
Christopher W. Kuzawa and Dan T.A. Eisenberg
INTRODUCTION
In humans, evolution has produced a species with a lifespan several decades longer than expected for a mammal or primate of its size. Evolutionary principles provide helpful clues to what likely facilitated this outcome (Hawkes et al., 1998; Kaplan et al., 2000). First, human ancestors must have found ways to limit extrinsic mortality sufficient to make it less risky to spread reproductive effort across a long adult life, thus favoring intensive investment in lifespan-extending maintenance effort (Kirkwood and Rose, 1991; Stearns, 1992; Ricklefs, 2008). Second, humans’ social units are unusual in the extent of the reliance upon intergenerational transfers of energy and knowledge from older to younger generations, which selected for increased longevity (Hawkes et al., 1998; Lee, 2003). Reduced mortality relative to other great apes, and the increased reproductive value of later human life stages, are among the forces that worked to favor a strategy of maintaining relatively healthy, functional bodies several decades beyond the ages of direct reproduction.
Although an unusually long lifespan is a defining characteristic of Homo sapiens, prehistoric and historic evidence for trends in human life expectancy, along with contemporary disparities in relation to ethnicity, class, and race (Keppel et al., 2002), demonstrate the enormous impact of environments as influences on heterogeneity in potential longevity among members of the species. Large historical improvements in life expectancy in Europe and elsewhere were due primarily to the control of common in-
fectious diseases, which greatly reduced infant and child survival (Omran, 1971). As one example, increases in life expectancy occurred in the United States as the germ theory of disease gained in acceptance and led to new public health measures like boiling bottles and milk, hand washing, and isolating sick children (Preston, 1996).
As early life infections are controlled in a population and individuals begin living to advanced ages, chronic degenerative processes come to predominate as determinants of mortality and life expectancy. Although it was traditionally believed that such post-demographic and epidemiologic-transition populations would eventually reach a ceiling in life expectancy, long-term demographic data suggest that high-income human populations are not yet approaching this ceiling, if in fact it exists (Oeppen and Vaupel, 2002). This is reflected in the fact that, despite pervasive disparities within societies (Keppel et al., 2002), lifespan in many industrialized nations has been consistently increasing, at a remarkably steady rate, for more than a century (Oeppen and Vaupel, 2002; Tuljapurkar et al., 2000; Vaupel, 2010). These ongoing secular trends in population life expectancy have generated much attention, and there is great interest in understanding their underlying causes. Particularly because the most pronounced changes have come about in a scant four generations, genetic change is an unlikely explanation for them (Burger et al., 2012).
Recent years have witnessed growing interest in a new potential explanation for some of these trends. An extensive literature now supports the hypothesis that part of what determines late-life function and mortality are experiences early in life, starting in utero, and potentially even tracing to the experiences and behaviors of recent generations of ancestors (Gluckman and Hanson, 2006; Kuzawa and Quinn, 2009). Early observational work in humans reported an inverse relationship between birth weight (BW) and future risk of cardiovascular and other chronic diseases, pointing to a likely role of prenatal nutrition in risk for diseases associated with late-life aging and mortality. Importantly, the elevation in cardiovascular disease (CVD) risk is not limited to abnormally low birth weight individuals, but is found in relation to increases in BW across the full BW spectrum, suggesting that the overall quantity of nutrition that a fetus receives may be important. In parallel, an extensive nonhuman literature has replicated many of these findings in model species using experimental designs, demonstrating that early environments, per se, are likely important drivers of many of these effects.
Inspired by this work, research by economists and demographers has been instrumental in demonstrating the likely causality of such relationships in humans (e.g., Almond, 2006; van den Berg et al., 2009). As a result of this work, the idea that long-term health traces in part to early-life experiences is increasingly accepted by researchers across many fields.
While this work has clearly been important, basic scientific understanding of the effects of early environments is rapidly expanding and now routinely demonstrates effects that linger not only into later life, but also that are transmitted to two, three, or even more generations of offspring, operating through several distinct biological inheritance pathways. For biodemography, this work implies that a full understanding of the determinants of human life expectancy, including the vast heterogeneity across and within societies, may require investigating not only the role of early rearing environments, but also historical environments experienced by ancestors, potentially extending back multiple generations.
In this paper, we begin by briefly tracing the history of scientific interest in the early-life determinants of late-life health. Then we review some of the mechanisms now understood to contribute to these relationships. This serves as a backdrop for the second section, in which we explore newer evidence that the effects of early environments can linger beyond adulthood to impact offspring, operating not only through several distinct maternal-offspring pathways, but also through direct parent-offspring epigenetic transfers. These emerging findings add new complexity to understanding of the determinants of aging and life expectancy and inspire a new generation of hypotheses. Our goal is to review the state of evidence for such effects in humans and other species and to outline testable hypotheses for the determinants of human health and life expectancy that emerge from this literature. As we will argue, we believe that demography and related population research fields are well situated to help refine understandings of the pathways for intergenerational transmission of environmental influences on health, which are coming to light through animal model research but have thus far proven challenging to evaluate in human populations.
DEVELOPMENTAL AND INTERGENERATIONAL PLASTICITY
History and Summary of Research Linking Gestational or Infancy Conditions to Adult Health
The hypothesis that undernutrition or early deprivation could compromise adult health was proposed in the early work of Kermack, McKendrick, and McKinlay (1934) documenting birth cohort effects on mortality in England, Sweden, and Wales (see Doblhammer, 2004). In a 1934 Lancet article, they noted that secular trends in mortality were predicted by year of birth and concluded (Kermack et al., 1934, p. 700) that “the expectation of life was determined by the conditions which existed during the child’s earlier years.” Forsdahl (1977, 1978) later provided additional support for this hypothesis from analyses of Norwegian demographic data.
Subsequently, researchers in the United Kingdom observed that the risk of dying from CVD, or of suffering from conditions that precede CVD like hypertension or diabetes, is highest among individuals who were light as newborns, thus linking these late-life outcomes with an early life proxy of nutrition (Barker et al., 1989; Barker, 1994). Human studies have since replicated similar findings relating lower BW to CVD risk factors and CVD mortality in populations across the globe (Leon et al., 1998; Adair et al., 2001; Law et al., 2001; Kuzawa et al., 2003; Yajnik, 2004; Tian et al., 2006; Gupta et al., 2007; Huxley et al., 2007).
Although BW is a complex phenotype, there is considerable evidence that the relationship between birth outcomes and adult chronic disease are not simply due to genetic influences or the result of BW indexing environmental status, which also has persistent effects on chronic disease risk later in life. For instance, among monozygotic twins, the twin born lighter has been shown to have elevated risk for obesity, diabetes, and hypertension later in life (Bo et al., 2000; Iliadou et al., 2004; but see Oberg et al., 2011, for an alternate view), showing that differences in birth size predict adult CVD risk among genetically identical siblings occupying the same postnatal environment. Animal model work provides true experimental tests of the hypothesis that early-life experiences shape future adult health. Such studies have replicated many of the disease outcomes found in relation to lower BW in human populations (Symonds et al., 2003). For instance, restricting the nutritional intake of pregnant rats, mice, or sheep, or directly restricting blood flow to the fetus, increases post-natal blood pressure, cholesterol, abdominal fat deposition, stress reactivity, and diabetes risk in offspring (Kind et al., 2003; Langley-Evans et al., 2003; McMillen and Robinson, 2005; De Blasio et al., 2007).
Developmental and Epigenetic Pathways Linking Early Environments with Adult Health
Several types of biological adjustment are made by the developing fetus in response to prenatal stimuli that contribute to these long-term changes in disease risk. All are examples of developmental plasticity, or the capacity of the developing body to modify its structure and function in response to environmental or behavioral experiences. The most straightforward mechanism of plasticity involves changes in growth of a tissue or organ as reflected in size or cell number. For instance, the kidneys of prenatally undernourished individuals tend to be smaller and have fewer nephrons, a trait that increases risk of hypertension and renal failure in adulthood (Lampl et al., 2002; Luyckx and Brenner, 2005). Similarly, changing the number or type of muscle cells can modify the body’s ability to clear glucose
from the blood stream, leading to changes in insulin sensitivity and diabetes risk (Jensen et al., 2007).
One increasingly well-studied set of mechanisms linking early environments with adult health involves epigenetic changes, defined as chemical modifications that alter gene expression in a specific tissue or organ without changing the nucleotide sequences of the DNA (Jenuwein and Allis, 2001; Waterland and Michels, 2007). Several epigenetic mechanisms have received attention for their likely role as links between early environments and adult health. Chemical modification of histone proteins that the DNA strands are wound around in the cell nucleus can lead to tighter or looser DNA packing in the region of specific genes, reducing or enhancing gene expression respectively. A second molecular mechanism of epigenetic marking involves noncoding RNA (e.g., microRNA or miRNA). These DNA transcripts do not code for proteins, but can influence biology and health by regulating gene expression at other protein-coding genes (Fabian et al., 2010). To date, most attention in human epidemiological studies has focused on a third mechanism, methylation, in which methyl groups are attached to DNA in regions adjacent to specific gene promoters. Although there are exceptions, increased methylation in the vicinity of a gene impedes binding by transcription factors and thereby silences gene expression in that cell (Berger, 2007). All three mechanisms of epigenetic marking are potentially responsive to environmental stimuli and stable for varying timeframes, thereby potentially helping link experiences across the lifecourse with altered biology and health.
Experimental animal model studies confirm that modifying nutritional or other characteristics of early environments can lead to durable epigenetic changes that persist into later life to influence biology and underlying disease processes (Gluckman et al., 2007; Waterland and Michels, 2007). Maternal experiences during or prior to pregnancy have similarly been shown to predict altered epigenetic markings in human offspring, supporting a role for such effects on human biology and disease (Heijmans et al., 2008; Waterland et al., 2010).
In summary, there is solid evidence that the widely documented relationships between early life measures such as BW and later CVD partly reflect the effects of the gestational and infancy environments on the development of biological systems, including effects on how the body manages glucose and lipids, deposits fat, regulates blood pressure, and responds to stress. Each of these is an important determinant of the pace of chronic disease development and thus life expectancy. These effects typically reflect changes in the growth and development of specific organs and tissues or modifications in the regulation of hormones, metabolism, or physiology. They are increasingly being traced to durable, environmentally induced epigenetic changes in the chromosomes that modify gene expression without modifying the DNA sequence.
Pathways for the Intergenerational Transmission of Environmental Effects
There is mounting evidence that environmental effects may be transmitted to future generations operating through several pathways (Drake and Liu, 2010; Daxinger and Whitelaw, 2012; Guerrero-Bosagna and Skinner, 2012; Benyshek, 2013). To date, these intergenerational effects have received little research attention among demographers, despite their potentially large impacts on patterns of health and life expectancy within and between societies.
There are two primary biological pathways by which environmental experiences or behaviors in one generation may impact health and lifespan in future generations. The first involves phenotype-to-phenotype transmission in which the lingering biological effects of early experiences (e.g., fetal programming leading to altered adult metabolism or physiology) impact the next generation by altering the gestational environment or milk composition experienced by offspring (Benyshek et al., 2001; Kuzawa and Sweet, 2009; Drake and Liu, 2010).
As a second pathway, there is now a compelling body of research documenting direct transmission of environmental influence across one or more offspring generations transmitted by epigenetic factors packaged in sperm or egg (Daxinger and Whitelaw, 2012; Guerrero-Bosagna and Skinner, 2012). These studies show that environments can have biological effects that linger across two, three, or more generations of offspring, while also showing that such effects reflect not only maternal but also paternal experience. Taken together, these intergenerational pathways greatly extend the “reach” of past environments as influences on health and aging, and provide new challenges and opportunities for researchers interested in explaining this heterogeneity.
Intergenerational Pathway #1: Phenotypic-to-Phenotype Transmission
Perhaps the most straightforward phenotypic pathways for intergenerational effects involve biological systems that, when modified, directly alter the gestational or lactational environments experienced by the developing fetus or infant. The long-term effects of an adverse gestational environment on traits like insulin resistance, high blood pressure, inflammation, or stress reactivity can all negatively impact the gestational environment experienced by the next generation. There are many such examples of mutually reinforcing early-life and adult biological change. For instance, not only does fetal growth restriction or low BW predict higher adult blood pressure (Adair et al., 2001), but also prepregnancy hypertension in turn leads to fetal growth restriction and lower BW (Kramer et al., 1999). Similarly, inflammation has been shown to be higher among individuals born with a lower BW
(McDade et al., 2010), while adult inflammation is associated with more adverse birth outcomes (Kuzawa et al., 2012).
One well-understood pathway for human phenotype-to-phenotype transmission involves the intergenerational effects of maternal obesity and metabolic dysfunction, which are increasingly important in societies faced with rising obesity rates (see Benyshek, 2013). Fetal growth and fat deposition are under control of hormones that are driven by insulin (Gluckman and Pinal, 2003). Because insulin is stimulated by circulating glucose and other nutrients, maternal circulating glucose levels during pregnancy are strong predictors of offspring BW and adiposity (Metzger et al., 2008). Longitudinal studies have shown that maternal glucose during pregnancy continues to be a strong predictor of adiposity and insulin sensitivity measured in childhood (Chandler-Laney et al., 2011), while adult obesity and diabetes have been linked back to prenatal exposure to gestational diabetes (Benyshek, 2013).
Evidence that maternal overweight can directly impact offspring adiposity and metabolic disease risk is seen in the excess estimated heritability for body mass index observed through mother-offspring pairs compared to father-offspring pairs (Murrin et al., 2012). Although a range of genetic (e.g., mitrochondrial DNA) or environmental explanations for such an effect are possible (Kuzawa and Eisenberg, 2012), among obese mothers electing for gastric bypass surgery to lose weight, offspring born after the surgery have been shown to have dramatically reduced body fat and normalized metabolic parameters when compared to their siblings born prior to the surgery (Smith et al., 2009). This suggests that the metabolic characteristics of the gestational environment associated with an obese mother, per se, are important influences on offspring metabolism and metabolic disease risk. These findings show how any change in glucose metabolism triggered by the prenatal or early post-natal environments that a woman experienced as a fetus or infant may also alter disease risk in her future offspring (Drake and Liu, 2010; Benyshek, 2013).
The effects of the adult phenotype on offspring are by no means limited to physiology and metabolism, and indeed, there are abundant opportunities for parental behavior (itself reflecting a lifetime of experience and development) to influence offspring epigenetic state and development, particularly during early periods of dependence and attachment. As a well-studied example, stress physiology is responsive to social interaction and stress, which can link early experiences with lasting changes in the stress response and downstream traits, including metabolism, memory, and affect regulation (Entringer et al., 2009; Flinn et al., 2011). In humans, maternal exposure to stress during pregnancy predicts changes in stress hormone regulation in early infancy (Lee et al., 2014) and adulthood (Entringer et al., 2009).
Experimental work in rats illustrates how epigenetic changes can contribute to the intergenerational perpetuation of the effects of stress on offspring biology and behavior. In a well-known study, Meaney and colleagues reported that highly attentive rat mothers who exhibit a nurturing grooming style raise pups that, as adults, exhibit changes in gene promoter methylation and histone acetylation that modify hypothalamic–pituitary–adrenal (HPA)-axis function (Weaver et al., 2004). These modifications include heightened negative feedback sensitivity of the stress hormone axis, lower stress hormone levels, and reduced anxiety. Intriguingly, in part as a result of these epigenetic modifications, female offspring are biased towards exhibiting the same style of maternal care that they experienced as pups, thereby contributing to the intergenerational transmission of an environmentally induced behavioral phenotype.
Intergenerational Pathway #2: Direct Germ Line Epigenetic Inheritance
In a second class of nongenetic inheritance, a variety of epigenetic factors capable of directly regulating gene expression are modified in the parent in response to their experiences and then transferred via sperm or egg to offspring (and potentially, grand and great-grandoffspring), among whom they can have lasting impacts on biology and health. In these examples, ancestral environmental experiences may be viewed as having direct access to, and influence over, gene expression in future generations of offspring.
During early development, most epigenetic markings are erased during the formation of gametes (sperm and egg) and again in the embryo prior to implantation (Reik et al., 2001). This epigenetic resetting is believed to be necessary to remove parent-of-origin imprinting at imprinted loci (in which genes are only expressed in offspring if inherited from one or the other parent) along with any accumulated epigenetic modifications or errors (Reik et al., 2001). Early epigenetic reprogramming events are crucial to establishing the cellular totipotency that allows the cells in the early embryo to differentiate into any of the body’s hundreds of cell types. Nonetheless, a growing body of work demonstrates that some environmentally induced epigenetic markings are not erased, but may be maintained in the germ line. This work links exposure to toxins, stressors, nutrients, or other factors to altered epigenetic marking, gene regulation, and phenotypic state across one or more generations of offspring (Daxinger and Whitelaw, 2012; Guerrero-Bosagna and Skinner, 2012).
Of particular interest in this literature are examples of “transgenerational inheritance” in which a phenotype is present despite that generation not having been exposed as a fetus or even as a gamete. In pregnant females (F0), environmental experiences during pregnancy potentially directly affect both the in utero offspring (F1) and their developing gametic precursor cells,
which will later form the F2 generation. Thus, clear evidence for transgenerational epigenetic inheritance in females (F0) requires demonstrating that the phenotype persists to the great-grandoffspring (F3) of the pregnant female. Because there is no male analogue to maternal-fetal phenotype-to-phenotype inheritance, demonstrating an effect in grandoffspring (F2) of males (F0) is evidence for transgenerational inheritance.
Early evidence for such transgenerational epigenetic transmission of environmental effects through the germ line came from animal models involving exposure to toxins and chemical compounds. In rats, exposure to endocrine-disrupting chemicals has been shown to impair the fertility of offspring (F1), operating through both the matriline and patriline (Anway et al., 2005; Nilsson et al., 2012). In the case of males exposed to the endocrine disrupter vinclozilin, male offspring show evidence for reduced sperm quality, attenuated fertility, and a host of related disorders (e.g., pancreatic cancer) for at least four generations (F4) (Anway et al., 2005). When female rats were exposed to similar toxicants during the period of gonadal sex determination, offspring (F1) and great-grandoffspring (the F4 generation) had impaired fertility and ovarian health as indicated by 35-60 percent reductions in primordial follicle counts along with symptoms of early polycystic ovary syndrome (PCOS; Nilsson et al., 2012). Alterations in methylation and gene transcription were found in ovarian cells in the F3 animals, pointing to an epigenetic basis to the phenotype. Because the phenotype persisted to the great-grandoffspring (F3) of the exposed animal in both the male and female experiments above, these studies provide strong evidence of transgenerational epigenetic inheritance, that is, expression of an environmentally induced phenotype in a generation that was entirely unexposed.
In addition to transgenerational effects induced by chemical compounds, a growing list of studies show that “natural” day-to-day exposures, such as psychosocial stress or nutrition, can lead to similar patterns of epigenetic inheritance across one or more generations of offspring. For instance, in an experiment in mice in which maternal separation stress was imposed during the first 2 weeks of post-natal life (Franklin and Mansuy, 2010), methylation was modified in the vicinity of the cannabinoid receptor and corticotrophin-releasing factor receptor in the sperm of the males exposed to stress as newborn pups. A similar pattern of changed methylation and downstream changes in mRNA levels was found in the neurons of female offspring of the stress-exposed males, strongly suggesting direct germ line epigenetic inheritance. Thus, in this example, a stressor experienced by one generation modified the regulation of genes involved in modulating anxiety and stress response in offspring neurons. Another similar study found that chronic (6-week) exposure of male mice to stress either during puberty or in adulthood led to reduced stress hormone (HPA axis) reactivity in offspring (Rodgers et al., 2013), thus suggesting that exposures later in the lifecycle
may also have intergenerational phenotypic effects. In the case of the second mouse experiment, there were large changes in microRNA content in the sperm of the exposed males, pointing to a possible molecular basis for the inherited effect.
Other studies demonstrate epigenetic transmission of the effects of diet on offspring operating through direct germ line inheritance. In one recent experiment (Carone et al., 2010), among male mice fed a low-protein diet, there were large reductions in cholesterol in offspring livers, along with coordinated changes in the expression of genes involved in lipid biosynthesis. The authors found changes in methylation of the PPARα gene, which encodes a transcription factor that regulates expression of multiple genes involved in lipid biosynthesis. This study provides a strong precedent for the concept that diet among males may have intergenerational epigenetic effects on cholesterol and lipid metabolism (and, by extension, cardiometabolic disease risk and longevity) in offspring. As another related example of an intergenerational effect of paternal diet on offspring lipid metabolism, male pigs fed a diet enriched in methyl donors (e.g., folate) had grandoffspring with reduced body fat along with altered gene methylation and gene transcription in the liver (Braunschweig et al., 2012). In a similar study, female offspring of male mice fed a high-fat diet were found to be lighter but more insulin resistant and with reduced beta cell function, contributing to heightened diabetes risk (Ng et al., 2010).
Human Evidence for Germ Line Epigenetic Inheritance of Environmental Effects
Although animal experiments provide important biological precedents illustrating how the environment can impact subsequent generations, the relevance of these findings for understanding human biology and health remains unclear. To date, only a handful of studies provide evidence for possible intergenerational epigenetic environmental inheritance in humans. In a series of well-known studies of a cohort born in 1905 in Överkalix, Northern Sweden (for review, see Pembrey et al., 2013), lifespan was longer when the paternal grandfather experienced poor nutrition during late childhood (9-12 years). Subsequent analyses expanded to three Överkalix birth cohorts and stratified on gender found that paternal grandfather experiences only predicted mortality in grandsons, while paternal grandmother experiences only predicted mortality in granddaughters (Pembrey et al., 2006). They report that findings consistent with this interpretation were present in two of the three cohorts. Specifically, mortality was increased in grandchildren if the corresponding same-sex paternal grandparent was exposed to abundant nutrition during late childhood, compared to individuals exposed to moderate nutrition. In addition, mortality was substantially
reduced in grandoffspring of same-sex paternal grandparents who experienced favorable nutritional conditions from birth to 3 years of age, in this case suggesting an intergenerational benefit of favorable early life nutrition that is passed on to the next generation through sons. This set of findings, which has not been replicated in other populations, provides the strongest human evidence to date for germ-line epigenetic inheritance of nutritional experiences on offspring mortality.
In the same paper (Pembrey et al., 2006), this group used data from the ALSPAC cohort1 in Britain to demonstrate a possible intergenerational effect of paternal smoking on offspring body mass index (BMI). Offspring BMI showed a modest, but significant, graded relationship with the age when the father first smoked, with earlier ages of smoking (all in late childhood) predicting higher BMI in male, but not female offspring. The authors concluded that these findings, viewed alongside those of Överkalix, provide evidence that the “slow-growth period” of late childhood is a critical period in epigenetic programming in humans.
One additional study suggests an intergenerational effect of paternal diet on offspring health. Studies have shown that chronic betel nut chewing can increase risk for obesity, diabetes, and the metabolic syndrome. In a study of more than 5,000 Taiwanese men, offspring of fathers who chewed betel nut had increased risk of the metabolic syndrome (Chen et al., 2006). The effects were dose-dependent and strong, and consistent with prior findings of hyperglycemia in offspring of male mice fed betel nut (Boucher et al., 1994).
Plasticity in Telomere Length as a Potential Transgenerational Influence on Longevity?
The above examples involve transfer of epigenetic marks that modify gene expression without modifying the DNA sequence itself. Recent findings point to an unusual mechanism of plasticity in which experiences and behaviors in prior generations—in particular, mortality and reproduction—alter the length of telomere DNA that offspring inherit. Telomeres are repeating sequences of DNA found at the ends of chromosomes that shorten as cells divide, leading to a decrease in telomere length (TL) with age. When TL becomes too short, the cell lineage is no longer able to replicate, which is thought to contribute to senescence. Correspondingly, individuals with shorter TL in immune cells show compromised immune function and increased mortality rates (Cawthon et al., 2003; Cohen et al., 2013).
_______________
1ALSPAC (Avon Longitudinal Study of Parents and Children), see http://www.bristol.ac.uk/alspac/ [June 2014].
Telomere-based constraints on cell proliferation likely contribute to functional decline in cell-proliferation dependent tissues throughout the body.
Contrary to the telomere shortening that occurs with age in somatic cells, older men have longer sperm TL than younger men, and as such offspring of older fathers inherit longer TL (Kimura et al., 2008). This is believed to result from a high level of a telomere-lengthening enzyme (telomerase) in the testes. We recently demonstrated that this paternal age at conception effect persists across at least two generations: the paternal grandfather’s age at conception of the father predicts the TL of grandchildren, independent of and additive to the effect of the father’s age at his own conception (Eisenberg et al., 2012). These findings raise the intriguing and testable hypothesis that societal trends toward delayed age at male reproduction may themselves lead to the transmission of longer telomeres that contribute to a lengthening of late-life function and life expectancy—and conversely that earlier ages at male reproduction will lead to shorter telomeres (Eisenberg and Kuzawa, 2013).
Evolutionary Significance of Intergenerational Inheritance: Damage or Design?
The above examples show that the biological and health impacts of environments often do not end in adulthood, but may be passed on to offspring generations. These findings thus link the patterns of biology and aging-related chronic disease processes in today’s adults to the environments, experiences, and behaviors of their recent ancestors. This leads to two important questions: Why do these effects exist, and do they have a function?
Some of the lingering effects of early experiences on adult health simply reflect a failure of the mother’s body, or the placenta, to fully buffer the developing body from nutritional or other forms of stress, which can lead to impaired function (Ellison and Jasienska, 2007). For example, babies born smaller tend to have nephron-deficient kidneys that elevate risk for kidney disease and hypertension in adulthood (Lampl et al., 2002; Luyckx and Brenner, 2005). Fetal life is also marked by rapid brain development, and fetal growth restriction can lead to lasting cognitive impairments as reflected in lower childhood or adolescent school achievement (Low et al., 1992). From the perspective of epigenetic inheritance, the transgenerational disease impacts of exposure to artificial toxicants like vinclozilin in rats suggests an error in epigenetic marking that is passed across multiple offspring generations (Anway et al., 2005), as are human examples of the infertility and immune disorders seen in the children of women who were exposed in utero to the synthetic estrogen agonist diethylstilbesterol (Titus-Ernstoff et al., 2006). Whether scenarios such as these reflect maladaptive responses, a shortage of resources leading to developmental “damage,” or adaptive
responses made by the fetus to achieve short-term aims (like surviving gestation), they clearly illustrate that some of the post-natal or adult effects of early environments are nonfunctional by-products that require no adaptive explanation in their own right (Kuzawa and Quinn, 2009).
Other examples are not so straightforward, such as fat deposition, which is a key link between early experience and adult health. Lower BW individuals are more likely to develop CVD, but this is not because they become obese. In fact, they tend to have lower BMIs as adults, but as they gain weight, they preferentially deposit fat in the visceral (abdominal) depot, which is metabolically active as a result of being innervated by sympathetic nerve fibers originating in the brain. When the body experiences stress, these nerves rapidly secrete adrenaline in visceral fat cells, which release fats for use as energy to help the body overcome the stressor. Not only do low-BW individuals deposit more fat in this depot, but also their fat cells mobilize more stored fats when exposed to the same adrenaline dose (Boiko et al., 2005). These changes in fat metabolism are not suggestive of damage. Rather, these findings suggest that the body has a capacity to prioritize depositing fat in a rapidly usable depot in response to early life nutritional stress (Kuzawa, 2010).
Many of the examples of transgenerational epigenetic inheritance discussed above exhibit a level of complexity that is similarly not easily reconciled with a model of simple impairment. Among the findings reviewed was evidence that the diet a male consumes influences how his offspring and grandoffspring metabolize and synthesize nutrients within the body (Carone et al., 2010). Another study showed that the experience of early post-natal separation stress leads to intergenerational epigenetic changes that modify how offspring and grandoffspring respond to similar stressors (Franklin and Mansuy, 2010). Although the specific pathways are only partially understood, such findings are unlikely due to simple impairment or error. In instances such as these, an experience must influence the body and then be communicated in molecular form to the germ line. In cases of transgenerational epigenetic inheritance, any induced changes must survive the normal erasing of germ line epigenetic marks that occurs during gametogenesis and at implantation. In offspring generations, those marks then alter a behavior or biological system that responds to that same environmental stimulus or stressor that initially triggered this cascade several generations prior. The mechanistic basis for such effects is only partially understood in animal models and has scarcely been investigated in humans. But what seems clear is that such examples are improbable and also require a complex multigenerational molecular cascade to sustain. They are unlikely due to chance or damage (Kuzawa and Thayer, 2011).
Because some of the biological changes induced by early-life cues appear to reflect a change in biological regulation, rather than developmental
impairment, it has been speculated that early-life developmental plasticity and epigenetic sensitivity could help the fetus prepare for conditions likely to be experienced after birth (Bateson et al., 2004; Gluckman and Hanson, 2005; Kuzawa, 2005). Some of the adjustments made by the nutritionally stressed fetus in utero, such as a tendency to deposit more abdominal body fat and the glucose-sparing effect of muscle insulin resistance, could help save scarce glucose for use in more essential functions like brain metabolism and immunity (while also heightening risk for diabetes and CVD; Kuzawa, 2010). In addition, other systems that change biological settings in response to early environments, such as stress reactivity (Weaver et al., 2004), immunity (McDade et al., 2010), and reproductive biology (Kuzawa et al., 2010), also hint at capacities to use early-life cues to “fine-tune” how systems function and are regulated. By this reasoning, nutrition, hormones, and other gestational stimuli experienced by the developing fetus might convey information about local ecological conditions, thereby allowing the fetus to adjust priorities in anticipation of these future experiences.
Intergenerational Phenotypic Inertia
One challenge to the hypothesis of long-term anticipatory adaptation comes from the long duration of the human lifespan. Because humans typically live many decades, the ecological conditions experienced during a few months of early development, such as gestation or early infancy, may not serve as reliable cues of environments likely to be experienced in adult life (Kuzawa, 2005). We have argued that it is precisely the brief and early timing of many of the body’s periods of heightened developmental sensitivity that paradoxically helps the developing organism reliably predict its future (Kuzawa and Quinn, 2009). Here the idea is that the mother’s biology buffers the fetus against the vagaries of day-to-day, month-to-month, or seasonal environmental fluctuations, while passing along integrative information about local conditions that is more stable and reliable. Because the mother’s biology and behavior have been modified by her lifetime of experiences, the nutrients and hormones that she transfers to the fetus in utero, or to her infant via breast milk, could correlate with her average experiences more than what she is experiencing during any week or month of gestation itself (Kuzawa, 2005; Wells, 2003).
Evidence for this capacity to convey average, rather than transient, ecological information comes from studies of the effects of a mother’s nutrition on offspring BW (Kuzawa, 2005). While BWs tend to be lighter in populations in which nutrition has been marginal for multiple generations, supplementing the dietary intake of pregnant women often has minimal effects on offspring BW (Kramer and Kakuma, 2003). In contrast, the mother’s body composition and weight prior to conception, reflecting her energy balance in
the years in the run-up to pregnancy, tends to be an important predictor of BW (Institute of Medicine, 1990). Other studies show that the mother’s leg length is among the strongest of predictors of her offspring’s BW (Lawlor et al., 2003; Martin et al., 2004; Chung and Kuzawa, 2014). Because leg growth is more sensitive than other stature components to infancy and childhood nutrition (Frisancho, 2007), this finding is evidence for an intergenerational effect of the mother’s cumulative nutrition during the early years of her own development on the nutrients transferred in utero to her future offspring.
Thus, it appears that long-term nutritional history in an environment may be an important influence on the resources transferred in utero in support of offspring growth, but that fluctuations in intake during pregnancy itself—reflected, for instance, in dietary supplementation—have comparably modest effects. This “phenotypic inertia”—reflecting the lingering biological but nongenetic effects of the mother’s average experiences in the past—could allow the fetus to track those dynamic features of environments that are relatively stable on the timescale of decades or several generations (see Figure 4-1; see Kuzawa, 2005).
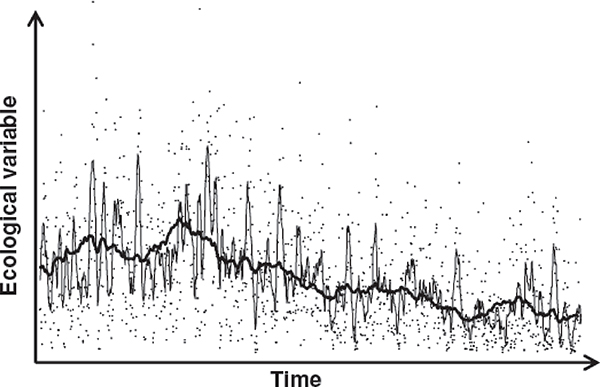
FIGURE 4-1 The intergenerational phenotypic inertia hypothesis.
NOTE: Dots represent the fluctuating availability of a hypothetical ecological resource (e.g., nutrition). The two lines are running averages calculated across 10 time units (thin line) and 100 time units (dark line). As the window of averaging increases, an underlying long-term trend is uncovered. It has been hypothesized that the intergenerational influences of maternal and grandmaternal nutritional history on fetal nutrition help achieve a similar feat.
SOURCE: Kuzawa and Quinn (2009), used with permission.
Above, we reviewed evidence for direct germ line epigenetic inheritance reflecting experiences across multiple generations and through both the matriline and patriline. Although their role in human populations is far less understood, these findings hint at additional means by which the developing body takes multiple samples or assays of local ecological conditions over time, when setting developmental trajectories and biological settings. Because maternal physiology buffers the effects of transient, short-term nutritional fluctuations, these deeper signals may serve as reliable bases for adjusting characteristics such as growth rate, body composition, or nutritional requirements as environmental conditions gradually shift across decades or several generations (Kuzawa and Quinn, 2009). From an applied perspective, the phenotype inertia model underscores the need to envision the goal of interventions as not simply alleviating stress temporarily, but finding creative ways to mimic cues of sustained environmental improvement in the recent past (Kuzawa and Thayer, 2011).
INTERGENERATIONAL PLASTICITY: HYPOTHESES FOR BIODEMOGRAPHY
The pathways for phenotypic and epigenetic inheritance that we reviewed could have substantial impacts on individual and population heterogeneity in morbidity and mortality, including ongoing secular trends in life expectancy documented in high-income populations. Specifically, they suggest that improvements in living conditions, reflecting factors like nutrition, control of infections, or reduced psychosocial stress, will have complex relationships with patterns of health and disease in offspring generations. These pathways should be testable with demographic and cohort databases in which phenotypes are available across two or more generations. To date, the majority of biodemographic research investigating these effects has focused on the late-life effects of stressors experienced during infancy or early childhood, or the role of maternal experience, particularly during pregnancy, on offspring adult outcomes (e.g., Doblhammer and Vaupel, 2001; Almond, 2006; van den Berg et al., 2009; Gagnon, 2012; Gagnon and Bohnert, 2012). The evidence that we reviewed shows that focusing on these lifecourse pathways alone underestimates the potential impact of developmental and epigenetic contributions to health and aging, many of which may have intergenerational and transgenerational components. Building on our review, we next outline testable hypotheses for biodemography inspired by the literature on intergenerational and transgenerational inheritance.
Hypotheses: Phenotype-to-Phenotype Transmission Across Two Generations
In some ways the most straightforward tests of intergenerational effects include examples in which early environments influence adult characteristics that have similar downstream effects on fetal or infant development in the next generation, which alters the gestational environment of offspring and thereby increases risk of these same phenotypes in grandchildren. In such cases, early-life and adult female phenotypes may reinforce each other across generations (Drake and Liu, 2010). Environmental or lifestyle change could thus drive not only phenotypic transmission, but also cumulative intergenerational phenotypic change (Benyshek, 2013). Although there are other possible interpretations (see Kuzawa and Eisenberg, 2012), excess estimated heritability of the phenotype through the matriline than through the patriline would be evidence consistent with matrilineal phenotype-to-phenotype transmission. In contrast to a conventional gene X environment interaction model, in which each generation only inherits its genotype from the prior generation, this model assumes that the phenotype is partly heritable as well, and, thus, a different pattern of phenotypic development within and across generations can be expected (see Figure 4-2).
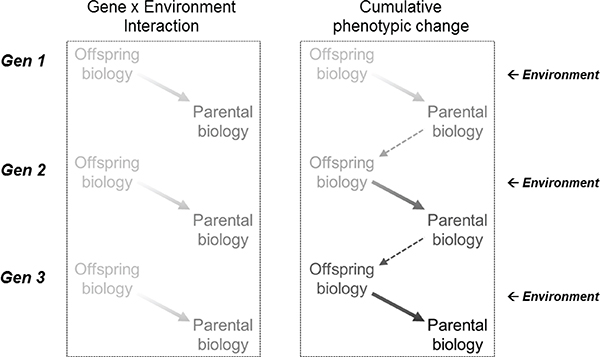
FIGURE 4-2 Conventional gene x environment model of phenotype and a cumulative phenotypic change model.
Hypotheses: Direct Germ Line Epigenetic Inheritance
Examples of germ line epigenetic inheritance do not involve a direct effect of an altered phenotype on offspring and thus can be transmitted through both the matriline and patriline. It is important to note that the relative abundance of animal model studies reporting patrilineal inheritance speaks more to the simplicity of demonstrating transgenerational epigenetic inheritance in males (among whom gestational, lactational, and other direct phenotype-to-phenotype pathways are not present) than to any inherently stronger effects through the patriline.
As we reviewed above, there is now evidence for epigenetic inheritance of phenotypes through both the matriline and patriline that is truly transgenerational, meaning that the phenotype is present in a generation that was never exposed (even as germ cells in their in utero parents). Animal model studies are providing some clues about the likely timing of critical periods. In male and female rats, true transgenerational inheritance has been demonstrated when the environmental factor is experienced during gametogenesis (soon after conception), which is a critical period in the establishment of epigenetic marks in both sexes. In contrast, exposures during spermatogenesis (adulthood) have been shown to only have effects on the next generation (intergenerational inheritance). It is presently not clear whether similar critical periods apply to humans, but these findings represent the best starting points for hypothesis testing with human databases. These findings lead to the prediction that exposures or stressors around the time of conception will have the potential to influence multiple generations of offspring. If a pregnant woman experiences a stressor at this time, her fetus and the fetus’s gametes (future grandoffspring) may also be exposed. Thus, demonstrating the induced phenotype in great-grandoffspring of the originally exposed pregnancy generation would be necessary to demonstrate transgenerational epigenetic inheritance of the phenotype with more confidence. On the other hand, if a male is exposed in utero or after birth and the phenotype persists to grandoffspring, this would be strong evidence for transgenerational epigenetic inheritance.
Hypotheses: Replicating and Extending the Överkalix Findings
The Swedish Överkalix findings provide among the best evidence for transgenerational epigenetic inheritance in humans. Despite this, this pattern of findings is limited to a single population, and we are aware of no attempts to replicate them. The Överkalix findings lead to the prediction that favorable nutrition during the prepubertal “slow-growth period” of late childhood will increase risk for chronic disease, and reduce life expectancy, among same-sexed grandoffspring. In addition, if the paternal grand-
mother (PGM) experienced favorable early life nutrition (birth ~3 years) granddaughters had improved health and survival, pointing to possible epigenetic transmission of early grandmaternal experience that is relayed through her son to her granddaughter (i.e., is not phenotype-to-phenotype transmission).
The sex-specificity of the Överkalix findings, wherein the childhood experience of PGM only shows an association with granddaughters’ (GDs) disease risk and paternal grandfathers’ (PGFs) late childhood experience only show an association with grandsons’ (GSs) disease risk, is puzzling. Extending the adaptive logic of intergenerational inertia (see above), it is possible (although speculative) that same-sex ancestors’ environmental experience is more predictive of the environment a descendant will experience than those of opposite-sex ancestors. This adaptationist perspective leads to the prediction that maternal grandparents experience should similarly influence same-sex offspring but not opposite-sex offspring. To the extent that this is true, the pattern might be more extreme in highly sexually dimorphic species among whom each sex occupies more discrete ecological niches and/or in which reproductive strategies differ more.
Pembrey and colleagues however suggest an alternative explanation for the same-sex ancestors finding—mainly that they are mediated by X- and Y-chromosome2 linked epigenetic mechanisms (Grossniklaus et al., 2013). The unusual inheritance of the sex chromosomes (males are XY and inherit one X chromosome from their mother and one Y chromosome from their father, whereas girls are XX and inherit one X from each of their parents) leads to some specific, testable hypotheses about the nature and strength of transgenerational effects in humans. PGMs pass on ~50 percent of their two X chromosomes (equivalent to one X chromosome) to their GDs and none to their GSs. In contrast, PGFs do not pass on their X chromosomes to their sons, GDs, or GSs at all, but do have a 100 percent certainty of passing on their Y chromosome to their sons and GSs and 0 percent certainty of passing their Y chromosome along to their GDs.
On the matrilineal side, maternal grandmothers (MGM) pass on ~25 percent of their two X chromosomes (equivalent to half an X chromosome) to their GS or GD, and maternal grandfathers (MGF) pass on 0 percent of their Y chromosomes to GS or GD but ~50 percent of their one X chromosome to their GS or GD (see Figure 1 in Fox, 2009, for an illustration). These mechanistic peculiarities of the mammalian sex inheritance/determination patterns, together with Pembrey and colleagues’ hypothesis, lead to a very particular set of predictions about the magnitude of effects if additively mediated via X-chromosomes: (1) MGM and MGF effects on
_______________
2When we refer to Y-chromosome here, we are only referring to the nonrecombining portion of the physical Y-chromosome (which makes up ~ 95 percent of the physical Y-chromosome).
grandchildren should be equal and (2) effect GS and GD equally; however, (3) MGM/MGF effects on grandchildren will be half that of PGM effects on GD, (4) PGM will have no effect on GSs, and (5) PGF will have no effect on GSs or GDs.
We note that there is also some evidence that grandmothers provide care to grandoffspring in a fashion predicted by their degree of sex-chromosome relatedness (Fox et al., 2010). It is thus possible that differential grandparental care is a mediator of the Överkalix findings. One test for this hypothesis would involve an examination of whether the grandparent was alive and living nearby the grandchild (e.g., same village or household) as a marker of the potential provisioning of care. Care-mediated effects will only be evident with grandparents who live nearby grandchildren, while epigenetic effects should be invariant to this.3
Hypotheses: Intergenerational Phenotypic Inertia
The phenotypic inertia model, discussed above, leads to several testable hypotheses. This model proposes that fetal development is buffered against transient ecological fluctuations, allowing transfer of cues reflecting more stable average trends. Focusing on the determinants of fetal nutrition and growth, the model leads to the hypothesis that offspring birth outcomes will be relatively refractory to the mother’s macronutrient nutrition during pregnancy, but will relate to chronic nutrition as experienced across several matrilineal generations.
Because what is “transient” or “stable” for individual organisms is inherently relative, the model also leads to the prediction that species will vary in the magnitude of response to similar maternal pregnancy stimuli depending on generation time or lifespan. Because humans typically live decades rather than months or years, this implies that the types and magnitudes of environmental change that are relevant when orienting an individual human life will be different than for a member of a short-lived species. Consistent with this prediction, we recently showed that offspring of longer-lived species tend to experience comparably small changes in BW and adult disease risk in response to maternal diet restriction in pregnancy compared to offspring of smaller and shorter-lived species (see Kuzawa and Thayer, 2011).
Hypotheses: Paternal Age at Conception Effect on Telomere Length
The finding of cumulative, multigenerational increases in offspring telomere length in relation to delayed paternal and paternal grandfather ages
_______________
3Future studies should also consider possible bias from nonpaternity or paternal/grandparental uncertainty.
at conception leads to relatively straightforward and testable hypotheses (Eisenberg et al., 2012): that telomere length and thus late-life function will be positively and cumulatively related to age at conception of direct patrilineal ancestors. At present there is evidence from one small study that these effects are cumulative across two generations (Eisenberg et al., 2012), but no study has looked for effects beyond this. Historical databases would provide an ideal means of testing these effects across multiple generations. Because telomeres are believed to influence health primarily by limiting cell division, it will be important to test this model using an appropriate endpoint that is plausibly downstream of shortened telomeres, such as infectious disease or cancer secondary to impaired immunity (Eisenberg, 2011).
Hypotheses: How Important Is Selection?
As we have emphasized in this review, there is compelling evidence that a variety of nongenetic mechanisms can link early-life environments with adult phenotype and the phenotypes of successive generations. However, to date, few studies have considered the potential role of early-life mortality/selection effects, which may contribute to some of these findings. The probability of human conception under ideal circumstances is estimated at only 33 percent (Wilcox et al., 1995). After conception, pregnancy loss rates (many silent) are estimated to be 55 percent for 20-year-old women and to increase progressively with age to 84 percent at age 30 and 96 percent at age 40 (Holman and Wood, 2001). Many of these fetal losses are believed to arise from chromosomal abnormalities, which are unlikely to account for the types of phenotypic variation seen in studies of early origins or intergenerational effects. However, environmental chemicals, endocrine factors including stress hormone levels, infections, and other immunological causes are also implicated in fetal loss (Holman and Wood, 2001; Nepomnaschy et al., 2006). If there are genotypes that, under some environmental conditions, have a higher likelihood of being conceived and/or not aborted and have pleiotropic effects on later-life health, this would be difficult to distinguish from some of the postulated nongenetic inheritance patterns.
Evolutionary models point to the likelihood of selective fetal loss based on the phenotype of the conceptus and maternal environment (reviewed in Haig, 2008). Empirical evidence for conception/prenatal selection in humans comes, for instance, from evidence that some genotypes vary depending upon season and photoperiod at birth (Lucock et al., 2010; Gonda et al., 2012) and evidence for increased fetal loss of male conceptuses in poor environmental conditions (Bruckner and Catalano, 2007). An expanding list of genes that pleiotropically influence early- and late-life phenotypes and could lead to selection of this sort are being discovered. For instance, alleles in the IGF1 and ADCY5 genes have been associated with both low
BW and type 2 diabetes risk (Vaessen et al., 2002; Horikoshi et al., 2013). Thus, selective mortality and survival as an influence on fetal loss should be considered as a potential explanation in studies linking early environments with adult health outcomes. This is a potential issue not only in observational research, but also in quasi-experiments and laboratory based experiments. If such selection effects are found to mediate some of the associations between parental experiences and offspring health, it will be important to consider whether this reflects evolved adaptive mechanisms to improve the reproductive success of the parent or nonadaptive constraints.
CONCLUDING POINTS
Among early biologists, including Darwin, there was a prominent and uncritical assumption that environmental experience led to biological changes that were transmitted to offspring (now widely referred to as “Lamarckian inheritance”). In response to these unsubstantiated assumptions, and in the wake of subsequent insights into the genetic basis of heredity, mainstream biology adopted a firm view that environments do not meaningfully influence the phenotype of descendants except via conventional genetic inheritance. The research that we review is posing important challenges to these assumptions, for it illustrates a range of molecular pathways by which environments in one generation can impact the biology of one or more generations of offspring (Jablonka and Lamb, 2005). There is a long research tradition demonstrating the importance of phenotypic and developmental plasticity to human adaptation (Lasker, 1969). Recent findings of inter- and transgenerational epigenetic inheritance are revealing new levels of complexity in the nature of human developmental and epigenetic adaptations to environmental change (Kuzawa and Bragg, 2012).
Despite these recent advances, much more is known about the nature and health effects of these responses in animal model species than in humans, and indeed, there is only minimal evidence for transgenerational epigenetic inheritance in human populations. As we emphasize in our review, the likely pathways of environmental inheritance are not limited to the better-studied effects linking early or gestational environments to adult health, but can also include patrilineal and matrilineal effects that linger across one or more offspring generations. These are inherently challenging to study in a long-lived species such as humans, for they require information on environments and experiences across multiple generations, with extreme care taken to minimize confounding and other factors that constrain causal inference.
Given these challenges, biodemographic research, and population research more generally, is well situated to advance an understanding of the basic biology of nongenetic inheritance and its impacts on human longevity.
Strengths of this field, including access to historical databases and multigeneration cohorts, a focus on exogenous shocks and quasi-experimental designs, and careful attention to issues of selection and endogeneity, will be critical to better understand how current health reflects not only one’s own experiences, but those recent ancestors. We hope that this review, and the hypotheses that we outline, will help inspire new studies focused on exploring these fascinating effects and their impacts on human aging and longevity.
ACKNOWLEDGMENTS
Michael Skinner, Marcus Pembrey, and Zaneta Thayer provided references or conversations that benefitted the manuscript.
REFERENCES
Adair, L.S., Kuzawa, C.W., and Borja, J. (2001). Maternal energy stores and diet composition during pregnancy program adolescent blood pressure. Circulation, 104(9), 1034-1039.
Almond, D. (2006). Is the 1918 Influenza pandemic over? Long-term effects of in utero Influenza exposure in the post-1940 US population. Journal of Political Economy, 114(4), 672-712.
Anway, M.D., Cupp, A.S., Uzumcu, M., and Skinner, M.K. (2005). Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science, 308(5727), 1466-1469.
Barker, D.J. (1994). Mothers, Babies, and Disease in Later Life. London, UK: BMJ.
Barker, D.J., Osmond, C., Golding, J., Kuh, D., and Wadsworth, M.E. (1989). Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. British Medical Journal, 298(6673), 564-567.
Bateson, P., Barker, D., Clutton-Brock, T., Deb, D., D’Udine, B., Foley, R.A., Gluckman, P., Godfrey, K., Kirkwood, T., Lahr, M.M., et al. (2004). Developmental plasticity and human health. Nature, 430(6998), 419-421.
Benyshek, D.C. (2013). The “early life” origins of obesity-related health disorders: New discoveries regarding the intergenerational transmission of developmentally programmed traits in the global cardiometabolic health crisis. American Journal of Physical Anthropology, 152(Suppl. 57), 79-93.
Benyshek, D.C., Martin, J.F., and Johnston, C.S. (2001). A reconsideration of the origins of the type 2 diabetes epidemic among Native Americans and the implications for intervention policy. Medical Anthropology, 20(1), 25-64.
Berger, S.L. (2007). The complex language of chromatin regulation during transcription. Nature, 447(7143), 407-412.
Bo, S., Cavallo-Perin, P., Scaglione, L., Ciccone, G., and Pagano, G. (2000). Low birthweight and metabolic abnormalities in twins with increased susceptibility to Type 2 diabetes mellitus. Diabetic Medicine, 17(5), 365-370.
Boiko, J., Jaquet, D., Chevenne, D., Rigal, O., Czernichow, P., and Levy-Marchal, C. (2005). In situ lipolytic regulation in subjects born small for gestational age. International Journal of Obesity, 29(6), 565-570.
Boucher, B.J., Ewen, S.W., and Stowers, J.M. (1994). Betel nut (Areca catechu) consumption and the induction of glucose intolerance in adult CD1 mice and in their F1 and F2 offspring. Diabetologia, 37(1), 49-55.
Braunschweig, M., Jagannathan, V., Gutzwiller, A., and Bee, G. (2012). Investigations on transgenerational epigenetic response down the male line in F2 pigs. PLOS ONE, 7(2), e30583.
Bruckner, T., and Catalano, R. (2007). The sex ratio and age-specific male mortality: Evidence for culling in utero. American Journal of Human Biology, 19(6), 763-773.
Burger, O., Baudisch, A., and Vaupel, J.W. (2012). Human mortality improvement in evolutionary context. Proceedings of the National Academy of Sciences of the United States of America, 109(44), 18210-18214.
Carone, B.R., Fauquier, L., Habib, N., Shea, J.M., Hart, C.E., Li, R., Bock, C., Li, C., et al. (2010). Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell, 143(7), 1084-1096.
Cawthon, R.M., Smith, K.R., O’Brien, E., Sivatchenko, A., and Kerber, R.A. (2003). Association between telomere length in blood and mortality in people aged 60 years or older. The Lancet, 361(9355), 393-395.
Chandler-Laney, P.C., Bush, N.C., Rouse, D.J., Mancuso, M.S., and Gower, B.A. (2011). Maternal glucose concentration during pregnancy predicts fat and lean mass of prepubertal offspring. Diabetes Care, 34(3), 741-745.
Chen, T.H., Chiu, Y.H., and Boucher, B.J. (2006). Transgenerational effects of betel-quid chewing on the development of the metabolic syndrome in the Keelung Community-based Integrated Screening Program. The American Journal of Clinical Nutrition, 83(3), 688-692.
Chung, G., and Kuzawa, C. (2014). Intergenerational effects of early life nutrition: Maternal leg length predicts offspring placental weight and birth weight among women in rural Luzon, Philippines. American Journal of Human Biology. doi: 10.1002/ajhb.22579. Available: http://onlinelibrary.wiley.com [July 2014].
Cohen, S., Janicki-Deverts, D., Turner, R.B., Casselbrant, M.L., Li-Korotky, H.S., Epel, E.S., and Doyle, W.J. (2013). Association between telomere length and experimentally induced upper respiratory viral infection in healthy adults. Journal of the American Medical Association, 309(7), 699-705.
Daxinger, L., and Whitelaw, E. (2012). Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nature Reviews Genetics, 13(3), 153-162.
De Blasio, M.J., Dodic, M., Jefferies, A.J., Moritz, K.M., Wintour, E.M., and Owens, J.A. (2007). Maternal exposure to dexamethasone or cortisol in early pregnancy differentially alters insulin secretion and glucose homeostasis in adult male sheep offspring. American Journal of Physiology Endocrinology and Metabolism, 293(1), E75-E82.
Doblhammer, G. (2004). The Late Life Legacy of Very Early Life. Berlin, Germany: Springer-Verlag.
Doblhammer, G., and Vaupel, J.W. (2001). Lifespan depends on month of birth. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2934-2939.
Drake, A.J., and Liu, L. (2010). Intergenerational transmission of programmed effects: Public health consequences. Trends in Endocrinology & Metabolism, 21(4), 206-213.
Eisenberg, D.T. (2011). An evolutionary review of human telomere biology: The thrifty telomere hypothesis and notes on potential adaptive paternal effects. American Journal of Human Biology, 23(2), 149-167.
Eisenberg, D.T., and Kuzawa, C.W. (2013). Commentary: The evolutionary biology of the paternal age effect on telomere length. International Journal of Epidemiology, 42(2), 462-465.
Eisenberg, D.T., Hayes, M.G., and Kuzawa, C.W. (2012). Delayed paternal age of reproduction in humans is associated with longer telomeres across two generations of descendants. Proceedings of the National Academy of Sciences of the United States of America, 109(26), 10251-10256.
Ellison, P.T., and Jasienska, G. (2007). Constraint, pathology, and adaptation: How can we tell them apart? American Journal of Human Biology, 19(5), 622-630.
Entringer, S., Kumsta, R., Hellhammer, D.H., Wadhwa, P.D., and Wust, S. (2009). Prenatal exposure to maternal psychosocial stress and HPA axis regulation in young adults. Hormones and Behavior, 55(2), 292-298.
Fabian, M.R., Sonenberg, N., and Filipowicz, W. (2010). Regulation of mRNA translation and stability by microRNAs. Annual Review of Biochemistry, 79, 351-379.
Flinn, M.V., Nepomnaschy, P.A., Muehlenbein, M.P., and Ponzi, D. (2011). Evolutionary functions of early social modulation of hypothalamic-pituitary-adrenal axis development in humans. Neuroscience & Biobehavioral Reviews, 35(7), 1611-1629.
Forsdahl, A. (1977). Are poor living conditions in childhood and adolescence an important risk factor for arteriosclerotic heart disease? British Journal of Preventive & Social Medicine, 31(2), 91-95.
Forsdahl, A. (1978). Living conditions in childhood and subsequent development of risk factors for arteriosclerotic heart disease. The cardiovascular survey in Finnmark 1974-75. Journal of Epidemiology and Community Health, 32(1), 34-37.
Fox, M., Sear, R., Beise, J., Ragsdale, G., Voland, E., and Knapp, L.A. (2010). Grandma plays favourites: X-chromosome relatedness and sex-specific childhood mortality. Proceedings Biological Sciences/The Royal Society, 277(1681), 567-573.
Franklin, T.B., and Mansuy, I.M. (2010). Epigenetic inheritance in mammals: Evidence for the impact of adverse environmental effects. Neurobiology of Disease, 39(1), 61-65.
Frisancho, A.R. (2007). Relative leg length as a biological marker to trace the developmental history of individuals and populations: Growth delay and increased body fat. American Journal of Human Biology, 19(5), 703-710.
Gagnon, A. (2012). Effect of birth season on longevity: Thrifty and hopeful phenotypes in historical Quebec. American Journal of Human Biology, 24(5), 654-660.
Gagnon, A., and Bohnert, N. (2012). Early life socioeconomic conditions in rural areas and old-age mortality in twentieth-century Quebec. Social Science & Medicine, 75(8), 1497-1504.
Gluckman, P.D., and Hanson, M.A. (2005). The Fetal Matrix: Evolution, Development, and Disease. New York: Cambridge University Press.
Gluckman, P.D., and Hanson, M.A. (2006). Developmental Origins of Health and Disease. Cambridge, UK: Cambridge University Press.
Gluckman, P.D., and Pinal, C.S. (2003). Regulation of fetal growth by the somatotrophic axis. Journal of Nutrition, 133(5, Suppl. 2), 1741S-1746S.
Gluckman, P.D., Hanson, M.A., and Beedle, A.S. (2007). Non-genomic transgenerational inheritance of disease risk. Bioessays, 29(2), 145-154.
Gonda, X., Fountoulakis, K.N., Csukly, G., Dome, P., Sarchiapone, M., Laszik, A., Bedi, K., Juhasz, G., Siamouli, M., Rudisch, T., et al. (2012). Star-crossed? The association of the 5-HTTLPR s allele with season of birth in a healthy female population, and possible consequences for temperament, depression and suicide. Journal of Affective Disorders, 143(1-3), 75-83.
Grossniklaus, U., Kelly, W.G., Ferguson-Smith, A.C., Pembrey, M., and Lindquist, S. (2013). Transgenerational epigenetic inheritance: How important is it? Nature Reviews Genetics, 14(3), 228-235.
Guerrero-Bosagna, C., and Skinner, M.K. (2012). Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Molecular and Cellular Endocrinology, 354(1-2), 3-8.
Gupta, M., Gupta, R., Pareek, A., Bhatia, R., and Kaul, V. (2007). Low birth weight and insulin resistance in mid and late childhood. Indian Pediatrics, 44(3), 177-184.
Haig, D. (2008). Intimate relations: Evolutionary conflict of pregnancy and childhood. In S.C. Stearns and J.C. Koella (Eds.), Evolution in Health and Disease. 2nd ed. (pp. 65-76). New York: Oxford University of Press.
Hawkes, K., O’Connell, J.F., Jones, N.G., Alvarez, H., and Charnov, E.L. (1998). Grandmothering, menopause, and the evolution of human life histories. Proceedings of the National Academy of Sciences of the United States of America, 95(3), 1336-1339.
Heijmans, B.T., Tobi, E.W., Stein, A.D., Putter, H., Blauw, G.J., Susser, E.S., Slagboom, P.E., and Lumey, L.H. (2008). Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences of the United States of America, 105(44), 17046-17049.
Holman, D.J., and Wood, J.W. (2001). Pregnancy loss and fecundability in women. In P.T. Ellison (Ed.), Reproductive Ecology and Human Evolution (pp. 15-38). New Brunswick, NJ: Transaction.
Horikoshi, M., Yaghootkar, H., Mook-Kanamori, D.O., Sovio, U., Taal, H.R., Hennig, B.J., Bradfield, J.P., St. Pourcain, B., Evans, D.M,. Charoen, P., et al. (2013). New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nature Genetics, 45(1), 76-82.
Huxley, R., Owen, C.G., Whincup, P.H., Cook, D.G., Rich-Edwards, J., Smith, G.D., and Collins, R. (2007). Is birth weight a risk factor for ischemic heart disease in later life? The American Journal of Clinical Nutrition, 85(5), 1244-1250.
Iliadou, A., Cnattingius, S., and Lichtenstein, P. (2004). Low birthweight and Type 2 diabetes: A study on 11 162 Swedish twins. International Journal of Epidemiology, 33(5), 948-953.
Institute of Medicine. (1990). Nutrition During Pregnancy. Committee on Nutritional Status During Pregnancy and Lactation. Washington, DC: National Academy Press.
Jablonka, E., and Lamb, M.J. (2005). Evolution in Four Dimensions: Genetic, Epigenetic, Behavioral, and Symbolic Variation in the History of Life. Cambridge, MA: MIT Press.
Jensen, C.B., Storgaard, H., Madsbad, S., Richter, E.A., and Vaag, A.A. (2007). Altered skeletal muscle fiber composition and size precede whole-body insulin resistance in young men with low birth weight. The Journal of Clinical Endocrinology and Metabolism, 92(4), 1530-1534.
Jenuwein, T., and Allis, C.D. (2001). Translating the histone code. Science, 293(5532), 1074-1080.
Kaplan, H., Hill, K., Lancaster, J., and Hurtado, A.M. (2000). A theory of human life history evolution: Diet, intelligence and longevity. Evolutionary Anthropology: Issues, News, and Reviews, 9, 156-185.
Keppel, K.G., Pearcy, J.N., and Wagener, D.K. (2002). Trends in racial and ethnic-specific rates for the health status indicators: United States, 1990-98. Healthy People 2000 Statistical Notes/National Center for Health Statistics, 23, 1-16.
Kermack, W., McKendrick, A., and McKinlay, P. (1934). Death rates in Great Britain and Sweden: Some general regularities and their significance. Lancet, 226, 698-703.
Kimura, M., Cherkas, L.F., Kato, B.S., Demissie, S., Hjelmborg, J.B., Brimacombe, M., Cupples, A., Hunkin, J.L., Gardner, J.P., Lu, X., et al. (2008). Offspring’s leukocyte telomere length, paternal age, and telomere elongation in sperm. PLOS Genetics, 4(2), e37.
Kind, K.L., Clifton, P.M., Grant, P.A., Owens, P.C., Sohlstrom, A., Roberts, C.T., Robinson, J.S., and Owens, J.A. (2003). Effect of maternal feed restriction during pregnancy on glucose tolerance in the adult guinea pig. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 284(1), R140-R152.
Kirkwood, T.B., and Rose, M.R. (1991). Evolution of senescence: Late survival sacrificed for reproduction. Philosophical Transactions of the Royal Society B: Biological Sciences, 332(1262), 15-24.
Kramer, M.S., and Kakuma, R. (2003). Energy and protein intake in pregnancy. Cochrane Database of Systematic Reviews, 4, CD000032.
Kramer, M.S., Platt, R., Yang, H., McNamara, H., and Usher, R.H. (1999). Are all growth-restricted newborns created equal(ly)? Pediatrics, 103(3), 599-602.
Kuzawa, C.W. (2005). Fetal origins of developmental plasticity: Are fetal cues reliable predictors of future nutritional environments? American Journal of Human Biology, 17(1), 5-21.
Kuzawa, C.W. (2010). Beyond feast-famine: Brain evolution, human life history, and the metabolic syndrome. In M. Muehlenbein (Ed.), Human Evolutionary Biology (pp. 518-527). Cambridge, UK: Cambridge University Press.
Kuzawa, C.W., and Bragg, J.M. (2012). Plasticity in human life history strategy: Implications for contemporary human variation and the evolution of genus Homo. Current Anthropology, 53(Suppl. 6), S369-S382.
Kuzawa, C.W., and Eisenberg, D.T. (2012). Intergenerational predictors of birth weight in the Philippines: Correlations with mother’s and father’s birth weight and test of maternal constraint. PLOS ONE, 7(7), e40905.
Kuzawa, C.W., and Sweet, E. (2009). Epigenetics and the embodiment of race: Developmental origins of US racial disparities in cardiovascular health. American Journal of Human Biology, 21(1), 2-15.
Kuzawa, C.W, and Thayer, Z. (2011). Timescales of human adaptation: The role of epigenetic processes. Epigenomics, 3(2), 221-234.
Kuzawa, C.W., and Quinn, E. (2009). Developmental origins of adult function and health: Evolutionary hypotheses. Annual Review of Anthropology, 38, 131-147.
Kuzawa, C.W., Adair, L.S., Avila, J.L., Cadungog, J.H., and Le, N.A. (2003). Atherogenic lipid profiles in Filipino adolescents with low body mass index and low dietary fat intake. American Journal of Human Biology, 15(5), 688-696.
Kuzawa, C.W., McDade, T.W., Adair, L.S., and Lee, N. (2010). Rapid weight gain after birth predicts life history and reproductive strategy in Filipino males. Proceedings of the National Academy of Sciences of the United States of America, 107(39), 16800-16805.
Kuzawa, C.W., Tallman, P.S., Adair, L.S., Lee, N., and McDade, T.W. (2012). Inflammatory profiles in the non-pregnant state predict offspring birth weight at Cebu: Evidence for inter-generational effects of low grade inflammation. Annals of Human Biology, 39(4), 267-274.
Lampl, M., Kuzawa, C.W., and Jeanty, P. (2002). Infants thinner at birth exhibit smaller kidneys for their size in late gestation in a sample of fetuses with appropriate growth. American Journal of Human Biology, 14(3), 398-406.
Langley-Evans, S.C., Langley-Evans, A.J., and Marchand, M.C. (2003). Nutritional programming of blood pressure and renal morphology. Archives of Physiology and Biochemistry, 111(1), 8-16.
Lasker, G. (1969). Human biological adaptability: The ecological approach in physical anthropology. Science, 166, 1480-1486.
Law, C.M., Egger, P., Dada, O., Delgado, H., Kylberg, E., Lavin, P., Tang, G.H., von Hertzen, H., Shiell, A.W., and Barker, D.J. (2001). Body size at birth and blood pressure among children in developing countries. International Journal of Epidemiology, 30(1), 52-57.
Lawlor, D.A., Davey Smith, G., and Ebrahim S. (2003). Association between leg length and offspring birthweight: Partial explanation for the trans-generational association between birthweight and cardiovascular disease: Findings from the British Women’s Heart and Health Study. Paediatric and Perinatal Epidemiology, 17(2), 148-155.
Lee, J., Fried, R., Thayer, Z., and Kuzawa, C.W. (2014). Preterm delivery as a predictor of diurnal cortisol profiles in adulthood: Evidence from Cebu, Philippines. American Journal of Human Biology. doi: 10.1002/ajhb.22569. Available: http://www.ncbi.nlm.nih.gov/pubmed/24898414 [August 2014].
Lee, R.D. (2003). Rethinking the evolutionary theory of aging: Transfers, not births, shape senescence in social species. Proceedings of the National Academy of Sciences of the United States of America, 100(16), 9637-9642.
Leon, D.A., Lithell, H.O., Vagero, D., Koupilova, I., Mohsen, R., Berglund, L., Lithell, U.B., and McKeigue, P.M. (1998). Reduced fetal growth rate and increased risk of death from ischaemic heart disease: Cohort study of 15 000 Swedish men and women born 1915-29. British Medical Journal, 317(7153), 241-245.
Low, J.A., Handley-Derry, M.H., Burke, S.O., Peters, R.D., Pater, E.A., Killen, H.L., and Derrick, E.J. (1992). Association of intrauterine fetal growth retardation and learning deficits at age 9 to 11 years. American Journal of Obstetrics and Gynecology, 167(6), 1499-1505.
Lucock, M., Glanville, T., Ovadia, L., Yates, Z., Walker, J., and Simpson, N. (2010). Photoperiod at conception predicts C677T-MTHFR genotype: A novel gene-environment interaction. American Journal of Human Biology, 22(4), 484-489.
Luyckx, V.A., and Brenner, B.M. (2005). Low birth weight, nephron number, and kidney disease. Kidney International. Supplement, 97, S68-S77.
Martin, R.M., Smith, G.D., Frankel, S., and Gunnell, D. (2004). Parents’ growth in childhood and the birth weight of their offspring. Epidemiology, 15(3), 308-316.
McDade, T.W., Rutherford, J., Adair, L., and Kuzawa, C.W. (2010). Early origins of inflammation: Microbial exposures in infancy predict lower levels of C-reactive protein in adulthood. Proceedings Biological Sciences/The Royal Society, 277(1684), 1129-1137.
McMillen, I.C., and Robinson, J.S. (2005). Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiological Reviews, 85(2), 571-633.
Metzger, B.E., Lowe, L.P., Dyer, A.R., Trimble, E.R., Chaovarindr, U., Coustan, D.R., Hadden, D.R., McCance, D.R., Hod, M., McIntyre, H.D., et al. (2008). Hyperglycemia and adverse pregnancy outcomes. The New England Journal of Medicine, 358(19), 1991-2002.
Murrin, C.M., Kelly, G.E., Tremblay, R.E., and Kelleher, C.C. (2012). Body mass index and height over three generations: Evidence from the Lifeways Cross-Generational Cohort Study. BMC Public Health, 12, 81.
Nepomnaschy, P.A., Welch, K.B., McConnell, D.S., Low, B.S., Strassmann, B.I., and England, B.G. (2006). Cortisol levels and very early pregnancy loss in humans. Proceedings of the National Academy of Sciences of the United States of America, 103(10), 3938-3942.
Ng, S.F., Lin, R.C., Laybutt, D.R., Barres, R., Owens, J.A., and Morris, M.J. (2010). Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature, 467(7318), 963-966.
Nilsson, E., Larsen, G., Manikkam, M., Guerrero-Bosagna, C., Savenkova, M.I., and Skinner, M.K. (2012). Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PLOS ONE, 7(5), e36129.
Oberg, S., Cnattingius, S., Sandin, S., Lichtenstein, P., and Iliadou, A.N. (2011). Birth weight predicts risk of cardiovascular disease within dizygotic but not monozygotic twin pairs: A large population-based co-twin-control study. Circulation, 123(24), 2792-2798.
Oeppen, J., and Vaupel, J.W. (2002). Demography. Broken limits to life expectancy. Science, 296(5570), 1029-1031.
Omran, A.R. (1971). The epidemiologic transition. A theory of the epidemiology of population change. Milbank Memorial Fund Quarterly, 49(4), 509-538.
Pembrey, M.E., Bygren, L.O., Kaati, G., Edvinsson, S., Northstone, K., Sjostrom, M., and Golding, J. (2006). Sex-specific, male-line transgenerational responses in humans. European Journal of Human Genetics, 14(2), 159-166.
Pembrey, M.E., Bygren, L.O., and Golding, J. (2013). The nature of human transgenerational responses. In R.L. Jirtle and F.L. Tyson (Eds.), Environmental Epigenomics in Health and Disease (pp. 257-271). Berlin, Germany: Springer-Verlag.
Preston, S.H. (1996). American Longevity: Past, Present, and Future. Paper 36. Center for Policy Research, Maxwell School of Citizenship and Public Affairs, Syracuse University. Available: http://surface.syr.edu/cpr/36 [June 2014].
Reik, W., Dean, W., and Walter, J. (2001). Epigenetic reprogramming in mammalian development. Science, 293(5532), 1089-1093.
Ricklefs, R.E. (2008). The evolution of senescence from a comparative perspective. Functional Ecology, 22, 379-392.
Rodgers, A.B., Morgan, C.P., Bronson, S.L., Revello, S., and Bale, T.L. (2013). Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33(21), 9003-9012.
Smith, J., Cianflone, K., Biron, S., Hould, F.S., Lebel, S., Marceau, S., Lescelleur, O., Biertho, L., Simard, S., Kral, J.G., et al. (2009). Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. The Journal of Clinical Endocrinology and Metabolism, 94(11), 4275-4283.
Stearns, S.C. (1992). The Evolution of Life Histories. Oxford, UK and New York: Oxford University Press.
Symonds, M.E., Mostyn, A., Pearce, S., Budge, H., and Stephenson, T. (2003). Endocrine and nutritional regulation of fetal adipose tissue development. Journal of Endocrinology, 179(3), 293-299.
Tian, J.Y., Cheng, Q., Song, X.M., Li, G., Jiang, G.X., Gu, Y.Y., and Luo, M. (2006). Birth weight and risk of type 2 diabetes, abdominal obesity and hypertension among Chinese adults. European Journal of Endocrinology/European Federation of Endocrine Societies, 155(4), 601-607.
Titus-Ernstoff, L., Troisi, R., Hatch, E.E., Wise, L.A., Palmer, J., Hyer, M., Kaufman, R., Adam, E., Strohsnitter, W., Noller, K., et al. (2006). Menstrual and reproductive characteristics of women whose mothers were exposed in utero to diethylstilbestrol (DES). International Journal of Epidemiology, 35(4), 862-868.
Tuljapurkar, S., Li, N., and Boe, C. (2000). A universal pattern of mortality decline in the G7 countries. Nature, 405(6788), 789-792.
Vaessen, N., Janssen, J.A., Heutink, P., Hofman, A., Lamberts, S.W., Oostra, B.A., Pols, H.A., and van Duijn, C.M. (2002). Association between genetic variation in the gene for insulin-like growth factor-I and low birthweight. Lancet, 359(9311), 1036-1037.
van den Berg, G.J., Doblhammer, G., and Christensen, K. (2009). Exogenous determinants of early-life conditions, and mortality later in life. Social Science & Medicine, 68(9), 1591-1598.
Vaupel., J.W. (2010). Biodemography of human ageing. Nature, 464(7288), 536-542.
Waterland, R.A., and Michels, K.B. (2007). Epigenetic epidemiology of the developmental origins hypothesis. Annual Review of Nutrition, 27, 363-388.
Waterland, R.A., Kellermayer, R., Laritsky, E., Rayco-Solon, P., Harris, R.A., Travisano, M., Zhang, W., Torskaya, M.S., Zhang, J., Shen, L., et al. (2010). Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLOS Genetics, 6(12), e1001252.
Weaver, I.C., Cervoni, N., Champagne, F.A., D’Alessio, A.C., Sharma, S., Seckl, J.R., Dymov, S., Szyf, M., and Meaney, M.J. (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7(8), 847-854.
Wells, J.C. (2003). The thrifty phenotype hypothesis: Thrifty offspring or thrifty mother? Journal of Theoretical Biology, 221(1), 143-161.
Wilcox, A.J., Weinberg, C.R., and Baird, D.D. (1995). Timing of sexual intercourse in relation to ovulation. Effects on the probability of conception, survival of the pregnancy, and sex of the baby. The New England Journal of Medicine, 333(23), 1517-1521.
Yajnik, C.S. (2004). Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. Journal of Nutrition, 134(1), 205-210.






























