
3
Supportive Policy Environment for Biomarker Tests for Molecularly Targeted Therapies
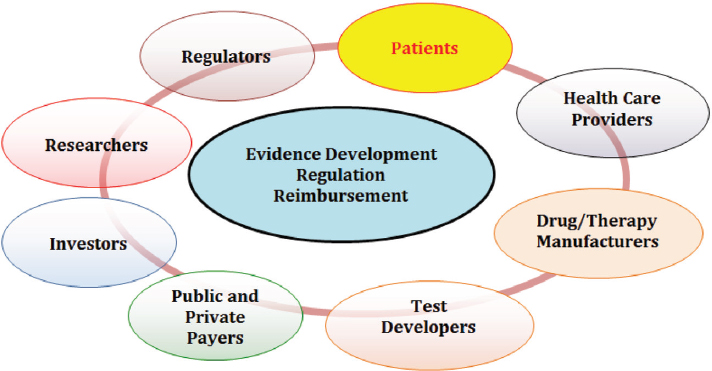
The committee’s vision of a rapid learning system encompasses three key components—supportive policy environment, supporting data infrastructure, and processes to improve patient care—as discussed in the previous chapter. This chapter explores the first of the three components by examining the key policy challenges that influence the integration of biomarker tests for molecularly targeted therapies into routine clinical practice. These challenges—including issues related to standards of evidence for the clinical utility of biomarker tests, alignment of regulatory and reimbursement decision processes, and test coverage and reimbursement1—are located at the intersection of interests of an array of stakeholders, including patients, health care providers, payers, regulators, test developers, device manufacturers, and pharmaceutical companies (see Figure 3-1).
These policy challenges must be viewed within the larger context
___________________
1 This report uses the term “reimbursement” as an umbrella term encompassing coverage and payment. Coverage refers to the ways in which public and private plans outline the services and products they will cover and under what circumstances they will reimburse for certain services and products. A coverage decision may be favorable, unfavorable, or limited; coverage decisions typically are based on clinical evidence. Reimbursement is the payment given to a provider or facility for a covered service or product. Coding is often included under reimbursement, as it refers to the systems that detail information about the nature of health care services provided, the technologies used, and the patient’s illness. Payment for medical services is based on the code(s) associated with a particular service and the dollar amount(s) assigned to the code (SACGHS, 2006). Coding related to biomarker tests for molecularly targeted therapies is discussed further in Appendix B of this report.
of the current system for biomarker test regulation as well as the U.S. health care reimbursement system, which while still predominantly fee-for-service, is in flux as alternative payment models are introduced that shift risk to health care providers and are aligned more closely with health care value (Anderson et al., 2015; Bipartisan Policy Center, 2015; Burwell, 2015).2
Developing appropriate and effective regulatory and reimbursement frameworks responsive to rapidly evolving technologies is critical to ensuring that health care providers and their patients have access to—and the ability to benefit from—the potential of biomarker tests for molecularly targeted therapies to optimize care. At the same time, it is important that regulatory and reimbursement pathways support an environment in which manufacturers and investors continue to see potential economic value in developing such tests and treatments (IOM, 2013b, 2015; PMC, 2015). Indeed, regulatory and reimbursement policy affects all new medical technologies, and has a direct impact on how medical product industries evolve and grow (Deverka and Dreyfus, 2014). To make evidence-based decisions on whether to cover and reimburse the cost of biomarker tests, payers require clarity about the types of information required to establish clinical utility, or the test’s usefulness in terms of its impact on clinical outcomes. Thus, policy challenges involve balancing the competing demands of the patient’s need and desire for access to tests
___________________
2 Alternative payment models include accountable care organizations, bundled or episode-based payments, capitated payments, and patient-centered medical homes (NASEM, 2015).
to direct novel therapies against the need for sufficient evidence to assess the potential risks and benefits of the tests (IOM, 2013d).
The discussion in this chapter is divided into four parts. The first part explores the issue of evidentiary standards for clinical utility, which is a cross-cutting issue that influences regulatory, reimbursement, and clinical practice areas. The discussion then shifts to an overview of the current regulatory structure for biomarker tests for molecularly targeted therapies and discusses communication and information related to test performance and intended use. The third part of the chapter explores the key reimbursement-related challenges facing clinical implementation of biomarker tests for molecularly targeted therapies. Regulatory and reimbursement challenges are interrelated, and the committee’s integrated approach to addressing those challenges forms the fourth and final part of the chapter.
EVIDENTIARY STANDARDS OF CLINICAL UTILITY
The Secretary’s Advisory Committee on Genetics, Health, and Society (SACGHS),3 which produced a number of detailed reports and recommendations related to the integration of genetic and genomic technologies into health care, specified that information on clinical utility is critical on several levels: for managing the treatment of patients, for developing clinical guidelines to assist health care providers in providing the best available treatment, and for coverage determinations by payers. SACGHS also specified that the lack of evidence of clinical utility is a significant challenge for developing clinical guidelines and ensuring access to tests through coverage and reimbursement decisions (SACGHS, 2008). Achieving consensus on the evidentiary standards for clinical utility has proven to be an elusive goal, however, with the earliest efforts dating back 20 years to the development of a grading system to define levels of evidence for tumor markers (CMTP, 2013; Hayes et al., 1996; IOM, 2012b; Parkinson et al., 2014). Health care providers, payers, and test developers widely perceive the lack of a common evidentiary framework to assess clinical utility to be a limiting factor in the development and use of biomarker tests. Payers increasingly expect evidence of clinical utility of new tests
___________________
3 SACGHS, which was in operation for nearly 10 years until its charter expired in 2011, examined a wide range of topics, including the integration of genetic and genomic technologies into health care and public health; the clinical, public health, ethical, economic, legal, and societal implications of these technologies; gaps in research and data collection; the impact of patent policy and licensing practices on their accessibility and availability; and how genetic and genomic technologies are used in other settings such as education, employment, insurance, and law. (For more on SACGHS see http://osp.od.nih.gov/office-clinical-researchand-bioethics-policy/genetics-health-and-society/sacghs-archives [accessed June 6, 2016].)
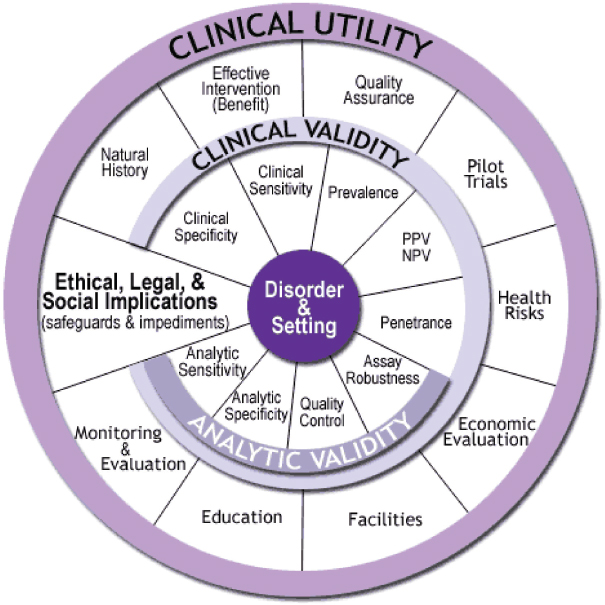
NOTE: NPV = negative predictive value; PPV = positive predictive value.
SOURCE: http://www.cdc.gov/genomics/gtesting/ACCE (accessed June 4, 2015.
while test developers are hesitant to invest in evidence development without reasonable certainty such expenditures of resources will be supported by a level of market access that produces a reasonable return on investment. Clearer and more consistent evidence requirements would significantly improve manufacturers’ incentives to develop biomarker tests (Faulkner, 2009; Goldman et al., 2013).
The Centers for Disease Control and Prevention (CDC) developed a useful framework known as the ACCE Model,4 which presents a process for evaluating scientific data on emerging genetic tests. ACCE refers to four main evaluation criteria: analytic validity, clinical validity, clinical utility, and ethical/legal/social implications (see Figure 3-2). The ACCE Model process is based on a standard set of 44 targeted questions related
___________________
4 See http://www.cdc.gov/genomics/gtesting/ACCE (accessed June 4, 2015).
to the four main evaluation criteria.5 According to this model, the clinical utility of a biomarker test always rests on established analytic validity and clinical validity. A test’s analytic validity reveals how well the test detects the specific analytes it was designed to detect and includes assessment of the test’s range, accuracy, precision, bias, and reproducibility when used by different operators or instruments across different settings. Clinical validity is a measure of the accuracy of a test for a specific clinical purpose, such as correlation with the presence of a disease or prediction of response to a targeted therapy in a specific patient population. Clinical validity involves assessment of the clinical sensitivity, specificity, and other parameters of a test (Febbo et al., 2011; IOM, 2015; Parkinson et al., 2014; Teutsch et al., 2009). Demonstration of a test’s clinical validity is critical to reducing patients’ risk of harm from false-positive or false-negative results (Hwang et al., 2015). Both analytic validation and clinical validation lay the foundation for demonstration of clinical utility. The committee’s definitions for these terms, as used throughout this report, are outlined and clarified in Box 3-1.
CDC’s Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group, building on the ACCE Model, defined clinical utility as a test’s “evidence of improved measurable clinical outcomes, and usefulness and added value to patient management decision making compared with current management without genetic testing” (Teutsch et al., 2009, p. 11). Clinical utility encompasses evidence of utility in clinical settings as well as the balance of benefits and harms of the test (Teutsch et al., 2009). This understanding of clinical utility has been broadly used to explore policy, research, and clinical care issues related to the use of biomarker tests and corresponding molecularly targeted therapies (CMTP, 2013; Hayes et al., 2013; IOM, 2012a; Parkinson et al., 2014). No consensus currently exists, however, about the evidentiary standards that should be applied to assess the clinical utility of new technologies such as biomarker tests for molecularly targeted therapies (Woodcock, 2010).
Assessing Clinical Utility
Establishing a linkage to improved patient outcomes requires clear, consistent, and reasonable standards of evidence. The lack of such common standards of evidence of clinical utility represents a significant challenge in the implementation of effective biomarker tests for molecularly targeted therapies into routine clinical practice, with significant consequences for patient access to tests, as well as potential for patient harm if
___________________
5 See list of questions at http://www.cdc.gov/genomics/gtesting/ACCE/acce_proj.htm (accessed June 4, 2015).
tests with limited evidence of clinical utility are used to direct treatment, or if effective tests are not being used clinically due to lack of reimbursement. Such evidentiary standards are also critical for test development as they enable test developers and investors to assess the risk and potential returns on investment in biomarker tests; the lack of such standards may constrain the development of biomarker tests (IOM, 2013d).
The concept of reasonable evidentiary standards hinges on the importance of designing valid studies that can generate evidence in a timely manner that is broadly applicable to real-world clinical situations. Thus, the development of clear, consistent, and reasonable evidentiary standards should focus on evidence that is adequate for rational decision making instead of “ideal” or “best” evidence approaches. At the same time, such standards must be sufficient to address payers’ decision-making
requirements and ensure that patients are not harmed by the introduction into clinical use of new tests that lack adequate evidence (Faulkner, 2009).
Various approaches to systematically establish clinical utility have been proposed over the past two decades, including conducting prospective clinical trials that assess the test or by conducting prospective or retrospective studies using archived specimens collected from previous clinical trials or registries (IOM, 2012a) (see Table 3-1). The assessment of the clinical utility of a biomarker test generally requires applying the test to a large number of patients or patient samples and generation of data on the association of the test result with clinical decisions that lead to improved patient outcomes. Less rigorous studies can result in bias, or conclusions of limited generalizability (IOM, 2011b). The collection of such data requires long-term patient follow-up and collection of outcomes data from an adequate number of patients, which can be challenging
TABLE 3-1 Examples of Study Designs to Assess the Clinical Utility of a Biomarker Test
| Type of Trial Design | Description | Advantages and Limitations |
|---|---|---|
| Prospective, test-directed randomized controlled studies | ||
| Test-guided versus non-guided, with randomization (“All-comers”) | Patients are randomly assigned to the test-guided arm or the non-guided arm. Patients randomized to the test-guided arm are directed to therapy as dictated by the test (test-positive to new therapy, test-negative to standard of care). Patients randomized to the non-guided arm undergo a second randomization to receive either new therapy or standard of care. |
Advantages: Can be used to evaluate complex test-directed treatment strategies using a large number of treatment options or test categories. Limitations: Requires a larger number of patients to be enrolled relative to other designs. |
| Enrichment studies | The test is applied to all patients, but only test-positive patients are randomized and/or treated. Test-negative patients are either off study or followed prospectively in a registry. |
Advantages: Useful when clinical utility of some of the test-designated categories is already established or assumed and need not be re-evaluated, while other categories require prospective evaluation in a clinical trial. Acceptable in circumstances where a certain subgroup of patients is thought so unlikely to experience an event with standard of care, or to benefit from the new therapy, that it would be unethical to randomize those patients. Limitations: Cannot assess the treatment benefit in test-negative patients. Cannot definitively establish the predictive ability of a marker. |
| Type of Trial Design | Description | Advantages and Limitations |
|---|---|---|
| Other pragmatic/adaptive studies | ||
| Prospective–retrospective studies | Uses archived specimens from a previously conducted clinical trial with treatment(s) that are relevant to the intended clinical use of the test. | Advantages: Despite being less resource- and time-intensive, study design may have evidentiary value close to a prospective study under certain conditions (Simon et al., 2009). Study design can be used if prospective trial is not feasible for ethical or other reasons. Limitations: Archived specimens may be unavailable; uncertainty in collection, storage, or processing methods may render test assessment unreliable. Statistical inference concerns due to original trial likely not pre-specifying a plan to study treatment effect in subgroups. |
| Single-arm studies | Test is applied to all patients, and test-positive patients are uniformly treated with the existing, approved therapy believed to provide a differential benefit based on the test result. Test-negative patients are either off study or followed prospectively in a registry. |
Advantages: Can be used to identify a subset of patients who may benefit differentially from an existing FDA-approved therapy (with a disease-specific indication in the labeling that would make subsequent RCTs unethical), where archival tissue from the original trial is not available. Limitations: It must be feasible to use complete or overall response as an endpoint, and comparable data must be available from a suitable noncontemporaneous cohort. Single-arm studies only provide data on test-positive patients. Due to a lack of a control arm, test-negative patients cannot be assumed not to benefit from the treatment. |
| Type of Trial Design | Description | Advantages and Limitations |
|---|---|---|
| Longitudinal observational studies | A variety of possible designs, which do not include any study-directed testing or intervention and instead prospectively collect clinical and outcomes data, including “Quasi-experimental”: collection of clinical outcomes data pretest and posttest for at least two groups under investigation. Prospective cohort: broad enrollment criteria to facilitate examination of heterogeneity in test- or treatment-related effects. | Advantages: Can help generate hypotheses related to large hypothesized effect sizes, evolving treatment strategies, real-world clinical use of tests and multiple corresponding therapies, patient/provider preferences related to test use, long-term outcomes, and very large required sample sizes. Limitations: Careful consideration of the rationale for performing an observational study instead of other study types, as well as methods to address bias and confounders, is needed. Observational studies are particularly vulnerable to time-varying confounders. A fully defined research protocol (including hypotheses, intervention groups, outcome and subgroup definitions, power calculations, and analysis plan) is required to approximate a randomized study’s objective of causal inference. |
| Type of Trial Design | Description | Advantages and Limitations |
|---|---|---|
| Detection-analytic modeling techniques | For tests meeting an agreed upon plausibility threshold for clinical utility, modelling techniques can estimate overall downstream outcomes. Such models would include all relevant risks and benefits related to remaining survival and quality of life. |
Advantages: In cases where explicit evidence of clinical utility is absent, these techniques can estimate the effect of tests on patient outcomes using a variety of sources of evidence as input, through metrics including quality of life and life expectancy. Limitations: Tests to be assessed need to meet an established threshold for plausible evidence of clinical utility. Good modeling techniques are labor- and time-intensive to develop and validate (including establishing links between surrogate and final outcome measures), and are not recommended when there is a high degree of uncertainty about the underlying disease process or the link between test results and treatment effectiveness. |
NOTE: FDA = Food and Drug Administration; RCT = randomized controlled trial.
SOURCES: CMTP, 2013; Freidlin et al., 2010; IOM, 2012a; McShane, 2011; Sargent et al., 2005; Simon et al., 2009. Adapted from Freidlin et al., 2010; IOM, 2012a.
in light of the rarity of some of the mutations the tests are designed to identify. For example, BRAF gene mutations occur in 1 percent or less of lung cancer patients. For one study, researchers screened more than 11,000 lung cancer patients in order to enroll 23 patients with a specific BRAF mutation (IOM, 2015). Randomized controlled trials, considered the gold standard of evidence, typically focus on more restricted patient populations and thus do not capture broader patient groups more representative of “real-world” patients with co-morbidities and other issues that render treatment of the patient’s condition more complex (Lewis et al., 2015; Perlmutter, 2015; Sniderman and Furberg, 2009; Treweek et al., 2015), as discussed further in Chapter 5.
Currently no agency or organization in the United States is charged with the responsibility of developing evidentiary standards for the clinical
utility of biomarker tests (Schott et al., 2015). As discussed further in the next section, the current regulatory oversight structure for biomarker tests for molecularly targeted therapies does not include assessment of clinical utility. Given the critical need for evidentiary standards of clinical utility to ensure the implementation of appropriate biomarker tests into routine clinical practice to improve patient care, and the absence of a dedicated body responsible for developing such standards, many have called for public–private collaborations to take the lead (Parkinson et al., 2014; Sawyers, 2008). Organizations such as the Center for Medical Technology Policy (CMTP) have worked to fill the void by bringing together a range of public and private stakeholders to work together toward consensus on evidentiary standards for clinical utility (CMTP, 2014; IOM, 2013d, 2015).
REGULATORY CHALLENGES
The regulatory framework for biomarker tests for molecularly targeted therapies presents a number of challenges. First, there is concern over the adequacy of the current approach to the regulation of biomarker tests for molecularly targeted therapies. Second, the processes for regulatory and reimbursement decisions are not currently aligned. Finally, there is a lack of clearly communicated information about the performance characteristics and intended use of biomarker tests, particularly given the availability of multiple tests for the same purpose. All these challenges cause uncertainty and confusion among health care providers, patients, test manufacturers, and payers, and in some cases may potentially expose patients to harm.
Regulatory Oversight of Biomarker Tests for Molecularly Targeted Therapies
The current regulatory structure for biomarker tests for molecularly targeted therapies features key oversight authority by two federal agencies: the Food and Drug Administration (FDA) and the Centers for Medicare & Medicaid Services (CMS). Numerous state regulatory bodies and professional and accreditation organizations also are involved and provide complementary oversight of diagnostic tests and laboratory operations (see Table 3-2).
FDA is charged with overseeing the safety and effectiveness of drugs and medical devices under the Federal Food, Drug, and Cosmetic Act of 1938 and the Medical Device Amendments of 1976. FDA’s Center for Drug Evaluation and Research (CDER) regulates drugs, while the Center for Devices and Radiological Health (CDRH) regulates medical devices, including in vitro diagnostics (IVDs). FDA has defined IVDs as tests,
| Entity | Role in Quality Improvement or Oversight |
|---|---|
| Centers for Medicare & Medicaid Services (CMS) | CMS regulates laboratories under the Clinical Laboratory Improvement Amendments (CLIA) (CMS, 2015a). To ensure CLIA compliance, laboratories undergo review of results reporting, laboratory personnel credentialing (i.e., competency assessment), quality control efforts, and procedure documentation. Laboratories are also required to perform proficiency testing (PT), a process in which a laboratory receives an unknown sample to test and report the findings back to the PT program, which evaluates the laboratory’s performance. CMS also approves programs to perform PT. |
| CMS grants states or accreditation organizations the authority to deem a laboratory as CLIA-compliant. Approved accreditation organizations include: American Association of Blood Banks, American Association for Laboratory Accreditation, American Osteopathic Association/Healthcare Facilities Accreditation Program, American Society for Histocompatibility and Immunogenetics, Commission on Office Laboratory Accreditation, College of American Pathologists, and The Joint Commission. | |
| Centers for Disease Control and Prevention (CDC) | CDC performs research on laboratory testing processes, including quality improvement studies, and develops technical standards and laboratory practice guidelines. CDC also manages the Clinical Laboratory Improvement Advisory Committee, a body that offers guidance to the federal government on clinical laboratory quality improvement and revising CLIA standards (CDC, 2014). |
| Food and Drug Administration (FDA) | FDA reviews and assesses the safety, efficacy, and intended use of in vitro diagnostic tests (IVDs) (FDA, 2014c). FDA assesses the analytic validity (i.e., analytic specificity and sensitivity, accuracy, and precision) and clinical validity (i.e., the accuracy with which the test identifies, measures, or predicts the presence or absence of a clinical condition or predisposition), and it develops rules and guidance for CLIA complexity categorization. FDA has stated that it has statutory authority for the regulatory oversight of all tests used in patient care, but has used its enforcement discretion (meaning it has chosen not to exercise that authority) for the oversight of laboratory-developed tests (LDTs). However, FDA signaled its intent to begin regulating LDTs in draft guidance released in 2014 (FDA, 2014a). |
| Entity | Role in Quality Improvement or Oversight |
|---|---|
| College of American Pathologists (CAP) | CAP accreditation ensures the safety and quality of laboratories and satisfies CLIA requirements. CAP also offers an inter-laboratory peer PT program, which includes (1) Q-Tracks: a continuous quality monitoring process; (2) Q-Probes: a short-term study that provides a time-slice assessment of performance, and (3) Q-Monitors: customized programs that address process-, outcome-, and structure-oriented quality assurance issues. |
| American Academy of Family Physicians (AAFP) | AAFP offers a number of CMS-approved PT programs (AAFP, 2015). |
| American Society for Clinical Pathology (ASCP) | ASCP certifies medical laboratory professionals. ASCP also manages a CMS-approved PT program for gynecologic cytology (ASCP, 2014). |
| New York State Department of Health (NYSDOH) | NYSDOH’s Clinical Laboratory Evaluation Program (CLEP) seeks to ensure the accuracy and reliability of results of laboratory tests on specimens obtained within the state through on-site inspections, proficiency testing and evaluation of the qualifications of personnel of state permit-holding clinical laboratories and blood banks (NYSDOH, 2015a). |
SOURCE: Adapted from NASEM, 2015.
reagents, instruments, and systems used to diagnose medical conditions. FDA’s jurisdiction does not cover laboratory facilities or functions; rather, it focuses on individual IVD safety and effectiveness.
The regulatory pathways for clinical laboratory tests differ from that for drugs. Clinical laboratory tests such as biomarker tests for molecularly targeted therapies are introduced into standard clinical practice through several pathways. Medical device manufacturers may market a commercial “test kit,” which typically includes the necessary reagents, instructions, and statements regarding the intended use of the test. Such test kits are sold to laboratories, health care providers, or hospitals in interstate commerce, and they must be approved or cleared by FDA using the premarket approval or 510(k) process, respectively (see Box 3-2).
A test also may be developed for exclusive use within a specific laboratory; this is known as a laboratory-developed test (LDT), which FDA defines as an IVD manufactured, developed, validated and offered by a single laboratory. Although the uses of an LDT are often the same
as an FDA-cleared or approved IVD, many clinical laboratories choose to offer their own test. Indeed, the LDT pathway is a more commonly used route to market, enabling rapid test development and implementation into clinical use (IOM, 2015).6 Any clinical laboratory that reports tests for clinical management of patients falls under the purview of CMS’s Clinical Laboratory Improvement Amendments of 1988 (CLIA, as in a CLIA-certified laboratory) which provides a baseline level of oversight with respect to test performance and the quality of laboratory operations, discussed further below.
Importantly, review of tests by FDA focuses on demonstration of analytic validity and clinical validity; according to FDA guidelines, a “safe and effective” IVD has established both analytic and clinical validity. FDA’s authority extends to regulate the design, manufacturing quality, labeling, and legitimacy of manufacturer claims concerning “intended use” of diagnostics and drugs designed for clinical use. FDA does not require evidence of a test’s clinical utility prior to clinical use. Thus, FDA approval or clearance does not necessarily imply that the test improves clinical outcomes. LDTs performed in CLIA-certified laboratories also are not required to demonstrate clinical utility prior to use (IOM, 2010, 2012a, 2015).
FDA has stated that it has statutory authority for the regulatory oversight of all tests used in patient care, but has used its enforcement discretion (meaning it has chosen to not exercise that authority) for the oversight of LDTs.7 While an LDT developed by a CLIA-certified laboratory currently does not require FDA approval or clearance, FDA has indicated in a draft guidance released in October 2014 its intention to gradually phase in regulation of high-risk LDTs, followed by moderate-risk LDTs, over a 9-year period, as discussed further below (FDA, 2014c).
Companion Diagnostics
Biomarker tests that will be used to identify patients likely to benefit from a specific investigational targeted therapy may be co-developed with the drug; the biomarker and drug are both tested simultaneously in clinical trials, and the safety and efficacy of the test and the drug are evaluated in the same trial. Biomarker tests that are co-developed with a drug and co-approved by FDA are known as companion in vitro diag-
___________________
6 The alternative LDT pathway is not possible for drug development; premarket approval by FDA is required for all drugs.
7 The majority of biomarker tests for molecularly targeted therapies currently in clinical use are LDTs.
nostics8 (IOM, 2015). In oncology, for example, FDA has approved nearly two dozen companion diagnostics, most of which target the HER2 gene, but also include tests for BRAF gene mutation, EGFR gene mutations, and other targets.9 Companion diagnostics provide information that FDA considers necessary for the safe and effective use of a corresponding therapy and approved drugs and their companion diagnostics refer to each other in their labels (IOM, 2015). As Parkinson points out, “The approval in recent years of companion diagnostics developed with targeted therapies serve as the best available examples of successful clinical utility efforts” (Parkinson et al., 2014, p. 1439).
For a biomarker test that is co-developed with a drug (e.g., HER2 for trastuzumab), the regulatory pathway enables concurrent approval of the test and the drug (Frueh, 2013). Although establishment of clinical utility for the drug–diagnostic combination would be expected to ensure reimbursement of the test, this is not always the case; one study found limited and variable reimbursement of drug–diagnostic combinations, stating that “even in cases of co-developed combinations, drug reimbursement does not necessarily imply diagnostic reimbursement” (Cohen and Felix, 2014, p. 171).
An important note is that challenges to the companion diagnostics model include the fact that LDTs can be used in place of an FDA-approved companion diagnostic. Moreover, when FDA initially developed the companion diagnostics model, single-analyte tests predominately were used to indicate treatment with a specific companion drug. Given the increasing use of newer technologies such as next-generation sequencing (NGS), the companion diagnostic model of single test for single drug may not be feasible for assessing a single small cancer biopsy for potential response to multiple drugs using multiple individual tests on different testing platforms (Blumenthal et al., 2016; IOM, 2015; Mansfield, 2014).
___________________
8 According to FDA, “a companion diagnostic device can be an in vitro diagnostic device or an imaging tool that provides information that is essential for the safe and effective use of a corresponding therapeutic product. The use of an IVD companion diagnostic device with a particular therapeutic product is stipulated in the instructions for use in the labeling of both the diagnostic device and the corresponding therapeutic product, as well as in the labeling of any generic equivalents and biosimilar equivalents of the therapeutic product.” http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htm (accessed May 22, 2015).
9 See complete list at http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htm (accessed May 18, 2015).
Laboratory Oversight Under CLIA
All laboratories operating in the United States and involved in “the diagnosis, prevention, or treatment of any disease or impairment of, or the assessment of the health of, human beings” fall under the jurisdiction of CMS laboratory oversight under CLIA.10 Passed by Congress to ensure the quality of all clinical laboratories in the United States, the amendments were motivated in large part to target unregulated laboratories that were operating outside the authority of the earlier Clinical Laboratory Improvement Act of 1967 (CMS, 2006). Any laboratory performing biomarker testing used to guide treatment selection falls within the regulatory purview of CLIA.
The requirements for CLIA certification vary depending on the nature and complexity of the tests performed by a laboratory. This system is based on the degree of harm that an incorrect test could cause for a patient. Tests determined to be low-complexity tests such as urine pregnancy tests or fecal occult blood tests (for colorectal cancer screening) are granted waived status under CLIA (CMS, 2015c).
Laboratories performing medium- and high-complexity tests, including those for biomarker tests for molecularly targeted therapies, are subject to additional quality control and proficiency testing (PT) requirements in order to obtain the required CLIA Certificate of Compliance. These laboratories must participate in one of 13 national PT programs, which in turn are overseen by CMS or its approved accreditation organizations (see Table 3-2). Laboratories performing specialized testing (e.g., for hematology or toxicology) are further subject to specialty PT to ensure additional rigor in the assessment of the laboratory performance and quality (Astles et al., 2013).
The PT process assesses the analytic validity of laboratory tests: PT specimens are provided and the laboratory processes the samples in accordance with its standard operating procedures to detect certain analytes, then reports the results to the PT agency. To determine the quality of the laboratory, test results are evaluated according to CLIA’s criteria for acceptable performance. These criteria are often based on the mean performance of other laboratories performing the tests by the same methods, or are tied to a reference value (SACGHS, 2008).
CMS11 currently does not define specific standards for molecular pathology or genetics/genomics tests under CLIA, though requests for inclusion of specialty PT related to genetics have been made to CMS and
___________________
10 42 U.S.C. 263a.
11 See https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Downloads/lccodes.pdf (accessed May 19, 2015).
the Clinical Laboratory Improvements Advisory Committee (Murphy et al., 2006).12
The current level oversight of laboratories under CLIA raises concerns in the context of the clinical use of biomarker tests for molecularly targeted therapies, given the minimum standards required to maintain compliance with CLIA. CLIA requires that laboratories analytically validate their tests prior to use; the criteria for adequate test validation and performance are defined by the laboratory director (IOM, 2015). CLIA regulations do not explicitly require laboratories to verify clinical validity of LDTs, although the regulations may be interpreted to mandate such verification (Ferreira-Gonzalez et al., 2014).
Given CLIA regulations only explicitly require demonstration of analytic validity, concerns have been raised that a CLIA-certified laboratory potentially could develop an analytically valid test for clinical use without demonstrating the clinical validity or clinical utility of the test to positively impact patient care. Moreover, CLIA’s specialty areas that require a higher degree of oversight do not currently include genetic, genomic—or other omics—testing, raising concerns over the level of oversight of laboratories conducting highly complex biomarker tests (Hudson et al., 2007). Many laboratories are more highly regulated under frameworks defined by state laws and other accreditation authorities as discussed further below, but currently CLIA is the minimum national legal standard for clinical laboratories, including laboratories developing LDTs.
Evolving Regulatory Framework
Appropriate oversight and validation of LDTs has continued to be debated, driven by the need to ensure safe, accurate, and reliable biomarker tests without limiting patient access and quality of care. While FDA recognizes the importance of CLIA oversight of laboratories, concern over whether CLIA provides a sufficient regulatory framework for high-risk testing has arisen (FDA, 2014a). In the past, FDA attributed exercising enforcement discretion for LDTs to their simple, low-risk nature. FDA recognizes that current LDTs are:
- Manufactured with components that are not legally marketed for clinical use;
___________________
12 Additional CMS testing oversight measures include educational publications in the CDC’s Morbidity and Mortality Weekly Report, training of state surveyors in relevant technical and procedural issues, and requests for FDA assistance in validation of complex tests. https://www.genome.gov/Pages/About/OD/ReportsPublications/June2008_YostHoL.pdf (accessed June 21, 2015).
- Offered beyond local populations and manufactured in high volume;
- Used widely to screen for common diseases rather than rare diseases;
- Used to direct critical treatment decisions (e.g., prediction of drug response); and
- Highly complex (e.g., automated interpretation, multisignal devices, use of nontransparent algorithms and/or complex software to generate device results) (FDA, 2014b, p. 8).
The evolving complexity of LDTs propelled FDA to signal its intention to exercise its regulatory authority through issuance of draft guidance in October 2014 (FDA, 2014b). Certain stakeholder groups have reacted strongly to the proposed guidance (see Box 3-3 for an overview of selected stakeholder perspectives on increased oversight for laboratories and LDTs). Stakeholders’ reaction focuses on the concern about the FDA’s ability to regulate LDTs in a way that will not hamper innovation, concern that its processes are too slow to keep pace with the rapidly changing medical knowledge regarding the genetic and genomic causes of disease, as well as the lack of evidence that LDTs cause patient harm (Evans et al., 2015; Terry, 2014). FDA released a White Paper in November 2015 that examined 20 LDTs, which due to false-positive or false-negative results may have caused or did cause actual harm to patients. Other LDTs examined by FDA were found to have provided information that did not have any proven relevance to the disease or condition for which the test was intended for use (FDA, 2015c).
Once the FDA LDT guidance is finalized, all laboratories will have a 6-month grace period to notify FDA of all LDTs in clinical use, and begin to report significant adverse events to FDA. Notification details will include laboratory and test name, intended use, clinical use (e.g., prognostic, predictive), monthly test volume, disease or patient population, test method and analyte detected, and sample type(s) (FDA, 2014a). Test manufacturers and clinical laboratories developing tests would be required to continue to remove products from the market at any time if they are found to be unsafe.
FDA has stated that LDTs should be subject to further risk-based regulation requiring sufficient evidence to deem them safe and effective. The initial focus after the LDT guidance is finalized will be on premarket review of LDTs that have the same intended use as existing FDA-cleared or -approved companion diagnostics or high-risk medical devices (laboratories providing these LDTs will have a 12-month grace period to file for premarket approval [PMA] with FDA). FDA will determine further prioritization for review based on the notification data received for all
LDTs, through the use of an advisory panel. The first phase of the regulatory process is expected to last 5 years (with an additional 4 years for moderate-risk devices) (FDA, 2014b).
FDA will continue to exercise enforcement discretion for three categories of LDTs: (1) those intended for use with rare diseases or conditions (“rare” as defined by FDA refers to cases where the number of persons who may be tested with the device is less than 4,000 per year); (2) “traditional LDTs,” defined by FDA as being manufactured by a health care facility laboratory, interpreted by a qualified laboratory professional, used
in the treatment of a patient within the same health care system, composed only of components and instruments that are legally marketed for clinical use, and interpreted by qualified laboratory professionals without the use of automated instrumentation or software for interpretation; and (3) LDTs for unmet needs for which no FDA-approved diagnostic exists for its intended use. LDTs used in law enforcement and in determining transplant histocompatibility would also remain under enforcement discretion (FDA, 2014b).
This regulatory approach seeks to balance the dual goals of making LDTs available to patients in need, while also requiring accurate and reliable test performance. In an effort to improve transparency and efficiency in the regulatory process, FDA encourages laboratories to provide notification that they are developing tests. This early collaboration would enable advisory panels working in conjunction with FDA to better understand and prioritize tests in the pipeline according to risk (FDA, 2014a).
Newer technologies such as NGS also pose significant regulatory challenges. Such technologies are producing an unprecedented amount of omics data, which have the potential for both research and clinical use. However, variability in platforms, assay design, and bioinformatics approaches can complicate the validation of these vast amounts of often novel biological data (Boland et al., 2013; Ewing et al., 2015; Pant et al., 2014). FDA has proposed a collaborative approach to ensuring the accuracy and reproducibility of NGS tests and continues to work with the broader scientific community to refine its regulatory framework regarding these complex new technologies (Blumenthal et al., 2016; FDA, 2015a; Kass-Hout and Litwack, 2015).
REIMBURSEMENT CHALLENGES
As noted at the beginning of this chapter, the policy challenges to biomarker tests for molecularly targeted therapies must be viewed within the context of the broader U.S. health care system. Although the Affordable Care Act contained incentives to adopt alternative reimbursement models (Blumenthal et al., 2015)—such as global, bundled, or value-based payments, and accountable care organizations—the predominant reimbursement method in the U.S. health care system remains fee-for-service. Reimbursement systems have a significant impact on how care is delivered; fee-for-service reimbursement encourages volume of services and does not provide a strong framework for linking reimbursement to health outcomes or value (Patel et al., 2015). The Institute of Medicine’s (IOM’s) Best Care at Lower Cost report concluded that fee-for-service does not reward health care providers for quality of care and actually encourages wasteful and ineffective care (IOM, 2013a). In addition to an evolving
reimbursement structure, reimbursement for biomarker tests also must be viewed against the backdrop of ongoing broad-based efforts by payers to constrain health care spending (Deverka and Dreyfus, 2014).
The U.S. health care system features an array of private and public payers with different coverage and reimbursement policies (Chambers et al., 2015). Medicare is a key public payer, providing insurance coverage for more than 50 million beneficiaries (CMS, 2015b). The Medicare reimbursement system is governed by the “reasonable and necessary” standard. It cannot reimburse for experimental treatments, or most screening tests, and will not reimburse for biomarker tests for molecularly targeted therapies that are not the standard of care (IOM, 2015). Medicare at times makes coverage decisions that are applicable nationwide, known as national coverage determinations (NCDs) or more commonly, works with regional claims administrators known as Medicare Administrative Contractors (MACs) to make local coverage determinations (LCDs). The joint federal–state Medicaid program involves significant state-level flexibility and variability in benefits and reimbursement policies, and thus operationally is closer to 51 separate programs than one monolithic program13 (Schneider and Wachino, 2013).
Private payers insure approximately two-thirds of the U.S. population (U.S. Census Bureau, 2014) and there are hundreds of different coverage and reimbursement policies and decisions—what is often likened to a crazy quilt of policies—underscoring the absence of a standardized approach to coverage and reimbursement of tests such as biomarker tests for molecularly targeted therapies (Graf et al., 2013; Gustavsen et al., 2010; Meckley and Neumann, 2010; Trosman et al., 2010).
Recent moves among large health care payers indicate the continuation of a consolidation trend, leaving three large corporations that dominate the U.S. health insurance market.14 Once the proposed merger between Anthem and Cigna is finalized, one insurer will be responsible for coverage and reimbursement decisions for more than 50 million members. Such payer consolidation increases the large insurers’ ability to negotiate fees with health care providers (Caffrey and Joszt, 2015), while intensifying consumers’ and providers’ concerns about the impact on competition (Japsen, 2015).
Decisions by payers—private insurers, private health plans, and public programs—are critical to the integration of biomarker tests for molecu-
___________________
13 States are responsible for making Medicaid coverage decisions. Consequently there are significant state-by-state differences in Medicaid coverage for genetic tests and services (SACGHS, 2006), which can serve as a source of disparity in access to biomarker tests for molecularly targeted therapies, discussed further in Chapter 5 of this report.
14 Aetna announced an agreement to acquire Humana in July 2015, and Anthem subsequently announced the purchase of Cigna in August 2015. Currently, the top three largest publicly traded U.S. health insurers are Anthem, UnitedHealth Group, and Aetna.
larly targeted therapies into routine clinical practice. Public and private health insurers and health plans seek to ensure that biomarker tests for molecularly targeted therapies provide information that is beneficial to the selection of treatment and leads to improved patient outcomes, and expect to use evidence of clinical utility to determine coverage and reimbursement decisions. However, evidence of clinical utility is often lacking, as discussed earlier (Frueh and Quinn, 2014; Parkinson et al., 2014; Schott et al., 2015; Simonds et al., 2013). Indeed, coverage decisions are often made in the context of a seemingly contradictory environment of a large volume of genetic and genomic information and relatively limited evidence of clinical utility (IOM, 2014). The lack of evidentiary standards for clinical utility influences payers’ willingness to cover and reimburse the cost of biomarker tests for molecularly targeted therapies (Cohen and Felix, 2014; Hresko and Haga, 2012). Test developers, for their part, require adequate reimbursement levels to ensure sufficient return on their investment (Deverka et al., 2014).
Value of Biomarker Tests for Molecularly Targeted Therapies
New molecular test codes that took effect in 2013 (see Appendix B for a discussion of coding issues related to biomarker tests for molecularly targeted therapies) provide some clarity for biomarker tests manufacturers and payers, but the ongoing development and subsequent implementation of biomarker tests for molecularly targeted therapies hinges on the ability of test developers and investors to capture the value of their innovations through adequate reimbursement levels. As one observer points out, even with the assignment of the new Molecular Pathology codes, such cost-based reimbursement is “not grounded in systematic measurement of the value of diagnostic tests” and results in a significant imbalance between the high value of the tests and limited amount of reimbursement (Goldman et al., 2013, p. 130).
A biomarker test’s clinical value lies in its potential to improve patient care by directing effective targeted therapy, or determining that such treatment would not prove to be beneficial to the patient. A biomarker test’s financial value is linked to its ability to direct treatment to a specific subpopulation of patients most likely to respond to a specific and often expensive molecularly targeted therapy, optimizing patient care while containing costs that would otherwise be spent on ineffective treatments. The total financial cost of developing a biomarker test is typically calculated by adding the cost of discovery research and test validation as well as the cost of developing evidence of clinical utility. For test manufacturers to recoup the significant costs of these research efforts, a test needs to be reimbursed at a sufficient level, which varies depending on the volume of test use. Although the market historically has rewarded discovery and
development of molecularly targeted therapies with high levels of reimbursement, this has not necessarily been the case for biomarker tests to guide selection of those therapies, which has slowed their adoption into clinical practice (Hayes et al., 2013).
Indeed, reimbursement levels for biomarker tests are much lower than for targeted treatments, which impacts the ability to generate the high levels of evidence needed to demonstrate clinical utility, resulting in what Hayes et al. (2013) have termed a “vicious cycle” of undervaluation of biomarker tests by payers, leading to few biomarker tests with established clinical utility on the one hand, and adoption of tests into clinical practice without sufficient evidence of clinical utility, on the other (Hayes et al., 2013). Given the potential of biomarker tests to identify patients that are likely to respond to treatment, thereby avoiding ineffective treatments, many have called for policies to “reconcile the potential mismatch between innovator incentives and social value” (Goldman et al., 2013, p. 132). Moreover, the current focus on the cost of expensive drug therapies underscores the value of tools such as biomarker tests that are capable of identifying which patients are most likely to benefit from expensive therapies (Faulkner et al., 2012; Fugel et al., 2014).
Provisions in the Protecting Access to Medicare Act (PAMA) of 2014 (see discussion in Appendix B) call for the introduction of a market-based payment system for tests under the Clinical Laboratory Fee Schedule (CLFS). The law requires “applicable laboratories”15 to report to CMS the rates paid by private payers for each clinical diagnostic laboratory test and the volumes of each test provided over a specified period of time. CMS will use this rate information to calculate a weighted median payment amount for each test. Applicable laboratories will have to begin to collect payment rates from private payers from July to December 2015 and report the rate information to CMS during the first quarter of 2016. CMS published its 2016 CLFS fee schedule, using two different fee-setting processes depending on the type of test (discussed further in Appendix B) and PAMA is scheduled to take effect on January 1, 2017 (Ray, 2015). It remains to be seen if the proposed market-based pricing approach for advanced laboratory tests, such as biomarker tests for molecularly targeted therapies, represents progress toward the goal of value-based reimbursement (Carey, 2014). There is concern, for example, that potential aggressive pricing of tests by laboratories may influence the final reimbursement rate calculated by CMS (Newcomer, 2015).
___________________
15 An applicable laboratory is defined by CMS as a lab that receives more than 50 percent of its Medicare revenues as paid under the CLFS or physician fee schedule. This would exclude hospital labs. In addition, labs that have Medicare revenues of less than $50,000 would be excluded.
Information That Payers Use to Support Coverage and Reimbursement Decisions
Health plans, insurers, and other payers seek certainty and value for their health care reimbursement dollars when making coverage and reimbursement determinations, but such certainty is difficult to attain, particularly in the field of precision medicine with its promising, but complex and rapidly evolving, tests and associated targeted treatments. The explosion in the number of tests leaves payers and providers to navigate what has been referred to as a “wild West” environment of ever-increasing numbers and complexity of tests without the necessary evidentiary support required to make decisions about clinical use and coverage (IOM, 2013d). As discussed earlier in this chapter, the lack of common evidentiary standards for biomarker tests for molecularly targeted therapies may limit health care providers’ and patients’ access to such tests.
Payers’ coverage and reimbursement policies are informed by evidence of analytic and clinical validity of a test, as well as evidence of clinical utility (Deverka and Dreyfus, 2014; Deverka et al., 2014; Meckley and Neumann, 2010). Payers expect to make coverage and subsequent reimbursement decisions based on evidence that the use of a biomarker test is: (1) medically necessary; (2) linked to improved outcomes for patients; and (3) better than the tests currently used in standard care or no test at all (IOM, 2015). Studies have found a significant variability of coverage of genomic testing among payers (Hresko and Haga, 2012; Meckley and Neumann, 2010; Trosman et al., 2010). One study found that lack of evidence or limited published studies demonstrating a test’s clinical utility is a key factor in insurers’ decisions to not provide coverage of disease-related genomic tests (Hresko and Haga, 2012).
In the absence of evidence of clinical utility, or consensus regarding evidentiary standards of clinical utility, payers rely on a variety of information sources to develop their coverage policies (Graf et al., 2013; Trosman et al., 2011). In addition to peer-reviewed studies published in medical journals, payers consider:
- Reviews of published studies on a particular topic, such as those conducted by the Agency for Healthcare Research and Quality (AHRQ), Blue Cross/Blue Shield Technology Evaluation Center, or Duke Evidence-based Practice Center.16
- Evidence-based consensus statements or guidelines from professional societies or other nationally recognized health care organiza-
___________________
16 AHRQ: http://www.ahrq.gov (accessed June 20, 2015); BCBS: http://www.bcbs.com/blueresources/tec/tec_staff.html (accessed June 20, 2015); Duke: http://guides.mclibrary.duke.edu/c.php?g=158201&p=1036021 (accessed June 20, 2015).
-
tions, such as the American Society of Clinical Oncology (ASCO) or the National Comprehensive Cancer Network (NCCN) (IOM, 2015).
- Guidance documents developed by multistakeholder groups such as CMTP (McDonough, 2015).
Private payers are generally believed to often follow Medicare’s coverage determinations. However, a recent study found that the coverage decisions for medical devices by 16 private payers aligned with Medicare decisions only half the time (Chambers et al., 2015). Most large health insurers have clinical policy divisions responsible for evaluating the evidence associated with medical technologies. Payers also consider a number of evidentiary frameworks in an effort to guide coverage and reimbursement decisions. For example, the ACCE process, supported by CDC’s EGAPP initiative discussed earlier in this chapter, is considered useful for examining clinical validity and utility (McDonough, 2015; Veenstra et al., 2013).17
Randomized controlled trials are widely considered the gold standard for generating evidence, as noted earlier in this chapter, but they are costly and require tracking large numbers of patients over time; as a result they are not typically conducted to evaluate biomarker tests for molecularly targeted therapies. This is due in part to the thinner profit margins of IVD test developers relative to pharmaceutical companies, which makes it less likely they would be able to support clinical trials on the scale of those funded by pharmaceutical companies (Faulkner, 2009). Other types of studies that payers may consider in evaluating clinical utility to support reimbursement include prospective–retrospective and observational studies (McDonough, 2015). In cases where compendia, which are summaries of drug information based on expert reviews of clinical data, indicate the need for a specific test to use a drug for a specific indication, payers tend to cover those tests. However, research indicates that information contained in compendia is of variable quality and often not supported by sufficient evidence (Abernethy et al., 2010).
Payer Use of Clinical Guidelines
Health care providers often rely on clinical practice guidelines (CPGs) to translate research findings into actionable steps for providing care (IOM, 2011a, 2013b). Payers, for their part, often turn to CPGs to help
___________________
17 Other initiatives include a recently developed tool to help payers assess the clinical and economic evidence for companion diagnostics associated with targeted drug therapies (Canestaro et al., 2015).
inform coverage and reimbursement decisions (Graf et al., 2013; Meckley and Neumann, 2010). CPGs are important sources of information for health care providers as well as payers, but there is significant variability in the quality of the underlying evidence base for the guidelines, and as the IOM has pointed out, “the CPG development process is often fragmented, lacking in transparency, and plagued by potential conflicts of interest in the membership of the CPG panels that might bias the resulting product” (IOM, 2013b, p. 294).
The IOM convened a consensus committee to develop CPG standards. The committee concluded that to be trustworthy, CPGs should be based on a systematic review of the evidence; be transparently developed by a knowledgeable and multidisciplinary panel in conjunction with patients and reflects patient preferences; provide ratings of evidence and strength of recommendations; and be updated regularly (IOM, 2011a). Further discussion of the development of CPGs, as well as the committee’s recommended approach to CPGs related to biomarker tests for molecularly targeted therapies, is presented in Chapter 5 of this report.
Payer Use of Clinical Pathways
In addition to clinical guidelines, payers have increasingly turned to the use of clinical pathways to guide care decisions. Such clinical or treatment pathways are developed by health insurers or provider organizations and tend to present fewer options than guidelines. Pathways reflect the evidence base as well as the total cost of care (IOM, 2013b). Given significant variation in clinical practice and differences in treatment costs, payers view clinical pathways as a way to control costs, with the potential to also improve quality (DeMartino and Larsen, 2012; IOM, 2013b,c, 2015).
Private health insurance plans provide financial incentives to health care providers for adherence to prescribed pathways, generally in the form of a per member/per month case management fee. Studies have shown that pathways can limit variation in treatment use, in the case of chemotherapy, for example, contributing to lower overall costs with no significant difference in overall survival rates (Patel et al., 2015). One of the largest U.S. health insurers, Anthem, manages a clinical pathways program for treatment of cancer that it developed through a process that includes review of national guidelines (e.g., NCCN, ASCO) and peer-reviewed evidence from clinical trials, supplemented by input by an external group of experts. The process results in the development of treatment pathways specific to tumor type, biomarkers, and patient characteristics. These pathways are updated quarterly to reflect new information, changes to guidelines, and new FDA-approved indications. The pathway treatment information is made available to health care providers when
they enter biomarker test results into the patient’s electronic health record (EHR). Though adherence to the pathways is not mandatory, health care providers receive an additional monthly reimbursement of $350 for each cancer patient treated through a recommended pathway. The insurer estimates a 3 to 4 percent annual reduction in the cost of treatment by using treatment pathways (PMC, 2015).
Patient advocates have raised concerns about payer use of clinical pathways and the linkage to financial incentives, pointing to significant differences in the evidence used and the processes by which payer organizations develop and update treatment pathways, and the limited transparency of pathway development. Patients and health care providers are concerned that such programs could have a negative impact on patient access to treatments designated as off pathway, and that such pathways may prevent health care providers from customizing care plans to individual patient needs (Avalere Health, 2015; Balch et al., 2015). The Personalized Medicine Coalition notes that clinical pathways and other decision support tools will be “challenged to keep pace” with changes in evidence and, importantly, what works for an individual patient (PMC, 2015).
Medicare’s Coverage with Evidence Development
As noted earlier in the chapter, CMS requires that tests be “reasonable and necessary,” which CMS links to improvement in health outcomes. Medicare’s coverage with evidence development (CED) program is a policy tool through which CMS agrees conditionally to cover new medical technologies provided that sponsors/manufacturers collect additional data to support more informed coverage decisions (CMTP, 2010, 2013). CED offers conditional coverage—in essence a third option between denial and approval of coverage—for promising medical tests, technologies, etc., that would not otherwise meet Medicare’s standards of evidence for “reasonable and necessary” treatment. CED represents an approach to balancing the competing priorities of—and inherent tensions between—access to emerging technologies and evidence-based medical policy to protect patients from harm.
CED first appeared on a formal basis in 2005 with CMS draft guidance describing its new CED approach, developed in the context of the use of CED for Medicare’s coverage of implantable cardioverter defibrillators (Tunis and Pearson, 2006). CMS released revised guidance in 200618 clari-
___________________
18 See https://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/ced.pdf (accessed May 1, 2015).
fying the specifics of the new policy, and issued revised draft guidance as recently as 201419 to better define CED in an effort to expand its use.
Essentially, CED creates a direct link between coverage of certain new technologies and the development of evidence through collection and analysis of clinical data to assess more fully the potential risks and benefits to patients. CED provides temporary reimbursement as the evidence is further developed by requiring patients to participate in a registry or clinical trial20 to qualify for coverage of the technology. This is particularly important for biomarker tests for molecularly targeted therapies, given the time and financial resources required to collect data on the impact of testing on patient management and clinical outcomes. Indeed, high research costs and extended timelines needed to conduct clinical trials are often viewed as major obstacles by manufacturers developing novel diagnostic tests (Schulman and Tunis, 2010). CED facilitates discussion and collaboration between payers and product developers about clinical trial design with the goal of efficiently developing data with which to assess clinical utility. Thus, CED offers a pathway to enable coverage for promising new technologies that are still considered experimental or investigational under CMS evidence requirements, and thus are excluded from traditional coverage (CMTP, 2010).
However, despite its potential public health benefits, many private payers are reluctant to embrace CED. Though CED has been used in a number of different areas, including Positron emission tomography imaging and treatment of localized prostate cancer, it has faced numerous challenges ranging from legal and limited funding to lack of sufficient data and problems with reaching consensus on clinical study designs (Tunis et al., 2011). As a result, CED has had limited application, with CMS implementing CED policies fewer than two dozen times to date, though the majority of recent NCDs issued by CMS have applied the CED approach. CMS has only twice used evidence generated through the CED process to revise an NCD (Health Affairs, 2015). Some observers argue that additional support for CED studies from research funding organizations such as the Patient-Centered Outcomes Research Institute (PCORI) and the National Institutes of Health (NIH), as well as collaboration with other federal agencies such as the FDA, is necessary in order for CED to transition successfully from a “one-off” tool to a more broadly and systematically
___________________
19 See https://www.cms.gov/medicare-coverage-database/details/medicare-coveragedocument-details.aspx?MCDId=27 (accessed May 19, 2015).
20 The statutory basis for requiring these studies is not under CMS’s authority, but that of AHRQ. AHRQ is authorized under the Social Security Act to conduct and support research on outcomes, effectiveness, and appropriateness of services for Medicare beneficiaries (Health Affairs, 2015).
used approach to developing evidence for novel technologies (Daniel et al., 2013).
CMTP has done extensive work in the area of CED, conducting research and analysis, and convening several multistakeholder working meetings on the topic, through which they have identified a number of improvements that could enhance the ability of CED to achieve its stated objectives. Given an intensified focus on constraining health care spending, the use of CED could help to meet the greater demand for evidence of comparative effectiveness of promising new technologies. CMTP cautions, however, that despite the potential role for CED application by private health plans, CED studies will often need to be coordinated across multiple health plans, requiring an independent party to align the approach across plans, as well as stakeholder groups to focus on study design and implementation (CMTP, 2010).
A recent IOM report suggests that the CED policy lever could be applied to create incentives for device industries to participate in evidence generation comparable to the pharmaceutical industry’s research efforts related to new drug development (IOM, 2013b). Others have suggested CED be used to pay for innovative therapies such as proton beam treatment for patients with prostate and other cancers, providing they are enrolled in a randomized trial to compare outcomes to other treatment approaches (Emanuel and Pearson, 2012).
CED holds the promise of an effective approach to providing coverage while simultaneously strengthening the evidence base for emerging technologies such as biomarker tests for molecularly targeted therapies. The application of CED to date, however, has yet to realize its full potential. CED studies have been found to have significant design flaws, including lack of rigorous data collection, which can fail to produce evidence that is of sufficient quality to inform sound coverage policy (Tunis et al., 2011). Other implementation challenges include significant staff reductions within CMS’s Coverage and Analysis Group that is responsible for CED implementation (Jacques, 2014) and lack of funding for CED studies (Health Affairs, 2015). While some private insurers, such as Priority Health of Michigan, are developing policies similar to CED to cover biomarker test–directed, off-label use of targeted therapies in the context of clinical trials (IOM, 2015), many private insurers are unwilling to pay for what they perceive to be research activities that should be covered by the test manufacturers (Newcomer, 2015). CED does offer payers an effective approach to cost savings; however, if the evidence that is being collected indicates that a specific test is not useful, payers may then have a strong basis to no longer pay for that test.
Palmetto GBA, a MAC, implemented a pilot program in 2011 called MolDX to identify molecular diagnostic tests and determine coverage and
reimbursement. The program currently covers laboratory tests in states that fall within Palmetto GBA’s geographic jurisdiction, and manufacturers and laboratories seeking coverage for their tests must provide Palmetto evidence of their test’s analytic and clinical validity, as well as clinical utility.21 Although several other MACs have followed MolDX’s decisions, CMS has not determined how or whether MolDX will be expanded on a national basis (Hughes, 2014). Such programs may be viewed as vehicles to facilitate discussion between payers and test developers about trial design, with the goal of ongoing data collection to support the development of evidence of clinical utility (Radensky, 2015). As with any study approach, CED has its advantages as well as its limitations. Data obtained are more broadly representative of patient populations compared to clinical trials, though the observational nature of CED studies may serve as a potential limit to the generalizability of study conclusions.
Challenges Related to Next-Generation Sequencing
Though single-analyte tests currently are the most commonly used biomarker tests for molecularly targeted therapies, test technology is rapidly advancing with the introduction of next-generation sequencing. NGS includes a number of advanced sequencing technologies that are distinct from Sanger sequencing, which has been in use for 40 years. In contrast to what is viewed as conventional molecular diagnostics tests, which typically involve single-test/single-result assays and return a single biomarker result (Trosman et al., 2015), NGS platforms “perform massively parallel sequencing during which millions of fragments of DNA from a single sample are sequenced simultaneously” (Schott et al., 2015, p. 1930). NGS offers the benefit of performing DNA sequencing more extensively, quickly, and less expensively than Sanger sequencing, and uses a smaller amount of DNA compared to multiple separate molecular diagnostic tests. Though NGS technologies typically seek to identify DNA-based variations, they may also be used to probe for other omics variations22 (Schott et al., 2015), which are likely to be integrated on an increasing basis with genomic data to refine precision medicine.
Laboratory use of NGS technologies (and related billing claims) may raise additional coverage and reimbursement challenges. Although NGS offers a relatively low incremental cost of assessing many molecular bio-
___________________
21 See http://www.palmettogba.com/palmetto/MolDX.nsf/DocsCat/MolDx%20Website~MolDx~Browse%20By%20Topic~General~8R5QUL0858?open&navmenu=Browse^By^Topic (accessed August 1, 2015).
22 Such omics variations may include those in the proteome (protein), transcriptome (RNA), metabolome (metabolites), and the microbiome (organisms living within and on humans).
markers, payers typically evaluate and reimburse tests on a per-analyte basis and reimburse only the medically necessary components of gene panels. For this reason, a laboratory seeking reimbursement on a per-panel level may see those claims denied or pared back. In this context, some payers consider NGS technology to be investigational, while others are in the process of developing specific NGS coverage policies. According to a survey of large private health plans about next-generation tumor sequencing (NGTS), 80 percent of health plan representatives indicated that such tests do not meet their criteria for medical necessity. Seventy percent said they view gene panels as bundles of individual gene tests, and each gene marker needs to be evaluated separately (Trosman et al., 2015).
NGTS can be viewed as disruptive to the current coverage and reimbursement framework in that it supports an integrated approach to patient care, one that includes both standard and experimental elements. Though offering a potential broader choice of therapy options (which are often not grounded in a high level of evidence), NGTS runs against the grain of current approaches to reimbursement—most notably the separation of standard of care from experimental activities for reimbursement purposes. Large cancer centers such as MD Anderson Cancer Center have developed a sophisticated process to report NGS results to distinguish standard of care from investigational use for reimbursement purposes (Mendelsohn, 2013; Meric-Bernstam et al., 2013; Trosman et al., 2015).
CMTP recently released initial guidelines for coverage of NGS testing in oncology that were developed by the Center’s Green Park Collaborative, a multistakeholder, public–private group. The guidelines represent a framework for coverage of targeted NGS gene panels that recognizes the unique evidentiary challenges of demonstrating clinical utility for a panel of tests rather than evaluating a specific diagnostic test for a well-defined clinical use. The initial draft coverage guidelines include two specific recommendations for payers: the first is that payers cover NGS panels with 5 to 50 genes when those panels include a subset of genes considered to be medically necessary,23 and the second recommendation is for payers to rely on the College of American Pathologists’ accreditation program and proficiency testing to assure the analytic validity of NGS tests (CMTP, 2015).
The National Institute of Standards and Technology recently developed and made available reference material to facilitate the validation of clinical NGS tests by laboratories, which may support coverage decisions. This new DNA reference material will enable clinical laboratories to
___________________
23 The guidelines recommend payment for these panels at a rate not to exceed the cost of individual sequencing of the medically necessary genes by other methods.
determine whether they are providing accurate genomic analyses, which may in turn increase the confidence of health insurers in the accuracy and quality of such test results, thereby increasing the likelihood that payers will approve payment for NGS tests (Pear, 2015).
Lack of Alignment of Regulatory and Reimbursement Decision Processes
In addition to multiple regulatory pathways, the policy environment for biomarker tests for molecularly targeted therapies is further complicated by the existence of separate processes for regulatory and reimbursement decisions. FDA and CMS both play critical roles in the adoption of biomarker tests into clinical practice. Each agency has its distinct statutory mandate that in turn defines its evidentiary standards to guide approval decisions: FDA evaluates drugs and devices based on evidence that the product is safe and effective; CMS coverage determinations are based on whether the product is reasonable and necessary for Medicare beneficiaries (Health Affairs, 2015). In broad terms, FDA oversight is intended to ensure that the benefits of marketed products are greater than the risks under carefully defined circumstances. CMS looks for evidence that medical interventions are likely to improve outcomes for patients when available for broad clinical use.
The Medicare program is the largest single payer for laboratory tests in the United States, and therefore influences Medicaid and private payer coverage and reimbursement decisions (OIG, 2013). Medicare is required by law to pay only for items and services that are “reasonable and necessary” for its beneficiaries, which is interpreted generally as improving clinically meaningful health outcomes, although determining the precise definition of these terms has “proven to be an enduring challenge” (Neumann and Chambers, 2012, p. 1775). For example, in 1989, Medicare released a proposed regulation that defined reasonable and necessary as “safe, effective, non-investigational, appropriate, and cost-effective.” The proposal was withdrawn after criticism from external stakeholders—including some medical professional societies and the medical device industry—who argued that such a definition would result in patients being denied necessary care. In addition, the use of “least costly alternative” in CMS reimbursement policy was successfully challenged in the courts in 2008. Efforts to clarify the terms continue, with some calling for a legislative remedy to provide definitional clarity (Neumann and Chambers, 2012).
The evidentiary requirements for regulatory decisions by FDA are viewed as less stringent than the evidence requirements for reimbursement decisions (Deverka and Dreyfus, 2014). As Walcoff and Pfeifer note
(2012, p. 305), “some stakeholders have suggested that FDA clearance or approval be the litmus test for meeting the reimbursement standard of reasonable and necessary” (Walcoff and Pfeifer, 2012, p. 305). Indeed, the findings of a 2013 study suggest that compared to FDA, CMS took a more restrictive approach to coverage of medical devices than of drugs, representing a significant challenge for device manufacturers (Health Affairs, 2015).
Given the different agencies’ requirements, two separate evidentiary standards exist, requiring product developers to generate two separate sets of data. Clinical trials designed to generate the necessary evidence for FDA that a product is safe and effective tend to be conducted, for example, with tightly controlled patient populations. CMS, for its part is interested in knowing how the product performs in “real-world” Medicare patients who tend to be more diverse and have more comorbidities than the typical clinical trial patient. Given the two agencies’ different statutory mandates and respective evidentiary requirements, FDA may approve or clear a product that does not meet Medicare’s reasonable and necessary requirements. On the other hand, Medicare may approve a product for payment that FDA has not approved or cleared as safe and effective (as in off-label use of a product already on the market) (Health Affairs, 2015). These two separate review processes can lead to delays in getting a test on the market or limit the success of market entry—both of which potentially can have a negative impact on patients’ access to new tests. Coordination of evidentiary standards would facilitate a streamlined decision process for biomarker tests and their associated targeted therapies.
Communication About Biomarker Test Performance Characteristics and Use
The availability of two paths to market for test developers discussed above creates challenges for health care providers who are faced with the difficulty of selecting the appropriate biomarker test and most effective treatment for their patients (IOM, 2010). Simply put, “it is clear that the performance of many new tests has not been well established or well documented” (Parkinson et al., 2014, p. 1430).
The increasing number of complex biomarker tests for molecularly targeted therapies being implemented in clinical practice has raised concerns about insufficient access to information about test performance and intended use. Health care providers need accurate information to support effective clinical decision making. In particular, they need such information to avoid use of biomarker tests with poor clinical validity or insufficient evidence of clinical utility that could result in harm to the patient—either through under- or overtreatment (Yu et al., 2015). Indeed,
given the complex nature of biomarker tests for molecularly targeted therapies, the continuous evolution of biomarker knowledge and emergence of new tests, and the impact of evolving evidence about new uses of existing tests, health care providers and patients as well as payers need information on test performance and intended use that is more transparent than currently available.
CREATING A SUPPORTIVE POLICY ENVIRONMENT FOR BIOMARKER TESTS FOR MOLECULARLY TARGETED THERAPIES
The committee identified a number of key regulatory and reimbursement challenges related to biomarker tests for molecularly targeted therapies, including the absence of common standards of evidence of clinical utility; the lack of alignment between the regulatory and reimbursement approval processes; the information gaps and communication challenges on the part of health care providers and patients; and the need for ongoing assessment of the clinical utility of biomarker tests. The committee subsequently developed a recommended set of associated policy measures. These recommendations represent important steps in the development of a supportive policy environment as one component of a rapid learning system for biomarker tests for molecularly targeted therapies. Progress toward achieving the promise of precision medicine is likely to be significantly enhanced by timely implementation of these recommendations.
Establishing Common Standards of Evidence
Clear, consistent, and reasonable standards of evidence for biomarker tests for molecularly targeted therapies are required in order for these promising technologies to realize their potential to improve patient outcomes (Faulkner, 2009). To ensure patient access to effective technologies, renewed focus must be brought to bear on the existing lack of common standards of evidence to assess clinical utility:
Given that demonstration of clinical utility is currently often a missing element in the evaluation of new laboratory tests, there is a critical need to raise the consciousness about its importance, to encourage the use of better planning and methodologies in addressing the search for utility, and to create guidance that will be of value to all stakeholders working to medical care more effective. (Parkinson et al., 2014, p. 1440)
Of critical importance is that the federal government needs to play a key leadership role in raising awareness of the urgency of this need by bringing together a broad group of public and private stakeholders to work in a collaborative manner to achieve the establishment of clear, consistent, and reasonable evidentiary standards. As noted by Ginsburg
and Kuderer: “No organization has owned the evaluation of genetic testing; it has been a distributed process with widespread heterogeneity among stakeholders” (Ginsburg and Kuderer, 2012, p. 4237). Given the complexity of issues involved in establishing evidentiary standards for the clinical utility for biomarker tests for molecularly targeted therapy, “the convening power of a central body . . . will be required to achieve meaningful consensus” among diverse stakeholders (Callahan and Darzi, 2015, p. 1566).
The federal government should champion the effort, clearly articulating the urgency and value of the work to be done. This leadership role would align with and support the federal government’s launch of its Precision Medicine Initiative (Collins and Varmus, 2015). Thus, the committee recommends that the Secretary of HHS should facilitate the development of common clinical utility evidentiary standards that are applied for initial and ongoing coordinated regulatory, coverage, and reimbursement decisions for biomarker tests for molecularly targeted therapies. One mechanism for development of these evidentiary standards could be convening one or more independent, public–private, multistakeholder bodies (Recommendation 1).
Furthermore, the process for developing evidentiary standards should recognize that the standards being developed will be used as the basis for different types of decisions, including clinical, regulatory, coverage, and reimbursement, as well as guideline recommendations, and will evolve over time as more evidence accumulates through further research and clinical use of biomarker tests for molecularly targeted therapies. The development of a common evidentiary framework should focus on “sufficient” evidence for rational decision making instead of “ideal” or “best” evidence approaches, yet at the same time such standards must be strong enough to address payers’ decision-making requirements and to ensure that patients are not harmed by the introduction into clinical use of new tests that lack adequate evidence (Faulkner, 2009). As a result, HHS should ensure the development of consistent and coordinated evidentiary standards and study design approaches, including rapid learning systems, that simultaneously accommodate the various types of decisions (including clinical, regulatory, coverage/reimbursement, and guideline recommendations), and facilitate the ongoing development of evidence of clinical utility.
The stakeholder community involved in biomarker tests for molecularly targeted therapies is a diverse group encompassing patients, health care providers, academia, industry, government agencies, and payers, each with their own perspective on what qualifies as “adequate” evidence for clinical utility. As Ginsburg and Kuderer further note: “more dialogue and coordination among stakeholders is needed to facilitate the develop-
ment of the necessary evidence base. It is equally apparent that test development and reimbursement need to focus on the clinical utility of the test and the net benefit to patients” (Ginsburg and Kuderer, 2012, p. 4237). Establishing common evidentiary standards would help ensure that all stakeholders—patients, health care providers, payers, test developers, and policy makers—are all “working from the same playbook” and have a clear understanding of expectations for evidence development (Faulkner, 2009). This type of multistakeholder dialogue to develop evidentiary standards for coverage and reimbursement can be viewed as “reimbursement science,” which focuses on the development of new tools, standards, and approaches for comparative effectiveness research and the assessment of the value of products covered by public and private health plans. Thus it is similar to “regulatory science,” which develops evidentiary standards to guide clinical research intended to inform regulatory decision making (IOM, 2015).
To serve as an effective forum for discussion and collaboration, the process for developing evidentiary standards must include the diverse range of stakeholder perspectives and policy priorities, as is also the case for regulatory science discussions. In encompassing a broad array of participants, such a process provides a framework for public engagement; “transparency and accountability can be expressed in a dialogue between those who determine policy and those who are affected by it” (Callahan and Darzi, 2015, p. 1567). Collaborative, multistakeholder approaches are critical to the integration of complex biomarker tests for molecularly targeted therapies into clinical practice. The broadest possible coalition of public and private perspectives—government, policy makers, payers, test developers, health care providers, and patients—will ensure balanced, pragmatic, and viable approaches to the development of a common evidentiary framework (Faulkner, 2009; Hoffman et al., 2010). The committee recognizes that the involvement of a variety of stakeholders will be critical to ensure that clinical utility studies are designed to reflect a range of decision-making needs and to strike an acceptable balance between ideal utility assessment and study feasibility. Stakeholders participating in these initiatives should include patients, health care providers, clinical practice guideline developers, public and private payers (including CMS), FDA, test developers, pharmaceutical companies, molecular pathologists, clinical laboratory geneticists, informaticians, and research funders (e.g., PCORI, NIH, AHRQ).
The committee acknowledges the considerable challenges involved in bringing together diverse perspectives to establish common standards of clinical utility across all stakeholders, but believes it is imperative that this initiative be undertaken to enable biomarker tests for molecularly targeted therapies to achieve the promise of precision medicine. This is
consistent with the long-term approach required to establish clinical utility: the need to plan from the early stages of development and anticipate the different types of data that will be necessary to build on evidence of analytic validity and clinical validity (Deverka et al., 2014; Parkinson et al., 2014), as well as ensuring that new evidence thresholds are sensitive to the value of ongoing research (Veenstra et al., 2013). The committee recommends that analytic and clinical validity of biomarker tests should be assured prior to assessing clinical utility.
In addition, the committee recognizes that evidentiary standards for clinical utility may vary across different diseases. For example, although most biomarker tests are currently used to guide treatment for cancer, the rapid rate of advances in understanding the molecular basis of disease likely will lead to their increased use to direct therapies for a range of diseases, potentially requiring distinct evidentiary standards. Therefore, HHS could determine that more than one advisory body may be necessary to develop disease-specific evidentiary standards of clinical utility for biomarker tests for molecularly targeted therapies.
The evidentiary standards for clinical utility developed in collaboration with multiple stakeholders will be used to guide the creation of new labels for biomarker tests and corresponding therapies (see Recommendation 3). Such standards will also be useful in the context of the collaborative development of clinical guidelines (see Recommendation 10).
The committee stressed in its deliberations the progressive nature of establishing evidentiary standards of clinical utility. The committee strongly views this as an ongoing, iterative process with standards evolving over time as evidence strengthens, methods and data infrastructure are improved, and the field of precision medicine itself evolves. This may initially appear counterintuitive as standards are often considered to be fixed in time, and to have some degree of stability over time in order to be useful. In light of the evolving nature of the process of establishing evidentiary standards, the committee recommends that HHS continue to support the ongoing refinement of common evidentiary standards as they evolve. Establishment of standards will require ongoing and sustained discussion across all stakeholders as new scientific discoveries and technological advances influence the development of new biomarker tests as well as new uses for existing tests.
Many effective multistakeholder partnerships exist in the genomics domain that may serve as models for, or provide input to, the process for developing evidentiary standards for biomarker tests for molecularly targeted therapies. The Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children (SACHDNC) exemplifies a multistakeholder effort to facilitate consensus, and has as its focus the devel-
opment of standards for newborn screening for genetic disorders.24 Consistent with this IOM Biomarkers consensus committee’s recommendation, the SACHDNC brings together multiple stakeholders with different perspectives, including state health officials, laboratory medicine experts, researchers, academics, private foundations, and representatives from federal agencies including CDC, the Health Resources and Services Administration, NIH, AHRQ, and FDA to discuss which disorders should be included in the Recommended Uniform Screening Panel (RUSP).
The SACHDNC meets approximately three times per year to discuss and vote on whether additional disorders should be added to the panel. Once a new disorder has been nominated to be added to the RUSP, the committee reviews the nomination to see if there is sufficient evidence to support further investigation. If there is, they send the nomination to an external evidence review board. If the nomination moves on, then work groups conduct a full review of the research on the disorder in question. If they find sufficient evidence to recommend screening, they propose that the Secretary of HHS include the specific disorder in the panel (Kemper et al., 2014).
The SACHDNC developed an evidence-based approach to recommending whether a condition or disorder should be included in the RUSP, which could prove informative for this committee’s recommendation. An external group of experts is consulted for each condition being considered, and provides the SACHDNC with an evidence-based assessment of the risks and benefits of screening for each condition. The SACHDNC reviews the expert report and assigns one of five ratings based on the evidence presented and the consensus of the committee. The SACHDNC collaborated to develop a RUSP that includes screening for 32 core disorders and 26 secondary disorders (HRSA, 2015; Kemper et al., 2014). This group’s experience developing standards and evaluating tests on a case-by-case basis in the context of variability in state-level regulations serves as a relevant model for the committee’s recommended process for developing standards.
Other examples of independent, multistakeholder collaborations exist, such as the partnership that was formed between Friends of Cancer Research and the Engelberg Center for Health Care Reform at the Brookings Institution. These two organizations joined forces to convene an annual conference on clinical cancer research, which brought “together a diverse group of experts in cancer drug development from academic and clinical research centers, federal health and regulatory agencies, patient advocacy organizations, and the private sector to develop practi-
___________________
24 See http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders (accessed July 22, 2015).
cal, consensus-driven solutions to critical challenges in the development of drugs for cancer.”25 Importantly, many of the conference panels contributed to significant changes in cancer drug development, including the Advancing Breakthrough Therapies for Patients Act of 2012, which facilitated the expedited development of exceptional new drugs, and the development of the Lung-MAP trial, a biomarker-driven clinical trial that matches patients with investigational new treatments.26
Another multistakeholder collaborative group is CMTP’s Green Park Collaborative (GPC), a multistakeholder forum that brings together patients, health care providers, payers, researchers, regulators, life sciences companies, and other stakeholders to develop recommendations to guide the generation of evidence needed to inform both clinical and reimbursement decisions (CMTP, 2013). GPC’s most recent effort focuses on guidelines for clinical use of NGS in oncology (CMTP, 2015). GPC’s multistakeholder process and proposed policy framework could serve as a model for the recommended process to develop standards of evidence required for biomarker tests for molecularly targeted therapies.
A final example is the Molecular Evidence Development Consortium (MED-C), a nonprofit public–private partnership of stakeholder representatives—including patients, payers, providers, regulators, industry, laboratories, and pharmaceuticals—collaborating to establish policy on the validation of genomic tests by creating a standardized outcome registry. The overarching goal of MED-C is to apply a systematic approach to large-scale data collection to advance evidence of clinical utility for molecular diagnostic tests and treatment (Dickson, 2015). The initial pilot program, a registry for NGS in patients with non-small cell lung cancer, has recently been funded. As this program expands, it is hoped that the registry can serve to drive accrual on clinical trials and other precision medicine-related research efforts (MED-C, 2015).
These are only a few examples of the multistakeholder approach recommended by the committee. These and other working partnerships may serve as a guide for HHS as it addresses the long-standing and increasingly urgent need for a common evidentiary framework for current and emerging biomarker tests for molecularly targeted therapies—to facilitate patient access to appropriate tests, and to protect them from potential harm as well.
___________________
25 See http://www.focr.org/conference-clinical-cancer-research (accessed October 11, 2015).
26 See http://www.focr.org/about-us (accessed October 11, 2015).
Creating an Integrated Review Process for Regulatory and Reimbursement Decisions
Another significant challenge to the appropriate and effective clinical adoption of biomarker tests for molecularly targeted therapies is the lack of alignment between the regulatory and reimbursement decision processes. This misalignment creates significant inefficiencies, and “if the practice of clinical medicine is to benefit from the biological and technical advances resulting from the new ‘omics’ era, regulatory and reimbursement expectations need to be more aligned” (Parkinson et al., 2014, p. 1440). Thus, the committee recommends that the Secretary of HHS should facilitate the development of a new integrated federal review process involving FDA and CMS, as a pathway for coordinated regulatory, coverage, and reimbursement decisions for IVD and LDT biomarker tests for molecularly targeted therapies, including multianalyte tests performed using current or new technologies, and any corresponding molecularly targeted therapies27 (Recommendation 2).
In crafting its recommended FDA28–CMS integrated review process for biomarker tests for molecularly targeted therapies, the committee was cognizant of the different statutory authority of the two agencies, as discussed earlier in this chapter. The committee is not calling for statutory reconciliation of the two agencies, which would require congressional action and would go beyond the narrow context of the committee’s charge: biomarker tests for molecularly targeted therapies. Rather, the committee is calling for the two agencies to work closely together, to coordinate their review approaches with the goal of creating a streamlined decision-making process for biomarker tests for molecularly targeted therapies that developers could voluntarily choose to undergo.
The committee’s recommended integrated review process is a voluntary, multifaceted process encompassing regulatory, coverage, and reimbursement review, which applies to a limited subset of all clinical tests: biomarker tests for molecularly targeted therapies. This integrated review process is, by design, sufficiently flexible to accommodate a variety of scenarios, as depicted in Figure 3-3. For example, tests could be reviewed simultaneously with an associated targeted therapy: This could be the case when a sponsor is seeking initial approval for a novel drug, or seeking expansion of intended clinical use of an approved drug. Tests could also be submitted separately for integrated regulatory and reimbursement
___________________
27 This coordinated pathway is designed to reflect the current predominant fee-for-service reimbursement system for clinical tests.
28 Given that the committee’s recommended review process could incorporate both test and drug review, both the drug (CDER) and device (CDRH) centers of FDA would be involved.

NOTE: CMS = Centers for Medicare & Medicaid Services; FDA = Food and Drug Administration.
* See Table 3-1 for descriptions of various approaches to evidence generation.
review. For example, where regulatory approval of an intended use of a drug already exists based on an existing biomarker test, a test to demonstrate further or differential benefit may be submitted in order to secure regulatory and reimbursement approval. Multianalyte tests using technologies such as NGS not associated with a specific therapy, or linked to multiple therapies, could be submitted separately. Thus, the integrated review process recommended by the committee acknowledges the expanding use of newer testing technologies such as NGS that are typically not linked to a single treatment, and in so doing, offers a more comprehensive framework than FDA’s current companion diagnostic model. The committee’s recommended integrated review process does not replace that model—which as discussed earlier in this chapter is likely to evolve given the increased use of newer technologies such as NGS—rather it serves as a complementary review pathway.
Indeed, while a version of this integrated process exists in a somewhat related form in the companion diagnostic model, as noted earlier
in this chapter, drug–test co-development may facilitate, but does not guarantee, reimbursement. Of critical importance, developers of biomarker tests for molecularly targeted therapies—whether IVDs or LDTs, and including multianalyte tests using technologies such as NGS—that choose to undergo the committee’s proposed integrated review process (and successfully reach the approval stage) would be guaranteed reimbursement by CMS through a national uniform coverage decision. In contrast, tests that do not go through this integrated review process will not have the benefit of guaranteed reimbursement and will need to seek reimbursement through traditional avenues. Application of common evidentiary standards of clinical utility will be required for coordinated regulatory, coverage and reimbursement decisions; these standards should be developed through the process as described in Recommendation 1 above.
The committee is not expecting FDA to become a payment organization, nor for CMS to become a regulatory authority. CMS will maintain its authority to set payment levels through its usual mechanisms, but it will work closely with FDA to conduct a coordinated review for coverage, reimbursement and regulatory decisions. Consistent, transparent evidentiary standards should span regulatory and reimbursement considerations and lay the groundwork for a more streamlined, efficient review process.
Once FDA and CMS have made their coordinated decision regarding regulatory and reimbursement approval for a test or test–drug combination, other payers are encouraged to make a similar decision regarding test reimbursement on a national basis. As noted earlier in this chapter, CMS covers a large patient population and private payers often follow the lead of CMS in terms of reimbursement decisions, and the committee hopes this continues to be the case in regard to its recommended integrated review process. For this to occur, it will be important for private payers to have confidence in the rigor of the integrated review process, including the standards of evidence that are being applied when making such decisions.
The process for developing evidentiary standards described in Recommendation 1 above will determine a minimum level of evidence of clinical utility to enable a biomarker test for molecularly targeted therapy to enter into clinical use, with the understanding that stronger evidence may be generated through the monitoring of the test’s clinical use and patient outcomes, including potential changes to patient management, over time. Incentives to continue to refine tests, in the form of increased reimbursement as evidence of clinical utility is generated, may be an option if considered feasible and effective.
The committee’s recommended integrated review process is designed
to address the lack of clarity and consistency of reimbursement decisions by payers, which are considered to be variable and opaque. The committee’s recommended coordinated pathway would enable concurrent regulatory and reimbursement decisions for test–drug combinations, thus developing a more efficient, streamlined review process for both regulators and payers, and creating greater certainty for test and drug manufacturers and more rapid reimbursement decisions for health care providers and patients needing to make treatment decisions. Consistent with the committee’s envisioned rapid learning system, the committee recommends that summary data—upon which approval decisions are made within the context of the integrated review process—will be made publicly available.
The integrated review process also includes requirements for standardized test labeling and data collection efforts. As discussed further below, a new type of test label will include standardized test information to enhance provider and patient communication and understanding of test performance and intended use. Moreover, labels for biomarker tests and their associated therapies will be updated in a coordinated fashion.
Finally, as an important initial step in the committee’s vision of a rapid learning system for biomarker tests for molecularly targeted therapies, the integrated review process will include financial incentives designed to encourage health care providers and health systems to submit test data on patient use and outcomes to a national data repository, as discussed further below.
Thus, in summary, this coordinated pathway should accomplish all of the following through application of common evidentiary standards (as described in Recommendation 1):
- Primary (and follow-on) biomarker test review and approval with detailed test labeling requirements (Recommendation 3).
- Drug review and approval with detailed labeling that includes standardized biomarker test information (Recommendation 3), when occurring concurrently with biomarker test review.
- A national uniform coverage decision for a biomarker test and molecularly targeted therapy in specific clinical uses, including financial incentives for data submission on use and outcomes (Recommendation 7).
- A defined process for coordinated updates of biomarker test and drug labels (Recommendation 3).
- Public sharing of the summary data upon which the review process based the approval and coverage decisions for a biomarker test and drug combination.
An important note is that the committee crafted this recommendation to reflect the reality of the current dominance of the fee-for-service reimbursement approach in the U.S. health care system. If alternative payment arrangements and mechanisms such as accountable care organizations or global, bundled, and value-based payments make deeper inroads into the U.S. health care system, such an integrated regulatory and reimbursement review process would need to be reassessed, based on a clearer understanding of the impact of the new payment models.
The committee’s call for FDA and CMS to work closely together in an effort to streamline the decision-making process for biomarker tests for molecularly targeted therapies builds upon existing precedent. Indeed, FDA and CMS are well positioned to work together to support timely review, coverage, and reimbursement of new advanced tests; a key example of interagency collaboration is the FDA–CMS parallel review pilot program.29 Historically, FDA and CMS have worked independently, with separate staff focused on different points of a product’s development lifecycle, and with different evidentiary expectations from FDA’s focus on safe and effective, to CMS’s focus on reasonable and necessary. In 2010, FDA and CMS introduced the concept of a parallel review process and officially launched a pilot program the following year (Gaffney, 2014).
The pilot parallel review program was developed in response to the fact that attaining FDA approval of a product based on safety and efficacy does not necessarily result in a timely determination by Medicare that the product is medically necessary and therefore should be covered (Pomager, 2014a,b). The stated goal of the program was to reduce the time between FDA approval and CMS national coverage determinations, which are important for products to be integrated broadly into clinical practice (Messner and Tunis, 2012).
The pilot parallel review program is only available for certain qualifying technologies30 and offers three key advantages: (1) it targets products deemed innovative by developers, FDA, and CMS, with the capacity to improve patient care; (2) it provides product developers with a clearer sense of the evidence expectations of both regulators and payers; and (3) it promotes better evidence generation for clinical decision makers about novel technologies (Messner and Tunis, 2012). A notable success
___________________
29 See http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm274833.htm (accessed September 11, 2015).
30 The parallel review program is only available for qualifying new medical device technologies that must meet certain criteria. See http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHInnovation/ucm456149.htm#parallelreview (accessed September 9, 2015).
that emerged from the parallel review pilot program is Cologuard,31 the first non-invasive colorectal DNA screening test, and the first product to receive simultaneous decisions from FDA (premarket approval) and CMS (national coverage decision) (Pomager, 2014a,b).
FDA’s CDRH established the Payer Communication Task Force (PCTF) to manage the parallel review program with CMS and to examine potential ways to reduce the time between FDA approval or clearance and payer (public and private) coverage decisions. PCTF has incorporated key lessons learned from the parallel review program to identify ways for payers to interact with FDA and medical device manufacturers earlier in the development process: “The PCTF believes … that early engagement between device manufacturers, CDRH, and payers will allow for the design of clinical trials that may produce required outcomes for both regulatory approval or clearance and positive coverage determinations” (FDA, 2015b).
The pilot program was designed to run through the end of 2015, and it is unclear at this time whether it will be extended. Significant staff reductions within CMS’s Coverage and Analysis Group that oversees the parallel review program have had a limiting impact on the implementation of the program (Health Affairs, 2015; Jacques, 2014). Increased CMS staff resources may be necessary to adequately support the coordination required to implement the committee’s recommended integrated review process.
Another existing group that could help to inform the FDA–CMS integrated review process recommended by the committee is the FDA–CMS Task Force on LDT Quality Requirements. The interagency task force was established in the spring of 2015 to leverage the resources of the two agencies involved in the oversight of LDTs, with the goal of avoiding duplication of efforts and increasing efficiency through greater collaboration (Shuren and Conway, 2015). Such collaborative efforts between FDA and CMS are critically important and should be expanded.
The development of a pathway for coordinated regulatory and reimbursement decisions has the potential to create a more harmonized process for market entry for biomarker tests for molecularly targeted therapies. Such FDA–CMS integrated review with simultaneous regulatory and reimbursement decisions will serve to reduce delays inevitable with dual decision-making processes, and by clarifying requirements for coverage and reimbursement will enable test and drug manufacturers to plan accordingly, which in turn will help to increase the rate at which new
___________________
31 See http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm409021.htm (accessed September 9, 2015).
technologies are made available to health care providers and their patients (Hwang et al., 2015).
In summary, the integrated review process recommended by the committee is a voluntary process that applies to a small subset of all clinical tests—biomarker tests for molecularly targeted therapies—represents a complementary pathway to the current companion diagnostic model, and builds on an existing precedent of cooperation between FDA and CMS. The committee recognizes that its recommended approach may entail implementation challenges, but the current lack of coordination between the two agencies is detrimental to patients and test and drug manufacturers. The coordinated review process would provide benefits to both patients and manufacturers: test and drug manufacturers could receive FDA approval and meet evidentiary standards for payment in parallel, providing patients with faster access to appropriate biomarker tests for molecularly targeted therapies. Broader health care system and societal benefits from such a coordinated review process also should be acknowledged as some biomarker tests in clinical use and covered by insurers have been found to be ineffective and/or harmful to patients (FDA, 2015c).
Enhancing Communication and Information About Tests
Given the proliferation and complexity of biomarker tests for molecularly targeted therapies, it is challenging for health care providers to understand fully the performance characteristics and evidence for specific intended uses of biomarker tests and molecularly targeted therapies. Detailed information should be contained in the biomarker test’s label, which should state in clear language the test’s use, the specific actions to be taken, the relevant clinical condition as well as information on analytic validity, clinical validity, and clinical utility, if available. Moreover, given the speed at which predictive tests are evolving, test information needs to be updated regularly and reflect new information as the tests are continuously evaluated for their impact on patient outcomes (IOM, 2010).
Transparent labeling of biomarker tests for molecularly targeted therapies designed to provide useful information will help physicians make effective decisions, enable patients to be engaged in the decision-making process, and help payers seeking information on which to base coverage and reimbursement decisions. Thus, the committee recommends that FDA should develop a patient- and provider-friendly standardized label for IVD and LDT biomarker tests to facilitate transparency of test performance characteristics and the level of evidence for the intended use(s) of the test. FDA or laboratory accrediting bodies should approve the label for each biomarker test, including tests not reviewed through
the integrated process (specified above) (Recommendation 3). The committee recommends a two-step process. First, FDA develops a uniform, transparent labeling system, defining the specific contents of the biomarker test label. Second, although the committee recognizes the product label as the property of the drug or device manufacturer or developer, the contents of the label are to be approved by either FDA or laboratory accrediting bodies. This standardized labeling system will apply to all biomarker tests for molecularly targeted therapies—regardless of whether the tests undergo the integrated review process described above.
The committee recommends that labels should prominently feature an easily understood ranking system (e.g., 4-star scales) separately for the evidence to support the clinical validity and clinical utility for each intended clinical use of a test. The evidence ranking standards could be developed by the process described in Recommendation 1. The analytic validity of a biomarker test is assumed as part of the labeling process; however, the analytic sensitivity and specificity will be included on the label. The proposed star system is similar to the system used to rank variants that are reported to NIH’s ClinVar database (Rehm et al., 2015).32 A single star would signify limited or single-source validation of clinical validity or utility for a given intended use, while 3 or 4 stars would indicate widespread acceptance of clinical validity or clinical utility commensurate with an expert panel recommendation or clinical practice guideline. This would enable the labels to be flexible enough to accommodate emerging intended uses (while maintaining transparency as to their level of validation) as well as enabling analytically valid “follow-on” panels and other multianalyte tests, such as NGS tests of well-established single-analyte tests to label themselves with 3 or 4 stars, as appropriate. Such evidence ranking standards could be developed in conjunction with the standards for clinical utility.33
___________________
32 ClinVar is a freely accessible, public archive of reports of the relationships among human variations and phenotypes, with supporting evidence. ClinVar facilitates access to and communication about the relationships asserted between human variation and observed health status, and the history of that interpretation. ClinVar collects reports of variants found in patient samples, assertions made regarding their clinical significance, information about the submitter, and other supporting data. The alleles described in submissions are mapped to reference sequences, and reported according to the Human Genome Variation Society standard. ClinVar then presents the data for interactive users as well as those wishing to use ClinVar in daily workflows and other local applications. See http://www.ncbi.nlm.nih.gov/clinvar (accessed June 6, 2016).
33 Other ranking systems relevant to biomarker tests for molecularly targeted therapies exist, such as the Clinical Pharmacogenetics Implementation Consortium. See https://www.pharmgkb.org/page/cpic (accessed June 6, 2016) for guidelines for gene/drug pairs. The strength of these recommendations (strong, moderate, or optional) is based on a consideration of the quality of supporting evidence and any additional clinical implications,
The committee’s labels provide opportunity for manufacturers to specify potential intended uses of their biomarker tests for those tests that have not undergone integrated review. For this reason, these claims of clinical validity, or clinical relevance, will need to be evaluated. Multiple methods of clinical validation exist; some interpret current CLIA regulations to require clinical validation of tests prior to their use (Ferreira-Gonzalez et al., 2014), the New York State Department of Health requires such information as part of their approval process for tests, and the IOM and other multistakeholder groups have discussed the need for tests to have a defined intended use population and associated outcomes (Deverka et al., 2015; IOM, 2012a). These sources stress the need to clinically validate a test using a number of specimens commensurate with the complexity of the test, and which are representative of the population to be treated in real-world clinical scenarios (e.g., not using primarily Caucasian specimens when the underlying disease overwhelmingly affects African Americans).
The committee recognizes that it will be important for the proposed labeling process to include not only the development of labels, but the periodic review and updates of labels for biomarker tests and any corresponding molecularly targeted therapies. The committee suggests that developers of biomarker tests and manufacturers of the drugs with which they are paired, CMS, other payers, and PCORI could potentially collaborate to fund rapid learning approaches to assessing the clinical utility of biomarker tests. Thus, the committee recommends that labeling should be subject to expedited revision as further evidence develops, providing an incentive for developers to establish the clinical utility of their products.
The proposed labels would provide information on test performance and clinical use that is intelligible and useful to each group of stakeholders, namely patients, health care providers, and directors of laboratories and institutions that administer such tests, the vendors of such tests, payers, and health technology assessors. Thus, the committee recommends that labels should use standardized terminology and should be clear enough for patients to understand as well as sufficiently useful to inform clinical decision making and to provide a basis for reimbursement.
The structure of such a label should be uniform for all biomarker tests for molecularly targeted therapies used in clinical practice, regardless of their status (i.e., whether evaluated and labeled by FDA or not). The label could include multiple “intended uses,” each with different levels
___________________
including morbidity, potential for harm, and impact on current practice (Relling and Klein, 2011). This system is similar to those used in other guidelines, as discussed in Chapter 5.
of evidence. Linkage to these labels should be built into the patient-facing portals of EHRs, as well as laboratory websites and existing national test information sites such as NIH’s Genetic Test Registry.34 Moreover, training about the new labels should be included in educational efforts for providers (e.g., health care providers and genetic counselors) who will become resources to patients for using the information.
Two illustrative example labels are provided in Figures 3-4a and 3-4b. Figure 3-4a depicts a label for a test directing cancer therapy with a well-established evidentiary base, and Figure 3-4b depicts a label for a test directing multiple emerging therapies related to cystic fibrosis. In addition to briefly describing the purpose, accuracy, limitations, and methodology, the ranking system provided on the label would enable providers and patients easily to determine the relevance to their clinical condition and outcomes. The label also would specify whether or not the test was approved or cleared by FDA. The committee appreciates that the process for developing standards may necessitate modification to the rating system for clinical utility in the proposed labels, and offers the label examples below for illustrative purposes only.
The committee proposes that the test labeling process be initiated as a pilot program for biomarker tests for molecularly targeted therapies, given that such tests are the focus of the committee’s statement of task. The impact of the pilot labeling program would be monitored and tracked in a formal evaluation process designed to determine the program’s effectiveness and whether it has the potential to improve transparency of test information and could be expanded beyond biomarker tests for molecularly targeted therapies and implemented on a broader scale to apply to all biomarker tests. The stakeholders described in Recommendation 1 above could be instrumental in developing specific evaluation criteria, which would be tailored to the different target audiences, including health care providers, patients, and payers.
The evaluation should determine, for example, if the labels are achieving the stated goal of patient friendliness by testing patient comprehension of the label information with patient focus groups and surveys. Other stakeholder-specific assessments should be undertaken to determine whether the labels are improving provider, payer, and other stakeholders’ understanding about the performance and use of biomarker
___________________
34 The Genetic Testing Registry (GTR®) provides a central location for voluntary submission of genetic test information by providers. The scope includes the test’s purpose, methodology, validity, evidence of the test’s usefulness, and laboratory contacts and credentials. The overarching goal of the GTR is to advance the public health and research into the genetic basis of health and disease. See http://www.ncbi.nlm.nih.gov/gtr (accessed June 6, 2016).
a Analytic Validity, or the accuracy of a test to detect the specific entity that it was designed to detect.
b Clinical Validity, or the accuracy of a test for disease diagnosis or predicting response to specific drug therapy.
c Ratings indicate:
****: Single source criteria provided;
****: Multisource consistency;
****: Expert panel consensus;
****: Practice guideline.
d Clinical Utility, or evidence of improved measurable clinical outcomes, usefulness, or added value.
tests for molecularly targeted therapies. The evaluation should identify specific areas where adjustments may be needed.
Update and Strengthen Laboratory Oversight and Accreditation
The fourth recommendation proposed by the committee to create a supportive policy environment for biomarker tests for molecularly targeted therapies is intended to enhance laboratory accreditation standards
a Analytic Validity, or the accuracy of a test to detect the specific entity that it was designed to detect.
b Analytic sensitivity refers to the ability for a test to discern a specific low concentration from background noise; analytic specificity refers to how well a test only detects the entity of interest instead of other, similar entities.
c Clinical Validity, or the accuracy of a test for disease diagnosis or predicting response to specific drug therapy.
d Ratings indicate:
****: Single source criteria provided;
****: Multisource consistency;
****: Expert panel consensus;
****: Practice guideline.
e Clinical Utility, or evidence of improved measurable clinical outcomes, usefulness, or added value.
to a level appropriate for complex biomarker tests, such as NGS tests. As noted earlier in this chapter, given the current environment of highly complex biomarker tests, CLIA is not perceived to be up to date. Other organizations have developed laboratory accreditation standards that exceed CLIA standards and have developed voluntary laboratory quality control accreditation programs and provide further quality assurance to address CLIA’s limitations.
The specific standards of these accrediting programs may deviate from the CLIA requirements as long as they meet or exceed CLIA standards, as determined by CMS; as noted earlier in the chapter, such accrediting organizations are granted “deemed status” to review laboratories in lieu of CLIA inspection.35 The College of American Pathologists (CAP), for instance, has instituted a comparatively stringent accreditation program, which has been granted authority by CMS to inspect laboratories in lieu of a CMS inspection. CAP also requires documentation in its Molecular Pathology Inspection Checklist that is related to clinical validity (American Clinical Laboratory Association, 2009). CAP’s authority also is recognized by The Joint Commission (a nonprofit U.S. health care accrediting organization) and certain state certification programs.
States traditionally have taken responsibility for regulating medical practice, and some states have developed independent laboratory certification programs that entail licensure, and in some cases, inspection. The New York State Department of Health (NYSDOH), for example, operates a state certification program that currently has been deemed by CMS to meet or exceed CLIA standards, rendering laboratories under its jurisdiction exempt from specific CLIA oversight.36
NYSDOH’s Clinical Laboratory Evaluation Program (CLEP) entails regular inspection and PT, and additionally reviews all non-FDA-cleared or -approved tests for evidence of analytic and clinical validity (SACGHS, 2008). Through this rigorous test approval policy, CLEP has issued specific requirements for molecular genetic testing and guidelines for the NGS testing for germline genetic variants (NYSDOH, 2013, 2015b). Moreover, NYSDOH’s Clinical Laboratory Reference System (of which CLEP is a part) oversees both clinical and forensic testing on all specimens originating in the state of New York. Therefore, even laboratories outside of New York that handle specimens originating in the state require New York licensure.37
Regardless of FDA’s final guidance regarding regulation of certain
___________________
35 See https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Accreditation.html (accessed May 15, 2015).
36 63 FR 26732 § 493.551.
37 See http://www.wadsworth.org/docs/clrs.shtml (accessed November 4, 2015).
LDTs, the committee’s view is that although CLIA functions as a minimal standard for the operation of a clinical laboratory, it is not up to date and is inadequate for the oversight of more complex biomarker tests to guide selection of molecularly targeted therapies. Moreover, CLIA standards are inadequate for current NGS tests and other emerging technologies. Thus, the committee recommends that the Secretary of HHS should establish and enforce up-to-date laboratory accreditation standards for biomarker tests for molecularly targeted therapies, either through CMS’s CLIA or in collaboration with an existing up-to-date accreditation organization. Reimbursement for such biomarker testing should be dependent on meeting these standards (Recommendation 4). These standards should exceed current CLIA standards, similar to those of existing voluntary accreditation programs and other state-level regulatory bodies, as described above. These standards should comply with test labeling requirements as discussed earlier (see Recommendation 3).
Ongoing Assessment of the Clinical Utility of Biomarker Tests
The committee highlights the importance of viewing evidence generation for biomarker tests for molecularly targeted therapies as an ongoing process that takes place along a continuum (depicted in Figure 3-5), where
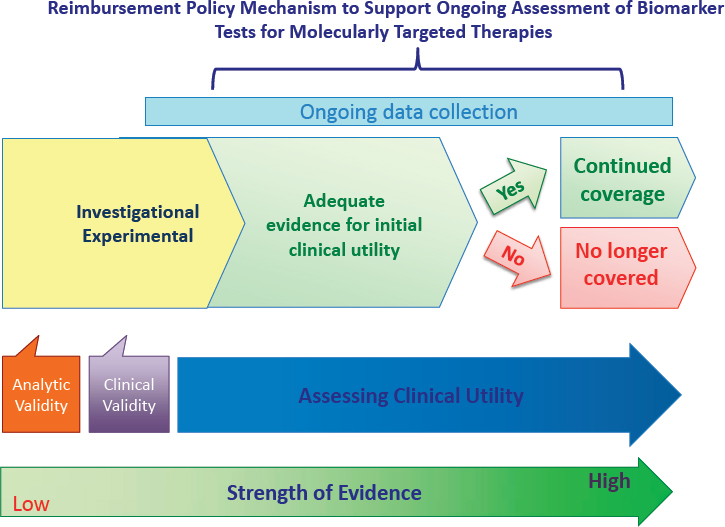
evidence increases in amount and quality through additional studies and well-documented clinical use over time. The continuous accumulation of evidence enables a change in designation from investigational/experimental to adequate to make an initial determination regarding the clinical utility of the biomarker test for molecularly targeted therapy. This initial determination is subsequently supported and strengthened by additional clinical studies as well as ongoing data collection to provide greater certainty about the optimal use of the biomarker test for molecularly targeted therapy.
The process for developing evidentiary standards described above could be relied on to determine what level of evidence of clinical utility is sufficient for initial determination that a biomarker test is reasonably likely to benefit patients, and thus could be used effectively in clinical practice and be reimbursed by payers. In most cases, however, additional evidence will be needed to confirm the impact of the test based on longer-term clinical outcomes, to define the patient subgroups for whom testing is most useful, and to compare the outcomes achieved with use of the test to alternative approaches to patient management. Thus, it will be extremely valuable for payers to apply policy mechanisms designed to promote the ongoing assessment of evidence of the clinical utility of biomarker tests for molecularly targeted therapies.
An important note is that initial clinical use of biomarker tests should only proceed if basic assessments of test validity have been conducted, and there is sufficient evidence to demonstrate that these tests are reasonably likely to benefit patients. Ongoing collection and evaluation of data may be the most effective way to fully assess biomarker tests. In the words of one expert: “If you get stuck with either the highest level of evidence or nothing at all, genomics will never really come to light” (IOM, 2012b, p. 55).
Importantly for payers, assessment of postmarket data could support decisions for continued reimbursement of a biomarker test or alternatively, provide the rationale for discontinued reimbursement. In the former case, payers will have reassurances that coverage of the test is beneficial to patients. In the latter case, payers will benefit from cost savings associated with no longer paying for a test that has been determined to not be effective in directing targeted therapy. Thus, the committee recommends that when existing evidence of clinical utility is sufficient for initial use of a biomarker test for a molecularly targeted therapy, CMS and other payers should develop reimbursement models that support the ongoing collection of data within a rapid learning system. Such data will be used to further assess the evidence of clinical utility (Recommendation 5a). It is critical that CMS takes the lead regarding these CED efforts, assuming primary responsibility for the data registries
and analysis associated with CED. Other payers should follow CMS’s lead and support data collection efforts because the data will be used to refine clinical use and reimbursement policy decisions related to biomarker tests for molecularly targeted therapies.
The committee recommends that payers could develop several potential approaches to support such ongoing assessment of clinical utility. For example, payers could pay for biomarker tests that meet predefined clinical and evidentiary criteria (Recommendation 1), with the requirement for ongoing postmarket data collection and assessment (through the national database as proposed in Recommendation 7, discussed in Chapter 4).
Consistent with the committee’s vision of a rapid learning system, and recognition of the evolving nature of a biomarker test’s ability to direct therapy to different targets than originally intended, the committee recommends that payers support reimbursement for biomarker tests with data collection for patient populations for which the evidence is less substantial, such as rare diseases or underrepresented populations and less studied groups. Also consistent with the committee’s proposed rapid learning system, the committee further recommends that payers consider innovative incentives to promote the submission of data to a national repository for biomarker tests and molecularly targeted therapies that have initial evidence of clinical utility (discussed further in Chapter 4).
Coverage with Evidence Development
As noted at the outset of this chapter, the current fee-for-service reimbursement system does not provide incentives to improve quality, nor does it reward value. Alternative reimbursement methods are emerging that focus on improving quality, rewarding value, and containing health care costs. Such reimbursement models that focus on value are needed, particularly as precision medicine becomes more widely implemented, for as McClellan and Thoumi note: “As care becomes more personalized, fee-for-service reimbursements are likely to become increasingly misaligned with high-value treatment and service combinations for particular patients” (McClellan and Thoumi, 2015, p. 225). Some observers caution, however, that the shift from traditional fee-for-service to risk-based reimbursements may have an impact on care delivery, particularly for newer technologies such as biomarker tests for molecularly targeted therapies, in that the increasing emphasis on achieving quality and cost benchmarks may make health care professionals reluctant to use newer, not yet fully proven technologies (PMC, 2015).
The adoption of innovative reimbursement approaches such as
performance-based risk-sharing arrangements (PBRSAs) would be consistent with the committee’s recommendations. Such approaches have the potential to support continuous collection of evidence to enable progressive approval while the strength of the evidence base becomes more robust (Carlson et al., 2010; Ramsey and Sullivan, 2014). The International Society for Pharmacoeconomics and Outcomes Research Task Force on PBRSAs has defined two general types of arrangements: (1) reimbursement is linked to product performance in the real world, and (2) limited access to a promising therapy is based on strategies to collect and evaluate additional evidence (Garrison et al., 2013). Under the latter of the two arrangements, known alternately as managed entry or coverage with evidence development, the payer provides coverage and reimbursement of a biomarker test for molecularly targeted therapies if the patients receiving the test agree to enroll in a clinical study to generate the additional evidence required for ongoing assessment of clinical utility. The ongoing collection of data is viewed as the best solution to resolve the uncertainty regarding the clinical utility of the test; critical evidence would not be made available without such an approach. A number of ongoing efforts to capture data for evidence development are discussed further in Chapter 4 of this report.
As discussed earlier in this chapter, CED is a policy tool that ties coverage of an emerging technology with a requirement that patients receiving the service are enrolled in clinical studies—either patient registries or clinical trials with the express purpose of systematically gathering data to inform future coverage decisions (Health Affairs, 2015; Neumann and Chambers, 2013; Tunis et al., 2011). In this way, CED is consistent with the rapid learning system envisioned by the committee, as research and clinical practice are interrelated and reinforce one another (Jacques, 2014; Lewis et al., 2015).
CED can be viewed as representing a win–win for all stakeholders: Patients would be able to access promising new biomarker tests before full evidence is available as to their clinical utility; payers share responsibility for generating evidence necessary to make informed coverage decisions; and manufacturers gain reimbursement for their products during the period when they are developing further evidence to support full coverage decisions by payers (CMTP, 2009; Lewis et al., 2015). Importantly, CED can facilitate the collection of clinical data of those patients underrepresented in traditional clinical trials (e.g., minorities or the elderly), and allow Medicare and other payers to develop more relevant, evidence-based coverage policies (MedPAC, 2010). CED is also beneficial for patients with rare diseases where a strong evidence base has not yet, and may never be, developed due to the lack of clinical trials when only a limited number of patients would be eligible (Lewis et al., 2015).
Policy makers continue to see value in CED as a policy tool as evidenced by the Medicare Payment Advisory Commission’s (MedPAC’s) recommendation to grant Medicare clear statutory authority to implement innovative strategies such as CED (MedPAC, 2010), and the Obama Administration’s highlighting of CED as a viable approach to providing incentives for innovative technologies in its 2012 National Bioeconomy Blueprint38 (Neumann and Chambers, 2013; OSTP, 2012). Moreover, an initial draft of the 21st Century Cures Act contained provisions to revise the statutory language relating to CED, though such provisions are not included in the final version of the bill (Health Affairs, 2015).39
Significant challenges limiting the effective implementation of CED include the requirement of CMS’s “reasonable and necessary” authority to implement CED. CMS interprets reasonable and necessary under its standard statutory authority to mean there is adequate evidence to conclude that the item of service improves health outcomes, yet this is in opposition to the intended aim of CED to generate adequate evidence for promising new technologies and treatments, creating somewhat of a Catch-22 situation. Moreover, under current statute, CMS’s implementation of CED depends on AHRQ’s legal authority40 to conduct research related to the outcomes, effectiveness, and appropriateness of health care services and procedures for the Medicare population (Tunis et al., 2011).
Experts note that the challenges facing CED are not design flaws; rather, the major limiting factor is the lack of a clear statutory basis to enable CMS to appropriately implement this critical policy tool (Jacques, 2014; Tunis et al., 2011). As noted by MedPAC, a solid statutory foundation could enable CED to overcome its current implementation challenges and realize its full potential as an effective approach to coverage of emerging technologies (MedPAC, 2010).
The committee recommends that CMS should seek to clarify and expand appropriate implementation of CED, which has potential to be an effective policy lever to generate evidence to support reimbursement decisions for promising technologies, such as biomarker tests for molecularly targeted therapies. Importantly, CED studies of biomarker tests should include coverage of treatment directed by those tests.
Finally, the committee recognizes the importance of rapid learning approaches to support the assessment of clinical utility for biomarker tests
___________________
38 See https://www.whitehouse.gov/sites/default/files/microsites/ostp/national_bioeconomy_blueprint_april_2012.pdf (accessed September 1, 2015).
39 See http://energycommerce.house.gov/sites/republicans.energycommerce.house.gov/files/114/Analysis/Cures/20150127-Cures-Discussion-Document.pdf (accessed May 4, 2015).
40 Section 1862(a)(1)(E) of the Social Security Act references AHRQ’s authority to conduct research for Medicare.
for molecularly targeted therapies and encourages funders to develop granting mechanisms to support such efforts. A number of existing programs within NIH examine issues related to clinical utility, and could potentially implement the committee’s research recommendation. For example, the NCI’s Implementation Science Program, under the Division of Cancer Control and Population Sciences, supports research grants examining the dissemination and implementation of evidence-based treatment in clinical practice settings (NCI, 2014). In addition, the Implementation Science Research Program under NIH’s Fogarty International Center seeks to “understand the behavior of healthcare professionals and other stakeholders as a key variable in the sustainable uptake, adoption, and implementation of evidence-based interventions” (NIH, 2013). NCI’s Community Oncology Research Program performs multidisciplinary cancer care delivery research on outcomes (including patient health, costs, quality of care, and access), as a result of financial, organizational, and behavioral factors related to adoption of new health care technology (NCI, 2015). The Biomarkers Consortium, a broad multistakeholder initiative managed under the Foundation for the National Institutes of Health, currently has a wide variety of active and upcoming research projects specifically related to evaluating the use of biomarkers in clinical practice (Biomarkers Consortium, 2015). The Consortium may therefore serve as an ideal organization to assist with research into clinical utility for biomarker tests for molecularly targeted therapies.
The independent PCORI was created to fund comparative effectiveness research (CER).41 According to a 2014 analysis by the Center for American Progress, few PCORI studies focus on CER priority areas identified by the IOM, and PCORI has not funded any CER studies of medical devices (Tanden et al., 2014). Notably, research on “biomarker testing versus usual care in preventing and treating cancers and other conditions for which promising biomarkers exist” is among the top quartile42 CER priority areas identified by the IOM (IOM, 2009, p. 4). The Center for American Progress called on PCORI to increase CER funding to at least 80 percent of its total research funding by fiscal year (FY) 2016, and
___________________
41 PCORI’s activities are financed through the PCOR Trust Fund, which includes funds from general appropriations, transfers from the CMS trust fund, and fees (approximately $2 for each covered individual) assessed on private insurance and self-insured health plans. PCORI’s annual funding more than doubled from $320 million in FY 2013 to approximately $650 million for FY 2014. This level continues until the organization’s funding expires in 2019 (PCORI, 2015).
42 The IOM CER committee grouped the 100 individual CER topics into quartiles according to the number of votes each received during the committee’s voting process. Topics within the first quartile were considered higher priority than those in the fourth quartile, but the order within quartiles does not signify rank (IOM, 2009).
to prioritize CER that focuses on the top quartile topics identified by the IOM (Tanden et al., 2014).
PCORI-funded CER studies of promising biomarkers for molecularly targeted therapies would be a natural complement to clinical efficacy research funded by NIH. Therefore, the committee recommends that PCORI and NIH, as well as other funding groups, should develop granting mechanisms that support the assessment of the clinical utility of biomarker tests for molecularly targeted therapies using rapid learning approaches (Recommendation 5b).
SUMMARY AND RECOMMENDATIONS
The committee examined a number of key regulatory and reimbursement policy challenges influencing the adoption of biomarker tests for molecularly targeted therapies into clinical use, and crafted an interrelated set of recommended approaches to tackling those challenges. In so doing, the committee laid the foundation for one component of its vision of a rapid learning system for biomarker tests for molecularly targeted therapies: the development of a supportive policy environment.
The committee’s interrelated policy recommendations are expressly designed to develop a rapid learning system to promote the development, assessment, and use of precision medicine’s powerful tools—biomarker tests for molecularly targeted therapies. Such tests have the potential to improve health outcomes and lower overall health care costs by avoiding the use of therapies that are not effective and may in fact cause harm to the patient. A supportive regulatory and reimbursement policy environment—such as the one recommended here—is required for biomarker tests to realize their full potential.
Goal 1: Establish common evidentiary standards of clinical utility—using evidence generated both within and outside the context of clinical trials—across all stakeholders.
Recommendation 1: The Secretary of the Department of Health and Human Services (HHS) should facilitate the development of common clinical utility evidentiary standards that are applied for initial and ongoing coordinated regulatory, coverage, and reimbursement decisions for biomarker tests for molecularly targeted therapies. One mechanism for the development of these evidentiary standards could be convening one or more independent, public–private, multistakeholder bodies.
- Consistent and coordinated evidentiary standards and study design approaches, including rapid learning systems, should be
-
developed that simultaneously accommodate the various types of decisions (including clinical, regulatory, coverage/reimbursement, and guideline recommendations), and facilitate the ongoing development of evidence of clinical utility.
- Involvement of a variety of stakeholders will be critical to ensure that clinical utility studies are designed to reflect a range of decision-making needs and to strike an acceptable balance between ideal utility assessment and study feasibility. Stakeholders participating in these initiatives should include patients, health care providers, clinical practice guideline developers, public and private payers (including the Centers for Medicare & Medicaid Services), the Food and Drug Administration, test developers, pharmaceutical companies, molecular pathologists, clinical laboratory geneticists, informaticians, and research funders (e.g., the Patient-Centered Outcomes Research Institute, the National Institutes of Health, and the Agency for Healthcare Research and Quality).
- Recognizing that evidentiary standards for clinical utility may vary across diseases, HHS could determine that more than one advisory body may be necessary to develop such disease-specific standards.
- Standards for ongoing development of clinical utility evidence will be used to guide the creation of new labels for biomarker tests and corresponding therapies (see Recommendation 3), and for guideline development (see Recommendation 10).
- Analytic and clinical validity of biomarker tests should be assured prior to assessing clinical utility.
- HHS should continue to support ongoing refinement of common evidentiary standards as they evolve.
Goal 2: Establish a more coordinated and transparent federal process for regulatory and reimbursement decisions for biomarker tests for molecularly targeted therapies.
Recommendation 2: The Secretary of the Department of Health and Human Services should facilitate the development of a new integrated federal review process involving the Food and Drug Administration and the Centers for Medicare & Medicaid Services, as a pathway for coordinated regulatory, coverage, and reimbursement decisions for biomarker tests for molecularly targeted therapies (including in vitro diagnostics, laboratory developed tests and multianalyte tests performed using current or new technologies,
and any corresponding molecularly targeted therapies).43 This coordinated pathway should accomplish all of the following through application of common evidentiary standards (as described in Recommendation 1):
- Primary (and follow-on) biomarker test review and approval with detailed test labeling requirements (as described in Recommendation 3).
- Drug review and approval with detailed labeling that includes standardized biomarker test information (as described in Recommendation 3), when occurring concurrently with biomarker test review.
- A national uniform coverage decision for a biomarker test and molecularly targeted therapy in specific clinical uses, including financial incentives for data submission on use and outcomes (see Recommendation 7).
- A defined process for coordinated updates of biomarker test and drug labels.
- Public sharing of the summary data upon which the review process based the approval and coverage decisions for a biomarker test and drug combination.
Goal 3: Enhance communication to patients and providers about the performance characteristics and evidence for use of specific biomarker tests for molecularly targeted therapies.
Recommendation 3: The Food and Drug Administration (FDA) should develop a patient- and provider-friendly standardized label for biomarker tests (including in vitro diagnostics and laboratory developed tests) to facilitate transparency of test performance characteristics and the level of evidence for the intended use(s) of the test. FDA or laboratory accrediting bodies should approve the label for each biomarker test, including tests not reviewed through the integrated process specified in Recommendation 2.
- Labels should prominently feature an easily understood ranking system (e.g., 4-star scales) separately for the evidence to support the clinical validity and clinical utility for each intended clinical use of a test. The evidence ranking standards would be developed by the process described in Recommendation 1.
- Labeling should be subject to expedited revision as further evidence develops, providing an incentive for developers to establish the clinical utility of their products.
___________________
43 This coordinated pathway is designed to reflect the current predominant fee-for-service reimbursement system for clinical tests.
- Labels should use standardized terminology and should be clear enough for patients to understand as well as sufficiently useful to inform clinical decision making and to provide a basis for reimbursement.
Goal 4: Update and strengthen oversight and accreditation of laboratories providing biomarker tests for molecularly targeted therapies.
Recommendation 4: The Secretary of the Department of Health and Human Services should establish and enforce up-to-date laboratory accreditation standards for biomarker tests for molecularly targeted therapies, either through the Centers for Medicare & Medicaid Services’ Clinical Laboratory Improvement Amendments (CLIA) or in collaboration with an existing up-to-date accreditation organization. Reimbursement for such biomarker testing should be dependent on meeting these standards.
- Current CLIA standards are inadequate for current advanced biomarker tests performed using next-generation sequencing and other emerging technologies.
- These standards should comply with test labeling requirements (see Recommendation 3).
Goal 5: Ensure ongoing assessment of the clinical utility of biomarker tests for molecularly targeted therapies.
Recommendation 5a: When existing evidence of clinical utility is sufficient for initial use of a biomarker test for a molecularly targeted therapy, the Centers for Medicare & Medicaid Services (CMS) and other payers should develop reimbursement models that support the ongoing collection of data within a rapid learning system. Such data will be used further to assess the evidence of clinical utility.
Potential approaches that payers could use to support this data collection include the following:
-
Reimbursement for biomarker tests that meet predefined clinical and evidentiary criteria (see Recommendation 1), with the requirement for ongoing postmarket data collection and assessment (through the national database as proposed in Recommendation 7).
- – These data could support decisions for continued reimbursement or provide the rationale for discontinued reimburse-
ment for a specific biomarker test and its molecularly targeted therapy for specific patient groups.
- Reimbursement for biomarker tests with data collection for patient populations for which the evidence is less substantial, such as rare diseases or underrepresented populations and less studied groups.
- Consider innovative incentives to promote the submission of data to the national repository for biomarker tests and molecularly targeted therapies that have initial evidence of clinical utility.
- CMS should seek to clarify and expand appropriate implementation of CED, which has potential to be an effective policy lever to generate evidence to support reimbursement decisions for promising technologies, such as biomarker tests for molecularly targeted therapies.
Recommendation 5b: The Patient-Centered Outcomes Research Institute and the National Institutes of Health, as well as other funding groups, should develop granting mechanisms that support the assessment of the clinical utility of biomarker tests for molecularly targeted therapies using rapid learning approaches.
REFERENCES
AAFP (American Academy of Family Physicians). 2015. About the AAFP proficiency testing program. www.aafp.org/practice-management/labs/about.html (accessed September 30, 2015).
Abernethy, A., R. R. Coeytaux, K. Carson, D. McCrory, S. Y. Barbour, M. Gradison, R. J. Irvine, and J. L. Wheeler. 2010. Report on the evidence regarding off-label indications for targeted therapies used in cancer treatment. Rockville, MD: Agency for Healthcare Research and Quality.
American Clinical Laboratory Association. 2009. Comments to Genentech citizen petition.
Anderson, G. F., K. Davis, and S. Guterman. 2015. Medicare payment reform: Aligning incentives for better care. New York: The Commonwealth Fund.
ASCP (American Society for Clinical Pathology). 2014. Patient access to test results. www.ascp.org/Advocacy/Patient-Access-to-Test-Results.html (accessed September 30, 2015).
Astles, J. R., H. Stang, T. Alspach, G. Mitchell, M. Gagnon, and D. Bosse. 2013. CLIA requirements for proficiency testing: The basics for laboratory professionals. Medical Laboratory Observer 45(9):8-10, 12, 14-15; quiz 16.
Avalere Health. 2015. Clinical pathways: Overview of current practices and potential implications for patients, payers, and providers. http://avalere-health-production.s3.amazonaws.com/uploads/pdfs/1438012880_AH_Pathways_final.pdf (accessed September 8, 2015).
Balch, A., C. Balch, A. B. Benson III, D. Morosini, R. M. Rifkin, and L. A. Williams. 2015. Clinical pathways: Barrier or benefit to patient access and personalized medicine? http://cqrcengage.com/npaf/file/tTiSOowxuVB/Clinical%20Pathways%20White%20Paper%20July%202015.pdf (accessed September 1, 2015).
Biomarkers Consortium. 2015. Projects overview. http://www.biomarkersconsortium.org/projects.php (accessed January 21, 2016).
Bipartisan Policy Center. 2015. Transitioning from volume to value: Accelerating the shift to alternative payment models. Washington, DC.
Blumenthal, D., M. Abrams, and R. Nuzum. 2015. The Affordable Care Act at 5 years. New England Journal of Medicine 372(25):2451-2458.
Blumenthal, G. M., E. Mansfield, and R. Pazdur. 2016. Next-generation sequencing in oncology in the era of precision medicine. Journal of the American Medical Association Oncology 2(1):13-14.
Boland, J. F., C. C. Chung, D. Roberson, J. Mitchell, X. Zhang, K. M. Im, J. He, S. J. Chanock, M. Yeager, and M. Dean. 2013. The new sequencer on the block: Comparison of life technology’s proton sequencer to an illumina HiSeq for whole-exome sequencing. Human Genetics 132(10):1153-1163.
Burwell, S. M. 2015. Setting value-based payment goals—HHS efforts to improve U.S. health care. New England Journal of Medicine 372(10):897-899.
Caffrey, M. K., and L. Joszt. 2015. Massive consolidation among healthcare payers. http://www.ajmc.com/journals/evidence-based-oncology/2015/august-2015/massive-consolidation-among-healthcare-payers- (accessed November 10, 2015).
Callahan, R., and A. Darzi. 2015. Five policy levers to meet the value challenge in cancer care. Health Affairs 34(9):1563-1568.
Canestaro, W. J., D. E. Pritchard, L. P. Garrison, R. Dubois, and D. L. Veenstra. 2015. Improving the efficiency and quality of the value assessment process for companion diagnostic tests: The companion test assessment tool (cat). Journal of Managed Care Pharmacy 21(8):700-712.
Carey, B. 2014. Summary of CLFS reform provisions in the Protecting Access to Medicare Act of 2014. http://www.foleyhoag.com/publications/alerts-and-updates/2014/april/clinical-laboratory-fee-schedule-reform-provisions (accessed May 5, 2015).
Carlson, J. J., S. D. Sullivan, L. P. Garrison, P. J. Neumann, and D. L. Veenstra. 2010. Linking payment to health outcomes: A taxonomy and examination of performance-based reimbursement schemes between healthcare payers and manufacturers. Health Policy 96(3):179-190.
CDC (Centers for Disease Control and Prevention). 2014. Clinical Laboratory Improvement Amendments (CLIA). wwwn.cdc.gov/clia (accessed September 30, 2015).
Chambers, J. D., M. Chenoweth, T. Thorat, and P. J. Neumann. 2015. Private payers disagree with Medicare over medical device coverage about half the time. Health Affairs 34(8):1376-1382.
Clement, P. D., and L. H. Tribe. 2015. Laboratory testing services, as the practice of medicine, cannot be regulated as medical devices. New York: American Clinical Laboratory Association.
CMS (Centers for Medicare & Medicaid Services). 2006. Informational supplement. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/downloads/Informational_Supplement.pdf (accessed September 30, 2015).
CMS. 2015a. Clinical Laboratory Improvement Amendments (CLIA). www.cms.gov/RegulationsandGuidance/Legislation/CLIA/index.html?redirect=/clia (accessed September 30, 2015).
CMS. 2015b. On its 50th anniversary, more than 55 million Americans covered by Medicare. https://www.cms.gov/Newsroom/MediaReleaseDatabase/Press-releases/2015-Press-releases-items/2015-07-28.html (accessed August 14, 2015).
CMS. 2015c. Tests granted waived status under CLIA. Regulations and guidance. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/downloads/waivetbl.pdf (accessed June 4, 2015).
CMTP (Center for Medical Technology Policy). 2009. Coverage for evidence development: A conceptual framework. Baltimore, MD: Center for Medical Technology Policy.
CMTP. 2010. Coverage with evidence development (CED) in the private sector: Lessons in design and implementation. Baltimore, MD: Center for Medical Technology Policy.
CMTP. 2013. Evaluation of clinical validity and clinical utility of actionable molecular diagnostic tests in adult oncology. Baltimore, MD: Center for Medical Technology Policy.
CMTP. 2014. Demonstrating the clinical utility of next generation sequencing in clinical oncology. Baltimore, MD: Center for Medical Technology Policy.
CMTP. 2015. Initial medical policy and model coverage guidelines for clinical next generation sequencing in oncology: Report and recommendations. Baltimore, MD: Center for Medical Technology Policy.
Cohen, J. P., and A. E. Felix. 2014. Personalized medicine’s bottleneck: Diagnostic test evidence and reimbursement. Journal of Personalized Medicine 4(2):163-175.
Collins, F. S., and H. Varmus. 2015. A new initiative on precision medicine. New England Journal of Medicine 372(9):793-795.
Daniel, G. W., E. K. Rubens, and M. McClellan. 2013. Coverage with evidence development for Medicare beneficiaries: Challenges and next steps. Journal of the American Medical Association Internal Medicine 173(14):1281-1282.
DeMartino, J., and J. K. Larsen. 2012. NCCN white paper equity in cancer care: Pathways, protocols, and guidelines. Journal of the National Comprehensive Cancer Network 10(Suppl 1).
Deverka, P. A., and J. C. Dreyfus. 2014. Clinical integration of next generation sequencing: Coverage and reimbursement challenges. The Journal of Law, Medicine & Ethics 42(Suppl 1):22-41.
Deverka, P. A., D. Kaufman, and A. L. McGuire. 2014. Overcoming the reimbursement barriers for clinical sequencing. Journal of the American Medical Association 312(18):1857-1858.
Deverka, P. A., D. A. Messner, R. McCormack, G. H. Lyman, M. Piper, L. Bradley, D. Parkinson, D. Nelson, M. L. Smith, L. Jacques, T. Dutta, and S. R. Tunis. 2015. Generating and evaluating evidence of the clinical utility of molecular diagnostic tests in oncology. Genetics in Medicine December 3, 2015 (Epub ahead of print).
Dickson, D. J. 2015. The Molecular Evidence Development Consortium (MED-C): A shared method to advance personalized medicine. https://www.med-c.org/the-molecular-evidence-development-consortium-med-c-a-shared-method-to-advance-personalized-medicine (accessed July 12, 2015).
Emanuel, E. J., and S. D. Pearson. 2012. It costs more, but is it worth more? The New York Times, January 2, 2012.
Evans, B. J., W. Burke, and G. P. Jarvik. 2015. The FDA and genomic tests—getting regulation right. New England Journal of Medicine 372(23):2258-2264.
Ewing, A. D., K. E. Houlahan, Y. Hu, K. Ellrott, C. Caloian, T. N. Yamaguchi, J. C. Bare, C. P’ng, D. Waggott, V. Y. Sabelnykova, ICGC-TCGA DREAM Somatic Mutation Calling Challenge. participants, M. R. Kellen, T. C. Norman, D. Haussler, S. H. Friend, G. Stolovitzky, A. A. Margolin, J. M. Stuart, and P. C. Boutros. 2015. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nature Methods 12(7):623-630.
Faulkner, E. 2009. Clinical utility or impossibility? Addressing the molecular diagnostics health technology assessment and reimbursement conundrum. Journal of Managed Care Medicine 12(4):42-54.
Faulkner, E., L. Annemans, L. Garrison, M. Helfand, A. P. Holtorf, J. Hornberger, D. Hughes, T. Li, D. Malone, K. Payne, U. Siebert, A. Towse, D. Veenstra, J. Watkins, Personalized Medicine Development and Reimbursement Working Group. 2012. Challenges in the development and reimbursement of personalized medicine-payer and manufacturer perspectives and implications for health economics and outcomes research: A report of the ISPOR Personalized Medicine Special Interest Group. Value in Health 15(8):1162-1171.
FDA (Food and Drug Administration). 2014a. Draft guidance for industry, Food and Drug Administration staff, and clinical laboratories: FDA notification and medical device reporting for laboratory developed tests (LDTs). http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM416684.pdf (accessed March 3, 2015).
FDA. 2014b. Framework for regulatory oversight of laboratory developed tests (LDTs). http://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416685.pdf (accessed March 3, 2015).
FDA. 2014c. In vitro companion diagnostic devices guidance: Draft guidance for industry and Food and Drug Administration staff. http://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm262327.pdf (accessed January 15, 2015).
FDA. 2015a. Optimizing FDA’s regulatory oversight of next generation sequencing diagnostic tests—preliminary discussion paper. http://www.fda.gov/downloads/MedicalDevices/NewsEvents/WorkshopsConferences/UCM427869.pdf (accessed April 10, 2015).
FDA. 2015b. Payer Communication Task Force (PCTF). http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHInnovation/ucm456149.htm#parallelreview (accessed August 4, 2015).
FDA. 2015c. The public health evidence for FDA oversight of laboratory developed tests: 20 case studies. Silver Spring, MD: Office of Public Health Strategy and Analysis.
Febbo, P. G., M. Ladanyi, K. D. Aldape, A. M. De Marzo, M. E. Hammond, D. F. Hayes, A. J. Iafrate, R. K. Kelley, G. Marcucci, S. Ogino, W. Pao, D. C. Sgroi, and M. L. Birkeland. 2011. NCCN task force report: Evaluating the clinical utility of tumor markers in oncology. Journal of the National Comprehensive Cancer Network 9(Suppl 5):S1-S32; quiz S33.
Ferreira-Gonzalez, A., R. Emmadi, S. P. Day, R. F. Klees, J. R. Leib, E. Lyon, J. A. Nowak, V. M. Pratt, M. S. Williams, and R. D. Klein. 2014. Revisiting oversight and regulation of molecular-based laboratory-developed tests: A position statement of the Association for Molecular Pathology. Journal of Molecular Diagnostics 16(1):3-6.
Freidlin, B., L. M. McShane, and E. L. Korn. 2010. Randomized clinical trials with biomarkers: Design issues. Journal of the National Cancer Institute 102(3):152-160.
Frueh, F. W. 2013. Regulation, reimbursement, and the long road of implementation of personalized medicine—a perspective from the United States. Value Health 16(6 Suppl):S27-S31.
Frueh, F. W., and B. Quinn. 2014. Molecular diagnostics clinical utility strategy: A six-part framework. Expert Review of Molecular Diagnostics 14(7):777-786.
Fugel, H. J., M. Nuijten, and E. Faulkner. 2014. The application of economics concepts to stratified medicine—use of health economics data to support market access for stratified medicine interventions. Journal of Medical Economics 17(5):305-311.
Gaffney, A. 2014. FDA announces first-ever approval under CMS parallel review program. http://www.raps.org/Regulatory-Focus/News/2014/08/12/20009/FDA-Announces-First-Ever-Approval-Under-CMS-Parallel-Review-Program (accessed August 25, 2015).
Garrison, L. P., Jr., A. Towse, A. Briggs, G. de Pouvourville, J. Grueger, P. E. Mohr, J. L. Severens, P. Siviero, and M. Sleeper. 2013. Performance-based risk-sharing arrangements-good practices for design, implementation, and evaluation: Report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value Health 16(5):703-719.
Ginsburg, G. S., and N. M. Kuderer. 2012. Comparative effectiveness research, genomics-enabled personalized medicine, and rapid learning health care: A common bond. Journal of Clinical Oncology 30(34):4233-4242.
Goldman, D. P., C. Gupta, E. Vasudeva, K. Trakas, R. Riley, D. Lakdawalla, D. Agus, N. Sood, A. B. Jena, and T. J. Philipson. 2013. The value of diagnostic testing in personalized medicine. Forum for Health Economics and Policy 16(2):121-133.
Graf, M. D., D. F. Needham, N. Teed, and T. Brown. 2013. Genetic testing insurance coverage trends: A review of publicly available policies from the largest US payers. Personalized Medicine 10(3):235-243.
Gustavsen, G., K. Phillips, and K. Pothier. 2010. The reimbursement landscape for novel diagnostics. Weston, MA: Health Advances.
Hayes, D. F., R. C. Bast, C. E. Desch, H. Fritsche, Jr., N. E. Kemeny, J. M. Jessup, G. Y. Locker, J. S. Macdonald, R. G. Mennel, L. Norton, P. Ravdin, S. Taube, and R. J. Winn. 1996. Tumor marker utility grading system: A framework to evaluate clinical utility of tumor markers. Journal of the National Cancer Institute 88(20):1456-1466.
Hayes, D. F., J. Allen, C. Compton, G. Gustavsen, D. G. Leonard, R. McCormack, L. Newcomer, K. Pothier, D. Ransohoff, R. L. Schilsky, E. Sigal, S. E. Taube, and S. R. Tunis. 2013. Breaking a vicious cycle. Science Translational Medicine 5(196):196cm.
Health Affairs. 2015. Health policy briefs: Aligning FDA and CMS review. http://healthaffairs.org/healthpolicybriefs/brief_pdfs/healthpolicybrief_143.pdf (accessed September 9, 2015).
Hoffman, A., R. Montgomery, W. Aubry, and S. R. Tunis. 2010. How best to engage patients, doctors, and other stakeholders in designing comparative effectiveness studies. Health Affairs 29(10):1834-1841.
Hresko, A., and S. B. Haga. 2012. Insurance coverage policies for personalized medicine. Journal of Personalized Medicine 2(4):201-216.
HRSA (Health Resources and Services Administration). 2015. Recommended uniform screening panel (as of March 2015). http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/recommendedpanel (accessed December 6, 2015).
Hudson, K., G. Javitt, W. Burke, P. Byers, and American Society of Human Genetics Social Issues Committee. 2007. ASHG statement on direct-to-consumer genetic testing in the United States. Obstetrics & Gynecology 110(6):1392-1395.
Hughes, K. 2014. MolDX may be the norm, but is it the future? http://avalere.com/expertise/life-sciences/insights/moldx-may-be-the-norm-but-is-it-the-future (accessed November 13, 2015).
Hwang, T. J., L. S. Lehmann, and A. S. Kesselheim. 2015. Precision medicine and the FDA’s draft guidance on laboratory-developed tests. Nature Biotechnology 33(5):449-451.
IOM (Institute of Medicine). 2009. Initial national priorities for comparative effectiveness research. Washington, DC: The National Academies Press.
IOM. 2010. Policy issues in the development of personalized medicine in oncology: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2011a. Clinical practice guidelines we can trust. Washington, DC: The National Academies Press.
IOM. 2011b. Generating evidence for genomic diagnostic test development: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2012a. Evolution of translational omics: Lessons learned and the path forward. Washington, DC: The National Academies Press.
IOM. 2012b. Genome-based diagnostics: Clarifying pathways to clinical use: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2013a. Best care at lower cost: The path to continuous learning health care in America. Washington, DC: The National Academies Press.
IOM. 2013b. Delivering high-quality cancer care: Charting a new course for a system in crisis. Washington, DC: The National Academies Press.
IOM. 2013c. The economics of genomic medicine: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2013d. Genome-based diagnostics: Demonstrating clinical utility in oncology: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2014. Assessing genomic sequencing information for health care decisions: Workshop summary. Washington, DC: The National Academies Press.
IOM. 2015. Policy issues in the development and adoption of biomarkers for molecularly targeted cancer therapies: Workshop summary. Washington, DC: The National Academies Press.
Jacques, L. 2014. U.S. House of Representatives. Energy and Commerce Subcommittee on Health Hearing. 21st century cures: Barriers to ongoing communication and evidence development. July 22, 2014. http://docs.house.gov/meetings/IF/IF14/20140722/102524/HHRG-113-IF14-Wstate-JacquesL-20140722.pdf (accessed May 22, 2015).
Japsen, B. 2015. Antitrust showdown next after Aetna, Humana shareholders approve merger. Forbes, October 19, 2015.
Kass-Hout, T. A., and D. Litwack. 2015. Advancing precision medicine by enabling a collaborative informatics community. http://blogs.fda.gov/fdavoice/index.php/2015/08/advancing-precision-medicine-by-enabling-a-collaborative-informatics-community (accessed September 20, 2015).
Kemper, A. R., N. S. Green, N. Calonge, W. K. Lam, A. M. Comeau, A. J. Goldenberg, J. Ojodu, L. A. Prosser, S. Tanksley, and J. A. Bocchini, Jr. 2014. Decision-making process for conditions nominated to the Recommended Uniform Screening Panel: Statement of the US Department of Health and Human Services Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children. Genetics in Medicine 16(2):183-187.
Lewis, J. R., I. Kerridge, and W. Lipworth. 2015. Coverage with evidence development and managed entry in the funding of personalized medicine: Practical and ethical challenges for oncology. Journal of Clinical Oncology 33(34):4112-4117.
Mansfield, E. A. 2014. FDA perspective on companion diagnostics: An evolving paradigm. Clinical Cancer Research 20(6):1453-1457.
McClellan, M. B., and A. I. Thoumi. 2015. Oncology payment reform to achieve real health care reform. Journal of Oncology Practice 11(3):223-230.
McDonough, R. 2015. Biomarkers for molecularly targeted therapies: Payment and reimbursement issues. Presentation to the Committee on Policy Issues in the Clinical Development and Use of Biomarkers for Molecularly Targeted Therapies, May 4, 2015, Washington, DC.
McShane, L. 2011. Challenges in the development and validation of biomarker-based tests for personalized therapeutic decision making in oncology. Paper presented at Accelerating Anticancer Agent Development and Validation Workshop, May 19, 2011, Bethesda, MD.
Meckley, L. M., and P. J. Neumann. 2010. Personalized medicine: Factors influencing reimbursement. Health Policy 94(2):91-100.
MED-C (Medical Evidence Development Consortium). 2015. Press release: Med-C secures cornerstone funding. http://www.prnewswire.com/news-releases/med-c-secures-cornerstone-funding-300165204.html (accessed November 15, 2015).
MedPAC (Medicare Payment Advisory Commission). 2010. Report to Congress: Aligning incentives in Medicare. http://www.medpac.gov/documents/congressional-testimony/20100623_EandC_Testimony_AligningIncentivesinMedicare.pdf?sfvrsn=0 (accessed August 5, 2015).
Mendelsohn, J. 2013. Personalizing oncology: Perspectives and prospects. Journal of Clinical Oncology 31(15):1904-1911.
Meric-Bernstam, F., C. Farhangfar, J. Mendelsohn, and G. B. Mills. 2013. Building a personalized medicine infrastructure at a major cancer center. Journal of Clinical Oncology 31(15):1849-1857.
Messner, D. A., and S. R. Tunis. 2012. Current and future state of FDA-CMS parallel reviews. Clinical Pharmacology & Therapeutics 91(3):383-385.
Murphy, J., G. Javitt, and K. Hudson. 2006. Creating a genetic testing specialty under CLIA: What are we waiting for? Washington, DC: Pew Charitable Trusts.
NASEM (The National Academies of Sciences, Engineering, and Medicine). 2015. Improving diagnosis in health care. Washington, DC: The National Academies Press.
NCI (National Cancer Institute). 2014. Implementation science—funding. http://cancercontrol.cancer.gov/IS/funding.html (accessed January 22, 2016).
NCI. 2015. Research areas. http://ncorp.cancer.gov/research (accessed January 21, 2016).
Neumann, P. J., and J. D. Chambers. 2012. Medicare’s enduring struggle to define “reasonable and necessary” care. New England Journal of Medicine 367(19):1775-1777.
Neumann, P. J., and J. D. Chambers. 2013. Medicare’s reset on “coverage with evidence development.”http://healthaffairs.org/blog/2013/04/01/medicares-reset-on-coverage-with-evidence-development (accessed October 7, 2015).
Newcomer, L. 2015. Reimbursement issues for biomarkers. Presentation to the Committee on Policy Issues in the Clinical Development and Use of Biomarkers for Molecularly Targeted Therapies, April 1, 2015, Washington, DC.
NIH (National Institutes of Health). 2013. Frequently asked questions about implementation science. http://www.fic.nih.gov/News/Events/implementation-science/Pages/faqs.aspx (accessed January 21, 2016).
NYSDOH (New York State Department of Health). 2013. Genetic testing molecular checklist. http://www.wadsworth.org/labcert/TestApproval/forms/Genetic_Testing_Molecular_Checklist.pdf (accessed September 30, 2015).
NYSDOH. 2015a. Clinical laboratory evaluation program. http://www.wadsworth.org/labcert/clep/clep.html (accessed November 2, 2015).
NYSDOH. 2015b. Guidelines for validation submissions of next generation sequencing (NGS) assays under the NYS testing category of genetic testing—molecular. http://www.wadsworth.org/labcert/TestApproval/forms/Germline_NextGen_Validation_Guidelines.pdf (accessed September 30, 2015).
OIG (Office of the Inspector General). 2013. Comparing lab test payment rates: Medicare could achieve substantial savings. Washington, DC: Office of the Inspector General.
OSTP (Office of Science and Technology Policy). 2012. National Bioeconomy Blueprint. Washington, DC: The White House.
Pant, S., R. Weiner, and M. J. Marton. 2014. Navigating the rapids: The development of regulated next-generation sequencing-based clinical trial assays and companion diagnostics. Frontiers in Oncology 4:78.
Parkinson, D. R., R. T. McCormack, S. M. Keating, S. I. Gutman, S. R. Hamilton, E. A. Mansfield, M. A. Piper, P. Deverka, F. W. Frueh, J. M. Jessup, L. M. McShane, S. R. Tunis, C. C. Sigman, and G. J. Kelloff. 2014. Evidence of clinical utility: An unmet need in molecular diagnostics for patients with cancer. Clinical Cancer Research 20(6):1428-1444.
Patel, K., A. Thoumi, J. Nadel, J. O’Shea, and M. McClellan. 2015. Transforming oncology care: Payment and delivery reform for person-centered care. American Journal of Managed Care 21(5):388-393.
PCORI (Patient-Centered Outcomes Research Institute). 2015. How we’re funded. http://www.pcori.org/about-us/annual-reports-and-financials/how-were-funded (accessed August 25, 2015).
Pear, R. 2015. U.S. introduces new DNA standard for ensuring accuracy of genetic tests. The New York Times, May 14, 2015.
Perlmutter, J. 2015. Commentary: Do participants in adjuvant breast cancer trials reflect the breast cancer population? Breast Diseases: A Year Book Quarterly 26(4).
PMC (Personalized Medicine Coalition). 2015. How alternative payment models could help or hinder the field. Washington, DC: Personalized Medicine Coalition.
Pomager, J. 2014a. Bridging the FDA-CMS divide: An optimized route to market for medical devices—part 1. http://www.meddeviceonline.com/doc/bridging-the-fda-cms-divide-an-optimized-route-to-market-for-medical-devices-part-1-0001 (accessed June 23, 2015).
Pomager, J. 2014b. Bridging the FDA-CMS divide: An optimized route to market for medical devices—part 2. http://www.meddeviceonline.com/doc/bridging-the-fda-cms-divide-an-optimized-route-to-market-for-medical-devices-part-2-0001 (accessed June 23, 2015).
Radensky, P. 2015. Policy issues in the clinical development and use of biomarkers for molecularly targeted therapies—reimbursement webinar. Presentation to the Committee on Policy Issues in the Clinical Development and Use of Biomarkers for Molecularly Targeted Therapies, May 4, 2015, Washington, DC.
Ramsey, S. D., and S. D. Sullivan. 2014. A new model for reimbursing genome-based cancer care. Oncologist 19(1):1-4.
Ray, T. 2015. CMS plan to implement lab test pricing regulation gets critical first response from industry. GenomeWeb, September 28, 2015.
Rehm, H. L., J. S. Berg, L. D. Brooks, C. D. Bustamante, J. P. Evans, M. J. Landrum, D. H. Ledbetter, D. R. Maglott, C. L. Martin, R. L. Nussbaum, S. E. Plon, E. M. Ramos, S. T. Sherry, M. S. Watson, and ClinGen. 2015. ClinGen-—the clinical genome resource. New England Journal of Medicine 372(23):2235-2242.
Relling, M. V., and T. E. Klein. 2011. CPIC: Clinical Pharmacogenetics Implementation Consortium of the pharmacogenomics research network. Clinical Pharmacology & Therapeutics 89(3):464-467.
SACGHS (Secretary’s Advisory Committee on Genetics, Health, and Society). 2006. Coverage and reimbursement of genetic tests report. Washington, DC: Department of Health and Human Services.
SACGHS. 2008. U.S. system of oversight of genetic testing: A response to the charge of the Secretary of Health and Human Services. Washington, DC: Department of Health and Human Services.
Sargent, D. J., B. A. Conley, C. Allegra, and L. Collette. 2005. Clinical trial designs for predictive marker validation in cancer treatment trials. Journal of Clinical Oncology 23(9):2020-2027.
Sawyers, C. L. 2008. The cancer biomarker problem. Nature 452(7187):548-552.
Schneider, A., and V. Wachino. 2013. Chapter 4: Medicaid administration. In The Kaiser Commission on Medicaid and the Uninsured: Menlo Park, CA: Kaiser Family Foundation.
Schott, A. F., C. M. Perou, and D. F. Hayes. 2015. Genome medicine in cancer: What’s in a name? Cancer Research 75(10):1930-1935.
Schulman, K. A., and S. R. Tunis. 2010. A policy approach to the development of molecular diagnostic tests. Nature Biotechnology 28(11):1157-1159.
Shuren, J., and P. H. Conway. 2015. FDA and CMS form task force on LDT quality requirements. http://blogs.fda.gov/fdavoice/index.php/2015/04/fda-and-cms-form-task-force-on-ldt-quality-requirements (accessed June 23, 2015).
Simon, R. M., S. Paik, and D. F. Hayes. 2009. Use of archived specimens in evaluation of prognostic and predictive biomarkers. Journal of the National Cancer Institute 101(21):1446-1452.
Simonds, N. I., M. J. Khoury, S. D. Schully, K. Armstrong, W. F. Cohn, D. A. Fenstermacher, G. S. Ginsburg, K. A. Goddard, W. A. Knaus, G. H. Lyman, S. D. Ramsey, J. Xu, and A. N. Freedman. 2013. Comparative effectiveness research in cancer genomics and precision medicine: Current landscape and future prospects. Journal of the National Cancer Institute 105(13):929-936.
Sniderman, A. D., and C. D. Furberg. 2009. Why guideline-making requires reform. Journal of the American Medical Association 301(4):429-431.
Tanden, N., Z. Emanuel, T. Spiro, E. O. Lee, and T. Huelskoetter. 2014. Comparing the effectiveness of health care: Fulfilling the mission of the Patient-Centered Outcomes Research Institute. https://www.americanprogress.org/issues/healthcare/report/2014/01/24/82775/comparing-the-effectiveness-of-health-care (accessed October 10, 2015).
Terry, S. F. 2014. A call for participatory oversight. Genetic Testing and Molecular Biomarkers 18(2):71-72.
Teutsch, S. M., L. A. Bradley, G. E. Palomaki, J. E. Haddow, M. Piper, N. Calonge, W. D. Dotson, M. P. Douglas, A. O. Berg, and the EGAPP Working Group. 2009. The EGAPP initiative: Methods of the EGAPP working group. Genetics in Medicine 11(1):3-14.
Treweek, S., R. Dryden, C. McCowan, A. Harrow, and A. M. Thompson. 2015. Do participants in adjuvant breast cancer trials reflect the breast cancer patient population? European Journal of Cancer 51(8):907-914.
Trosman, J. R., S. L. Van Bebber, and K. A. Phillips. 2010. Coverage policy development for personalized medicine: Private payer perspectives on developing policy for the 21-gene assay. Journal of Oncology Practice 6(5):238-242.
Trosman, J. R., S. L. Van Bebber, and K. A. Phillips. 2011. Health technology assessment and private payers’ coverage of personalized medicine. Journal of Oncology Practice 7(3 Suppl):18s-24s.
Trosman, J. R., K. A. Phillips, C. B. Weldon, and R. K. Kelley. 2015. Challenges of coverage policy development for next-generation tumor sequencing panels: Experts and payers weigh in. Journal of the National Comprehensive Cancer Network 13(3):311-318.
Tunis, S. R., and S. D. Pearson. 2006. Coverage options for promising technologies: Medicare’s “coverage with evidence development.” Health Affairs 25(5):1218-1230.
Tunis, S. R., R. A. Berenson, S. E. Phurrough, and P. E. Mohr. 2011. Improving the quality and efficiency of the Medicare program through coverage policy. Washington, DC: The Urban Institute.
U.S. Census Bureau. 2014. Health insurance—Highlights 2013. http://www.census.gov/hhes/www/hlthins/data/incpovhlth/2013/highlights.html (accessed August 19, 2015).
Veenstra, D. L., M. Piper, J. E. Haddow, S. G. Pauker, R. Klein, C. S. Richards, S. R. Tunis, B. Djulbegovic, M. Marrone, J. S. Lin, A. O. Berg, and N. Calonge. 2013. Improving the efficiency and relevance of evidence-based recommendations in the era of whole-genome sequencing: An EGAPP methods update. Genetics in Medicine 15(1):14-24.
Walcoff, S. D., and J. D. Pfeifer. 2012. Modernizing US regulatory and reimbursement policy to support continued innovation in genomic pathology. Personalized Medicine 9(3):295-308.
Woodcock, J. 2010. Assessing the clinical utility of diagnostics used in drug therapy. Clinical Pharmacology & Therapeutics 88(6):765-773.
Yu, P. P., M. A. Hoffman, and D. F. Hayes. 2015. Biomarkers and oncology: The path forward to a learning health system. Archives of Pathology & Laboratory Medicine 139(4):451-456.










































































