
5
Microbiological and Genetic Analyses of Material in the Letters
As discussed in Chapter 2, the bacterium Bacillus anthracis (B. anthracis) is the causative agent of the disease anthrax. Anthrax generally affects grazing mammals (e.g., cattle, sheep, horses). Anthrax in humans, particularly inhalational anthrax, is rare and occurs mainly in individuals with occupations involving the handling of hides, hair, or bone from infected animals. Inhalational anthrax can also be a sign of a biological attack with B. anthracis spores, especially in individuals without likely exposure to infected animals or their products. This was the case in 2001, when a number of cases of both cutaneous and inhalational anthrax were diagnosed among media and postal employees and others after the delivery of letters containing suspicious powders in several places.
In October 2001, clinical reporting of human anthrax cases spurred a broad epidemiological investigation to identify the source of the illnesses as well as any other infected parties (see Chapter 3 for a timeline and a more detailed discussion of this epidemiological investigation). Identification of B. anthracis as the cause of the 22 cases of illness and five deaths in 2001 was determined by clinical laboratory means (CDC, 2001a, b, c). Because these illnesses and deaths appeared to have resulted from a bioterrorist attack, immediate efforts were undertaken to identify a common source for the outbreak, including molecular genetic analysis of the causative agent.
5.2 IDENTIFICATION OF THE B. ANTHRACIS STRAIN
The first step in the search for a source of the anthrax-causing powders in the 2001 mailings was to identify which “strain” (or strains) of B. anthracis was used in the attack. Disparate cases of human anthrax in Connecticut, New York, Florida, and Washington, D.C., and contaminated environmental locations in the last three of these sites were all linked when a single B. anthracis
strain, the Ames strain, was identified in all of these cases and associated locations (Keim et al., 2008). As noted in Chapter 2, B. anthracis is one of the most genetically homogeneous microorganisms known (Keim et al., 2000). Nonetheless, even in the most homogeneous species there are usually some differences in genome sequences among populations. These sequence differences, although few in number, are sufficient to characterize subgroups, or “strains.” Strains are members of the same species, but their differences reflect the divergence of sublineages as they evolved over time (Keim et al., 2000). Among B. anthracis populations, a variety of strains had already been recognized even before sequencing technology enabled detailed characterizations of genome differences among strains.
Work performed by Paul Keim and others well before the 2001 anthrax attacks had resulted in the development of several molecular methods to detect genetic differences among Bacillus species as well as among isolates of B. anthracis. In the mid-1990s, work by Hendersen and colleagues (1995) and Anderson and colleagues (1996) led to the identification of a 12-nucleotide variable number tandem repeat (VNTR) sequence (called vrrA for “variable repeat region A”) that provided the first molecular marker that distinguished among B. anthracis strains. The basis of this marker was shown to be differences in the number of repeated sections of this genetic sequence, and five different variations were detected. Subsequently, VNTR analysis at multiple genetic loci (Multiple Locus VNTR Analysis or MLVA) enabled the characterization of 426 B. anthracis isolates with 89 distinct genotypes (Keim et al., 2000).
Another approach, amplified fragment length polymorphism (AFLP) analysis, has been particularly useful for examining differences between B. anthracis and close relatives, such as B. cereus and B. thuringiensis (Keim et al., 1999, 2008; Hoffmaster et al., 2002; Keim et al., 2008). The AFLP technique had been used to identify about 30 variable regions and provided an ability to profile portions of the genome of a large number of diverse B. anthracis strains. In addition, the pagA gene, which encodes the protective antigen (PA) protein (one of the three anthrax toxin proteins discussed in Chapter 2) also had been sequenced (Price et al., 1999). Because of the importance of pagA in the development of immunity to anthrax, this gene was of interest in determining whether a particular strain might have been genetically altered, or “engineered,” for increased effectiveness as a weapon (Hoffmaster et al., 2002).
These new molecular approaches, combined with the creation of a collection of strains from many of the world’s geographic regions, greatly enhanced scientific capabilities for identifying genetic variations among anthrax strains at that time. Using these methods, Keim and colleagues (1999, 2000) had established a picture of the evolutionary lineages of B. anthracis. These methods for rapid, reliable molecular subtyping were also critical in determining the identity of the clinical and environmental isolates in the 2001 anthrax attacks. Although a complete genome sequence provides the most effective genetic signature
for identifying an organism, at the time of the attack mailings no B. anthracis genomes had yet been completely sequenced and published (Keim et al., 2008).
Using MLVA at eight loci identified by Keim and his colleagues, scientists from the Centers for Disease Control and Prevention’s (CDC’s) Laboratory Response Network subtyped 135 B. anthracis isolates (samples) collected from the attack victims, letter powders, and environmental samples, and determined that all these isolates were likely to have been derived from a common source (Hoffmaster et al., 2002). In addition, CDC scientists sequenced the pagA genes from a subset of these isolates and concluded that none of the isolates appeared to have been engineered (Hoffmaster et al., 2002). (The use of additional tests performed under the aegis of the FBI to assess the possibility of genetic engineering is discussed below.) All attack-associated isolates were identified as MLVA genotype 62 and PA genotype I. Genotype 62 is the genotype of the Ames strain commonly used worldwide for laboratory research for vaccine development. The PA I genotype was also identical to the Ames strain PA genotype. These results led CDC to conclude that the B. anthracis strain used in the attacks was indistinguishable from the Ames strain (Hoffmaster et al., 2002).
As early as October 2001, samples from the spore-laden envelopes and clinical isolates (including one from Robert Stevens, the deceased Florida patient and index case) were also sent to Paul Keim’s laboratory at Northern Arizona University. The Keim laboratory had already established the B. anthracis MLVA sequence database that contained information on more than 1,000 samples from around the world. This database proved useful for identifying the B. anthracis strain in the forensic samples.
Beginning in January 2002, Keim also began conducting genetic testing at the request of the FBI on isolates of B. anthracis provided by the United States Army Medical Research Institute for Infectious Diseases (USAMRIID), which had received evidentiary samples from the FBI. The first 18 evidentiary samples (designated Batch E0001) received by the Keim laboratory were handled according to FBI chain of custody requirements and were initially not identified. An initial MLVA-8 analysis found that all but two of the samples (“Connecticut samples”) provided by USAMRIID were identical to and consistent with the Ames strain genotype (Keim, 2002a). An expanded analysis of 15 MLVA loci (Keim, 2002b) yielded similar results, with all but 3 forensic samples matching the Ames strain. One sample differed from the Ames strain in that it had lost the pXO2 plasmid (see Chapter 2), but was otherwise identical to the others. Keim noted that plasmids are commonly lost during culture and that this loss may have occurred prior to shipment of the sample to his laboratory, but was not to be interpreted as necessarily indicating that the original forensic sample was pXO2 negative. The two Connecticut samples proved to be distinct from the other evidence, but were identical to each other. Their genotypes matched 10 isolates from China in the Keim laboratory database that did not have a genotype or strain designation at that time, but most
closely resembled reference strain Genotype 61. At the time, Keim noted that these samples clearly did not match the Ames strain. These samples proved to be from a separate case (Malakoff, 2002) and not from the elderly Connecticut patient who died from inhalational anthrax. The FBI was initially interested in these samples until it was determined that they were not related to the anthrax letter investigation. The Keim laboratory continued to receive and strain type the B. anthracis samples submitted to the FBI Repository (see Chapter 6) over the next six years.
5.3 WAS THE B. ANTHRACIS IN THE LETTERS GENETICALLY ENGINEERED?
An important investigative issue was whether the B. anthracis strain used in the mailings had been genetically engineered. For example, the FBI was interested in whether antibiotic resistance genes or virulence factors had been introduced into the attack strain’s genome from other strains or species, and whether there were any other mutations in the sample, engineered or otherwise, that might help investigators determine its source.
To address some of these questions, Paul Jackson and colleagues at the Los Alamos National Laboratory (LANL) analyzed samples from the attack envelopes from October 2001 through mid-2002. The materials tested included samples of the progenitor Ames strain isolated from the dead cow in 1981 (Ames “Ancestor”); an isolate from the deceased Florida patient, Robert Stevens; isolates from the Brokaw, New York Post, and Daschle letters; and several stocks from USAMRIID, including a sample from flask RMR-1029. The samples were tested for possible indications of genetic engineering using DNA sequencing (see Box 5-1) and polymerase chain reaction (PCR) amplification (see Box 5-2) to look for (1) the presence of genes encoding resistance against the antibiotics ciprofloxacin, tetracycline, erythromycin, bleomycin, kanamycin, and chloramphenicol; (2) modifications of the protective antigen, edema factor, and lethal factor protein genes; and (3) inserted sequences derived either from cloning vectors (plasmids) known from the literature to have been used to engineer B. anthracis or from the insertion of the cereolysin genes of B. cereus, reported (Pomerantsev et al., 1997) to have conferred upon the strain an ability to evade protective immunity induced by some anthrax vaccines (FBI Documents, B1M4D2). The LANL scientists reported that “none of the isolates assayed showed any indication of genetic manipulation based on the presence of markers normally associated with genetic manipulation of B. anthracis.”
It should be noted that the LANL investigators did not look for other less obvious alterations that also might have indicated that the organisms in the evidentiary samples had been genetically engineered. Indeed, they acknowledged that a well-financed laboratory could exploit or develop other cloning vectors and other methods for genetic manipulation without leaving clear molecular
Until the 1970s, DNA was the most difficult molecule in biology to analyze because of its enormous length and its “monotonous” chemical structure. The DNA molecule is a string of chemical building blocks—the nucleotides or “bases” adenine (A), thymine (T), cytosine (C), and guanine (G). DNA sequencing is the process of determining the exact order of these building blocks in a piece of DNA. For example, “ATCGGCTAA” is part of a DNA “sequence.” Today, DNA sequencing has become indispensable for basic research and for numerous applications, such as disease diagnostics, biotechnology, systematics, and forensic biology. The earliest DNA sequencing methods were developed in the 1970s and were laborious and very slow. But the Human Genome Project, which began in 1990 and was largely completed in 2003, greatly stimulated the development of new sequencing technologies. These are largely automated and are orders of magnitude faster than earlier efforts. For example, in 2001 it could take about a year to sequence the genome of a bacterium like B. anthracis, whereas today such a process requires only a few hours.
Genome sequencing generally requires that the genome being studied first be broken into smaller pieces. This process is usually carried out by enzymes that “cut” the DNA into short fragments. In an alternate approach called “shotgun” sequencing, the DNA of interest is mechanically broken into random overlapping fragments. Each fragment is sequenced numerous times, and the genome is reassembled using computational methods to order the fragments based on the regions of overlap. If there is sufficient overlap, the genome sequence can be considered complete or “closed.” In bacteria, which usually have circular chromosomes, this means that the entire circle of the sequence is known. This technique works particularly well for small genomes, such as those of bacterial species that do not have extensive regions of repetitive nucleotide sequences. For the much larger genomes of animals and plants, the DNA of interest may also be broken into pieces and then cloned by inserting the fragments into bacteria, which make copies of the DNA when they divide and reproduce. The cloned fragments are then mapped against the genome being sequenced. The mapping reduces the likelihood that regions containing repetitive sequences (much more common in eukaryotic genomes) will be assembled incorrectly.
The enormous increases in speed and efficiency of DNA sequencing have led to a revolution in scientists’ ability to identify mutations quickly and precisely. Recently, the term “deep sequencing” was coined to describe this approach of simultaneous sequencing of massive numbers of short fragments derived from a mixture of genomes, such as might exist in an evolving population derived from a single cell that has, over time, accumulated genetic variants. These millions of short sequences are then ordered by computer programs, enabling the identification of single nucleotide polymorphisms (SNPs) and other genetic variants.
BOX 5-2
The Polymerase Chain Reaction Technique
The PCR technique provides a rapid means to amplify (increase the number of copies of) DNA segments of interest. Knowledge of the DNA sequence to be amplified is used to design two specific, but fairly short, synthetic DNA strands, or oligonucleotides, that are complementary to the sequence of DNA to be amplified. These oligonucleotides serve as “primers” for DNA synthesis and determine the segment of DNA amplified. In PCR, the original double-stranded DNA is first heated to separate the strands. The separated strands are then cooled in the presence of an excess of the two primers, which hybridize with the complementary sequences in the strands of DNA being studied. The mixture is then incubated with the nucleotides that are the raw materials for DNA synthesis and an enzyme called “DNA polymerase.” This enzyme synthesizes new DNA starting from the primers and copying the DNA strand to which the primer is bound. The result of the first cycle of PCR produces two new double-stranded DNA molecules that each contains one strand of the original DNA and one strand from the primer. This cycle of denaturation, hybridization, and synthesis is then repeated many times, increasing the number of copies of the DNA sequence of interest. Each cycle generates fresh templates for further amplification, and only the sequences bracketed by the primers are amplified, while regions that lack priming sites are not. After a number of these cycles, a substantial proportion of the reaction mixture corresponds to the amplified DNA.
SOURCE: Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and P. Walter. (2002). Molecular Biology of the Cell, 4th ed. New York: Garland Science.
signatures, or at least the signatures that the investigators sought. However, the subsequent completion of the genomic sequences of the Ames Ancestor and later of the letter isolates (see Section 5.5.5 below) strongly supported the findings of the LANL group (see also Box 5-1 on genome sequencing). As noted by Read and colleagues (2002), identification of genes that have been altered or inserted deliberatively in potential bioweapons agents is facilitated by complete genome sequencing. No further testing related to the issue of genetic engineering of the attack powders was performed after mid-2002 aside from the genome sequencing.
Prior to the 2001 letter attacks, the Institute for Genomic Research (TIGR) had begun sequencing the genome of the “Porton Down” isolate of B. anthracis (Read et al., 2002). The genomes of many bacteria have multiple parts, typically including a single large circular chromosome and one or more smaller plasmids. In particular, most B. anthracis strains carry two plasmids, called pXO1 and pXO2, that encode proteins required for virulence but that are not essential for the bacteria to grow under laboratory conditions. However, the Porton
isolate had been rendered avirulent by “curing” (eliminating) both plasmids from the Ames strain (Read et al., 2003), making it a less than optimal choice for a reference genome. Shortly after the letter attacks, the National Science Foundation provided critical funding that allowed TIGR to sequence to draft quality the isolate from the spinal fluid of the deceased Florida patient, Robert Stevens (Ames “Florida”). Here, “draft quality” refers to a genome sequence for which a small fraction of the nucleotide bases remains uncertain, there are remaining small gaps in the coverage of the complete genome, or both. This work was described in a paper by Read and colleagues in 2002, and included a comparison of the Florida and Porton isolates and an examination of a group of isolates of the Ames strain obtained from various laboratories prior to 2001 as well as an isolate from a Texas goat obtained in 1997. The latter was the only other isolate of the Ames strain known to have been collected in the field aside from the 1981 dead cow “Ancestor” isolate. In this initial set of comparisons, 11 sequence differences were found between the chromosomes of the Porton and Florida isolates (Read et al., 2002); however, thesel differences were also found in all the other Ames isolates tested. From these data, and the understanding that the Porton Down strain was derived from earlier isolates at USAMRIID (Read et al., 2002), it was inferred that the mutations in the Porton strain occurred after the 1982 transfer of the strain to Porton Down. No sequence differences appeared to distinguish the isolate of the Florida victim from many of the isolates of the Ames strain present in various laboratories before the attack.
In spring 2003 TIGR also initiated the sequencing of the Ames Ancestor and completed this work in October 2003 (Ravel, 2010). Unlike the Florida isolate, which was sequenced only to draft quality, the Ames Ancestor sequence was “closed,” that is, assembled into one contiguous sequence. Annotation and analysis of the Ames Ancestor sequence continued until mid-2004 and it was released to GenBank on June 1, 2004. (The paper announcing the Ames Ancestor sequence was not, however, published until 2009; see Ravel et al., 2009). The Ames Ancestor sequence served as the high-quality reference genome needed for the comparative genomics work that TIGR later performed on colony morphotypes identified from the attack letter materials (see below).
The apparent absence of chromosomal differences between the attack strain and Ames strains had important implications, both positive and negative, for the investigation. On the positive side, the findings strongly supported the inference that the attack strain had come—directly or indirectly—from a laboratory that possessed the Ames strain. Also on the positive side, the findings supported the conclusion that the attack strain had not been engineered to make it resistant to treatment or more virulent. On the negative side, the absence of distinctive sequences in the attack strain seemed to mean that it would not be possible to use genetic markers to trace the attack material to one particular source among the various institutions (and laboratories within the institutions) that possessed the Ames strain.
Nonetheless, an important clue as to the source of the strain in the letters came from microbiologists at USAMRIID who discovered that the attack material contained at low to moderate frequency several subtypes of B. anthracis that produced morphologically distinct colonies. These colony morphological variants (or “morphotypes”) retained their distinctive appearance when single cells from the colonies were regrown into new colonies. This persistence meant that the morphotypes were genetically distinct from the standard (wild-type) Ames strain in the samples; that is, they apparently contained a mutation or mutations that caused them to produce their distinctive-looking colonies. That several morphotypes could be distinguished indicated that different morphotypes contained different mutations. The morphotypes appeared to be spontaneous mutants that arose during the preparation of the batches of spores that were eventually used in the attacks.
As described and discussed later in this chapter and in Chapter 6, TIGR also sequenced the genomes of several of these morphotypes to identify the mutations that distinguished them from the wild-type Ames strain and from each another (FBI Documents, B1M5D1-2). These mutations played a central role in the investigation (USAMRIID, 2005; FBI Documents, B1M2D12; Worsham, 2009), helping the FBI to trace attack materials to a possible source.
The investigation summaries provided to the committee in December 2010 refer to the presence of a pE03 vector referred to as an ‘Israeli cloning vector’ among certain repository isolates (B3D1). On the January 11, 2011 meeting the FBI was asked to clarify what was known about the vector. The committee was told that this vector was a derivative of the commonly used cloning vector pBR322 that was found in some isolates, and that it had no forensic value to the investigation (FBI/USDOJ, 2011).
5.4 B. SUBTILIS CONTAMINATION OF THE NEW YORK SAMPLES
A finding that was initially of high forensic interest was the discovery, based on cultivation techniques, that the powder from the letters sent on September 18 to two New York City addresses (the New York Post and Tom Brokaw at NBC) contained a mixture of B. anthracis and, at a frequency of about 1 to 5 percent, a non-B. anthracis bacterium. The contaminating bacterial species was identified by the CDC as B. subtilis on the basis of 16S rRNA gene sequencing and later whole genome sequencing (CDC, 2001a; FBI Documents, B2M1D2). B. subtilis is a ubiquitous bacterial species that is readily isolated from environmental samples from around the world. The identification of the contaminant as B. subtilis was at first considered of potential importance because certain strains of B. subtilis are widely used in academic and industrial laboratories. Hence, if the contaminant had proved to be a particular laboratory strain, it might have provided a clue to the origin of the New York City powders.
Whole genome sequencing analysis carried out and reported in 2004 by TIGR of an isolate referred to as GB22 from the New York Post letter showed high (98 percent) similarity, but not identity, to the published sequence of the standard laboratory strain B. subtilis 168 (Kunst et al., 1997). In 2006, TIGR developed 95 PCR assays for 23 B. subtilis loci in the evidentiary sample that differed from the reference (B. subtilis 168) genome. The amplified DNA regions were compared using gel electrophoresis. (DNA sequencing of these amplified regions would have been a more definitive approach.) The B. subtilis isolates from the New York Post and Brokaw letters were identical to each other at all 23 loci, indicating that they were the same strain. (The whole genome sequence of the B. subtilis from the Brokaw letter was not determined, however, so their identity was not definitively demonstrated.)
Because the 95 PCR assays would have been cumbersome to perform on larger collections of samples, the FBI Laboratory next identified four genetic markers in the GB22 letter strain, three of which (designated ID 65, ID 91, and ID 107 by TIGR) were rare in a survey of 72 B. subtilis strains isolated from around the world. These strains were obtained from the NRRL (formerly the Northern Regional Research Laboratory) collection of the U.S. Department of Agriculture and the American Type Culture Collection, and they were meant to represent a geographically and genetically diverse collection (FBI Documents, B2M4D2). The three rare markers distinguished the GB22 strain from the other strains. The fourth marker (sboA) was common to all the B. subtilis strains examined. This combination of markers was designed first to determine whether any B. subtilis was present in additional samples based on the presence of the sboA marker and, second, to determine whether such samples contained a strain that might be similar to or the same as the GB22 strain from the New York letters.
The FBI Laboratory developed TaqMan (see Box 5-3) real-time PCR (RT-PCR) assays for the four markers, and these assays were provided to the Naval Medical Research Center (NMRC) and the National Bioforensic Analysis Center (NBFAC) at the Department of Homeland Security’s National Biodefense Analysis and Countermeasures Center, where the assays were validated by blind testing. NMRC and NBFAC used the assays to evaluate over 300 evidentiary samples. Only two B. subtilis strains from these samples were found that matched GB22 at all four loci. But when TIGR followed up by further characterizing these two samples using the complete set of 95 PCR assays, they proved to be genetically different from GB22 (FBI, 2009). NBFAC later (2007) also tested all Ames strain samples in the FBI Repository (see Chapter 6) for the presence of B. subtilis contamination (see NBFAC analytical result reports, November 2006-December 2007, FBI Documents,
The TaqMan® technique is highly sensitive (Easterday et al., 2005a) and reliable as a diagnostic tool (Easterday et al., 2005b), and allows the detection of genetic differences between samples even at the level of SNPs (Van Ert et al., 2007b). It uses an oligonucleotide probe that anneals to both the wild-type and mutant target DNA. The probe is labeled with both a fluorescent tag and a fluorescence quencher and binds tightly to the exact complementary sequence in the target DNA. PCR is initiated using primers that anneal nearby. One of these primers is designed such that it anneals only to a template containing the specific allele to be detected. Two primer sets are generally used, one that is specific for the wild-type allele and a second that is specific for the mutant allele. If the primer anneals and Taq polymerase synthesizes a new strand along the template, then the bound fluorescent TaqMan probe will be digested by the exonuclease activity of the advancing polymerase, thereby releasing the fluorophore and producing a signal that indicates the presence of the particular allele. If the primer does not anneal and Taq polymerase does not synthesize DNA, then the oligonucleotide probe will remain bound and intact, and the fluorescent tag will not emit detectable fluorescence due to the close proximity of the quencher.
B2M4D3-15). Although 322 out of 1057 repository samples tested positive for the sboA nucleic acid sequence, further testing showed that none of these 322 samples was positive for the rare ID 65 marker in GB22 (NBFAC, 2007; FBI Documents, B2M4D13). (If any samples had been positive for the presence of the ID 65 marker, analyses for the ID 91 and ID 107 markers would have also been performed.) In short, many repository samples were contaminated with B. subtilis, although apparently not by the same strain as in the New York Post letter. Ultimately, the FBI concluded that the testing for B. subtilis did not provide useful information leading to the source of the New York letter materials—GB22 is apparently an environmental strain of unknown origin that could not be traced to any particular source.
5.5 IDENTIFICATION AND CHARACTERIZATION OF COLONY MORPHOLOGICAL VARIANTS IN THE EVIDENTIARY MATERIAL
5.5.1 Why Was the FBI Interested in Colony Morphotypes?
Any microbial geneticist can attest to the fact that close scrutiny reveals unusual individual variants in a population of microbes. Such variants often carry genetic alterations that produce noticeable phenotypic changes in physiology, behavior, or morphology. When the variants are observed at the level of the physical appearance of colonies as they grow on agar plates, those variants are often referred to as “morphotypes.” As the selective pressures for rapid growth under laboratory conditions may differ from those experienced by
organisms in their natural environment, genetic variants (mutants) may arise that replicate faster and, over time, replace the “wild-type” strain during repeated cycles of laboratory culture. This process represents, in effect, rapid evolution leading to a population becoming better adapted to its particular laboratory conditions.
Analyses of the spore samples from the attack letters from as early as November 2001 (FBI Documents, B1M2D7) revealed the presence of colony morphotypes whose stability suggested that they resulted from the presence of genetically distinct subpopulations. The specific set of genetic alterations in a population might provide a useful profile for that population and if it can be demonstrated to be present in two different sample populations, might suggest that the two were derived from a common source. With sufficient knowledge about the identities of such genetic variations and the frequencies with which they arise in the population under specific culture conditions, the statistical significance of the similarities between the two populations might, in principle, be calculated. Under some circumstances, the chance of two independent populations containing the same genetic variant subpopulations might be so small that it could be concluded with high confidence that they were derived from a single source. With this goal, the FBI pursued detailed characterization of the phenotypic and genetic variation among the evidence samples.
5.5.2 Background Information on Morphotypes
Given the rapid generation times (many generations per day) and large populations (often billions of cells) typically observed in laboratory bacterial cultures, it is highly likely that genetic variants will arise in cultured populations. If a genetic variant in a population is able to initiate growth more quickly, grow more rapidly, or sustain growth longer as conditions become less favorable, it tends to increase its frequency in the population. This process can lead to cultures with multiple genetically distinct subpopulations. While this basic process of selection drives evolution in nature, it can present problems for genetic studies in the laboratory, as the characteristics of an organism may change over the time frame of a scientific study (Elena and Lenski, 2003).
Microbiologists usually seek to avoid this phenomenon using two primary methods. First, stock cultures are stored under nongrowth conditions in either a dried or frozen state, since most mutations do not arise in the absence of growth. Fresh samples of that nonvarying stock are then used to initiate each new set of analyses. Second, cultures retrieved from the stock are streaked on growth medium in such a way that individual colonies are obtained and each colony contains a population derived from a single cell (i.e., a clonal population). Some genetic variants that make up a minor proportion of the stock population may be recognizable based on their atypical colony morphology, and those variants can be avoided in the generation of cultures for further uses.
It is well known in laboratories that study bacterial spore formation (sporulation) that low-frequency sporulating (oligosporogenous) variants appear in a bacterial population at a readily observable frequency (Velicer et al., 1998; Michel et al., 1968). Furthermore, laboratory selective pressures can result in these variants becoming dominant members of the population and supplanting the sporulation-proficient parent strain. Two phenomena may increase the abundance of asporogenous (nonsporulating) or oligosporogenous (sporulating with reduced frequency) variants above that of other types of variants. When a population encounters a decline in nutrients, sporulation-proficient cells cease growth and enter the dormant spore state. Sporulation-deficient variants may continue growth to some degree at the expense of their neighbors, increasing their own relative abundance in the population. If this aged population of cells and spores is eventually transferred to fresh medium, the nonsporulated cells may also resume growth more rapidly than the spores, again increasing their abundance in the resulting population. These may be selective factors leading to the frequent observation of oligosporogenous variants in laboratory cultures.
Studies of B. anthracis phenotypic variants, many of which were shown to be oligosporogenous, were previously carried out by Patricia Worsham and Michele Sowers at USAMRIID (Worsham and Sowers, 1999) as part of an effort to identify useful strains for vaccine research. Working with multiple strains (e.g., Ames, Sterne, Vollum, and others) of B. anthracis, Worsham and Sowers identified numerous phenotypic variants (Worsham and Sowers, 1999). Their initial screen for phenotypic variation was growth on medium containing a dye called Congo red. The dye accumulated by wild-type B. anthracis colonies results in the colonies assuming a salmon-pink color. Poorly sporulating colonies do not accumulate the dye, resulting in a white appearance that can be easily identified by eye. Further study of the white colony morphotypes revealed that they were sporulation deficient. While there is no clear physiological explanation for the coincidence of the absence of Congo red binding and sporulation deficiencies, no such explanation is required for the practical use of this morphotype screening method. One of these phenotypic variants was carefully characterized as carrying a mutation in spo0A, a primary regulatory gene for entry into sporulation (Hoch, 2000). Indeed, mutants of spo0A and other genes governing entry into sporulation were historically recognized in experiments in which cultures of cells sporulating in rich medium were maintained for prolonged periods of time (Michel et al., 1968).
Reinforcing the view that laboratory conditions frequently enrich for mutants defective in sporulation, a recent publication by Sastalla and colleagues (2010) reports that repeated passage of B. anthracis on rich medium results in the accumulation of sporulation mutants, which were recognized by their distinctive colony phenotype. Many of the mutants contained mutations in the sporulation regulatory gene spo0A, as had been observed by Patricia
Worsham (Worsham and Sowers, 1999). These included point mutations as well as deletions and insertions. Sastalla and colleagues further showed that the spo0A mutants exhibited a shorter lag time than did the wild type when inoculated on rich medium. Thus, one factor contributing to the enrichment of sporulation-deficient mutants upon repeated passage is that at least some sporulation mutants resume rapid (exponential phase) growth more quickly than does the wild type.
5.5.3 Detection and Characterization of Morphotypes in the Anthrax Letter Samples
Part of the analysis of the anthrax letter evidence samples involved the preparation of relatively low-density liquid suspensions of spores recovered from the letters and the spreading of these suspensions on the surface of a solid growth medium. This procedure allowed each viable spore to germinate, grow, and divide in isolation, producing a clonal population of millions of cells—all derived from a single cell—in the form of a colony. This is a standard microbiological practice used for the enumeration of viable spores in a sample. The method also allows the visual detection of variants in the population that may carry genetically heritable traits responsible for altered phenotypic properties, although the sensitivity and discriminatory capacity of this method are low. Relatively early in the investigation (fall 2001), Terry Abshire, a microbiologist at USAMRIID, observed the presence of multiple phenotypic variants (morphotypes) among the colonies generated from the anthrax letter spore samples. The analysis of these variants eventually became a major feature in the FBI’s effort to identify the most likely source of the specific culture used to produce the letter samples. To the advantage of the FBI, Patricia Worsham was one of the USAMRIID scientists that assisted with the analysis of the letter spore samples. Due to her extensive experience with B. anthracis and her specific experience studying B. anthracis phenotypic variants, Worsham and her colleagues were readily able to apply methods for the study and characterization of phenotypic variants in the letter samples.
In early analysis of the Leahy letter sample, spores were plated on sheep blood agar and incubated for three days. On these plates, Abshire observed the presence of morphotypes with more defined colony edges and a yellow tint relative to the majority of the colonies. Based on this observation, Worsham was asked to develop a protocol to characterize morphotypes in the multiple evidentiary samples. The goal of this analysis was the identification of phenotypic and eventually genetic signatures of the letter samples that might contribute to determining the source of the letter material.
Spore samples from the Daschle, Leahy, and New York Post letters were suspended in water at a low density, such that they could be spread to give rise to between 10 and 50 colonies per plate, allowing the observation of individual
colony phenotypes. Thousands of colonies from each sample were visually inspected. The initial plating was on sheep blood agar plates to allow the most efficient colony recovery. The plates were incubated at either 26°C or 37°C with room air, or at 37°C with an atmosphere containing 20 percent CO2. These conditions were employed to allow the detection of variants that might be apparent only under particular growth conditions. While the characteristics of colony morphotypes are most obvious following incubation for 48 hours or more after nutrient exhaustion and sporulation, these starvation conditions were avoided in order to diminish the likelihood of the selection of new variants that were not present in the initial evidentiary samples. Plates were inspected after 18 to 22 hours of incubation, which was sufficient to allow the development of sizable colonies. Most classes of colony morphotypes were identified predominantly by the presence of a yellow tint (although the committee notes that this tint is not apparent in Figures 5-1, 5-2, and 5-3, while one class exhibited a smaller, more opaque colony appearance.
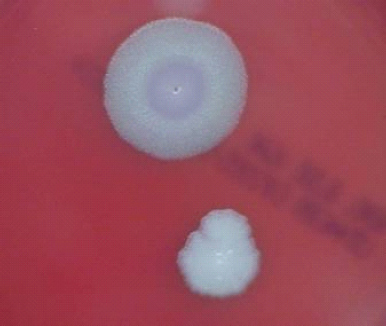
FIGURE 5-1 B. anthracis Colony Morphotype “A.” Photograph of colonies formed by growth of B. anthracis cells on blood agar. The colony on the top displays the morphology designated “Type A.” The colony on the bottom displays the typical wild-type morphology.
SOURCE: USAMRIID. This image is a work of the United States Army Medical Research Institute for Infectious Diseases, taken or made during the course of an employee’s official duties. As a work of the U.S. federal government, the image is in the public domain.
FIGURE 5-2 B. anthracis Colony Morphotype “B.” Photograph of colonies formed by growth of B. anthracis cells on blood agar. The colony on the top displays the morphology designated “Type B.” The colony on the bottom displays the typical wild-type morphology.
SOURCE: USAMRIID. This image is a work of the United States Army Medical Research Institute for Infectious Diseases, taken or made during the course of an employee’s official duties. As a work of the U.S. federal government, the image is in the public domain.
Upon the identification of each candidate variant colony, the colony was scraped from the culture plate. To produce an archival stock of the variant, 50 percent of the recovered material was stored at −70°C. This is a standard microbiological practice for the maintenance of strains under conditions that prevent the further accumulation of genetic variation. The remainder of the recovered material was again streaked onto a fresh sheep blood agar plate and incubated at 37°C for 18 to 22 hours in order to produce numerous colonies for each candidate morphotype. This new growth was inspected and compared to a nonvariant isolate to determine whether the phenotype of each candidate morphotype was stable over multiple generations, indicating the presence of an underlying genetic change (mutation), and whether the recovered material produced a uniform colony appearance, indicating a pure (clonal) culture of the morphotype. Colonies were collected from this second plate to produce an additional stock at −70°C that could be used for multiple analyses without sacrifice of the archival stock.
FIGURE 5-3 B. anthracis Colony Morphotype “E.” Photograph of colonies formed by growth of B. anthracis cells on blood agar. The colony on the top displays the morphology designated “Type E.” The colony on the bottom displays the typical wild-type morphology.
SOURCE: USAMRIID. This image is a work of the United States Army Medical Research Institute for Infectious Diseases, taken or made during the course of an employee’s official duties. As a work of the U.S. federal government, the image is in the public domain.
Each morphotypic variant recovered was subjected to numerous phenotypic tests that had been used in earlier studies (Worsham and Sowers, 1999) to differentiate variants from the wild-type strain. In each case, candidate variants were observed side by side, on the same plate of medium, with colonies of the wild-type strain as well as isolates of several previously identified morphotype classes. Variants were examined for Congo red binding, degree of colony spreading, degree of colony pigmentation, and “bull’s-eye” appearance of the colony (which might be due to variation between concentric rings in colony thickness, pigmentation, or other physiological traits). Sporulation was scored by microscopic observation of material recovered from colonies on two different media:
TABLE 5-1 Phenotypic Characteristics of the Morphotypes
| Colony size | Colony spread | Pigmentation | Hemolysis | |
| Morphotype A | Large | Yes | Bull's eye | ++ |
| Morphotype B | Large | Yes | Altered | ++ |
| Morphotype C/D | Large | Yes | Bull's eye | + |
| Morphotype E | Small | No | Opaque | − |
sheep blood agar, which is a rich medium not conducive to efficient sporulation, and Leighton-Doi medium, a minimal medium that is normally used for spore production. Hemolysis, the rupturing of red blood cells and alteration of the red heme color, can be detected after growth on sheep blood agar for 48 hours at 37°C followed by refrigeration for two weeks, during which the hemolytic reaction becomes more visibly pronounced.
Following extensive testing, four predominant morphotype classes were identified in the Leahy letter material, although there were also other less abundant types. The four were named A, B, C/D (two initial similar classes, C and D, were eventually merged), and E (also called opaque) (see Table 5-1). All of the morphotypes were oligosporogenous, producing significantly fewer spores than the wild type on all media tested. Morphotypes A, B, and C/D exhibited large zones of hemolysis that were not produced by the wild-type strain. The defining characteristic of morphotype A was a large spreading colony with a bull’s-eye center. Morphotype B was similar to A in colony size but did not produce the bull’s eye and produced slightly different pigmentation on some media. Morphotype C/D was also similar in appearance to A, but produced a smaller zone of hemolysis and was somewhat more proficient at spore production. Morphotype E was unique in producing colonies that were smaller than those of the wild type, had more regular edges, and were more opaque. Morphotype E also exhibited temperature-sensitive sporulation, producing significantly more spores at 26°C than at 37°C, yet still fewer than the wild type at either temperature. Screening of samples from the Daschle and New York Post letters revealed morphotype classes that were phenotypically similar to those from the Leahy letter. (Due to a scarcity of material from the Brokaw letter, no screening of samples for colony morphotypes was performed on that material [Hassell, 2010].)
5.5.4 Selection Criteria for Genetic Variations Used in Screening
Classes of morphotypes present in the letter samples had been characterized by the scientists at USAMRIID as early as January 2002, and the FBI began to consider methods for screening the FBI Repository (FBIR) (see Chapter 6) of B. anthracis isolates to identify a source culture. Phenotypic screening of the
FBIR samples was deemed an impractical method for several reasons. First, phenotypic screens of the types described above are relatively slow, labor intensive, and highly dependent on the trained eyes of the investigator to identify variant colonies. Second, similar phenotypic variations can be associated with different genetic alterations located in either the same or widely separated genetic loci. The presence of similar colony morphotypes in two samples would not provide direct genetic evidence to link the two sample populations. Third, phenotypic screens are insensitive and do not reliably detect rare variants. Identification of the specific mutations associated with each phenotypic variation was required for the development of definitive assays to detect the presence of shared mutations in multiple strains within the repository. Such DNA-based assays are rapid, sensitive, compatible with high-throughput methods, and definitive to the level of nucleotide sequence.
Scientists selected representative morphotype isolates as well as control wild-type isolates from each of three letter samples for detailed genetic analysis. Several criteria were used for this selection. First, the scientists needed to be able to distinguish the variants from the wild-type colonies on plates. Second, these particular morphotypes must have been present at a high enough frequency for the scientists to identify them repeatedly. The third essential criterion was the apparent presence of the morphotype in each of the three letter samples (Leahy, Daschle, New York Post) that were subjected to this analysis. The final selection of morphotypes focused on four variants: A, B, C/D, and E. There were other morphotypes found in the letter materials, but they were not used for further forensic testing. Worsham and colleagues at USAMRIID quantified the percentages of variants by randomly picking about 370 isolated colonies from plates made using dilutions from the Leahy letter. These colonies were 79 percent wild-type morphology, 6.7 percent C/D morphotype, 1.1 percent B morphotype, 1.3 percent A morphotype, and 4.9 percent E morphotype (other morphotypes accounted for the remaining fraction) (FBI Documents, B1M2D12). It is important to note that two identical-looking morphotypes need not, and often did not, have the same genotype. Indeed, as discussed in the next section, two independent isolates exhibiting similar colony morphotypes might have mutations in different genes or even different mutations in the same gene. Also, some colonies identified as morphotypic variants may not have had any mutation, as the distinction between genetic and nongenetic variation is not always clear. Thus, it was crucial to identify differences in nucleotide sequence as an unambiguous signature of different mutant subpopulations.
5.5.5 Whole Genome Sequencing of Morphotype Isolates
To determine whether the genetic alterations associated with each colony morphotype might be suitable for use as forensic markers, the genome sequences of multiple morphotype isolates were determined (Table 5-2). Genomic DNA
TABLE 5-2 B. anthracis Isolates Analyzed by the Institute for Genomic Research (TIGR)
| TIGR ID | FBI ID | Origin | Morphotype | Sequencing status |
| GBA | Ames Porton | Porton Down | Wild type | 12X - closed |
| GB6 | Ames Ancestor | Texas/USAMRIID | Wild type | 12X - closed |
| GB8 | LL10/E3 | Leahy letter | A | 8X - closed |
| GB9 | LL9/E2 | Leahy letter | B | 8X |
| GB10 | LL1/E1 | Leahy letter | Wild type | 8X |
| GB11 | PL10/E6 | New York Post letter | A | 8X |
| GB12 | PL9/E7 | New York Post letter | B | 8X - closed |
| GB13 | PL1/E9 | New York Post letter | Wild type | 8X - closed |
| GB15 | DL10/E4 | Daschle letter | A | NS |
| GB16 | DL9/E5 | Daschle letter | B | NS |
| GB17 | DL1/E8 | Daschle letter | Wild type | NS |
| GB18 | LL6/E10 | Leahy letter | C | 12X |
| GB19 | LL7/E11 | Leahy letter | D | NS |
| GB23 | LL18 | Leahy letter | E | 12X |
| GB24 | LL19 | Leahy letter | E | NS |
NS = not sequenced
SOURCE: FBI Documents, B1M5D1-2.
extracted from the colony morphotypes identified by USAMRIID was provided by Paul Keim to TIGR, where it was prepared for genome sequence analysis. Plasmid libraries were produced from the genomic DNAs and shotgun sequencing was carried out to produce approximately 8X (in some cases 12X) average coverage of the genome. However, most of these genome sequences were not closed (i.e., assembled into one contiguous sequence). In some cases (e.g., the samples from the Daschle letter), no sequencing was performed. In these cases the TIGR scientists used PCR to test whether the same genetic differences in the sequenced samples were also present in the unsequenced ones (FBI Documents, B1M5D1). Again, as previously noted, efforts were not undertaken to identify these or other morphotypes in the Brokaw letter.
Control wild-type isolates from each letter were found to possess no genetic differences from the Ames Ancestor strain (FBI Documents, B1M5D1-2). The genome sequences of each of the chosen morphotype isolates did, however, exhibit differences from the wild-type isolates in particular genetic regions of the B. anthracis chromosome or, in the case of the E morphotype, in the pXO1 plasmid. Some samples of morphotype A contained a single nucleotide
polymorphism (SNP) while others carried a duplication. SNPs were also found for morphotypes B and C, while D contained a chromosomal deletion and E had a deletion in the pXO1 plasmid. Following the identification of these sequence differences, unsequenced morphotype isolates were further tested for the presence of the same sequence differences using PCR amplification and sequencing of the amplified DNA. The results are summarized in Table 5-3. These data revealed that multiple isolates of some of the morphotypes (e.g., B) were associated with a single genetic change while others (e.g., isolates of the A morphotype) exhibited several different sequence variations in the same chromosomal region.
The genotypes of the A morphotype isolates from three letters (Leahy, New York Post, and Daschle) were of two general kinds. The first was a SNP
TABLE 5-3 Further Genetic Characterization of the Morphotype Isolates
| Morphotype class | Affected locus | Type of mutation | Genotype examined in greater detail | Letter source | Assay method developed |
| A | One copy of 16S rRNA gene | Insertions in different sites overlapping a 16S rRNA gene | A1, 2024 bp | Leahya Daschleb New York Postb |
qPCR |
| A2, 2608 bp | New York Posta | Not used | |||
| A3, 823 bp | Leahy b Daschlea New York Post b |
qPCR | |||
| B | spo0F promoter | SNP in intergenic region | B | Leahya New York Posta |
Not used |
| C | Sensor his kinase | SNP producing stop codon | C | Leahya | Not used |
| D | Sensor his kinase | 258 bp deletion | D | Leahya | Taqman + PCR |
| E | Putative response regulator (plasmid) | 9 or 21 bp deletion, or SNP | E, 9 bp deletion | Leahya Daschlec New York Postc |
Taqman + PCR |
NOTE: bp = base pair; qPCR = quantitative polymerase chain reaction; SNP = single nucleotide polymorphism.
a Source of original morphotype isolate with this genotype.
b Sample subsequently found to contain DNA carrying this genotype.
c Source of subsequent morphotype isolate demonstrated to have this genotype.
SOURCE: FBI Documents, B1M5D1-2.
in a gene encoding a K+ uptake protein. This SNP was found only in the New York Post letter material, but not in the Porton Down, Ames Ancestor, or Leahy genomes, and no further forensic use was made of this mutation. The second kind of A morphotype genotype involved large insertions in one of the eleven copies of the B. anthracis 16S rRNA gene. A 2024 bp (base pair) insertion (later termed the A1 genotype) and an 823 bp insertion (A3 genotype) were both found in the materials from the Leahy, Daschle, and New York Post letters, and both were subsequently chosen for the development of two separate assays to be used to screen the FBI Repository of Ames strain samples (see Chapter 6). A 2608 bp insertion (A2 genotype) was found only in the New York Post letter and an assay was developed for the detection of this genotype by Commonwealth Biotechnologies, Inc., (CBI) in Richmond, Virginia. In validation testing, the performance of the A2 assay was surpassed by that of the A1 and A3 assays. Consequently, the FBI decided not to use the A2 assay for evidence analyses. Eventually, many more variants ranging from 822 to 2608 bp were found among other samples provided to TIGR (FBI Documents, B1M5D4).
B morphotype isolates from the Leahy and New York Post were fully sequenced (Table 5-2), while the morphotype isolate from the Daschle letter was studied using PCR. The sequencing revealed that the Leahy and New York Post letters contained an identical SNP in the noncoding region between the spo0F gene and an adjacent gene, and this SNP was not present in the Ames Ancestor or Ames Porton reference samples. PCR amplification and resequencing of the amplicon confirmed the presence of the same SNP in the same region of the Daschle morphotype B isolate. The SNP was the replacement of a thymine (T) with a cytosine (C). The two open-reading frames are divergently transcribed, so this intergenic region likely contains the promoters for these genes, one of which (spo0F) plays a key role in governing entry into sporulation. This mutation may explain why the morphotype is sporulation deficient. The gene expression patterns of these mutants were not examined because these kinds of experiments were considered outside the scope of the investigation. The consistent presence of the B SNP provided a second potential genotypic signature for comparing FBIR samples to the letter materials, but it was not used by the FBI to screen repository samples because multiple efforts by contractors to develop assays for this SNP failed (see below), nor were any other SNPs representing single base pair mutations used to that end (see Chapter 6).
The morphotype C and D isolates shared a very similar phenotype. For the C morphotype, only material from the GB18 sample from the Leahy letter was sequenced (Table 5-2). Again, the TIGR team’s reasoning was that other samples did not need to be sequenced because any polymorphisms found in GB18 could be tested by PCR later. The GB18 C morphotype analysis found one SNP corresponding to a nucleotide change from G to A, creating a stop codon in a histidine sensor kinase (“his kinase”) gene, a member of a family of proteins that regulate gene expression. When the D morphotype sample
GB19 from the Leahy letter was subsequently examined for the presence of the C SNP in the same gene sequence, a 258 bp deletion was found instead. This genetic deletion resulted, in turn, in a deletion of 86 amino acids in the same his kinase protein. The SNP found in GB18 is located in the chromosomal region that is deleted in GB19. Thus the similarity in the C and D phenotypes could be explained since both the C SNP and the D deletion likely produced a nonfunctioning protein from the same his kinase gene, which plays a role in sporulation. The New York Post and Daschle letters were not tested for the C and D morphotypes.
The morphotype E isolate from the Leahy letter (GB23) was sequenced and had no chromosomal mutations. Instead, this strain had a 21 bp deletion in the pXO1 plasmid. The deletion was located in a gene encoding a putative gene expression regulator. A PCR/resequencing assay was used to test for the presence of the same deletion in the GB24 Leahy letter sample. This sample contained a 9 bp deletion in the same genetic locus. The PCR/resequencing analysis was run on a series of other blind samples relevant to the investigation according to TIGR’s 2005 report (FBI Documents, B1M5D4). Some of these additional strains carried the 9 or 21 bp deletions, but others contained a SNP representing a single point mutation (CGT → TGT) in the same locus that appeared to create defects in the corresponding proteins severe enough to interfere with normal function, although it was beyond the scope of TIGR’s work to test this.
Table 5-4 provides a summary of the distribution among the case letters of the morph genotypes that were ultimately used for screening of the FBIR.
TIGR completed this stage of the study by analyzing a set of samples (listed in Table 7, FBI Documents, B1M5D4) that included additional evidentiary samples as well as noncase strains from the Keim laboratory’s scientific collections. TIGR tested these samples using the assays developed for the various genotypes. These additional analyses showed that only the colony morphotype samples themselves contained the specific polymorphisms identified by the TIGR team, which was interpreted to mean that these genotypes represented
TABLE 5-4 Distribution Among the Anthrax Letters of the Genotypes Selected for FBIR Screening
| Letter | Genotype A1 | Genotype A3 | Genotype D | Genotype E |
| Leahy | + | + | + | + |
| Daschle | + | + | NT | + |
| New York Post | + | + | NT | + |
| Brokaw | NT | NT | NT | NT |
NT = not tested
SOURCE: Hassell (2010).
unique markers suitable for further forensic use. The A1, A3, D, and E genotypes were employed for the development of validated assays that were used to screen samples of the Ames strain collected by the FBI from all domestic and foreign sources that it was able to identify. The details of this screening are provided in Chapter 6.
5.5.6 Development and Application of Assays for the Genotypes
CBI was the contractor selected by the FBI to develop the genetic assays for the A1 and A3 morphotypes and test the FBI Repository samples (Chapter 6). CBI began work for the FBI in mid-2002 (FBI Documents, B2M5D2). The A morphotypes that were analyzed most thoroughly were found to contain large insertions overlapping a 16S rRNA gene (FBI Documents, B1M5D1). Although the insertion was of a different size in each of the three A morphotypes, all three had insertions in the same locus. The A1 genotype had a 2024 bp insertion and was originally identified in an isolate from the Leahy letter. During assay development by CBI this allele was also detected in DNA derived from bulk Daschle and New York Post letter spores (Hassell, 2010). The A3 genotype contained an 823 bp insertion that was originally identified in an isolate from the Daschle letter, but during assay development by CBI this allele was also detected in DNA derived from bulk Leahy and New York Post letter spores (Hassell, 2010). The A2 genotype had a 2608 bp insertion and was originally identified in an isolate from the New York Post letter. No acceptable assay for A2 was developed, and it is not known whether the A2 allele was present in spore material from the other letters. The assays developed by CBI used the TaqMan analytical technique (Didenko, 2001), which is an adaptation of PCR (see Boxes 5-2 and 5-3).
CBI completed its validation studies in February 2004. Limits of detection were estimated at 0.005 percent for the A1 genotype assay and 0.001 percent for the A3 assay in a background of 20 nanograms (ng) of Ames Ancestor DNA. Appropriate reaction controls were also developed. Sequencing of amplicons served as a final confirmatory step. The A1 and A3 genotype assays were chosen by the FBI for use in analyzing the FBIR samples (see Chapter 6) as problems (e.g., high number of false positives) with the validation of the assay for genotype A2 ultimately caused this assay to be abandoned. It should be noted that assay development and validation took almost two years.
Three companies—CBI, IIT Research Institute (IITRI), and Midwest Research Institute (MRI)—were hired to develop assays for the B and D geno-
types. None of the contractors was successful in developing a reliable B assay. In addition, the FBI expressed a preference not to use assays directed at SNPs (FBI, 2009). Consequently, the FBIR samples were not screened for the B genotype. The IITRI and MRI assays for the D genotype were both accepted by the FBI and used to screen the FBIR samples. The D genotype had a 258 bp deletion and was originally identified in an isolate from the Leahy letter. It was never determined whether this allele was present in the spore populations in the other evidentiary letter samples. Assay development and validation in each case took almost one year.
IITRI assay (FBI Documents, B2M7): A technical proposal for assay development for the D genotype was submitted by IITRI in July 2004 and validation of this assay was completed in April 2005. Using TaqMan/PCR (Boxes 5-2 and 5-3) this assay detected the genotype D when it was present at levels as low as 0.01 percent relative to the Ames Ancestor background. The repository screenings using the IITRI assay for the D genotype began in May 2005 and were completed in early 2007.
MRI assay (FBI Documents, B2M8): MRI submitted its technical proposal for development of the D deletion assay to the FBI in July 2004 and assay development was completed in June 2005. It used RT-PCR and had a detection limit of 0.01 percent in the Ames Ancestor background. Three approaches were used to increase sensitivity: closely spaced primers, short annealing time (15 seconds), and confirmation of reaction amplicon with melt curve and fragment size analysis. This second D assay also went forward for screening the repository and other samples. Screening began in December 2005 and was completed in October 2007.
The E morphotype was identified from the Leahy, Daschle, and New York Post letters (FBI Documents, B1M5D4). Although there were apparently several different mutations that produced the “opaque” phenotype, all appeared to involve the same gene on the pXO1 plasmid. One isolate from the Leahy letter material carried a 21 bp deletion in this gene, and another isolate also from the Leahy letter had a 9 bp deletion in the same region of that gene. Both of these deletions were also found in E isolates from the Daschle and New York Post letters using PCR and sequencing of this locus (Hassell, 2010). Other E isolates contained a single bp substitution in the same gene.
The 9 bp deletion was chosen for the development of an assay for use in screening the FBIR samples. The assay was developed in 2005 using TaqMan technology, validated, and applied to the repository by TIGR under contract from the FBI in 2007 (FBI Documents, B2M9). Preparations of purified mutant
and wild-type DNA were mixed in amounts covering a 1,000,000-fold range of ratios of mutant-to-wild-type DNA, and the assay was shown to reliably detect the mutant genome when present at 0.01 percent of the total DNA in the sample. In addition, the assay did not produce false positives for the presence of the mutant allele using varying amounts of wild-type DNA. This assay was approved by the FBI for testing repository samples, which was performed from June to August of 2007.
Finding 5.1: The dominant organism found in the letters was correctly and efficiently identified as the Ames strain of B. anthracis. The science performed on behalf of the FBI for the purpose of Bacillus species and B. anthracis strain identification was appropriate, properly executed, and reflected the contemporary state of the art.
Finding 5.2: The initial assessment of whether the B. anthracis Ames strain in the letters had undergone deliberate genetic engineering or modification was timely and appropriate, though necessarily incomplete. The genome sequences of the letter isolates that became available later in the investigation strongly supported the FBI’s conclusion that the attack materials had not been genetically engineered.
In the first few months following the attacks, isolates from the letters and other sources were examined only for the presence of some obvious and expected signs of genetic engineering. This examination was not exhaustive and would have missed less obvious or less well recognized signatures of deliberate genetic alteration. Had the case not involved the Ames strain of B. anthracis, with its relatively brief history and high degree of characterization, this limitation could have been a serious one.
Finding 5.3: A distinct Bacillus species, B. subtilis, was a minor constituent of the New York Post and Brokaw (New York) letters, and the strain found in these two letters was probably the same. B. subtilis was not present in the Daschle and Leahy letters. The FBI investigated this constituent of the New York letters and concluded, and the committee concurs, that the B. subtilis contaminant did not provide useful forensic information. While this contaminant did not provide useful forensic information in this case, the committee recognizes that such biological contaminants could prove to be of forensic value in future cases and should be investigated to their fullest.
Although the B. subtilis isolates in the two New York letters appeared to be closely related, the B. subtilis isolate in the Brokaw letter was not fully
sequenced, and therefore the presumed identity of the two isolates was not definitively demonstrated. Although B. subtilis was found in several hundred repository samples, the strains in these samples did not match the isolates found in the New York letters. Biological contaminants could prove to be of great forensic value and should be investigated to their fullest in future cases.
Finding 5.4: Multiple colony morphotypes of B. anthracis Ames were present in the material in each of the three letters that were examined (New York Post, Leahy, and Daschle), and each of the phenotypic morphotypes was found to represent one or more distinct genotypes.
This important discovery greatly facilitated the subsequent laboratory investigation and is a testament to the critical importance of attentive, thoughtful scientists who were prepared to explore unexpected results in the setting of a forensic investigation.
Finding 5.5: Specific molecular assays were developed for some of the B. anthracis Ames genotypes (those designated A1, A3, D, and E) found in the letters. These assays provided a useful approach for assessing possible relationships among the populations of B. anthracis spores in the letters and in samples that were subsequently collected for the FBI Repository (see also Chapter 6). However, more could have been done to determine the performance characteristics of these assays. In addition, the assays did not measure the relative abundance of the variant morphotype mutations, which might have been valuable and could be important in future investigations.
In the course of developing the assays that were used to screen the FBIR samples for the four genotypes, procedures were employed to examine both the specificity and sensitivity of the assays, including analyses of defined mixtures of genotypes at known proportions. However, the repository included both homogeneous and heterogeneous samples, in unknown proportions, and the extent of genetic diversity in the heterogeneous samples was also unknown. More could have been done to determine the performance characteristics, including reproducibility of results, under the actual conditions associated with the repository samples.
In addition, these assays were not used to quantify the relative abundance of the genotypes in the FBIR samples and the evidentiary materials. Measurement of relative abundance of genotypes might have helped clarify the relationship between the evidentiary spore samples and whether they were derived from the same or different cultivation events.
Finding 5.6: The development and validation of the variant morphotype mutation assays took a long time and slowed the investigation. The committee
recognizes that the genomic science used to analyze the forensic markers identified in the colony morphotypes was a large-scale endeavor and required the application of emerging science and technology. Although the committee lauds and supports the effort dedicated to the development of well-validated assays and procedures, looking toward the future, these processes need to be more efficient.
Future cases may not allow for a time frame as lengthy as that of the anthrax letters investigation. Assay development and validation took almost two years in some cases, for reasons that are not clear to the committee. The committee recognizes that the experience gained in the case, as well as faster and greatly improved technologies, could help speed future investigations. These factors alone, however, may not be sufficient for all contingencies. In particular, future cases could involve less well documented or less easily grown species and strains, and precious investigation time could be lost because of the need to establish basic information about the relevant organism’s biology and population genetics. In addition, original attack material (in this case, the powder in the anthrax letters) may not be available in all bioterrorism scenarios. Also, in some future cases of bioterrorism the attacks may continue until the perpetrators are identified and apprehended.




























