
3
Detection and Diagnostics
OVERVIEW
Workshop presentations on infectious disease detection and diagnostics surveyed current capacity, needs, and challenges; anticipated forthcoming developments; and imagined a future in which diseases can be diagnosed prior to the appearance of symptoms (see Summary and Assessment).
Diagnostics for Developing Countries
The session began with a reminder from Mark Perkins of the Foundation for Innovative New Diagnostics (FIND) that while emerging diseases and bioterrorism threaten public health, infectious diseases such as tuberculosis and malaria have long imposed a severe burden on the developing world. In their contribution to this chapter, Perkins and Peter Small of the Gates Foundation discuss the need for rapid, accurate, inexpensive, robust diagnostics in developing countries—a need that could be met by recent advances in genomics, proteomics, and materials science if there was a profitable market. To fill this gap, FIND guides the development and adoption of novel diagnostic products for diseases of the developing world in much the same way as public–private partnerships have been established to produce drugs and vaccines for low-resource settings. With FIND’s support, companies that produce low-cost diagnostics for use in developing countries realize sufficient cost savings (in manufacturing, approval procedures, and marketing) to sustain profits.
Rapid Diagnostics
Soldiers at risk of contracting infectious disease—either from the natural environment or from bioweapons—need diagnostics that are rugged, rapid, and easy to use, according to speaker Mark Wolcott of the Diagnostic Systems Division at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID). In their contribution to this chapter, Wolcott and co-authors discuss the rationale, design, and development of rapid diagnostic assays for infectious diseases. They offer brief, comparative descriptions of a variety of platform technologies that in the future may be combined to produce comprehensive, integrated diagnostic systems—perhaps in the guise of miniaturized “labs on chips” that process samples, perform assays, and automatically report their results. “As technologies mature and new technologies are developed, rapid infectious disease diagnostics will become available and practical,” the authors predict.
Rapid diagnostic tools are also improving infectious disease surveillance in animals. Workshop presenter Alex Ardans, who directs the California Animal Health and Food Safety Laboratory System, described the development of polymerase chain reaction-based (PCR-based) assays to screen for diseases that have caused devastating outbreaks in livestock, such as exotic Newcastle disease (END) in poultry and foot-and-mouth disease (FMD) in cattle. California also developed a highly efficient tuberculosis testing program after the disease was detected in several of the state’s large dairies.
Based on such experiences, Ardans argued that the state’s laboratory system plays its most crucial role when recognizing and responding to unusual disease events. For example, following a recent END outbreak among fighting cocks, whose handlers worked in and spread the disease to commercial poultry operations, the laboratory optimized an existing real-time PCR assay for END that was used to perform more than 85,000 tests (Crossley, 2005). Such emergencies present unique opportunities to improve disease diagnosis, Ardans said, although not necessarily with the latest technology. He noted that laboratory researchers, in pursuit of the source of E. coli O157:H7 following a recent outbreak in spinach, discovered that a gauze swab used to sample irrigation waters for contaminants performed better than newer concentration devices.
Emerging Diagnostics
Although Koch’s postulates remain diagnostic standards, adapting them to a vastly expanded understanding of disease states has become increasingly problematic, observed presenter Ian Lipkin and co-author Thomas Briese of Columbia University’s Jerome L. and Dawn Greene Infectious Disease Laboratory. Their paper discusses contemporary problems in proving causality, and illustrative case studies that reveal how these challenges are shaping pathogen surveillance and discovery. The authors also provide a taxonomy and comparative guide to proven
and proposed methods for characterizing infectious agents without recourse to cultivation, including two platforms of their own creation: MassTag PCR and the GreeneChip. In the future, Lipkin and Briese predict, substantial advances against chronic disease will occur “not from technical improvements but from investments in prospective serial sample collections and an appreciation that many diseases reflect intersections of genes and environment in a temporal context.”
Pre-Symptomatic Diagnosis
Imagining a future in which bioterrorism agents are continually reengineered to elude standard detection and diagnostic methods as well as therapeutics, speaker and Forum member Stephen Johnston offers a model of diagnosis for exposure to a pathogen before symptoms appear: a host-based detection system, capable of analyzing hundreds to thousands of components in samples of blood, sputum, or urine, and thereby capable of detecting any type of engineered or natural threat agent. In the final paper of this chapter, Johnston discusses the feasibility of developing such a system and its potential not merely to detect biothreats, but to “convert standard health practice from one that treats symptoms to one that detects disease very early—even presymptomatically.”
PARTNERING FOR BETTER MICROBIAL DIAGNOSTICS1
Mark D. Perkins, M.D.2
Foundation for Innovative New Diagnostics
Peter M. Small, M.D.3
Bill and Melinda Gates Foundation
Timely and accurate diagnosis is critical to the global efforts to prevent and treat infectious diseases. And yet, those on the front lines of this battle struggle to make do with inadequate and antiquated testing technology. For example, a 100-year old test is used to diagnose tuberculosis, a disease that kills someone every 16 seconds, and precious new antimalarial drugs are being rolled out with the same diagnostic imprecision that currently mistreats several hundred million cases every year. The tragic reality is that diagnostic uncertainty exacts a huge toll in morbidity and mortality. Reliance on underperforming diagnostic technologies limits the control of the world’s greatest killers, especially in settings with high
human immunodeficiency virus (HIV) prevalence. We contend that innovative mechanisms are needed to produce, develop and deploy new and better diagnostic tools for infectious diseases in developing countries.
Global Public Health Goals at Risk
Acknowledging the impact of the global tuberculosis epidemic in the early 1990s, the World Health Assembly of the World Health Organization (WHO; Geneva) declared tuberculosis a global emergency and ratified goals for case detection and cure under the DOTS (directly observed therapy shortcourse) strategy by the year 2005. Although important successes in fighting tuberculosis have been achieved in recent years, reliance on weak diagnostic tools has slowed progress. Case detection targets for smear-positive tuberculosis have not been met, and fewer than 25 percent of all cases are now detected and reported as smear positive (WHO, 2004). The data available suggest that the Millennium Development Goal of halving tuberculosis prevalence by 2015 also cannot be achieved universally without improved methods for diagnosing tuberculosis (Dye et al., 2005).
The weaknesses of standard diagnostic tests for tuberculosis are well documented. Even in controlled research settings, the average sensitivity of sputum microscopy for pulmonary tuberculosis is only 60 percent in immunocompetent populations, and it is substantially lower among people infected with HIV. Conventional culture methods are so slow that testing often loses clinical relevance, and the poor predictive value of the tuberculin skin test renders it essentially worthless in disease-endemic areas. The weaknesses of the available diagnostic technologies are only amplified in high-burden countries, which typically have insufficient infrastructure and inadequate staffing.
Reliance on inadequate diagnostic tools cripple TB control efforts. Because of limited access to diagnostic services and the low sensitivity of conventional testing, patients in many high-burden countries remain undiagnosed for three to six months (Madebo and Lindtjørn, 1999; Liam and Tang, 1997). These delays result in increased morbidity and mortality, mounting costs combined with loss of work, and continuing tuberculosis transmission to families and communities.
Unlike tuberculosis, which requires months of treatment to cure, malaria can be treated with a few doses of unsupervised treatment. This dramatically reduces the motivation to confirm the diagnosis. Microscopy for malaria is notoriously difficult, and experienced microscopists give substantially different results on up to a third of all slides. In most settings where malaria is endemic, quality microscopy is poorly available and malaria treatment is given by default to almost all patients with fever. Fever is an exceedingly common symptom in the tropics, and an estimated 800 million malaria treatments are given each year for fevers, the great majority of which are not caused by malaria (Amexo et al., 2004). This massive mistreatment of hundreds of millions of people results in
the fatal under-treatment of other diseases, such as pneumonia and sepsis, which present with similar symptoms.
Having watched at least two generations of malaria medicines fall to mounting drug resistance, the international malaria community has called for greater diagnostic accuracy before treatment, especially as expensive artemisinin-based therapies are introduced. In 2004, the WHO recommended that malaria should be confirmed by parasitologic examination before treatment in all patients older than five years of age. In this setting, the development of simple and rapid diagnostic tests (RDTs) that can detect circulating Plasmodium antigens in a drop of finger-prick blood is a key recent development.
The success of RDTs in improving the targeting of drug therapy, and their acceptance in malaria management by remote health workers and patients, will depend on the reliability and accuracy of the tests. There are now more than three dozen manufacturers of such tests, many of which show inadequate sensitivity, thermostability and geographic applicability. Though RDTs are now in wide use in some areas, the lack of true performance data on most of these tests, the variability in published performance of others and the lack of a global quality assurance mechanism has generated chaos and confusion with regard to test selection and has resulted in many end-users rejecting test results in favor of presumptive treatment.
The lethal convergence of these diseases and HIV exacerbates the negative impact of weak diagnostic tools. The rise of HIV in tuberculosis-endemic settings dramatically increases tuberculosis incidence, the number of symptomatic individuals and the pressures on already overburdened health systems. HIV coinfection decreases the sensitivity of microscopy for TB at the same time that it increases the urgency for rapid diagnosis and treatment. From South Africa to Brazil (Pronyk et al., 2004; Gutierrez et al., 2002), 30 to 50 percent of HIV-infected people die with undiagnosed tuberculosis, and Mycobacterium tuberculosis is now a leading cause of bacteremia in febrile patients visiting emergency rooms in sub-Saharan Africa (Archibald et al., 1998). Fever in HIV endemic areas cannot be assumed to be benign if nonmalarial. Thus, for many countries burdened by HIV, the need for improved diagnostic tests is increasingly urgent.
New Opportunities
Recent trends in science and technology, and in the diagnostics industry, indicate that there may be important new opportunities to improve diagnostic tests suitable for developing countries. Availability of the complete genomic sequence of M. tuberculosis allows a comprehensive assessment of potential diagnostic targets. Massive investment in biodefense has generated a range of diagnostic technologies intended for front-line use. The growing diagnostics industry can develop new diagnostic tests at a fraction of the cost and time needed to bring drugs and vaccines to licensure.
Motivated primarily by the small but significant market in industrialized countries, the tuberculosis diagnostics industry has produced several new tests in recent years. For example, shortcuts around the slow growth of M. tuberculosis using phage-based or molecular methods allow tuberculosis detection and screening for rifampin resistance within 48 hours (Albert et al., 2002; Johansen et al., 2003). Other new tests exploit tuberculosis-specific proteins to detect latent infection with much improved specificity, especially in BCG (BacilleCalmette Guérin)-vaccinated populations (Lalvani et al., 2001; Mori et al., 2004). Likewise, for malaria diagnosis, several rapid immunochromatographic tests detecting Plasmodium antigens in blood have been developed over the past 15 years, and they now reach a market of some 25 million people.
Forging a Public–Private Initiative
Market forces alone, however, will not yield the diagnostic tools needed to improve global health. Private companies often avoid developing products that will primarily be used in developing countries out of skepticism about the return on their investment. Developing countries have little capacity to pay the higher prices typically attached to new products, even when these costs result in overall savings to health care systems. The processes by which these countries license, purchase, and distribute products are often inadequately developed and poorly understood by industry.
The drive to develop new diagnostics for the developing world is unlikely to succeed without the private sector, with its expertise in product development, manufacturing capacity, product distribution and quality control. Unless measures are put in place to address current market dynamics, the number of companies engaged in diagnostics development will likely remain limited, and most will continue to tailor their products to markets in industrialized countries. The resulting products, such as the molecular amplification systems and automated systems for early detection of mycobacterial growth—which have markedly improved the diagnosis of tuberculosis in industrialized countries—may be little used in developing countries and thus have no impact on the global tuberculosis problem. Most of the companies manufacturing rapid malaria tests are small and do not have the resources to redevelop their assays to address important deficiencies in sensitivity and shelf life, especially at tropical temperatures.
Goal-driven public sector action is needed across the development pathway to forge a strong and sustainable partnership with industry to generate new diagnostics (Figure 3-1). Public sector actors must be prepared to sponsor basic research, partner equitably with industry on product development, evaluate products in a regulatory-quality fashion (Small and Perkins, 2000), demonstrate the efficacy of implementation, change technical and financial policies to foster new diagnostics, and actively facilitate the latter’s distribution and use. In pursuit of these goals, the public sector should explore such innovative approaches as the
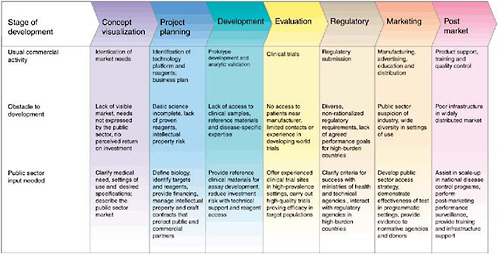
creation of novel financing mechanisms and distribution strategies to increase industry confidence that a viable market will exist in resource-limited settings.
There are many examples of innovative public–private partnership for the development of drugs and vaccines. Few are, however, focused on diagnostics. The Foundation for Innovative New Diagnostics (FIND; Geneva, Switzerland; of which Mark D. Perkins is Chief Scientific Officer), is one such entity. Launched in 2003, FIND aims to develop a model for public sector action to drive the development of diagnostic products for diseases of the developing world, using the search for new diagnostics as the test case for the model’s development. FIND seeks to identify the most promising product candidates and accelerate the process of development, testing, approval, distribution and incorporation into routine public health policy. Although motivated by the desire to create new public goods, FIND has many of the attributes of a private company, pursuing a clear business plan and using rigorous scientific criteria to identify priority product candidates.
RAPID INFECTIOUS DISEASE DIAGNOSTIC ASSAYS4
Mark J. Wolcott, Ph.D.5
U.S. Army Medical Research Institute of Infectious Diseases
Randal J. Schoepp, Ph.D.5
U.S. Army Medical Research Institute of Infectious Diseases
David A. Norwood, Ph.D.5
U.S. Army Medical Research Institute of Infectious Diseases
David R. Shoemaker, Ph.D.5
U.S. Army Medical Research Institute of Infectious Diseases
Rapid disease diagnostics (“serving to identify a particular disease or pathogen”) for many infectious agents are not as well developed as other laboratory technologies. Laboratory tests for many infectious agents still rely on decades-old technologies and techniques. Culture remains the gold standard for identifying organisms, but not all pathogens can be cultured, making alternative tests necessary.
When culture is difficult or not available (virus cultures in field laboratories), serological diagnosis of the antibody response to the organism is typically used.
However, a problem with both traditional culture and immunodiagnostics is the time required to obtain results. Culture may take several days and immunodiagnosis is limited by the time required to mount an antibody response, often a week or more (Figure 3-2). Current efforts in rapid diagnostics are shifting the window of detection closer to the point at which clinical disease symptoms become evident. Ultimately, future rapid diagnostics will shift the window to a point soon after exposure, giving the clinician the greatest opportunity to intervene in the disease process.
Orthogonal diagnostic testing is the key to improving the reliability of rapid diagnostic technologies. Orthogonal testing refers to tests that are statistically independent or non-overlapping but, in combination, provide a higher degree of certainty of the final result. Although orthogonal testing is not a standard perspective in the clinical diagnostic industry, the concept and its application are paramount when investigating some infectious agents. Any single detection technology has a set of limits with regard to sensitivity and, most importantly, specificity. Orthogonal testing seeks to overcome the inherent limitations of individual test results with the strength of data combinations (Henchal et al., 2001). The application of orthogonal diagnostic testing uses an integrated testing strat-
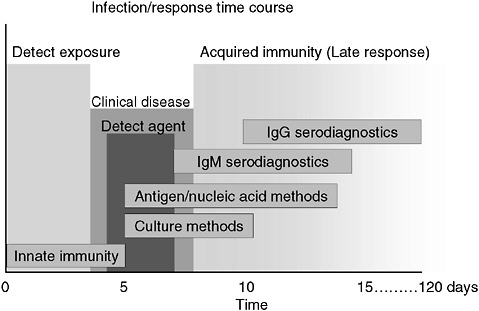
FIGURE 3-2 Infection and response time course. Various detection methodologies have highly different entry points in their use on human disease. As the time points extend out, the ability of medical interventions have less success. The earlier the time of medical intervention, the more successful the prognosis is for most diseases.
SOURCE: Wolcott (2006).
egy where more than one technology, technique, or biomarker is used to produce diagnostic results, which are then interpreted collectively (Figure 3-3).
The Department of Defense has an acquisition program to acquire quality diagnostic products that satisfy the needs of commanders with missions to support the warfighter. This acquisition program is designed to be timely with fair and reasonable associated costs. The acquisition program includes design, engineering, test and evaluation, production, and operations and support of defense systems (Table 3-1). To simplify and expedite the acquisition timeline for the fielding of a rapid diagnostic system, commercial off-the-shelf technology is evaluated and a formal selection process is used to select a system for further development and fielding. The Joint Biological Agent Identification and Diagnostic System (JBAIDS) acquisition program was formally launched in September 2003 with the award of the first phase, a molecular diagnostic system, in fall 2005 (Figure 3-4).
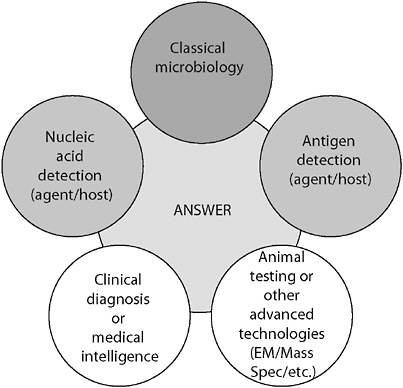
FIGURE 3-3 Orthogonal diagnostic testing. Although each method provides an independent assessment, together the power of the diagnostic becomes large. The failure of any one independent assessment does not fail the system.
SOURCE: Wolcott (2006).
TABLE 3-1 Department of Defense (DoD) Acquisition Program for Diagnostic Devices
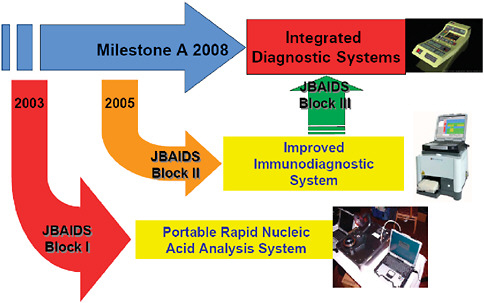
FIGURE 3-4 Acquisition program—evolutionary strategy. The acquisition process for developing and fielding a rapid infectious disease diagnostic assays system is designed around an evolutionary strategy. By leveraging commercial technologies that currently exist in the commercial market, and furthering development on those platforms, the final field-deployable system will be quicker and cheaper than trying to obtain the final product up front.
SOURCE: Wolcott (2006).
Current molecular diagnostic technologies are based on the amplification of specific DNA sequences from extracted nucleic acids, DNA or RNA. Amplification techniques take tiny amounts of nucleic acid material and replicate them many times through enzymatic reactions, some that occur through cycles of heating and cooling. These include methods that involve target amplification (e.g., polymerase chain reaction [PCR], reverse transcriptase–PCR [RT-PCR], strand displacement amplification, transcription amplification), signal amplification (e.g., branched DNA assays, hybrid capture), probe amplification (e.g., ligase chain reaction, cleavase-invader, cycling probes), or postamplification analysis (e.g., sequencing the amplified product or melting curve analysis as is done in real-time PCR).
Nucleic acid-based methods are generally specific and highly sensitive and can be used for all categories of microbes (Christensen et al., 2006; Emanuel et al., 2003a). Amplification methods can identify minute traces of the genetic material of an organism in a specimen, avoiding the need for culture. These techniques are particularly useful for organisms that are difficult to culture or identify using other methods (e.g., viruses, obligate intracellular pathogens), or are present in very low numbers. Results can be provided more rapidly than through most conventional methods, especially culture. However, because amplification methods are so sensitive, false positives from trace contamination of the specimen or equipment can easily occur. In addition, because these techniques depend on enzymatic activity, false-negatives also occur when a sample contains contaminants that inhibit enzyme activity (Hartman et al., 2005). Nucleic acid-based tests are also limited in that they do not provide information on the viability of the detected organism.
Immunodiagnostics is the standard against which many agent detection, identification, and diagnostic technologies are compared. Antibody-based assays continue to serve as preliminary and confirmatory diagnostic formats for many infectious and noninfectious diseases. These assays are typically rapid, sensitive, specific, reliable, and robust. Immunodiagnostic technologies are relatively unsophisticated, making them available to nearly any laboratory.
Hand-held assays (HHAs) are immunoassays that are based on immunochromatography or lateral flow assay format. Generally, a sample is applied to the testing unit and by flowing along a membrane, an indicator line forms where antibodies to the analyte of interest are bound. The presence of a line indicates the presence of the analyte, while the absence of a line denotes a negative result. Applying a sample solubilizes the tagged antibodies and initiates the first binding of the target by the tagged antibodies. As the sample continues migrating down the filter paper, the analyte of interest encounters a set of antibodies bound to the membrane and an antibody-analyte-antibody sandwich is formed. While early HHAs incorporated enzymes as labels to yield a visible signal, advances have done away with the multistep enzyme immunoassay format and have incorporated reporter molecules such as colloidal gold or colored latex spheres that yield
a direct signal. These physical signal generators rely on the aggregation of a large number of tags to enhance signal visualization by the naked eye. HHAs, like all analytic systems, have inherent limitations in their use and interpretation; they require a relatively large amount of sample, their sensitivity is limited, and they have a potential for false positives as a result of “dirty” environmental samples that form a confounding “dirt” line in the antibody capture zone. Although HHAs have limitations, their overall ease of use and quickness make them useful in certain situations.
Time-resolved fluorescence (TRF) is an immunoassay application that employs the basic immunoassay analyte sandwich capture format, but with detector antibodies that are directly labeled with a lanthanide chelate, such as europium, samarium, terbium, and dysprosium. The strengths of TRF are its increased sensitivity and the potential for multiplexing. TRF uses the differential fluorescence life span of lanthanide chelate labels compared to background fluorescence. The long-lived fluorescence signal and the difference in wavelength between absorbed and emitted light results in a very high signal-to-noise ratio and excellent sensitivity (Hemmila et al., 1984; Soini and Kojola, 1983). The long fluorescence decay time allows the measurement of immunoassay fluorescence after any background fluorescence has decayed. By pulsing the excitation light repeatedly, in 1 second the fluorescent material can be excited more than 100 times with an accumulation of the generated signal that improves both the overall signal and the reduction of background signals. TRF assays are particularly useful in clinical immunoassays, but have limitations with environmental samples where europium or other lanthanides naturally occur. The contaminating compounds behave much like labeled lanthanides, prolonging the background fluorescence and lowering TRF sensitivity.
Electrochemiluminescence (ECL) is immunoassay technology in which a detector antibody is tagged with a chemical that emits light (luminescence) when it is excited by an electrical stimulus. There are several electrochemiluminescent chemical moieties, but ruthenium is the most common. Ruthenium, in the form tris (2,2’ - bipyridine) ruthenium (Ru), is relatively small, allowing easy conjugation to antibodies. The technology relies on two components: the ECL-label (Ru) coupled to an antibody and tripropylamine (TPA) present in the reaction buffer. When an electrical current is applied to an electrode, both components are activated by oxidation. The oxidized TPA is transferred into a highly reducing agent, which reacts with activated Ru to create an excited-state form of Ru. This form returns to its ground state with emission of a photon at 620 nm wavelength. An advantage of the Ru-TPA methodology is that the measurement of a single sample can be repeated multiple times because the electron-transfer photon-release reaction regenerates the Ru resulting in signal amplification. Although ECL assays are simple, rapid, and sensitive (Kijek et al., 2000; Smith et al., 2001), the sample matrix can affect the assay sensitivity. The sample matrix will influence the sensitivity by varying positive cut-off values; therefore, matrix-
specific positive and negative control samples are used to establish standard curves and cutoff values.
Several diagnostic systems are using a technology to analyze microsphere-based multiplex protein assays. The advantage of multiplex assays is that multiple results are available from one sample without individual testing. Up to 100 different biomolecules (proteins, peptides, or nucleic acids) can be analyzed in a single test. A microplate platform allows the automated analysis of a 96-well plate in 30 minutes yielding a throughput of 1,920 assays in a 20-plex system. Currently kits for simultaneous quantitative measurement of up to 25 to 30 proteins are available, including cytokines, phosphoproteins, growth factors, kinases, and transcription factors. Several investigators are using these systems to develop multiplexed assays for biological warfare agents. One system was evaluated by the U.S. Army with extremely good results, but the equipment is currently not rugged enough for use by the warfighters.
The key to future rapid diagnostic systems is the development of a completely and fully integrated system. Previous diagnostic research efforts were only concerned with the development of an assay technique and failed to address the full spectrum of an integrated system. To fully address an integrated system, protocols, sample processing, reagents, assays, platforms, and evaluations need to be completely explored. Protocols are equivalent to an intended-use statement. Without addressing how and why the assay or system is to be used, misapplication will result in incorrect and potentially serious testing reliability issues.
The single most important aspect of rapid testing is sample processing. The sample is the most important component in a system, and an inappropriate or improperly handled sample will jeopardize an otherwise robust assay. For example, detection of Bacillus anthracis is highly problematic. The spores of Bacillus anthracis are very refractile to easy and rapid sample preparation. Alternate methods are required to produce the highest quality sample, which include concentrating the sample (if possible) and methods to release either the nucleic acids or specific proteins from the spore. These include techniques such as germination, sonication, or mechanical disruption (“bead-beating”). Another consideration of sample preparation, especially for many molecular methods, is the removal or neutralization of inhibitors of amplification.
Systems consist of more than just assays (Figure 3-5). Developers need to be cognizant of all the details. While most commercial manufacturers have appropriate production systems and quality manufacturing practices in place for producing consistent, reliable, and appropriate reagents that are compliant with Food and Drug Administration (FDA) requirements, research-derived systems often fall short. In addition, integration of assays with various platforms is often overlooked in initial system development. While some assays perform well on multiple platforms, many assays suffer optimization issues when moved from one platform to another. Unless provisions are made for multi-platform development, and shown to be equally effective through validation, platform equivalency
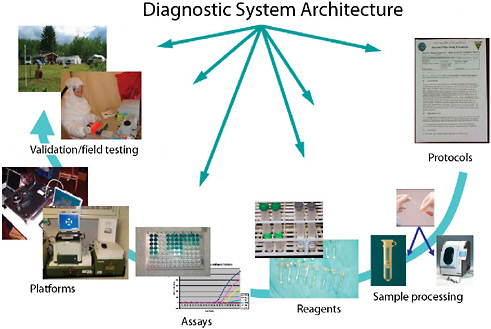
FIGURE 3-5 Diagnostic system architecture. Systems-based architecture needs to include the full gamut of functions from protocols through validation.
SOURCE: Wolcott (2006).
should not be assumed. Another consideration in system development for systems developed by professional scientists working in modern laboratory facilities is the inherent expectation that assays and systems will work in the hands of less trained personnel outside of the pristine laboratory facilities. Often, this is not the case. Field evaluation, under conditions of actual employment, is critical before assays and systems can be confidently deployed and used.
Validation of the appropriateness and effectiveness of assays and systems is paramount in the development process (Emanuel et al., 2003b). Development of assays and systems needs to include assay validation parameters such as linearity, limits of detection, inclusivity and exclusivity testing, ruggedness, robustness, and repeatability. Validation parameters are detailed in Box 3-1.
A critical and often overlooked issue is that diagnostic systems and tests intended to be used to test clinical samples must be approved by the U.S. Food and Drug Administration in order to legally be distributed and used in the United States. Many of the technologies discussed in this article are mature enough to produce clinically useful diagnostic products. However, companies that may have the capability to manufacture these diagnostic tests, and to gain FDA approval for them, typically are not interested in doing so for tests to diagnose tropical
|
BOX 3-1 Example of Diagnostic Systems Validation Parameters
SOURCE: Wolcott (2006). |
diseases or biological threat agents because the commercial demand is low. This is a chronic problem with no easy solution.
To help support the deployment of rapid agent identification systems, especially those that do not have enough commercial value to be fully supported by commercial manufacturers, the Department of Defense relies on the Joint Program Executive Office–Critical Reagents Program (CRP). The CRP is a national resource for the biological defense community, whose mission is production of
detection reagents, standardization of procedures and training, and optimization and transition of detection technologies. Their commodity areas include the production of antigenic and genomic materials for test and evaluation purposes, antibodies, to include the manufacturing of hand-held devices, molecular detection reagents, and sampling kits. Because of the confined nature of these materials and the lack of commercialization due to the limited customer base, CRP provides a vital link to the defense community to ensure harmonization of tests and evaluations and as an avenue for advanced development.
In the course of development of newer, faster, better, and cheaper rapid diagnostic devices, the Department of Defense program is looking at potential future platforms. Many characteristics of those future systems are discussed above, but one that is showing some promise is DNA microarrays. Microarrays or DNA chips are one of the latest methods for rapid infectious disease diagnostics. Microarrays are a recent adaptation of Northern blot technology (Grunstein and Hogness, 1975; Schena et al., 1995). The ability to label nucleotide sequences with fluorescent tags, much like fluorescent antibody technology, has increased their use in diagnostics. Microarrays are small, solid supports (typically glass slides) on which DNA sequences are attached, or spotted, at fixed, orderly, addressable locations. The DNA is composed of short, single-stranded fragments, typically 5 to 50 nucleotides long. Microarrays can have up to tens of thousands of spots, allowing for a large amount of data collected for each sample tested. Microarrays depend on the annealing of two nucleic acid strands to function. When sample DNA is prepared, usually through polymerase-based amplification, fluorescent dyes are incorporated into the amplicon so that hybridization can be detected. The kind of information required from microarrays drive how the arrays are developed and used. Microarrays can be spotted with known sequences of a variety of oligonucleotides for basic genomic investigation. Gaining wider acceptance is the use of microarrays to “resequence” organisms. Utilizing known sequences from already sequenced organisms and hybridizing genomic material from organisms not previously sequenced, sequence differences can be determined. With more than 10,000 sequences (and growing as automated systems improve) to interrogate on a single chip, variation in genomic sequences can provide accurate species and subspecies determination. Finally, one of the earliest applications of microarrays is their use in “transcriptomics” or gene expression studies. Gene expression-based measurements of mRNA levels, and the differences between these levels in various states of organism growth (i.e., aerobic versus anaerobic growth), has provided significant insights in gene regulation of various organism functions.
Although microarrays have the demonstrated potential for diagnostics, routine use is hampered by several considerations. The first hurdle for microarrays is the availability of high-quality, validated, and standardized arrays and processes. A key limitation to implementation of routine diagnostic microarrays is identification of appropriate targets. Although ribosomal RNA gene targets are widely
used, they are limited in their ability to resolve bacteria below the species level (Saliba et al., 1966). Other bacterial target genes, including housekeeping genes, are potentially useful, but data across the full breath of organisms are limited or nonexistent. Even with good targets, optimal hybridization conditions for all the probes on a single array are still challenging. Redundant variations in probes help compensate to a degree. Another challenge to microarray routine use is the sensitivity of most systems. To obtain appropriate sensitivity, polymerase amplification is necessary. In most systems, this requires a multitude of specific primers for the genes of interest. Because multiplexed PCR is limited to a dozen or so reactions, several hundred iterations of PCR could be required to completely cover all the potential probes on an array, which is not practical in routine use. Until a good on-chip amplification or signal detection method is developed, the use of diagnostic microarrays will be limited.
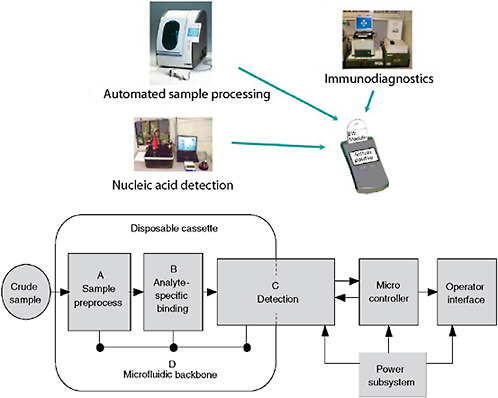
Ultimately, to meet the needs of users, rapid infectious disease diagnostic assays need a comprehensive integrated system. This includes automated sample processing and the use of multiple technologies to obtain results that can be interpreted against the clinical picture or medical intelligence. Currently, immunoassays and molecular assays are the most mature technologies. Immunoassays are a maturing technology that has improving sensitivity and specificity. With improvements in signal amplification and the use of monoclonal antibodies, immunoassays are fast, robust, and approaching the sensitivity of some molecular methods. Molecular methods are rapidly developing but are not at the full maturity level yet. Amplification methods achieve exquisite sensitivity, but at the risk of potential contamination events. Together, immunoassays and molecular techniques are very complementary and a powerful set of techniques for an integrated system (Henchal et al., 2001) (Figure 3-6).
The future for rapid infectious disease diagnostics is the lab-on-a-chip approach, where all sample processing, assay technologies, detection, and reporting are fully integrated into one unit. Miniaturized, disposable, and cost-effective units will evolve from our current systems. As technologies mature and new technologies are developed, rapid infectious disease diagnostics will become available and practical.
Ramping Up to Success
Although we believe that the products of strategic, adequately supported public–private partnerships to develop diagnostics could transform approaches to control infectious diseases in poor countries, progress inevitably will be incremental, especially in the near term. Because new tests are likely to be imperfect, we will need flexibility and creativity to ensure that these tools are used to maximum effect. For example, rather than discard a rapid tuberculosis test with high sensitivity but low specificity, we should consider incorporating it into a diagnostic algorithm to quickly rule out tuberculosis in most patients present-
ing with chronic cough—a step that could dramatically decrease the workload of tuberculosis clinics. Rather than develop a single test to replace the sputum microscopy, we should embrace the concept of market segmentation and develop a range of new tools suitable for different diagnostic environments.
We must also accept that new technology may require changes to longstanding public health practices. For example, tuberculosis epidemiology has long been tracked by monitoring the number of smear-positive patients. If microscopy is replaced with a more sensitive test, tracking of tuberculosis trends could be disrupted. But this is a small price to pay for better serving patients and strengthening the world’s ability to bring tuberculosis under control. Similarly, microscopy offers quantitative estimates of parasite burden, which is often used by clinicians to estimate the severity of illness or to monitor the effectiveness of treatment. Replacement with qualitative testing will force a change of practice, even as it brings the power of confirmatory diagnosis out of referral laboratories and into the community.
Finally, we emphasize that the impact of a new diagnostic test ultimately will be determined by the extent to which it is used. Expanding use of a new technology, as with any global health intervention, ultimately will depend on political will. Integration of improved diagnostics into national programs in the same structured fashion that has been used for standard and second-line tuberculosis drugs is possible, but only if leaders confront a range of issues that will make implementation possible—from lifting import taxes to improving laboratory capacity to modifying disease control guidelines. Is this too much to ask to give our health-care practitioners the tools they need to do their jobs?
EMERGING TOOLS FOR MICROBIAL DIAGNOSIS, SURVEILLANCE, AND DISCOVERY
W. Ian Lipkin, M.D.6
Columbia University
Thomas Briese, Ph.D.6
Columbia University
Introduction
Here we describe methods and perspectives for pathogen surveillance and discovery, and discuss challenges associated with proving causality. We provide examples from our own experience to illustrate the complexity of pursuing
research in this arena and to provide the reader with insights into the process that led to the implementation of particular strategies.
Proof of Causation
Discovery of an organism in association with disease is only the first step in understanding its role in pathogenesis. Many have wrestled with the challenge of codifying the process of proving causation. Based on the germ theory of disease formulated by Pasteur, Koch, and Loeffler proposed precise criteria that define a causative relationship between agent and disease: The agent should be present in every case of a disease, it should be specific for a disease (i.e., present in none other), and it should be propagated in culture and proven capable of causing the same disease upon inoculation into a naïve host. Known as Koch’s postulates (Koch, 1891), these criteria were subsequently modified by Rivers for specific application to viruses (Rivers, 1937), and by Fredricks and Relman to reflect the advent of molecular methods (Fredricks and Relman, 1996) (Table 3-2). Nonetheless, Koch’s postulates remain the ideal standard by which causality is considered to be proven. There are problems with holding to this standard. Some agents cannot be propagated in culture. Additionally, for many human viral pathogens, there may be no animal model. In many acute viral diseases, the responsible agent can be readily implicated because it replicates at high levels in the affected tissue at the time the disease is manifest, morphological changes consistent with infection are evident, the agent is readily identified with classical or molecular methods, and there is evidence of an adaptive immune response. However, implication of viruses in chronic diseases may be confounded because persistence requires restricted gene expression, classical hallmarks of infection are absent, and/or mechanisms of pathogenesis are indirect or subtle. In the final analysis, investigators are occasionally left with what amounts to an assessment of strength of epidemiological association based on the presence of the agent, its footprints (nucleic acid, and preferably, an immune response), and biological plausibility based on analogy to diseases with related organisms where linkage is persuasive.
Many Routes to Microbial Pathogenesis
Implication of an infectious agent is most straightforward in instances where it is present at the site of disease at the time the disease is manifest. Two classic examples where effects are readily appreciated at the infection site are poliomyelitis, where virus replicates in motor neurons of the brain and spinal cord, causing cell loss and paralysis, and cholera, where Vibrio cholerae replication and local elaboration of toxin in the intestine alters ion transport, resulting in diarrhea. A more complex example of intoxication is botulism where replication of Clostridium botulinum in the skin or the gastrointestinal tract leads to local
TABLE 3-2 Criteria for Proof of Causation
|
Robert Koch (1890)a |
Thomas R. Rivers (1937) |
Fredricks and Relman (1996) |
|
A microbe must be:
NOTE: The pathogen should be present at the proper time in specific regions and the disease should be produced with some regularity by serious inoculation of infected material into a susceptible host. |
NOTE: The possibility of a viral carrier state was recognized and Koch’s requirements of propagation in media or cell culture was abandoned.
NOTE: This fourth postulate though not required by Koch, logically follows his other conditions and so has been added by some reviewers. |
|
|
aAlthough Koch included basic points already in earlier papers, especially his 1884 paper on the etiology of tuberculosis, his most explicit presentation was given at the 1890 International Congress of Medicine; the proceedings of which were published in 1891. SOURCE: Koch (1891); Rivers (1937); Fredricks and Relman (1996). |
||
expression of a toxin that traffics to the neuromuscular junction to interfere with motor function. Host responses to infection may contribute to pathogenesis. Acute infection with influenza virus or severe acute respiratory syndrome (SARS) coronavirus elicits cytokines and chemokines that cause pulmonary dysfunction. Chronic inflammation in hepatitis B and hepatitis C infections can result in hepatic failure and hepatocellular carcinoma. Infection can also lead to inhibition of immune function. The capacity of viruses to enhance susceptibility to opportunistic agents is now best known in the context of HIV/AIDS; however, the observation of virus-induced immunosuppression dates back to the early 1900s when von Pirquet noted the loss of skin reactivity to tuberculin in association with measles infection. The effects of infection may depend on the age and maturation status of the host. Individuals at either extreme of life are at increased risk for acute morbidity and mortality with a wide variety of infections. Encephalitis
is far more common in individuals infected with West Nile virus after the age of 50 years than in other adults or children. Infection during organogenesis may have different consequences than at other times. Congenital rubella infection, for example, can be associated with characteristic cardiac and central nervous system defects. Persistent viral infections are described in animal models where subtle effects on cellular physiology result in alterations in the expression of neurotransmitters or hormones that have profound effects including cognitive impairment, hypothyroidism, or diabetes mellitus. Whether similar mechanisms can be implicated in human disease remains to be determined; nonetheless, these preclinical studies indicate biological plausibility. Infection can break tolerance for “self,” resulting in autoimmune disease. A classical example is molecular mimicry in group A beta-hemolytic streptococcus infection where cross-reactivity to heart and brain results in valvular disease and chorea, respectively. The capacity for infections to cause disease via myriad mechanisms, direct and indirect, short and long term, pose challenges for pathogen discovery.
Molecular Strategies for Pathogen Discovery
Methods for cloning nucleic acids of microbial pathogens directly from clinical specimens offer new opportunities to investigate microbial associations in chronic diseases (Relman, 1999). The power of these methods is that they can succeed where methods for pathogen identification through serology or cultivation may fail due to absence of specific reagents or fastidious requirements for agent replication. Over the past decade, the application of molecular pathogen discovery methods resulted in identification of novel agents associated with both acute and chronic diseases, including Borna disease virus, hepatitis C virus, Sin Nombre virus, HHV-6, HHV-8, Bartonella henselae, Tropheryma whippelii, West Nile virus, and SARS coronavirus (Challoner et al., 1995; Chang et al., 1994; Choo et al., 1989; Lipkin et al., 1990; Nichol et al., 1993; Relman et al., 1990, 1992; VandeWoude et al., 1990).
Various methods are employed or proposed for cultivation-independent characterization of infectious agents. These can be broadly segregated into methods based on direct analysis of microbial nucleic acid sequences (e.g., complementary DNA [cDNA] microarrays, consensus polymerase chain reaction [cPCR], representational difference analysis [RDA], differential display [DD]), direct analysis of microbial protein sequences (e.g., mass spectrometry), immunological systems for microbe detection (e.g., expression libraries, phage display), and host response profiling.
The decision to employ a specific method is guided by the clinical features, epidemiology, and spectrum of potential pathogens to be implicated. Expression libraries, composed of cDNAs or synthetic peptides, may be useful tools in the event that large quantities of acute and convalescent sera are available for screening purposes; however, the approach is cumbersome and labor-intensive,
and success depends on the presence of a specific, high-affinity humoral immune response. Mass spectrometry is an intriguing approach to pathogen discovery (Dalluge, 2000; van Baar, 2000); however, potential confounds include mutations in flora that alter spectra without clinical correlation; the requirement for establishment of large libraries of spectra representing flora of thousands of organisms propagated in vitro and isolated in vivo; and the difficulties associated with extending this technology to viruses, where disease may occur without robust protein expression, and pathogenicity may be correlated with single base substitutions. The utility of host response messenger RNA (mRNA) profile analysis has been demonstrated in several in vitro paradigms and some inbred animal models (Diehn and Relman, 2001; Taylor et al., 2000; Zhu et al., 1998); nonetheless, a variety of organisms may activate similar cascades of chemokines, cytokines, and other soluble factors that influence host gene expression to produce what are likely to be convergent gene expression profiles. RDA is an important tool for pathogen identification and discovery. However, RDA is a subtractive cloning method for binary comparisons of nucleic acid populations (Hubank and Schatz, 1994; Lisitsyn et al., 1993). Thus, although ideal for analysis of cloned cells or tissue samples that differ only in a single variable of interest, RDA is less well suited to investigation of syndromes wherein infection with any of several different pathogens results in similar clinical manifestations, or infection is not invariably associated with disease. An additional caveat is that because the method depends on the presence of a limited number of restriction sites, RDA is most likely to succeed for agents with large genomes. Indeed, in this context, it is noteworthy that the two viruses detected by RDA were herpesviruses (Challoner et al., 1995; Chang et al., 1994).
Consensus PCR also has been a remarkably productive tool for biology. In addition to identifying pathogens, this method has facilitated identification of a wide variety of host molecules, including cytokines, ion channels, and receptors. One difficulty in applying cPCR to pathogen discovery in virology has been that it is difficult to identify conserved viral sequences of sufficient length to allow cross-hybridization, amplification, and discrimination in a traditional PCR format. Although this may not be problematic when one is targeting only a single virus family, the number of assays required becomes infeasible when preliminary data are insufficient to permit a more directed, efficient analysis. To address this problem, we adapted cPCR to differential display, a PCR-based method for simultaneously displaying the genetic composition of multiple sample populations in acrylamide gels (Liang and Pardee, 1992). This hybrid method, known as domain-specific differential display (DSDD), employs short, degenerate primer sets designed to hybridize to viral genes that represent larger taxonomic categories than can be resolved in cPCR. Although this modification allowed us to identify West Nile virus as the causative agent of the 1999 New York City encephalitis outbreak (Briese et al., 1999), it did not resolve issues of low throughput with cPCR due to limitations in multiplexing.
To address the need for sensitive, facile, highly multiplexed pathogen surveillance, we established two new platforms for viral detection, MassTag PCR and the GreeneChip. MassTag PCR is a multiplex PCR method that can accommodate in excess of 20 genetic targets with sensitivity in the range of 10 to 1,000 RNA copies (variability is a function of primer degeneracy). The GreeneChip is a comprehensive viral microarray that addresses all vertebrate viruses in the International Committee on Taxonomy of Viruses (ICTV) database. Both methods rely on the presence of an agent related to one already known. In instances where agents are novel or sufficiently distant in sequence to related agents to confound hybridization it may be necessary to resort to subtractive cloning or high-throughput unbiased sequencing. Our algorithm for characterization of clinical materials is illustrated in Figure 3-7. Where the list of candidates to be considered can be addressed using MassTag PCR this is our method of choice due to low cost, speed, and sensitivity. Where MassTag PCR fails or the list of candidates exceeds 30 targets, we move to GreeneChips (viral or panmicrobial). In the event that GreeneChips fail we shift to unbiased high-throughput sequencing or subtractive cloning.
MassTag PCR
Although singleplex PCR assays are well established in clinical microbiology and have proved indispensable in management of HIV and hepatitis C virus (HCV), and in control of outbreaks where an agent is identified, multiplex assay applications have lagged behind. Fluorescence reporter systems in real-time PCR achieve quantitative detection with sensitivity similar to nested amplification; however, their capacity to simultaneously query multiple targets is limited to the number of fluorescent emission peaks that can be unequivocally separated. At present up to four fluorescent reporter dyes are detected simultaneously. To address the need for highly multiplexed assays, we created MassTag PCR, a platform wherein digital mass tags rather than fluorescent dyes serve as reporters (Figure 3-8). The first description of this method was published in the context of a panel that distinguishes 22 different viral and bacterial respiratory pathogens (Briese et al., 2005). It allowed us to identify viral and bacterial sequences in respiratory samples as well as cultured materials, and to recognize instances of coinfection not appreciated in reference laboratories using established diagnostics assays. We later expanded the repertoire to include causative agents of hemorrhagic fever, and to subtype influenza viruses. Between October and December 2004, an increased incidence of influenza-like illness (ILI) was recorded by the New York State Department of Health that tested negative for influenza virus by molecular testing, and negative for other respiratory viruses by culture. Concern that a novel agent might be implicated led us to investigate clinical materials (Lamson et al., 2006). MassTag PCR resolved 26 of 79 previously negative samples, revealing the presence of rhinoviruses in a large proportion of samples,
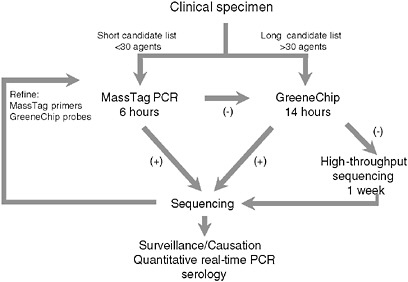
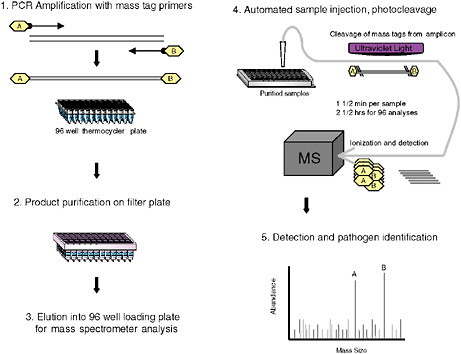
about half of which belonged to a previously uncharacterized genetic clade. The 2004 New York ILI study confirmed the utility of MassTag PCR for surveillance, outbreak detection, and epidemiology by demonstrating its potential to rapidly query samples for the presence of a wide range of candidate viral and bacterial pathogens that may act alone or in concert.
MassTag PCR may not suffice in instances where either larger numbers of known pathogens must be considered or sequence divergence may impair binding of PCR primers. The limitations of MassTag PCR (and other PCR platforms) were poignantly demonstrated during analysis of samples from Marburg hemorrhagic fever outbreaks in the Democratic Republic of Congo during 1998–1999 wherein two of five subjects were negative. The explanation for failure became clear after cPCR amplification and sequencing revealed three mismatches in the forward and one in the reverse primer (Palacios et al., 2006). If we had enjoyed our current access to unpublished, proprietary filovirus sequences at the time primers were designed, we would have averted difficulty in this instance. Nonetheless, despite access to sequences in World Health Organization (WHO) network laboratories, this experience reinforced the need for a complementary tool with higher tolerance for sequence divergence, and led us to develop the GreeneChip, a DNA microarray system.
Establishment of the Greene Microbial Database
A critical early step in the development of the MassTag PCR and microarray tools was the establishment of a viral sequence database. This effort was facilitated in 2002 by the move of the ICTVdB (International Committee on Taxonomy of Viruses Database)7 and its director, Cornelia Büchen-Osmond, from Biosphere 2 (Earth Institute) in Oracle, Arizona, to the Greene Laboratory; and the establishment of a Northeast Biodefense Center Biomedical Informatics Core. Because vertebrate viruses are highest priority for human disease, we focused on them first, with a plan to extend the database to viruses of invertebrates, plants, and prokaryotes as resources permitted. To ensure comprehensive coverage, we included every vertebrate virus listed in the ICTVdB, a taxonomic database that describes viruses at the levels of order, family, genus, and species. Efforts to identify cognate sequences for members of each of these taxa in the National Center for Biotechnology Information (NCBI) sequence database proved to be more difficult than anticipated. The NCBI database is not exhaustively curated; thus, it contains many entries where annotation is missing, outdated, or inaccurate. An additional confound is that only incomplete sequence is available for many viruses, bacteria, and parasites, particularly some relevant to this project, where genomic sequencing efforts are less advanced. To circumvent limitations in curation and nomenclature in the NCBI database, and to minimize computa-
tional costs in establishment of multiple alignments at the nucleotide (nt) level, we began construction of the Greene Viral Database (GreeneVrdB) by using the Protein Families database of alignments (Pfam)8 and Hidden Markov Models (HMM). Sequences for the design of oligonucleotide probes and MassTag PCR primers were selected based on biological parameters, including the degree of conservation of proteins or domains, their expression level during infection, and the amount of data available for the respective region.
The GreeneVrdB was established by integrating the taxonomy database of ICTV and the sequence database of NCBI (Figure 3-9).9 The majority of viral protein coding sequences in the NCBI database (84 percent) were represented in the Pfam database; the remainder were mapped using pair-wise Basic Local Alignment Search Tool (BLAST) alignments. A panmicrobial database (GreenePmdB) was established by supplementing the GreeneVrdB with ribosomal RNA (rRNA) sequences of fungi, bacteria, and parasites obtained from the Ribosomal Database Project (RDP)10 or the NCBI database. At the time of this writing the GreenePmdB comprises the 382,512 viral sequences of the GreeneVrdB, representing both complete and partial viral genomes; 41,790 bacterial 16S rRNAs; 4,109 fungal 18S rRNAs; and 2,626 18S parasitic rRNAs. These sequences represent all 2,011 vertebrate virus species and 135 bacterial, 73 fungal, and 63 parasite genera.
GreeneChips
DNA microarrays have potential to provide a platform for highly multiplexed differential diagnosis of infectious diseases. The number of potential features far exceeds that with any other known technology. Furthermore, probes of up to 70 nt are not uncommon. Thus, unlike PCR where short primer sequences demand precise complementarity between probe and target, DNA arrays are less likely to be confounded by minor sequence mismatches. Lastly, one can incorporate both microbial and host gene targets. This affords an opportunity to both detect microbes and assess host responses for signatures consistent with various classes of infectious agents. Despite these advantages, DNA arrays have not been widely employed because of limited sensitivity. Although a viral array was helpful in identifying the causative agent of SARS in 2003, critical to its success was the discovery that the agent could be propagated to high titer and had cytopathic effect in Vero cells (Ksiazek et al., 2003). Once this advance was shared, several investigators rapidly and independently identified the agent by electron microscopy, differential display, cDNA cloning, microarray, and cPCR. The challenge of array sensitivity has now been addressed with improved methods for sample
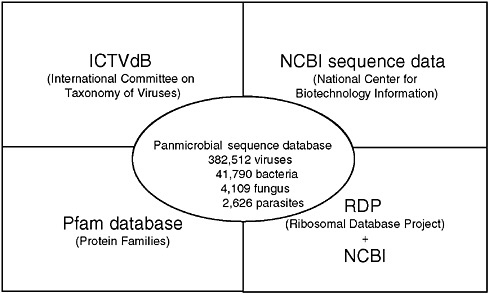
FIGURE 3-9 Greene pathogen database.
SOURCE: Lipkin (2006).
preparation, amplification, labeling, and printing. Together with Agilent Technologies, we created a DNA array platform suited to analysis of clinical materials without amplification in culture. Investigation by MassTag PCR and viral DNA microarray of blood collected during the 2005 Angola Marburg virus outbreak from an individual who died of hemorrhagic fever failed to yield a pathogen; however, implementation of a panmicrobial DNA array, GreeneChipPm, implicated Plasmodium falciparum infection (Palacios et al., 2007).
Microbial Probes
Viral probes were designed to represent a minimum of three distinct genomic target regions for every family or genus of vertebrate virus in the ICTVdB. Where possible, we chose highly conserved regions within coding sequence for an enzyme such as a polymerase, and two other regions corresponding to more variable structural proteins. Our reasoning was that RNAs encoding structural proteins may be present at higher levels than those encoding proteins needed only in catalytic amounts, and that use of probes representing noncontiguous sites along the genome might allow detection of naturally occurring or intentionally created chimeric viruses. The viral array has been through several iterations as the database evolved and technology allowed increases in probe density. The first release, GreeneChipVr1.0, comprised a total of 9,477 viral probes. The second release, GreeneChipVr1.1, added 6,271 more typing probes for influenza virus A hemagglutinin and neuraminidase genes. Recent releases,
GreeneChipVr1.5 (15,700 probes) and GreeneChipVr2.0 (86,300 probes), are the result of higher printing density on the Agilent array platform and a new generation of probe design algorithms. The process for identifying bacterial, fungal, and parasitic probes was similar, although restricted to 16S and 18S rRNA sequences. GreeneChipPm1.0 contained a total of 29,495 probes, including the probes comprising GreeneChipVr1.1 as well as 11,479 16S rRNA bacterial, 1,120 18S rRNA fungal, and 848 18S rRNA parasite probes.
Host Gene Markers
Identification of signal(s) representing a microbe in samples from affected subjects is a primary objective in pathogen discovery. Nonetheless, evidence of infection is bolstered by coterminous evidence of gene expression consistent with an activated host immune response. Furthermore, gene expression profiles may be helpful in implicating specific strains or serotypes (e.g., Th1 cytokine responses are more robust with H5N1 than H1N1 influenza infection) (Cheung et al., 2002). Finally, in cases where we fail to find clear evidence of a known pathogen, a profile consistent with immune activation may be helpful in determining whether to pursue additional studies focused on pathogen discovery. Thus, GreeneChips include probes for genes associated with cytokines, chemokines, and their receptors; components of the interferon-inducible signaling pathways; immunoglobulins (Igs) and Ig receptors; toll-like receptors and their downstream signaling pathways; complement components; major histocompatibility complex (MHC) molecules; and heat shock proteins from a set of validated oligonucleotides (Wright and Church, 2002).
GreeneLAMP Analysis Software and GreeneChip Validation
GreeneLAMP (Log-transformed Analysis of Microarrays using P-values) version 1.0 software was created to assess results of GreeneChip hybridizations. Common analysis software focuses on the differential two-color analysis used in gene expression arrays, which is not applicable to the GreeneChip. GreeneLAMP has a robust and generalized framework for microarray data analysis, including: flexible data loading, filtering, and control experiment subtraction. Probe intensities are background corrected, log2-transformed, and converted to Z-scores (and their corresponding p-values). Where available, control matched experiments from uninfected samples are used and spots >2 standard deviations (SD) from the mean are subtracted. In instances where matched control samples are not available, the background distribution of signal fluorescence is calculated using fluorescence associated with 1,000 random 60-mers (Null probes). In both scenarios, positive events are selected by applying a false-positive rate of 0.01 (the rate at which Null probes are scored as significant) and a minimum p-value per probe of 0.1 (in cases with a matching control) and 0.023 (2 SD; in cases without
a matching control). A map, built from a Basic Local Alignment Search Tool for nucleotides (BLASTN) alignment of probes to the Greene Pathogen Database, is used to connect probe sequences to the respective entries in the Greene Pathogen Database. Each of those sequences corresponds to an NCBI Taxonomy ID (TaxID). The individual TaxIDs are mapped to nodes in a taxonomic tree built based on ICTV virus taxonomy or NCBI taxonomic classification for other organisms. The program output is a ranked list of candidate TaxIDs. Candidate TaxIDs are ranked by combining the p-values for the positive targets for that TaxID using the QFAST method of Bailey and Gribskov (Bailey and Gribskov, 1998).
The specificity of the viral GreeneChip was assessed using extracts of cultured cells infected with adeno-, alpha-, arena-, corona-, entero-, filo-, flavi-, herpes-, orthomyxo-, paramyxo-, pox-, reo-, and rhabdoviruses (a total of 49 viruses). All were accurately identified by GreeneLAMP analysis. To assess sensitivity, viral RNA extracted from infected cell supernatants (adeno-, West Nile, St. Louis encephalitis, respiratory syncytial, entero-, SARS corona-, and influenza viruses) was quantitated by real-time PCR, serially diluted, and subjected to GreeneChip analyses. The threshold of detection for adenovirus was 10,000 RNA copies; the threshold of detection for the other reference viruses was 1,000 RNA copies per reverse transcription (RT)-reaction. The respiratory GreeneChip was tested for detection and typing with 31 influenza virus A and B reference strains of human and animal origin and, because reference strains represent only a limited fraction of the genetic variability, with numerous circulating human influenza virus strains isolated worldwide since 1999. In summary, a total of 69 viruses comprising 54 influenza virus A and B isolates of human, avian, and porcine origin and 15 non-influenza human respiratory viruses were tested, identified, and subtyped.
GreeneChips were also validated with clinical samples from patients with respiratory disease, hemorrhagic fever, tuberculosis, and urinary tract infections, and were demonstrated to identify human enterovirus A, human respiratory syncytial virus A, influenza A virus, Lake Victoria marburgvirus, SARS coronavirus, lactobacillus, mycobacteria, and gammaproteobacteria in various specimen types, including cerebrospinal fluid, nasopharyngeal swabs, sera/plasma, stools, and urine.
Recovery of Hybridized Sequences from GreeneChips
Arrays can facilitate cloning and sequence analysis as well as pathogen identification. Hybridized products typically range from 200 nt to >1,000 nt. Because GreeneChips display three or more probes representing different genomic regions for each virus, one can rapidly recover sequence not only for hybridized products but also for sequences between those products through use of PCR.
Unbiased High-Throughput Sequencing
The advent of high-throughput sequencing technology affords unique opportunities for pathogen discovery. Unlike consensus PCR or array methods where investigators are limited by known sequence information and must make choices regarding the range of pathogens to consider in a given experiment, high-throughput sequencing is unbiased. Several systems are in development. We have experience with the pyrosequencing system of 454 Life Sciences; however, the principles for sample preparation and data analysis are broadly applicable across platforms. Because all nucleic acid in a sample (whether host or pathogen) is amplified and sequenced, elimination of host nucleic acid can be critical to boosting pathogen signal toward the threshold for detection. Our approach is to apply a similar sample preparation and random PCR amplification protocol as developed for the GreeneChip including extensive DNase I treatment of the RNA template to remove host chromosomal DNA. This process obviates the potential for detecting DNA genomes of pathogens; however, our reasoning is that an active infection should be associated with transcription. After amplification and sequencing reads typically range in size from 40 to 400 base pairs. Raw sequence reads are trimmed to remove sequences derived from the amplification primer and filtered to eliminate highly repetitive sequences. After trimming and eliminating repeats, sequences are clustered into nonredundant sequence sets. Unique sequence reads are assembled into contiguous sequences, which are then compared to the nonredundant sequence databases using programs that examine homology at the nucleotide and amino acid levels (using all six potential reading frames with adjustments for sequence gaps). Specific PCR tests are then designed to examine association with disease, measuring burden, and obtaining additional sequence for phylogenetic characterization.
Vignettes in Pathogen Discovery
Borna Disease Virus and Neuropsychiatric Disease
In 1985, Rott and Koprowski reported that serum from patients with bipolar disorder reacted with cells infected with Borna disease virus (BDV), an unclassified infectious agent named after a town in Saxony (eastern Germany) that had large outbreaks of equine encephalitis in the late 1800s. Intrigued both by the concept that infection might be implicated in a neuropsychiatric disease, and that established methods for virus isolation had failed, we and others began to pursue characterization of this elusive neurotropic virus using molecular tools. BDV nucleic acids were isolated by subtractive hybridization in 1989, the first successful application of subtractive cloning in pathogen discovery (Lipkin et al., 1990). This effort relied on cDNA cloning with home brew kits as it preceded the advent of polymerase chain reaction and ready access to sequencing technologies.
The correlation between cloned materials and disease was achieved by demonstrating that (1) candidate cDNAs competed with RNA template from brains of infected rats for transcription and translation of a protein biomarker present in brain (hybrid arrest experiments), (2) the distribution of candidate nucleic acid correlated with pathology in brains of experimentally infected rats and naturally infected horses (in situ hybridization), and (3) no signal was obtained in Southern hybridization experiments, wherein normal brain was probed with candidate clones. Based on northern hybridization experiments the genome was variously reported as a 8.5 kb negative polarity RNA or an 11 kb positive polarity RNA. Over the next 5 years, the genome was cloned, and the virus was visualized and classified as the prototype of a new family of nonsegmented negative-strand (NNS) RNA virus with unusual properties: nuclear replication/transcription, posttranscriptional modification of selected mRNA species by splicing, low-level productivity, broad host range, neurotropism, and capacity for persistence (Briese et al., 1992, 1994; Cubitt et al., 1994; de la Torre, 1994; Schneemann et al., 1995; Schneider et al., 1994). It was widely held that the introduction of specific reagents such as recombinant proteins and nucleic acid probes would allow rapid assessment of the role of BDV in human disease. However, in a classic example of the pitfalls of PCR diagnostics, particularly using nesting methods, BDV was implicated in a wide variety of disorders that included unipolar depression, bipolar disorder, schizophrenia, chronic fatigue syndrome, AIDS encephalopathy, multiple sclerosis, motor neuron disease, and brain tumors (glioblastoma multi-forme) (Lipkin et al., 2001; Schwemmle et al., 1999). At the time of this writing, there is no conclusive evidence that BDV infects humans. BDV is nonetheless a fascinating virus, and its discovery has yielded intriguing models of viral pathogenesis, and provided guidance regarding methods for rigorously investigating the role of infection in chronic disease with sensitive molecular tools. It is worth noting that the two years of molecular gymnastics required to identify BDV could be collapsed into a few weeks with current art. However, even with the explosion in viral sequence data over the past decade, BDV is sufficiently different that it could not be identified by consensus PCR or microarrays based on sequences other than those representing Bornaviridae. To our knowledge it is unique in this respect.
West Nile Virus Encephalitis
In late August 1999, health officials reported an outbreak of encephalitis accompanied by profound weakness in Queens, New York. There was neither an apparent increase in the frequency of encephalitis in New York, nor an automatic reporting event that resulted in detection of the outbreak. Thus, the recognition of the syndrome was due to the clinical acumen of Deborah Asnis, an infectious diseases physician at Flushing Hospital Medical Center, and Marcelle Layton, Assis-
tant commissioner, Communicable Disease Program, New York City Department of Health, and their associates.
On September 3, serology for the presence of antibodies to North American arboviruses yielded results consistent with infection with St. Louis encephalitis virus (SLEV) (Asnis et al., 2000). St. Louis encephalitis (SLE) had not been reported previously in New York although mosquito vectors competent for transmission of SLE were present. Investigation of the outbreak epicenter revealed sites of active mosquito breeding and early victims of the outbreak had histories consistent with mosquito exposure. Thus, on September 3, a mosquito eradication program was adopted by the state and by the city of New York. Concurrently, wildlife observers independently noted increased mortality of avian species, including free-ranging crows and exotic birds housed in the Bronx Zoo. Tracy McNamara, a veterinary pathologist at the Wildlife Conservation Society, performed histologic analysis of birds and found meningoencephalitis, gross hemorrhage of the brain, splenomegaly, and myocarditis (Steele et al., 2000). Although 70 percent of emerging infectious diseases are zoonoses and the coincidence between the human and nonhuman outbreaks was striking, McNamara was unable to persuade her colleagues in human infectious disease surveillance to review materials. She forwarded tissue samples from diseased birds to the U.S. Department of Agriculture (USDA) National Veterinary Service Laboratory in Ames, Iowa, where virus was cultured and electron micrographs reported to be consistent with the presence of either a togavirus or a flavivirus. Thereafter the avian virus was forwarded from USDA to the Centers for Disease Control and Prevention (CDC) in Fort Collins, Colorado, for molecular analysis (Lanciotti et al., 1999).
On September 13–15, the CDC Encephalitis Project (composed of centers in California, New York, and Tennessee) held its annual meeting in Albany, New York. Data emerging from both California and New York over an 18-month survey period indicated that an etiological agent was never identified in 70 percent of cases of encephalitis despite culture, serology, and molecular analyses. In this context, our group was invited to discuss methods for identification of unknown pathogens and to consider application to project samples of a new method for amplifying viral nucleic acids, domain-specific differential display (DSDD). Sherif Zaki at CDC Atlanta had demonstrated the presence of flavivirus protein in brains of human victims of the New York City outbreak; however, efforts to amplify SLEV or other flaviviral sequences by conventional reverse transcription PCR (RT-PCR) had been unsuccessful. Employing several degenerate primer sets designed to target in DSDD highly conserved domains in the NS3, NS5, and 3’-untranslated regions of flaviviruses, we obtained positive results for four of the five New York patients in only a few hours. Sequence analysis confirmed the presence of a lineage one West Nile virus (Briese et al., 1999; Jia et al., 1999). Concurrently, our colleagues at CDC in Fort Collins reported West Nile-like
sequences in cell lines infected with homogenates from New York birds. In concert these findings confirmed that the outbreak in New York City was a zoonosis due to West Nile virus (WNV).
Subsequently, we established quantitative real-time PCR assays for sensitive high-throughput detection of virus in clinical materials and mosquito pools. Analysis of blood samples from infected humans revealed the presence of WNV sequences in late 1999 (Briese et al., 2000); however, the significance of human-human transmission was not appreciated until 2002, when transmission through organ transplants and blood transfusion led to implementation of blood screening by nucleic acid amplification tests (CDC, 2003, 2004). This outbreak illustrates the power of molecular methods for addressing the challenges of emerging infectious diseases. As an example of an emerging zoonosis it also underscores the significance of enhancing communication between the human and veterinary medicine communities.
Enteroviruses and Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a disorder characterized by progressive loss of motor neurons and muscle atrophy. An inherited form caused by mutations in the superoxide dismutase gene has been described; however, the majority of cases are idiopathic. In 2000 Berger and colleagues, using nested PCR, sequencing, and in situ hybridization methods, reported the striking finding that 15 of 17 French subjects with ALS, and only 1 of 29 subjects with other neurologic diseases had sequences of a novel echovirus in the spinal cord (Berger et al., 2000). Although other enteroviruses such as poliovirus and human enterovirus 71 have been unequivocally implicated in acute motor neuron disease, this publication was the first to provide compelling evidence that enteroviruses could cause slowly progressive chronic neurologic disease. Given the potential utility of antiviral treatment of this devastating neurodegenerative disorder we were encouraged by the National Institute of Neurological Disorders and Stroke (NINDS) to try to independently replicate the echovirus data. Our experience in the BDV field, where problems with PCR hygiene had led to spurious links to disease, was invaluable in directing experimental design. Whereas the Berger group had used RNA template extracted from sections cut on cryostats and analyzed by nested PCR in the same laboratory, we collected frozen tissues from two tissue banks, extracted RNA in a laboratory with no history of virus research, and performed blinded real-time PCR analyses in yet another laboratory. Real-time PCR is similar in sensitivity to nested PCR but is less sensitive to false-positive results because assays are performed in a closed system wherein signal is read as fluorescent signal. Analysis of spinal cord and motor cortex from 20 subjects with ALS and 14 controls revealed no echovirus sequences (Walker et al., 2001). These results were well received by colleagues but elicited less positive correspondence from some individuals who noted that
our publication was foreclosing a promising research lead and clinical trials with antiviral drugs.
Future Perspectives
Technologies will continue to evolve, allowing faster, more sensitive, and less expensive methods for pathogen surveillance and discovery. Although multiplex PCR is relatively mature, microarray technology is still in its infancy; near-term modifications already in development include microfluidic sample processing and direct measurement of conductance changes associated with hybridization. We have only touched the surface of proteomics and host response profiling. It is conceivable that biomarkers will be found that are specific for classes of infectious agents and/or provide insights that can guide clinical management. In chronic diseases the most substantive advances are likely to come not from technical improvements, but from investments in prospective serial sample collections and an appreciation that many diseases reflect intersections of genes and environment in a temporal context.
Acknowledgments
We thank our colleagues at the Scripps Research Institute, the University of California–Irvine, and Columbia University who have enabled our work in pathogen discovery over a period of more than 20 years. Current efforts are supported by National Institutes of Health awards AI062705, AI070411, HL083850-01, AI51292, AI056118, AI55466, AI57158 (Northeast Biodefense Center-Lipkin), NS047537, and EY017404.
THE POTENTIAL IMPORTANCE OF PRESYMPTOMATIC, HOST-BASED DIAGNOSIS IN BIODEFENSE AND STANDARD HEALTH CARE
Stephen Albert Johnston, Ph.D.11
Arizona State University
Abstract
Through programs such as BioShield, BioWatch, and BioSense we have created a first line of defense against traditional biothreats—our Bio-Maginot Line. However, the biotechnology revolution is driving the potential to create engineered pathogens that could circumvent these barriers. The increased risk inherent in this revolution is unstoppable, and efforts to control the risk through
regulation are probably unwise. Fortunately, for historical and technical reasons we may have a window of opportunity to get ahead of this threat curve. Key to this opportunity is the development of a diagnostic capability that could detect infections before symptoms appeared. This host-based detection system would be capable of detecting any type of threat agent—engineered or natural. Key aspects of this diagnostic system are that it would be capable of reading hundreds to thousands of blood, sputum, or urine components; rely on self-normalization by regular testing of individuals; and be widely distributed in homes. Evidence to date indicates that it may be possible to develop such a system. Obviously, its cost could not be justified by the unpredictable probability of a biothreat attack. Fortunately, the need for such a capability for biodefense is exactly in line with the need for the same capability to transform traditional medicine, most obviously for detecting natural outbreaks. The current health-care system is economically unsustainable. One solution to this crisis is to convert standard health practice from one that treats symptoms to one that detects disease very early—even presymptomatically. The convergence of the need for presymptomatic diagnosis capability, both for biodefense and standard medical practice, justifies an Apollo-like effort to create this technology.
Particularly since 9/11 there has been increasing concern about biological attacks. In the area of detection of attacks, we are relying on two basic strategies. One strategy, BioWatch, would have enough detectors distributed throughout the country to pick up airborne releases of pathogens. The hope is that a biothreat release would be detected before people develop symptoms. The problems with this strategy have been widely debated, but largely come down to the cost–benefit ratio of sustaining a system that would be effective. There is also the concern that engineered organisms would not be detected. The second major strategy is based on sufficient, organized surveillance of health-related data to detect early evidence of symptomatic people. The BioSense program is one example of this effort. Unlike BioWatch, this type of approach has a clear crossover advantage to standard medical practice. However, in the specific application to biothreat detection, it is dependent on detecting sick people.
Programs such as BioWatch (defined on page 4), BioShield,12 and BioSense13 have created a certain level of defense, largely against pathogens and scenarios based on analysis from the past century. This Bio-Maginot Line would provide a measure of defense against the obvious attack. As with the real Maginot Line, the concern is that the attack would go around the fortifications (Figure 3-10). Although no one can predict the risk of a future attack, let alone one that would

FIGURE 3-10 The Bio-Maginot Line.
SOURCE: Johnston (2006).
involve an engineered organism, there is little doubt that the threat will increase over time. As cartooned in Figure 3-11, this increase is largely due to the biotechnology revolution. The ability to both understand and manipulate life is increasing exponentially. Most measures of technological capability in biotechnology, like microchips, are obeying Moore’s Law.14 Whether it be growth in sequence deposits to GenBank (Figure 3-12), ability to sequence DNA, or facility at synthesizing genes, the revolution is amazing.
This revolution will drive remarkable change, but with it will come new opportunities for ill application. For example, the science of interfering RNAs started with some strange observations in plants in the 1990s. It progressed quickly to study in animal systems and now is standard technique for manipulating gene expression. The technology is offered as kits, and several biotechnology companies are pursuing its medical applications. A Nobel Prize was given for its discovery in 2006, a record time from discovery to prize. Yet it takes relatively little imagination to see how the incorporation of RNA interference (RNAi) constructs into viruses might augment their virulence. Almost every new technology in biotechnology and almost every new understanding of immunology and host–pathogen interactions could be configured to ill ends.
The same revolution that will drive dramatic new opportunities for contributions from biotechnology will increase the prospects for bad applications,
FIGURE 3-12 Growth of GenBank, 1982–2005.
SOURCE: NCBI (2007).
or even accidental events. The opportunities are dynamic. The sequence of any pathogen can be determined in one day. We are rapidly increasing our knowledge of host–pathogen interactions and the human immune response to infection. As stated above new technologies are being developed at a rapid pace. In addition, the ability to set up high-throughput screens is becoming more common. The combination of these trends will create the potential to create new pathogens (Figure 3-13).
The impact of these trends is clear. We are moving from a relatively simple threat space involving a list of potential pathogens and likely scenarios to one that has much higher dimensionality (Figure 3-14). The implications are that in the future, lists of relative importance of pathogens (e.g., Select Agent lists) and likely scenarios of attack are going to become, if they are not already, less useful.
That is the bad news. There is good news. For all the foreboding, a bioattack has not occurred since October 11, 2001. Why not if the risk is increasing? There are probably many explanations. One is that the Soviet biothreat program was
decommissioned before the real revolution in biotechnology had penetrated their operation. Second, Islam has placed biology as a low science. This was reflected in the number of graduate students in biology versus engineering, though this trend may be changing. Finally, making biothreat agents is still not easy. It would involve multiple steps and specific reagents. Anything of this nature can easily have one step go wrong. Of course if enough attempts are made, one will likely succeed—but the odds are now in our favor.
We may now be in a grace period relative to preparing for a biological attack—the valley of the shadow of death (Figure 3-11). If we continue to base our preparedness on protection against specific pathogens or scenarios, we may be in trouble. An alternative is to invest in developing platform technologies and strategies that offer broad-based defense. Examples include developing systems to generate and validate vaccines rapidly, and creating strategies to quickly produce new therapeutics to new pathogens from preexisting modules. However, I think the most important shift in emphasis would be to host-based diagnosis to allow presymptomatic detection of infections. The ability to detect infections before they are symptomatic has obvious value to strategies from quarantine to antibiotic treatment. It also addresses the problem of detecting the release of a new pathogen as the sensing is the host itself. Detection is not dependent on knowing what pathogens might be used.
The premise for this concept is presented in Figure 3-15. All of the factors that determine a person’s health status could be monitored in near-real time by
FIGURE 3-15 Biosignature pattern recognition in human diseases. Host-based presymptomatic detection of events.
SOURCE: Johnston (2006).
profiling all the components of blood. Blood chemistry, or that of sputum or urine, reflects changes in health, specifically early effects of infection. Continuous monitoring of blood components of healthy individuals would create their own biosignatures of health and disease (Figure 3-16), the ultimate in personalized diagnostics. Devices capable of generating such biosignatures are already in development. These units are aimed at a clinical setting largely for application to early detection or characterization of a specific illness, such as cancer. A DocInBox diagnostic device relevant to biodefense would have to be capable of detecting the early events of infection against the background of all other causes of change in health status.
This type of biosignature diagnosis has two distinctive features. Approximately 45 biomarkers are FDA approved. In contrast, biosignatures would involve measuring hundreds or possibly thousands of blood components. If the basis of disease could be anything, one has to sample broadly. Second, real-time and frequent monitoring of individuals would allow normalizing each person’s bio-
FIGURE 3-16 Personalized medicine based on biosignatures.
SOURCE: Johnston (2006).
signature to himself or herself. This is in contrast to traditional biomarkers where diagnosis is made based on values established in the population (Figure 3-17).
To meet these expectations the basic specifications for such a DocInBox are clear. To detect a pathogen release, the unit must work in near-real time. Most respiratory infections have a presymptomatic period of a few days at most (Figure 3-18). An assay system that takes a week to process has little value relative to infections, but would for other chronic ailments. If the goal is presymptomatic diagnosis obviously well people need to be monitored. Particularly for infectious disease, it follows that the diagnostic devices should be in the homes or places of work. Having these units in the physician’s office or emergency room will do little good in detecting a biothreat release. If the units are to be dispersed in homes, their operation must be rugged and inexpensive.
There are two issues relative to the possibility of attaining this goal. First, is presymptomatic diagnosis of infections biologically feasible? The evidence is scant in this regard. This topic will be the focus of a more extensive review, but there are some positive indications. In vitro studies have shown that the transcription pattern of dendritic cells changes on exposure to pathogens and that different pathogens elicit different patterns (Huang et al., 2001). This is important as dendritic cells are in the first line of exposure to pathogens. Microarray analysis of human blood cells has shown that individual patterns can be monitored over time (Whitney et al., 2003). Finally, it appears that microarrays of gene expres-
FIGURE 3-17 Biosignatures versus biomarkers.
SOURCE: Johnston (2006).
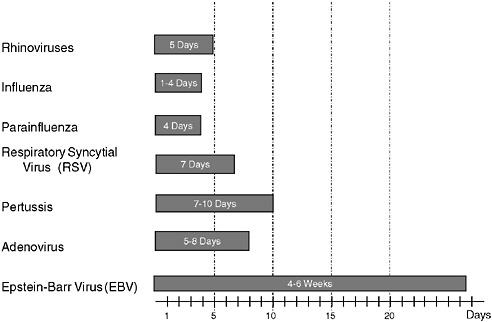
FIGURE 3-18 Upper respiratory disease incubation periods.
SOURCES: Adapted from Meneghetti (2006); Basu (1998); Smith et al. (2006).
sion can also detect presymptomatic responses in primates (Rubins et al., 2004). We have preliminary evidence (Johnston and Magee, unpublished) from a model of cowpox infection in mice that the infected mice can be distinguished from mock-infected mice three hours after infection, also by microarray analysis of blood cells. Clearly, more definitive studies of the limitations of presymptomatic diagnosis are needed.
The other issue is the technological challenge of creating the diagnostic system. This will be a formidable challenge. It will involve a coordinated, highly interdisciplinary effort that will include new instrumentation, modeling/algorithm development, data handling and transmission as well as judicious use of animal models and clinical testing (Figure 3-19). One challenge we have been addressing is how to develop the binding agents to measure thousands of blood components.
Though the technological challenges are great, such a diagnostic system is probably feasible. If developed it would be a major factor in preventing large-scale loss from a biothreat attack and may serve as a serious deterrent. However, the effort and cost to put such a system in place could not be justified based solely on the probability of a biothreat attack. Though its application to detection of natural outbreaks could be more easily supported, even this use would probably not drive an economic imperative to initiate this development program.
FIGURE 3-19 Program to create DocInBox diagnosis.
SOURCE: Johnston (2006).
Fortunately, a presymptomatic diagnostic system is also needed for another more easily justifiable application—the impending crisis in standard health care.
The cost of U.S. health care was approximately $2.2 trillion in 2006. This cost is estimated to be approximately $4 trillion by 2015 (Figure 3-20). Currently this cost accounts for approximately 19 percent of our Gross Domestic Product (GDP), rising to 25 percent or more by 2015 (Figure 3-21). By comparison, health-care costs have outpaced energy costs since the 1980s (Figure 3-22). Because most health costs are in the later years of life, with an aging population these trends are expected to continue (Figure 3-23). Clearly, we spend an enormous amount of our wealth on health care. If this investment contributes substantially to the productivity and creative output of the population it is money well spent. However, approximately 85 percent of this expenditure is on taking care of sick people and only about 15 percent on drugs and diagnostics. Our health-care system costs so much because it is largely postsymptomatic focused, and therefore centered on taking care of sick people. This system is clearly unsustainable economically. It will require either reducing care or revolutionizing medicine. If we opt for the latter, the key aspect will be converting medicine to a focus on presymptomatic diagnosis. A corollary of this transition will be improvement in quality of life. This will afford a “squaring” of the life curve (Figure 3-24) such that we not only live longer, but better.
We are fortunate that a key technology required for being prepared for the biothreats of the future is also the exact capability we have basically no choice but to develop for standard health care of the future, as well as for more prob-
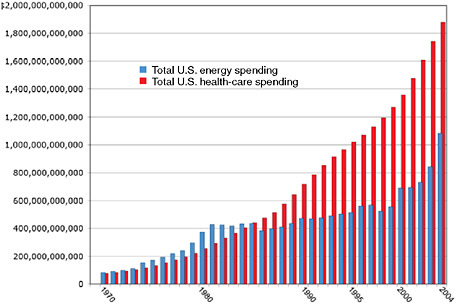
FIGURE 3-22 Comparison of U.S. spending on energy and health care, 1970–2004.
NOTE: The 2001 to 2004 numbers were projected based on oil prices. Total energy costs 2002–2004: Numbers are estimates based on extrapolation of energy price increase based on increases in petroleum prices, applied to Department of Defense known energy use figures. OPEC basket price averaged $50.71 per barrel in 2005, $36.05 per barrel in 2004, $28.10 per barrel in 2003, $24.36 per barrel in 2002, $23.12 per barrel in 2001, and $27.60 per barrel in 2000 (DoE, 2006).
SOURCES: EIA (2005); HHS (2007).

FIGURE 3-23 Average annual health-care expenditures by age, 2005.
SOURCE: DoL (2007).
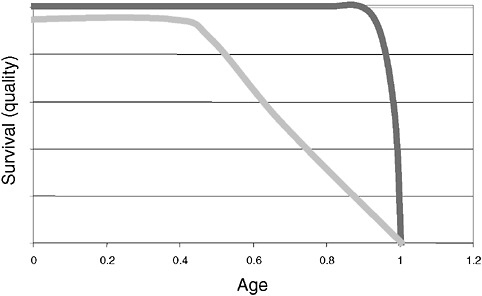
FIGURE 3-24 Human species needs to square life’s curve: Higher quality = less cost.
SOURCE: Johnston (2007).
able threats from natural infections. From the perspective of being prepared for engineered biothreats, we should take advantage of the valley of the shadow of death (Figure 3-11) to get ahead of the threat curve. Presymptomatic diagnosis should be a key element in this preparedness. From the perspective of standard of care, this same technology could be key to revolutionizing us as a species. Such potential merits an Apollo-like effort to complete.
REFERENCES
Albert, H., A. Heydenrych, R. Brookes, R. J. Mole, B. Harley, E. Subotsky, R. Henry, and V. Azevedo. 2002. Performance of a rapid phage-based test, FastplaqueTB™, to diagnose pulmonary t-berculosis from sputum specimens in South Africa. International Journal of Tuberculosis and Lung Disease 6(6):529-537.
Amexo, M., R. Tolhurst, G. Barnish, and I. Bates. 2004. Malaria misdiagnosis: Effects on the poor and vulnerable. Lancet 364(9448):1896-1898.
Archibald, I. K., M. O. den Dulk, K. J. Pallangyo, and L. B. Reller. 1998. Fatal Mycobacterium tuberculosis bloodstream infections in febrile hospitalized adults in Dar es Salaam, Tanzania. Clinical Infectious Diseases 26(2):290-296.
Asnis, D. S., R. Conetta, A. A. Teixeira, G. Waldman, and B. A. Sampson. 2000. The West Nile Virus outbreak of 1999 in New York: The Flushing Hospital experience. Clinical Infectious Diseases 30(3):413-418.
Bailey, T. L., and M. Gribskov. 1998. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 14(1):48-54.
Basu, S. J. 1998. The adenovirus family, http://virus.stanford.edu/adeno/adeno.html (accessed May 15, 2007).
Berger, M. M., N. Kopp, C. Vital, B. Redl, M. Aymard, and B. Lina. 2000. Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS. Neurology 54(1):20-25.
Briese, T., J. C. de la Torre, A. Lewis, H. Ludwig, and W. I. Lipkin. 1992. Borna disease virus, a negative-strand RNA virus, transcribes in the nucleus of infected cells. Proceedings of the National Academy of Sciences 89(23):11486-11489.
Briese, T., A. Schneemann, A. J. Lewis, Y. S. Park, S. Kim, H. Ludwig, and W. I. Lipkin. 1994. Genomic organization of Borna disease virus. Proceedings of the National Academy of Sciences 91(10):4362-4366.
Briese, T., X. Y. Jia, C. Huang, L. J. Grady, and W. I. Lipkin. 1999. Identification of a Kunjin/West Nile-like flavivirus in brains of patients with New York encephalitis. Lancet 354(9186):1261-1262.
Briese, T, W. G. Glass, and W. I. Lipkin. 2000. Detection of West Nile virus sequences in cerebrospinal fluid. Lancet 355(9215):1614-1615.
Briese, T., G. Palacios, M. Kokoris, O. Jabado, Z. Liu, N. Renwick, V. Kapoor, I. Casas, F. Pozo, R. Limberger, P. Perez-Brena, J. Ju, and W. I. Lipkin. 2005. Diagnostic system for rapid and sensitive differential detection of pathogens. Emerging Infectious Diseases 11(2):310-313.
CDC (Centers for Disease Control and Prevention). 2003. Detection of West Nile virus in blood donations—United States, 2003. Morbidity and Mortality Weekly Report 52(32):769-772.
CDC. 2004. Update: West Nile virus screening of blood donations and transfusion-associated transmission—United States, 2003. Morbidity and Mortality Weekly Report 53(13):281-284.
CDC. 2007. BioSense, http://www.cdc.gov/biosense/ (accessed July 31, 2007).
Challoner, P. B., K. T. Smith, J. D. Parker, D. L. MacLeod, S. N. Coulter, T. M. Rose, E. R. Schultz, J. L. Bennett, R. L. Garber, M. Chang, P. A. Schad, P. M. Stewart, R. C. Nowinski, J. P. Brown, and G. C. Burmer. 1995. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proceedings of the National Academy of Sciences 92(16):7440-7444.
Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266(5192):1865-1869.
Cheung, C. Y., L. L. Poon, A. S. Lau, W. Luk, Y. L. Lau, K. F. Shortridge, S. Gordon, Y. Guan, and J. S. Peiris. 2002. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet 360(9348):1831-1837.
Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244(4902):359-362.
Christensen, D. R., L. J. Hartman, B. M. Loveless, M. S. Frye, M. A. Shipley, D. L. Bridge, M. J. Richards, R. S. Kaplan, J. Garrison, C. D. Baldwin, D. A. Kulesh, and D. A. Norwood. 2006. Detection of biological threat agents by real-time PCR: Comparison of assay performance on the R.A.P.I.D., the LightCycler, and the Smart Cycler platforms. Clinical Chemistry 52(1):141-145.
Crossley, B. M., S. K. Hietala, L. M. Shih, L. Lee, E. W. Skowronski, and A. A. Ardans. 2005. High-throughput real-time PCR assay to detect the exotic Newcastle Disease Virus during the California 2002–2003 outbreak. Journal of Veterinary Diagnostic Investigation 17(2):124-132.
Cubitt, B., C. Oldstone, J. Valcarcel, and J. C. de la Torre. 1994. RNA splicing contributes to the generation of mature mRNAs of Borna disease virus, a non-segmented negative strand RNA virus. Virus Research 34(1):69-79.
Dalluge, J. J. 2000. Mass spectrometry for direct determination of proteins in cells: Applications in biotechnology and microbiology. Fresenius’ Journal of Analytical Chemistry 366(6-7):701-711.
de la Torre, J. C. 1994. Molecular biology of borna disease virus: Prototype of a new group of animal viruses. Journal of Virology 68(12):7669-7675.
Diehn, M., and D. A. Relman. 2001. Comparing functional genomic datasets: Lessons from DNA microarray analyses of host–pathogen interactions. Current Opinion in Microbiology 4(1):95-101.
DoE (Department of Energy). 2006. OPEC, http://www.eia.doe.gov/cabs/opec.html (accessed May 15, 2007).
DoL (Department of Labor). 2007. Consumer expenditures in 2005, http://www.bls.gov/cex/csxann05.pdf (accessed May 15, 2007).
Dye, C., C. J. Watt, D. M. Bleed, S. M. Hosseini, and M. C. Raviglione. 2005. Evolution of tuberculosis control and prospects for reducing tuberculosis incidence, prevalence, and deaths globally. Journal of the American Medical Association 293(22):2767-2775.
EIA (Energy Information Administration). 2005. State energy data 2001: Prices and expenditures report. Washington, DC: EIA.
Emanuel, P. A., R. Bell, J. L. Dang, R. McClanahan, J. C. David, R. J. Burgess, J. Thompson, L. Collins, and T. Hadfield. 2003a. Detection of Francisella tularensis within infected mouse tissues by using a hand-held PCR thermocycler. Journal of Clinical Microbiology 41(2):689-693.
Emanuel, P. A., C. Chue, L. Kerr, and D. Cullin. 2003b. Validating the performance of biological detection equipment: The role of the federal government. Biosecurity and Bioterrorism: Biodefense Strategy, Practice, and Science 1(2):131-137.
Fredricks, D. N., and D. A. Relman. 1996. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clinical Microbiology Reviews 9(1):18-33.
Grunstein, M., and D. S. Hogness. 1975. Colony hybridization: A method for the isolation of cloned DNAs that contain a specific gene. Proceedings of the National Academy of Sciences 72(10):3961-3965.
Gutierrez, E. B., D. M. Zanetta, P. H. Saldiva, and V. L. Capelozzi. 2002. Autopsy-proven determinants of death in HIV-infected patients treated for pulmonary tuberculosis in São Paulo, Brazil. Pathology, Research, and Practice 198(5):339-346.
Hartman, L. J., S. R. Coyne, and D. A. Norwood. 2005. Development of a novel internal positive control for Taqman-based assays. Molecular and Cellular Probes 19(1):51-59.
Hemmila, I., S. Dakubu, V. M. Mukkala, H. Siitari, and T. Lovgren. 1984. Europium as a label in time-resolved immunofluorometric assays. Analytical Biochemistry 137(2):335-343.
Henchal, E. A., J. D. Teska, G. V. Ludwig, D. R. Shoemaker, and J. W. Ezzell. 2001. Current laboratory methods for biological threat agent identification. Clinics in Laboratory Medicine 21(3):661-678.
HHS (Department of Health and Human Services). 2007. National health expenditure. Centers for Medicare and Medicaid Services, Office of the Actuary.
Huang, Q., D. Liu, P. Majewski, L. C. Schulte, J. M. Korn, R. A. Young, E. S. Lander, and N. Hacohen. 2001. The plasticity of dendritic cell responses to pathogens and their components. Science 294(5543):870-875, http://www.sciencemag.org/cgi/content/full/294/5543/870 (accessed May 15, 2007).
Hubank, M., and D. G. Schatz. 1994. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Research 22(25):5640-5648.
Jia, X. Y., T. Briese, I. Jordan, A. Rambaut, H. C. Chi, J. S. Mackenzie, R. A. Hall, J. Scherret, and W. I. Lipkin. 1999. Genetic analysis of West Nile New York 1999 encephalitis virus. Lancet 354(9194):1971-1972.
Johansen, I. S., B. Lundgren, A. Sosnovskaja, and V. O. Thomsen. 2003. Detection of multi-drug resistant Mycobacterium tuberculosis in clinical specimens in low- and high-incidence countries by line probe assay. Journal of Clinical Microbiology 41(9):4454-4456.
Johnston, S. A. 2006. Presymptomatic diagnosis. Paper presented at the Institute of Medicine Forum on Microbial Threats, Washington, DC, December 12-13.
Kijek, T. M., C. A. Rossi, D. Moss, R. W. Parker, and E. A. Henchal. 2000. Rapid and sensitive immunomagnetic-electrochemiluminescent detection of staphyloccocal enterotoxin B. Journal of Immunological Methods 236(1-2):9-17.
Koch, R. 1891. Ueber bakteriologische Forschung, Verhandlungen des X. Internationalen Medicinischen Congresses. Berlin, Germany: August Hirschwald. P. 35.
Ksiazek, T. G., D. Erdman, C. S. Goldsmith, S. R. Zaki, T. Peret, S. Emery, S. Tong, C. Urbani, J. A. Comer, W. Lim, P. E. Rollin, S. F. Dowell, A. E. Ling, C. D. Humphrey, W. J. Shieh, J. Guarner, C. D. Paddock, P. Rota, B. Fields, J. DeRisi, J. Y. Yang, N. Cox, J. M. Hughes, J. W. LeDuc, W. J. Bellini, and L. J. Anderson. 2003. A novel coronavirus associated with severe acute respiratory syndrome. New England Journal of Medicine 348(20):1953-1966.
Lalvani, A., A. A. Pathan, H. McShane, R. J. Wilkinson, M. Latif, C. P. Conlon, G. Pasvol, and A. V. Hill. 2001. Rapid detection of Mycobacterium tuberculosis infection by enumeration of antigen-specific T cells. American Journal of Respiratory and Critical Care Medicine 163(4):824-828.
Lamson, D., N. Renwick, V. Kapoor, Z. Liu, G. Palacios, J. Ju, A. Dean, K. St. George, T. Briese, and W. I. Lipkin. 2006. MassTag polymerase-chain-reaction detection of respiratory pathogens, including a new rhinovirus genotype, that caused influenza-like illness in New York state during 2004–2005. Journal of Infectious Diseases 194(10):1398-1402.
Lanciotti, R. S., J. T. Roehrig, V. Deubel, J. Smith, M. Parker, K. Steele, B. Crise, K. E. Volpe, M. B. Crabtree, J. H. Scherret, R. A. Hall, J. S. MacKenzie, C. B. Cropp, B. Panigrahy, E. Ostlund, B. Schmitt, M. Malkinson, C. Banet, J. Weissman, N. Komar, H. M. Savage, W. Stone, T. McNamara, and D. J. Gubler. 1999. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286(5448):2333-2337.
Liam, C. K., and B. G. Tang. 1997. Delay in the diagnosis and treatment of pulmonary tuberculosis in patients attending a university teaching hospital. International Journal of Tuberculosis and Lung Disease 1(4):326-332.
Liang, P., and A. B. Pardee. 1992. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257(5072):967-971.
Lipkin, W. I. 2006. Emerging tools for pathogen surveillance and discovery. Presentation at the Institute of Medicine Forum on Microbial Threats, Washington, DC, December 12-13.
Lipkin, W. I., G. H. Travis, K. M. Carbone, and M. C. Wilson. 1990. Isolation and characterization of Borna disease agent cDNA clones. Proceedings of the National Academy of Sciences 87(11):4184-4188.
Lipkin, W. I., M. Hornig, and T. Briese. 2001. Borna disease virus and neuropsychiatric disease—a reappraisal. Trends in Microbiology 9(7):295-298.
Lisitsyn, N., N. Lisitsyn, and M. Wigler. 1993. Cloning the differences between two complex genomes. Science 259(5097):946-951.
Madebo, T., and B. Lindtjørn. 1999 (June 18). Delay in treatment of pulmonary tuberculosis: An analysis of symptom duration among Ethiopian patients. Medscape General Medicine:E6.
Meneghetti, A. 2006. Upper respiratory infection, http://www.emedicine.com/med/topic2339.htm (accessed May 15, 2007).
Mori, T., M. Sakatani, F. Yamagishi, T. Takashima, Y. Kawabe, K. Nagao, E. Shigeto, N. Harada, S. Mitarai, M. Okada, K. Suzuki, Y. Inoue, K. Tsuyuguchi, Y. Sasaki, G. H. Mazurek, and I. Tsuyuguchi. 2004. Specific detection of tuberculosis infection: An interferon-gamma-based assay using new antigens. American Journal of Respiratory and Critical Care Medicine 170(1):59-64.
NCBI (National Center for Biotechnology Information). 2007. Growth of GenBank, http://www.ncbi. nlm.nih.gov/Genbank/genbankstats.html (accessed May 15, 2007).
Nichol, S. T., C. F. Spiropoulou, S. Morzunov, P. E. Rollin, T. G. Ksiazek, H. Feldmann, A. Sanchez, J. Childs, S. Zaki, and C. J. Peters. 1993. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 262(5135):914-917.
Palacios, G., T. Briese, V. Kapoor, O. Jabado, Z. Liu, M. Venter, J. Zhai, N. Renwick, A. Grolla, T. W. Geisbert, C. Drosten, J. Towner, J. Ju, J. Paweska, S. T. Nichol, R. Swanepoel, H. Feldmann, P. B. Jahrling, and W. I. Lipkin. 2006. MassTag polymerase chain reaction for differential diagnosis of viral hemorrhagic fever. Emerging Infectious Diseases 12(4):692-695.
Palacios, G., P. L. Quan, O. Jabado, S. Conlan, D. Hirschberg, Y. Liu, J. Zhai, N. Renwick, J. Hui, H. Hegyi, A. Grolla, J. Strong, J. Towner, T. W. Geisbert, P. B. Jahrling, C. Büchen-Osmond, H. Ellerbrok, M. Sanchez-Seco, Y. Lussier, P. Formenty, S. T. Nichol, H. Feldmann, T. Briese, and W. I. Lipkin. 2007. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerging Infectious Diseases 13(1):73-81.
Pronyk, P. M., K. Kahn, J. R. Hargreaves, S. M. Tollman, M. Collinson, H. P. Hausler, and J. D. Porter. 2004. Undiagnosed pulmonary tuberculosis deaths in rural South Africa. International Journal of Tuberculosis and Lung Disease 8(6):796-799.
Relman, D. A. 1999. The search for unrecognized pathogens. Science 284(5418):1308-1310.
Relman, D. A, J. S. Loutit, T. M. Schmidt, S. Falkow, and L. S. Tompkins. 1990. The agent of bacillary angiomatosis. An approach to the identification of uncultured pathogens. New England Journal of Medicine 323(23):1573-1580.
Relman, D. A., T. M. Schmidt, R. P. MacDermott, and S. Falkow. 1992. Identification of the uncultured bacillus of Whipple’s disease. New England Journal of Medicine 27(5):293-301.
Rivers, T. M. 1937. Viruses and Koch’s postulates. Journal of Bacteriology 33(1):1-12.
Rubins, K. H., L. E. Hensley, P. B. Jahrnling, A. R. Whitney, T. W. Geisberg, J. W. Huggins, A. Owen, J. W. LeDuc, P. O. Brown, and D. A. Relman. 2004. The host response to smallpox: Analysis of the gene expression program in peripheral blood cells in a nonhuman primate model. Proceedings of the National Academy of Sciences 101(42):15190-15195, http://www.pnas.org/cgi/reprint/0405759101v1.pdf (accessed May 15, 2007).
Saliba, G. S., F. C. Harmston, B. E. Diamond, C. L. Zymet, M. I. Goldenberg, and T. D. Chin. 1966. An outbreak of human tularemia associated with the American dog tick, Dermacentor variabilis. American Journal of Tropical Medicine and Hygiene 15(4):531-538.
Schena, M., D. Shalon, R. W. Davis, and P. O. Brown. 1995. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270(5235):467-470.
Schneemann, A., P. A. Schneider, R. A. Lamb, and W. I. Lipkin. 1995. The remarkable coding strategy of borna disease virus: A new member of the nonsegmented negative strand RNA viruses. Virology 210(1):1-8.
Schneider, P. A., A. Schneemann, and W. I. Lipkin. 1994. RNA splicing in Borna disease virus, a nonsegmented, negative-strand RNA virus. Journal of Virology 68(8):5007-5012.
Schwemmle, M., C. Jehle, S. Formella, and P. Staeheli. 1999. Sequence similarities between human bornavirus isolates and laboratory strains question human origin. Lancet 354(9194):1973-1974.
Small, P., and M. D. Perkins. 2000. Improved trials needed for TB diagnostics. Lancet 356:1048-1049.
Smith, D. R., C. A. Rossi, T. M. Kijek, E. A. Henchal, and G. V. Ludwig. 2001. Comparison of dissociation-enhanced lanthanide fluorescent immunoassays to enzyme-linked immunosorbent assays for detection of staphylococcal enterotoxin B, Yersinia pestis-specific F1 antigen, and Venezuelan equine encephalitis virus. Clinical and Diagnostic Laboratory Immunology 8(6):1070-1075.
Smith, N. M., J. S. Bresee, D. K. Shay, T. M. Uyeki, N. J. Cox, and R. A. Strikas. 2006. Prevention and control of influenza. Morbidity and Mortality Weekly Report 55(RR10):1-42, http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5510a1.htm (accessed May 15, 2007).
Soini, E., and H. Kojola. 1983. Time-resolved fluorometer for lanthanide chelates: A new generation of non-isotopic immunoassays. Clinical Chemistry 29(1):65-68.
Steele, K. E., M. J. Linn, R. J. Schoepp, N. Komar, T. W. Geisbert, R. M. Manduca, P. P. Calle, B. L. Raphael, T. L. Clippinger, T. Larsen, J. Smith, R. S. Lanciotti, N. A. Panella, and T. S. McNamara. 2000. Pathology of fatal West Nile virus infections in native and exotic birds during the 1999 outbreak in New York City, New York. Veterinary Pathology 37(3):208-224.
Taylor, L. A., C. M. Carthy, D. Yang, K. Saad, D. Wong, G. Schreiner, L. W. Stanton, and B. M. McManus. 2000. Host gene regulation during coxsackievirus B3 infection in mice: Assessment by microarrays. Circulation Research 87(4):328-334.
van Baar, B. L. 2000. Characterisation of bacteria by matrix-assisted laser desorption/ionisation and electrospray mass spectrometry. FEMS Microbiology Reviews 24(2):193-219.
VandeWoude, S., J. A. Richt, M. C. Zink, R. Rott, O. Narayan, and J. E. Clements. 1990. A borna virus cDNA encoding a protein recognized by antibodies in humans with behavioral diseases. Science 250(4985):1278-1281.
Walker, M. P., R. Schlaberg, A. P. Hays, R. Bowser, and W. I. Lipkin. 2001. Absence of echovirus sequences in brain and spinal cord of amyotrophic lateral sclerosis patients. Annals of Neurology 49(2):249-253.
White House. 2004. Project Bioshield, http://www.whitehouse.gov/infocus/bioshield/ (accessed July 31, 2007).
Whitney, A. R., M. Diehn, S. J. Popper, A. A. Alizadeh, J. C. Boldrick, D. A. Relman, and P. O. Brown. 2003. Individuality and variation in gene expression patterns in human blood. Proceedings of the National Academy of Sciences 100(4):1896-1901, http://www.pnas.org/cgi/reprint/100/4/1896.pdf (accessed May 15, 2007).
WHO (World Health Organization). 2004. Global tuberculosis control, http://www.who.int/tb/publications/global_report/2004/en/ (accessed May 29, 2007).
Wolcott, M. 2006 (December 12). Rapid infectious disease diagnostic assays. Paper presented at the Institute of Medicine Forum on Microbial Threats, Washington, DC, December 12-13.
Wright, M. A., and G. M. Church. 2002. An open-source oligomicroarray standard for human and mouse. Nature Biotechnology 20(11):1082-1083.
Zhu, H., J. P. Cong, G. Mamtora, T. Gingeras, and T. Shenk. 1998. Cellular gene expression altered by human cytomegalovirus: Global monitoring with oligonucleotide arrays. Proceedings of the National Academy of Sciences 95(24):14470-14475.























































