
In the first session, speakers were asked to highlight broad issues that arise when incorporating genetics into the drug development process. Robert Nussbaum, chief medical officer at Invitae, summarized key themes and recommendations from a National Academies consensus study report on the clinical development and use of biomarkers for molecularly targeted therapies. Jane Perlmutter, president and founder of the Gemini Group, shared her perspective as a patient and patient advocate on engaging patients in clinical research. Michael Pacanowski, associate director for genomics and targeted therapy at the Center for Drug Evaluation and Research of the U.S. Food and Drug Administration (FDA), discussed FDA’s regulatory pathway for in vitro diagnostics (IVDs) in clinical drug development. The session was moderated by Nisenbaum.
BIOMARKER TESTS FOR MOLECULARLY TARGETED THERAPIES: HIGHLIGHTS FROM A NATIONAL ACADEMIES CONSENSUS STUDY
The National Institutes of Health (NIH) defines precision medicine as an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person. One key feature of precision medicine is the use of biomarker tests that identify molecular variations within an individual patient. Biomarker tests can be used for disease screening, diagnosis, treatment, and post-treatment monitoring, as well as for predicting a patient’s response to treatment. Biomarker tests can therefore be applied within the drug development process, and the workshop planning committee agreed that it is important to discuss the challenges and opportunities associated with developing, validating, regulating, and integrating biomarker tests for molecularly targeted therapies. Therefore, a brief overview of the recent National Academies consensus study report Biomarker Tests for Molecularly Targeted Therapies: Key to Unlocking Precision Medicine was provided by Nussbaum, who was a member of the study committee (NASEM, 2016b).1 The consensus study committee was charged with examining policy issues related to the clinical development and use of biomarker tests (including genomics-based tests) for targeting therapies to patients, reviewing the opportunities for and challenges to the use of biomarker tests to select an optimal therapy, and formulating recommendations to accelerate progress in the field. The
___________________
1 The full text of the consensus study report is available at https://www.nap.edu/catalog/21860 (accessed April 26, 2017).
report, Nussbaum said, provided a high-level review of a very fragmented and complex ecosystem involving health care providers, third-party payers, diagnostics developers, and patients, with the goal of improving the understanding of and ultimately providing better biomarkers and therapies.
Nussbaum highlighted four key themes from the report:
- Accurate, reliable, clinically useful, and appropriately implemented biomarker tests for molecularly targeted therapies are key to realizing the full potential of precision medicine.
- Substantial variation in the evidence used to inform regulatory, reimbursement, and treatment decisions ultimately limits the broader adoption of potentially useful biomarker tests for molecularly targeted therapies into clinical practice.
- In this rapidly changing field, regulation, coverage, reimbursement, and practice guidelines will continue to evolve as evidence is generated and new information becomes available. Data sharing is essential.
- A rapid learning system represents a framework for collecting and analyzing data and information and enables continuous learning from research and clinical practice.
Nussbaum also reviewed the ten main goals identified by the report committee, which were crafted around three main topic areas that the committee identified as forming the key components of a rapid learning system framework for biomarker tests for molecularly targeted therapies. The three topic areas were developing a more supportive policy and regulatory environment, advancing clinical practice processes to improve patient care, and supporting a data infrastructure to better integrate and analyze data (see Figure 2-1). Nussbaum referred workshop participants to the full report, which provides more detailed information on these goals and the accompanying recommendations for how they could be achieved, and discusses the rapid learning system for continual improvement of biomarkers for molecularly targeted therapies.
PERSPECTIVE OF A PATIENT AND PATIENT ADVOCATE
Central to the concept of precision medicine is the well-being of patients. The overarching goal for most patients is to live long and live well, Perlmutter said. However, each patient is unique with regard to biology, age, ethnicity, culture, geography, and other characteristics that interact and potentially affect whether and how the patient participates in clinical trials. At the time a patient is diagnosed with a genetic disease, he or she might lack the scientific or health literacy needed to be an effective partner in the
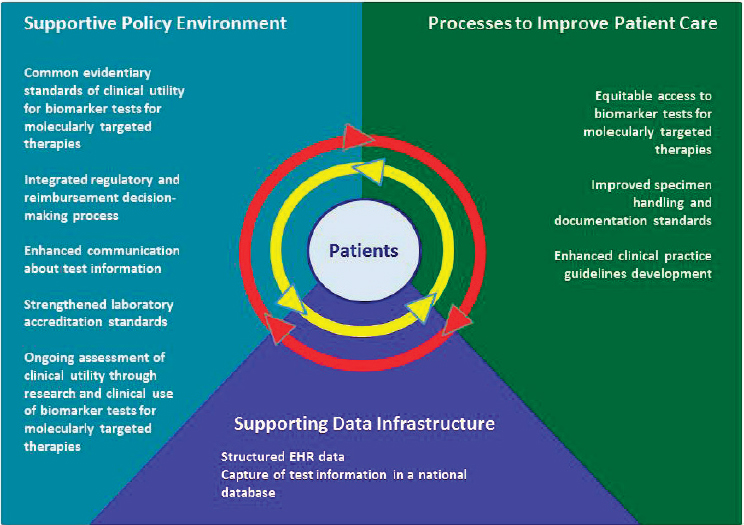
NOTE: EHR = electronic health record.
SOURCE: Robert Nussbaum, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017, adapted from NASEM (2016c).
drug development process. Perlmutter went on to share her perspectives on genetics-enabled drug development from her experiences as a patient, a patient advocate, and a scientist.
Patient Concerns: Data Privacy and Security
Principle concerns for patients, particularly in the context of genetics and genomics, are data privacy and security and the timely return of their genetic testing results.2 While much attention is paid to privacy and security in medicine (for example, the protections afforded by the Health Insurance
___________________
2 Perlmutter referred workshop participants to recent guidelines from the Multi-Regional Clinical Trials initiative at Harvard University, which describe the principles for returning aggregate and individual results to trial participants. More information is available at http://mrctcenter.org/wp-content/uploads/2015/11/2015-03-19_mrct_ror_guidance_1.0.pdf (accessed May 5, 2017).
Portability and Accountability Act), many patients share personal medical information extensively on social media and the Internet, Perlmutter pointed out. She suggested that the medical field might be overemphasizing data privacy. Data sharing and return of results to patients and trial participants need more emphasis, she said, but researchers often lack the funding needed to develop the requisite standards for sharing data with participants or other researchers, or to translate genetic testing or trial summary results into lay-accessible formats.
Educating and Engaging Potential Clinical Trial Participants
To better engage patients and encourage participation in clinical trials, Perlmutter noted that patients could be more effectively educated on the trial process before they are acutely in need of a clinical trial. She noted that more effective education could help people understand the genetic underpinnings of disease and also the medical research and development processes, including protections afforded to patients involved in clinical trials. It is not advisable to create undue hype around investigational drugs, she added, and it is important to be careful when articulating both the promises and limitations of investigational drugs. To this end, scientists and providers are increasingly careful to make clear that investigational medical products follow a lengthy developmental trajectory, which could likely result in the termination of development. However, misperceptions still exist. For example, when a provider says that an investigational product is going to be “in the clinic” within 2 years, the provider might actually mean that it will be entering Phase 1 clinical trials. The patient, however, may interpret this to mean that the product will be available from his or her doctor in 2 years and may become disheartened upon learning that it could be 10 to 15 years, or longer, if the drug succeeds in trials and is approved. Perlmutter also shared her “bill of rights” for what she believes clinical trial participants deserve (see Box 2-1).
In summary, Perlmutter said, patients and patient advocates should be included in the development of clinical trials to ensure that their design, implementation, and follow-up are patient-centric. Patient-centric design elements could include, for example, selecting beneficial endpoints, minimizing patient burden, and maximizing the potential patient benefit. Implementing trials in a patient-centric manner could include developing patient-friendly informed consent and educational materials and training health care staff in patient communication. Follow-up could include disseminating both individual genetic testing and trial summary results to trial participants and sharing a plain language, accessible summary with the public.
NAVIGATING THE REGULATORY PATHWAY FOR IN VITRO DIAGNOSTIC TESTS AND BIOMARKERS IN CLINICAL DRUG DEVELOPMENT
Pacanowski provided an overview of key regulatory issues associated with the identification of biomarkers and the development of companion IVDs. The use of genomics in drug development, he said, can be preemptive (e.g., to validate molecular targets, predict adverse events or toxicities, or define target population), retrospective (e.g., to explain variability, identify responders, or characterize drug interactions), or prospective (e.g., to enrich clinical trials or adapt dosages to increase the probability of responders or event rates). Prospective use of genomics in drug development may result in what is generally termed as precision medicines—drugs or biologics intended for use with a genomic, proteomic, or other specific biomarker that identifies patients within a clinically defined disease who are eligible for treatment, that aids in determining the appropriate dose, or that allows for the monitoring of responses in order to individualize therapy. Genetic
biomarkers are generally diagnostic, prognostic, or predictive, and, in precision medicine, there is generally some mechanistic relationship between the biomarker and the drug that is being developed. In support of this concept, Pacanowski highlighted data from a recent study that suggests that the use of biomarkers to enrich clinical trial populations is associated with lower rates of attrition and a higher probability of success (BIO, 2016).
An increasing number of drugs in recent years have been approved by FDA for molecularly defined subsets of patients, Pacanowski said. This has been facilitated, he added, by increased clarity in the regulatory pathway for developing molecularly targeted therapies and by FDA’s ability to coordinate the review of drugs and devices (i.e., IVDs). Pacanowski highlighted several factors that might accelerate targeted drug development in a molecularly defined subset of patients:
- The pharmacology or basic biology of the biomarker is known.
- The biomarker being pursued for development is in the causal pathway of the disease or is the primary drug target.
- Non-clinical studies show limited or paradoxical activity of a drug in a certain subset of gene or protein variants.
- Early-stage clinical trials show large treatment effects in certain biomarker-defined subgroups.
Considerations for the approval of biomarker-based indications include
- confidence in the biomarker (e.g., predictive utility, whether it is routinely measured in clinical practice, or other intrinsic properties that could impact reliable measurement in the clinic);
- nature of the disease (e.g., morbidity, mortality, available treatments); or
- therapeutic properties of the drug (e.g., magnitude and nature of benefit, safety profile, dosing approach).
A Discussion of FDA Lessons Learned and Guidance Documents for Industry
FDA’s experience in reviewing biomarkers and diagnostic assays for precision drug development has imparted some important lessons, shared by Pacanowski. The first insight was that relying on retrospective data analysis to identify potential biomarkers depends highly on context and could be affected by incomplete biospecimen sampling. Instead, he said, a better approach could entail using prospectively planned biomarker analysis accompanied by a more complete biospecimen collection. Furthermore, unreliable IVDs can adversely alter the representativeness of a clinical trial population and therefore the rigor of the statistical analysis, compromising
the ability to interpret trial results. Assuming that the drug is approved by FDA, a reliable companion IVD is also needed to identify the target populations in the clinical setting and to provide useful information to assess individual benefits and risks for prospective patients. Finally, trials in marker-negative patients, who may not have met the eligibility criteria for a molecularly defined cohort of a genetics-enabled trial, can be conducted in the postmarket setting, he said, adding that such trials can provide a better sense of risks and benefits in the broader, real-world population.
Over the years, FDA has drafted and released a number of guidance documents for industry related to the collection of biospecimens in clinical trials for biomarker development, the use of enrichment strategies in clinical trials, and, recently, the codevelopment of companion diagnostics in the context of a therapeutic trial.3 Pacanowski highlighted select key points from the recent draft codevelopment guidance (see Pacanowski presentation, March 8, 2017):
- Determine what investigational device exemptions apply to the IVD and what the responsibility of investigators is for complying with the investigational device regulations.
- Complete analytical validation studies before using the IVD in the therapeutic trial.
- Use an IVD with “market-ready” performance characteristics in the pivotal trials.
- Establish pre-analytical operating procedures, and qualify trial sites (particularly in a clinical trial setting where there is a lot of shipping, movement, and retesting of samples).
- Characterize potential prescreening bias and evaluate the target population (i.e., how does preselection of patients affect the analytical and clinical validity of the clinical trial assay?).
- Bank specimens in sufficient quantity to support analytical validation and bridging studies.
- Submit and review co-developed IVDs in a contemporaneous fashion with the drug so that they can be approved simultaneously.
Addressing Molecular Diversity in Rare Genetic Diseases
The molecular diversity of genetic diseases presents a number of challenges for developing targeted therapeutics, Pacanowski said (see Figure 2-2). For genetic diseases with unknown molecular etiology, targeted drug development is not an option (scenario 1). For genetic diseases
___________________
3 Pacanowski referred participants to the FDA website on precision medicine for additional information. See https://www.fda.gov/ScienceResearch/SpecialTopics/PrecisionMedicine/default.htm (accessed April 26, 2017).
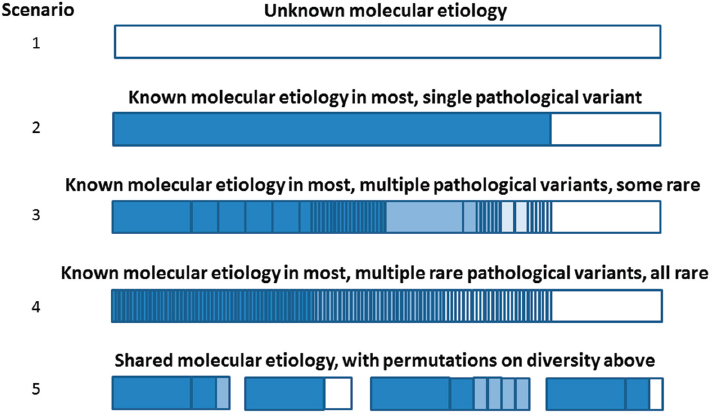
SOURCE: Michael Pacanowski, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
where the molecular etiology is known, there might be a single pathological variant that is responsible for the observed pathological phenotype, and, therefore, it might be possible to make or confirm a clinical diagnosis based on that variant (scenario 2). More commonly, the molecular etiology is known in the majority of cases, but many different pathological variants might be responsible for the disease phenotype, some of which could be quite rare (scenario 3, e.g., cystic fibrosis). In other cases, there might be hundreds or thousands of different, rare defects that could result in a genetic disease (scenario 4, e.g., Fabry disease). Finally, it is also possible that different diseases could result from mutations within the same gene (scenario 5). In all cases, the observed mutations could have widely varying effects on transcription, translation, or protein function, and the resulting disease phenotype depends on the specific mutation(s) and the epistatic interactions at play.4
The potential molecular complexity presented by genetic diseases also raises the question of how trial eligibility criteria should be defined to enroll molecularly defined subsets of patients in confirmatory trials, Pacanowski
___________________
4 Epistasis is a circumstance where the expression of one gene is affected by the expression of one or more independently inherited genes. For more information on reduced penetrance and variable expressivity, see https://ghr.nlm.nih.gov/primer/inheritance/penetranceexpressivity (accessed May 6, 2017).
said. He discussed factors to consider when addressing molecular diversity in clinical trials, including enrollment strategies, data analysis and interpretation, and the identification of additional indications in the postmarket setting. There should be a robust conversation about the trial enrollment strategy from the outset, considering what is known about the mechanistic, clinical, or nonclinical data to best inform how patients might best be grouped by molecular subtype, he said.
Diverse molecular subtypes that are expected to respond similarly to the same drug could be grouped together, and the benefit–risk profile could be better elucidated across subtypes, noted Pacanowski. In general, it would be favorable to enroll as broad a subset as possible, given that it is generally not possible to draw meaningful conclusions about treatment effects in small (i.e., rare) populations. The case for FDA approval is built on the totality of evidence, he said, and approved indications could be based on individual mutations, codons, genes, pathways, or functional groupings thereof. Accordingly, the breadth of the approved indication(s)—and therefore the relevant clinical population for which the drug can be marketed—depends on the trial’s enrollment criteria, the benefit–risk profile of the product, and the IVD design. Ideally, postmarketing studies would be conducted to monitor additional outcomes in certain patient subsets, Pacanowski said, which FDA has the authority to require under certain circumstances.
Targeted Drug Development in Common Complex Diseases
In general, genetically enabled drug development for common complex diseases remains infrequent and, when pursued, tends to focus on the stratification of patient populations using well-established markers of drug metabolism and disease, such as the association between the apolipoprotein E gene (APOE) and the progression of Alzheimer’s disease, Pacanowski said. Reasons for the limited translation of genomic biomarker discovery efforts (e.g., GWASs) into successful drug/diagnostic codevelopment programs, said Pacanowski, may include issues such as small or incomplete sampling, sampling bias, lack of heterogeneity across populations, marginal p-values and contrived analyses, inconsistency between early- and late-phase trial endpoints, and a lack of mature efficacy and safety data. However, it is possible to leverage GWAS data for targeted drug development in complex diseases to strengthen the validity of these trials, Pacanowski said. He offered several possible ways to strengthen the validity of these trials:
- Identify the causal locus and perform functional validation of the gene.
- Recruit a study population large enough to provide a robust signal to indicate that the genetic marker of interest is predicting clinical response.
- Enroll biomarker-negative patients in the study to help understand how well the biomarker discriminates responders from non-responders.
- Enroll a diverse patient population because a particular single nucleotide polymorphism marker in one geographic area or ethnicity may not be proportionally represented in another.
The use of biomarkers to drive targeted drug development can result in a range of outcomes with differing benefit–risk profiles, which could influence whether the approval of an IVD is pursued for a biomarker in a given indication, Pacanowski said (see Figure 2-3). For example, there may be no discernible difference in the benefit–risk profile between biomarker-positive and biomarker-negative populations (scenario 1); some difference in the benefit–risk profile, where the biomarker-positive population realizes greater benefit on average, but uncertainty remains as to the utility of the IVD overall (scenario 2); wide separation in the benefit–risk profile between the subpopulations, with a greater net benefit for the biomarker-positive population, which might support the use of an IVD (scenario 3); marginal difference in the benefit–risk profile, but with some net benefit observed in the biomarker-positive population (scenario 4); a wide separation in the benefit–risk profile, with net benefit observed in the biomarker-positive population (scenario 5); and, finally, a clear untoward effect in the biomarker-negative subgroup and a beneficial effect in the positive subgroup, a scenario in which the development and approval of an IVD is most clearly supported (scenario 6).
While targeted drug development has been relatively uncommon outside of oncology, a variety of issues arise during the review of drugs and IVDs for common, complex medical diseases to which FDA attends closely, Pacanowski said. What biomarkers or other genetic factors should be prospectively assessed? Are additional studies needed to resolve potential genetic variability in drug exposure or response? Do genetics-enabled studies indicate a potential for target-based toxicities in certain molecular subpopulations that should be addressed with enhanced safety monitoring in the postmarket setting (e.g., risk evaluation and mitigation strategies)? Is the target population appropriate according to the known population-level distribution of the genetic variant(s) or other molecularly defined subsets? Should different dosing, dosing durations, target populations, conditions, or other factors be recommended on the basis of differences in exposure or response across biomarker subgroups? In consideration of these issues, Pacanowski referred participants to recent FDA guidance
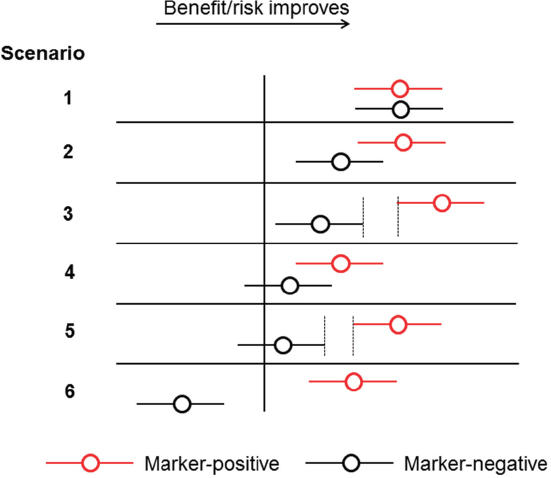
SOURCE: Michael Pacanowski, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
documents released for public comment in 2016 related to the qualification process for biomarkers.5
Regulatory Issues to Consider
In summary, Pacanowski said that there are three key regulatory issues to be considered in precision drug development:
- The strength of the evidence provided to support biomarker-directed precision drug development.
- The identification and management of microheterogeneity.
- Evidence concerning the effectiveness and clinical need for an IVD.
To address these issues, he suggested that trials be designed such that they gather evidence for both the drug and the biomarker test; that product sponsors plan ahead for the development of the IVD; and that sponsors
___________________
5 See https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM337169.pdf (accessed June 5, 2017).
engage FDA early. With regard to enrolling biomarker-negative populations, some data are desirable, particularly when the merits and limitations of the biomarker are unknown, Pacanowski said. Finally, when moving from a GWAS finding to a Phase 3 trial, using robust methods, performing supportive experiments, and addressing ethnic diversity are all important considerations, he said.
This page intentionally left blank.














