
Chemical Ecology: A View from the Pharmaceutical Industry
LYNN HELENA CAPORALE
Effective medical treatments are available for many diseases. Recent discoveries include drugs that treat hypertension, ulcers, and many bacterial infections (for examples, see ref. 1). For other devastating diseases, such as diabetes, cancer, AIDS, stroke, and Alzheimer disease, at best only inadequate therapy is available; new approaches to treat these diseases are sought intensively by researchers within the pharmaceutical industry and elsewhere. In the future, new infectious agents may be expected to emerge as important threats, much as human immunodeficiency virus (HIV) has challenged us. Because of the magnitude of these challenges, we continue to seek to improve the strategies by which we discover new drugs.
Most of the effort to create and discover medicinal molecules occurs in the research laboratories of the pharmaceutical industry. Drug discovery demands a coordinated effort among people with diverse talents. Researchers who elucidate disease mechanisms (which point to macromolecules that would make good drug discovery targets) work with those skilled in isolating or synthesizing new molecules and with those who can determine the safety and effectiveness of these molecules. Pharmaceutical company researchers begin the search for clues to the type of molecules that, for example, inhibit a targeted enzyme, by testing diverse structures in their compound collections. These collections consist of
Lynn Caporale is vice president of strategic development at CombiChem, Inc., La Jolla, California.
compounds synthesized previously in the course of other research programs. Discovery is optimized by scanning as wide a range of structural types as possible (within resource constraints), including compounds found in nature.
Drug discovery has benefited significantly from the study of compounds that organisms have evolved for their own purposes. All of the drugs discovered at the Merck Research Laboratories that became available to patients in the last decade emerged from programs that benefited from knowledge of biological diversity. Some drugs were discovered via natural product screens (2, 3). Others emerged from medicinal chemistry efforts that drew on knowledge such as how the venom of a Brazilian viper lowers blood pressure (4-6). Another program was inspired by a human genetic variation that indicated that a particular enzyme would be a good drug target (7).
SELECTION OF DRUG DISCOVERY TARGETS
A key step in the process of selecting a molecular target for a drug discovery program involves a demonstration that altering the activity of the proposed target should affect the disease. This is illustrated by the choice of the HIV protease as a target. When the HIV genome was sequenced, Asp-Thr-Gly was recognized, based on previous work with other organisms, as a sequence found in aspartyl proteases. Viral-encoded proteases were known to free active proteins from a viral polyprotein precursor. Therefore, it was considered possible that inactivating the proposed HIV aspartyl protease might prevent the formation of viable virus and, thus, that the HIV protease would be an important target for a drug discovery effort (8). To test this hypothesis, a genetic approach was taken to inactivate the aspartyl protease, changing the proposed active site Asp to Asn. This mutation did indeed inactivate the HIV protease; further, mutant HIV, lacking an active protease, was not viable (Figure 1). With this proof of concept in hand, an intensive effort was mounted to find an HIV protease inhibitor. This proof of concept has been reinforced as small molecule inhibitors of the HIV protease have subsequently been found to block the spread of the virus in tissue culture (9).
The search for inhibitors of the HIV protease began with compounds that had been prepared during prior work to inhibit related proteases, notably renin, an aspartyl protease involved in blood pressure regulation (9, 10). While new structures that inhibit the HIV protease have emerged from natural product screens (11, 12) [one structure that inhibits aspartyl proteases, the statine moiety, had been discovered through screening bacterial natural products before the emergence of HIV (10)], the most significant advances in this program have come through an extensive
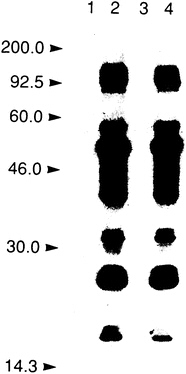
FIGURE 1 Selection of the HIV protease as a drug discovery target. Demonstration that inactivation of the HIV protease interferes with HIV infectivity. Immunoblot of HIV proteins in mock-transfected cells (lane 1), cells transfected with HIV containing active protease (lanes 2 and 4), and cells transfected with HIV in which the protease was inactivated. Reprinted with permission from ref. 8 (copyright Kohl et al.).
medicinal chemistry effort that built upon prior work with aspartyl proteases. Inhibitors had to be found that spared other important proteases (such as renin). A more difficult challenge was to discover nontoxic molecules that are bioavailable and have a half-life long enough to be able to block the virus not only in a laboratory assay but also in a person (9, 13, 14).
It has proven extraordinarily difficult to obtain an inhibitor of sufficient potency (15), bioavailability, and safety to be used in the clinic. The ability of HIV to mutate, and thus to develop resistance to many inhibitors, is an additional hurdle. In spite of the difficulties, chemists persisted, motivated by the knowledge that inactivating this HIV-encoded enzyme inactives the virus. Their efforts have provided HIV protease inhibitors that are being evaluated for clinical efficacy (16).
Examples of successful natural product screens directed against proven targets include screens designed to look for inhibitors of bacterial cell wall biosynthesis. These screens were established once it became clear
that certain very useful antibiotics of low toxicity inhibit this pathway. This strategy led to the discovery of a variety of potent broad-spectrum antibiotics (17). Advances in analytical methodology facilitated the isolation and characterization of unstable molecules present in minute quantities in source organisms, such as the potent broad-spectrum antibiotic thienamycin (3).
As our understanding of biology deepens, we are increasingly able to select (and obtain in sufficient quantities) a specific protein target for a drug discovery effort (18). Identification of human genes that predispose to diseases of complex etiology, such as Alzheimer disease (19), may provide clues to biochemical sites of intervention. Thus, many future therapies may be targeted to (i.e., discovered by screening against) not the most common human sequence at a locus but disease-associated alleles. Progress in cloning DNA has placed in our hands an array of receptor subtypes and of isozymes. [For example, there are at least 14 human serotonin receptor subtypes (20).] Different receptor subtypes may couple the action of the signaling ligand (which might be a hormone or a neurotransmitter) to different second messenger systems (e.g., activating adenyl cyclase or opening a potassium channel). Subtypes typically have distinct tissue distribution (i.e., one may be found in the brain, another in the heart, or both may be in the same region of the brain, but one may be postsynaptic and the other presynaptic). Consequently, the effects of drugs designed to selectively activate or inhibit one subtype can be confined to a subset of the functions of the endogenous ligand. Many side effects of older drugs are caused by binding to nontarget receptor subtypes. Therefore, selecting one subtype as a target and counterscreening against nontarget subtypes provides a rational approach to broaden the window between efficacy and side effects. Targeting an isozyme provides a similar advantage. For example, the nonselective phosphodiesterase inhibitor theophylline, which is widely used to treat asthma, has undesired cardiovascular and gastrointestinal side effects. A selective inhibitor of one of the phosphodiesterase isozymes might provide an improved treatment for asthma [if theophylline's desired and undesired effects arise from inhibition of different phosphodiesterase isozymes (21)].
It is intriguing to reflect on why there might be more than one receptor subtype acting through the same second messenger. As receptor subtypes with distinct amino acid sequences interact with distinct sets of drugs, are there endogenous molecules (perhaps metabolites of the ligand for which we have named the receptor) to which receptor subtypes are differentially sensitive or might subtypes have evolved to bind to different conformations of the same ligand? A subtype that binds to a low-energy conformation with a higher affinity would sense a lower concentration of the endogenous ligand than that detected by other receptors; the latter would
be activated only when ligand concentrations rise. Sensitivity of histamine receptor subtypes to different concentrations of histamine has been proposed to allow triggering of the H1 subtype by low concentrations; higher concentrations, sensed by the H2 subtype, can feed back to shut down an inflammatory response (22). If this is so, once we understand the biological rationale for assigning a given sensitivity to a given subtype, we will be better able to design drugs for a specific purpose. We would select, from the array of possible conformations of the natural ligand, the appropriate conformation to mimic with our drug.
Drug discovery targets can be suggested by the study of organisms that have evolved the ability to regulate our biochemistry. For example, there is much we can learn from a study of infectious organisms. Parasites and viruses often manipulate key regulatory steps in host defenses (23, 24). Pathogenic Yersinia, a genus that includes Yersinia pestis, which causes bubonic plague, interferes with the cellular immune response of a mammalian host (25). The entry of Salmonella into a cell appears to be aided by its use of a leukotriene D4-activated calcium channel that Salmonoella activates by triggering host leukotriene D4 synthesis. Salmonella activates leukotriene D4 synthesis via the epidermal growth factor receptor, which activates the mitogen-activated protein kinase, which in turn activates phospholipase A2 (25). Some viruses have evolved the ability to override cell death programs that otherwise can eliminate virus-infected cells (26, 27). Shigella flexneri, on the other hand, eliminates our first line of defense against invasion by triggering a cell death program in mammalian macrophages (28). If we can learn to manipulate key biochemical control points that trigger or block cell death as well as these pathogens do, then we could save neurons in neurodegenerative disease or kill cells in a tumor.
Cyclosporin (originally discovered due to its antifungal activity) and, more recently, FK506 are used clinically to protect against transplant rejection. Elucidation of the biochemical mechanism of action of these microorganism-derived immunosuppressants [complexes of these ligands with the cyclophilins and FK506 binding proteins, respectively], inhibit the calcium/calmodulin-dependent phosphatase calcineurin (29-31) suggested routes to related drug discovery targets. This work highlighted the important role of this phosphatase in immune regulation and in other widespread calcium-activated signaling pathways, ranging from the recovery from cell cycle arrest by a-mating factor in yeast to the opening of an inward potassium channel involved in stomatal opening in plant guard cells (32). The broader concept of a small molecule acting through complexing large molecules recalls the recognition of antigen bound to histocompatibility molecules by the T-cell receptor (33).
Other organisms are good sources of agents that selectively block the
action of coagulation proteases. Not surprisingly, these include leeches (34) and ticks (35). The plasminogen activator evolved by vampire bats appears to be even more fibrin-dependent (and, therefore, more clot selective) than that evolved by humans and used clinically (36, 37).
While the usefulness of anticoagulants and fibrinolytic agents to blood-sucking organisms appears obvious, it is less obvious what the selective advantage of opium alkaloid synthesis is to the poppy plant. Whatever its role in nature, morphine's interaction with the vertebrate central nervous system played an important role in the birth of molecular pharmacology. It led to the identification and isolation of human receptors that bind to opioids and to the identification of endogenous ligands for these receptors (38, 39). Some organisms provide not drugs or even leads but research tools (18, 40). For example, a screen can be designed to identify small molecules that block the binding of toxins from spiders, scorpions, or cone snails to ion channels. Potential uses for such small molecule receptor blockers range from limiting the extent of damage after a stroke (41) to treating asthma (42).
SELECTING SOURCES TO BE SCREENED FOR DRUGS AND LEADS
If organisms were selected for screening by focusing solely on taxa that had previously yielded interesting structures (43), the potential of as yet unexplored groups of organisms would be missed. Our screens span a broad range of diseases and, therefore, many molecular targets. To increase the chance of finding drugs or at least leads for a medicinal chemistry effort, we gather a diverse array of molecular structures for screens. Molecular diversity is sampled through our chemical collections and by collecting organisms from a broad range of sources. Fungi that live in complex environments and synthesize molecules that regulate the growth of neighboring insects, plants, bacteria, and/or other fungi are likely to be a potentially rich source of leads (44). A program established to maximize molecular diversity by screening microorganisms from a wide range of phylogenetic and ecological origins, such as endophytic (45) and coprophilic fungi (46), contributed to the discovery of a new class of metabolites, the zaragozic acids (Figure 2), which inhibit the first committed step in the isoprenoid pathway dedicated to sterol biosynthesis in fungi and mammals (48). Thus, zaragozic acids are broad-spectrum fungicides and also can lower mammalian cholesterol levels. Other interesting fungal metabolites identified by this approach include the antifungal viridofungans (49), selective nonpeptidyl inhibitors of the antitumor target Ras farnesyl-protein transferase (50), and an inhibitor of the HIV protease (51) (Figure 2).
Plants are another source of molecules with dramatic activity on both
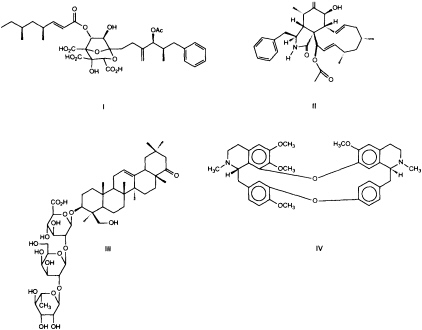
FIGURE 2 Examples of the variety of structures obtained in natural product screens. I, zaragozic acid A, is an inhibitor of mammalian and fungal sterol synthesis, obtained from fungi (48); II, L-696,474, is an inhibitor of the HIV protease, obtained from fungi (51); III, dehydrosoyasaponin I, is an agonist of the calcium-activated potassium channel, obtained from a medicinal plant (58); IV, tetrandrine, is an inhibitor of L-type calcium channels, obtained from a plant (78).
invertebrates and vertebrates. Some of these activities may have evolved under selective pressure from foraging animals. For example, plants that produce phytoecdysones can disrupt the development of insects that might devour them. The ecdysone mimic of Cycas, cycasterone, is more potent and less susceptible to metabolic inactivation than a-ecdysone itself (52). Bruchid beetles have been observed to avoid seeds of the legume Mucuna and may do so because of the high concentrations of L-dopa contained in these seeds (53). (However, L-dopa, used to treat Parkinson disease, was not originally discovered by following beetle behavior.)
A fungus that grows on wheat expends resources to produce ergot alkaloids, which affect vertebrate monoamine neurotransmission. Do ergot alkaloids have an internal plant/fungus purpose or have they evolved as compounds that protect the plant host and, thus, also the fungus from
foraging vertebrates (54)? A plant growth hormone analogue, an insect antifeedant, and a tremorgenic compound that affects sheep and cattle are synthesized by ryegrass when it is infected with the endophyte Acremonium loliae (55). This may explain the observation that infected ryegrass is taller and more disease-resistant than uninfected ryegrass (56). However, while plants that resist foragers are potential sources of compounds that interact with human receptors and enzymes, many of these compounds are toxic agents, not drugs. Plants also can be selected for screening based on the advice of indigenous peoples (43, 57); for example, knowledge that Desmodium adscendens is used as a medicinal herb in Ghana for treatment of asthma provided a long-sought agonist of the maxi-K channel (58). This biochemical activity is consistent with this herb's use in other conditions associated with smooth muscle contraction (59).
Because compounds produced by one species may be modified by another species, knowledge of the natural history of an organism from which a lead was obtained may prove useful to medicinal chemists. For example, a male Danaid butterfly modifies Senecio alkaloids to synthesize pheromones (60); the female pine bark beetle Dendoctonus brevicomus and microorganisms in her gut alter the monoterpenes of the ponderosa pine (52). Thus, to assist medicinal chemists in their exploration of how modifications in the structure of the lead affect its activity, ecologists could suggest potential sources of structures derived from the lead. Understanding the biochemical basis of an ecological interaction has the potential to guide us to drugs (61). For example, the enzyme hydroxymethylglutaryl (HMG)-CoA reductase catalyzes a key step in sterol biosynthesis, the conversion of acetyl-CoA to mevalonate. As mevalonate has been shown to overcome catabolite repression of gibberellin synthesis (62), it seems likely that an inhibitor of HMG-CoA reductase plays a role in regulation of gibberellin synthesis. (Gibberellins, first isolated from the phytopathogenic fungus Gibberella fujikuroi, are plant growth hormones that stimulate stem elongation, induce flowering, and overcome seed dormancy.) Thus, if asked where to look for an inhibitor of HMG-CoA reductase, a chemical ecologist studying gibberellins might have suggested screening such phytopathogenic fungi. [The importance of HMG-CoA reductase inhibition in medicine is demonstrated by lovastatin (2, 63), discovered by random screening of fungi, developed as a drug, and now used by several million people in the United States alone to lower plasma cholesterol (64). Lovastatin may regulate sterol biosynthesis in the fungi that synthesize it or interfere with sterol biosynthesis in a fungal competitor, but its role in nature is not known.]
The leaf-cutter ant Atta cephalotes, which cultivates a fungus as food, produces compounds that increase the growth of the fungal food and compounds that block the germination of undesired fungal spores,
bacteria, and pollen (52, 65, 66). This ant avoids leaves of Hymenaea courbaril, which contain a terpenoid harmful to the fungus that the ant cultivates for food (67). Thus, collecting leaves avoided by an ant can point us to antifungal compounds.
The antiparasitic agent Ivermectin, which can prevent river blindness, was discovered in the microorganism Streptomyces avermitilis (68). S. avermitilis may synthesize a nematocide to kill nearby nematodes as a source of food (69), to eliminate a nematode that might otherwise eat the fungus or compete with it for food, or for a biologically unrelated purpose involving a target structurally related to Ivermectin's molecular target. An improved understanding of chemical ecology might allow us to predict where to find such valuable molecules.
Certain compounds appear in nature only when specific organisms interact. For example, the wild tobacco plant Nicotiana sylvestris increases its synthesis of alkaloids when under attack from larvae of Manduca sexta (70). Pathogens may alter plant gene expression (71) and trigger the synthesis of compounds that help resist pathogen attack (72, 73). While plants can respond to fungus-derived signals by synthesizing protective phytoalexins, fungi can respond by preventing phytoalexin accumulation or by detoxifying a specific phytoalexin by enzymatic conversion to a new structure (52, 73). Such a metabolite is missed by routine high throughput screens, which do not evaluate the plant and its fungus together.
In the laboratory, cells from the plant Digitalis lanata do not produce cardiac glycosides unless they differentiate (74). In the field, the concentration of codeine in the opium poppy varies with the hour of the day (75). A dramatic example of the influence of the environment on our ability to find potent compounds in an organism is seen with the poison dart frog Phyllobates terribilis. While a lethal dose of the voltage-dependent sodium channel agonist batrachotoxin can be harvested by rubbing the tip of a blow dart across the back of a field-caught specimen, batrachotoxin could not be detected in second generation terrarium-reared P. terribilis. Does batrachotoxin, or an essential precursor, come from the frog's diet, and/or is there an environmental trigger of its synthesis (76)? In either case, if the poison arrow frog had been studied only in the laboratory, we would be completely unaware of batrachotoxin. Similarly, if an organism that interacts with one we evaluate becomes extinct, we may miss finding potentially valuable molecules (72, 77).
INTEGRATING INFORMATION FROM MANY ORGANISMS TO GAIN INSIGHT FOR DRUG DESIGN
Natural product screening may capture a compound, such as an antibiotic, insecticide, or HMG-CoA reductase inhibitor, designed for the
very purpose of interacting with the target of the screen. Since diverse organisms, from a quetzal to a Sequoia or from a fungus to human beings, share a great deal of biochemical architecture, it is not surprising that small molecules from species as distant as plants, fungi, and bacteria can interact with macromolecules in our bodies. Natural product screening allows us to sift through the extraordinary diversity that overlays these common molecular features. It may be that an interaction is fortuitous, given the number and diversity of natural products examined in pharmaceutical industry screens. However, a compound, discovered in a screen, that interacts with a human protein may have evolved to interact with a protein bearing a homologous domain, active site, or regulatory region in the organism within which it is made (or in a neighboring organism). For example, might tetrandrine (found in a Chinese medicinal herb, Stepania tetrandra, used to treat angina and hypertension), which blocks human L-type calcium channels (78), have evolved to regulate plant calcium channels (79)? Information about related proteins from different organisms can help us to understand how a target protein's function emerges from its structure. Amino acids conserved in related receptors produced by different species can guide us to residues important, for example, in activating the receptor (80). Sequences of variant receptors or enzymes with unique pharmacology point to key residues of a target important for drug recognition. Thus, the Monarch butterfly Dannus plexippus is able to feed on plants rich in cardiac glycosides that are toxic for most animals. The substitution His-122 → Asn in its Na+ /K+-ATPase has been proposed to explain the butterfly's decreased sensitivity to the cardiotonic agent ouabain (81).
As masses of DNA sequences accumulate for protein family members from a wide range of organisms, we can learn to recognize binding motifs that define active and regulatory sites. A study of homologous proteins from diverse organisms can provide a new level of insight into their activity. For example, the exact distance between the leucine-zipper and the basic region proposed as a DNA contact surface was noted to be conserved in DNA binding proteins derived from a range of species including plants, fungi, and mammals. This observation led to a helical model for the binding of this family of transcription regulatory proteins to DNA and to an understanding of how dimers of such proteins could specifically recognize directly abutted dyad-symmetric DNA sequences (82) (Figure 3). A new science, bioinformatics, is evolving to extract information, with increasing levels of abstraction, from a burgeoning list of DNA sequences (84). Even otherwise unrelated proteins may have related stretches of amino acid sequence (motifs) (85, 86) and/or similar backbone structure (87) at the binding site. While most computer searches for such motifs focus on the primary sequence of the encoded protein, it
is beginning to be feasible to search for homology in the three-dimensional structures that linear sequences can form (84, 86, 89). While certain of these homologous regions may interact with the same compound [e.g., ATP (85)], others may interact with structurally homologous but distinct ligands such as individual members of a protein family.
Comparing the sequences of two families of macromolecules that interact, such as DNA sequences and the proteins that recognize them, helps us to perceive the molecular basis of specificity (90). Variant amino acids that provide an individual sequence's functional specificity are intermixed with consensus amino acids, which provide a motif's structural framework. A compound (whether discovered in a screen or designed by medicinal chemists) that binds to a given sequence in one protein may provide a lead for a compound that can bind to that motif in other proteins. Such information combined with advances in analytical (91) and molecular (92) modeling methodology dramatically improves our ability to visualize how recognition is framed in three dimensions. This insight can reveal a rational route to improved potency and specificity. Because members of protein families have related three-dimensional structures, small molecules that interact with the members of a given protein family also may have common underlying three-dimensional structures, which may be viewed as ''scaffolds." Specificity of a small molecule for an individual protein family member may be overlayed on a common scaffold by groups that provide specific binding interactions (hydrophobic bonds, salt bridges, hydrogen bonds, etc.). For example, scaffolds designed to interact with an important family of signal transducing molecules, G-protein-linked receptors, provide an approach to a novel set of potentially selective ligands (47) (Figure 4). Even when the biological ligand of our target is another protein or peptide, we are beginning to be able to select or design scaffold structures that direct interactions to the right places in space (93, 94). Favored scaffolds upon which to introduce specificity-defining binding interactions include classes of molecules known to have generally good bioavailability, such as steroids and benzodiazepines (95, 96) (much as the steroid nucleus itself serves, in nature, as a scaffold on which different substituents are responsible for selective interaction with individual members of the steroid receptor protein family); other scaffolds may be found through screening natural products.
EFFICIENT ROUTES TO THE DISCOVERY OF USEFUL COMPOUNDS
Life generates possibilities by providing opportunities for randomness, upon which selection acts. However, untethered randomness is not the most efficient route to success in a competitive world. Organisms have
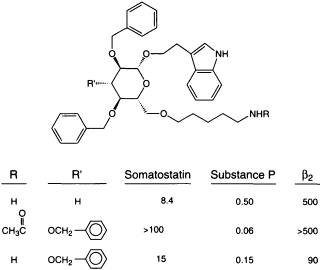
FIGURE 4 Scaffold that interacts selectively with different members of a protein family. These ß-D-glucose derivatives (47) can compete with each G-protein-linked receptor's natural ligand for binding, whether the natural ligand is a peptide or a catecholamine. (Note that the natural ligands do not compete with each other for binding.) By varying the substituents, the ability to discriminate between members of a protein family can be overlayed on a common scaffold. ß2, ß2 receptor.
evolved a variety of efficient mechanisms to acquire new traits, such as passing around antibiotic resistance genes (97) or copying and modifying useful structures (98, 99). In the development of an immune response against a pathogen, DNA sequences encoding variable regions, which provide the specificity for the pathogen, are moved next to constant regions, which provide for different effector functions (such as crossing the placenta, binding to phagocytes, or degranulating mast cells) (100). While this occurs during the life of an individual, it appears that a similar process occurs during evolution (98, 99, 101, 102). This assembling of DNA sequences from different sources generates patchwork genes, such as that encoding the low density lipoprotein receptor (103). Similarly, new serine proteases appear to have been created by duplication of DNA encoding a functional serine protease domain, followed by mutation (104) and attachment of modules with varied functions (105). Thus, long before the industrial revolution, nature discovered the efficiency of interchangeable parts (106). Mechanisms that direct genetic variation away from a useful framework and toward sites where such variation can generate
novel functions rather than destroy useful ones are likely to have been evolutionarily selected (107). For example, after duplication of a serine protease gene, a functional serine protease that is specific for a new substrate arises through mutations that alter amino acids located in those regions of the protease that determine substrate specificity (108). The most efficient route to new active serine proteases would tend to avoid mutations likely to inactivate the enzyme, such as those in the active site or framework of the three-dimensional serine protease structure. Exploration could be directed to specific stretches of DNA, for example, by modulating the fidelity of replication (109) and employing the degeneracy of the genetic code to generate sequence-specific effects on DNA structure (107). Another suggestion of directed variation involves cone snails, which move at a snail's pace yet prey on fish, which move quickly. The snail must paralyze the fish instantly. To meet this challenge, cone snails have evolved a family of peptides (110) with a conserved ion-channel active framework. It has been proposed that the genus Conus uses a strategy involving cassette mutagenesis to conserve the ion channel active scaffold while generating specificity for diverse channels (111). A recombination-based mechanism for targeted diversity also has been proposed for histocompatibility antigens (112). Somatic mutation appears to be directed to the variable (antigen binding) region of immunoglobulin molecules (113). The better we understand how specificity is overlaid on conserved frameworks in biochemical targets, the more efficient our design of potent and specific drugs will be.
Once we obtain a structure that interacts with a target (whether through computerized searches and assays of a compound collection or through natural product screening), variants of this structure are synthesized to optimize potency and specificity. It is becoming possible to explore variations of some leads rapidly through combinatorial synthesis (114) in which, typically, solid-phase synthesis (115) is adapted to perform a mixed sequence of reactions simultaneously, generating hundreds or hundreds of thousands of products in a single synthesis. Such a combinatorial approach to drug discovery is dependent upon both the availability of a rapid screening method to select active molecules and a rapid method of defining their structures and potencies. One source of creativity for combinatorial chemistry is the set of synthons used in combination. Enzymes obtained from microorganisms are likely to be enlisted to help us perform challenging synthetic steps, increasing the diversity of structures that are accessible to combinatorial chemistry.
In the absence of any information about the structure of a target, combinatorial synthesis can be used to explore a set of diverse scaffolds that direct potential binding interactions to different angles and distances from each other. The structure-activity relationship that emerges from
such a study helps to define a three-dimensional array that provides successful ligands for the target. A scaffold that fits the target may exist in nature fortuitously, having a different function but just the right shape to interact with our target, and/or may have evolved when nature confronted the same structural problem as that before us. Scaffolds previously discovered to interact with a particular receptor family or motif can be used as a starting point for combinatorial chemistry. As our ability to carry out combinatorial chemistry evolves, libraries of molecules will emerge that include compounds selective for each variant of a protein motif. Whether a useful compound emerges from variation and natural selection or through combinatorial chemistry and laboratory screening, our ability to capture compounds selective for a target protein, combined with increased understanding of the structure and function of conserved and variant regions of protein targets, is enabling us to evolve more efficient strategies for discovering useful molecules.
LEARNING TO ASK BETTER QUESTIONS
A key step in the drug discovery process is the identification of a target and demonstration that its activity affects the disease process. We can learn to identify key points of biological regulation by studying the fruits of evolution, such as cone snail toxins, vampire bat fibrinolytics, viruses that interfere with cell death, fungal and bacterial immunosuppressants, and human genetic variation. We can be inspired in our search for novel drugs by the diverse interactions revealed by research in chemical ecology.
Evolution is a process of generating possibilities and selecting among them. The value of a new biological activity is not an absolute and not an intrinsic property of the new structure; rather, value depends on the possibilities that the new property generates in the context of other compounds or organisms in the environment in which the activity emerged (116). Life is challenged by interactions to evolve. Natural selection may favor not only those organisms that produce molecules optimal for their purposes but also those lineages that have evolved the most efficient processes by which to generate optimal pathways and molecules. Molecular mechanisms have evolved that enlist and modify useful structures for new purposes. We are now beginning to search for efficient strategies by which to discover new molecules to protect us against faltering biochemical regulatory mechanisms and evolving infectious agents. The more information we have about the structure of a drug discovery target and its relationship to other proteins, the more efficiently we can optimize potency and selectivity in our drug discovery process. Drug design efforts can begin with a structure that was previously
designed or one that was discovered through natural product screening. Combinatorial chemistry will allow us to add rapidly to the rich variety of biologically active molecules that has emerged from molecular evolution.
In the use of natural product screening for drug discovery, our descendants may better understand why a given activity and/or structure can be found in this molecule of this cell of this organism at this stage in its life cycle. A higher level of understanding should emerge from studying many compounds and many organisms in many complex settings. As more species are lost, our biochemical reference library becomes poorer. As we study enough threads and their connections, we can begin to see patterns. These patterns form a framework into which we can place observations and identify gaps in our knowledge. The loss of the shamans' knowledge of the medicinal uses of plants, as their cultures disappear, has been compared to the burning of the library at Alexandria (117). But for those of us who are beginning to understand how to read the molecules within living things, the loss of biological diversity itself is also the loss of a library, a library that contains answers to questions we have not yet learned to ask.
SUMMARY
Biological diversity reflects an underlying molecular diversity. The molecules found in nature may be regarded as solutions to challenges that have been confronted and overcome during molecular evolution. As our understanding of these solutions deepens, the efficiency with which we can discover and/or design new treatments for human disease grows. Nature assists our drug discovery efforts in a variety of ways. Some compounds synthesized by microorganisms and plants are used directly as drugs. Human genetic variations that predispose to (or protect against) certain diseases may point to important drug targets. Organisms that manipulate molecules within us to their benefit also may help us to recognize key biochemical control points. Drug design efforts are expedited by knowledge of the biochemistry of a target. To supplement this knowledge, we screen compounds from sources selected to maximize molecular diversity. Organisms known to manipulate biochemical pathways of other organisms can be sources of particular interest. By using high throughput assays, pharmaceutical companies can rapidly scan the contents of tens of thousands of extracts of microorganisms, plants, and insects. A screen may be designed to search for compounds that affect the activity of an individual targeted human receptor, enzyme, or ion channel, or the screen might be designed to capture compounds that affect any step in a targeted metabolic or biochemical signaling pathway.
While a natural product discovered by such a screen will itself only rarely become a drug (its potency, selectivity, bioavailability, and/or stability may be inadequate), it may suggest a type of structure that would interact with the target, serving as a point of departure for a medicinal chemistry effort—i.e., it may be a "lead." It is still beyond our capability to design, routinely, such lead structures, based simply upon knowledge of the structure of our target. However, if a drug discovery target contains regions of structure homologous to that in other proteins, structures known to interact with those proteins may prove useful as leads for a medicinal chemistry effort. The specificity of a lead for a target may be optimized by directing structural variation to specificity-determining sites and away from those sites required for interaction with conserved features of the targeted protein structure. Strategies that facilitate recognition and exploration of sites at which variation is most likely to generate a novel function increase the efficiency with which useful molecules can be created.
I am grateful to Thomas Eisner and Jerrold Meinwald for organizing this thought-provoking Colloquium; to Gerald Bills, Ralph Hirschmann, Joel Huff, Chris Sander, Catherine Strader, Gary Stoecker, and especially, Jerrold Meinwald for their very helpful comments on earlier versions of this manuscript; and to Marianne Belcaro for assistance with its preparation.
REFERENCES
1. Merck & Co. (1993) Annual Report (Merck & Co., Rahway, NJ).
2. Alberts, A. W., Chen, J., Kuron, G., Hunt, V., Huff, J., et al. (1980) Proc. Natl. Acad. Sci. USA 77, 3957-3961.
3. Kahan, J. S., Kahan, F. M., Goegelman, R., Currie, S. A., Jackson, M., Stapley, E. O., Miller, T. W., Miller, A. K., Hendlin, D., Mochales, S., Hernandez, S., Woodruff, H. B. & Bimbaum, J. (1978) J. Antibiot. 32, 1-12.
4. Ferreira, S. H. (1965) Br. J. Pharmacol. 24, 163-169.
5. Ondetti, M. A., Rubin, B. & Cushman, D. W. (1977) Science 196, 441-444.
6. Patchett, A. A., Harris, E., Tristam, E. W., Wyvratt, M., Wu, M. T., et al. (1980) Nature (London) 288, 280-283.
7. Imperato-McCinley, J., Guerrero, L., Gautier, T., & Petersen, R. (1974) Science 186, 1213-1215.
8. Kohl, N. E., Emini, E. A., Schleif, W. A., Davis, L. J., Heimbach, J. C., Dixon, R. A. F., Scolnick, E. M. & Sigal, I. S. (1988) Proc. Natl. Acad. Sci. USA 85, 4686-4690.
9. Huff, J. R. (1991) J. Med. Chem. 34, 2305-2314.
10. Wiley, R. A. & Rich, D. H. (1993) Med. Res. Rev. 13, 327-384.
11. Lingham, R. B., Hsu, A., Silverman, K.C., Bills, G. F., Dombrowski, A., Goldman, M. E., Darke, P. L., Huang, L., Koch, G., Ondeyka J. G. & Goetz, M. A. (1992) J. Antibiot. 45, 686-691.
12. Kashman, Y., Gustafson, K. R., Fuller, R. W., Cardellina, J. H., McMahon, J. B.,
Currens, M. J., Buckheit, R. W., Jr., Hughes, S. H., Cragg, G. M. & Boyd, M. R. (1992) J. Med. Chem. 35, 2735-2743.
13. Lam, P. Y. S., Jadhav, P. K., Eyermann, C. J., Hodge, C. N., Ru, Y., Bacheler, L. T., Meek, J. L., Otto, M. J., Rayner, M. M., Wong, Y. N., Chang, C., Weber, P. C., Jackson, D. A., Sharpe, T. R. & Erickson-Viitanen, S. (1994) Science 263, 380-384.
14. Thompson, W. J., Fitzgerald, P. M. D., Holloway, M. K., Emini, E. A., Darke, P. L., McKeever, B. M., Schleif, W. A., Quintero, J. C., Zugay, J. A., Tucker, T. J., Schwering, J. E., Homnick, C. F., Nunberg, J., Springer, J. P. & Huff, J. R. (1992) J. Med. Chem. 35 , 1685-1701.
15. Stinson, S. (1994) C & E News5/16/94, 6-7.
16. Vacca, J. P., Dorsey, B. D., Schleif, W. A., Levin, R. B., McDaniel, S. L., et al. (1994) Proc. Natl. Acad. Sci. USA 91, 4096-4100.
17. Gadebusch, H. H., Stapley, E. O. & Zimmerman, S. B. (1992) Crit. Rev. Biotechnol. 12, 225-243.
18. Monaghan, R. L. & Tkacz, J. S. (1990) Annu. Rev. Microbiol. 44 , 271-301.
19. Tanzi, R., Gaston, S., Bush, A., Romano, D., Pettingell, W., Peppercorn, J., Paradis, M., Gurubhagavatula, S., Jenkins, B. & Wasco, W. (1994) Genetica 91, 255-263.
20. Watson, S. & Girdlestone, D., eds. (1994) 1994 Receptor and Ion Channel Nomenclature Supplement (Elsevier, London).
21. Torphy, T. J. & Undem, B. J. (1991) Throax 46, 512-523.
22. Busse, W. W. & Sosman, J. (1976) Science 194, 737-738.
23. Gooding, L. R. (1992) Cell 71, 5-7.
24. Kierszenbaum, F. (1994) Parasitic Infections and the Immune System (Academic, San Diego), pp. 163-164.
25. Bliska, J. B., Galan, J. E. & Falkow, S. (1993) Cell 73, 903-920.
26. Henderson, S., Rowe, M., Gregory, C., Croom-Carter, D., Wang, F., Longnecker, R., Kieff, E. & Rickinson, A. (1991) Cell 65, 1107-1115.
27. Crook, N. E., Clem, R. J. & Miller, L. K. (1993) J. Virol. 67, 2168-2174.
28. Zychlinsky, A., Kenny, B., Menard, R., Prevost, M., Holland, I. B. & Sansonetti, P. J. (1994) Mol. Microbiol. 11, 619-627.
29. McKeon, F. (1991) Cell 66, 823-826.
30. Liu, J., Farmer, J. D., Lane, W. S., Friedman, J., Weissman, I. & Schreiber, S. L. (1991) Cell 66, 807-815.
31. Clardy, J. (1995) Proc. Natl. Acad. Sci. USA 92, 56-61.
32. Poole, R. J. (1993) Proc. Natl. Acad. Sci. 90, 3125-3126.
33. Jorgensen, J. L., Reay, P. A., Ehrich, E. W. & Davis, M. M. (1992) Annu. Rev. Immunol. 10, 835-873.
34. Kaiser, B., (1992) Thromb. Haemostasis 17, 130-136.
35. Mellott, M.J., Stranieri, M. T., Sitko, G. R., Lynch J. J., Jr., & Vlasuk, G. P. (1993) Fibrinolysis 7, 195-202.
36. Gardell, S. J., Duong, L. T., Diehl, R. E., York, J. D., Hare, T. R., Register, R. B., Jacobs, J.J., Dixon, R. A. F. & Friedman, P. A. (1989) J. Biol. Chem. 264, 17947-17952.
37. Pennica, D., Holmes, W. E., Kohr, W. J., Harkins, R. N., Vehar, G. A., Ward, C. A., Bennett, W. F., Yelverton, E., Seeburg, P. H., Heyneker, H. L., Goeddel, D. V. & Collen, D. (1983) Nature (London) 301, 214-221.
38. Pert, C. B. & Snyder, S. H. (1973) Science 179, 1011-1014.
39. Kosterlitz, H. W. & Hughes, J. (1977) Br. j. Psychiatry 130, 298-304.
40. Jones, T. R., Charette, L., Garcia, M. L. & Kaczorowski, G. J. (1990) J. Pharmacol. Exp. Ther. 255, 697-705.
41. Valentino, K., Newcomb, R., Gadbois, T., Singh, T., Bowersox, S., Bitner, S., Justice, A., Yamashiro, D., Hoffman, B. B., Ciaranello, R., Miljanich, G. & Ramachandran, J. (1993) Proc. Natl. Acad. Sci. USA 90, 7894-7897.
42. Huang, J., Garcia, M. L., Reuben, J. P. & Kaczorowski, G.J. (1993) Eur. J. Pharmacol. 235, 37-43.
43. Balick, M.J. (1990) in Bioactive Compounds from Plants, Ciba Foundation Symposium 154, eds. Chadwick, D. J. & Marsh, J. (Wiley, Chichester, U.K.), pp. 22-39.
44. Nisbet, L.J., Fox, F.M. & Hawksworth, D. L. (1991) The Biodiversity of Microorganisms and Invertebrates: Its Role in Sustainable Agriculture (CAB Int., Wallingford, U.K.), pp. 229-244.
45. Bills, G. F. (1994) in Systematics, Ecology and Evolution of Endophytic Fungi in Grasses and Woody Plants, ed. Redlin, S. C. (APS Press, St. Paul), in press.
46. Bills, G. F. & Polishook, J. D. (1993) Nova Hedwigia Z. Kryptogramenkd. 57, 195-206.
47. Hirschmann, R., Nicolau, K. C., Pietranico, S., Leahy, E. M., Salvino, J., et al. (1993) J. Am. Chem. Soc. 115, 12550-12568.
48. Bills, G. F., Palaez, F., Polishook, J. D., Diez-Matas, M. T., Harris, G. H., Clapp, W. H., Dufresne, C., Byrne, K. M., Nallin-Omstead, M., Jenkins, R. G., Mojena, M., Huang, L. & Bergstrom, J. D. (1994) Mycol. Res. 98, 733-739.
49. Harris, G. H., Jones, E. T. T., Meinz, M. S., Nallin-Omstead, M., Helms, G. L., Bills, G. F., Zink, D. & Wilson, K. E. (1993) Tetrahedron Lett. 34, 5235-5238.
50. Singh, S. B., Zink, D. L., Liesch, J. M., Goetz, M. A., Jenkins, R. G., Nallin-Omstead, M., Silverman, K. C., Bills, G. F., Mosley, R. T., Gibbs, J. B., AlbersSchonberg, G. & Lingham, R. B. (1993) Tetrahedron 49, 5917-5926.
51. Dombrowski, A. W., Bills, G. F., Sabnis, G., Koupal, L. R., Meyer R., Ondeyka, J. G., Giacobbe, R. A., Monaghan, R. L. & Lingham, R. B. (1992) J. Antibiot. 45, 671-678.
52. Harborne, J. B. (1988) Introduction to Ecological Biochemistry (Academic, New York), 3rd Ed.
53. Janzen, D. (1969) Evolution 23, 1-27.
54. Wink, M. (1993) in The Alkaloids: Chemistry and Pharmacology, ed. Cordell, G.A. (Academic, New York), pp. 1-104.
55. Rowan, D. D., Hunt, M. B. & Gaynor, D. L. (1986) J. Chem. Soc. Chem. Commun. 935-936.
56. Clay, K. (1991) in Microbial Mediation of Plant-Herbivore Interactions, eds. Barbosa, P., Krischik, V. A. & Jones, C. G. (Wiley, New York), pp. 199-226.
57. Cox, P. A. (1990) in Bioactive Compounds from Plants, Ciba Foundation Symposium 154, eds. Chadwick, D. J. & Marsh, J. (Wiley, Chichester, U.K.), p. 40.
58. McManus, O. B., Harris, G. H., Giangiacomo, K. M., Feigenbaum, P., Reuben, J. P., Addy, M.E., Burka, J. F., Kaczorowski, G.J. & Garcia, M.L. (1993) Biochemistry 32, 6128-6133.
59. Arvigo, R. & Balick, M. (1993) Rainforest Remedies (Lotus, Twin Lakes, WI).
60. Eisner, T. & Meinwald, J. (1995) Proc. Natl. Acad. Sci. USA 92 , 50-55.
61. Horn, W. S., Smith, J. L., Bills, G. F., Raghoobar, S. L., Helms, G. L., Kurtz, M. B., Marrinan, J. A., Frommer, B. R., Thornton, R. A. & Mandala, S. M. (1992) J. Antibiot. 45, 1692-1696.
62. Bruckner, B. (1992) Secondary Metabolites: Their Function and Evolution, Ciba
Foundation Symposium 171, eds. Chadwick D. J. & Whelan J. (Wiley, Chichester, U.K.), pp. 129-143.
63. Slater, E. E. & MacDonald, J. S. (1988) Drugs 36S3, 7-82.
64. Anonymous (1993) Stat. Bull., 10-17.
65. Iwanami, Y. (1978) Protoplasma 95, 267-271.
66. Cherrett, J. M., Powell, R. J. & Stradling, D. J. (1988) in Coevolution of Fungi with Plants and Animals, eds. Pirozynski, K.A. & Hawksworth, D.L. (Academic, New York), pp. 93-120.
67. Hubbell, S.P., Wiemer, D.F. & Adejare, A. (1983) Oecologia (Berlin) 60, 321-327.
68. Burg, R. W., Miller, B. M., Baker, E. E., Birnbaum, J., Currie, S. A., Hartman, R., Kong, Y., Monaghan, R. L., Olson, G., Putter, I., Tunac, J. B., Wallick, H., Stapley, E. O., Oiwa, R. & Omura, S. (1979) Antimicrob. Agents Chemother. 15, 361-367.
69. Giuma, A. Y. & Cooke, R. C. (1971) Trans. Br. Mycol. Soc. 56, 89-94.
70. Harborne, J. B. (1990) Bioactive Compounds from Plants, Ciba Foundation Symposium 154, eds. Chadwick, D. J. & Marsh, J. (Wiley, Chichester, U.K.), pp. 126-139.
71. Collinge, D. B. & Slusarenko, A. J. (1987) Plant Mol. Biol.9, 389-410.
72. Gaffney, T., Friedrich, L., Vernooij, B., Negrotto, D., Nye, G., Uknes, S., Ward, E., Kessmann, H. & Ryals, J. (1993) Science 261, 754-756.
73. Barz, W., Bless, W., Borger-Papendorf, G., Gunia, W., Mackenbrock, U., Meier, D., Otto, Ch. & Super, E. (1990) Bioactive Compounds from Plants, Ciba Foundation Symposium 154, eds. Chadwick, D. J. & Marsh, J. (Wiley, Chichester, U.K.), pp. 140-156.
74. Luckner, M. & Diettrich, B. (1989) in Primary and Secondary Metabolism of Plant Cell Cultures, ed. Kurz, W. G. W. (Springer, Berlin), pp. 117-124.
75. Grindley, J. (1993) Scrip Mag.12/93, 30-33.
76. Daly, J. W. (1995) Proc. Natl. Acad. Sci. USA92, 9-13.
77. Enyedi, A. J., Yalpani, N., Silverman, P. & Raskin, I. (1992) Cell 70, 879-886.
78. King, V. F., Garcia, M. L., Himmel, D., Reuben, J. P., Lam, Y. T., Pan, J., Han, G. & Kaczorowski, G. J. (1988) J. Biol. Chem. 263 , 2238-2244.
79. Graziana, A., Fosset, M., Ranjeva, R., Hetherington, A. M. & Lazdunski, M. (1988) Biochemistry 27, 764-768.
80. Strader, C. D., Sigal, I. S., Register, R. B., Candelore, M. R., Rands, E. & Dixon, R. A. F. (1987) Proc. Natl. Acad. Sci. USA 84, 4384-4388.
81. Holzinger, F., Frick, C. & Wink, M. (1992) FEBS Lett. 314, 477-480.
82. Vinson, C. R., Sigler, P. B. & McKnight, S. L. (1989) Science 246, 911-916.
83. Ellenberger, T. E., Brandl, C. J., Struhl, K. & Harrison, S.C. (1992) Cell 71, 1223-1237.
84. Taylor, W. R. (1992) Patterns in Protein Sequence and Structure, Springer Series in Biophysics (Springer, Berlin), Vol. 7.
85. Bairoch, A. (1992) Nucleic Acids Res. 20 (Suppl.), 2013-2018.
86. Johnson, M. S., Overington J. P. & Blundell, T. L. (1993) J. Mol. Biol. 231, 735-752.
87. Rizo, J. & Gierasch, L. M. (1992) Annu. Rev. Biochem. 61, 387-418.
88. Sheridan, R. P. & Venkataraghavan, R. (1992) Proteins 14, 16-28.
89. Ouzounis, C., Sander, C., Scharf, M. & Schneider, R. (1993) J. Mol. Biol. 232, 805-825.
90. Desjarlais, J. R. & Berg, J. M. (1993) Proc. Natl. Acad. Sci. USA 90, 2256-2260.
91. Roberts, G. C. K. (1993) NMR of Macromolecules (Oxford Univ. Press, New York).
92. Cohen, F. E. & Moos, W., eds. (1993) Perspect. Drug Discovery Des. 1, 391-417.
93. Olson, G. L., Bolin, D. R., Bonner, M. P., Bos, M., Cook, C. M., Fry, D. C., Braves, B. J., Hatada, M., Hill, D. E., Kahn, M., Madison, V. S., Rusiecki, V. K., Sarabu, R., Sepinwall, J., Vincent, G. P. & Voss, M. E. (1993) J. Med. Chem. 36, 3039-3049.
94. Smith, A. B., Hirschmann, R., Pasternak, A., Akaishi, R., Guzman, M. C., Jones, D. R., Keenan, T. P., Sprengeler, P. A., Darke, P. L., Emini, A., Holloway, M. K. & Schleif, W. A. (1994) J. Med. Chem. 37, 215-218.
95. Hirschmann, R., Sprengeler, P.A., Kawasaki, T., Leahy, J. W., Shakespeare, W. C. & Smith, A. B., III (1992) J. Am. Chem. Soc. 114 , 9699-9701.
96. Bunin, B. A., Plunkett, M. J. & Ellman, J. A. (1994) Proc. Natl. Acad. Sci. USA 91, 4708-4712.
97. Zahner, H., Anke, H. & Anke, T. (1983) in Secondary Metabolism and Differentiation in Fungi, eds. Bennett, J. W. & Ciegler, A. (Dekker, New York), pp. 153-171.
98. Doolittle, R. & Bork, P. (1993) Sci. Am. 269, 50-56.
99. Caporale, L. H. (1973) Ph.D. Thesis (Univ. of California, Berkeley).
100. Hozumi, N. & Tonegawa, S. (1976) Proc. Natl. Acad. Sci. USA 73, 3628-3632.
101. Rossman, M. G. (1981) Philos. Trans. R. Soc. London B. 293, 191-203.
102. Gilbert, W. (1978) Nature (London) 271, 501.
103. Sudhof, T. C., Goldstein, J. L., Brown, M. S. & Russell, D. W. (1985) Science 228, 815-822.
104. Patthy, L. (1993) Methods Enzymol. 222, 10-28.
105. Irwin, D. M., Robertson, K. A. & MacGillivray, R. T. A. (1988) J. Mol. Biol. 200, 31-45.
106. Hays, S. P. (1957) The Response to Industrialism 1885-1914 (Univ. of Chicago Press, Chicago).
107. Caporale, L. H. (1984) Mol. Cell. Biochem. 64, 5-13.
108. Graham, L. D., Haggett, K. D., Jennings, P. A., De Brocque, D. S. & Whittaker, R. G. (1993) Biochemistry 32, 6250-6258.
109. Kunkel, T. A. (1992) J. Biol. Chem. 267, 18521.
110. Woodward, S. R., Cruz, L. J., Olivera, B. M. & Hillyard, D. R. (1990) EMBOJ.9, 1015-1020.
111. Myers, R. A., Cruz, L. J., Rivier, J. E. & Olivera, B. M. (1993) Chem. Rev. 93, 1923-1930.
112. Pease, L. R., Schulze, D. H., Pfaffenbach, G. M. & Nathenson, S. G. (1983) Proc. Natl. Acad. Sci. LISA 80, 242-246.
113. Pech, M., Höchtl, J., Schnell, H. & Zachau, H. (1981) Nature (London) 291, 668-670.
114. Gordon, E. M., Barrett, R. W., Dower, W. J., Fodor, S. P. A. & Gallop, M. A. (1994) J. Med. Chem. 37, 1385-1401.
115. Merrifield, B., (1986) Science 232, 341-347.
116. Eiseley, L., (1959) The Immense Journey (Vintage, New York).
117. Linden, E. (1991) Time 9/3/91, 46-55.




















