
Panelists in the fourth session explored ways in which genetics-enabled clinical trials could improve the efficiency of the drug development pathway. Robert Plenge, vice president and head of translational medicine at Merck (at the time of the workshop), shared a model for enhancing the efficiency and productivity of drug discovery and development, including using genetics to identify potentially successful drug targets, thereby reducing late phase failures. Philip J. “P. J.” Brooks, program director in the Division of Clinical Innovation at the National Center for Advancing Translational Sciences at NIH, described applying the basket trial approach to shared molecular etiologies in rare genetic diseases. David Leventhal, director of clinical innovation at Pfizer, discussed determining the return on investment of incorporating patient input in the drug development process. The session and discussion were moderated by Bray Patrick-Lake.
NEW TARGETS, MODALITIES, AND CHALLENGES: THE INCONVENIENT PATH OF HUMAN GENETICS IN DRUG DISCOVERY
Precision medicine is about identifying those patient subsets for whom a therapeutic intervention will work more effectively than in the general population, Plenge said. He suggested that the greatest impact moving forward for precision medicine will be to guide new drug development from its earliest stages, rather than applying various “omics” technologies to approved drugs or compounds in late-stage development. The path to precision medicine may be difficult and inconvenient, he said, because it involves understanding both the biology of molecular pathways involved in human diseases and how to develop a targeted therapeutic that recapitulates that biology.
The biopharmaceutical industry is facing two major challenges: the increasing trajectory of drug development costs and, at the same time, medicines being delivered into the real world that are not delivering sufficient value or improving the cost of health care. Bending the cost trajectory of drug development will not be the result of minor improvements, Plenge said, but will instead require a comprehensive, disciplined approach to improving the efficiency of the system (Plenge, 2016).
New drugs may not provide real-world value or bend the cost curve of health care because they do not differentiate from, or add value above, existing therapies due to the lack of sound, novel therapeutics hypotheses, Plenge said. “As an industry, (we) spend too much time chasing what other people are chasing,” he said. “We need to come at this from a novel place of biology.” There are two main reasons for the steady rise in drug development costs, Plenge said. First, there are too many failures in Phases 2 and 3: “It is not the cost of the successes,” he said. “It is the cost of failures
(in Phases 2 and 3) that are driving the increased cost of drug research and development.” Second, he suggested that the cycle time between early discovery and proof-of-concept trials is too long. In general, it takes 5 to 7 years to advance a therapeutic hypothesis to first-in-human proof-of-concept trials, he said, adding that if the cost of drug development is to be significantly lessened, this early phase would need to be radically shortened. This would be a significant challenge, he said, but there are potential solutions to be leveraged through precision medicine approaches, such as selecting targets based on a deep understanding of human genetics and human biology and the concept of programmable therapeutics to test in proof-of-concept trials.
Multi-omic approaches could be used to find new targets, identify new pharmacodynamics biomarkers, and identify subsets of patients for which drugs are more likely to work, Plenge said. Geoffrey Ginsburg, director of the Duke Center for Applied Genomics and Precision Medicine, endorsed the potential of other omics technologies, including transcriptomics, proteomics, and metabolomics, in early-stage clinical development. The field of oncology has been particularly successful in applying omics approaches, like using omics data to identify gene expression signatures that might predict drug response. There are some challenges facing practical use of omics, however, such as the complexity of the requisite diagnostic platforms. For example, a community hospital setting might not have the resources or capabilities to test for an RNA expression signature.
Using Genetics Approaches to Select Drug Targets
Using human genetics for prioritizing drug targets is similar to a dose–response curve, Plenge said, because an increase in genetic perturbation has a corresponding effect on human phenotype (Plenge et al., 2013). The first step in using genetics to guide drug discovery is to choose a human phenotype that is relevant and consider the desired outcomes in response to therapeutic modulation. Next, a genetic study (such as a GWAS) is conducted to find those alleles that influence risk for the phenotype. Once the alleles that influence risk are identified, the next step is to assess the biological function of the alleles. Does the genetic variant result in a premature stop codon, for example, where the effect of the gene is then completely ablated? Or, as is often the case with common variants, is the genetic perturbation less clear? The data resulting from genetic studies of allele frequencies can be used to estimate an “efficacy response curve” (see Figure 5-1). Those same alleles can also be used as probes to assess pleiotropy1 to predict the effect(s) of modulating a particular target
___________________
1 One gene affecting multiple, seemingly unrelated phenotypes.
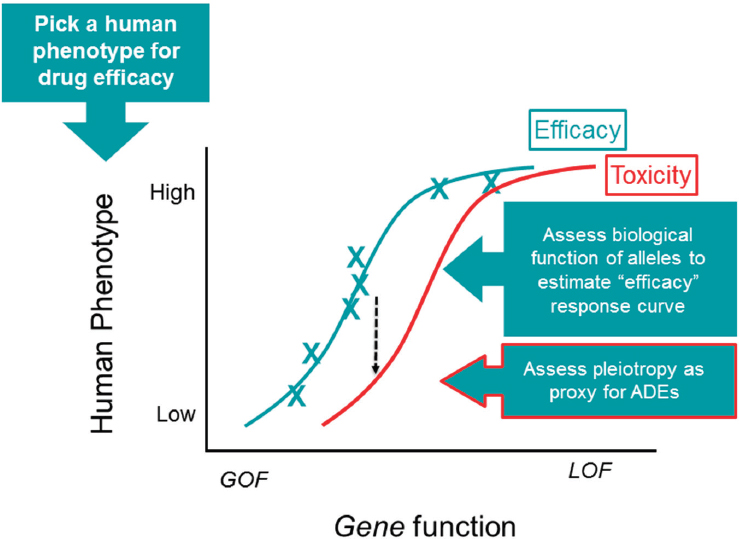
NOTES: By modulating the function of a target (x-axis), it is possible to assess its effect on a biological phenotype (y-axis), such as cellular signaling or receptor levels. The points on the graph indicate a dose-dependent relationship between target function and biological phenotype, where the gain of function of a target leads to a reduced (low) biological activity (phenotype) and loss of function leads to increased (high) biological activity; ADE = adverse drug event; GOF = gain of function; LOF = loss of function.
SOURCE: Robert Plenge, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
(beyond the intended target effect). This type of knowledge from genetics studies has the potential to reduce adverse drug events, such as toxicity, Plenge said.
As an example of this approach, Plenge cited a recent study that assessed whether heterozygous carriers of pathogenic mutations in the gene encoding lipoprotein lipase had increased odds of coronary artery disease (Khera et al., 2017). After researchers examined sequencing data from nearly 50,000 individuals, 188 participants were identified as having a high-risk mutation in the lipoprotein lipase gene. More than 100 different rare variants and 6 common variants were identified and tested for an association with plasma
triglyceride levels and the presence of coronary artery disease. The authors of this study noted that further research is needed to determine whether the variants identified are causally related to the observed deleterious phenotypes.
The concept of using human genetics to select novel drug targets can help improve the rates of success in Phase 2 and 3 trials, Plenge said; however, the approach does not shorten cycle time. “Programmable therapeutics” could potentially offer a solution for testing hypotheses more quickly, but Plenge acknowledged that the industry has a long way to go to realize this. He identified several factors that are currently limiting the field’s ability to decrease cycle times and the associated costs of drug development and to increase the value of medicines released to the market:
- The biologic function of the disease-associated genes, variants, and pathways are incompletely understood.
- Less than 20 percent of genetic targets can be modulated by existing conventional modalities (e.g., small molecules or monoclonal antibodies).
- New modalities are greatly needed (e.g., gene therapy, mRNA replacement, or innovative delivery vehicles), but they are limited by issues of delivery and pharmacologic properties.
For precision medicine to be successful, Plenge summarized, there are three main solutions that could be implemented:
- Anchor early-stage therapeutic development in an understanding of the molecular mechanisms of pathogenic genetic variants and pathways.
- Recapitulate disease mechanisms with credible therapeutic molecules.
- Shorten cycle time to testing therapeutic hypothesis in small proof-of-concept trials of molecularly defined subgroups.
GROUPING RARE DISEASE PATIENTS BY SHARED MOLECULAR ETIOLOGY TO ACCELERATE CLINICAL TRIALS
Another potential approach to streamlining genetics-enabled drug development and decreasing the amount of time required to bring new medicines to market was presented by Brooks. NCATS was established at NIH in 2011 to “catalyze the generation of innovative methods and technologies that will enhance the development, testing, and implementation of diagnostics and therapeutics across a wide range of human diseases and conditions.”2 In contrast to many other NIH institutes and centers,
___________________
2 See https://ncats.nih.gov/about/center (accessed April 26, 2017).
Brooks said, NCATS does not focus on specific diseases or organ systems, but rather on the process of translation itself. Underscoring the importance of NCATS is the fact that the number of human conditions with a known molecular basis is increasing rapidly, particularly with the implementation of new genetic diagnostic procedures, while the development of corresponding therapies is lagging significantly behind. As of March 2017, there were almost 6,000 disease phenotypes with a known molecular basis, but only approximately 500 approved therapies.3 A fundamental change is needed in the way that treatments are developed for these rare diseases, Brooks said, and this is a gap that NCATS aims to help address with a program called SaME Therapeutics (Targeting Shared Molecular Etiologies Underlying Multiple Diseases to Accelerate Translation).
Concept: Grouping Disease by Shared Molecular Etiology
The SaME Therapeutics approach proposes grouping patients on the basis of underlying shared molecular etiologies (SMEs), rather than by individual diseases defined by symptoms, to stimulate novel clinical trials of SME-targeted drugs (Brooks et al., 2014). Brooks said that the modern precedent for this approach is genetically enabled basket trials (e.g., the STARTRK-2 trial discussed by Hornby in Chapter 3). There are thousands of rare diseases, he said, but far fewer molecular etiologies. Genetic diseases are caused by two types of mutations: dominant (generally gain-of-function) and recessive (generally loss-of-function). There are a limited number of loss-of-function mutation types, Brooks said, the most common of which are nonsense mutations that result in premature stop codons and missense mutations that result in abnormal protein folding. Both of these are, in principle, amenable to therapeutics, he said.
To illustrate this approach, Brooks explained that in cystic fibrosis, Gaucher disease, and Tay-Sachs disease, a patients’ disease phenotype can be the result of either stop codon mutations or missense protein misfolding mutations. A traditional trial approach might study a stop codon read-through drug and a protein misfolding drug separately for each disease, requiring a total of six clinical trials. By contrast, grouping patients according to underlying SMEs could require only two trials. The approach could be further expanded and the results magnified by re-characterizing many diseases by their underlying molecular pathology, such as “premature stop codon disease” or “protein misfolding disease,” rather than by their organ system or clinical phenotype.
For missense mutations that cause abnormal protein folding, one therapeutic opportunity could be a drug that would allow the misfolded protein
___________________
3 See https://www.omim.org/statistics/geneMap (accessed May 15, 2017).
to fold correctly and carry out its normal function in the cell. Traditionally, Brooks said, development of such a drug has focused on small molecule chaperones that target the individual misfolded protein. A more efficient approach would be to target the cellular pathways active in essentially all cells that help maintain protein homeostasis (proteostasis). Some examples of proteostasis pathway regulators include heat shock proteins, the unfolded protein response or oxidative stress response, autophagy, and histone deacetylases. Such an approach has potential applicability to multiple diseases and has been adopted in several proof-of-concept studies, Brooks said (Das et al., 2015; Mu et al., 2008; Wang and Segatori, 2013).
There are four different stop codons, and stop codon read-through compounds can have sequence-specific effects, so drug development programs and trial planning should consider a range of compounds to account for this underlying variability, Brooks said. Furthermore, the effects of premature stop codon read-through compounds depend on the underlying biology of the affected proteins. For example, a premature stop codon located in a gene that encodes a structural protein will have a much different effect than one located in a gene encoding for an enzyme, which can have extensive functional effects. As such, it is important that trials of premature stop codon read-through compounds include multiple diseases, as it may not be possible to predict the potential clinical effects in each situation.
Shared Molecular Etiology-Driven Trials
It may be possible to apply the basket trial approach, which has been effective in genetically enabled oncology trials, to SMEs in rare genetic diseases, Brooks said. Though there is promise for using this approach in rare diseases, there are key differences in both enrollment criteria and clinical endpoints between basket trials for cancer and basket trials for rare diseases; these differences include
- Enrollment criteria—Oncology basket trials generally involve a standardized assay (e.g., genotyping or immunostaining) for a molecular marker in a comparable cell type, and patients with marker-positive tumors are enrolled. However, in rare disease SME basket trials, disease-specific cellular assays or biochemical measurements in patient-derived cells will likely be needed.
- Trial endpoints—Oncology basket trials generally have a primary endpoint that is a straightforward outcome, such as tumor size. The clinical endpoints in rare disease SME trials, however, might involve more complex pharmacodynamic or response biomarkers that could require specific and precise assays.
To address these issues, Brooks emphasized the utility of collaborating with rare disease organizations, including associated partners such as patient advocates, clinical specialists, and laboratory scientists who possess the specific expertise to carry out the requisite disease-specific assays. Establishing these partnerships could provide researchers with patient biospecimens, which could then be used by laboratory scientists to develop assays. For enrollment, an in vitro assay could be used to identify a patient-specific drug response and determine whether a patient meets the eligibility criteria. To track outcomes that will inform the clinical endpoint, such as a pharmacodynamic analyses or response biomarkers, it might be necessary to develop disease-specific cellular assays as well.
There are regulatory pathway considerations that should be addressed to fully leverage the power of SME-driven trials, Brooks said. For example, it is unlikely that there would be a common endpoint for all distinct disease entities within a basket trial, so would an approved, disease-specific endpoint be needed for each disease in the basket, or could endpoints be based on pharmacodynamics and response? Furthermore, would early-stage trials that are successful in specific disease states be required to move onto disease-specific registration trials, or would they be eligible for more “rapid” approval pathways, such as accelerated approval?
There are several possible options for funding SME clinical trials in the future, Brooks said. Participants were referred to the Collaborative Innovation Awards of the NCATS Clinical and Translational Science Award (CTSA) program.4 These awards were designed to stimulate collaboration with investigators from at least three different CTSA sites, and although it is a broad funding opportunity, the program has a vested interest in supporting clinical trials of drugs targeting SMEs underlying rare diseases. Small business funding opportunities are also available through the Small Business Innovation Research and Small Business Technology Transfer programs.5
RETURN ON INVESTMENT FROM PATIENT INPUT IN DRUG DEVELOPMENT: LESSONS FROM CTTI AND PFIZER
The final speaker in this session, Leventhal, presented a model for evaluating the potential financial benefits of engaging with patient groups during clinical drug development. There are many opportunities for patient groups and research sponsors to work together for mutual benefit, Leventhal said. The value of incorporating patient input throughout the drug development
___________________
4 See https://grants.nih.gov/grants/guide/notice-files/NOT-TR-17-004.html (accessed April 26, 2017).
5 See https://www.sbir.gov (accessed April 26, 2017).
process can be measured, he said, a process that was undertaken by the CTTI Patient Groups and Clinical Trials (PGCT) Project.6 Leventhal went on to describe the findings of the project; he noted that its recommendations are being implemented within drug development programs in Pfizer and detailed its approaches for patient engagement.
CTTI Patient Groups and Clinical Trials Project
In October 2015, the CTTI PGCT Project put forth recommendations that outlined best approaches for engaging patient groups across the research and development continuum and for managing sponsor–patient group relationships to ensure that the needs of all stakeholders are met and to improve the overall quality of clinical trials. As a follow-on to these recommendations, the PGCT commenced a second stage of the project to develop a model for assessing the economic value of conducting patient engagement in drug development programs, Leventhal said. He highlighted two conceptual approaches for evaluating the value of patient-centric initiatives, the second of which the CTTI working group adopted for its model:
- Return on engagement—The Drug Information Association and Tufts Center for the Study of Drug Development (CSDD) collaborated on a retrospective review of case studies and historical data on patient engagement efforts undertaken by the biopharmaceutical industry and other research sponsors to determine the impact of these patient-centered initiatives.7
- Value proposition—CTTI PGCT developed a prospective modeling approach to assess the expected net present value (ENPV) of patient engagement in drug development programs.
Leventhal discussed the major methodological and data components that CTTI used to develop a model to assess the value and financial impact of patient engagement initiatives. First, the CTTI team used common methods for modeling financial net present value (NPV)8 based on project development cost, time, and risk. Second, Phase 2 and 3 oncology development programs were used as “base cases” to compare the impact of patient engagement initiatives, as measured by a reduction in the number of protocol amendments and improvement in enrollment and retention. Finally, the
___________________
6 CTTI work was conducted by a multi-stakeholder project team. To access more information on the team members and projects within the CTTI PGCT Project, see https://www.ctti-clinicaltrials.org/projects/patient-groups-clinical-trials (accessed April 26, 2017).
7 To view the full DIA–Tufts CSDD Study, see http://www.diaglobal.org/en/resources/toolsand-downloads/dia-tufts-csdd-study (accessed May 14, 2017).
8 Future revenue stream adjusted for taxes and inflation.
model took into account published benchmark and source data from the literature—primarily from the Center for Information and Study on Clinical Research Participation and Tufts CSDD. The drivers of value that produce the NPV and the risk factors taken into account to produce the ENPV were described by Leventhal (see Box 5-1).
Leventhal shared a case study wherein the model was populated with the anticipated probabilities of success and failure for a drug development program and the ensuing NPV and ENPV was calculated (see Figure 5-2). In each phase of drug development, Leventhal noted, there are opportunities for a program to fail (indicated by probabilities assigned to each development pathway) that are based on scientific, operating, and regulatory risks. Each pathway also has an associated cost or reward expressed as cash flow in NPV. Based on the modeling, Leventhal said, for the case study presented in Figure 5-2 there was a 22 percent probability of launch, and the NPV, given technical and regulatory success, was $400 million. Accounting for potential failures, which are associated with a negative NPV, the average
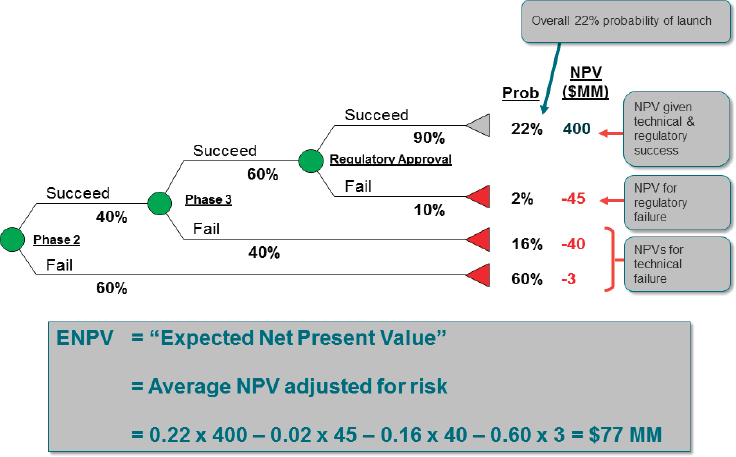
NOTES: Each line is a development path, circles represent studies or other key risk milestones, triangles represent failures or successes. $MM = millions of dollars; NPV = net present value; Prob = probability.
SOURCES: Figure developed by the Clinical Trials Transformation Initiative (CTTI) Patient Groups and Clinical Trials Project team. See more at https://www.ctti-clinicaltrials.org (accessed June 28, 2017). Presented by David Leventhal, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
ENPV was $77 million. Leventhal added that the model as presented did not account for pre-Phase 2 failures, which can be high.
As noted previously in this section, there are two inflection points in drug development where patient-centric initiatives could potentially have an impact: avoiding one or more protocol amendments, and improving feasibility and patient experience to increase enrollment and retention. Underscoring the importance of these impact measures, Leventhal said that 70 percent of Phase 2 and 3 trials have at least one amendment, and 22 percent of amendments are due to recruitment difficulty or feedback from sites or investigators. Implementing a single amendment adds approximately 3 months and as much as half a million dollars in direct costs to a trial (Getz et al., 2011, 2016). Increasing feasibility and patient experience in trials could include increasing the comprehensibility of informed consent forms, simplifying eligibility criteria, reducing participation burden,
and accelerating overall study cycle time (e.g., startup, enrollment, and completion). The model developed by CTTI suggests that, in the example case study, avoiding one protocol amendment could have a NPV impact of $24.5 and $32 million in Phase 2 and Phase 3, respectively, with an ENPV of $3.8 million and $15 million, respectively. The NPV of improving patient experience in Phase 2 and Phase 3 is $38 and $31 million, respectively, with $30 and $57 million in ENPV, respectively (see Figure 5-3).
This model provides compelling financial incentives for implementing patient engagement initiatives, Leventhal said. ENPV modeling can account for important drivers of value in a clear and well-accepted summary metric, he said, adding that for engagement activities resulting in the avoidance of an amendment or an improved patient trial experience, the benefits in cost and ENPV vastly outweigh the resources spent on engagement. Therefore, ENPV modeling and similar approaches can support sponsor decisions to increase patient engagement throughout development.
Patient Engagement at Pfizer
Pfizer has begun implementing the recommendations set forth by CTTI’S PGCT Project across its drug development programs as part of its Clinical Innovation Patient Engagement Framework, Leventhal said. Supporting this framework for engagement are playbooks, toolkits, policies and guidance, and external collaborations (e.g., CTTI). Leventhal highlighted select examples of the components of this framework:
- Find-a-Trial—A clinical trials resource on Pfizer’s website that was developed as an alternative to ClinicalTrials.gov, with simplified language and more participant-friendly, IRB-approved materials.
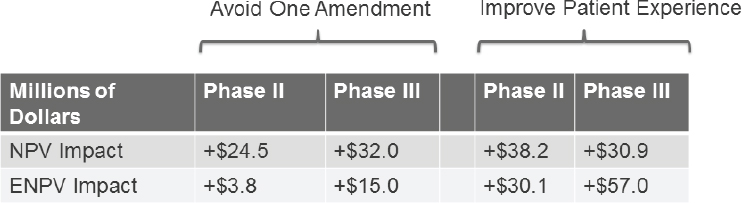
NOTE: ENPV = expected net present value; NPV = net present value.
SOURCE: David Leventhal, National Academies of Sciences, Engineering, and Medicine workshop presentation, March 8, 2017.
- Social Media Policy/Guidance—A social media policy and guidance for clinical development allows the use of social media channels for trial recruitment and retention.
- Live Trial Protocol Simulations—Patient insights from live simulations of clinical protocols are used to improve patient and investigator experience and to optimize protocols.
- Innovative Engagement and Retention—Electronic informed consent, mobile applications for scheduling and reminders, a participant dashboard, and telehealth capabilities engage participants in clinical trials and help increase trial retention.
- PfizerLink—A patient alumni community open to every U.S. clinical trial participant. Past participants can download clinical data collected during the conduct of the trial and access lay language summaries for all Phase 2 and Phase 3 U.S. studies.9
- Patients in Global Product Development Initiative—Soliciting input from patients about ongoing clinical development programs, including how to measure impact, identify gaps, and implement improvements.
During the discussion session, a workshop participant suggested that biopharmaceutical companies involve experienced patient advocates in external stakeholder meetings and foster conversations among stakeholders to better understand the patient perspective. The workshop participant expressed dismay that the value of including patients in the development must be demonstrated before it can be supported, whereas the input of clinicians into the process is assumed. These types of approaches are still in the early stages within many organizations, Leventhal said, and much of the path forward will depend on resources and funding. Expenditures must be justified at every step in industry, he said, but he reiterated that there is value in patient engagement.
___________________
9 Similar plain language summaries will be available for studies conducted in Europe in the near future according to regulations that will come into effect in 2018.
This page intentionally left blank.














