
11
Stress and Metabolic Disease
Karen K. Ryan
Persistent exposure to psychosocial stress is linked to an increased risk of metabolic disease, including obesity, cardiovascular disease (CVD), and Type-2 diabetes mellitus (T2DM). Delineating the behavioral, physiological, and molecular mechanisms by which stress adversely affects these endpoints may uncover important opportunities for therapeutic intervention. This paper explores the role of stress-induced activation of the sympatho-adrenomedullary system and the hypothalamic-pituitary-adrenocortical axis as key mechanisms linking chronic stress exposure to metabolic health and disease.
PHYSIOLOGICAL RESPONSES TO STRESS
Stress can be defined as a real or perceived threat to homeostasis or well-being, and it can be either psychological or physical in nature. Threatening stimuli are integrated in the central nervous system (CNS) and recruit downstream effectors to modify behavioral and physiological responses—thereby reducing the expected detriment to the individual. Specifically, the sympatho-adrenomedullary (SAM) system and hypothalamic-pituitary-adrenocortical (HPA) axis are activated, allowing for both immediate (SAM) and sustained (HPA) physiological and behavioral responses, as discussed below. (See Ulrich-Lai and Herman, 2009, and Ulrich-Lai and Ryan, 2014, for a more thorough review.)
The sympathetic nervous system is activated almost immediately upon presentation of an acute stress stimulus and provides for rapid, but tran-
sient, physiological responses via direct catecholaminergic innervation of peripheral organs. Additionally, sympathetic drive to the adrenal medulla elicits the release of catecholamines into systemic circulation (Ulrich-Lai and Engeland, 2002). Together, this increased SAM activity facilitates the mobilization of stored energy, increases heart rate and blood pressure, and redirects blood flow away from reproductive and digestive processes. For example, increased sympathetic drive to adipose tissue results in the release of free fatty acids (Bartness and Song, 2007). Likewise, increased sympathetic drive to the liver promotes glycogenolysis and increases hepatic glucose production (Yamaguchi, 1992). Together, these acute responses facilitate the immediate ability of an individual to flee a predator, fight an aggressive conspecific, mount an immune response to infection, or otherwise deal with the threat at hand.
HPA axis activation provides a relatively slower, but amplified and sustained response to stress. Corticotrophin releasing hormone (CRH) is synthesized in hypothalamic neurons, released at the median eminence, and travels via the portal circulation to the anterior pituitary, where it stimulates the release of adrenocorticotropic hormone (ACTH) into systemic circulation. ACTH stimulates the synthesis and secretion of glucocorticoids from the adrenal cortex. Glucocorticoids act at their receptors to exert a number of effects, including the sustained mobilization of fuels. For example, glucocorticoid-signaling increases lipolysis and the release of fatty acids from adipose tissue (Xu et al., 2009; Campbell et al., 2011) and increases hepatic gluconeogenesis (Baxter, 1976). Glucocorticoid-signaling both decreases pancreatic insulin production and increases insulin resistance, promoting the greater availability of plasma glucose (Lambillotte et al., 1997; Andrews and Walker, 1999). Importantly, glucocorticoids also act at their receptors in the hypothalamus and pituitary to exert negative feedback on CRH and ACTH release, allowing for the eventual resolution of this stress response. Thus, together with rapid SAM action, the somewhat slower and longer-lasting action of glucocorticoid-signaling promotes survival and restores homeostasis across multiple organ systems (Ulrich-Lai and Herman, 2009).
Under conditions of chronic stress exposure, frequent use of CNS stress-regulatory circuits adjusts gene expression and synaptic plasticity, resulting in persistent alterations to stress system function (Ulrich-Lai and Herman, 2009). The result is increased basal SAM and HPA tone. In addition, excitability is enhanced, such that stress-evoked SAM and HPA responses are exaggerated (Akana et al., 1992; Grippo et al., 2002; Ulrich-Lai et al., 2007). Such stress facilitation ensures the continued ability to exhibit appropriate responses to new stressors, despite basal increases in glucocorticoids and changes in immune and endocrine function (Akana et al., 1992).
CHRONIC STRESS AND METABOLIC DISEASE
Chronic stress is linked to increased morbidity and mortality, thought to occur at least in part because, although SAM and HPA responses may be adaptive in the short term, frequent and/or persistent exposure to these physiological processes leads to “wear and tear” on organs and tissues and can predispose to disease (McEwan and Stellar, 1993). Frequent and persistent override of metabolic homeostasis, required to mobilize fuels in response to stressful stimuli, may make metabolic regulatory systems particularly vulnerable to adverse consequences (Ulrich-Lai and Ryan, 2014). Consistent with this possibility, chronic stress alters feeding behavior and promotes obesity, and is thought to be an important risk factor for a number of metabolic diseases, including cardiovascular disease (CVD), Type-2 diabetes mellitus (T2DM), and polycystic ovarian syndrome (PCOS), discussed below. Additionally, stress exposure at earlier life history stages likewise can alter behavioral and physiological responses to acute and chronic stress in adulthood, thereby conditioning the later risk of disease.
Chronic Stress, Feeding Behavior, and Obesity
Glucocorticoids regulate food intake and adiposity via both central and peripheral mechanisms. Glucocorticoid receptor (GR) signaling in the CNS increases caloric intake and stimulates body weight gain (Green et al., 1992; Tempel et al., 1992; Tataranni et al., 1996; Zakrzewska et al., 1999). These effects are thought to be mediated, at least in part, by synaptic changes and altered excitability within canonical hypothalamic circuits important for the regulation of energy balance (Gyengesi et al., 2010). In addition, stress stimulates the selection of calorically dense, highly palatable foods over more nutritious less-rewarding options—further contributing to its adverse metabolic effects (McCann et al., 1990; Oliver and Wardle, 1999; Dallman et al., 2003; Laugero et al., 2011; Groesz et al., 2012; Kim et al., 2013; Tryon et al., 2013b). Stress increases the propensity of both rodents and humans to work for palatable food (Willner et al., 1998; Lemmens et al., 2011), and increases neuronal activation in brain motivation and reward circuitry following exposure to palatable foods (Rudenga et al., 2013; Tryon et al., 2013a). Importantly, this stress-induced consumption of palatable foods can occur even among the subset of people who decrease their total caloric intake in response to stress (Oliver and Wardle, 1999).
In addition to promoting the increased consumption of palatable foods, persistent exposure to circulating glucocorticoids during chronic stress facilitates the redistribution of body fat towards metabolically unhealthy visceral depots (Lönn et al., 1994; Dallman et al., 2003). For instance, chronic job strain was significantly associated with both increased general
obesity and increased central obesity in the Whitehall II study (Brunner et al., 2007). Likewise, chronic stress increased the intra-abdominal fat of cynomolgus monkeys, relative to unstressed controls (Jayo et al., 1993). Because visceral depots are thought to exhibit a greater lipolytic response to adrenergic stimulation (Ostman et al., 1979; Arner, 1995; Hoffstedt et al., 1997), fat redistribution associated with chronic stress may mirror stress facilitation in neural circuits, by ensuring the system can rapidly mobilize fatty acids in response to new acute stressors (Ulrich-Lai and Ryan, 2014). Unfortunately, however, such fat redistribution may also have long-term pathophysiological consequences, since increased visceral adiposity is also thought to contribute to various metabolic diseases, by promoting ectopic fat storage in liver and vascular tissues.
Chronic Stress and Cardiovascular Disease
A large literature now supports that psychosocial factors contribute significantly to the pathogenesis of CVD in human populations (Rozanski et al., 1999; Steptoe and Kivimäki, 2012). This relationship may represent an important target for the development of therapeutic interventions, since CVD remains the leading cause of death worldwide. Work-related stress, marital discord, and caregiver status are chronic life stressors that have been extensively studied with respect to cardiovascular outcomes. As just one example, a recent meta-analysis reports a 1.4 relative ratio of coronary heart disease for men working in jobs characterized by high demand together with low decision latitude, low reward, and/or minimal control (Kivimäki et al., 2006). Likewise, a high demand–low reward work environment is associated with significant 4-year progression of atherosclerosis (Lynch et al., 1997), and marital stress among working women is associated with a 2.9-fold increase in the risk of both recurrent cardiovascular events (Orth-Gomér et al., 2000) and accelerated progression of coronary heart disease (Wang et al., 2007). Moreover, spousal caregivers of patients with Alzheimer disease exhibited higher plasma triglycerides and increased blood pressure compared to non-caregiver controls (von Känel et al., 2011).
Chronic stress is also associated with indices of CVD in nonhuman primates and laboratory rodents. For example, dominant male cynomolgus monkeys housed in an unstable (i.e., stressful) social environment exhibit accelerated progression of coronary atherosclerosis and impaired endothelial function (Kaplan et al., 1983, 1987). Among female monkeys, social subordinates and singly housed individuals had 5 and 12 times the coronary artery atherosclerosis of social dominants, respectively (Shively et al., 1989). In laboratory rodents, both the chronic social-defeat stress and chronic mild stress paradigms induce a pro-atherosclerotic plasma lipid pro-
file (Neves et al., 2009; Chuang et al., 2010) and increase vascular indices of atherosclerosis (Neves et al., 2009).
Such adverse cardiovascular consequences may arise in part from stress-associated changes in food choice and increased visceral adiposity, discussed above. However, other physiological mechanisms likely play a role, since vascular pathology has been observed among stressed rodents and primates that are exclusively maintained on a low-fat, “healthy” laboratory chow (Kaplan et al., 1983; Neves et al., 2009). Classical physiologic adaptations to chronic stress, including increased basal and stress-evoked SAM and HPA tone, are also thought to contribute. For example, stress-induced vascular pathology in male cynomolgus monkeys can be abolished by concurrent treatment with the β-adrenergic antagonist propranolol, supporting that elevated SAM tone is a contributing mechanism (Kaplan et al., 1987). Likewise, chronic exposure to elevated glucocorticoids is associated with hypertension and atherosclerosis in Cushing disease patients (Orth, 1995), and this is recapitulated in a mouse model in which elevated corticosterone is provided chronically in the drinking water (Okutsu et al., 2014). Exaggerated vascular pathology in response to excess glucocorticoids is associated with dyslipidemia, although a thorough understanding of the molecular mechanisms remains to be determined (Okutsu et al., 2014).
Chronic Stress and Type-2 Diabetes Mellitus
Type-2 diabetes mellitus (T2DM) is a chronic metabolic disorder that results from defects in both insulin secretion and insulin action. While the nature of the relationship between chronic stress and diabetes is less-well understood, limited evidence supports that stress may precipitate T2DM or exacerbate its progression. For example, psychological abuse by cohabitating partners significantly increases the risk of T2DM (Mason et al., 2013). Likewise, acute exposure to psychosocial stress impairs post-prandial glucose clearance among diabetic subjects (Faulenbach et al., 2012). Chronic stress may facilitate insulin resistance and/or T2DM by the direct action of glucocorticoids to induce cellular insulin resistance (discussed above), and/or it may act indirectly by promoting unhealthy eating behavior, visceral adiposity, and hepatic steatosis. Duong et al. (2012), for example, report that poor glucose control in T2DM subjects was associated with both high circulating cortisol and “low-quality” dietary choices (Duong et al., 2012).
Chronic Stress and Polycystic Ovarian Syndrome
Polycystic Ovarian Syndrome (PCOS) is a complex endocrine and metabolic disorder characterized by increased abdominal obesity, hyperandrogenism, insulin resistance, hypertension, and ovulatory dysfunction,
and is a common cause of female infertility. The etiology of PCOS is unknown, but its symptoms have been associated with primary defects in the HPA axis and enhanced SAM activity (Reaven et al., 1996; Tsilchorozidou et al., 2004). While it is clear that women with PCOS suffer from increased social and other life stressors, the psychosocial implications of PCOS symptoms (e.g., hirsutism) make it difficult to resolve the cause/effect nature of this relationship in human subjects. Importantly, however, in a rat model of PCOS wherein such psychosocial factors are presumably less prevalent, PCOS females exhibit increased SAM and HPA activity together with ovarian pathology (Stener-Victorin et al., 2005). Such findings suggest that altered stress regulation in PCOS is not entirely secondary to human psychosocial factors and may indeed contribute to the etiology of this disorder.
EARLY LIFE STRESS AND METABOLIC DISEASE
In addition to promoting metabolic disease among individuals as they cope with ongoing chronic life stress, exposure to stress both perinatally and at other early life history stages may also impair metabolic health later in life. Indeed, an abundance of epidemiological, clinical, and experimental evidence now supports this possibility. A more thorough review of this “developmental origins” hypothesis can be found elsewhere (e.g., Bruce and Hanson, 2010; Tamashiro and Moran, 2010; Reynolds, 2013), but is discussed briefly below.
Fetal exposure to high concentrations of maternal glucocorticoids retards intrauterine growth in humans (Goedhart et al., 2010; Reinisch et al., 1978), nonhuman primates, and laboratory rodents, whereas low birth weight is associated with increased risk of developing cardiometabolic disease during adulthood (Edwards et al., 1993; Barker, 1995; Seckl and Meaney, 2004; Gluckman et al., 2008). For example, in a population based study of U.S. women, very low birth weight was associated with a significantly increased risk of obesity in adulthood (Leong et al., 2003). Similarly, in a study of 22,846 U.S. men, low birth weight was associated with increased risk of hypertension and diabetes (Curhan et al., 1996). Low birth weight may also predict the development of PCOS. In a cohort of 948 Australian females, for example, thinness at birth was a significant predictor of hyperandrogenism, menstrual dysfunction, and polycystic ovaries (Davies et al., 2012).
In addition to the effects of prenatal stress and low birth weight, exposure to stress during infancy and childhood also presents a risk for obesity, CVD, and T2DM. In a bonnet macaque model of early life stress, for example, a variable foraging demand (VFD) paradigm was imposed on monkey mothers for 16 weeks, beginning when their offspring were 3-5 months
of age. Despite that VFD had no effect on total food availability or early offspring growth, VFD monkeys exhibited greater body weight, body mass index, and abdominal circumference at puberty, compared to unstressed controls (Kaufman et al., 2007). These findings suggest the adverse metabolic consequences of early life stress are not strictly dependent on early growth retardation. Likewise, persons who reported several adverse childhood experiences exhibited a 1.4- to 1.6-fold increase in the incidence of severe obesity as adults (Felitti et al., 1998). Early life stress among participants in the Helsinki Birth Cohort, who were evacuated abroad during World War II and separated from their parents, were more likely to exhibit both cardiovascular morbidity and T2DM as adults (Alastalo et al., 2009). In a similar manner, a harsh childhood environment and low childhood socioeconomic status predicted hypertension in the Coronary Artery Risk Development in Young Adults sample (Lehman et al., 2009).
The molecular and physiological mechanisms linking early life stress to cardiometabolic disease have not been clearly elucidated, but may be the consequence of organizational changes to the developing HPA axis, which eventually result in altered basal and stress-evoked SAM and HPA tone during adulthood. In a model of maternal separation stress, for example, rats exposed to this early life stressor had exaggerated sympathetic responses in adulthood (Loria et al., 2013). Similarly, when pregnant rats are exposed to restraint stress during the third week of gestation, their adult male offspring exhibit a prolonged corticosterone response to challenge with a novel object (Henry et al., 1994). Likewise, receiving relatively poor maternal care results in lower glucocorticoid-receptor (GR) expression in key stress regulatory brain regions, facilitating enhanced HPA responses to stress in adult offspring (Liu et al., 1997). Altered GR expression likely results from altered epigenetic modification of the GR promoter, since infusing a histone deacetylase inhibitor directly into the brain eliminated these effects (Weaver et al., 2004). In this way, exposure to early life stress may elicit a chronic stress-like phenotype in adulthood—underscoring potential mechanisms that link early life stress to an increased risk of developing metabolic disease.
STRESS, INFLAMMATION, AND METABOLIC DISEASE
As discussed above, exaggerated and persistent SAM and HPA axis activity likely contribute to the development of chronic stress-associated metabolic disease, by driving increased “wear and tear” on metabolic regulatory systems (McEwen and Stellar, 1993). Downstream cellular and molecular mechanisms for this proposed wear and tear are not fully understood, but one important mediator is thought to be stress-induced activation of the innate immune system and inflammatory responses.
The innate immune system is the primary early barrier to infectious agents. It recognizes conserved molecular structures that are essential for the lifecycle of various pathogens, providing a nonspecific response (Kumar et al., 2011). For example, the toll-like receptor 4 (TLR4) is a type-I trans-membrane receptor that recognizes lipopolysaccharide (LPS) derived from bacterial cell walls. When it is bound by LPS, TLR4 signaling leads to activation of macrophages and increased production of inflammatory cytokines (Akira and Takeda, 2004; Selvarajoo, 2006; Milanski et al., 2009; Kawai and Akira, 2010). In response to infection (a physical stressor), this coordinated action leads to fever and other appropriate physiological reactions.
Intriguingly, the innate immune system is also activated by acute psychological stressors and exhibits ongoing low-level activity associated with chronic stress (Segerstrom and Miller, 2004). Stress reliably increases inflammation in a variety of contexts (Jaremka et al., 2013). For example, the Trier Social Stress Test, a well-characterized laboratory stressor involving anticipation, public speaking, and mental arithmetic, leads to increased indices of inflammation relative to baseline levels (Bierhaus et al., 2003); this is exaggerated among individuals with a history of adversity early in life (Carpenter et al., 2010). Similarly, low socioeconomic status is associated with both chronic stress and chronic low-grade inflammation (Hemingway et al., 2003; Cohen et al., 2006). Although the origins of this inflammation are not thoroughly understood, stress may facilitate activation of the immune system through direct innervations of lymphatic tissue, release of SAM and HPA hormones that bind to and alter the functions of immunologically active cells, and/or through stress-induced behavioral changes (Cohen et al., 2007). For example, social disruption stress increases spleen weight, macrophage activation, and circulating cytokines in laboratory mice. All of this could be prevented with concurrent administration of the β-adrenergic antagonist propranolol (Hanke et al., 2012), identifying a critical role for the SAM to mediate these effects.
Importantly, a growing body of evidence implicates chronic low-grade activation of the innate immune response in the pathogenesis of various metabolic diseases including CVD, obesity, and T2DM. It is well known, for example, that inflammation plays a key role in the development of atherosclerosis (Cole et al., 2013). Specifically, atherogenesis is thought to begin with an initial insult to endothelial function that promotes accumulation of inflammatory cells in the vessel wall. These cells produce cytokines and growth factors that evoke a cascade of morphological changes, eventually contributing to plaque formation and rupture (Libby et al., 2011). Conversely, therapies that reduce vascular inflammation are effective to reduce the incidence and progression of atherosclerosis. Inflammation in the central nervous system circuits that regulate feeding behavior contributes to the overconsumption of high-fat diets and increases weight gain (Thaler
and Schwartz, 2010; Ryan et al., 2012). Chronic low-grade systemic inflammation also contributes to the increased risk of peripheral insulin resistance associated with obesity (Donath and Shoelson, 2011). This depends at least in part on TLR4 signaling, since mice that lack TLR4 are protected from high-fat diet induced obesity and insulin resistance (Shi et al., 2006; Cani et al., 2007; Tsukumo et al., 2007). Consequently, there has been some interest in the use of anti-inflammatory agents to treat Type-2 diabetes mellitus (Larsen et al., 2007; Fleischman et al., 2008; Goldfine et al., 2008).
Gut Microbiota and the Innate Immune Response
Related to all this, vast communities of commensal microbes inhabit the alimentary tract of humans and other mammals (Dethlefsen et al., 2006). Indeed, gut microbiota outnumber human cells by a factor of 10 and, as a whole, possess 100-fold more genes than the human genome (Hooper and Gordon, 2001). A rapidly growing research enterprise now focuses on interactions between gut microbial ecology, inflammation, and human health (Caesar et al., 2010; Tremaroli and Bäckhed, 2012). One intriguing direction for future research, explored below, is the idea that stress-associated changes to gut microbial communities, and subsequent stimulation of the immune response, contribute to the origin of stress-associated chronic inflammation, providing an additional mechanistic link between stress and metabolic disease.
The innate immune system responds to conserved molecular structures that are essential for the lifecycle of various microbial species—both pathogenic and commensal. In this way, commensal gut microbiota may affect host physiology by modulating inflammatory signaling pathways (Caesar et al., 2010; Chu and Mazmanian, 2013). In addition, the composition of gut microbial communities is thought to significantly affect barrier function of the intestinal wall (Berg, 1999; Tlaskalová-Hogenová et al., 2011), which in turn would determine the infiltration of microbes and/or microbial particles to ultimately modulate innate immune responses. For example, modifying gut microbial communities, by administration of a probiotic, significantly reduced circulating cytokines among patients with rheumatoid arthritis (Vaghef-Mehrabany et al., 2013). Likewise, manipulating mouse gut microbial communities, by adding a pre-biotic fermentable carbohydrate to their diet, reduces intestinal permeability, circulating concentrations of LPS, and systemic inflammation (Cani et al., 2009).
GUT MICROBIOTA, STRESS, AND METABOLIC DISEASE
An explosion of recent research has begun to identify important links between humans’ commensal microbiota and metabolic disease. Impor-
tantly, an altered gut microbiota has now been linked with obesity, T2DM, and CVD (see Figure 11-1). Gut microbial communities are altered in obese individuals compared to lean counterparts and can be modified by dietary intake (Ley et al., 2005; Turnbaugh et al., 2009). These patterns may indicate that an altered gut microbiota contributes functionally to obesity, or conversely, that obesity and/or food selection per se alters the gut microbiota. A growing body of evidence from prospective human studies and experimental animal models supports the possibility of a functional association. For example, a prospective Finnish study of 49 individuals sampled as infants identified specific microbial groups that were associated with the development of overweight by 7 years of age (Kalliomäki et al., 2008). In addition, transplantation of the gut microbiota from obese mice into germ-free gnotobiotic mice is associated with weight gain (Turnbaugh et al., 2006, 2008). Finally, changes in gut microbial communities, secondary to an altered local environment, are implicated in the weight loss and other metabolic benefits achieved with bariatric surgery, in both humans (Furet et al., 2010; Graessler et al., 2012) and in rodents (Liou et al., 2013;
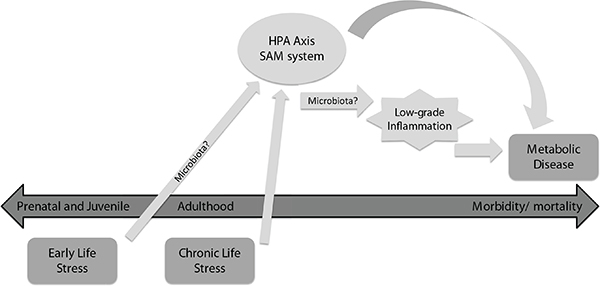
FIGURE 11-1 Potential mechanisms linking stress to increased risk of metabolic disease.
NOTE: Exposure to stress both early in life and chronically, during adulthood are significant risk factors for obesity, diabetes, and cardiovascular disease. This may result from frequent and/or persistent activation of hypothalamic-pituitary-adrenocortical (HPA) and sympatho-adrenomedullary (SAM) stress responses. HPA and/or SAM activity may contribute to the etiology of metabolic disease either directly, or by eliciting mild, persistent activation of the innate immune system. Finally, stress is known to alter the composition of gut microbial communities, whereas differences in gut microbial ecology has likewise been linked to both inflammation and the risk of metabolic disease.
Ryan et al., 2014). Gnotobiotic mice receiving a transplantation of gut microbiota from donors that had gastric bypass surgery gained less weight compared to those that receive a transplant from sham-operated controls, supporting the functional nature of this relationship (Liou et al., 2013).
Both human and rodent studies also implicate the gut microbiota as contributing to the pathogenesis of CVD. For example, commensal bacterial species previously thought to be restricted to the alimentary tract have now been found in atherosclerotic plaques (Koren et al., 2011), implicating increased leakiness of the gut mucosal barrier and likely contributing to increased inflammation associated with CVD. Consistent with this, patients who had experienced an atherosclerotic event had altered abundance of specific gut microbial species, associated with a decrease in the synthesis of the anti-inflammatory metabolite butyrate (Karlsson et al., 2012).
The gut microbiota may likewise link aspects of the Western diet to increased risk of CVD. Specifically, an omnivorous diet is associated with the increased abundance of specific gut microbes that metabolize dietary L-carnitine—a trimethylamine abundant in red meat. A metabolite of this microbial reaction, trimethylamine-N-oxide (TMAO), is associated with increased risk of CVD. Accordingly, the presence of specific microbial species, together with high plasma levels of L-carnitine and TMAO, predicted an increased risk of CVD and the occurrence of adverse cardiovascular events in human subjects. Moreover, supplementing the diet of laboratory mice with L-carnitine both altered cecal microbial composition and increased vascular pathology (Koeth et al., 2013). In this way, the gut microbiota provide an important mechanism underlying the well-established link between high levels of red meat consumption and the risk of CVD.
Gut microbial communities are responsive to host psychological stress. A number of studies now demonstrate that stress exposure, or exposure to stress hormones, significantly alters the composition of the gut microbiota and translocation of gut microbes across the intestinal wall (Tannock and Savage, 1974; Lyte and Bailey, 1997; Bailey et al., 2006, 2011). An altered microbiota is thought to be one link between stress responses and increased systemic inflammation, since stress-induced changes in both gut microbial composition and increased circulating cytokines could be abrogated by concurrent treatment with an antibiotic (Bailey et al., 2007). Thus, one interesting possibility is that activation of the SAM and HPA axis by psychosocial stress facilitates chronic metabolic disease by altering the gut microbiota, thereby activating the innate immune system, although this possibility has not yet been tested.
Furthermore, although the majority of mechanistic research has focused on how social defeat, isolation, and subordination promote disease (discussed above), social relationships may, of course, be both anxiolytic and/or anxiogenic. Therefore, social interactions may be either detrimental or
beneficial depending on the context. In fact, an abundance of epidemiological work indicates that positive social relationships may offer stress relief—contributing to improved health and longevity (Berkman and Syme, 1979). In wild baboons, for example, the degree of affiliation is related to HPA status (Sapolsky et al., 1997). Likewise, lack of attachment with intimate contacts is associated with high resting SAM hormones in young men (Knox et al., 1985). Consistent with the role of the HPA axis and SAM system in facilitating the incidence and progression of CVD, supportive social relationships are associated with better prognosis in coronary artery disease (Williams et al., 1992) and with reduced cardiovascular mortality (Welin et al., 1992). It is interesting to speculate that positive social interactions may blunt SAM and HPA activity in the face of stress, facilitating favorable changes to the gut microbiota, which thereby contribute to these benefits.
In addition to directly blunting activation of the SAM system and HPA axis, social interactions may alter stress system function and/or risk of metabolic disease by facilitating the inter-individual transmission of commensal microbes (Lombardo, 2007; Archie and Theis, 2011). Social transmission of gut microbes has been demonstrated in a variety of animals including iguana (Troyer, 1982), chimney swift (Kyle and Kyle 1993), and mice (Lombardo, 2007). Some evidence suggests this may occur in humans as well, since the gut microbiomes of unrelated adults (typically married partners) living in the same household were significantly more similar to each other than to unrelated individuals living in a different household (Yatsunenko et al., 2012). To the extent that frequent social partners share microbiota, one might hypothesize that individuals with stronger and more diverse social networks possess a more diverse microbiome. Thus, a reduction in the diversity of social relationships that may occur with aging could contribute to the observed aging-associated decline in microbial diversity (Claesson et al., 2011). Accordingly, increasing diversity of the gut microbiome is generally associated with improved health. Increased gut microbial diversity is thought to be protective against a number of diseases, including obesity (Ley et al., 2005), atherosclerosis (Karlsson et al., 2012), and Crohn disease (Manichanh et al., 2006). More directly, gut microbiota are transmitted from mother to offspring during birth (Fåk et al., 2008; Palmer et al., 2007) and lactation (Fernández et al., 2013). In light of the effects of microbiota on metabolic disease, this may provide an alternate mechanism by which maternal stress has long-term organizational effects on the metabolic health of her offspring.
SUMMARY AND FUTURE DIRECTIONS
Although the link between psychosocial stress and metabolic disease has been appreciated for decades, significant work remains to delineate
mechanistic details. A growing body of evidence implicates exaggerated and persistent stress-associated activation of the SAM system and HPA axis, as well as induction of the innate immune system and increased systemic inflammation.
Understanding the potential role of stress-associated changes in the gut microbiota as a source for systemic inflammation and disease is an important avenue for future research that may highlight important opportunities for intervention. For example, gut microbial composition is altered in pregnancy (Koren et al., 2012) and affects the inoculation of newborns. A better understanding of how maternal stress affects maternal gut microbial communities, and downstream physiological consequences for both mother and offspring, will be informative. Importantly, this may represent a critical period for therapeutic interventions aimed at disrupting the connections among maternal stress, low birth weight, and metabolic disease of adult offspring. In sum, this “bottom-up” approach to understanding the relationships among stress, the microbiome, physiology, and social structure/behaviors provides ample opportunity to inform behavioral, nutritional, and pharmacological interventions to combat metabolic disease.
REFERENCES
Akana, S.F., Dallman, M.F., Bradbury, M.J., Scribner, K.A., Strack, A.M., and Walker, C.D. (1992). Feedback and facilitation in the adrenocortical system: Unmasking facilitation by partial inhibition of the glucocorticoid response to prior stress. Endocrinology, 131, 57-68.
Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nature Reviews Immunology, 4, 499.
Alastalo, H., Raikkonen, K., Pesonen, A.-K., Osmond, C., Barker, D.J.P., Kajantie, E., Heinonen, K., Forsen, T.J., and Eriksson, J.G. (2009). Cardiovascular health of Finnish war evacuees 60 years later. Annals of Medicine, 41, 66-72.
Andrews, R.C., and Walker, B.R. (1999). Glucocorticoids and insulin resistance: Old hormones new targets. Clinical Science, 96(5), 513-523.
Archie, E.A., and Theis, K.R. (2011). Animal behaviour meets microbial ecology. Animal Behaviour, 82, 425-436.
Arner, P. (1995). Differences in lipolysis between human subcutaneous and omental adipose tissues. Annals of Medicine, 27, 435-438.
Bailey, M.T., Engler, H., and Sheridan, J.F. (2006). Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. Journal of Neuroimmunology, 171, 29-37.
Bailey, M.T., Engler, H., Powell, N.D., Padgett, D.A., and Sheridan, J.F. (2007). Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 293, R1180-R1190.
Bailey, M.T., Dowd, S.E., Galley, J.D., Hufnagle, A.R., Allen, R.G., and Lyte, M. (2011). Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain, Behavior, and Immunity, 25, 397-407.
Barker, D.J. (1995). Fetal origins of coronary heart disease. British Medical Journal, 311, 171-174.
Bartness, T.J., and Song, C.K. (2007). Thematic review series: Adipocyte biology. Sympathetic and sensory innervation of white adipose tissue. Journal of Lipid Research, 48, 1655-1672.
Baxter, J.D. (1976). Glucocorticoid hormone action. Pharmacology & Therapeutics, 2, 605-659.
Berg, R.D. (1999). Bacterial translocation from the gastrointestinal tract. Advances in Experimental Medicine and Biology, 473, 11-30.
Berkman, L.F., and Syme, S.L. (1979). Social networks, host resistance, and mortality: A nine-year follow-up study of Alameda County residents. American Journal of Epidemiology, 109, 186-204.
Bierhaus, A., Wolf, J., Andrassy, M., Rohleder, N., Humpert, P.M., Petrov, D., Ferstl, R., von Eynatten, M., Wendt, T., Rudofsky, G., et al. (2003). A mechanism converting psychosocial stress into mononuclear cell activation. Proceedings of the National Academy of Sciences of the United States of America, 100, 1920-1925.
Bruce, K.D., and Hanson, M.A. (2010). The developmental origins, mechanisms, and implications of metabolic syndrome. Journal of Nutrition, 140, 648-652.
Brunner, E.J., Chandola, T., and Marmot, M.G. (2007). Prospective effect of job strain on general and central obesity in the Whitehall II study. American Journal of Epidemiology, 165, 828-837.
Caesar, R., Fåk, F., and Bäckhed, F. (2010). Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. Journal of Internal Medicine, 268, 320-328.
Campbell, J.E., Peckett, A.J., D’souza, A.M., Hawke, T.J., and Riddell, M.C. (2011). Adipogenic and lipolytic effects of chronic glucocorticoid exposure. American Journal of Physiology: Cell Physiology, 300, C198-C209.
Cani, P.D., Amar, J., Iglesias, M.A., Poggi, M., Knauf, C., Bastelica, D., Neyrinck, A.M., Fava, F., Tuohy, K.M., Chabo, C., et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes, 56, 1761-1772.
Cani, P.D., Possemiers, S., Van de Wiele, T., Guiot, Y., Everard, A., Rottier, O., Geurts, L., Naslain, D., Neyrinck, A., Lambert, D.M., et al. (2009). Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut, 58(8), 1091-1103.
Carpenter, L.L., Gawuga, C.E., Tyrka, A.R., Lee, J.K., Anderson, G.M., and Price, L.H. (2010). Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology, 35, 2617-2623.
Chu, H., and Mazmanian, S.K. (2013). Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nature Immunology, 14, 668-675.
Chuang, J.-C., Cui, H., Mason, B.L., Mahgoub, M., Bookout, A.L., Yu, H.G., Perello, M., Elmquist, J.K., Repa, J.J., Zigman, J.M., et al. (2010). Chronic social defeat stress disrupts regulation of lipid synthesis. Journal of Lipid Research, 51, 1344-1353.
Claesson, M.J., Cusack, S., O’Sullivan, O., Greene-Diniz, R., de Weerd, H., Flannery, E., Marchesi, J.R., Falush, D., Dinan, T., Fitzgerald, G., et al. (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proceedings of the National Academy of Sciences of the United States of America, 108(Suppl. 1), 4586-4591.
Cohen, S., Doyle, W.J., and Baum, A. (2006). Socioeconomic status is associated with stress hormones. Psychosomatic Medicine, 68, 414-420.
Cohen, S., Janicki-Deverts, D., and Miller, G.E. (2007). Psychological stress and disease. Journal of the American Medical Association, 298, 1685-1687.
Cole, J.E., Kassiteridi, C., and Monaco, C. (2013). Toll-like receptors in atherosclerosis: A “Pandora’s box” of advances and controversies. Trends in Pharmacological Sciences, 34, 629-636.
Curhan, G.C., Willett, W.C., Rimm, E.B., Spiegelman, D., Ascherio, A.L., and Stampfer, M.J. (1996). Birth weight and adult hypertension, diabetes mellitus, and obesity in U.S. men. Circulation, 94, 3246-3250.
Dallman, M.F., Pecoraro, N., Akana, S.F., La Fleur, S.E., Gomez, F., Houshyar, H., Bell, M.E., Bhatnagar, S., Laugero, K.D., and Manalo, S. (2003). Chronic stress and obesity: A new view of “comfort food.” Proceedings of the National Academy of Sciences of the United States of America, 100, 11696-11701.
Davies, M.J., March, W.A., Willson, K.J., Giles, L.C., and Moore, V.M. (2012). Birthweight and thinness at birth independently predict symptoms of polycystic ovary syndrome in adulthood. Human Reproduction, 27, 1475-1480.
Dethlefsen, L., Eckburg, P.B., Bik, E.M., and Relman, D.A. (2006). Assembly of the human intestinal microbiota. Trends in Ecology & Evolution, 21, 517-523.
Donath, M.Y., and Shoelson, S.E. (2011). Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology, 11, 98.
Duong, M., Cohen, J.I., and Convit, A. (2012). High cortisol levels are associated with low quality food choice in type 2 diabetes. Endocrine, 41, 76-81.
Edwards, C.R., Benediktsson, R., Lindsay, R.S., and Seckl, J.R. (1993). Dysfunction of placental glucocorticoid barrier: Link between fetal environment and adult hypertension? Lancet, 341, 355-357.
Fåk, F., Ahrné, S., Molin, G., Jeppsson, B., and Weström, B. (2008). Microbial manipulation of the rat dam changes bacterial colonization and alters properties of the gut in her offspring. American Journal of Physiology: Gastrointestinal and Liver Physiology, 294, G148-G154.
Faulenbach, M., Uthoff, H., Schwegler, K., Spinas, G.A., Schmid, C., and Wiesli, P. (2012). Effect of psychological stress on glucose control in patients with type 2 diabetes. Diabetic Medicine, 29, 128-131.
Felitti, V.J., Anda, R.F., Nordenberg, D., Williamson, D.F., Spitz, A.M., Edwards, V., Koss, M.P., and Marks, J.S. (1998). Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. American Journal of Preventive Medicine, 14, 245-258.
Fernández, L., Langa, S., Martín, V., Maldonado, A., Jiménez, E., Martin, R., and Rodríguez, J.M. (2013). The human milk microbiota: Origin and potential roles in health and disease. Pharmacological Research, 69, 1-10.
Fleischman, A., Shoelson, S.E., Bernier, R., and Goldfine, A.B. (2008). Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care, 31, 289-294.
Furet, J.-P., Kong, L.-C., Tap, J., Poitou, C., Basdevant, A., Bouillot, J.-L., Mariat, D., Corthier, G., Doré, J., Henegar, C., et al. (2010). Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes, 59, 3049-3057.
Gluckman, P.D., Hanson, M.A., Cooper, C., and Thornburg, K.L. (2008). Effect of in utero and early-life conditions on adult health and disease. New England Journal of Medicine, 359, 61-73.
Goedhart, G., Vrijkotte, T.G.M., Roseboom, T.J., van der Wal, M.F., Cuijpers, P., and Bonsel, G.J. (2010). Maternal cortisol and offspring birthweight: Results from a large prospective cohort study. Psychoneuroendocrinology, 35, 644-652.
Goldfine, A.B., Silver, R., Aldhahi, W., Cai, D., Tatro, E., Lee, J., and Shoelson, S.E. (2008). Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clinical and Translational Science, 1, 36.
Graessler, J., Qin, Y., Zhong, H., Zhang, J., Licinio, J., Wong, M.-L., Xu, A., Chavakis, T., Bornstein, A.B., Ehrhart-Bornstein, M., et al. (2012). Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: Correlation with inflammatory and metabolic parameters. The Pharmacogenomics Journal, 13(6), 514-522.
Green, P.K., Wilkinson, C.W., and Woods, S.C. (1992). Intraventricular corticosterone increases the rate of body weight gain in underweight adrenalectomized rats. Endocrinology, 130, 269-275.
Grippo, A.J., Moffitt, J.A., and Johnson, A.K. (2002). Cardiovascular alterations and autonomic imbalance in an experimental model of depression. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 282, R1333-R1341.
Groesz, L.M., McCoy, S., Carl, J., Saslow, L., Stewart, J., Adler, N., Laraia, B., and Epel, E. (2012). What is eating you? Stress and the drive to eat. Appetite, 58, 717-721.
Gyengesi, E., Liu, Z.-W., D’Agostino, G., Gan, G., Horvath, T.L., Gao, X.-B., and Diano, S. (2010). Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons in adult mice. Endocrinology, 151, 5395-5402.
Hanke, M.L., Powell, N.D., Stiner, L.M., Bailey, M.T., and Sheridan, J.F. (2012). Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain, Behavior, and Immunity, 26, 1150-1159.
Hemingway, H., Shipley, M., Mullen, M.J., Kumari, M., Brunner, E., Taylor, M., Donald, A.E., Deanfield, J.E., and Marmot, M. (2003). Social and psychosocial influences on inflammatory markers and vascular function in civil servants (the Whitehall II study). American Journal of Cardiology, 92, 984-987.
Henry, C., Kabbaj, M., Simon, H., Moal, M., and Maccari, S. (1994). Prenatal stress increases the hypothalamo-pituitary-adrenal axis response in young and adult rats. Journal of Neuroendocrinology, 6, 341-345.
Hoffstedt, J., Arner, P., Hellers, G., and Lönnqvist, F. (1997). Variation in adrenergic regulation of lipolysis between omental and subcutaneous adipocytes from obese and non-obese men. Journal of Lipid Research, 38, 795-804.
Hooper, L.V., and Gordon, J.I. (2001). Commensal host-bacterial relationships in the gut. Science, 292, 1115-1118.
Jaremka, L.M., Lindgren, M.E., and Kiecolt-Glaser, J.K. (2013). Synergistic relationships among stress, depression, and troubled relationships: Insights from psychoneuroimmunology. Depression and Anxiety, 30, 288-296.
Jayo, J.M., Shively, C.A., Kaplan, J.R., and Manuck, S.B. (1993). Effects of exercise and stress on body fat distribution in male cynomolgus monkeys. International Journal of Obesity and Related Metabolic Disorders, 17(10), 597-604.
Kalliomäki, M., Collado, M.C., Salminen, S., and Isolauri, E. (2008). Early differences in fecal microbiota composition in children may predict overweight. American Journal of Clinical Nutrition, 87, 534-538.
Kaplan, J.R., Manuck, S.B., Clarkson, T.B., Lusso, F.M., Taub, D.M., and Miller, E.W. (1983). Social stress and atherosclerosis in normocholesterolemic monkeys. Science, 220, 733-735.
Kaplan, J.R., Manuck, S.B., Adams, M.R., Weingand, K.W., and Clarkson, T.B. (1987). Inhibition of coronary atherosclerosis by propranolol in behaviorally predisposed monkeys fed an atherogenic diet. Circulation, 76, 1364-1372.
Karlsson, F.H., Fåk, F., Nookaew, I., Tremaroli, V., Fagerberg, B., Petranovic, D., Bäckhed, F., and Nielsen, J. (2012). Symptomatic atherosclerosis is associated with an altered gut metagenome. Nature Communications, 3, 1245.
Kaufman, D., Banerji, M.A., Shorman, I., Smith, E.L.P., Coplan, J.D., Rosenblum, L.A., and Kral, J.G. (2007). Early-life stress and the development of obesity and insulin resistance in juvenile bonnet macaques. Diabetes, 56, 1382-1386.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nature Immunology, 11, 373.
Kim, Y., Yang, H.Y., Kim, A.-J., and Lim, Y. (2013). Academic stress levels were positively associated with sweet food consumption among Korean high-school students. Nutrition, 29, 213-218.
Kivimäki, M., Virtanen, M., Elovainio, M., Kouvonen, A., Väänänen, A., and Vahtera, J. (2006). Work stress in the etiology of coronary heart disease—a meta-analysis. Scandinavian Journal of Work, Environment & Health, 32, 431-442.
Knox, S.S., Theorell, T., Svensson, J.C., and Waller, D. (1985). The relation of social support and working environment to medical variables associated with elevated blood pressure in young males: A structural model. Social Science & Medicine, 21(5), 525-531.
Koeth, R.A., Wang, Z., Levison, B.S., Buffa, J.A., Org, E., Sheehy, B.T., Britt, E.B., Fu, X., Wu, Y., Li, L., et al. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature Medicine, 19, 576-585.
Koren, O., Spor, A., Felin, J., Fåk, F., Stombaugh, J., Tremaroli, V., Behre, C.J., Knight, R., Fagerberg, B., Ley, R.E., et al. (2011). Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America, 108(Suppl.), 4592-4598.
Koren, O., Goodrich, J.K., Cullender, T.C., Spor, A., Laitinen, K., Bäckhed, H.K., Gonzalez, A., Werner, J.J., Angenent, L.T., Knight, R., et al. (2012). Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell, 150, 470-480.
Kumar, H., Kawai, T., and Akira, S. (2011). Pathogen recognition by the innate immune system. International Reviews of Immunology, 30, 16-34.
Kyle, P.D., and Kyle, G.Z. (1993). An evaluation of the role of microbial flora in the salivary transfer technique for hand-rearing Chimney Swifts. Wildlife Rehabilitation, 8, 65-71.
Lambillotte, C., Gilon, P., and Henquin, J.C. (1997). Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. Journal of Clinical Investigation, 99, 414-423.
Larsen, C.M., Faulenbach, M., Vaag, A., Volund, A., Ehses, J.A., Seifert, B., Mandrup-Poulsen, T., and Donath, M.Y. (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. New England Journal of Medicine, 356, 1517-1526.
Laugero, K.D., Falcon, L.M., and Tucker, K.L. (2011). Relationship between perceived stress and dietary and activity patterns in older adults participating in the Boston Puerto Rican Health Study. Appetite, 56, 194-204.
Lehman, B.J., Taylor, S.E., Kiefe, C.I., and Seeman, T.E. (2009). Relationship of early life stress and psychological functioning to blood pressure in the CARDIA study. Health Psychology, 28, 338-346.
Lemmens, S.G., Rutters, F., Born, J.M., and Westerterp-Plantenga, M.S. (2011). Stress augments food “wanting” and energy intake in visceral overweight subjects in the absence of hunger. Physiology & Behavior, 103, 157-163.
Leong, N.M., Mignone, L.I., Newcomb, P.A., Titus-Ernstoff, L., Baron, J.A., Trentham-Dietz, A., Stampfer, M.J., Willett, W.C., and Egan, K.M. (2003). Early life risk factors in cancer: The relation of birth weight to adult obesity. International Journal of Cancer, 103, 789-791.
Ley, R.E., Bäckhed, F., Turnbaugh, P., Lozupone, C.A., Knight, R.D., and Gordon, J.I. (2005). Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America, 102, 11070-11075.
Libby, P., Ridker, P.M., and Hansson, G.K. (2011). Progress and challenges in translating the biology of atherosclerosis. Nature, 473, 317-325.
Liou, A.P., Paziuk, M., Luevano, J.-M.J.-M., Machineni, S., Turnbaugh, P.J., and Kaplan, L.M. (2013). Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Science Translational Medicine, 5, 178ra41.
Liu, D., Diorio, J., Tannenbaum, B., Caldji, C., Francis, D., Freedman, A., Sharma, S., Pearson, D., Plotsky, P.M., and Meaney, M.J. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science, 277, 1659-1662.
Lombardo, M.P. (2007). Access to mutualistic endosymbiotic microbes: An underappreciated benefit of group living. Behavioral Ecology and Sociobiology, 62, 479-497.
Lönn, L., Kvist, H., Ernest, I., and Sjöström, L. (1994). Changes in body composition and adipose tissue distribution after treatment of women with Cushing’s syndrome. Metabolism, 43, 1517-1522.
Loria, A.S., Brands, M.W., Pollock, D.M., and Pollock, J.S. (2013). Early life stress sensitizes the renal and systemic sympathetic system in rats. American Journal of Physiology: Renal Physiology, 305, F390-F395.
Lynch, J., Krause, N., Kaplan, G.A., Salonen, R., and Salonen, J.T. (1997). Workplace demands, economic reward, and progression of carotid atherosclerosis. Circulation, 96, 302-307.
Lyte, M., and Bailey, M.T. (1997). Neuroendocrine-bacterial interactions in a neurotoxininduced model of trauma. Journal of Surgical Research, 70, 195-201.
Manichanh, C., Rigottier-Gois, L., Bonnaud, E., Gloux, K., Pelletier, E., Frangeul, L., Nalin, R., Jarrin, C., Chardon, P., Marteau, P., et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut, 55, 205-211.
Mason, S.M., Wright, R.J., Hibert, E.N., Spiegelman, D., Jun, H.-J., Hu, F.B., and Rich-Edwards, J.W. (2013). Intimate partner violence and incidence of type 2 diabetes in women. Diabetes Care, 36, 1159-1165.
McCann, B.S., Warnick, G.R., and Knopp, R.H. (1990). Changes in plasma lipids and dietary intake accompanying shifts in perceived workload and stress. Psychosomatic Medicine, 52, 97-108.
McEwen, B.S., and Stellar, E. (1993). Stress and the individual. Mechanisms leading to disease. Archives of Internal Medicine, 153, 2093-101.
Milanski, M., Degasperi, G., Coope, A., Morari, J., Denis, R., Cintra, D.E., Tsukumo, D.M.L., Anhe, G., Amaral, M.E., Takahashi, H.K., et al. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. The Journal of Neuroscience, 29, 359-370.
Neves, V.J., Moura, M.J.C.S., Tamascia, M.L., Ferreira, R., Silva, N.S., Costa, R., Montemor, P.L., Narvaes, E.A.O., Bernardes, C.F., Novaes, P.D., et al. (2009). Proatherosclerotic effects of chronic stress in male rats: Altered phenylephrine sensitivity and nitric oxide synthase activity of aorta and circulating lipids. Stress, 12, 320-327.
Okutsu, M., Lira, V.A., Higashida, K., Peake, J., Higuchi, M., and Suzuki, K. (2014). Corticosterone accelerates atherosclerosis in the apolipoprotein E-deficient mouse. Atherosclerosis, 232, 414-419.
Oliver, G., and Wardle, J. (1999). Perceived effects of stress on food choice. Physiology & Behavior, 66, 511-515.
Orth, D.N. (1995). Cushing’s syndrome. New England Journal of Medicine, 332, 791-803.
Orth-Gomér, K., Wamala, S.P., Horsten, M., Schenck-Gustafsson, K., Schneiderman, N., and Mittleman, M.A. (2000). Marital stress worsens prognosis in women with coronary heart disease. Journal of the American Medical Association, 284, 3008.
Ostman, J., Arner, P., Engfeldt, P., and Kager, L. (1979). Regional differences in the control of lipolysis in human adipose tissue. Metabolism, 28, 1198-1205.
Palmer, C., Bik, E.M., DiGiulio, D.B., Relman, D.A., and Brown, P.O. (2007). Development of the human infant intestinal microbiota. PLOS Biology, 5, e177.
Reaven, G.M., Lithell, H., and Landsberg, L. (1996). Hypertension and associated metabolic abnormalities—the role of insulin resistance and the sympathoadrenal system. New England Journal of Medicine, 334, 374-381.
Reinisch, J.M., Simon, N.G., Karow, W.G., and Gandelman, R. (1978). Prenatal exposure to prednisone in humans and animals retards intrauterine growth. Science, 202, 436-438.
Reynolds, R.M. (2013). Glucocorticoid excess and the developmental origins of disease: Two decades of testing the hypothesis—2012 Curt Richter Award Winner. Psychoneuroendocrinology, 38(1), 1-11.
Rozanski, A., Blumenthal, J.A., and Kaplan, J. (1999). Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation, 99, 2192-2217.
Rudenga, K.J., Sinha, R., and Small, D.M. (2013). Acute stress potentiates brain response to milkshake as a function of body weight and chronic stress. International Journal of Obesity, 37, 309-316.
Ryan, K.K., Woods, S.C., and Seeley, R.J. (2012). Central nervous system mechanisms linking the consumption of palatable high-fat diets to the defense of greater adiposity. Cell Metabolism, 15, 137-149.
Ryan, K.K., Tremaroli, V., Clemmensen, C., Kovatcheva-Datchary, P., Myronovych, A., Karns, R., Wison-Perez, H., Sandoval, D.A., Kohli, R., Bäckhed, F., et al. (2014). FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature, 509(7499), 183-188.
Sapolsky, R.M., Alberts, S.C., and Altmann, J. (1997). Hypercortisolism associated with social subordinance or social isolation among wild baboons. Archives of General Psychiatry, 54, 1137-1143.
Seckl, J.R., and Meaney, M.J. (2004). Glucocorticoid programming. Annals of the New York Academy of Sciences, 1032, 63-84.
Segerstrom, S.C., and Miller, G.E. (2004). Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychological Bulletin, 130, 601-630.
Selvarajoo, K. (2006). Discovering differential activation machinery of the Toll-like receptor 4 signaling pathways in MyD88 knockouts. FEBS Letters, 580, 1457.
Shi, H., Kokoeva, M. V, Inouye, K., Tzameli, I., Yin, H., and Flier, J.S. (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of Clinical Investigation, 116, 3015.
Shively, C.A., Clarkson, T.B., and Kaplan, J.R. (1989). Social deprivation and coronary artery atherosclerosis in female cynomolgus monkeys. Atherosclerosis, 77, 69-76.
Stener-Victorin, E., Ploj, K., Larsson, B.-M., and Holmäng, A. (2005). Rats with steroid-induced polycystic ovaries develop hypertension and increased sympathetic nervous system activity. Reproductive Biology and Endocrinology, 3, 44.
Steptoe, A., and Kivimäki, M. (2012). Stress and cardiovascular disease. Nature Reviews Cardiology, 9, 360-370.
Tamashiro, K.L.K., and Moran, T.H. (2010). Perinatal environment and its influences on metabolic programming of offspring. Physiology & Behavior, 100, 560-566.
Tannock, G.W., and Savage, D.C. (1974). Influences of dietary and environmental stress on microbial populations in the murine gastrointestinal tract. Infection and Immunity, 9, 591-598.
Tataranni, P.A., Larson, D.E., Snitker, S., Young, J.B., Flatt, J.P., and Ravussin, E. (1996). Effects of glucocorticoids on energy metabolism and food intake in humans. American Journal of Physiology, 271, E317-E325.
Tempel, D.L., McEwen, B.S., and Leibowitz, S.F. (1992). Effects of adrenal steroid agonists on food intake and macronutrient selection. Physiology & Behavior, 52, 1161-1166.
Thaler, J.P., and Schwartz, M.W. (2010). Minireview: Inflammation and obesity pathogenesis: The hypothalamus heats up. Endocrinology, 151, 4109-4115.
Tlaskalová-Hogenová, H., Stěpánková, R., Kozáková, H., Hudcovic, T., Vannucci, L., Tučková, L., Rossmann, P., Hrnčíř, T., Kverka, M., Zákostelská, Z., et al. (2011). The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cellular & Molecular Immunology, 8, 110-120.
Tremaroli, V., and Bäckhed, F. (2012). Functional interactions between the gut microbiota and host metabolism. Nature, 489, 242-249.
Troyer, K. (1982). Transfer of fermentative microbes between generations in a herbivorous lizard. Science, 216, 540-542.
Tryon, M.S., Carter, C.S., Decant, R., and Laugero, K.D. (2013a). Chronic stress exposure may affect the brain’s response to high calorie food cues and predispose to obesogenic eating habits. Physiology & Behavior, 120, 233-242.
Tryon, M.S., DeCant, R., and Laugero, K.D. (2013b). Having your cake and eating it too: A habit of comfort food may link chronic social stress exposure and acute stress-induced cortisol hyporesponsiveness. Physiology & Behavior, 114-115, 32-37.
Tsilchorozidou, T., Overton, C., and Conway, G.S. (2004). The pathophysiology of polycystic ovary syndrome. Clinical Endocrinology (Oxford), 60, 1-17.
Tsukumo, D.M.L., Carvalho-Filho, M.A., Carvalheira, J.B.C., Prada, P.O., Hirabara, S.M., Schenka, A.A., Araújo, E.P., Vassallo, J., Curi, R., Velloso, L.A., and Saad, M.J. (2007). Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes, 56(8), 1986-1998.
Turnbaugh, P.J., Ley, R.E., Mahowald, M.A., Magrini, V., Mardis, E.R., and Gordon, J.I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature, 444, 1027-1031.
Turnbaugh, P.J., Bäckhed, F., Fulton, L., and Gordon, J.I. (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe, 3, 213-223.
Turnbaugh, P.J., Hamady, M., Yatsunenko, T., Cantarel, B.L., Duncan, A., Ley, R.E., Sogin, M.L., Jones, W.J., Roe, B.A., Affourtit, J.P., et al. (2009). A core gut microbiome in obese and lean twins. Nature, 457, 480-484.
Ulrich-Lai, Y.M., and Engeland, W.C. (2002). Adrenal splanchnic innervation modulates adrenal cortical responses to dehydration stress in rats. Neuroendocrinology, 76, 79.
Ulrich-Lai, Y.M., and Herman, J.P. (2009). Neural regulation of endocrine and autonomic stress responses. Nature Reviews Neuroscience, 10, 397-409.
Ulrich-Lai, Y.M., and Ryan, K.K. (2014). Neuroendocrine circuits governing energy balance and stress regulation: Functional overlap and therapeutic implications. Celularl Metabolism, 19(6), 910-925.
Ulrich-Lai, Y.M., Ostrander, M.M., Thomas, I.M., Packard, B.A., Furay, A.R., Dolgas, C.M., Van Hooren, D.C., Figueiredo, H.F., Mueller, N.K., Choi, D.C., et al. (2007). Daily limited access to sweetened drink attenuates hypothalamic-pituitary-adrenocortical axis stress responses. Endocrinology, 148, 1823-1834.
Vaghef-Mehrabany, E., Alipour, B., Homayouni-Rad, A., Sharif, S.-K., Asghari-Jafarabadi, M., and Zavvari, S. (2013). Probiotic supplementation improves inflammatory status in patients with rheumatoid arthritis. Nutrition, 30(4), 430-435.
Von Känel, R., Mausbach, B.T., Dimsdale, J.E., Mills, P.J., Patterson, T.L., Ancoli-Israel, S., Ziegler, M.G., Roepke, S.K., Chattillion, E.A., Allison, M., et al. (2011). Cardiometabolic effects in caregivers of nursing home placement and death of their spouse with Alzheimer’s disease. Journal of the American Geriatrics Society, 59, 2037-2044.
Wang, H.X., Leineweber, C., Kirkeeide, R., Svane, B., Schenck-Gustafsson, K., Theorell, T., and Orth-Gomér, K. (2007). Psychosocial stress and atherosclerosis: Family and work stress accelerate progression of coronary disease in women. The Stockholm Female Coronary Angiography Study. Journal of Internal Medicine, 261, 245.
Weaver, I.C.G., Cervoni, N., Champagne, F.A., D’Alessio, A.C., Sharma, S., Seckl, J.R., Dymov, S., Szyf, M., and Meaney, M.J. (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7, 847-854.
Welin, L., Larsson, B., Svärdsudd, K., Tibblin, B., and Tibblin, G. (1992). Social network and activities in relation to mortality from cardiovascular diseases, cancer and other causes: A 12 year follow up of the study of men born in 1913 and 1923. Journal of Epidemiology & Community Health, 46, 127-132.
Williams, R.B., Barefoot, J.C., Califf, R.M., Haney, T.L., Saunders, W.B., Pryor, D.B., Hlatky, M.A., Siegler, I.C., and Mark, D.B. (1992) Prognostic importance of social and economic resources among medically treated patients with angiographically documented coronary artery disease. Journal of the American Medical Association, 267, 520-524.
Willner, P., Benton, D., Brown, E., Cheeta, S., Davies, G., Morgan, J., and Morgan, M. (1998). “Depression” increases “craving” for sweet rewards in animal and human models of depression and craving. Psychopharmacology, 136, 272-283.
Xu, C., He, J., Jiang, H., Zu, L., Zhai, W., Pu, S., and Xu, G. (2009). Direct effect of glucocorticoids on lipolysis in adipocytes. Molecular Endocrinology, 23, 1161-1170.
Yamaguchi, N. (1992). Sympathoadrenal system in neuroendocrine control of glucose: Mechanisms involved in the liver, pancreas, and adrenal gland under hemorrhagic and hypoglycemic stress. Canadian Journal of Physiology and Pharmacology, 70, 167-206.
Yatsunenko, T., Rey, F.E., Manary, M.J., Trehan, I., Dominguez-Bello, M.G., Contreras, M., Magris, M., Hidalgo, G., Baldassano, R.N., Anokhin, A.P., et al. (2012). Human gut microbiome viewed across age and geography. Nature, 486, 222-227.
Zakrzewska, K.E., Cusin, I., Stricker-Krongrad, A., Boss, O., Ricquier, D., Jeanrenaud, B., and Rohner-Jeanrenaud, F. (1999). Induction of obesity and hyperleptinemia by central glucocorticoid infusion in the rat. Diabetes, 48, 365-370.






















