
The third session of the workshop considered novel ways of integrating genetics into the drug development process to increase efficiency and improve outcomes for complex diseases. Theresa Strong, director of research programs at the Foundation for Prader-Willi Research, provided a patient-focused perspective on genetics-enabled drug development from her experience as a researcher, patient advocate, and parent of a child with a genetic disorder. Matthew Nelson, the head of genetics at GlaxoSmithKline (GSK), discussed examples of how genetics can inform drug safety and efficacy in common complex diseases. Rebecca Blanchard, head of clinical pharmacogenomics and operations at Merck, discussed the incorporation of pharmacogenomic and genetics research into clinical programs across therapeutic areas at Merck and how to incorporate GWAS findings into clinical development. Michael Farkouh, professor and the vice-chair of research in the Department of Medicine at the University of Toronto, provided an overview of the Tailored Antiplatelet Therapy Following Percutaneous Coronary Intervention (TAILOR-PCI) clinical study, which was designed to evaluate the impact of genetic variants on clopidogrel pharmacodynamics and clinical outcomes.
Responses to the presentations and additional commentary was provided by G. Scott Chandler, vice president and global head of licensing and early development safety at Genentech/Roche; Jessica Langbaum, principal scientist at Banner Alzheimer’s Institute; Mark Trusheim, visiting scientist at the Massachusetts Institute of Technology Sloan School of Management; and L. Keoki Williams, associate director of the Center for Health Policy and Health Services Research, Henry Ford Health System. There was an open discussion among the reaction panel, session presenters, and audience, moderated by Russ Altman, forum co-chair and professor of bioengineering, genetics, and medicine at Stanford University.
PATIENT PERSPECTIVE ON GENETICS-ENABLED DRUG DEVELOPMENT: VIEW FROM THE PRADER-WILLI SYNDROME COMMUNITY
Strong discussed the pathology and clinical course of Prader-Willi syndrome (PWS) and the integration of genetic information into precision drug development for PWS treatments, including the challenges of and opportunities for engaging patients and their families in precision medicine.
Etiology and Pathophysiology of Prader-Willi Syndrome
PWS is a complex, genetic, multisystem disorder with a prevalence of about 1 in 15,000 births. It occurs spontaneously and in equal proportion in males and females and across races and ethnicities. The underlying genetic pathology of PWS is complex, stemming from a loss of function of genetic material in the “PWS region” of the paternally inherited chromosome 15. There is an accurate diagnostic genetic test for PWS, which detects more than 99 percent of cases, Strong said.
In infancy, PWS is associated with weak muscle tone (hypotonia), which leads to feeding difficulties and poor growth and delayed development. The onset of the defining features of PWS—hyperphagia (excessive appetite) and obesity—is variable, ranging from toddlerhood to adolescence. Parents and health care providers might employ environmental controls to maintain weight, which, Strong explained, can mean placing locks on refrigerators and other food sources and supervising the individual with PWS at all times. Individuals with PWS often exhibit intellectual disabilities and behavioral problems, including obsessive compulsive disorder, cognitive rigidity, temper outbursts, and anxiety. The most striking and important difference that occurs across genetic subtypes, however, relates to the risk of mental illness: the uniparental disomy subtype is highly susceptible to psychosis as individuals enter adulthood, where up to 65 percent have bipolar disorder or psychosis (Yang, et al., 2013). PWS is a life-threatening disease, Strong said.
PWS has a significant impact on quality of life for both patients and caregivers. A breakdown in structured weight control can lead to rapid weight gain, morbid obesity, and complications associated with near-constant and sometimes voracious eating in an attempt to find satiation. As individuals with PWS age, their families and caregivers tend to limit social engagement, which also limits opportunities for employment and independence. There is a high rate of depression associated with PWS, which Strong suggested could be in part due to the underlying genetics, but also in part to the social ramifications.
Precision Medicine Approaches for PWS
Many PWS phenotypes are observed across all patients (e.g., growth hormone deficiency, hypogonadotropic hypogonadism), while other features occur only in a minority (e.g., seizures, autism, central adrenal insufficiency). It could be of importance, Strong said, to understand how epistatic interactions outside of the PWS genes might influence the risk of developing these features so that the clinical care of patients can be better informed by patients’ underlying genetics.
Growth hormone is FDA-approved for the treatment of PWS, and though it positively affects development and body composition, it does not have a discernible effect on the hallmark hyperphagia or the other spectrum of symptoms observed in PWS. To address this unmet medical need, an increasing number of clinical trials for PWS have been conducted in recent years that focus on symptomatic relief, such as appetite control and behavioral modification. However, Strong said, conducting clinical trials in this population presents practical challenges, including
- The patient population from which to recruit and allocate to active trials is limited, an issue exacerbated in the context of genetic stratification.
- The underlying physiological mechanisms that drive the extreme hunger are not well understood, and there are no quantitative biomarkers of hunger; data are limited to the qualitative observation of behavior.
- There is a potential for phenotypic variability across genetic subtypes, which can affect the interpretation of pan-disease trials.
- Environmental factors can influence the severity of phenotypes (e.g., quality of care, weight management, access to interventions or treatments, such as growth hormone).
Genetics-enabled clinical trials with molecularly defined subpopulations could potentially inform drug efficacy and safety profiling for PWS treat-
ments, Strong said. Because PWS is a small population from which to draw patients—for example, a Phase 3 trial might have only 100 participants—a GWAS would not likely have enough power to detect previously unknown variants that could influence disease course or predict treatment response. It might be possible, however, to retrospectively observe how known genetic variants correlate with efficacy, Strong said. For example, oxytocin has been studied in a number of small PWS trials with initial signs of efficacy, and there are known genetic variants of the oxytocin receptor that might modify responsiveness to the drug.
Collecting DNA in clinical trials to assess potential underlying genetic factors related to drug safety is critical, Strong said. She described a Phase 3 clinical study designed to treat hyperphagia in PWS in which there was an unexpected serious adverse event (fatal pulmonary embolism). There is some indication, though insufficient data to support the claim, that the risk of thromboembolism is increased in PWS, Strong said. However, there are also known genetic variants that affect blood clot formation. The question arose: did the individual have a genetic variant that increased susceptibility to the fatal embolism, or was the event associated with the drug? Unfortunately, Strong said, the trial was not designed to consent participants for DNA collection and testing. If such genetic information had been available, it could have informed a safety evaluation for that trial and for future drug development. Ultimately, however, a second fatal incident led the company to suspend its development of the drug.
More genetic and genomic information about PWS is needed, Strong said. To further this goal, the Foundation for Prader-Willi Research is initiating a pilot PWS Genomes project which will conduct whole-genome sequencing of individuals with PWS. Genetic data will be linked to information in the Global PWS Registry, which has now enrolled 1,000 individuals.1 Together, these different sources of information could inform clinical management and drug selection, serve as a source of molecularly defined populations for the enrichment and stratification of clinical trials, and help develop a better understanding of the genetic variability in drug safety and efficacy characteristics of PWS treatments.
Engaging PWS Patients and Families in Precision Medicine
The engagement and education of PWS patients and their families is important to advancing precision medicine aimed at treating the disease, but there are challenges, Strong said, including
___________________
1 For more information about the PWS Genomes project see https://www.fpwr.org/global-pws-registry (accessed April 26, 2017).
- Breadth and depth of information—PWS is a multifaceted genetic disorder with an array of causes and symptoms, which themselves are associated with complex terminology (e.g., imprinting, uniparental disomy). In addition to and in light of the daily burden of managing PWS, this information can overwhelm patients and their families.
- Informed but unburdened participants—There is a need for patient and caregiver education about clinical trials, including education that informs the expectations of patients who participate in trials. There is also a need for an informed consent process that conveys useful information to participants without further increasing the information burden on patients and families.
- Spectrum of patient intellectual capacity—Cognitive disability is common in PWS, but there are patients with PWS who fall within normal cognitive range. So, the question becomes, At whom should educational initiatives be aimed? For PWS, the answer may be the parents of children or patients who are engaged but present with learning disabilities.
- Limited funding for a diverse population—Funds for outreach are limited and often directed towards families that seek out assistance.
Strong noted that there is a need across disease advocacy organizations for more patient-friendly, graphical representations of complex issues, such as genetic variation or benefit–risk assessment, from definitive sources that could be adapted to different disorders. Advocacy organizations may also benefit from best practices and “off-the-shelf” models that can be adapted and used for educating families and patients with rare diseases, she said. There is also a need for a data-sharing model where genetic information is returned to and stays with the individual, Strong emphasized, noting that it is important that patients own their genetic data, so that these data are accessible to them as they move between treatment and clinical trial settings over the course of their lifetime.
Despite these challenges, there are opportunities to reach out to and engage with the PWS community, Strong said, noting the following observations:
- The rare disease community is in general a “tight-knit group,” and advocacy organizations are often thought of by patients as unintimidating, trusted partners.
- The rare disease community embraces and is well connected through technology and social media, and patients are comfortable learning about PWS through these avenues.
- PWS patients and their families are highly motivated to find new information that might affect the clinical course of the disease.
Strong noted that patient advocacy groups often use multiple approaches to accomplish their goals of engaging and educating patients. For the PWS community, she said, these approaches include establishing a community advisory board; developing written materials aimed at different groups and education levels and available in multiple formats; and the leveraging of telecommunication technology such as webinars, videos, live stream/recordings of conferences, social media, and in-person opportunities to discuss the aspects of genomics in PWS, such as annual conferences and clinic visits.
In summary, Strong said, genetic information has tremendous potential beyond the diagnosis of rare diseases to improve clinical trial efficiency, particularly with regard to trial design, interpreting efficacy findings, and safety. However, there are challenges to be addressed, including educating the rare disease community in ways that are informative yet accessible, reporting back genetic data to patients and families in a timely manner and usable format, and limiting the overall burden on families who are already overwhelmed by complex, chronic diseases.
PHARMACOGENETICS FOR SAFETY AND EFFICACY ASSESSMENTS IN COMPLEX DISEASES: CHALLENGES AND LESSONS LEARNED—VIEWPOINT FROM INDUSTRY
The field of pharmacogenetics in complex disease is still in its infancy, Nelson said, with limited lessons learned from the small number of case studies in the field. However, he said, thousands of genetic variants that influence complex human traits have been identified by GWASs over the past 11 to 12 years, facilitated in part by rapid advances in genotyping technologies such as whole-genome genotyping chips and arrays, and to a lesser extent, exome and whole-genome sequencing. These technologies allow for genotyping at reduced cost and increased scale, which make feasible large genetic association studies such as GWASs. As a result, the number of unique associations identified, as well as the number of unique traits for which associations have been found, have increased exponentially. The field has not yet reached the limit of this trajectory, Nelson said, particularly in terms of uncovering the role of genetics in disease susceptibility.
Pharmacogenetics and Adverse Events: A Case Study of HLA and Abacavir
One of the early discoveries that bolstered support for using pharmacogenetics to inform precision drug development was a retrospective, case-control study that found a strong association between a relatively common human leukocyte antigen (HLA) variant, HLA-B*5701, and the risk for hypersensitivity reactions to the HIV treatment abacavir (Hetherington et
al., 2002). At the time, about 3 to 5 percent of patients taking abacavir experienced this very serious and potentially fatal adverse event.
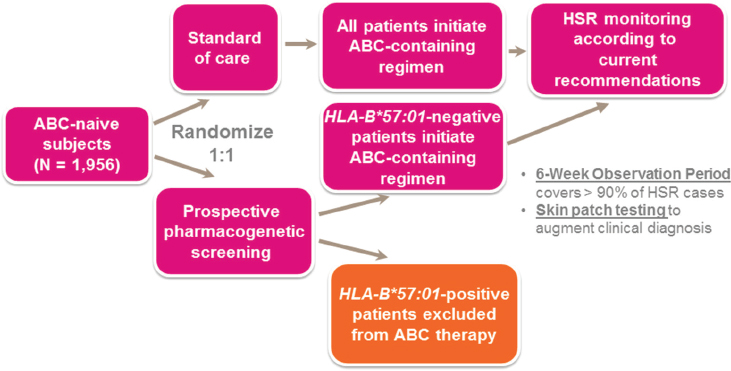
To study the association further, GSK launched the PREDICT-1 clinical trial, which was designed to prospectively test whether screening for HLA-B*5701 could manage and control the risk of hypersensitivity reaction in patients receiving abacavir (Hughes et al., 2008). In this two-arm study, one arm received the standard of care, abacavir, with no genetic prescreening, while the other arm was screened for HLA-B*5701. Enrollment for treatment with abacavir was open only to those participants who screened negative for HLA-B*5701 (see Figure 4-1).
This prospective, genetics-enabled trial confirmed findings from previous cross-sectional retrospective studies of existing clinical trial data, Nelson said, with HLA-B*5701 found to be highly predictive of hypersensitivity reactions in patients receiving abacavir. Approximately 50 percent of patients positive for HLA-B*5701 developed hypersensitivity reaction, whereas no patients who were HLA-B*5701-negative developed this adverse reaction (Mallal et al., 2008). This new screening information was added to the abacavir product label and became a regular part of decision making for abacavir treatment regimens.2 The association between HLA-B*5701 and abacavir represented a “perfect storm of conditions to make this an ideal poster child for pharmacogenetics in clinical drug development,” Nelson said, for the following reasons:
- Hypersensitivity to abacavir was a relatively common adverse event, affecting approximately 3–5 percent of patients receiving the treatment (Hetherington, et al., 2002).
- HLA-B*5701 is a relatively common genetic risk factor, present in approximately 5 percent of the general population (Orkin et al., 2010).
- There is a sensitive and specific relationship between the genetic variant and adverse reaction, with 50 percent of patients carrying the HLA-B*5701 variant likely to develop hypersensitivity reaction.
Taken together, the size of the odds ratio (>1,000) for the observed outcome coupled with the prevalence of the genetic variant in the general population meant that the pharmacogenetic risk factor had significant predictive value for the observed adverse event, Nelson said.
___________________
2 To read the full FDA alert about abacavir see https://www.fda.gov/Drugs/DrugSafety/ucm123927.htm (accessed May 18, 2017).
NOTE: ABC = abacavir; HLA = human leukocyte antigen; HSR = hypersensitivity reaction.
SOURCE: Matthew Nelson, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
Using Pharmacogenetics to Predict Safety and Efficacy in Clinical Drug Development
The abacavir study set a baseline expectation for the kind of impact that pharmacogenetics could have on the course of safety monitoring in clinical development, Nelson said. However, he continued, though successes have been realized in identifying genetic risk factors for adverse events, some of which have been incorporated into treatment guidelines (Martin et al., 2014), two main practical limitations have prevented the majority of this pharmacogenetic evidence from making it into treatment guidelines. First, within the current health care structure in the United States, genetic information is not prospectively collected and incorporated into the medical record, obviating the clinical utility of such guidelines. In addition, most of the observed adverse events are fairly rare and do not exhibit the characteristics observed by HLA-B–abacavir. Though some genetic risk factors exhibit fairly high odds ratios, the causative variants are also highly prevalent within the general population (1:100 to 1:20) and as such have poor prognostic and predictive characteristics.
Since the early days of abacavir hypersensitivity, Nelson said, GSK has incorporated genetics into many of its clinical development programs with reasonable levels of success. He described five associations that were identified either during or shortly after clinical development, including abacavir,
all of which were related to safety outcomes (i.e., adverse drug reactions). Among 20 clinical trials with efficacy outcomes that were accompanied by a GWAS, there have been no examples of genetic variants associated with efficacy outcomes, he said. For example, GSK and others have studied the potential impact of genes on steroid treatment response in asthma patients. GSK undertook the largest pharmacogenetic study to date of patient response to inhaled corticosteroids, Nelson said, evaluating data across seven GSK clinical trials with a total of 2,672 patients. There was no evidence of any genetic variant being associated with change in FEV-1 (forced expiratory volume in 1 second, a measure of pulmonary function) over the course of treatment (Mosteller et al., 2017). Furthermore, within the field, there has been no statistically significant evidence of genetic variants associated with efficacy of inhaled corticosteroids for asthma treatment.
To develop a better overall view of what is known about pharmacogenetics and drug efficacy for common, complex traits, GSK reviewed all GWASs (2,223 studies) reported in the National Human Genome Research Institute–European Bioinformatics Institute GWAS Catalog (as of July 2015). About 3.4 percent (n = 76) of all GWASs were related to drug efficacy, and, of those, 17 percent (n = 13) were reasonably robust and provided a high statistical confidence of association between a genetic factor and drug efficacy. Therefore, Nelson concluded, the success rate of GWASs to identify genetic factors that influence drug efficacy has been relatively low, and the use of GWASs thus far has not contributed significantly to prior candidate gene work. To further examine potential genetic effects on drug efficacy, GSK reviewed the literature more broadly to identify studies with findings of gene–drug associations identified through other mechanisms (not GWASs), for which there was sufficient evidence to be compelling. An additional 11 associations across seven drugs were identified. The review indicates that research on gene–drug efficacy associations has generally focused on three functional mechanisms: gene variants involved in absorption, distribution, metabolism, and excretion (ADME); variants encoding for the drug target; and variants involved in disease mechanism more broadly.
Lessons Learned from Pharmacogenetics Studies for Complex Diseases: Viewpoint from Industry
Nelson summarized lessons learned from studies examining the association between genetic variants and drug safety and efficacy.
Drug Safety—Using Genetic Screening to Identify Risk Factors for Adverse Events
- Mechanisms are primarily through HLA and ADME genes.
- Most HLA genetic risk factors are drug- and serious adverse event-specific (i.e., one cannot extrapolate across drugs or across adverse events for a drug).
- Common genetic risk factors alone generally do not accurately predict patient risk, particularly for rare serious adverse events.
- It is estimated that less than 50 percent of drugs that demonstrate serious adverse events are likely to have a major genetic contributor to that adverse event.
Drug Efficacy—Applying Pharmacogenetics
- Mechanisms of associations are primarily through ADME, the drug targets themselves, and genes that are involved in the disease process.
- Based on a systematic review of the literature, it is expected that approximately 10 percent of drugs have genetic predictors of efficacy that could influence clinical decision making.
Nelson suggested that the probability of success of pharmacogenetics in drug development could be improved with tailored analysis strategies, including the following:
- Where possible, take advantage of sensitive, quantitative biomarkers of response.
- Balance hypothesis-driven with hypothesis-free approaches.
- Combine data across clinical trials for the same or similar drugs.
- Do not shy away from investigating very effective drugs, for which there is the greatest power and best opportunity to identify subgroups that might differentially benefit.
With proper consent or secondary use, anonymized clinical trial data could help to advance the understanding of genetic diseases, Nelson said. For example, at GSK, all clinical trials at Phase 2 and beyond attempt to consent and collect DNA from research participants (unless there is a reason for exception, such as a specific country’s laws prohibit it), and samples are genotyped and screened prospectively for any genetic factors that might impact either the efficacy or safety of the drug under investigation.
CHALLENGES AND OPPORTUNTIES FOR LEVERAGING GWAS FINDINGS DURING DRUG DEVELOPMENT: A PHASE III CASE STUDY
Providing another perspective of genetics-enabled drug development in the biopharmaceutical industry, Blanchard described how Merck has operationalized relatively routine pharmacogenomics research into clinical development programs across almost all therapeutic areas and offered a specific case study of how a GWAS “hit” identified late in drug development was incorporated into the development program.
Incorporating Pharmacogenomics into Drug Development: A Case Study at Merck
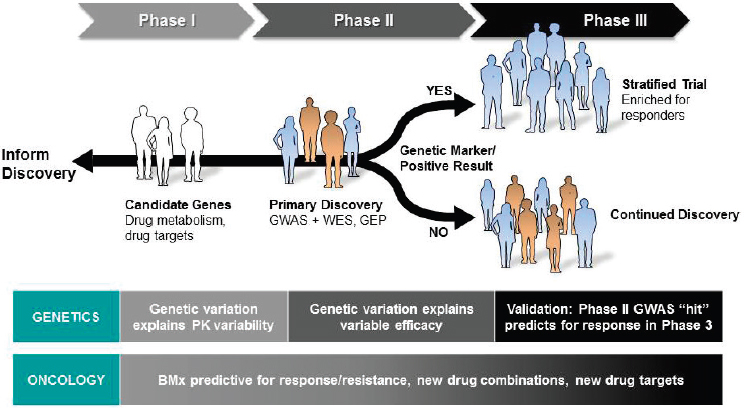
In the past 2 years, Merck has begun incorporating an exploratory pharmacogenomic objective into all of its therapeutic clinical trials, Blanchard said. DNA is collected and genotyped from all participants who enroll in a clinical trial, and the resulting data are stored until needed in order to test a specific hypothesis (see Figure 4-2). Hypotheses could be a part of testing candidate genes associated with drug metabolism or targets,
NOTE: BMx = biomarker; GEP = gene expression programming; GWAS = genome-wide association study; PK = pharmacokinetic; WES = whole exome sequencing.
SOURCE: Rebecca Blanchard, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
of conducting GWASs to perform primary discovery or detect large effect sizes in Phase 2 trials, or of validation during Phase 3 trials of GWAS hits through trial enrichment and patient stratification, in addition to potentially validating a companion diagnostic. For oncology trials, Blanchard noted, Merck also conducts thorough genomic (and other “-omic”) profiling of patients’ tumors.
A “dual-consenting” informed consent approach, Blanchard explained, is used to incorporate pharmacogenomics into immediate and future clinical trials, with the goal of maintaining patient privacy and consenting choice without delaying the trial. To address the planned exploratory pharmacogenomics objective in the trial for which the participant is enrolling, participants are consented for DNA collection and genotyping via the primary study informed consent process. This is a required element of every protocol, she said, except in those countries whose laws or regulations forbid it. There is also a separate, voluntary consent for future biomedical research that allows patients to choose to participate in broader research by permitting long-term sample retention. The consent form clearly informs potential enrollees that Merck does not return research data, including genetic data, to study participants, Blanchard said. There are a variety of reasons for this policy, she said, including the lack of infrastructure to be able to manage the return of these data, and that additional resources are needed within the larger clinical trial community to return results to participants.
Case Example: SNP Association with Primary Clinical Endpoint
Blanchard provided a case example in which a GWAS hit was identified as the result of a purposeful search late in a drug development program. Because the studies are still under way, Blanchard used non-specific details to explain the scenario. In this program, there were two randomized controlled Phase 3 studies that demonstrated the efficacy of a compound in the overall trial population. However, it was expected that if the drug being studied were approved, it would most likely be preferentially used within high-risk populations characterized by certain baseline, high-risk demographic factors. Therefore, the study team was interested in understanding whether there were genetic factors that might identify participants outside of this high-risk population who respond differentially better to the drug and thus increase the potential patient pool. Because the study was commenced before the mandatory pharmacogenomic objective for clinical trials had been implemented within the company, a GWAS of 1,000 study participants who voluntarily consented to DNA collection and testing was performed during Phase 3. The GWAS identified an SNP (single nucleotide polymorphism) for which the T allele was associated with improved outcomes relative to the primary clinical endpoint.
This GWAS result was an exploratory finding and needed to be replicated in order to be actionable, Blanchard said, adding that in this specific drug development program replication would require conducting a new clinical trial—a significant investment. To better inform that decision, Merck performed a thought experiment on how this pharmacogenomic information, if confirmed, might improve the care of these patients. First, the overall trial results were evaluated in the context of the presence or absence of two baseline risk factors among the participant population. This subcategorization of the participant population showed that the therapy was most beneficial in patients who presented with both baseline risk factors—in short, high-risk patients responded best to the drug.
Researchers next overlaid the pharmacogenomic information onto the subcategorized trial results. These data showed that the high-risk population responded to the treatment regardless of genotype (although those carrying a T allele might show increased response), and as such, a pharmacogenomics test might not be warranted in the high-risk patient population. However, two additional populations, defined by the presence of one baseline risk factor and the T allele, might benefit from the drug, but would not be indicated according to the current label. If this finding were replicable, she said, it could provide a means to modify the label to indicate additional patients beyond the high-risk cohort who could benefit from the drug. This, Blanchard emphasized, represents a pharmacogenomic opportunity.
An additional and important consideration, Blanchard said, is how this pharmacogenomic information might be used within the clinical setting, so Merck conducted a marketing assessment in three large global markets, with 50 physicians per market. The physicians were introduced to the product profile of the drug and the pharmacogenomic results and were asked how they would use an IVD test in two different scenarios: with the test being optional for prescribing (complementary diagnostic) or being required for prescribing (companion diagnostic). The results of this assessment indicated that the physicians would test more than 50 percent of patients in the mandatory scenario and more than 40 percent of the patients in the optional setting; however, in both scenarios, the group that was most likely to be tested was the high-risk population (in which the test was not required). This suggested that including a complementary or companion IVD with the drug would result in decreased prescribing, even among those high-risk patients who did not require the test but had high medical need. Therefore, it was concluded that having an IVD could potentially be a disservice to this patient population. At this point in the program, Blanchard said, there has been no decision to invest in a replication study, and any additional decisions around future investments will be driven, in part, by how this drug is actually used in real-world practice.
In closing, Blanchard highlighted some challenges to achieving impact with pharmacogenomics during drug development:
- It is difficult to obtain IRB and health authority approval to conduct genomic research in global settings, particularly given that some countries do not allow genomic research, and that some IRBs are not familiar with this topic and are therefore hesitant of approving this research, even though most patients are interested in participating when consented.
- The return of genetic data and incidental findings to participants is an important topic, and the ethical, legal, and social implications along with resource needs should be further discussed by the research community as a whole.
- Late-stage trials have the most power to detect a pharmacogenomic association, but it is difficult to adjust a development strategy in late-stage clinical research.
- Significant education is required—both to gain approval for conducting pharmacogenomic research globally and for using such information in the clinical community—of IRBs, health authorities, physicians, payers, and patients.
CLOPIDOGREL PHARMACOGENETICS IN CORONARY ARTERY DISEASE: THE TAILOR-PCI TRIAL
In the context of coronary artery disease (CAD),3 a common complex disease, Farkouh discussed the pharmacogenetic characteristics of clopidogrel, a treatment indicated for patients with CAD, and discussed the TAILOR-PCI trial, which is using genetic data to improve patient outcomes after percutaneous coronary intervention for CAD.
Background to TAILOR-PCI: Clopidogrel
Clopidogrel (brand name, Plavix) is used in patients with atherosclerotic vascular disease, and FDA-approved indications for clopidogrel include myocardial infarction, thrombotic stroke, peripheral arterial disease, and percutaneous coronary intervention (PCI) with stent implantation. Clopidogrel is metabolized through the cytochrome P450 system in the liver to its active metabolite, which then binds irreversibly to the P2Ry12 receptor on blood platelets. The CYP2C19 gene encodes for the CYP2C19 enzyme of the cytochrome P450 system. There are a host of CYP2C19 poly-
___________________
3 Also referred to as atherosclerosis, arteriosclerosis, coronary heart disease, or hardening of the arteries.
morphisms, including two loss-of-function alleles, *2 and *3, which can be tested for in the point-of-care setting, and there are ethnic variations in the prevalence of these polymorphisms.
A key question is whether genetic variation in CYP2C19, specifically the presence of the loss-of-function alleles, affects clopidogrel outcomes. A meta-analysis of all major clinical research studies of clopidogrel in which genetic polymorphisms were measured found a 57 percent increase in risk of cardiovascular death, myocardial infarction, or stroke in patients who had loss-of-function alleles (Mega et al., 2010). The same study also showed that 91 percent of patients in the analysis had undergone a PCI, many with a coronary stent implantation, and that one in three of these patients presented with an post-PCI ischemic event when treated with standard-of-care clopidogrel if they harbored a CYP2C19 loss-of-function allele.
The findings of this meta-analysis led to a black box warning being added to the clopidogrel label by FDA in March of 2010.4 The warning states that the effectiveness of clopidogrel depends on activation to an active metabolite, poor metabolizers exhibit higher cardiovascular events following PCI or acute coronary syndrome, and tests are available to identify a patient’s CYP2C19 genotype. The cardiovascular community grappled with how to make practical use of this new information and the FDA warning. A consensus document was developed by the American College of Cardiology (ACC) and the American Heart Association (AHA) stating that “the evidence base is insufficient to recommend either routine genetic or platelet function testing at the present time. There is no information that routine testing improves outcomes in large subgroups of patients” (Holmes et al., 2010). The issue of genetic testing for patients taking clopidogrel was debated in the popular press, and patients raised the issue with their doctors. Subsequent ACC and AHA guidelines have maintained that the routine clinical use of genetic testing to screen patients treated with clopidogrel who are undergoing PCI is not recommended (Levine et al., 2011; O’Gara et al., 2013). Complicating the issue, Farkouh said, is the fact that two new compounds, prasugrel and ticagrelor, have been developed and have shown in large clinical trials to be superior to clopidogrel in terms of major adverse cardiovascular events. Despite the evidence for the use of the newer agents, clopidogrel still holds about 70 percent of the market share, Farkouh said, and is regularly used for PCI with stent implantation.
The black box warning remains on the clopidogrel label, yet there is insufficient clinical evidence as to whether altering therapy based
___________________
4 See https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020839s048lbl.pdf (accessed April 26, 2017).
NOTE: BID = bis in die (twice per day); CV = cardiovascular; MI = myocardial infarction; PCI = percutaneous coronary intervention; WT = wild type.
SOURCE: Michael Farkouh, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
on CYP2C19 status affects clinical outcomes. To address this gap, the TAILOR-PCI clinical trial was launched in May of 2013 by the Mayo Clinic and the University of Toronto.5
The TAILOR-PCI Study Design
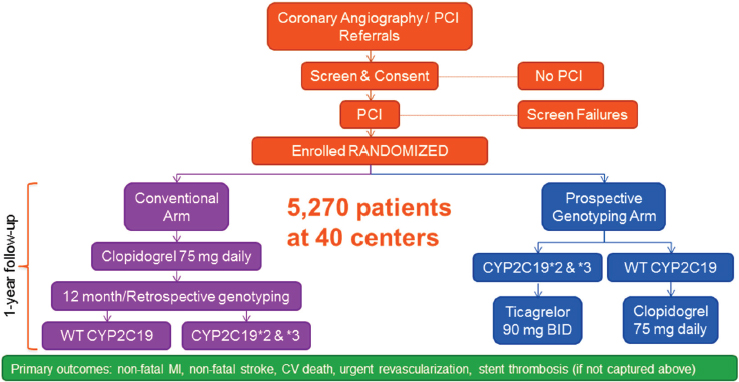
TAILOR-PCI is a large, randomized controlled trial that focuses on the loss-of-function alleles CYP2C19*2 and *3. Patients who will be undergoing PCI are screened, consented, and randomized to one of two arms (see Figure 4-3). Those in the conventional arm receive 75 milligrams of clopidogrel daily for 12 months, with genotyping performed at the end of the study. Those in the prospective genotyping arm are screened prior to treatment, and those with CYP2C19 *2/*3 loss-of-function alleles are given ticagrelor, 90 milligrams twice per day, for 12 months, while those with wildtype CYP2C19 receive clopidogrel, 75 milligrams daily. A key element of the trial is the rapid turnaround time of the Spartan Bioscience IVD assay, a novel genotyping platform used for screening at the point of enrollment for detection of CYP2C19 variants.
___________________
5 To read more about the TAILOR-PCI trial see https://clinicaltrials.gov/ct2/show/NCT01742117 (accessed April 26, 2017).
The endpoints for TAILOR-PCI are cardiovascular death, myocardial infarction, stroke, and urgent revascularization. It was anticipated that event rates for TAILOR-PCI would be about 10 percent overall, with 8 percent in the intervention (prospective genetic screening) arm and 12 percent in the control (conventional standard of care) arm. Farkouh said that the statistical analysis involves the comparison of the *2/*3 clopidogrel conventional arm and the *2/*3 ticagrelor prospective genotyping arm, rather than the combined comparison of the two randomized arms.
There is ethnic variability in the prevalence of CYP2C19 polymorphisms, Farkouh reiterated, and an important issue for this trial was the proportion of the participants who have the loss-of-function allele. The prevalence of the loss-of-function alleles is about 30 percent in North America and up to 50 percent in East Asian population. Accordingly, a U.S., Canadian, and Korean partnership was established to enrich the enrollment of participants with loss-of-function polymorphisms in the trial, which is currently at a 35 percent prevalence rate.
Finally, securing funding for the trial initially presented a challenge, Farkouh said. The Mayo Clinic provided an initial infusion of money for the pilot study (consisting of approximately 2,700 patients), before NIH contributed funding approximately halfway through enrollment in May 2016. The point of enrollment genotyping platform is provided by Spartan Bioscience. The trial has no direct industry funding, he said, adding that due to reservations about the possible loss of market share contingent on the study outcomes, the industry sponsor of ticagrelor did not participate as a partner in or provide the drug to the study, which instead had to be purchased by the study at market cost.
Potential Impact of TAILOR-PCI
The TAILOR-PCI trial has the potential to provide valuable information to the cardiovascular field, Farkouh said, including that it
- is a potentially high-impact study on a highly prevalent disease;
- addresses an unresolved practice issue by using research methodology;
- has immediate applicability of genotyping to clinical practice worldwide;
- has potential application to other disease states;
- creates a biobank for studies like GWASs; and
- creates an infrastructure for other pragmatic cost-effective multi-center studies.
In summary, the pharmacogenetic application of CYP2C19 for risk assessment of clopidogrel, one of the most commonly prescribed drugs in
the United States, remains unresolved, Farkouh said. FDA cautions providers on the use of clopidogrel in CYP2C19 poor metabolizers through its black box warning, but ACC and AHA have recommended against routine genotyping in the absence of a prospective clinical trial. TAILOR-PCI is a large pragmatic study designed and conducted to address this gap.
REACTION PANEL AND DISCUSSION
Following the speaker presentations, reaction panelists reflected on key ideas from the session and shared their perspectives on issues pertaining to the use of genetics for safety profiling, the economic considerations related to the implementation of genetics-enabled clinical trials, and the recruitment and retention of minority patients for clinical trials.
Precision Medicine as a Mechanism for Personalized Safety
Enabling precision medicine is important from the perspective of personalized safety, Chandler said. He defined precision medicine as providing the right medicine to the right patient at the right time, while maximizing the benefit–risk balance at the level of an individual patient. He defined personalized safety as identifying patients at risk for adverse events with certain therapies and using that information to manage toxicity. The abacavir and clopidogrel case studies (discussed previously in this chapter by Nelson and Farkouh, respectively) are two examples of personalized safety that pinpointed risk factors for toxicity. Furthermore, Chandler said, clinically relevant biomarkers may play an important role in identifying patients at risk, guiding therapy, and implementing appropriate risk management measures. The question raised is: How can a pharmacogenomics approach be used to affect product labeling and product use to better manage risk and toxicity? There is a need for an infrastructure and guidance on these issues, he said, and he referred participants to a recent European Medicines Agency (EMA) guidance.6 The EMA guidance outlines approaches for evaluating medicines with known pharmacogenomics associations as well as ideas for translating results of the evaluations into appropriate clinical actions.
Though precision medicine could afford providers and patients the opportunity to guide treatment decisions based on a patient’s genetic information, Nelson suggested that the current system of evidence generation does not produce enough evidence to make good decisions about treatment
___________________
6 See Guideline on key aspects for the use of pharmacogenomics in the pharmacovigilance of medicinal products. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/11/WC500196800.pdf (accessed May 17, 2017).
alternatives. If there is a known deleterious gene–drug (Drug A) association, and if a patient’s genetic information is available and indicates that he or she is at risk, questions raised include what are the risks for the patient if taking Drug B, and are there gene-specific risks to Drug B? This information could be pulled together into a single model, he said, to allow providers to make a rational decision about what maximizes the benefit–risk ratio for patients. A prospective approach to genetic data collection, along with new tools and emerging technologies, will to start to make those decisions possible, Chandler added.
Strategic Planning and Economies of Scale for Genetics-Enabled Drug Development
Strategic Planning: Translational Medicine Guide
Genetics-enabled drug development must begin as early in the drug development process as possible, Trusheim said, even as soon as lead optimization. The Translational Medicine Guide (TxM Guide) currently being employed by Merck KGaA/EMD Serono uses a question-based approach to decision making to provide the evidence needed to move a drug development program forward in three actionable areas: biological target, therapeutic window, and target patient population (Dolgos et al., 2016). The guide builds on the idea that systematically organizing drug development into continually better levels of evidence through addressing critical scientific questions and capabilities is a key to success. Furthermore, he added, biomarker and IVD strategy must evolve systematically throughout early development, and early knowledge of the potential target patient population can help inform precision drug development. To exemplify how the TxM Guide characterizes this principle, Trusheim highlighted a patient population-focused question from the guide that queries investigators in the lead optimization phase of development to consider: “Is the range of the target and (molecular) pathway variability in the intended indications suitable for a stratified medicine approach? Is this information incorporated into the biomarker and early clinical strategy?”
In response to Trusheim’s remarks, Altman agreed that genetic biomarkers should ideally be identified and developed early in the developmental pipeline of the investigational therapy, but observed that many of the workshop speakers had shared examples of biomarker successes from late-phase development. If there is enough known about the disease and the target, and if there is genetic validation in humans for that target early on, then stratification strategies can be built into early-phase studies, Blanchard added. However, in some cases unknown genetic variants can affect drug response, and there may be adverse events that could not have
been predicted up front. A treatment effect might be clinically significant, but the signal may not be apparent in a small population, Altman added, and it may take a large Phase 3 trial to detect the signal.
A workshop participant mentioned that a key finding from the Clinical Trials Transformation Initiative (CTTI) is the need for a plan to identify clinically relevant patient populations at the beginning of clinical drug development. It should not be an afterthought, the participant said, adding that FDA has issued an update to a guidance document that provides more explicit direction to commercial sponsors on having a clinically relevant patient population in trials and to plan for this no later than the end-of-Phase 2 meeting with FDA. Several workshop participants commented on ways in which their organizations are working to achieve this, such as
- launching trial sites in urban areas to encourage minority recruitment;
- using GWAS findings to recruit more diverse populations according to known allele frequencies in replication studies; and
- understanding the prevalence of allele frequencies in specific races and ethnicities in order to inform the planning and conducting of trials in geographic regions or countries where the allele frequency of interest is more highly represented.
Economies of Scale: Linked Clinical Trial Platforms
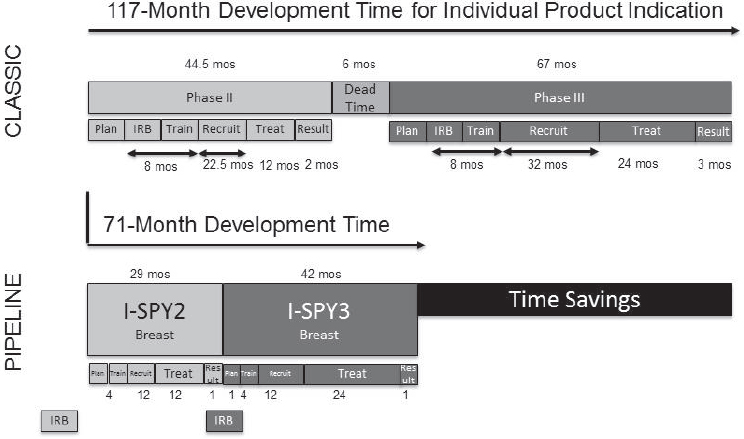
Genetics-enabled drug development will require the field to move from “individually designed evidence generation” (i.e., traditional one-phase, one-drug, one-indication, one-population, clinical trials) toward “evidence-generation engines” (i.e., linked clinical trial platforms) that can deliver economies of scale (Trusheim et al., 2016). Savings can be achieved through, for example, platform trials (e.g., adaptive, multi-center, multi-therapy trials) that leverage master protocols; central IRBs and standard informed consent procedures; prospective genotyping; standard entry criteria, regimens, and endpoints; reduced site training, management, and auditing; and faster recruitment (see Figure 4-4). There are real opportunities to move toward “industrial-scale evidence development” with proper strategic planning and the use of innovative trial platforms, Trusheim said.
Recruitment and Retention of Diverse, Representative Patient Populations
Some workshop participants discussed the challenges of gathering enough data to conclude whether there is an association between a genetic variant and a drug response, noting that key factors include statistical power and at what point a study has accrued sufficient participant sam-
NOTE: IRB = institutional review board; mos = months.
SOURCE: Mark Trusheim, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
ples and evidence to be able to reach the level of confidence needed to draw a definitive conclusion. Topics touched on by some workshop participants and speakers included case studies in asthma, the limitations of GWASs, and the importance of education and outreach to increasing trial representativeness.
Case Studies in Asthma
Williams shared lessons learned for recruiting diverse patient populations and retaining trial participants. The Study for Asthma Phenotypes and Pharmacogenomic Interactions by Race-Ethnicity (SAPPHIRE) is a study of genetic predictors of drug response across a diverse population. The study has enrolled more than 7,600 patients from the Detroit metropolitan area thus far, including more than 4,500 African Americans, 2,300 European Americans, and 240 Latinos. SAPPHIRE is a longitudinal study, and most of the patients have been followed for over 5 years. Follow-up for this study is efficient because all participants are part of the same health care plan, Williams said. The key lesson to be learned from this study, he said, is that researchers should engage health systems with diverse patient populations
when looking for representative patient populations from which to recruit and when seeking to follow trial participants longitudinally. Many of the data for the study are routinely collected as part of the participants’ EHRs.
Williams also discussed an observational study for inhaled corticosteroids that looked at measures of medication adherence, as estimated by prescription retrieval, and he noted two key takeaways (Williams et al., 2007). First, data showed a left shift in adherence, amounting to a 10 percent decrease, between African American and European American patient populations. The decreased adherence in the African American patient population could potentially be due to differences in perceptions or beliefs about treatment, he said, but it could also result from differences in treatment response and adverse effects experienced by the two populations. Second, patient adherence overall was quite poor. On average, adherence to the inhaled corticosteroid therapy in the overall patient population was approximately 50 percent. In clinical trials in general, Williams said, adherence is not often reported, and only patients who complete the trial are evaluated. When looking for genetic predictors of drug response, he said, not accounting for adherence could lead to a gross underpowering of a study.
In raising the point that social determinants of health can have an impact on treatment outcomes for asthma, a workshop participant asked Williams whether socioeconomic status was controlled for in the SAPPHIRE study. The participant suggested that epigenetic factors, such as the chronic stress that may accompany negative social determinants of health, could also impact outcomes. Factors affecting non-adherence have been considered using existing behavior models with different metrics of disease perception and belief in medications, Williams said. Some social and socioeconomic determinants have also been considered, and, even after adjusting for them, there is still a persistent difference in adherence. There may be other unmeasured factors contributing to the differences in adherence that are observed, he added. For example, he mentioned a study of crime rates in neighborhoods as a factor for determining medication adherence. It is a complex issue, and as yet, there is not one factor or a group of factors that explains the difference, he said.
In summary, Williams offered two take-home points:
- It is important not only to have a large enough patient population, such as that within a health system, to properly power a study, but also to calculate drug adherence and exposure.
- Health system data can be used to identify populations that are particularly at risk when observational study results are adjusted for drug adherence and exposure.
Current Limitations of Genome-Wide Association Studies
Many of the GWASs conducted to date are of a single population group, European Americans, and often the populations at highest risk for particular disease outcomes are not being targeted, Williams said. For example, the burden of diabetes and asthma rates are much higher in African Americans, yet GWASs for metformin, the drug of choice for the treatment of diabetes, and for inhaled corticosteroids for asthma, respectively, have been done almost exclusively in European Americans. If the groups at highest risk are not sufficiently included in the GWAS, it is not possible to conclude whether there is an overall genetic risk or to identify biomarkers that might be indicative of this risk in those populations.
Enhancing Recruitment Through Education and Outreach
A question was raised by a workshop participant about the systematic best practices for the education of providers and participants to improve recruitment outcomes and whether it would be helpful for a national group, perhaps in collaboration with other groups, to develop generalizable education materials as a resource for primary care providers. Patients most often desire hearing about clinical trials from their primary care physician, Langbaum said, yet the overwhelming majority of primary care physicians do not have the proper education or resources to provide that information. Specialized practices are typically the nexus for referring patients for clinical trials, she said, and the central database and registry for clinical trials, ClinicalTrials.gov, does not serve the purpose of a user-friendly, lay-accessible database of clinical trial information.
Rare disease advocacy groups have different levels of baseline literacy concerning genetics or clinical trials, Strong said, and accordingly their patient bases may or may not have all the information about the questions they should ask when approached about participation in a clinical trial. It is important to inform patients about what it means to participate in a clinical trial and to demystify the process, Langbaum said, so that when they are invited to participate in a study, they are prepared. Effort is needed on the front end to create educational materials, she said, and the preparation of materials should not be left solely to the study coordinators, who are often already overburdened.
It is important to include all stakeholders in the discussions of how to better recruit diverse, representative patient populations, including chief executive officers and leaders of health systems, EHR vendors, and informaticians, added Bray Patrick-Lake of the Duke University Clinical and Translational Science Institute. She observed that while clinical trials may struggle to recruit diverse populations, people representing a diversity
of ethnic and cultural backgrounds seek medical care in the health system every day, so there are opportunities to diversify these studies if the health care and research sectors work collaboratively.
Engaging and Recruiting Patients in Genetics-Enabled Clinical Trials: Case Studies in Alzheimer’s Disease
Langbaum offered some examples from the perspective of the Alzheimer’s Prevention Initiative (API) of successful mechanisms for engaging participants in precision-medicine clinical trials for Alzheimer’s disease.
Colombian Alzheimer’s Prevention Registry
A trial for a rare, autosomal-dominant form of Alzheimer’s disease is being conducted in Colombia, South America, where it is estimated that there are about 5,000 living family members of the world’s largest autosomal-dominant mutation kindred.7 The Colombian Alzheimer’s Prevention Registry was launched in anticipation of the trial and served as a pre-enrollment recruitment mechanism to identify potential study participants. The local investigator and his colleagues have enrolled more than 5,600 people in the registry to date, all of whom are distantly related to a common ancestor, Langbaum said. Of these enrollees, more than 1,100 mutation carriers and more than 4,000 kindred non-carriers have been identified.
A lesson from this example, she said, is that in this mixed community in Colombia, consisting of both rural and metropolitan areas, there is considerable interest in clinical research, but considerable community outreach was also required. Families were identified through reviews of church records, family meetings, and other outreach tactics. This was followed by meetings with families to provide information about the importance of clinical research, the genetic origin of their disease, and the potential hope provided through participation in research. The registry was essential to completing enrollment into the autosomal dominant trial, she said, noting that all participants for the trial were recruited from the registry.
GeneMatch
Another program, GeneMatch,8 was launched in November 2015 as an online, trial-independent genetic testing registry for the recruitment of
___________________
7 For more information about the PSEN1 E280A mutation and the Colombian kindred, see http://www.alzforum.org/mutation/psen1-e280a-paisa (accessed May 18, 2017).
8 For more information about GeneMatch see www.endALZnow.org/genematch (accessed April 26, 2017).
participants for the API’s Generation Study and to provide a resource for individuals who did not meet genetic eligibility criteria for the Generation Study to be re-contacted for future API studies. Langbaum said that participants do not receive their APOE (apolipoprotein E gene) test results upon enrolling in the registry; however, the studies to which they are referred may require APOE disclosure as part of their enrollment. In order to facilitate this, GeneMatch uses a CLIA-certified and CAP-accredited laboratory.
To date, more than 33,000 cognitively healthy adults, aged 55 to 75, have been enrolled in the GeneMatch program, suggesting that there is a high level of interest in participating in clinical research, Langbaum said. Implementing this registry has also required significant outreach and education, she said, in addition to continued engagement with participants, since it could be months to years until they receive a call to participate in a study. The most significant barrier to registration, however, is that potential participants want to receive their APOE test results.
The recruitment process for the Generation Study is similar to a funnel, Langbaum said. In the United States alone, more than 100,000 people likely would need to be registered into the GeneMatch program and genotyped in order to match and recruit the approximately 600 people needed for a randomized trial.


























