
3
Characterizing Interactions Between the Human Microbiome and Environmental Chemicals
Scientific research is beginning to illuminate the various ways in which the human microbiome can interact with environmental chemicals. As discussed earlier, multiple studies suggest that exposure to environmental chemicals can alter microbial composition and potentially affect function. Research has also indicated that the human microbiome can modulate exposure to environmental chemicals. The idea that microbiota in and on the host can contribute to host metabolism is deeply rooted in the field of drug metabolism. Early observations regarding the fate of the antibacterial prodrug1 Prontosil cemented the need to improve our understanding of how microorganisms metabolize chemicals and how these processes might affect the host, favorably or unfavorably (Spink et al. 1940). The concept of the microbiota and its contribution to host metabolism was further strengthened by the father of modern drug metabolism, R.T. Williams, and later expanded by his contemporaries who investigated the fates of simple aromatic chemicals, such as benzoic acid (Gingell et al. 1971; Williams 1972). However, technical limitations in identifying and cataloging the responsible microbiota have severely hindered progress in understanding underlying mechanisms. Only recently with the advent of high-throughput approaches, including sequence-based community profiling and metabolomics, has the contribution of microbiota to drug metabolism transitioned from basic observation to a more mechanistic understanding (Scheline 1968a,b; Patterson and Turnbaugh 2014; Spanogiannopoulos et al. 2016), although our understanding of its metabolic capacity remains limited (Idle and Gonzalez 2007).
Given that health risk is a function of both toxicity (dose-response) and exposure, a critical consideration for risk-assessment frameworks is how the activities encoded by the human microbiome influence the dose of toxicologically active chemicals at the ultimate target site (tissue, cell, or macromolecule). Knowledge of how the human microbiome modulates the pharmacokinetics and metabolism of environmental chemicals generally lags behind knowledge of how the microbiome modulates drugs. Still, there is compelling evidence on gut microbiome involvement in the metabolic transformation of environmental chemicals in broad chemical classes, including metals, polycyclic aromatic hydrocarbons (PAHs), pesticides and persistent organochlorines, nitroamines and aromatic amines, and other toxicant classes (Cerniglia et al. 1984; Van de Wiele et al. 2005; Van de Wiele et al. 2010; Claus et al. 2016).
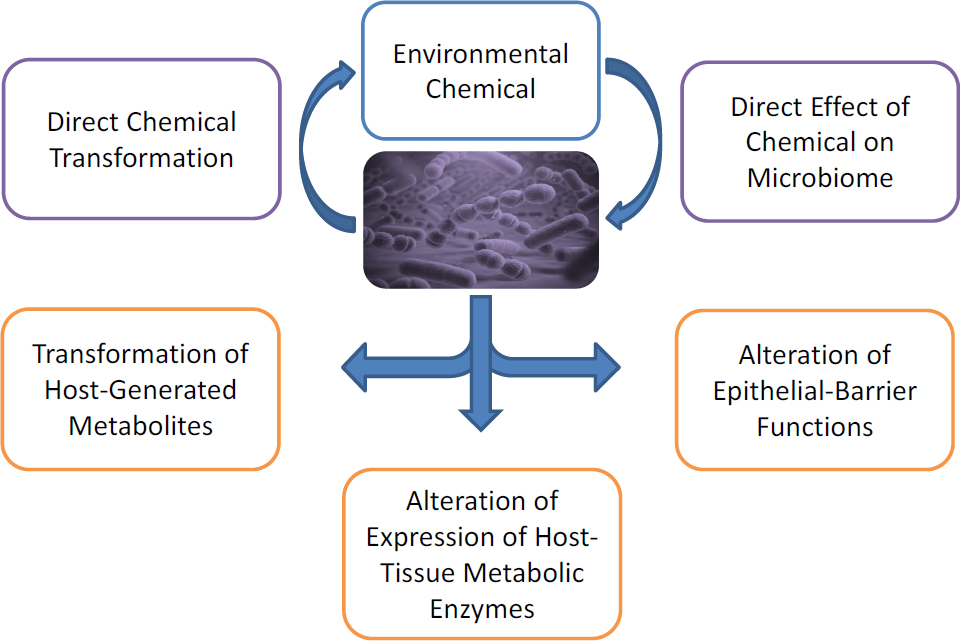
Many molecular mechanisms probably underlie microbiome interactions, and incorporating such molecular-level detail into the risk-assessment framework for each environmental chemical is a daunting challenge. Nonetheless, research suggests that the human microbiome might modulate the exposure–response relationships of environmental chemicals through a few general mechanisms, which might directly or indirectly influence the pharmacokinetics of the chemicals. The mechanisms include direct metabolic transformation of environmental chemicals and secondary transformation, such as deconjugation of host-generated metabolites; regulation of epithelial-barrier per-
___________________
1 A prodrug is a chemical whose metabolism forms a biologically active drug.
meability, with implications for transport or excretion of chemicals; and regulation of the expression or activity of endogenous host metabolic pathways (for example, in the host liver) via signaling processes that involve microbial products (Figure 3-1). As noted above, there is also a potential for direct effects of environmental chemicals on the composition of a microbiome itself. Although such changes might lead to adverse physiologic consequences through mechanisms that are independent of alterations of a chemical’s pharmacokinetics, disruptions in the composition or abundance of a microbial species has the potential to affect all other mechanisms that are mediated by the microbiome.
Conceptually, each interaction can have favorable or unfavorable influences on chemical exposure, and the role of the interactions in modifying susceptibility to toxicity in connection with environmentally relevant exposures remains uncertain. Furthermore, extensive metabolic cooperation and exchange of metabolites that occur between microbial species in a community and with the host is not apparent when species are studied in isolation (Phelan et al. 2011; Traxler et al. 2013). Understanding of the toxicologic significance of the interactions requires strategies that include the microbiome as an integrated part of a multiorgan host response. It should also be emphasized that research on the roles of microbiomes in metabolism of environmental chemicals has focused on the gut microbiome. Examples of the general mechanisms are provided here, but there is a general need to expand knowledge of potential mechanisms of interaction for other body sites. This chapter explores the mechanisms highlighted in Figure 3-1 and concludes with a discussion of interindividual variability and microbiome metabolism of environmental chemicals.
DIRECT EFFECTS OF A CHEMICAL ON MICROBIOME COMPOSITION
One way that interactions between the microbiome and an environmental chemical can influence host health is through direct chemical-induced changes in the microbiome. Such changes can be detected by assessing changes in community membership, relative abundance of existing members, spatial organization of the community, microevolution within particular member species, gene expression, or activity of particular metabolic pathways. It is well established that microbiomes of specific composition can have distinct causal effects on host biology. If exposure to an environmental chemical or any other factor leads to alterations in microbiome composition, the altered microbiome itself might have distinct direct effects on the host, although not all changes will contribute causally to host phenotype. It is also conceivable that changes induced by environmental chemical exposure will change the capacity of the microbiome to metabolize chemicals directly or indirectly. As described below, various experimental strategies can be used to test potential effects of chemicals and other environmental factors on the microbiome.
The capacity of environmental chemicals to induce microbiome changes in animals has been demonstrated with a variety of pesticides, metals, artificial sweeteners, and drugs (Patterson and Turnbaugh 2014; Claus et al. 2016). Most studies have relied on analysis of microbial community composition, but additional insights can be gained through combination with other assays. For example, exposure of mice to arsenic in drinking water at 10 ppm for 4 weeks induced consistent alterations in the gut microbiome, including changes within the Clostridiales order (reductions in Clostridiaceae and Catabacteriaceae families and increases in Family XIII Incertae Sedis). Fecal-metabolite analysis identified a distinct signature of arsenic treatment, including 370 molecular features, many of which—such as bile acid, indole, and isoflavone derivatives—are predicted to be directly generated or modified by gut bacteria. Correlations between affected bacterial taxa and fecal metabolites were also detected; for example, Family XIII Incertae Sedis was correlated negatively with indolelactic acid and dihydrodaidzein (Lu et al. 2014). It is important to note that the drinking-water arsenic concentrations used in Lu et al. (2014) far exceed the drinking-water standard for arsenic (10 ppb). Others have examined effects of low concentrations of arsenic on microbiome composition (Dheer et al. 2015), but administration of arsenic as a sodium salt without appropriately paired controls might be a confounding factor in the experiments. Despite a growing number of experimental studies that report that environmental chemicals can alter microbiome composition, the use of high doses that are of questionable relevance to human environmental exposures is a common limitation of the literature (Claus et al. 2016). However, such results do suggest potential metabolic functions of specific chemical-sensitive microorganisms.
The effects of environmental chemicals on the composition of host-associated microbiomes can be modulated by the host. For example, exposing mice to polychlorinated biphenyls (150 µmol/kg for 2 days) led to alterations in gut microbiota in sedentary animals but not in exercised animals (Choi et al. 2013). Effects of environmental chemicals on microbial composition and metabolite profiles can also be affected by sex, as demonstrated recently in mice exposed to diazinon (Gao et al. 2017). And host genotype contributes to microbial composition (Benson 2016). For example, dietary exposure to 2,3,7,8-tetrachlorodibenzofuran (24 μg/kg for 5 days) led to gut microbiome perturbations, inflammation, and alterations in bile-acid metabolism and signaling in wild-type mice but not in those lacking the aryl-hydrocarbon receptor (Zhang et al. 2015); this finding suggests that a host receptor-dependent mechanism is involved. However, our understanding of the role of host genotype in determining the effects of environmental chemicals on microbial composition is limited. To define the effects of chemicals on a microbiome independently of host effects, complex microbial communities (Joly et al. 2013; Maurice et al. 2013; Suez et al. 2014) or individual microbial strains (Shehata et al. 2013) have been cultured and exposed to chemicals in vitro to reveal effects on mi-
crobial growth, gene expression, and community composition.
Although those and other studies have shown that environmental chemicals can induce microbiome changes, the ability of the altered microbial communities to contribute causally to host phenotypes remains largely unknown. Studies that have analyzed other environmental factors provide instructive experimental strategies for addressing that question. For example, feeding mice a high-fat diet (Turnbaugh et al. 2008) or treating mice with low doses of penicillin from early in life (Cox et al. 2014) leads to changes in the gut microbiome and other host phenotypes. In each study, transplantation of the altered microbiome into germfree recipient mice induced phenotypes that were observed in the donor animals. Such microbiome-transplantation experiments are important because they can help to determine whether the microbiome changes have causal effects on host phenotypes. They can also help to answer the question of whether the host phenotypes are induced directly by the environmental factor or indirectly through the altered microbiome. Another experimental strategy for determining direct and indirect effects on the host is to compare the host phenotypic response to the environmental factor in the presence and absence of a microbiome. For example, administration of a high-dose broad-spectrum antibiotic cocktail in mouse models caused host responses (such as immune downregulation and mitochondrial-dependent epithelial-cell death) that could be explained by loss of antibiotic-sensitive microorganisms, by the remaining antibiotic-resistant microorganisms, or directly by the antibiotics in the absence of microorganisms (Morgun et al. 2015).
Several reports have shown that a chemical challenge can be sufficient to alter host physiology and microbiome composition and that the alteration of the microbiota is sufficient to change the physiology of germ-free recipient hosts after microbiome transplant (Cox et al. 2014; Suez et al. 2014; Chassaing et al. 2015). However, the reported experiments alone do not clearly distinguish between direct causal effects of the chemical on the microbiome and indirect effects of the chemical acting first on the host and altering selective pressures on the microbiome that change microbiome composition. Direct causal relationships between a chemical-induced change in the microbiome and host phenotype has been demonstrated only for noncaloric artificial sweeteners (Suez et al. 2014). That study demonstrated that drinking-water administration of saccharin at doses equivalent to the acceptable daily intake for humans (5 mg/kg-day) altered microbiome composition and induced glucose intolerance in mice. Fecal microbiomes from unexposed mice were also exposed to artificial sweeteners in vitro and then used to colonize germ-free mice; higher glucose intolerance was observed in the colonized mice.
ALTERATIONS IN THE FUNCTIONS OF EPITHELIAL BARRIERS
Epithelial barriers form the interface between many host tissues and the external environment. In addition to their roles as protective barriers, epithelia regulate sensory perception, absorption, surface transport, immune function, and excretion of molecules, ions, and water. Increasing evidence suggests that there are intimate bidirectional interactions between the microbiota and epithelial cells wherein the composition and activity of the gut microbiota, for example, modulate the structure and function of the intestinal epithelium and vice versa (Ulluwishewa et al. 2011; Peterson and Artis 2014; Kelly et al. 2015). Direct manipulations of the gut microbiota via gnotobiotic rearing, antibiotic treatment, or probiotic treatment have been causally linked to changes in intestinal permeability in animal models (van Ampting et al. 2010; Everard et al. 2013; Grover and Kashyap 2014; Leclercq et al. 2014; Tulstrup et al. 2015; Thevaranjan et al. 2017). Perturbations of the microbiota after exposure to such factors as infection, stress, and dietary changes have also been linked to changes in gut-barrier integrity. Clinical associations between microbial changes and “leaky gut syndrome” (increased intestinal permeability) in various gastrointestinal, immune, metabolic, and neurologic disorders raise the question of whether microbiota–epithelium interactions contribute to the cause and development of disease symptoms
(Hartmann et al. 2012; Marchesi et al. 2016; Richards et al. 2016). Overall, the gut microbiome is emerging as a key regulator of epithelial permeability and integrity with important implications for the absorption, transport, and excretion of environmental chemicals.
Exactly how the microbiota modifies epithelial-barrier integrity is poorly understood, but some evidence suggests that microbial regulation of tight-junction proteins, mucus-layer structures, and transport systems could contribute. Epithelia are comprised of a continuous layer of squamous, cuboidal, and columnar cells that are interconnected by tight-junction complexes that join adjacent cell membranes and regulate paracellular and transepithelial passage of solutes. Various probiotic treatments and microbiome manipulations have altered expression of tight-junction proteins concurrently with changes in intestinal permeability (Turner 2009). For example, in a mouse model of metabolic syndrome, probiotic administration of Akkermansia muciniphila increased small-intestine expression of the tight-junction proteins claudin 3 and occludins that correlated with decreases in concentrations of serum lipopolysaccharide, a surrogate measure of permeability (Everard et al. 2013; Plovier et al. 2017). Likewise, in a mouse model of autism spectrum disorder,2 treatment with the commensal Bacteroides fragilis altered colonic expression of claudins 8 and 15 that correlated with decreases in translocation of the fluorescent tag FITC-dextran, an indicator of enhanced barrier integrity (Hsiao et al. 2013).
Epithelia of many internal organs contain specialized mucus-secreting cells that cover the epithelia with protective layers of viscous colloidal fluid. Some studies suggest that the microbiota can influence mucus secretion, thickness, or density. For example, Akkermansia muciniphila-mediated improvements in intestinal barrier integrity, described above, also correlated with increases in intestinal mucus-layer thickness. In addition, biophysical forces resulting from microbial fermentation of complex polysaccharides can regulate physical compression of the mucus hydrogel (Datta et al. 2016). Such changes in mucus-layer structure would probably alter solute transport dynamics. Taken together, microbial influences on epithelial-barrier integrity could be mediated by various biologic pathways.
DIRECT CHEMICAL TRANSFORMATIONS
Databases arising from the bioremediation literature have cataloged over 1,500 chemical reactions that involve the biotransformation of chemicals by environmental microorganisms (Gao et al. 2010). Research relevant to environmental-chemical exposures of humans and animals, however, is largely limited to the gut microbiome, which probably has less complex pathways than environmental microbiomes because the gut is primarily an anaerobic environment and has less microbial diversity than environmental microbiomes (Thompson et al. 2017). In contrast with the mammalian liver, in which metabolism of environmental chemicals commonly involves oxidations by cytochrome P450 enzymes, chemical transformations mediated by the gut microbiome favor reactions that do not rely on oxygen or reactions whose products provide a substrate for microbial metabolism and growth. Accordingly, Spanogiannopoulos et al. (2016) broadly categorized the direct microbial metabolic transformations commonly observed for chemicals as reduction and hydrolysis reactions. Other investigators have classified the reactions further into at least five major core enzymatic families—azoreductases, nitroreductases, β-glucuronidases, sulfatases, and β-lyases—that are expressed widely by different phyla in the microbiome (Claus et al. 2016). Examples of major classes of metabolic transformation pathways of environmental chemicals in mammalian host-associated microbiomes are provided here to illustrate the current state of knowledge. Detailed descriptions and examples can be found in several comprehensive reviews (Sousa et al. 2008; Tralau et al. 2015; Claus et al. 2016; Spanogiannopoulos et al. 2016).
Much of the evidence on the direct actions of microbial enzymes on environmental chemicals is
___________________
2 Autism is associated with increased gut permeability and a higher incidence of gastrointestinal disorders, including irritable-bowel syndrome and disease (Coury et al. 2012).
derived from studies of drugs at high therapeutic concentrations. However, inasmuch as microbial enzymes often have broad substrate specificities, parallel examples can be drawn to illustrate the potential importance of the enzymes for classes of environmental chemicals of concern in relation to human exposure. For example, azoreductases that are found in several bacterial phyla in the human gut are associated with reduction and inactivation of azo-bonded prodrugs used in treatment of ulcerative colitis, such as 5-aminosalicylic acid (Sousa et al. 2014). Bacterial azoreductases are also implicated in production of mutagenic and carcinogenic aromatic amines via reduction of azo dyes that are common in foods, textiles, and other consumer products (Rafii et al. 1990; Xu et al. 2007). Considerable variability in azoreductase activity on different bacterial isolates has been reported (Rafii et al. 1990). However, the specific bacterial genera in the gut that are responsible for those activities are not clearly known.
Gut-microbiome involvement in the metabolism of mutagenic and carcinogenic chemicals that are commonly formed as byproducts of combustion, such as urban air-pollution emissions and emissions associated with flame-based food processing, has also been demonstrated in vitro and in vivo (Möller 1994; Möller et al. 1994). In the presence of human fecal bacteria in vitro, the direct mutagenic activity of 2-nitrofluorene (2-NF)3 is diminished, presumably because of reduction to a less mutagenic aminofluorene product (Hirayama et al. 2000). Such findings imply that gut microbiota might have a protective role against the toxicity of those chemicals. In contrast, studies that compared germ-free and conventional mice illustrated that the presence of gut microbiota enhances the potential of 2-NF to form DNA adduct and mutagens. The discrepancies might be explained by the more complex metabolism of aminofluorene that occurs in vivo, which involves additional systemic metabolism to mutagenic products that are not replicated in vitro. Studies that used simulated in vitro human gut microbiomes reported that gut microbiota can also convert PAHs, such as naphthalene and benzo[a]pyrene, into hydroxylated metabolites that have new estrogenic activity (Van de Wiele et al. 2005). The relative extent of the formation of those metabolites in vivo in the anaerobic environment of the gut compared with metabolic pathways that occur in other organ systems is not clear. Other early work suggests that the microbially mediated hydroxylation of naphthalene observed in vitro might occur through mechanisms different from those observed in vivo (Bakke et al. 1985).
Studies of human and rodent gut bacteria in vitro also show that gut microbiomes have the capacity to modify bioavailability and toxicity of metals in multiple complex ways (Diaz-Bone and Van de Wiele 2009). For example, methyl mercury can be demethylated to elemental form by fecal bacteria, and fecal excretion of mercury after administration of methylmercuric chloride is lower in germfree mice and mice treated with antibiotics than in control mice (Nakamura et al. 1977; Rowland et al. 1980). In humans, the complete methylation of inorganic arsenic to dimethyl arsenic is thought to be a key urinary elimination and detoxification pathway that is catalyzed by methyltransferase activity encoded by the host AS3MT gene, which is polymorphic in human populations (for review, see Hughes et al. 2011; Hall and Gamble 2012). However, in vitro studies that used human gut bacteria show that inorganic arsenic can be reduced and undergo methylation to intermediate forms that are more toxic, including monomethylarsonic and monomethylarsonous acids and other multi-methylated forms (Rowland and Davies 1981; Van de Wiele et al. 2010). Despite the transformations observed in vitro, the contribution of the methylated forms to arsenic toxicity in vivo is not clearly established. As noted by Hughes et al. (2011), the significance of gut microbiome-mediated metabolism of arsenic in human health risk depends on whether the bioavailability of the metabolites is different from that of the parent compounds, and this has yet to be resolved. It is noteworthy that physiologically based pharmacokinetic (PBPK) models for estimating tissue-level arsenic metabo-
___________________
3 2-NF is a common mutagen found in diesel-exhaust emissions and is formed during incomplete combustion processes (Moller 1994; Moller et al. 1994).
lism and dosimetry have been developed for multiple species (El-Masri and Kenyon 2008; Evans et al. 2008). However, the PBPK models do not explicitly distinguish between microorganism-specific metabolism and its influence on biodistribution and host-dependent processes, such as those mediated by gut enterocytes. Including microorganism-specific parameters in PBPK models could provide a framework for quantifying the specific role of the microbiome in modulating the pharmacokinetics of arsenic and would facilitate comparison of effects among species.
TRANSFORMATION OF HOST-GENERATED METABOLITES
Microbially mediated hydrolytic reactions can play important roles in modulating the pharmacokinetics and bioavailability of environmental chemicals. In particular, phase II conjugation reactions mediated by host liver enzymes, which often promote the detoxification and biliary elimination of environmental chemicals and drugs, can in some cases be reversed by microbial hydrolases in the gut. For example, the herbicide propachlor is conjugated with glutathione in the liver, which protects against hepatic toxicity of propachlor. Early studies have reported that the gut microbiota of rats can further metabolize the glutathione conjugates and thus potentially interfere with a key detoxification step (Bakke et al. 1980).
Deconjugation reactions by gut β-glucuronidases promote reabsorption of some drug metabolites, potentially altering pharmacokinetic profiles, toxicity, or efficacy of the parent drugs. A notable example is the colorectal cancer drug irinotecan (CPT-11), which is metabolized to an active ester that is later glucuronidated in the liver and eliminated by biliary excretion to the intestines. Microbial β-glucuronidases in the gut can cleave the glucuronide conjugate and promote enterohepatic recirculation of a parent drug molecule. The increased systemic drug concentrations and extended exposure in the gastrointestinal tract resulting from enterohepatic recirculation are thought to be responsible for gastrointestinal toxicity of CPT-11 observed in some cancer patients (Roberts et al. 2013; Wallace et al. 2015). Similar mechanisms have been associated with common nonsteroidal anti-inflammatory agents, such as indomethacin (Higuchi et al. 2009; Saitta et al. 2014). Intestinal β-glucuronidase activity has also been linked to metabolism of nitrated PAHs, which are common byproducts of incomplete combustion processes (Möller 1994). For example, 2-NF is metabolized after inhalation exposure to hydroxylated nitrofluorenes (OH-NFs) that have increased mutagenic potency. OH-NFs circulate systemically and can be further detoxified and excreted as glucuronide conjugates, but intestinal β-glucuronidase can regenerate OH-NFs and expose the intestine to increased mutagenic risk. In contrast, after oral exposure, 2-NF is reduced to the corresponding amine by intestinal microbiota and acetylated to form acetylaminofluorene, which can undergo further ring hydroxylation to products that have less mutagenic potency and are ultimately excreted. The broader influence of microbial β-glucuronidase activity on the toxicity of environmental chemicals is only beginning to be understood. However, because a wide variety of environmental chemicals might be subject to biliary elimination via β-glucuronidation, interactions with the gut microbiome through this mechanism might be more common than now appreciated.
There is a paucity of information on the potential for gut microbiota to catalyze conjugation reactions similar to that of phase II metabolism in the liver directly, such as glutathionylation, acetylation, and sulfation. However, the gut microbiome favors cleavage reactions that provide substrates for microbial growth (Spanogiannopoulos et al. 2016). A caveat to that observation is that metagenomic sequencing indicates the presence of homologues of phase II genes, such as glutathione S-transferases and N-acetyltransferases in human gut microbiomes, and this finding suggests a potential for such enzymatic activities (Das et al. 2016). Those metabolic pathways play important roles in detoxification and can vary substantially among individuals and human populations, so future research on their potential role in modifying chemical metabolism is warranted.
ALTERATIONS IN EXPRESSION OF HOST-TISSUE METABOLIC ENZYMES
In rodents and humans, metabolism (such as cytochrome P450 activity) is not fully developed at birth but continues to change throughout adolescence and after puberty (Hines 2013). Specifically, biotransformation reactions, including those associated with phase I and phase II metabolism, vary substantially throughout development. For example, substantial differences in protein concentrations and activity of cytochrome P450s (CYP), flavin monooxygenases, sulfotransferases (SULT), glutathione S-transferases, and uridine 5'-diphos-phoglucuronic acid glucuronosyltransferases have been reported in studies of fetal, postnatal, and adult liver tissue (reviewed in Hines 2008), and some members in each enzyme family are influenced by development more strongly than others or differently from others (for example, SULT1A1 vs SULT1E1 or CYP3A4 vs CYP3A7). Thus, a detailed understanding of the developmental events is critical for safe drug development, delivery, and dosing to neonates, infants, and young children: given the critical developmental windows, pharmacovigilance of these groups is essential (Fabiano et al. 2012). Similarly, early-life developmental changes in metabolism might constitute a critical window when risk of adverse responses to environmental chemicals is greatest; that observation is supported by gray baby syndrome, which results from the toxic effects of a lack of liver enzymes in newborns to metabolize the antibiotic chloramphenicol (Knight 1994). Important species and sex differences in the timing and expression of numerous chemical metabolizing enzymes should also be noted (Moscovitz and Aleksunes 2013).
Layered on top of developmental events are genetic influences that are the focus of pharmacogenomicists and their study of people who are poor, intermediate, extensive, and ultrarapid metabolizers identified through genetic screens (Ma and Lu 2011). Despite the extensive body of literature on the developmental and genetic influences on metabolism, gaps in understanding of how metabolism is developmentally regulated remain, and some have suggested that the gut microbiome is an important factor in this development (Selwyn et al. 2015). The discussion below deals largely with the relationship between chemical-metabolism development and the gut microbiome, but interactions between the skin microbiome and the lung microbiome might similarly influence the expression of host genes involved in chemical metabolism. Unlike the gut microbiome and chemical metabolism, however, how the skin or lung microbiome influences metabolism of chemicals has received little attention.
Observations of germ-free rats dating back to the 1960s provided some of the first evidence that the gut microbiota is an important contributor to host liver metabolism (Danielsson and Gustafsson 1959; Björkhem et al. 1970; Eriksson and Gustafsson 1970). Conventionally raised rats excrete much higher concentrations of free or unconjugated steroids (those lacking sulfate) than germfree rats because their gut microbiota has deconjugation enzymes (bile salt hydrolases) that are important for reducing bile salt toxicity (Ridlon et al. 2016). The early reports also provided some of the first evidence of the important role of the gut microbiota in the process of enterohepatic circulation (Dawson and Karpen 2015), a process of signaling and exchange of nutrients, chemicals, and other substances between the small intestine and the liver. Others have demonstrated that colonization of germ-free mice with microbiota derived from conventionally raised mice is associated with important changes in liver gene expression (CYP8b1), particularly through modification of bile acid synthesis (Claus et al. 2011). It is intriguing that the modification of bile acid pools by the gut microbiota regulates the community composition of the gut microbiome and host physiology.
Recent analyses based on comprehensive studies that used RNAseq profiling of the intestinal epithelium and liver show that the gut microbiota indeed contributes to the development and regulation of genes involved in chemical metabolism (Li et al. 2016; Selwyn et al. 2016). Comparison of gene expression from livers of conventionally raised mice and germ-free mice revealed significant differences in the expression of chemical metabolism genes in the liver (expression of 21 genes
increased, and expression of 34 genes decreased under germ-free conditions) (Selwyn et al. 2015). Most notably, CYP3a expression was significantly decreased under germ-free conditions; on colonization of germ-free mice with a probiotic cocktail, CYP3a expression could be restored to levels measured in conventionally raised mice (Selwyn et al. 2016). Those observations are important for two reasons: (1) CYP3a (and CYP2d6) enzymes are important for metabolizing over 50% of known drugs, and (2) regulation of CYP3a expression occurs via the pregnane X receptor, a nuclear receptor that is thought to serve as an important signaling conduit between the gut microbiota and the host (Björkholm et al. 2009). Additional research is needed to understand how the microbiome and its products interact with host nuclear receptors—including peroxisome proliferator-activated receptors α, β, and γ (Nicholson et al. 2005), constitutive androstane receptor (Björkholm et al. 2009), farnesoid X receptor (FXR) (Wahlström et al. 2017a), and the aryl hydrocarbon receptor (Zhang et al. 2015).
Recent developments in understanding FXR function have shed light on how host-gut microbiome interactions in the small intestine regulate gene expression in the liver. FXR is a ligand-activated nuclear receptor that is important for bile acid metabolism and for maintenance of glucose and lipid homeostasis (Gonzalez et al. 2016). Studies comparing germ-free mice and conventionally raised mice have identified FXR as a central mediator of the interactions between the liver, the small intestine, and the gut microbiota (Li et al. 2013; Sayin et al. 2013; Jiang et al. 2015a,b; Parséus et al. 2017; Wahlström et al. 2017b). Specifically, the gut microbiota can modulate liver metabolism by altering the composition of the intestinal bile acid pools (for example, FXR agonists include chenodeoxycholic acid and taurcholic acid, and FXR antagonists include tauro-β-muricholic acid) and thus influence intestinal FXR signaling back to the liver (Wahlström et al. 2017b). That process is critical for regulating bile acid secretion in the liver and uptake in the ileum that the microbiota tightly controls in such a way as to favor optimal growth conditions; strong evidence from rodent and human studies has implicated the gut microbiota–FXR signaling axis as a key contributor to metabolic disease (Gonzalez et al. 2016; Zhang et al. 2016; Xie et al. 2017). Observations from bariatric-surgery patients has provided additional support linking the gut microbiota, the small intestine, and changes in liver metabolism (Kuipers and Groen 2014).
Studies of other models of metabolism—including zebrafish (Danio rerio, Rawls et al. 2004), nematodes (Caenorhabditis elegans, Scott et al. 2017), and fruit flies (Drosophila melanogaster, Combe et al. 2014)—have similarly identified how microbiota colonization activates or contributes to development of chemical metabolism pathways. Important chemical metabolizing enzymes in the cytochrome P450 family were upregulated after microbiota colonization; however, these transcriptional changes were not well conserved and appear to be species-specific. Regardless, the microbiota-dependent upregulation of chemical metabolism genes in the model organisms further supports the evolutionary importance of the host–microbiota interaction in modulating environmental chemical exposures.
INTERINDIVIDUAL VARIABILITY AND MICROBIOME METABOLISM OF ENVIRONMENTAL CHEMICALS
As discussed in Chapter 2, many factors affect the human microbiome and lead to substantial differences in composition. How those compositional differences translate to functional variability in processes that influence the metabolism and disposition of environmental chemicals has received little attention. There are few experimental strategies to evaluate pharmacokinetic variability, and they have relied heavily on culture-based methods, which have limitations in their application to large human cohort studies. More recently, studies that leverage metagenomics sequence databases arising from the Human Microbiome Project have begun to identify microbial gene homologues for major families of chemical metabolism enzymes (Saad et al. 2012; Das et al. 2016). For example, a computational analysis of 397 individual gut metagenomes identified over 800 bacterial genera that
potentially can metabolize environmental chemicals, and it predicted individual variability in the abundance of metabolic enzymes on the basis of geography, age, and average drug use (Das et al. 2016). The authors suggested that differences in abundance patterns imply distinct roles of the microbiome in pharmacokinetic variations observed among individuals and predicted that gut microorganisms could be stratified into three groups on the basis of their capacity to metabolize drugs and environmental chemicals. Although the biologic implications of such genome-enabled strategies await future experimental validations, there is a need to develop similar analyses and databases for predicting environmental-chemical metabolic pathways in microbiomes at other body sites, such as oral, lung, and skin.
FINDINGS
- Although knowledge of how microbiomes modulate the pharmacokinetics and metabolism of environmental chemicals generally lags behind that of drugs, there is compelling evidence of gut microbiome involvement in the metabolic transformation of environmental contaminants in broad chemical classes.
- Research suggests that microbiomes might modulate the exposure–response relationships of environmental chemicals through a few general mechanisms, including regulation of epithelial-barrier permeability, with implications for transport or excretion of chemicals; direct metabolic transformation of environmental chemicals and secondary transformation, such as deconjugation, of host-generated metabolites; and regulation of the expression or activity of endogenous host metabolic pathways (such as in the host liver) via signaling processes involving microbial products. There is also a potential for direct effects of environmental chemicals on the composition of a microbiome itself.
- It is important to note that each interaction conceptually can increase or decrease chemical exposure and that the role of the interactions in modifying human susceptibility to toxicity of environmentally relevant exposures remains largely uncertain.
- Although research has provided important clues regarding microbial transformation of environmental chemicals and vice versa, there are substantial gaps in the understanding of how chemical exposure changes activity or function of a microbiome and of the breadth of potential metabolic pathways of environmental chemicals in a given microbiome.
- The community composition of the microbiome varies widely among species, individuals, and life stages, and how phylogenetic variability translates to functional variability in processes that influence the metabolism and disposition of environmental chemicals has received little attention.
- In vitro experiments have provided important information on microbial metabolism, but caution is needed in interpreting results solely from in vitro studies; the toxicologic significance of microbiome-mediated metabolism of chemicals needs to be evaluated as part of an integrated, multiorgan host response.
REFERENCES
Bakke, J.E., J.A. Gustafsson, and B.E. Gustafsson. 1980. Metabolism of propachlor by the germfree rat. Science 210(4468):433-435.
Bakke, J., C. Struble, J.A. Gustafsson, and B. Gustafsson. 1985. Catabolism of premercapturic acid pathway metabolites of naphthalene to naphthols and methylthiocontaining metabolites in rats. Proc. Natl. Acad. Sci. USA 82(3):668-671.
Benson, A.K. 2016. The gut microbiome-an emerging complex trait. Nat. Genet. 48(1):1301-1302.
Björkhem, I., J.A. Gustaffsson, and S.A. Gustafsson. 1970. Metabolism of steorids in germfree and conventional rats treated with a 3-beta-hydroxy-delta-5-steroid oxidoreductase inhibitor. Eur. J. Biochem. 16(3):557-566.
Björkholm, B., C.M. Bok, A. Lundin, J. Rafter, M.L. Hibberd, and S. Pettersson. 2009. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS ONE 4(9):e6958.
Cerniglia, C.E., P.C. Howard, P.P. Fu, and W. Franklin. 1984. Metabolism of nitropolycyclic aromatic hydrocarbons by human intestinal microflora. Biochem. Biophys. Res. Commun. 123(1):262-270.
Chassaing, B., O. Koren, J.K. Goodrich, A.C. Poole, S. Srinivasan, R.E. Ley, and A.T. Gewirtz. 2015. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519(7541):92-96.
Choi, J.J., S.Y. Eum, E. Rampersaud, S. Daunert, M.T. Abreu, and M. Toborek. 2013. Exercise attenuates PCB-induced changes in the mouse gut microbiome. Environ. Health Perspect. 121(6):725-730.
Claus, S.P., S.L. Ellero, B. Berger, L. Krause, A. Bruttin, J. Molina, A. Paris, E.J. Want, I. de Waziers, O. Cloarec, S.E. Richards, Y. Wang, M.E. Dumas, A. Ross, S. Rezzi, S. Kochhar, P. Van Bladeren, J.C. Lindon, E. Holmes, and J.K. Nicholson. 2011. Colonization-induced host-gut microbial metabolic interaction. MBio. 2(2):e00271-10.
Claus, S.P., H. Guillou, and S. Ellero-Simatos. 2016. The gut microbiota: A major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2:16003.
Combe, B.E., A. Defaye, N. Bozonnet, D. Puthier, J. Royet, and F. Leulier. 2014. Drosophila microbiota modulates host metabolic gene expression vis IMD/NF-kB signaling. PLoS ONE 9(4):e94729.
Coury, D.L., P. Ashwood, A. Fasano, G. Fuchs, M. Geraghty, A. Kaul, G. Mawe, P. Patterson, and N.E. Jones. 2012. Gastrointestinal conditions in children with autism spectrum disorder: Developing a research agenda. Pediatrics 130(Suppl. 2):S160-S168.
Cox, L.M., S. Yamanishi, J. Sohn, A.V. Alekseyenko, J.M. Leung, I. Cho, S.G. Kim, H. Li, Z. Gao, D. Mahana, Z. Rodriguez, A.B. Rogers, N. Robine, P. Loke, and M.J. Blaser. 2014. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158(4):705-721.
Danielsson, H., and B. Gustafsson. 1959. On serum-cholesterol levels and neutral fecal sterols in germ-free rats; bile acids and steroids 59. Arch. Biochem. Biophys. 83:482-485.
Das, A., M. Srinivasan, T.S. Ghosh, and S.S. Mande. 2016. Xenobiotic metabolism and gut microbiomes. PLoS ONE 11(10):e0163099.
Datta, S.S., A. Preska Steinberg, and R.F. Ismagilov. 2016. Polymers in the gut compress the colonic mucus hydrogel. Proc. Natl. Acad. Sci. USA 113(26):7041-7046.
Dawson, P.A., and S.J. Karpen. 2015. Intestinal transport and metabolism of bile acids. J. Lipid Res. 56(6):1085-1099.
Dheer, R., J. Patterson, M. Dudash, K.J. Bibby, D.B. Stolz, S. Shiva, Z. Wang, S.L. Hazen, A. Barchowsky, and J.F. Stolz. 2015. Arsenic induces structural and compositional colonic microbiome change and promotes host nitrogen and amino acid metabolism. Toxicol. Appl. Pharmacol. 289(3):397-408.
Diaz-Bone, R.A., and T.R. van de Wiele. 2009. Biovolatilization of metal(loid)s by intestinal microorganisms in the simulator of the human intestinal microbial ecosystem. Environ. Sci. Technol. 43(14):5249-5256.
El-Masri, H.A., and E.M. Kenyon. 2008. Development of a human physiologically based pharmacokinetic (PBPK) model for inorganic arsenic and its mono- and di-methylated metabolites. J. Pharmacokinet. Pharmacodyn. 35(1):31-68.
Eriksson, H., and J.A. Gustafsson. 1970. Steroid in germfree and conventional rats. Distribution and excretion of labelled pregnenolone and corticosterone in male and female rats. Eur. J. Biochem. 15(1):132-139.
Evans, S., S. Ferrando, M. Findler, C. Stowell, C. Smart, and D. Haglin. 2008. Mindfulness-based cognitive therapy for generalized anxiety disorder J. Anxiety Disord. 22(4):716-721.
Everard, A., C. Belzer, L. Geurts, J.P. Ouwerkerk, C. Druart, L.B. Bindels, Y.Guiot, M. Derrien, G.D. Muccioli, N.M. Delzenne, W.M. de Vos, and P.D. Cani. 2013. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 110(22):9066-9071.
Fabiano, V., C. Mameli, and G.V. Zuccotti. 2012. Adverse drug reactions in newborns, infants and toddlers: Pediatric pharmacovigilance between present and future. Expert Opin. Drug Saf. 11(1):95-105.
Gao, B., X. Bian, R. Mahbub, and K. Lu. 2017. Gender-specific effects of organophosphate diazinon on the gut microbiome and its metabolic functions. Environ. Health Perspect. 125(2):198-206.
Gao, J., L.B. Ellis, and L.P. Wackett. 2010. The University of Minnesota Biocatalysis/Biodegradation Database: Improving public access. Nucleic Acids Res. 38(Database issue):D488-D491.
Gingell, R., J.W. Bridges, and R.T. Williams. 1971. The role of the gut flora in the metabolism of prontosil and neoprontosil in the rat. Xenobiotica 1(2):143-156.
Gonzalez, F.J., C. Jiang, and A.D. Patterson. 2016. An intestinal microbiota-farnesoid X receptor axis modulates metabolic disease. Gastroenterology 151(5):845-859.
Grover, M., and P.C. Kashyap. 2014. Germ-free mice as a model to study effect of gut microbiota on host physiology. Neurogastroenterol. Motil. 26(6):745-748.
Hall, M.N., and M.V. Gamble. 2012. Nutritional manipulation of one-carbon metabolism: Effects on arsenic methylation and toxicity. J. Toxicol. (2012):Art.595307.
Hartmann, P., W.C. Chen, and B. Schnabl. 2012. The intestinal microbiome and the leaky gut as therapeutic targets in alcoholic liver disease. Front. Physiol. 3:402.
Higuchi, K., E. Umegaki, T. Watanabe, Y. Yoda, E. Morita, M. Murano, S. Tokioka, and T. Arakawa. 2009. Present status and strategy of NSAIDs-induced small bowel injury. J. Gastroenterol. 44(9):879-888.
Hines, R.N. 2008. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 118(2):250-267.
Hines, R.N. 2013. Developmental expression of drug metabolizing enzymes: Impact on disposition in neonates and young children. Int. J. Pharm. 452(1-2):3-7.
Hirayama, K., P. Baranczewski, J.E. Akerlund, T. Midtvedt, L. Möller, and J. Rafter. 2000. Effects of human intestinal flora on mutagenicity of and DNA adduct formation from food and environmental mutagens. Carcinogenesis 21(11):2105-2111.
Hsiao, E.Y., S.W. McBride, S. Hsien, G. Sharon, E.R. Hyde, T. McCue, J.A. Codelli, J. Chow, S.E. Reisman, J.F. Petrosino, P.H. Patterson, and S.K. Mazmanian. 2013. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155(7):1451-1463.
Hughes, M.F., B.D. Beck, Y. Chen, A.S. Lewis, and D.J. Thomas. 2011. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 123(2):305-332.
Idle, J.R., and F.J. Gonzalez. 2007. Metabolomics. Cell Metab. 6(5):348-351.
Jiang, C., C. Xie, F. Li, L. Zhang, R.G. Nichols, K.W. Krausz, J. Cai, Y. Qi, Z.Z. Fang, S. Takahashi, N. Tanaka, D. Desai, S.G. Amin, I. Albert, A.D. Patterson, and F.J. Gonzalez. 2015a. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest. 125(1):386-402.
Jiang, C., C. Xie, Y. Lv, J. Li, K.W. Krausz, J. Shi, C.N. Brocker, D. Desai, S.G. Amin, W.H. Bisson, Y. Liu, O. Gavrilova, A.D. Patterson, and F.J. Gonzalez. 2015b. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 6:10166.
Joly, C., J. Gay-Queheillard, A. Leke, K. Chardon, S. Delanaud, V. Bach, and H. Khorsi-Cauet. 2013. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ. Sci. Pollut. Res. Int. 20(5):2726-2734.
Kelly, J.R., P.J. Kennedy, J.F. Cryan, T.G. Dinan, G. Clarke, and N.P. Hyland. 2015. Breaking down the barriers: The gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front. Cell. Neurosci. 9:392.
Knight, M. 1994. Adverse drug reactions in neonates. J. Clin. Pharmacol. 34(2):128-135.
Kuipers, F., and A.K Groen. 2014. FXR: The key to benefits in bariatric surgey? Nat. Med. 20(4):337-338.
Leclercq, S., S. Metamoros, P.D. Cani, A.M. Neyrinck, F. Jamar, P. Stärkel, K. Windey, V. Tremaroli, F. Bäckhed, K. Verbeke, P. de Timary, and N.M. Delzenne. 2014. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 111(42):E4485-E4493.
Li, C.Y., H.J. Renaud, C.D. Klaassen, and J.Y. Cui. 2016. Age-specific regulation of drug-processing genes in mouse liver by ligands of zenobiotic-sensing transcription factors. Drug Metab. Dispos. 44(7):1038-1049.
Li, F., C. Jiang, K.W. Krausz, Y. Li, I. Albert, H. Hao, K.M. Fabre, J.B. Mitchell, A.D. Patterson, and F.J. Gonzalez. 2013. Microbiome remodeling leads to inhibition of intestinal farnesoid X receptor signaling and decreased obesity. Nat. Commun. 4:2384.
Lu, K., R.P. Abo, K.A. Schlieper, M.E. Graffam, S. Levine, J.S. Wishnok, J.A. Swenberg, S.R. Tannenbaum, and J.G. Fox. 2014. Arsenic exposure perturbs the gut microbiome and its metabolic profile in mice: An integrated metagenomics and metabolomics analysis. Environ. Health Perspect. 122(3):284-291.
Ma, Q., and A.Y. Lu. 2011. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol. Rev. 63(2):437-459.
Marchesi, J.R., D.H. Adams, F. Fava, G.D. Hermes, G.M. Hirschfield, G. Hold, M.N. Quraishi, J. Kinross, H. Smidt, K.M. Tuohy, L.V. Thomas, E.G. Zoetendal, and A. Hart. 2016. The gut microbiota and host health: A new clinical frontier. Gut 65(2):330-339.
Maurice, C.F., H.J. Haiser, and P.J. Turnbaugh. 2013. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152(1-2):39-50.
Morgun, A., A. Dzutsev, X. Dong, R.L. Greer, D.J. Sexton, J. Ravel, M. Schuster, W. Hsiao, P. Matzinger, and N. Shulzhenko. 2015. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut 64(11):1732-1743.
Möller, L. 1994. In vivo metabolism and genotoxic effects of nitrated polycyclic aromatic hydrocarbons. Environ. Health Perspect. 102(Suppl. 4):139-146.
Möller, L., M. Zeisig, T. Midtvedt, and J.A. Gustafsson. 1994. Intestinal microflora enhances formation of DNA adducts following administration of 2-NF and 2-AAF. Carcinogenesis 15(5):857-861.
Moscovitz, J.E., and L.M. Aleksunes. 2013. Establishment of metabolism and transport pathways in the rodent and human fetal liver. Int. J. Mol. Sci. 14(12):23801-23827.
Nakamura, I., K. Hosokawa, H. Tamura, and T. Miura. 1977. Reduced mercury excretion with feces in germfree mice after oral administration of methyl mercury chloride. Bull. Environ. Contam. Toxicol. 17(5):528-533.
Nicholson, J.K., E. Holmes, and I.D. Wilson. 2005. Gut microrganisms, mammalian metabolism and personalized health care. Nat. Rev. Microbiol. 3(5):431-438.
Parséus, A., N. Sommer, F. Sommer, R. Caesar, A. Molinaro, M. Ståhlman, T.U. Greiner, R. Perkins, and F. Bäckhed. 2017. Microbiota-induced obesity requires farnesoid X receptor. Gut 66(3):429-437.
Patterson, A.D., and P.J. Turnbaugh. 2014. Microbial determinants of biochemical individuality and their impact on toxicology and pharmacology. Cell Metab. 20(5):761-768.
Peterson, L.W., and D. Artis. 2014. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14(3):141-153.
Phelan, V.V., W.T. Liu, K. Pogliano, and P.C. Dorrestein. 2011. Microbial metabolic exchange--the chemotype-tophenotype link. Nat. Chem. Biol. 8(1):26-35.
Plovier, H., A. Everard, C. Druart, C. Depommier, M. Van Hul, L.Geurts, J. Chilloux, N. Ottman, T. Duparc, L. Lichtenstein, A. Myridakis, N.M. Delzenne, J. Klievink, A. Bhattacharjee, K.C. van der Ark, S. Aalvink, L.O. Martinez, M.E. Dumas, D. Maiter, A. Loumaye, M.P. Hermans, J.P. Thissen, C. Belzer, W.M. de Vos, and P.D. Cani. 2017. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 23(1):107-113.
Rafii, F., W. Franklin, and C.E. Cerniglia. 1990. Azoreductase activity of anaerobic bacteria isolated from human intestinal microflora. Appl. Environ. Microbiol. 56(7):2146-2151.
Rawls, J.F., B.S. Samuel, and J.I. Gordon. 2004. Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc. Natl. Acad. Sci. USA 101(13):4596-4601.
Richards, J.L., Y.A. Yap, K.H. McLeod, C.R. Mackay, and E. Marino. 2016. Dietary metabolites and the gut microbiota: An alternative approach to control inflammatory and autoimmune diseases. Clin. Transl. Immunol. 5(5):e82.
Ridlon, J.M., S.C. Harris, S. Bhowmilk, D.J. Kang, and P.B. Hylemon. 2016. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7(1):22-39.
Roberts, A.B., B.D. Wallace, M.K. Venkatesh, S. Mani, and M.R. Redinbo. 2013. Molecular insights into microbial beta-glucuronidase inhibition to abrogate CPT-11 toxicity. Mol. Pharmacol. 84(2):208-217.
Rowland, I.R., and M.J. Davies. 1981. In vitro metabolism of inorganic arsenic by the gastro-intestinal microflora of the rat. J. Appl. Toxicol. 1(5):278-283.
Rowland, I.R., M.J. Davies, and J.G. Evans. 1980. Tissue content of mercury in rats given methylmercuric chloride orally: Influence of intestinal flora. Arch. Environ. Health 35(3):155-160.
Saad, R., M.R. Rizkallah, and R.K. Aziz. 2012. Gut Pharmacomicrobiomics: The tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog. 4(1):16.
Saitta, K.S., C. Zhang, K.K. Lee, K. Fujimoto, M.R. Redinbo, and U.A. Boelsterli. 2014. Bacterial beta-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: Mode of action and pharmacokinetics. Xenobiotica 44(1):28-35.
Sayin, S.I., A. Wahlström, J. Felin, S. Jäntti, H.U. Marschall, K. Bamberg, B. Angelin, T. Hyötyläinen, M. Orešič, and F. Bäckhed. 2013. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17(2):225-235.
Scheline, R.R. 1968a. Drug metabolism by intestinal microorganisms. J. Pharm. Sci. 57(12):2021-2037.
Scheline, R.R. 1968b. The metabolism of drugs and other organic compounds by the intestinal microflora. Acta Pharmacol. Toxicol. 26(4):332-342.
Scott, T.A., L.M. Quintaneiro, P. Norvaisas, P.P. Lui, M.P. Wilson, K.Y. Leung, L. Herrera- Dominguez, S. Sudiwala, A. Pessia, P.T. Clayton, K. Bryson, V. Velagapudi, P.B. Mills, A. Typas, N.D.E. Greene, and F. Cabreiro. 2017. Host-microbe co-metabolism dictates cancer drug efficacy in C. elegans. Cell 169(3):442-456.
Selwyn, F.P., S.L. Cheng, T.K. Bammler, B. Prasad, M. Vrana, C. Klaassen, and J.Y. Cui. 2015. Developmental regulation of drug-processing genes in livers of germfree mice. Toxicol. Sci. 147(1):84-103.
Selwyn, F.P., S.L. Cheng, C.D. Klaassen, and J.Y. Cui. 2016. Regulation of hepatic drug-metabolizing enzymes in germ-free mice by conventionalization and probiotics. Drug Metab. Dispos. 44(2):262-274.
Shehata, A.A., W. Schrodl, A.A. Aldin, H.M. Hafez, and M. Kruger. 2013. The effect of glyphosate on potential pathogens and beneficial members of poultry microbiota in vitro. Curr. Microbiol. 66(4):350-358.
Sousa, T., R. Paterson, V. Moore, A. Carlsson, B. Abrahamssom, and A.W. Basit. 2008. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 363(1-2):1-25.
Sousa, T., V. Yadav, V. Zann, A. Border, B. Abrahamsson, and A.W. Basit. 2014. On the colonic bacterial metabolism of azo-bonded prodrugs of 5-aminosalicylic acid. J. Pharm. Sci. 103(10):3171-3175.
Spanogiannopoulos, P., E.N. Bess, R.N. Carmody, and P.J. Turnbaugh. 2016. The microbial pharmacists within us: A metagenomic view of xenobiotic metabolism. Nat. Rev. Microbiol. 14(5):273-287.
Spink, W.W., F.W. Hurd, and J. Jermsta. 1940. In vitro conversion of prontosil-soluble to sulfanilamide by various types of microorganism. Proc. Soc. Exp. Biol. Med. 43:172-175.
Suez, J., T. Korem, D. Zeevi, G. Zilberman-Schapira, C.A. Thaiss, O. Maza, D. Israeli, N. Zmora, S. Gilad, A. Weinberger, Y. Kuperman, A. Harmelin, I. Kolodkin-Gal, H. Shapiro, Z. Halpern, E. Segal, and E. Elinav. 2014. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514(7521):181-186.
Thevaranjan, N., A. Puchta, C. Schulz, A. Naidoo, J.C. Szamosi, C.P. Verschoor, D. Loukov, L.P. Schenck, J. Jury, K.P. Foley, J.D. Schertzer, M.J. Larché, D.J. Davidson, E.F. Verdú, M.G. Surette, and D.M. Bowdish. 2017. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21(4):455-466.
Thompson, L.R., J.G. Sanders, D. McDonald, A. Amir, J. Ladau, K.J. Locey, R.J. Prill, A. Tripathi, S.M. Gibbons, G. Ackermann, J.A. Navas-Molina, S. Janssen, E. Kopylova, Y. Vázquez-Baeza, A. González, J.T. Morton, S. Mirarab, Z. Zech Xu, L. Jiang, M.F. Haroon, J. Kanbar, Q. Zhu, S.J. Song, T. Kosciolek, N.A. Bokulich, J. Lefler, C.J. Brislawn, G. Humphrey, S.M. Owens, J. HamptonMarcell, D. Berg-Lyons, V. McKenzie, N. Fierer, J.A. Fuhrman, A. Clauset, R.L. Stevens, A. Shade, K.S. Pollard, K.D. Goodwin, J.K. Jansson, J.A. Gilbert, and R. Knight. 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551:457-463.
Tralau, T., J. Sowada, and A. Luch. 2015. Insights on the human microbiome and its xenobiotic metabolism: What is known about its effects on human physiology? Expert Opin. Drug Metab. Toxicol. 11(3):411-425.
Traxler, M.F., J.D. Watrous, T. Alexandrov, P.C. Dorrestein, and R. Kolter. 2013. Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. mBio 4(4):e00459-13.
Tulstrup, M.V., E.G. Christensen, V. Carvalho, C. Linninge, S. Ahrné, O. Højberg, T.R. Licht, and M.I. Bahl. 2015. Antibiotic treatment affects intestinal permeability and gut microbial composition in wistar rats dependent on antibiotic class. PLoS ONE 10(12):e0144854.
Turnbaugh, P.J., F. Bäckhed, L. Fulton, and J.I. Gordon. 2008. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3(4):213-223.
Turner, J.R. 2009. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9(11):799-809.
Ulluwishewa, D., R.C. Anderson, W.C. McNabb, P.J. Moughan, J.M. Wells, and N.C. Roy. 2011. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 141(5):769-776.
Van Ampting, M.T., A.J. Schonewille, C.Vink, R.J. Brummer, R. van der Meer, and I.M. Bovee-Oudenhoven. 2010. Damage to the intestinal epithelial barrier by antibiotic pretreatment of salmonella-infected rats is lessened by dietary calcium or tannic acid. J. Nutr. 140(12):2167-2172
Van de Wiele, T., L. Vanhaecke, C. Boeckaert, K. Peru, J. Headley, W. Verstraete, and S. Siciliano. 2005. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environ. Health Perspect. 113(1):6-10.
Van de Wiele, T., C.M. Gallawa, K.M. Kubachka, J.T. Creed, N. Basta, E.A. Dayton, S. Whitacre, G. Du Laing, and K. Bradham. 2010. Arsenic metabolism by human gut microbiota upon in vitro digestion of contaminated soils. Environ. Health Perspect. 118(7):1004-1009.
Wahlström, A., P. Kovatcheva-Datchary, M. Ståhlman, F. Bäckhed, and H.U. Marschall. 2017a. Crosstalk between bile acids and gut microbiota and its impact on farnesoid X receptor signaling. Dig. Dis. 35(3):246-250.
Wahlström, A., P. Kovatcheva-Datchary, M. Ståhlman, F. Bäckhed, and H.U. Marschall. 2017b. Induction of farnesoid X receptor signaling in germ-free mice colonized with a human microbiota. J. Lipid Res. 58(2):412-419.
Wallace, B.D., A.B. Roberts, R.M. Pollet, J.D. Ingle, K.A. Biernat, S.J. Pellock, M.K. Venkatesh, L. Guthrie, S.K. O’Neal, S.J. Robinson, M. Dollinger, E. Figueroa, S.R. McShane, R.D. Cohen, J. Jin, S.V. Frye, W.C. Zamboni, C. Pepe-Ranney, S. Mani, L. Kelly, and M.R. Redinbo. 2015. Structure and inhibition of microbiome beta-glucuronidases essential to the alleviation of cancer drug toxicity. Chem. Biol. 22(9):1238-1249.
Williams, R.T. 1972. Toxicologic implications of biotransformation by intestinal microflora. Toxicol. Appl. Pharmacol. 23(4):769-781.
Xie, C., C. Jiang, J. Shi, X. Gao, D. Sun, L. Sun, T. Wang, S. Takahashi, M. Anitha, K.W. Krausz, A.D. Patterson, and F.J. Gonzalez. 2017. An intestinal farnesoid X receptorceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes 66(3):613-626.
Xu, H., T.M. Heinze, S. Chen, C.E. Cerniglia, and H. Chen. 2007. Anaerobic metabolism of 1-amino-2-naphthol-based azo dyes (Sudan dyes) by human intestinal microflora. Appl. Environ. Microbiol. 73(23):7759-7762.
Zhang, L., R.G. Nichols, J. Correll, I.A. Murray, N. Tanaka, P.B. Smith, T.D. Hubbard, A. Sebastian, I. Albert, E. Hatzakis, F.J. Gonzalez, G.H. Perdew, and A.D. Patterson. 2015. Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ. Health Perspect. 123(7):679-688.
Zhang, L., C. Xie, R.G. Nichols, S.H. Chan, C. Jiang, R. Hao, P.B. Smith, J. Cai, M.N. Simons, E. Hatzakis, C.D. Maranas, F.J. Gonzalez, and A.D. Patterson. 2016. Farnesoid X receptor signaling shaptes the gut microbiota and controls hepatic lipid metabolism. mSystems 1(5):e00070-16.















