction; endocrine dysfunction (hormone dysregulation); and complications from surgery, chemotherapy, and radiation therapy (when utilized) (Ullrich, 2009).
Central Nervous System Germ Cell Tumors
Primary CNS germ cell tumors (GCTs) are a heterogeneous group of tumors that are more common in Asian than in other countries, the genetic and environmental reasons for which remain unknown. In the United States, CNS GCTs account for approximately 2–4 percent of all primary brain tumors, with a peak incidence from ages 10 to 19 years. They are more common in males than in females, and the pineal region is their most common anatomic location, followed by the suprasellar (pituitary) region (Fujimaki, 2009). Histologically (under a microscope), they appear similar to extra-CNS gonadal GCTs, such as testicular and ovarian tumors. CNS GCTs are broadly classified into germinomas and non-germinomatous GCTs (NGGCTs). These two types of GCT are discussed together below, with distinctions pointed out as appropriate.
The etiology of CNS GCTs is largely unknown. Historically, NGGCTs have had a worse prognosis than germinomas; however, with modern multimodal therapies, including both chemotherapy and radiotherapy, these survival differences have become less dramatic. A unique characteristic of CNS GCTs is that they may secrete tumor markers that can be detected and measured in the blood and CSF. In some circumstances, elevations in tumor markers paired with consistent imaging can enable diagnosis without the need for a biopsy. For this reason, as part of their evaluation, these patients require the measurement of tumor markers (beta-human chorionic gonadotropin and alfa-fetoprotein) in both the blood and CSF. Although rare, metastases into the spine and primary spinal GCTs can occur. For this reason, patients require both brain and spine MRI to identify the primary disease and any areas of metastasis. These patients also require a lumbar spinal tap to check CSF for metastatic tumor cells.
The treatment of CNS GCTs includes chemotherapy followed by radiation therapy. The duration of chemotherapy and the dose and volume of radiation differ for germinomas and NGGCTs, with NGGCTs typically being treated with more cycles of chemotherapy and more extensive radiotherapy. Common chemotherapy agents used in CNS GCT include carboplatin, ifosfamide, and etoposide. Recently completed and ongoing clinical trials are exploring reduction of the dose and volume of radiotherapy based on a good response to chemotherapy (COG, 2019; Fangusaro et al., 2019). With the use of these multimodal approaches, survival outcomes have improved.
Although most children with CNS GCTs will survive their disease, they frequently are left with numerous long-term impairments and functional deficits, which are often due to the tumor location and treatment interventions. Because these tumors are most commonly located in the pineal and pituitary regions, many survivors will suffer from vision dysfunction and endocrine dysfunction (hormone dysregulation) that will require long-term management. Patients may also develop paralysis; motor dysfunction; seizures; swallowing difficulties; rare psychiatric side effects; and complications from surgery, chemotherapy, and radiation therapy.
Choroid Plexus Tumors
Choroid plexus tumors (CPTs) are rare CNS tumors that arise from a structure within the brain called the choroid plexus, which lines the ventricles of the brain and normally functions to produce CSF. CPTs are most common in children within the first year of life; however, they can be seen in older children and rarely in adults. WHO divides them into three main categories: grade I choroid plexus papilloma (CPP), grade II atypical choroid plexus papilloma (aCPP), and grade III choroid plexus carcinoma (CPC). The risk and survival outcomes vary from grade I (best outcomes) to grade II (intermediate outcomes) to grade III (worst outcomes). The etiology of CPT is largely unknown; however, there is some evidence that patients with the cancer predisposition syndrome—Li-Fraumeni syndrome—are at higher risk for developing CPC (Orr et al., 2019). CPP is a localized disease; however, both aCPP and CPC can metastasize, and for this reason, both a spine MRI and a lumbar CSF collection to evaluate for tumor cells are commonly obtained in patients with these two types of CPT.
The standard of care for CPP is surgical resection. With a gross total resection, these patients have excellent long-term survival. The management of aCPP and CPC is less standardized. A maximum surgical resection is the first-line treatment for all CPT; however, adjuvant treatment in aCPP and CPC varies depending on patient age, residual tumor, and metastases. Many patients with CPC, in particular, are treated with combinations of standard chemotherapy followed by radiotherapy (Lafay-Cousin et al., 2011). Commonly utilized chemotherapy for CPC includes ifosfamide, carboplatin, and etoposide. The dose and volume of radiation vary.
Most impairments caused by CPT are due to the tumor location and treatment interventions. Patients may also develop paralysis; motor dysfunction; seizures; swallowing difficulties; and complications from surgery, chemotherapy, and radiation therapy. Because many children with CPT are very young at diagnosis, the tumor location, surgical intervention, and potential radiotherapy may lead to long-term neurocognitive dysfunction.
Embryonal Tumors
Embryonal tumors are malignant WHO grade IV cancers that occur primarily in children. The recent revision of the WHO Classification of Tumors of the Central Nervous System, published in 2016 (Louis et al., 2016), significantly changed the classification of these tumors, so that they now have a molecular classification in addition to traditional histologic criteria. The current classification includes medulloblastoma (subtypes SHH TP53 wild type, SHH TP53 mutant, WNT, MYC amplified Group 3 [non-SHH/WNT]), atypical teratoid rhabdoid tumor, and embryonal tumor with multilayered rosettes (ETMR). The diagnoses of ependymoblastoma and primitive neuroectodermal tumor have been discontinued. These tumors grow as a mass in the CNS (medulloblastomas grow in the infratentorial compartment, specifically), and have high potential to disseminate throughout the CNS, and rarely to organs outside of the CNS.
The treatment of embryonal tumors includes attempted surgical resection, fractionated external beam radiation to the entire brain and spine with a focal boost to the tumor bed for approximately 6 weeks, and multidrug chemotherapy for an additional 12–15 months for children older than 3 years. Children younger than 3 years undergo surgical resection followed by chemotherapy for 12 months (Rutkowski et al., 2005). The survival prognosis for the medulloblastoma WNT subtype is excellent. The survival prognosis for the medulloblastoma SHH TP53 mutant and MYC amplified Group 3 (non-SHH/WNT) subtypes and ETMR is extremely poor (Kool et al., 2012; Spence et al., 2014; Taylor et al., 2012). The other embryonal tumors have a variable prognosis, as shown in Annex Table 6-2 (Slavc et al., 2014).
Multimodal therapy for embryonal tumors is of high intensity, typically requiring at least 6 months’ recovery after treatment has been completed before the child has recovered sufficiently to resume activities, including full-time attendance at school. Specific neurologic impairments are highly related to the location of the primary tumor in the brain. The chemotherapy regimen typically includes cisplatin, associated with a high incidence of hearing loss, and vincristine, associated with peripheral neuropathy. Radiation to the whole brain, especially for children younger than 5 years, is associated with long-term cognitive deficiencies, increased risk of cerebrovascular disorders, cataracts, second malignancies, shortened truncal height, hormonal insufficiencies, and epilepsy (Walter et al., 1999).
Ependymal Tumors
Ependymal tumors are a class of glial tumors that can occur in the brain or spine. As noted above, the recent revision of the WHO Classification of Tumors of the Central Nervous System (Louis et al., 2016) includes
molecular classification in addition to traditional histologic criteria. The current classification of ependymal tumors includes WHO grade II and III ependymoma and ependymoma RELA fusion-positive, and myxopapillary ependymoma (WHO grade I). For children, the majority of ependymomas occur in the posterior fossa. There are two principal subtypes of posterior fossa ependymomas in children. Posterior fossa A tumors occur in younger children often off midline, have a balanced genotype, and have a poor clinical outcome. Posterior fossa B tumors occur in the midline fourth ventricle of older children, have many somatic DNA copy number abnormalities, and have an overall better prognosis (Archer and Pomeroy, 2011; Witt et al., 2011). Supratentorial and spinal ependymomas account for approximately 40 percent of tumors. Metastasis throughout the CNS occurs in fewer than 10 percent of cases and is associated with a poor prognosis.
Treatment of ependymomas consists of surgical resection followed by focal external beam radiation to the tumor and surrounding tissue. Complete resection is associated with the best outcomes. In some cases, radiation may be omitted for patients with completely resected WHO grade II supratentorial tumors or for infants below the age of 1 year. Treatment with chemotherapy is not the standard of care in most cases, although it may be administered for incompletely resected tumors prior to radiation or for infants less than 1 year old to delay radiation treatment. The prognosis is variable, as shown in Annex Table 6-2 (Massimino et al., 2016). The treatment for subependymomas and myxopapillary ependymomas is surgical resection alone. Both of these tumors have an excellent prognosis.
Neurologic impairments are related to the site of the tumor. Long-term effects of treatment with focal radiation are related to the site of the tumor and include increased risk of second malignancy and cerebrovascular effects within the field of radiation.
Pineal Tumors
Pineal tumors are rare tumors that arise in the pineal parenchyma. There are three primary types of pineal tumors in children: pineocytoma (WHO grade I), pineal parenchymal tumor (WHO grade II or III), and pineoblastoma (WHO grade IV). Pineocytomas remain in the pineal region and rarely spread, whereas pineal parenchymal tumors spread in 10–15 percent of cases, while pineoblastomas spread throughout the CNS in up to 35 percent of cases (PDQ Pediatric Editorial Board, 2020d).
Treatment for pineal tumors starts with attempted surgical resection. Completely resected pineocytomas often require no additional therapy. For pineal parenchymal tumors and pineoblastomas, surgical resection is typically followed by fractionated external beam radiation to the entire brain and spine with a focal boost to the tumor bed for approximately 6 weeks,
and multidrug chemotherapy for an additional 12–15 months for children older than 3 years. Children younger than 3 years undergo surgical resection followed by chemotherapy for 12 months (Raleigh et al., 2017). The survival prognosis for pineocytomas and pineal parenchymal tumors is variable, whereas pineoblastomas have an overall poor prognosis.
Multimodal therapy for pineal tumors is of high intensity, typically requiring at least 6 months’ recovery after treatment has been completed before the child has recovered sufficiently to resume activities, including full-time attendance at school. Specific neurologic impairments are highly related to the location of the primary tumor in the pineal gland, the severity of hydrocephalus at presentation, and craniospinal radiation. The chemotherapy regimen typically includes cisplatin, associated with a high incidence of hearing loss, and vincristine, associated with peripheral neuropathy. Radiation to the whole brain, especially for children younger than 5 years, is associated with long-term cognitive deficiencies, increased risk of cerebrovascular disorders, cataracts, second malignancies, shortened truncal height, hormonal insufficiencies, and epilepsy (Walter et al., 1999).
Craniopharyngiomas
Craniopharyngiomas are considered benign tumors of epithelial origin. The majority of childhood craniopharyngiomas arise in the suprasellar region and involve the region behind the chiasm, and may extend into the third ventricle and along the hypothalamus. There are two types of craniopharyngiomas, the most common in children being adamantinomatous craniopharyngioma. These tumors are typically calcified, cystic, solid tumors that can often include complex cysts and fluid containing “crankcase oil.” The second type of craniopharyngioma is papillary craniopharyngioma, which occurs almost exclusively in adults.
Children with craniopharyngiomas can present with visual field deficits or decreased vision as well as headaches, nausea, and vomiting (with increased intracranial pressure). More than 50 percent will present with endocrine deficits at diagnosis (Merchant et al., 2002).
Treatment generally entails surgical removal or partial surgical removal followed by local radiation therapy to the tumor site. Because of the complex location of these tumors, surgical removal and radiation therapy can both cause significant long-term impairments, making treatment of this “benign” disease difficult. When radiation therapy is administered, it is given in a fractionated (daily) course over 5–6 weeks. Disease control rates may be as high as 80 percent 5 years after treatment (Zacharia et al., 2012). Cognitive impairment, visual deficits, endocrine deficits, and neurologic deficits all contribute to the complex long-term management of this disease (Eveslage et al., 2019).
FINDINGS AND CONCLUSIONS
Findings
6-1 Classification of pediatric central nervous system (CNS) tumors is based on microscopic examination of the size, shape, structure, and staining characteristics of tumor cells/tissue, as well as identification of genetic and molecular abnormalities found in the tumor (i.e., biologic markers).
6-2 The World Health Organization (WHO) CNS tumor classification scheme uses a grading system to describe certain features in the tumor that are linked with specific outcomes. WHO divides brain and spinal cord tumors into four grades.
6-3 The basis for a definitive diagnosis in pediatric CNS tumors is a biopsy procedure to remove a sample of the tissue for microscopic evaluation.
6-4 In general, pediatric CNS tumors are classified as localized, meaning confined to one area of the brain or spine, or metastatic, meaning there is spread to other, distant parts of the brain and spine.
6-5 The treatment strategies for pediatric CNS tumors vary depending on the tumor type and its location, whether the tumor is newly diagnosed or recurrent, the tumor’s grade and molecular features, and specific patient characteristics (such as age). The standard therapies are surgery, radiation therapy, and chemotherapy, sometimes in combination.
6-6 More than 70 percent of children diagnosed with brain tumors will survive for more than 5 years after diagnosis; however, survival rates are wide-ranging depending on the tumor type and stage.
6-7 Long-term sequelae related to the effects of both the tumor and its treatment are common.
6-8 Optimal therapeutic management of children with primary brain or spinal cord tumors requires the coordinated efforts of a team of pediatric specialists who have expertise in the care of patients with these diseases.
Conclusions
6-1 Classification of pediatric CNS tumors is not static and will continue to evolve. It will need to be updated as molecular and genetic features are better described and validated.
6-2 In general, the lower the WHO grade assigned to a CNS tumor, the better the prognosis.
6-3 Although in the vast majority of cases, a definitive diagnosis of a specific pediatric CNS tumor is made by biopsy and pathologic
evaluation, diagnoses are also made, though rarely, on the basis of imaging and clinical characteristics alone, as is the case for diffuse intrinsic pontine glioma and neurofibromatosis.
6-4 Children with metastatic CNS tumors, denoting spread to other parts of the brain and spine (or rarely, outside of the CNS), have a worse prognosis relative to nonmetastatic CNS tumors.
6-5 Although the overall survival rate for pediatric CNS tumors is promising, most children experience impairment as a result of the tumor and/or its treatment.
6-6 The morbidity associated with pediatric CNS tumors, including acute, chronic, and late effects, is contingent on the tumor type; the tumor location; the patient’s age; and associated treatments, including surgery, chemotherapy, and radiation. These adverse effects are often greater in children with CNS tumors than in those with other solid tumors because the treatment (surgery, radiation) alters normal brain development.
6-7 The committee estimates that the duration of treatment for CNS tumors and its acute effects can range from less than 1 month to 2 years, depending on the aforementioned characteristics (tumor type and location, patient characteristics, and treatment).
REFERENCES
Al-Hussaini, M., I. Sultan, N. Abuirmileh, I. Jaradat, and I. Qaddoumi. 2009. Pineal gland tumors: Experience from the SEER database. Journal of Neuro-Oncology 94(3):351–358.
Archer, T. C., and S. L. Pomeroy. 2011. Posterior fossa ependymomas: A tale of two subtypes. Cancer Cell 20(2):133–134.
Bandopadhayay, P., G. Bergthold, W. B. London, L. C. Goumnerova, A. Morales La Madrid, K. J. Marcus, D. Guo, N. J. Ullrich, N. J. Robison, S. N. Chi, R. Beroukhim, M. W. Kieran, and P. E. Manley. 2014. Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: An analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatric Blood & Cancer 61(7):1173–1179.
Braunstein, S., D. Raleigh, R. Bindra, S. Mueller, and D. Hass-Kogan. 2017. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. Journal of Neuro-Oncology 134(3):541–549.
Buscariollo, D. L., H. S. Park, K. B. Roberts, and J. B Yu. 2012. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118:4212–4219.
Calaminus, G., D. Frappaz, R. D. Kortmann, B. Krefeld, F. Saran, T. Pietsch, A. Vasiljevic, M. L. Garre, U. Ricardi, J. R. Mann, U. Göbel, C. Alapetite, M. J. Murray, and J. C. Nicholson. 2017. Outcome of patients with intracranial non-germinomatous germ cell tumors-lessons from the SIOP-CNS-GCT-96 trial. Neuro-oncology 19(12):1661–1672.
Cho, Y. J., A. Tsherniak, P. Tamayo, S. Santagata, A. Ligon, H. Greulich, R. Berhoukim, V. Amani, L. Goumnerova, C. G. Eberhart, C. C. Lau, J. M. Olson, R. J. Gilbertson, A. Gajjar, O. Delattre, M. Kool, K. Ligon, M. Meyerson, J. P. Mesirov, and S. Pomeroy. 2011. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 29(11):1424–1430.
Clark, A. J., M. E. Sughrue, M. E. Ivan, D. Aranda, M. J. Rutkowski, A. J. Kane, S. Chang, and A. T. Parsa. 2010. Factors influencing overall survival rates for patients with pineocytoma. Journal of Neuro-Oncology 100(2):255–260.
COG (Children’s Oncology Group). 2019. Chemotherapy followed by radiation therapy in treating younger patients with newly diagnosed localized central nervous system germ cell tumors. Bethesda, MD: National Library of Medicine. https://clinicaltrials.gov/ct2/show/NCT01602666 (accessed March 23, 2020).
Cohen, K. J., I. F. Pollack, T. Zhou, A. Buxton, E. J. Holmes, P. C. Burger, D. J. Brat, M. K. Rosenblum, R. L. Hamilton, R. S. Lavey, and R. L. Heideman. 2011. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro-Oncology 13(3):317–323.
de Blank, P., P. Bandopadhayay, D. Hass-Kogan, M. Fouladi, and J. Fangusaro. 2019. Management of pediatric low-grade glioma. Current Opinion in Pediatrics 31(1):21–27.
Eveslage, M., G. Calaminus, M. Warmuth-Metz, R. D. Kortmann, F. Pohl, B. Timmermann, M. U. Schuhmann, J. Flitsch, A. Faldum, and H. L. Müller. 2019. The postoperative quality of life in children and adolescents with craniopharyngioma. Deutsches Arzteblatt International 116(18):321–328.
Fangusaro, J., and K. E. Warren. 2013. Unclear standard of care for pediatric high grade glioma patients. Journal of Neuro-Oncology 113(2):341–342.
Fangusaro, J., S. Wu, S. MacDonald, E. Murphy, D. Shaw, U. Bartels, S. Khatua, M. Souweidane, H. Lu, D. Morris, A. Panigrahy, A. Onar-Thomas, M. Fouladi, A. Gajjar, and G. Dhall. 2019. Phase II trial of response-based radiation therapy for patients with localized CNS nongerminomatous germ cell tumors: A Children’s Oncology Group study. Journal of Clinical Oncology 37(34):3283–3290.
Felker, J., and A. Broniscer. 2020. Improving long-term survival in diffuse intrinsic pontine glioma. Expert Review of Neurotherapeutics 20(7):647–658.
Fujimaki, T. 2009. Central nervous system germ cell tumors: Classification, clinical features, and treatment with a historical overview. Journal of Child Neurology 24(11):1439–1445.
Goldman, S., E. Bouffet, P. G. Fisher, J. C. Allen, P. L. Robertson, P. J. Chuba, B. Donahue, C. S. Kretschmar, T. Zhou, A. B. Buxton, and I. F. Pollack. 2015. Phase II trial assessing the ability of neoadjuvant chemotherapy with or without second-look surgery to eliminate measurable disease for nongerminomatous germ cell tumors: A Children’s Oncology Group study. Journal of Clinical Oncology 33(22):2464–2471.
Gupta, A., and T. Dwivedi. 2017. A simplified overview of World Health Organization classification update of central nervous system tumors. 2016. Journal of Neurosciences in Rural Practice 8(4):629–641.
Hosmann, A., F. Hinker, C. Dorfer, I. Slavc, C. Haberler, K. Dieckmann, E. Knosp, and T. Czech. 2019. Management of choroid plexus tumors—An institutional experience. Acta Neurochirurgica 161(4):745–754.
Jakacki, R. I., K. J. Cohen, A. Buxton, M. D. Krailo, P. C. Burger, M. K. Rosenblum, D. J., Brate, R. L. Hamilton, S. P. Eckel, T. Zhou, R. S. Lavey, and I. F. Pollack. 2016. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children’s Oncology Group ACNS0423 study. Neuro-Oncology 18(10):1442–1450.
Jones, D. T. W., M. W. Kieran, E. Bouffet, S. Alexandrescu, P. Bandopadhayay, M. Bornhorst, D. Ellison, J. Fangusaro, M. J. Fisher, N. Foreman, M. Fouladi, D. Hargrave, C. Hawkins, N. Jabado, M. Massimino, S. Mueller, G. Perilongo, A. Y. N. Schouten Van Meeteren, U. Tabori, K. Warren, A. J. Waanders, D. Walker, W. Weiss, O. Witt, K. Wright, Y. Zhu, D. C. Bowers, S. M. Pfister, and R. J. Packer. 2018. Pediatric low-grade gliomas: Next biologically driven steps. Neuro-Oncology 20(2):160–173.
Kool, M., A. Korshunov, M. Remke, D. T. W. Jones, M. Schlanstein, P. A. Northcott, Y. Cho, J. Koster, A. Schouten-Van Meeteren, D. Van Vuurden, S. C. Clifford, T. Pietsch, A. O. Von Bueren, S. Rutkowski, M. McCabe, P. V. Collins, M. L. Backlund, C. Haberler, F. Bourdeaut, O. Delattre, F. Doz, D. W. Ellison, R. J. Gilbertson, S. L. Pomeroy, M. D. Taylor, P. Lichter, and S. M. Pfister. 2012. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathologica 123(4):473–484.
Kristensen, B. W., L. P. Priesterbach-Ackley, J. K. Petersen, and P. Wesseling. 2019. Molecular pathology of tumors of the central nervous system. Annals of Oncology 30(8):1265–1278.
Lafay-Cousin, L., D. Keene, A. Carret, C. Fryer, J. Brossard, B. Crooks, D. Eisenstat, D. Johnston, V. Larouche, M. Silva, B. Wilson, S. Zelcer, U. Bartels, and E. Bouffet. 2011. Choroid plexus tumors in children less than 36 months: The Canadian Pediatric Brain Tumor Consortium (CPBTC) experience. Child’s Nervous System 27:259–264.
Louis, D. N., A. Perry, G. Reifenberger, A. Von Deimling, D. Figarella-Branger, W. K. Cave-nee, H. Ohgaki, O. D. Wiestler, P. Kleihues, and D. D. Ellison. 2016. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathologica 131(6):803–820.
Massimino, M., R. Miceli, F. Giangaspero, L. Boschetti, P. Modena, M. Antonelli, P. Ferroli, D. Bertin, E. Pecori, L. Valentini, V. Biassoni, M. L. Garre, E. Schiavello, I. Sardi, A. Cama, E. Viscardi, G. Scarzello, S. Scoccianti, M. Mascarin, L. Quaglietta, G. Cinalli, B. Diletto, L. Genitori, P. Peretta, A. Mussano, A. Buccoliero, G. Calareso, S. Barra, A. Mastronuzzi, C. Giussani, C. E. Marras, R. Balter, P. Bertolini, E. Giombelli, M. La Spina, F. R. Buttarelli, B. Pollo, and L. Gandola. 2016. Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro-Oncology 18(10):1451–1460.
Matsutani, M., and Japanese Pediatric Brain Tumor Study Group. 2001. Combined chemotherapy and radiation therapy for CNS germ cell tumors: The Japanese experience. Journal of Neuro-Oncology 54 (3):311–316.
Merchant, T. E., O. Goloubeva, D. L. Pritchard, M. W. Gaber, X. Xiong, R. K. Danish, and R. H. Lustig. 2002. Radiation dose-volume effects on growth hormone secretion. International Journal of Radiation Oncology, Biology, Physics 52(5):1254–1270.
Ogiwara, H., A. J. Dipatri Jr., T. D. Alden, R. M. Bowman, and T. Tomita. 2012. Choroid plexus tumors in pediatric patients. British Journal of Neurosurgery 26(1):32–37.
Orr, B. A., M. R. Clay, E. M. Pinto, and C. Kesserwan. 2019. An update on the central nervous system manifestations of Li-Fraumeni syndrome. Acta Neuropathologica 139(4):669–687.
Packer, R. J., A. Iavarone, D. T. W. Jones, J. O. Blakeley, E. Bouffet, M. J. Fisher, E. Hwang, C. Hawkins, L. Kilburn, T. Macdonald, S. M. Pfister, B. Rood, F. J. Rodriguez, U. Tabori, V. Ramaswamy, Y. Zhu, J. Fangusaro, S. S. Johnston, and D. H. Gutmann. 2020. Implication of new understandings of gliomas in children and adults with NF1: Report of a consensus conference. Neuro-Oncology 22(6):773–784.
Pajtler, K. W., H. Witt, M. Sill, D. T. W. Jones, V. Hovestadt, F. Kratochwil, K. Wani, R. Tatevossian, C. Punchihewa, P. Johann, J. Reimand, H. J. Warnatz, M. Ryzhova, S. Mack, V. Ramaswamy, D. Capper, L. Schweizer, L. Sieber, A. Wittman, Z. Huang, P. van Sluis, R. Volckmann, J. Koster, R. Versteeg, D. Fults, H. Toledano, S. Avigad, L. Hoffman, A. Donson, N. Foreman, E. Hewer, K. Zitterbart, M. Gilbert, T. Armstrong, N. Gupta, J. Allen, M. Karajannis, D. Zagzag, M. Hasselblatt, A. Kulozik, O. Witt, V. Collins, K. von Hoff, S. Rutkowski, T. Pietsch, G. Bader, M.-L. Yapso, A. von Deimling, P. Lichter, M. Taylor, R. Gilbertson, D. Ellison, K. Aldape, A. Korscunov, M. Kool, and S. Pfister. 2015. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27(5):728–743.
PDQ Pediatric Editorial Board (Physician Data Query® Pediatric Treatment Editorial Board). 2020a. PDQ childhood astrocytomas treatment. Bethesda, MD: National Cancer Institute. Updated January 16, 2020. https://www.cancer.gov/types/brain/hp/child-astrocytoma-treament-pdq (accessed June 24, 2020).
PDQ Pediatric Editorial Board. 2020b. PDQ childhood brain and spinal cord tumors treatment overview. Bethesda, MD: National Cancer Institute. Updated March 17, 2020. https://www.cancer.gov/types/brain/hp/child-brain-treatment-pdq (accessed March 30, 2020).
PDQ Pediatric Editorial Board. 2020c. PDQ childhood brain and spinal cord tumors treatment overview. Bethesda, MD: National Cancer Institute. Updated May 7, 2020. https://www.cancer.gov/types/brain/patient/child-brain-treatment-pdq (accessed May 26, 2020).
PDQ Pediatric Editorial Board. 2020d. Childhood medulloblastoma and other central nervous system embryonal tumors treatment. https://www.cancer.gov/types/brain/hp/child-cns-embryonal-treatment-pdq (accessed June 24, 2020).
Picard, D., S. Miller, C. E. Hawkins, E. Bouffet, H. A. Rogers, T. S. Chan, S. K. Kim, Y. S. Ra, J. Fangusaro, A. Korshunov, H. Toledano, H. Nakamura, J. T. Hayden, J. Chan, L. Lafay-Cousin, P. Hu, X. Fan, K. M. Muraszko, S. L. Pomeroy, C. C. Lau, H-K. Ng, C. Jones, T. Va Meter, S. Clifford, C. Eberhart, A. Gajjar, S. Pfister, R. Grundy, and A. Huang. 2012. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: An integrative genomic analysis. The Lancet Oncology 13(8):838–848.
Pollack, I. F., S. Agnihotri, and A. Broniscer. 2019. Childhood brain tumors: Current management, biological insights, and future directions. Journal of Neurosurgery Publishing Group 23(3):261–273.
Raleigh, D. R., B. Tomlin, B. D. Buono, E. Roddy, K. Sear, L. Byer, E. Felton, A. Banerjee, J. Torkildson, D. Samuel, B. Horn, S. E. Braunstein, D. A. Haas-Kogan, and S. Mueller. 2017. Survival after chemotherapy and stem cell transplant followed by delayed craniospinal irradiation is comparable to upfront craniospinal irradiation in pediatric embryonal brain tumor patients. Journal of Neuro-Oncology 131(2):359–368.
Rutkowski, S., U. Bode, F. Deinlein, H. Ottensmeier, M. Warmuth-Metz, N. Soerensen, N. Graf, A. Emser, T. Pietsch, J. E. A. Wolff, R. D. Kortmann, and J. Kuehl. 2005. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. New England Journal of Medicine 352(10):978–986.
Slavc, I., M. Chocholous, U. Leiss, C. Haberler, A. Peyrl, A. A. Azizi, K. Dieckmann, A. Woehrer, C. Peters, G. Widhalm, C. Dorfer, and T. Czech. 2014. Atypical teratoid rhabdoid tumor: Improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012. Cancer Medicine 3(1):91–100.
Spence, T., P. Sin-Chan, D. Picard, M. Barszczyk, K. Hoss, M. Lu, S. Kim, Y. Ra, H. Nakamura, J. Fangusaro, E. Hwang, E. Kiehna, H. Toledano, Y. Wang, Q. Shi, D. Johnston, J. Michaud, M. La Spina, A. M. Buccoliero, D. Adamek, S. Camelo-Piragua, V. Peter Collins, C. Jones, N. Kabbara, N. Jurdi, P. Varlet, A. Perry, D. Scharnhorst, X. Fan, K. M. Murasko, C. G. Eberhart, H. Ng, S. Gururangan, T. Van Meter, M. Remke, L. Lafay-Cousin, J. A. Chan, N. Sirachainan, S. L. Pomeroy, S. C. Clifford, A. Gajjar, M. Shago, W. Halliday, M. D. Taylor, R. Grundy, C. C. Lau, J. Phillips, E. Bouffet, P. B. Dirks, C. E. Hawkins, and A. Huang. 2014. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathologica 128(2):291–303.
Tahiri Elousrouti, L., M. Lamchahab, N. Bougtoub, H. Elfatemi, L. Chbani, T. Harmouch, M. Maaroufi, and A. J. Amarti Riffi. 2016. Subependymal giant cell astrocytoma (SEGA): A case report and review of the literature. Journal of Medical Case Reports 10(35).
Taylor, M. D., P. A. Northcott, A. Korshunov, M. Remke, Y. Cho, S. C. Clifford, C. G. Eberhart, D. W. Parsons, S. Rutkowski, A. Gajjar, D. W. Ellison, P. Lichter, R. J. Gilbertson, S. L. Pomeroy, M. Kool, and S. M. Pfister. 2012. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathologica 123(4):465–472.
Tian, Y., R. Liu, J. Qin, J. Wang, Z. Ma, J. Gong, and C. Li. 2018. Retrospective analysis of the clinical characteristics, therapeutic aspects, and prognostic factors of 18 cases of childhood pineoblastoma. World Neurosurgery e162–e168.
Ullrich, N. J. 2009. Neurologic sequelae of brain tumors in children. Journal of Child Neurology 24(11):1446–1454.
Vitanza, N. A., and M. Monje. 2019. Diffuse intrinsic pontine glioma: From diagnosis to next-generation clinical trials. Current Treatment Options in Neurology 21(8).
Walter, A. W., R. K. Mulhern, A. Gajjar, R. L. Heideman, D. Reardon, R. A. Sanford, X. Xiong, and L. E. Kun. 1999. Survival and neurodevelopmental outcome of young children with medulloblastoma at St. Jude Children’s Research Hospital. Journal of Clinical Oncology 17(12):3720–3728.
Waszak, S. M., P. A. Northcott, I. Buchhalter, G. Robinson, C. Sutter, S. Groebner, K. Grund, L. Brugières, D. Jones, K. Pajtler, A. Morrissy, M. Kool, D. Sturm, L. Chavez, A. Ernst, S. Brabetz, M. Hain, T. Zichner, M. Segura-Wang, J. Weischenfeldt, T. Rausch, B. Mardin, X. Zhou, C. Baciu, C. Lawerenz, J. Chan, P. Varlet, L. Guerrini-Rousseau, D. Fults, W. Grajkowska, P. Hauser, N. Jabado, Y.-S. Ra, K. Zitterbart, S. Shringarpure, F. De La Vega, C. Bustamante, H.-K. Ng., A. Perry, T. MacDonald, P. Driever, A. Bendel, D. Bowers, G. McCowage, M. Chintagumpala, R. Cohn, T. Hassall, G. Fleischhack, T. Eggen, F. Wesenberg, M. Feychting, B. Lannering, J. Schüz, C. Johansen, T. Andersen, M. Röösli, C. Kuehni, M. Grotzer, K. Kjaerheim, C. Monoranu, T. Archer, E. Duke, S. Pomeroy, R. Shelagh, S. Frank, D. Sumerauer, W. Scheurlen, M. Ryzhova, T. Milde, C. Kratz, D. Samuel, J. Zhang, D. Solomon, M. Marra, R. Eils, C. Bartram, K. von Hoff, S. Rutkowski, V. Ramaswamy, R. Gilbertson, A. Korshunov, M. Taylor, P. Lichter, D. Malkin, A. Gajjar, J. Korbel, and S. Pfister. 2018. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncology 19(6):785–798.
Witt, H., S. C. Mack, M. Ryzhova, S. Bender, M. Sill, R. Isserlin, A. Benner, T. Hielscher, T. Milde, M. Remke, D. T. W. Jones, P. A. Northcott, L. Garzia, K. C. Bertrand, A. Wittmann, Y. Yao, S. S. Roberts, L. Massimi, T. Van Meter, W. A. Weiss, N. Gupta, W. Grajkowska, B. Lach, Y. J. Cho, A. Deimling, A. E. Kulozik, O. Witt, G. D. Bader, C. E. Hawkins, U. Tabori, A. Guha, J. T. Rutka, P. Lichter, A. Korshunov, M. D. Taylor, and S. M. Pfister. 2011. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20(2):143–157.
Zacharia, B. E., S. S. Bruce, H. Goldstein, H. R. Malone, A. I. Neugut, and J. N. Bruce. 2012. Incidence, treatment and survival of patients with craniopharyngioma in the Surveillance, Epidemiology and End Results Program. Neuro-Oncology 14(8):1070–1078.
ANNEX TABLE 6-1
Selected Central Nervous System (CNS) Tumors: Diagnostic and Prognostic Information
| Cancer Type | Subtype | Diagnostic Evaluation |
|---|---|---|
| Glial and astrocytic tumor | High-grade glioma (World Health Organization [WHO] grades III and IV)c(1) | Brain magnetic resonance imaging (MRI), spine MRI, surgical resection or biopsy |
| Diffuse midline glioma/H3K27M+ (WHO grade IV) | Brain MRI, spine MRI, with or without biopsy | |
| Low-grade glioma (WHO grades I and II)c(2) | Brain MRI, with or without spine MRI, surgical resection or biopsy | |
| Subependymal giant-cell astrocytoma | Brain MRI and sometimes surgical resection | |
| Ependymal tumor | Ependymomac(3) | Brain MRI, spine MRI, surgical resection or biopsy, lumbar puncture |
| Subependymoma | Brain MRI and surgical resection or biopsy | |
| Pineal tumor | Pineocytoma | Brain MRI, with or without spine MRI, surgical resection or biopsy |
| Pineal parenchymal | Brain MRI, plus spine MRI, surgical resection or biopsy | |
| Pineoblastoma | Brain MRI, spine MRI, surgical resection or biopsy, lumbar puncture |
| Stage | Locationa | 5-Year Overall Survival (OS) Rateb | References |
|---|---|---|---|
| Localized or metastatic | Most commonly supratentorial, but can occur in infratentorial region and spine | <20% | Braunstein et al., 2017; PDQ Editorial Board, 2020a |
| Localized or metastatic | Brainstem, thalamus, and rarely spine | <20% | Felker and Broniscer, 2020 |
| Localized or metastatic | Most commonly optic pathway, hypothalamic region, and posterior fossa | >90% | de Blank et al., 2019 |
| Localized | Most commonly supratentorial, lateral walls of lateral ventricles | >90% | Tahiri Elousrouti et al., 2016 |
| Localized or metastatic | Most commonly posterior fossa (infratentorial), but can occur in supratentorial region and spine | 70–80% Myxopapillary subtype (spine) >90% |
Pajtler et al., 2015 |
| Localized | Most commonly 4th ventricle | >90% | |
| Localized or metastatic | Pineal region | >85% | Clark et al., 2010 |
| Localized or metastatic | Pineal region | 40–75% | Al-Hussaini et al., 2009 |
| Localized or metastatic | Pineal region | Children <3 years old, <20% | Tian et al., 2018 |
| Cancer Type | Subtype | Diagnostic Evaluation |
|---|---|---|
| Embryonal tumor | c(4) | Brain MRI, spine MRI, surgical resection or biopsy, lumbar puncture |
| CNS germ cell tumor | Germinoma | Brain MRI, spine MRI, surgical resection or biopsy, lumbar puncture, tumor markers in blood and cerebrospinal fluid (CSF) |
| Non-germinomatous | Brain MRI, spine MRI, surgical resection or biopsy, lumbar puncture, tumor markers in blood and CSF | |
| Choroid plexus tumor | c(5) | Brain MRI, with or without spine MRI, with or without lumbar puncture, surgical resection or biopsy |
| Craniopharyngioma | c(6) | Brain MRI and surgical resection or biopsy |
a With rare exceptions, almost all of these tumor types can be located in the brain or spine.
b With the exception of low-grade glioma, almost all recurrent tumors, regardless of type, have poor long-term survival.
c Specific histologic diagnosis:
1. Anaplastic astrocytoma; glioblastoma multiforme; oligodendroglioma (WHO grade III); oligoastrocytoma (WHO grade III); anaplastic pleomorphic xanthoastrocytoma; high-grade glioma not otherwise specified (NOS); and gliosarcoma.
2. Pilocytic astrocytoma; diffuse astrocytoma (WHO grade II); pleomorphic xanthoastrocytoma; neuronal and mixed neuronal-glial tumors (WHO grade I and II); and low-grade glioma NOS (WHO grade I and II).
| Stage | Locationa | 5-Year Overall Survival (OS) Rateb | References |
|---|---|---|---|
| Localized or metastatic | Medulloblastoma: cerebellum Atypical teratoid rhabdoid tumor (AT/RT): most commonly supratentorium, but can be seen in infratentorium or spine |
Medulloblastoma >85%
AT/RT 30–40% Embryonal tumors with multilayered rosettes <20% |
Cho et al., 2011; Kool et al., 2012; Waszak et al., 2018 Buscariollo et al., 2012 Picard et al., 2012 |
| Localized or metastatic | Most commonly pituitary and pineal region, but can be seen in thalamus, ventricles, and spine | >90% | Matsutani and Japanese Pediatric Brain Tumor Study Group, 2001 |
| Localized or metastatic | Most commonly pituitary and pineal region, but can be seen in thalamus, ventricles, and spine | >80% | Calaminus et. al., 2017; Goldman et al., 2015 |
| Localized or metastatic | Most commonly ventricular system | Papilloma, >90% Atypical varies 70–80% Carcinoma, 5-year 50–60% |
Hosmann et al., 2019; Ogiwara et al., 2012 |
| Localized | Suprasellar region | 80–90% | Zacharia et al., 2012 |
3. Classic ependymoma; anaplastic ependymoma; tanycytic ependymoma; myxopapillary ependymoma (located in the spine); ependymoma RELA fusion positive; and ependymoma posterior fossa group A and group B.
4. Medulloblastoma (includes sonic hedgehog [SHH], wingless [WNT], Group 3, and Group 4); AT/RT; and embryonal tumor with multilayered rosettes.
5. Choroid plexus papilloma; choroid plexus carcinoma; and atypical choroid plexus papilloma.
6. WNT in adamantinomatous craniopharyngioma and BRAF in papillary craniopharyngioma.
ANNEX TABLE 6-2
Selected Central Nervous System (CNS) Tumors: Treatment Information
| Cancer Type | Subtype |
|---|---|
| Glial tumor | High-grade glioma (World Health Organization [WHO] grades III and IV)*a |
| Diffuse midline glioma/H3K27M+ (WHO grade IV) | |
| Low-grade glioma (WHO grades I and II)*b | |
| Subependymal giant-cell astrocytoma | |
| Ependymal tumor | Ependymoma*c |
| Subependymoma | |
| Pineal tumor | Pineocytoma |
| Pineal parenchymal | |
| Pineoblastoma | |
| Embryonal tumor | *d |
| CNS germ cell tumor | Germinoma |
| Non-germinomatous | |
| Choroid plexus tumor | *e |
| Craniopharyngioma | *f |
* Specific histologic diagnosis:
a Anaplastic astrocytoma; glioblastoma multiforme; oligodendroglioma (WHO grade III); oligoastrocytoma (WHO grade III); anaplastic pleomorphic xanthoastrocytoma; high-grade glioma not otherwise specified (NOS); and gliosarcoma.
b Pilocytic astrocytoma; diffuse astrocytoma (WHO grade II); pleomorphic xanthoastrocytoma; neuronal and mixed neuronal-glial tumors (WHO grade I and II); and low-grade glioma NOS (WHO grade I and II).
c Classic ependymoma; anaplastic ependymoma; tanycytic ependymoma; myxopapillary ependymoma (located in the spine); ependymoma RELA fusion positive; and ependymoma posterior fossa group A and group B.
| Treatment | Duration of Treatment |
|---|---|
| Surgical resection or biopsy, tumor field radiation with concurrent chemotherapy | 2 months, possible prolonged chemotherapy thereafter |
| Focal tumor radiation | 2 months, possible prolonged chemotherapy thereafter |
| Surgical resection/biopsy with or without chemotherapy | With chemotherapy, 18–24 months postsurgery |
| Surgical resection with or without motor inhibitor medication | Variable if on motor inhibitor |
| Surgical resection, usually followed by focal radiation and possibly chemotherapy | 2 months; variable if on chemotherapy |
| Surgical resection alone | <1 month (surgery alone) |
| Surgical resection | <1 month |
| Surgical resection and sometimes focal radiation | <1 month; 2–3 months if radiation is added |
| Surgical resection, followed by (1) for children >3–5 years old, craniospinal radiation with tumor field boost and multidrug chemotherapy; (2) for children <3–5 years old, multidrug chemotherapy and often high-dose chemotherapy with autologous stem cell rescue | 12–15 months |
| Surgical resection, followed by (1) for children >3–5 years old, craniospinal radiation with tumor field boost and multidrug chemotherapy; (2) for children <3–5 years old, multidrug chemotherapy and often high-dose chemotherapy with autologous stem cell rescue | 12–15 months |
| Surgical resection/biopsy, plus chemotherapy and radiation | 6 months |
| Surgical resection/biopsy, plus chemotherapy and radiation | 6 months |
| Surgical resection alone for choroid plexus papilloma (CPP) and often atypical CPP (aCPP); multimodal chemotherapy and focal radiation for choroid plexus carcinoma (CPC) | CPC and aCPP: variable with chemotherapy |
| Surgical resection alone, sometimes followed by focal radiation if residual or progressive after surgery | 2 months with radiation |
d Medulloblastoma (includes sonic hedgehog [SHH], wingless [WNT], Group 3, and Group 4); atypical teratoid rhabdoid tumor; and embryonal tumor with multilayered rosettes.
e CPP, CPC, and aCPP.
f WNT in adamantinomatous craniopharyngioma and BRAF in papillary craniopharyngioma.
This page intentionally left blank.


























