
D
Case Study on Assessment of New Chemistries
The case study in this appendix describes a hypothetical scenario in which there are three choices of “new” chemicals for use in the manufacture of a product that will result in human exposure. Initial testing shows that the chemicals in question will most likely leach out of the product and possibly end up in food or water that will be ingested by people. In addition, contact with skin during the regular handling of the product is a possible route of human exposure. Finally, the chemical might become aerosolized and inhaled by workers in the manufacturing facility or as a result of indoor consumer use of the product. Therefore, chemical exposure is possible through inhalation, ingestion, and dermal pathways, and the chemical could pose a threat to human health.
For illustrative purposes, the committee chose to use three related drugs (weak acids)—ibuprofen, ibufenac and diclofenac—on which various amounts of in vitro data are publicly available. Table D-1 provides the chemical structures and selected physicochemical properties. To reflect a possible real-world scenario, a key assumption of this case study is that only the in silico and in vitro data presented here are available for the screening assessment; in vivo and clinical data are presumed to be “not yet available.” However, because the adverse effects of the chemicals on people have been studied, one can compare the results of the approach with actual human-safety outcomes. The example is intended to illustrate how available and emerging screening-level tools and data (read-across, screening-level models, and available high-throughput in vitro data) could be applied to inform decision-making and to identify some of the key data gaps and sources of uncertainty that are relevant to risk assessments. The committee notes that most practical approaches for assessing chemical similarity would exclude diclofenac from this comparison because of the chlorine and amine moieties that are not present in the other two chemicals. The committee includes it here for the sake of illustration, but it should be noted that there are limits to how dissimilar chemicals can be used in a read-across scenario.
STRUCTURAL ALERTS
All molecules that contain an arylacetic acid group can undergo acyl glucuronidation, a major metabolic conjugation pathway in mammals for chemicals that contain these groups. Acyl glucuronides have been implicated—although it is not definitively proved—as a cause of adverse effects in humans because they form protein adducts (Shipkova et al. 2003) (see Figure D-1). Common risk concerns are liver injury and hypersensitivity reactions (Regan et al. 2010). The relative reactivity and half-life of the acyl glucuronide has been suggested as a differentiating factor between chemicals that cause adverse events and ones that are of less concern. Other researchers suggest that arylacetic acids can undergo coenzyme A (CoA) conjugation, and interference of the CoA conjugates with lipid metabolism and other cellular processes can lead to the observed toxicity (Darnell and Weidolf 2013). The metabolic scheme might need to be confirmed experimentally to reduce uncertainty (Patlewicz et al. 2015).
IN VITRO DATA
To ensure data consistency among the chemicals in question, in vitro data were gathered only from the ToxCast website, and they are summarized in Table D-2 (EPA 2016). Only assays that yielded activity below a 10 µM threshold are considered because they constitute 20% of the observed assay activity for diclofenac and would most likely be the cause of the toxicity used to set assay doses. It is important to note that the assays used in the ToxCast program do not represent the entire spectrum of biological processes that might be relevant to human health (that is, all possible adverse effects of exposure to chemicals); therefore, there are likely to be gaps in knowledge of how the three chemicals would interact in a biological system. To give some context to the values in Table D-2, diclofenac was tested in a zebrafish toxicity screen and had a lowest effect level of 64 μM (Truong et al. 2014).
Data on ibufenac are not available, but given its structural similarity to ibuprofen and comparable physicochemical properties, one would expect ibufenac to have an in vitro activity profile similar to that of ibuprofen.
TABLE D-1 Chemical Structures and Selected Measured and Predicted Propertiesa
Chemical structure and name |
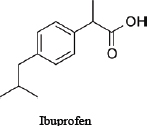 |
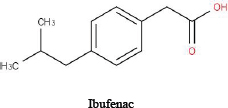 |
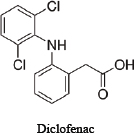 |
|---|---|---|---|
| Molar mass (g/mol) | 206.3 |
192.3 |
296.2 |
| log KOWb | 3.97 |
3.35 |
4.51 |
| log KAWc | -5.21 |
-5.33 |
-9.71 |
| pKad | 4.4 |
4.4 |
4.2 |
| logD (pH 7.4)e | 0.45 |
0.22 |
1.37 |
| Air half-life (h) | 10.8 |
12.7 |
0.78 |
| Predicted whole-body biotransformation half-life (h) (chemical similarity score) | 3.6 (0.36, low similarity) |
2.1 (0.24, low similarity) |
14.9 (0.36, low similarity) |
aPhysicochemical properties are from EPA’s EPI Suite™ (EPA 2011) and ACD Labs (ACD 2015). The whole-body biotransformation half-lives shown here are predicted from structure by using a screening-level quantitative structure–activity relationship (QSAR) model (Arnot et al. 2014a). Various methods can be used to determine the applicability domain of a QSAR prediction. Here, the chemical similarity score is a measure of the similarity, in structure and properties, of a predicted chemical to chemicals in the training dataset on the basis of a nearest-neighbors approach (Brown et al. 2012). The three chemicals have similar molar mass and partitioning and dissociation properties, and absorption efficiencies are expected to be similar in the chemicals but different for each exposure pathway.
blog KOW (or logP) is the log10 of the octanol–water partition coefficient of the neutral species.
clog KAW is log10 of the air–water partition coefficient of the neutral species.
dpKa is the log10 of the acid dissociation constant.
elogD is the log10 of the distribution coefficient of neutral and ionic species between octanol and water at pH 7.4.
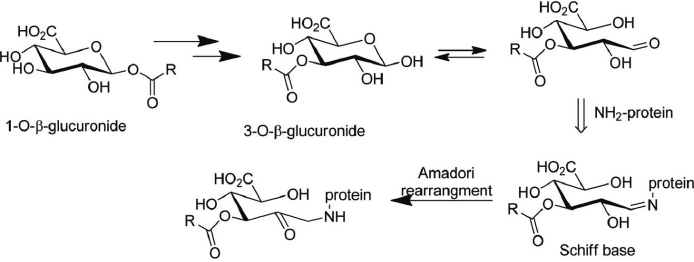
TABLE D-2 Data from In Vitro Assay in Which Chemicals Had Activity Below 10 µM
| Assay Activity | Platform | Diclofenac AC50 (µM) | Ibuprofen AC50 (µM) |
| Decrease in interleukin 8 (IL-8) | BioSeek | – | 0.002 |
| Decrease in matrix metalloproteinase-1 (MMP-1) | BioSeek | – | 0.003 |
| Suppression of prostaglandin E2 secretion (PGE2) | BioSeek | 0.010 | 1.203 |
| Inhibition of cyclooxygenase 1 (COX1) | NovaScreen | 0.163 | 3.0 |
| Inhibition of cyclooxygenase 2 (COX2) | NovaScreen | 0.215 | 30.0 |
| Increase in cell proliferation | BioSeek | – | 0.251 |
| Binding of peroxisome proliferator-activated receptor gamma (PPAR-γ) | NovaScreen | 0.523 | – |
| Decrease in collagen III | BioSeek | 26.108 | 3.509 |
| Decrease in interleukin 6 (IL-6) | BioSeek | – | 3.977 |
| Increase in thrombomodulin | BioSeek | 4.742 | 17.674 |
| Activation of pregnane X receptor (PXR) | Attagene | 7.438 | – |
| Decrease in low-density lipoprotein (LDL) receptor | BioSeek | – | 7.637 |
| Increase in macrophage colony-stimulating factor (M-CSF) | BioSeek | – | 7.639 |
| Decrease in monocyte chemotactic protein 1 (MCP1) | BioSeek | 7.704 | – |
| Activation of PPAR-γ | Attagene | 8.256 | 39.710 |
| Activation of glucocorticoid receptor (GR) | NovaScreen | 8.671 | – |
| Activation of estrogen receptor element (ERE) | Attagene | – | 9.566 |
Source: Data from PubChem. Available at https://pubchem.ncbi.nlm.nih.gov/.
Suppression of Prostaglandin Synthesis
Diclofenac is a potent inhibitor of cyclooxgenase 1 and 2 (COX1 and COX2), and inhibition of these enzymes can decrease prostaglandin biosynthesis (Vane 1971). Decreased secretion of prostaglandin E2 (PGE2) was observed in the Bioseek platform. Ibuprofen is a weak nonspecific inhibitor of COX1 (IC50, about 18 µM) and COX2 (IC50, about 370 µM) (Noreen et al. 1998) but also showed a similar suppression of PGE2 in the BioSeek platform. PGE2 is linked to suppression of T-cell receptor signaling and inflammation responses (Wiemer et al. 2011). However, PGE2 is also a vasodilator, so suppression of its secretion might lead to an increase in blood pressure and to cardiac toxicity (Strong and Bohr 1967).
Drugs that inhibit COX1 or COX2, such as celecoxib and rofecoxib, have been linked with causing cardiovascular events (Johnsen et al. 2005), and rofecoxib, a selective COX2 inhibitor, was withdrawn from the US market after being linked to heart attacks and strokes. Inhibitors of COX1 have been linked to causing ulceration and bleeding in the gastrointestinal tract as a result of suppressing the secretion of the protective prostaglandins PGE2 and PGI2 (Süleyman et al. 2007). Inhibitors of COX1 might also affect renal function by changing the role of prostaglandins on renal hemodynamics and glomerular filtration rate (GFR) (DuBois et al. 1998; Morita 2002).
Diclofenac and ibuprofen increase thrombomodulin (TM) in the BioSeek platform. TM is a cell-surface receptor for thrombin on endothelial cells that is involved in blood coagulation (Gerlitz et al. 1993). Increases in TM might increase clotting times but similarly reduce the risk of stroke and myocardial infarction (Esmon et al. 1982).
The low-density lipoprotein (LDL) receptor mediates the endocytosis of LDL. The accumulation of LDL in the blood is involved in the development of atherosclerosis, which is the process responsible for most cardiovascular diseases (Hobbs et al. 1992). A decrease in the LDL receptor might lead to an increased risk of cardiovascular events in people who are predisposed to atherosclerosis or who have cardiovascular conditions.
Liver Effects
Diclofenac is shown to increase the activity of the pregnane X receptor (PXR). PXR is a nuclear receptor that has important roles in integrating pathways related to fatty acid, lipid, and glucose metabolism (Wada et al.
2009). It also senses the presence of foreign substances and responds by upregulating proteins involved in their oxidation and others involved in their clearance (Kliewer 2003), and it is a transcriptional regulator of the cytochrome P450 gene CYP3A4, a major metabolizing enzyme for many drugs that is highly expressed in the liver.
Both diclofenac and ibuprofen activate the peroxisome proliferator–activated receptor gamma (PPAR-γ) that regulates fatty acid storage and glucose metabolism, although only at relatively high concentrations in the case of ibuprofen. The genes activated by PPAR-γ increase lipid uptake and adipogenesis by fat cells (Zou et al. 2016). PPAR-γ agonists have been used in the treatment of hyperlipidemia and hyperglycemia and therefore might induce hypoglycemia in healthy subjects (Spiegelman 1998; Rangwala and Lazar 2004). Some drugs that were designed to activate PPAR-γ have been linked with hepatotoxicity (troglitazone: Watkins 2005), cardiovascular events (rosiglitazone: Singh et al. 2007), and an increased incidence of in bladder cancer (pioglitazone: Ferwana et al. 2013). However, no direct link has been established between the activation of PPAR-γ and those adverse events.
Immune-Response Effects
Diclofenac and ibuprofen have effects on various cellular processes that are involved in inflammation and tissue repair. For example, diclofenac decreases the expression of monocyte chemotactic protein 1 (MCP1). MCP1 promotes movement of monocytes, memory T cells, and dendritic cells to sites of inflammation (Mukaida et al. 1998; Xue et al. 2015).
Similarly, diclofenac is an agonist of the glucocorticoid receptor (GR), which is expressed in almost every cell in the body and regulates genes that control development, metabolism, and immune response (Rhen and Cidlowski 2005; Lu et al. 2006). The activated GR complex prevents the movement of transcription factors from the cytosol into the nucleus, resulting in changes in expression of nuclear anti-inflammatory proteins and cytosolic proinflammatory proteins.
Ibuprofen decreases the secretion of Interleukin 8 (IL-8) as measured in the BioSeek platform. IL-8 is a chemokine that is produced by macrophages and other cell types, such as epithelial cells, airway smooth muscle cells (Hedges et al. 2000), and endothelial cells. IL-8 induces chemotaxis in neutrophils and causes them to migrate toward sites of infection and promotes phagocytosis at the infection site. It is also a potent promoter of angiogenesis and an important mediator of the immune reaction in the innate immune system response. Ibuprofen decreases the secretion of IL-6, which acts as a pro-inflammatory cytokine and an anti-inflammatory myokine (Schöbitz et al. 1994).
HAZARD IDENTIFICATION
On the basis of the available in vitro data, structural comparisons, and knowledge of structural alerts, a key safety concern about all three chemicals would be liver injury through the formation of reactive acyl glucuronides or acyl coenzyme A conjugates that would cause tissue damage and impaired organ function. Relative reactivity of the acyl conjugates would play an important role in determining the risk of liver injury. Chemicals that have alkyl substitutions at the α-carbon atom have been shown to have lower reactivity with protein nucleophiles; this suggests that inherent electronic and steric effects affect the overall rate of acyl glucuronide rearrangement (Stepan et al. 2011) and so could have a profound effect on the reactivity of the conjugates in the case of ibuprofen (Wang et al. 2004; Walker et al. 2007; Baba and Yoshioka 2009). The risk of liver injury could be increased by induction of cytochrome P-450s through activation of PXR and by lipid dysfunction as a result of activation of PPAR-γ.
Cardiovascular toxicity in the form of increased blood pressure and increased clotting times and renal damage or gastrointestinal bleeding caused by the suppression of prostaglandin secretion are also of concern with diclofenac and ibuprofen and by inference, ibufenac.
As discussed in Chapter 3, for inhibitors of G-protein–coupled receptors, the anticipated pharmacological response is often observed in vivo at plasma concentrations up to 3 times the measured IC50 of the chemical in question (McGinnity et al. 2007). As a general rule of thumb, a 100-fold difference between the measured IC50 or the inhibition constant in a cell-free assay and the circulating plasma Cmax free1 concentration could be considered to be adequate to pose minimal risk of toxicity from a pharmacological interaction. It is worth noting that for more phenotypic cellular responses, such as those measured by the BioSeek platform, more research is required to establish an appropriate translation from in vitro to in vivo.
EXPOSURE ASSESSMENT
In this hypothetical case study, the three chemicals of interest have not been used in commercial products; therefore, there are no monitoring data, and there are no emissions and use data on which to formulate a typical risk-based evaluation. However, the available premarket toxicity or bioactivity data identified above can be used to develop parameters for exposure models that can “back-calculate” the rates of chemical use for various scenarios that correspond to specific hazard thresholds. The selected threshold for such simulations could be a concentration from a bioassay in the case of ibuprofen or diclofenac or a read-across value in the case of ibufenac determined from
___________________
1 Cmax free is the maximum measured or observed concentration of the fraction of the chemical that is unbound to plasma proteins.
in vivo, in vitro, or computational methods or a no-effect threshold method, such as one that uses a threshold of toxicological concern.
The general exposure-assessment approach outlined here is analogous to the critical-emission-rate concept that has been applied in ecological assessments (Arnot et al. 2006) and to concepts applied in reverse toxicokinetics that is used to calculate intake rates expressed as oral equivalent doses (OEDs;2 mg/kg-day) from the in vitro testing data (Rotroff et al. 2010). In the present case, toxicokinetic models are combined with indoor-fate models to back-calculate the rates of chemical use that correspond to illustrative exposure scenarios. The simulations can consider various assumptions and contexts for exposure and chemical or product scenarios. The exposure models used in such simulations can vary in complexity according to the amount of data needed to satisfy all the parameter requirements. In that regard, tiered modeling strategies might be helpful.
In this example, a one-compartment, whole-body toxicokinetic model that considers primary routes of exposure and intake (dermal, ingestion, and inhalation) and routes of elimination (for example, exhalation, renal excretion, biotransformation, egestion, and desquamation) is linked to a representative indoor environment (Arnot et al. 2014b) to back-calculate the rates of chemical use for three hypothetical exposure scenarios:
- Scenario 1. The chemical is released to air in a defined indoor environment. Exposure pathways include inhalation, dermal permeation (from passive diffusion in air), and nondietary ingestion (from hand-to-surface and surface-to-mouth contact).
- Scenario 2. The chemical is applied directly to skin and assumed to be left on indefinitely. Exposure pathways include dermal permeation and inhalation (from volatilization of the chemical from dermal application).
- Scenario 3. The chemical is ingested.
Simplifying assumptions are steady-state calculations and no charged species (that is, no explicit calculation for charged species; only the neutral form is simulated). The latter assumption is similar to recent hazard and risk-based calculations that used ToxCast data in which the potential for chemical dissociation was ignored; that is, acids and bases were treated as nondissociating neutral organics (Rotroff et al. 2010; Wetmore et al. 2012; Shin et al. 2015).
The first step is to translate the in vitro bioassay concentrations (Cin vitro) that correspond to the observed bioactivity to in vivo concentrations (Cin vivo). Here, the committee uses the same assumptions as in recent applications of ToxCast data for OED calculations: Cin vivo, blood = Cin vitro (Rotroff et al. 2010; Wetmore et al. 2012; Shin et al. 2015). However, more explicit calculations should be used to account for differences in the in vitro and in vivo systems; for example, free dissolved concentrations rather than assumed nominal in vitro concentrations could be used (see discussion in Chapter 2). The steady-state volume of distribution is assumed to be 0.5 L/kg (35 L) for the three chemicals to relate blood concentrations to whole-body concentrations. Models for volume of distribution and other methods to address differential concentrations among and within tissues could be considered. The lowest AC50 from the available ToxCast assays is selected as the hazard threshold on which to base parameter values for the exposure model. That value for ibuprofen and ibufenac is 0.002 µM, and the selected threshold for diclofenac is 0.01 µM (see Table D-2).
The second step is to select the parameters needed for the exposure models to calculate chemical fate in various environments. For the sake of illustration, the committee assumes an adult human in a single room, although infants and children have greater breathing rates relative to body weight; the evaluative model requires the following chemical-specific information: KOW, KAW, and degradation half-lives in air (see Table D-1). Quantitative structure–activity relationship (QSAR) models are used here to predict whole-body biotransformation half-life data. Half-lives could also be determined by scaling in vitro assay data derived from hepatocytes to liver (see, for example, Rotroff et al. 2010) or whole-body half-life estimates. In addition, hepatic, renal, or other compartment-specific QSAR models could be used to provide parameter values for pharmacokinetic models that are used for exposure assessment. Ideally, multiple lines of evidence (for example, various measured and predicted estimates) will show concordance in key information used in the model simulations (chemical partitioning properties and reaction half-lives), and this concordance will foster confidence in the assessment results. If chemicals are shown to have high environmental persistence, adding far-field human-exposure models to the assessment is warranted to account for possible far-field exposure pathways (chemical dispersed and diffused into food and water).
RISK CHARACTERIZATION
The results of the back-calculation simulations are summarized in Table D-3. The calculations yield the indoor air release (Scenario 1), application (Scenario 2), and ingestion (Scenario 3) rates in milligrams per day corresponding to the selected in vitro bioactivity-assay data (assumed threshold values). The results could be used for interim guidance on use scenarios for each chemical and for comparative analyses between the three candidate chemicals. If one assumes that all three chemicals are
___________________
2 The OED is the chemical intake rate that corresponds to an assumed steady-state blood concentration related to the in vitro bioactivity.
| Chemical | Scenario 1: Release to Indoor Air | Scenario 2: Application Directly to Skin | Scenario 3: Ingestion |
| Diclofenac | 10 | 1.1 | 0.14 |
| Ibufenac | 1.8 | 0.15 | 0.13 |
| Ibuprofen | 1.9 | 0.16 | 0.08 |
used in the same quantity, the chemicals and scenarios with the lowest rates correspond to the greatest potential to achieve the in vitro bioactivity threshold. For example, for diffuse release to air in an indoor environment (Scenario 1), diclofenac shows the highest emission or release rate and, so could pose the lowest potential concern of the three chemicals. Of the three exposure scenarios, Scenario 3 results in the lowest use and application rates for all chemicals. Overall, the ranges of values are not large because the chemicals have similar properties for partitioning, reaction, and bioactivity (that is, the same bioactivity value is used for ibuprofen and ibufenac on the basis of structural read-across).
The values in Table D-3 do not show the uncertainty in the calculations and do not account for interindividual variability in the pharmacokinetics and pharmacodynamics that one would expect in a large diverse population. The results of this example are illustrative, but the general concept can be helpful in determining putative use and release scenarios for premarket chemicals. The application of exposure models to back-calculate emission and use rates corresponding to a toxic threshold or bioactivity can also be useful for evaluating commercial chemicals when emission and use rates are unknown or highly uncertain. Ultimately, confidence in the calculated emission and use rates depends on the confidence in and suitability of the toxicity (threshold) data and the exposure-model estimates. For the three chemicals in this example, measured volumes of distribution are about one-third to one-half the assumed values, and half-lives in adults are one-seventh to one-half the values used in this premarket assessment (Obach et al. 2008). For risk-based decision-making, additional analyses for various life stages and alternative use scenarios should be considered as warranted.
To put the exposure estimates in Table D-3 into context, the typical over-the-counter medicines that contain ibuprofen recommend an oral dose of 200–400 mg every 4 hours with a maximum dose of 1,200 mg in any 24-hour period for persons over 12 years old.3 However, doctors can prescribe ibuprofen to be given orally at up to 3,200 mg/day in doses of up to 800 mg at any one time.4 Similarly, ibuprofen has been approved in Europe for administration to children 3 to 6 months of age at a starting dose of 50 mg taken orally three times a day. Ibuprofen is contraindicated in pregnant women in their third trimester, and doctors recommend that women during the first 6 months of pregnancy not take it, if that is possible.
Diclofenac is approved for use by prescription, and the maximum recommended daily oral dose is 150 mg in adults; it is not recommended for use in children under 12 years old. It is also contraindicated for use by pregnant women. Ibufenac was withdrawn from the market because of severe hepatotoxicity and jaundice in patients taking the drug. At the time, the maximum recommended daily oral dose of ibufenac was 750 mg.
In Europe, both ibuprofen and diclofenac were approved for use as a topical gel (ibuprofen, 5% w/w gel; diclofenac, 2.32% w/w gel). A maximum daily application of the diclofenac gel was 8 g, which is equivalent to 160 mg of the active ingredient. Similarly, the recommended application of the ibuprofen gel was up to 125 mg, four times per day, which is equivalent to 25 mg of the active ingredient. However, only 22% of the dose is absorbed through the skin. Compared with the oral route of administration of ibuprofen, the plasma exposure is considered to be much lower and unlikely to cause systemic side effects.
The estimated oral and dermal exposures in the present example would be substantially below the therapeutic doses for most populations, including children. However, it should be noted that at therapeutic doses, some side effects and adverse events are observed with various, albeit relatively low, frequencies that might not necessarily be considered tolerable in an environmental or occupational risk assessment in which long-term, low-level exposures of a broad population demographic have to be considered. Similarly, conclusions cannot be drawn at this time about whether the estimated doses would ensure the protection of the most sensitive group—pregnant women.
___________________
3 See https://www.medicines.org.uk/emc/medicine/15681.
4 See http://www.accessdata.fda.gov/drugsatfda_docs/anda/2001/76-112_Ibuprofen.pdf.
REFERENCES
ACD (Advanced Chemistry Development). 2015. ACD/Percepta Suite. ACD, Toronto, ON, Canada.
Arnot, J.A., D. Mackay, E. Webster, and J.M. Southwood. 2006. Screening level risk assessment model for chemical fate and effects in the environment. Environ. Sci. Technol. 40(7):2316-2323.
Arnot, J.A., T.N. Brown, and F. Wania. 2014a. Estimating screening-level organic chemical half-lives in humans. Environ. Sci. Technol. 48(1):723-730.
Arnot, J.A., X. Zhang, I. Kircanski, L. Hughes, and J. Armitage. 2014b. Develop Sub-module for Direct Human Exposures to Consumer Products. Technical report for the US Environmental Protection Agency, by ARC Arnot Research & Consulting Inc., Toronto, Canada.
Baba, A., and T. Yoshioka. 2009. Structure-activity relationships for the degradation reaction of 1-beta-O-acyl glucuronides. Part 3: Electronic and steric descriptors predicting the reactivity of aralkyl carboxylic acid 1-beta-O-acyl glucuronides. Chem. Res. Toxicol. 22(12):1998-2008.
Brown, T.N., J.A. Arnot, and F. Wania. 2012. Iterative fragment selection: A group contribution approach to predicting fish biotransformation half-lives. Environ. Sci. Technol. 46(15):8253-8260.
Darnell, M., and L. Weidolf. 2013. Metabolism of xenobiotic carboxylic acids: Focus on coenzyme a conjugation, reactivity, and interference with lipid metabolism. Chem. Res. Toxicol. 26(8):1139-1155.
DuBois, R.N., S.B. Abramson, L. Crofford, R.A. Gupta, L.S. Simon, L.B. Van De Putte, and P.E. Lipsky. 1998. Cyclooxygenase in biology and disease. FASEB J. 12(12):1063-1073.
EPA (US Environmental Protection Agency). 2011. Estimation Programs Interface (EPI) Suite for Microsoft® Windows, Version 4.1. US Environmental Protection Agency, Washington, DC.
EPA (US Environmental Protection Agency). 2016. Toxicity Forecasting: Advancing the Next Generation of Chemical Evaluations [online]. Available: https://www.epa.gov/chemical-research/toxicity-forecasting [accessed July 26, 2016].
Esmon, C.T., N.L. Esmon, and K.W. Harris. 1982. Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor V activation. J. Biol. Chem. 257(14):7944-7947.
Ferwana, M., B. Firwana, R. Hasan, M.H. Al-Mallah, S. Kim, V.M. Montori, and M.H. Murad. 2013. Pioglitazone and risk of bladder cancer: A meta-analysis of controlled studies. Diabet. Med. 30(9):1026-1032.
Gerlitz, B., T. Hassell, C.J. Vlahos, J.F. Parkinson, N.U. Bang, and B.W. Grinell. 1993. Identification of the predominant glycosaminoglycan-attachment site in soluble recombinant human thrombomodulin: Potential regulation of functionality by glycosyltransferase competition for serine474. Biochem. J. 295(Pt 1):131-140.
Hedges, J.C., C.A. Singer, and W.T. Gerthoffer. 2000. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am. J. Respir. Cell Mol. Biol. 23(1):86-94.
Hobbs, H.H., M.S. Brown, and J.L. Goldstein. 1992. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1(6):445-466.
Johnsen, S.P., H. Larsson, R.E. Tarone, J.K. McLaughlin, B. Nørgård, S. Friis, and H.T Sørensen. 2005. Risk of hospitalization for myocardial infarction among users of rofecoxib, celecoxib, and other NSAIDs: A population-based case-control study. Arch. Intern. Med. 165(9):978-984.
Kliewer, A. 2003. The nuclear pregnane X receptor regulates xenobiotic detoxification. J. Nutr. 133(7 Suppl.):2444S2447S.
Lu, N.Z., S.E. Wardell, K.L. Burnstein, D. Defranco, P.J. Fuller, V. Giguere, R.B. Hochberg, L. McKay, J.M. Renoir, N.L. Weigel, E.M. Wilson, D.P. McDonnell, and J.A. Cidlowski. 2006. International union of pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: Glucocorticoid, mineralocorti-coid, progesterone, and androgen receptors. Pharmacol. Rev. 58(4):782-797.
McGinnity, D.F., J. Collington, R.P. Austin, and R.J. Riley. 2007. Evaluation of human pharmacokinetics, therapeutic dose and exposure predictions using marketed oral drugs. Curr. Drug Metab. 8(5):463-479.
Morita, I. 2002. Distinct functions of COX-1 and COX-2. Prostag. Oth. Lipid M. 68-69:165-175.
Mukaida, N., A. Harada, and K. Matsushima. 1998. Interleukin-8 (IL-8) and monocyte chemotactic and activating factor (MCAF/MCP-1), chemokines essentially involved in inflammatory and immune reactions. Cytokine Growth Factor Rev. 9(1):9-23.
Noreen, Y., T. Ringbom, P. Perera, H. Danielson, and L. Bohlin. 1998. Development of a radiochemical cyclooxygenase-1 and -2 in vitro assay for identification of natural products as inhibitors of prostaglandin biosynthesis. J. Nat. Prod. 61(1):2-7.
Obach, R.S., F. Lombardo, and N.J. Waters. 2008. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dispos. 36(7):1385-1405.
Patlewicz, G., N. Ball, P.J. Boogaard, R.A. Becker, and B. Hubesch. 2015. Building scientific confidence in the development and evaluation of read-across. Regul. Toxicol. Pharmacol. 72(1):117-133.
Rangwala, S.M., and M.A. Lazar. 2004. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol. Sci. 25(6):331-336.
Regan, S.L., J.L. Maggs, T.G. Hammond, C. Lambert, D.P. Williams, and B.K. Park. 2010. Acyl glucuronides: The good, the bad and the ugly. Biopharm. Drug Dispos. 31(7):367-395.
Rhen, T., and J.A. Cidlowski. 2005. Antiinflammatory action of glucocorticoids- new mechanisms for old drugs. N. Engl. J. Med. 353(16):1711-1723.
Rotroff, D.M., B.A. Wetmore, D.J. Dix, S.S. Ferguson, H.J. Clewell, K.A. Houck, E.L. Lecluyse, M.E. Andersen, R.S. Judson, C.M. Smith, M.A. Sochaski, R.J. Kavlock, F. Boellmann, M.T. Martin, D.M. Reif, J.F. Wambaugh, and R.S. Thomas. 2010. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci. 117(2):348-358.
Schöbitz, B., E.R. de Kloet, and F. Holsboer. 1994. Gene expression and function of interleukin I, interleukin 6 and tumor necrosis factor in the brain. Prog. Neurobiol. 44(4):397-432.
Shin, H.M., A. Ernstoff, J.A. Arnot, B.A.Wetmore, S.A. Csiszar, P. Fantke, X. Zhang, T.E. McKone, O. Jolliet, and D.H. Bennett. 2015. Risk-based high-throughput chemical screening and prioritization using exposure models and in vitro bioactivity assays. Environ. Sci. Technol. 49(11):6760-6771.
Shipkova, M., V.W. Armstrong, M. Oellerich, and E. Wieland. 2003. Acyl glucuronide drug metabolites: Toxicological and analytical implications. Ther. Drug Monit. 25(1):1-16.
Singh, S., Y.K. Loke, and C.D. Furberg. 2007. Long-term risk of cardiovascular events with rosiglitazone: A meta-analysis. JAMA 298(10):1189-1195.
Spiegelman, B.M. 1998. PPAR-gamma: Adipogenic regulator and thiazolidinedione receptor. Diabetes 47(4):507-514.
Stepan, A.F., D.P. Walker, J. Bauman, D.A. Price, T.A. Bail-lie, A.S. Kalgutkar, and M. Aleo. 2011. Structural alert/reactive metabolite concepts as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicitiy: A perspectove based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 24(9):1345-1410.
Strong, C.G., and D.F. Bohr. 1967. Effects of prostaglandins E1, E2, A1, and F1-alpha on isolated vascular smooth muscle. Am. J. Physiol. 213(3):725-733.
Süleyman, H., B. Demircan, and Y. Karagöz. 2007. Anti-inflammatory and side effects of cyclooxygenase inhibitors. Pharmacol. Rep. 59(3):247-258.
Truong, L., D.M. Reif, L. St Mary, M.C. Geier, H.D. Truong, and R.L. Tanguay. 2014. Multidimensional in vivo hazard assessment using zebrafish. Toxicol. Sci. 137(1):212-233.
Vane, J.R. 1971. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 231(25):232-235.
Wada, T., J. Gao, and W. Xie. 2009. PXR and CAR in energy metabolism. Trends Endocrinol. Metab. 20(6):273-279.
Walker, G.S., J. Atherton, J.Bauman, C. Kohl, W. Lam, M. Reily, Z. Lou, and A. Mutlib. 2007. Determination of degradation pathways and kinetics of acyl glucuronides by NMR spectroscopy. Chem. Res. Toxicol. 20(6):876-886.
Wang, J., M. Davis, F. Li, F. Azam, J. Scatina, and R. Talaat. 2004. A novel approach for predicting acyl glucuronide reactivity via Schiff base formation: Development of rapidly formed peptide adducts for LC/MS/MS measurements. Chem. Res. Toxicol. 17(9):1206-1216.
Watkins, P.B. 2005. Insight into hepatotoxicity: The troglitazone experience. Hepatology 41(2):229-230.
Wetmore, B.A., J.F. Wambaugh, S.S. Ferguson, M.A. Sochaski, D.M. Rotroff, K. Freeman, H.J. Clewell, III, D.J. Dix, M.E. Andersen, K.A. Houck, B. Allen, R.S. Judson, R. Singh, R.J. Kavlock, A.M. Richard, and R.S. Thomas. 2012. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci. 125(1):157-174.
Wiemer, A.J., S. Hegde, J.E. Gumperz, and A. Huttenlocher. 2011. A live imaging cell motility screen identifies prostaglandin E2 as a T Cell stop signal antagonist. J. Immun. 187(7):3663-3670.
Xue, J., F. Chen, J. Wang, S. Wu, M. Zheng, H. Zhu, Y. Liu, J. He, and Z. Chen. 2015. Emodin protects against concanavalin A-induced hepatitis in mice through inhibiting activation of the p38 MAPK-NF-ĸB signaling pathway. Cell Physiol. Biochem. 35:1557-1570.
Zou, Q., W. Hong, Y. Zhou, Q. Ding, J. Wang, W. Jin, J. Gao, G. Hua, and X. Xu. 2016. Bone marrow stem cell dysfunction in radiation-induced abscopal bone loss. J. Orthop. Surg. Res. 11:3.








