
Regulatory Perspectives on Technologies for the Heart
TINA M. MORRISON
US Food and Drug Administration
With advances in materials science, manufacturers are able to develop medical devices1 from stronger, superelastic materials and tissue (patient-specific or otherwise), opening the door for less invasive surgical therapies and personalized medicine. Moreover, access to computers with substantial processing power enables manufacturers to use computational tools paired with patient-specific diagnostic images to simulate treatment options, almost in real time. In addition, with the increasing cost of health care alongside the aging baby boomer population, there is a need to improve quality of life, decrease the number of doctor visits and length of hospital stays, and provide more efficient treatment options that reduce costs for people living with heart disease, as highlighted in Box 1.
The objectives of this paper are to highlight the regulatory process for medical devices from an engineering perspective, to discuss how manufacturers of medical devices can leverage different tools and techniques to get their devices to the
_____________
Note: The symposium presentation on regulatory perspectives was given by Sonna Patel-Raman of Halloran Consulting Group, Inc.
1 The FDA (www.fda.gov/aboutfda/transparency/basics/ucm211822.htm) defines a medical device as “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
- intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
- intended to affect the structure or any function of the body of man or other animals, and which does not achieve any of its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes.”
BOX 1
America’s Heart Disease Burden
- About 600,000 people die of heart disease in the United States every year—that’s 1 in every 4 deaths (Murphy et al. 2013).
- Heart disease is the leading cause of death for both men and women. More than half of the deaths due to heart disease in 2009 were in men (Murphy et al. 2013).
- Coronary heart disease is the most common type of heart disease, killing nearly 380,000 people annually (Murphy et al. 2013).
- Every year about 720,000 Americans have a heart attack; of these, 205,000 have already had a heart attack (Go et al. 2014).
- Coronary heart disease alone costs the United States $108.9 billion each year (Heidenreich et al. 2011) in healthcare services, medications, and lost productivity.
market, and how regulators might evaluate innovative medical technologies for the heart.
BACKGROUND
The US Food and Drug Administration’s Center for Devices and Radiological Health (CDRH) is responsible for regulating medical devices that are manufactured, repackaged, relabeled, and/or imported to be sold in the United States. Its mission “is to protect and promote the public health. We facilitate medical device innovation by advancing regulatory science, providing industry with predictable, consistent, transparent, and efficient regulatory pathways, and assuring consumer confidence in devices marketed in the U.S.”2 The center’s purview includes regulation of technologies for the heart, cardiovascular devices that treat a range of diseases that affect the heart.
Most implantable devices to treat heart disease are classified as the highest risk, Class III, because they are life sustaining and/or life supporting. Class III implantable devices include pacemakers, defibrillators, heart valves, coronary stents, ventricular assist devices, and artificial hearts. Manufacturers that wish to market Class III devices in the United States need to demonstrate that there is a reasonable assurance of both safety (i.e., the probable benefits to health outweigh
_____________
2 Information about the Center for Devices and Radiological Health is available at www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/ucm300639.htm.
TABLE 1 Comparison of Models Used to Assess High-Risk Medical Devices
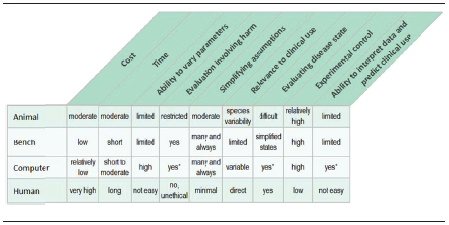
* There is a recognized potential. Computational modeling in medical devices, as compared to other industries, is nascent and is the one model with the most potential for refinement and improvement because other models are fairly mature.
any probable risks) and effectiveness (i.e., the device will provide clinically significant results).
Comprehensive evaluation of a premarket submission for a therapeutic, high-risk medical device is typically supported by a combination of valid scientific evidence from four types of models: animal, bench, computational, and human. These models can be leveraged at different stages of a medical device’s life cycle to demonstrate attributes of performance. Because each model has different strengths and limitations for predicting real-world clinical outcomes, the data portfolio for different devices and use conditions will vary. Some advantages of each model are shown in Table 1.
REGULATORY EVALUATION
Selecting the Appropriate Model for Evaluation
A firm that decides to manufacture a medical device should consider the regulatory pathway that will allow the device to be marketed in the United States. For many implantable devices to treat heart disease, a premarket approval (PMA) application is the appropriate pathway; PMA is “the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices.”3
_____________
3 Information about the PMA is available at www.fda.gov/Medicaldevices/Deviceregulationandguidance/Howtomarketyourdevice/Premarketsubmissions/Premarketapprovalpma/Default.Htm.
BOX 2
Questions from a Failure Modes and Effects Analysis
- What is the device intended for?
- What could go wrong?
- Why would the failure happen?
- What would be the consequences of failure?
- What is the likelihood of occurrence?
- What is the likelihood of detection?
- What is the severity of the failure mode?
Source: ISO (2007).
The firm must also develop a plan to gather the necessary valid scientific evidence to demonstrate a reasonable assurance of safety and effectiveness. The basis of this plan will depend on the indications for use—the disease to be treated, the affected patient population, the location of the implanted device, the expected duration and in vivo conditions of the implant, and the surgical procedure. With this information, the firm can use tools such as the Device Evaluation Strategy (FDA 2013, section 6.3) and Failure Modes and Effects Analysis (FMEA) for Medical Devices (ISO 2007) to address fundamental questions about device failure and potential consequences (see Box 2).
Depending on the function of the device, the firm identifies an attribute, the potential failure mode of that attribute, potential device and clinical effects, the design characteristic intended to mitigate the risk of the failure mode, and the model (animal, bench, computational, or human) that will be used to demonstrate that the function of the device will be attained and/or that the failure mode will not likely occur.
In vivo animal studies provide anatomic and clinical pathologic information of the local and systemic responses to device use. Larger animal models, such as pigs and sheep, are typically used for cardiovascular applications because the size and response of their anatomy more closely match those of human anatomy. Bench and computational models can act as surrogates for the in vivo environment and are useful because they can challenge an isolated feature of the device (e.g., implant integrity after deployment, long-term durability). Clinical trials are used for a variety of purposes, but for Class III devices they are mainly to demonstrate safety and effectiveness in the clinical setting and evaluate the device in the in vivo human environment.4
_____________
4 For other applications of clinical evaluations see FDA (2013).
Unique Material Considerations
When a firm uses traditional materials (e.g., stainless steel, polyurethane), whose behavior is well understood, the regulatory expectations tend to be straightforward. Additional engineering and regulatory questions may arise when complex materials are introduced and their behaviors are not well established. For example, there has been a shift from bare metal stents to drug-eluting and, more recently, absorbable stents.
Questions about drug-eluting stents have focused on understanding the elution and absorption rates of the drug, in addition to the mechanical performance of the stent. With absorbable devices, a major concern is the rate of degradation: absorbable devices are not intended to be permanent implants like metallic stents, but they do need to maintain a certain amount of structural integrity. Computational methods can be used for stress analysis, but they require more complex constitutive models. Other challenges arise for drug-eluting and absorbable products when the manufacturing process changes, because this can affect the elution and absorption rates for the drug or the degradation time frame for the absorbable material, which could result in additional testing. Identification of byproducts and their biological effects is another common consideration, and can involve complex in vivo (animal model) evaluations.
For medical devices that are tissue-engineered or regenerative medicine products, the regulatory framework is a bit different. Reviewers have to consider “purity, potency, and identity” for biologically derived products,5 and this requirement can pose limitations to traditional testing. For example, the long-term durability of permanent metallic implants (e.g., stents and heart valve frames) can be evaluated using accelerated durability bench testing and computational modeling, and the resulting data complement the outcomes from the clinical study regarding mechanical performance. This is not the case for tissue- and cell-based materials because the bench model does not allow for the cell and tissue adaptation process that occurs in vivo (e.g., cell infiltration and extracellular matrix deposition) and that may enable the product to repair itself in a normal timed setting in vivo. Therefore, manufacturers of biologic products must rely on extensive in vivo animal testing for performance evaluation.
Changes to Surgical Approaches
Another aspect of device design that can affect regulatory questions is a change in surgical technique or approach. For example, a recently introduced percutaneous (transcatheter) approach for implanting heart valves for high-risk patients engendered new questions about deliverability, deployment accuracy,
_____________
5 Information about this requirement is available at www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CBER/ucm133072.htm.
migration, integrity, and durability. The latter two are especially important with the new approach and are specifically assessed through a process called preconditioning: The heart valve is loaded onto a delivery system and guided through the arterial system, a process that subjects it to stresses and strains that do not occur in the traditional open surgical approach, in which the heart valve is directly implanted in the annulus. Preconditioning can greatly affect the integrity and durability of the implant and determine whether it is suitable for clinical use.
Unlike surgical bioprosthetic heart valves, transcatheter heart valves vary greatly in design, so the effects of preconditioning can be different for each design. Moreover, unlike surgically implanted mechanical heart valves, transcatheter valves do not usually remain circular upon implantation because the diseased leaflets and the calcium nodules are not removed, so the frame experiences noncircular deformations in vivo. The computational model is the only tool that can be used to determine changes in the stress (or strain) state of a device under different preconditioning states or implantation configurations. It can also predict the effects of preconditioning and implantation on fatigue performance (Duraiswamy et al. 2013). These predictions are then confirmed through accelerated durability testing.
Summary
Firms must provide valid scientific evidence from animal, bench, computational, and human models to support their marketing applications, and the amount of data collected from each model depends on the disease to be treated, the affected patient population, the location of the implanted device, the expected duration of the implant, and the surgical procedure. The data portfolio may change even more as companies expand their use of high-performance scientific computing to reduce time and cost in their efforts to bring safe and effective devices to patients in the United States.
TREATMENT PLANNING IN THE 21ST CENTURY
The practice of medicine is being shaped by powerful imaging capabilities, high-performance computation, wireless transmission of data, and massive storage of information. Physicians are now able to continuously monitor a patient’s health from a distance and determine whether a coronary lesion is relevant and treatment necessary, whether a patient is at risk of losing heart rhythm, and whether a patient will benefit from cardiac pacing (Miller 2014). In the near future, they will be able to select the optimal heart valve size and placement and to assess treatment options within a matter of hours (Simulia Community News 2014).
Several companies make such options available in the United States. Graphium Health uses cloud computing and mobile technology to help physicians, administrators, and patients make better pre- and postsurgery decisions
about care. HeartFlow uses patient-specific anatomy and physiological conditions to computationally estimate the amount of coronary burden due to a stenosis, alleviating in moderate cases the need for catheterization, an invasive procedure that is currently the standard of care.
Thanks to these and other tremendous advances, doctors have access to more data, information, and knowledge, and the potential to offer more clinical benefit to their patients. However, regulators are challenged with trying to determine which advances in computing and software are medical devices and, for those that are, what data are needed to support their entrance to the market in the United States (FDA, FCC, and HIT 2014).
From an engineering perspective, scientific computing is mature enough to simulate multiple design parameters and use conditions, and to visualize complex processes to revolutionize the way medical devices are investigated, treatments planned, and patient data utilized. With access to “digital patients,” device designers can download anatomic and physiologic computer models of patients with a given disease.6 They can then take their new device concepts and “deploy” them in the digital patients to simulate device performance, leading to more effective bench testing, in vivo animal studies, and (actual) clinical trials. The simulations enable detection of “soft failures,” failures that occur virtually before the devices are implanted in patients.
Finally, from a clinical perspective, physicians will soon be able to use simulation to predict the safety and effectiveness of a given medical product for an individual patient, thereby truly realizing personalized medicine. However, the regulatory burden for medical devices that have the potential to predict patient-specific outcomes remains to be determined.
CONCLUSIONS
New materials and surgical approaches are generating more treatment options for patients with heart disease. Moreover, there is a huge opportunity for imaging and high-performance computing to improve net health outcomes in the United States through treatment planning and better patient understanding of options. FDA’s engagement with industry and academia early on in the development of innovative products can help accelerate the field. The agency can provide the structure to help guide firms to determine appropriate models and data portfolio needs for evaluating their products. Early engagement also enables FDA to share its regulatory experience and to raise important questions that will protect patients and promote the overall health of the US population.
_____________
6 Two such resources are available from the Virtual Physiological Human (www.vph-institute.org) and the Foundation for Research on Information Technologies in Society (IT’IS) (www.itis.ethz.ch/services/anatomical-models).
ACKNOWLEDGMENTS
The author is grateful for the assistance of numerous contributors: Maureen Dreher, Carmen Gacchina-Johnson, Victor Krauthamer (in developing Box 1), Ken Skodacek, Jeremiah Wille, and Changfu Wu.
REFERENCES
Duraiswamy N, Ekrami Y, Weaver J, Retta S, Wu C. 2013. Non-circular configurations increase heart valve leaflet stresses and alter leaflet kinematics: A computational approach. Proceedings of the ASME 2013 Conference on Frontiers in Medical Devices: Applications of Computer Modeling and Simulation, September 11–13, Washington.
FDA [US Food and Drug Administration]. 2013. Investigational Device Exemptions (IDEs) for Early Feasibility Medical Device Clinical Studies, Including Certain First in Human (FIH) Studies. Center for Biologics Evaluation and Research. Bethesda. Available at www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm279103.pdf.
FDA, FCC, and HIT [US Food and Drug Administration, Federal Communications Commission, and Office of the National Coordinator for Health Information Technology]. 2014. FDASIA Health IT Report: Proposed Strategy and Recommendations for a Risk-Based Framework. Available at www.fda.gov/aboutfda/centersoffices/officeofmedicalproductsandtobacco/cdrh/cdrhreports/ucm390588.htm.
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, and 41 others. 2014. Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation 129:e28–e292.
Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, and 11 others. 2011. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation 123:e933–e944.
ISO [International Organization for Standardization]. 2007. 14971:2007 Medical devices—Application of risk management to medical devices. Geneva. Available at www.iso.org/iso/catalogue_detail?csnumber=38193.
Miller K. 2014. Doing the heart good: Translating models to the clinic. Biomedical Computation Review, Spring: 20–28. Available at http://biomedicalcomputationreview.org/content/doing-heart-good-translating-models-clinic.
Murphy SL, Xu JQ, Kochanek KD. 2013. Deaths: Final data for 2010. National Vital Statistics Reports 61(4). Hyattsville, MD: Division of Vital Statistics, Centers for Disease Control and Prevention. Available at www.cdc.gov/nchs/data/nvsr/nvsr61/nvsr61_04.pdf.
Simulia Community News. 2014. Looking deep into heart valve replacement. May, pp. 14–15. Available at http://feops.com/sites/default/files/simulia-scn-1405.pdf.








