
4
Current Methods for Studying the Human Microbiome
The human microbiome has evolved to be a remarkably diverse, finely balanced, and highly environment-specific ecosystem (Lloyd-Price et al. 2016). Each body site constitutes a specific habitat that can include trillions of microbial cells and hundreds of strains that differ nearly completely from one site to another throughout the body (HMP Consortium 2012a,b). Using techniques from molecular epidemiology, microbial ecology, and microbiology, researchers have demonstrated that changes in typical immune interactions, biomolecular activities, or pathogen exclusion are associated with such diseases as inflammatory bowel disease, autism, and cancer (Bäckhed et al. 2012; Hsiao et al. 2013; Petersen and Round 2014; Trompette et al. 2014; Garrett 2015). Culture-independent population studies of the human microbiome follow an approach similar to that of Franzosa et al. (2015), which includes collecting stabilized microbial biomass specimens at various times from people who have various exposures or phenotypes of interest; assaying the collected samples with one or more molecular profiling technologies (Segata et al. 2013); bioinformatically profiling the resulting raw data to quantify microbial features of interest, such as species abundances, strains, and biomolecular functional elements; and statistically associating changes in those features with population phenotypes or exposures. That approach is similar to other types of molecular epidemiology studies, such as gene-expression biomarker discovery or genome-wide association studies, and can be combined with experimental approaches that change or challenge the microbiome.
Because of the nature of human microbiome studies, the resulting associations are most often correlative rather than clearly causal; however, additional targeted assays can be used to establish causality and mechanism. The most common targeted assays might involve gnotobiotic transfer of human microbiome samples into controlled model organisms (such as mice) or change-inducing treatments, such as administration of antibiotics, to knock down or alter the composition of the microbiome (Morgun et al. 2015). In addition to transferring whole communities, individual microbial strains that are identified from whole-community profiling can be targeted for isolation (Faith et al. 2010) by using classical microbiology techniques or engineered systems, such as microfluidics. That approach allows the microbial physiology or biochemistry of individual strains of interest (such as secretion products or biomolecular repertoires) to be finely measured and manipulated. Such in vitro systems can be scaled up to include laboratory profiling of entirely synthetic communities, particularly in continuous-culture systems. Detailed properties of host immune sensing and control of a microbiome can be profiled from human tissues (Honda and Littman 2016) by measuring T-cell and B-cell populations, immunoglobulins, cytokine pools, small molecules, and gene expression. The profiling is most often conducted on microbial communities in the gut but can be done for any site-specific community, such as the oral cavity or the skin (Belkaid and Segre 2014). Computational analyses can complement any of the approaches discussed.
Each method for human microbiome profiling—epidemiology, animal, or in vitro studies—has benefits and drawbacks, generally similar to those of other methods in translational molecular research. Human population studies are expensive and difficult to control experimentally at each stage (sample collection, data generation, and data analysis), and they are not generally amenable to interventional studies to establish causality. However, direct measurements of exposures and health risks are possible. Animal models can rarely precisely recapitulate human-associated microbial community structure (Chung et al. 2012), and gnotobiotic facilities can be expensive and difficult to maintain. However, various gnotobiotic systems—including ones that use mice, fish, pigs, and even fruit flies—are now available for modeling different aspects of the human microbiome (Fritz et al. 2013); each can be colonized and perturbed in a targeted experimental manner. In vitro microbial systems, including ones that contain host cells in the microbial culture, have the longest history, are widely available, and present one of the most controllable environments for mechanistic and molecular profiling. However, continuous culture of many anaerobic organisms presents challenges, and in vitro systems are physiologically the least relevant.
This chapter continues the discussion and provides greater detail on the approaches and methods used today to study the human microbiome. The discussion is divided into three parts. First, systems for studying the human microbiome are described; aspects of sampling the human microbiome are considered, and then animal models, engineered in vitro and ex vivo systems, and culture systems are described. Second, technologies for assaying the microbiome—nucleotide sequencing, other molecular profiling techniques, and direct observation methods—are addressed. Third, methods and approaches for analyzing the data are discussed. The chapter concludes with a discussion of strengths, weaknesses, and gaps in the technologies.
SYSTEMS FOR STUDYING THE HUMAN MICROBIOME
Considerations in Sampling the Human Microbiome
The first step in a microbiome study typically involves the collection of stabilized microbial biomass specimens that will be used and analyzed in various assays. Each sampling method for human-associated microbial community types has strengths and weaknesses that are driven by the dramatically different microbial ecologies in or on the body. The methods that have been established for gathering a sample of sufficient biomass (referring to the quantity of microorganisms needed for an assay) for each major body site are described here, and limitations of each approach are noted.
The gut microbiome is most commonly sampled from stool, which represents well the microbial community of the colonic lumen and to a smaller degree that of the small intestine (Yasuda et al. 2015). Stool is easily obtained for sampling, has extreme microbial density and minimal human genetic contamination (HMP Consortium 2012a,b), and contains material that can be assayed with a variety of molecular techniques. Because microbial characteristics can change rapidly with environmental conditions (such as a sudden decrease in temperature and exposure to air), it is important to take steps to preserve samples by, for example, immediately freezing them or using various laboratory protocols and commercial kits to fix them (Franzosa et al. 2014; Song et al. 2016). It is possible to culture many microorganisms from frozen stool samples, whereas fixatives typically kill microorganisms (preventing culture) and might not be compatible with conducting some molecular assays at a later time. Fixatives do, however, allow convenient collection and shipping of samples. In a clinical setting, mucosal biopsies are common and provide a more precise and biogeographically resolved snapshot of the mucosally associated microbial community (Morgan et al. 2012), but they are more challenging to obtain and can be assayed only with technologies that are not affected by
the presence of human cells in the sample. Other sample types, such as mucosal brushing or rectal swabs, are also possible but are less well studied with respect to protocol consistency and community representation (Tong et al. 2014).
Skin sampling is limited primarily by the low microbial biomass that is found on typical surfaces. The moist, dry, and sebaceous sites across the body can have substantially different ecologies that are difficult to differentiate without detailed profiling (Grice and Segre 2011). Swab sampling is easiest but retrieves the smallest biomass, and microbial adhesion can be surprisingly affected by the type and material of swabs used (Aagaard et al. 2013). A combination of razor scraping and swabbing is the most practical for retrieving samples with greater biomass but requires training and care to perform safely (Oh et al. 2014). Biopsies obtain the greatest microbial and human biomass and, as in the gut, are typically amenable only to assays that are not affected by the inclusion of human genetic material. However, skin microbiome samples in general are often characterized as having high human nucleotide fractions—as much as about 90% of the sample (HMP Consortium 2012a,b)—and require more extensive sequencing and care during analysis. Because of the low biomass of skin microbiome samples and the challenges associated with collecting them, assays that evaluate skin microbiome samples must include special consideration of negative controls to ensure appropriate interpretation of sampling results (Oh et al. 2014).
Similar issues are encountered in connection with sampling methods for the respiratory microbiome. Clinically, the respiratory tract is divided into the upper and lower regions relative to the epiglottis; each region experiences different exposures to the external environment and has different mucosal-epithelial barrier properties (Wolff 1986). Given the variation, it is not surprising that different sampling approaches can provide different readouts and information. Varied clinical approaches and sampling tools have been used to obtain material from the nasal passages, sinus cavities, oral cavity and pharyngeal region, and the tracheobronchial tree. Although surgical specimens, such as those collected during sinus surgeries or from explanted lungs, offer the greatest opportunity for detailed sampling, less invasive approaches are necessary for larger studies (Perez-Losada et al. 2016; Dickson et al. 2017). Swabs, aspirates, sputum, lavage, and brushings have all been used in respiratory microbiome studies. Swabs—most often used to sample the upper respiratory tract—recover different amounts of material compared with aspirates, sputum, lavage, and brushings. Sputum can be spontaneous or collected via induction protocols, such as inhalation of hypertonic saline. Aspirates tend to collect secretions already present, whereas lavage involves instillation of saline into an airway passage and withdrawal of the fluid with suction. In the lungs, the volume returned from bronchoalveolar lavage can be highly variable and depend on disease state; for example, less volume is returned in cases of severe obstruction or emphysema. Thus, measurements based on lavage fluid need to consider dilution as a factor. Small brushes can also be inserted to obtain cells and secretions from the mucosal surface, but care is required to perform this method. Finally, as above with the skin, the respiratory tract is less microbially dense, and it is essential to use protocols that have carefully controlled elements to minimize sample contamination by nontarget tissue (Charlson et al. 2012; Salter et al. 2014; Lauder et al. 2016). Such elements include proper staff training; preparation of work materials, surfaces, and instruments; and collection of controls, including within-subject biologic controls (such as paired upper-airway and lower-airway samples) for accurate interpretation of microbial sequence data.
All human microbiome sampling protocols are sensitive to batch effects—technical, not biologic, differences that arise from many stages of the sampling and data-generation process (Salter et al. 2014). Such effects can make data from multiple studies difficult to compare and, in the worst case, can introduce subtle differences that result in misleading conclusions. Gross differences in population structure, geography, or environmental conditions can change measured microbial communities. Differences in how samples are collected and processed can strongly influence microbiome
assays. Differences in the protocols used to assay the samples can obfuscate biologic effects. And differences in data handling, quality control, and taxonomic, functional, or molecular profiling techniques can contribute to unwanted technical artifacts (Sinha et al. 2015). All those factors are important considerations during study design and data analysis when one dataset is compared with others (Sinha et al. 2017). It is difficult today to compare multiple microbiome datasets reliably because not all datasets can be combined. To enhance comparability, research programs need to make every effort to standardize protocols in advance, run cross-protocol controls throughout, and statistically meta-analyze any remaining systematic differences between datasets.
Understanding the Human Microbiome by Using Model Organisms
Insights into the microbiome and its interactions with human hosts and their chemical environments can be obtained or refined by using diverse nonhuman model systems. Although no nonhuman model system will fully recapitulate all aspects of the human microbiome, each has distinctive strengths that can be leveraged selectively to address scientific questions that would be difficult or impossible to answer with human studies alone. Overall, nonhuman models provide valuable opportunities to gain insights into molecular pathways, physiologic processes, host microbial genotypes, and microbial–chemical stimuli that might be relevant and translatable to the human microbiome and human health.
Animal models are widely used to investigate the human microbiome for several reasons. First, it is much easier to manipulate animal models than human subjects experimentally. Animal studies allow the careful control of experimental variables, scalability, and reproducibility that is often impossible in human studies. Second, ecologic and physiologic attributes of the animal body are highly complex and dynamic and cannot be comprehensively recapitulated in in vitro or in silico models. Finally, the common ancestry of humans and other animals has resulted in the conservation of many genomic, molecular, cellular, and physiologic traits across animal lineages and allows many (not all) findings derived from animal studies to be extrapolated to humans. The advantages of using animal models are counterbalanced by important caveats, including salient differences among animal lineages in anatomy, physiology, and microbiomes (Ley et al. 2008). Although the caveats might limit the relevance of animal models for understanding some aspects of the human microbiome, animal models are important in the larger field of microbiome science.
Several fundamental experimental strategies can be used to study microbiomes in animal models. First, animals can be used to test whether the microbiome composition and function correlate with such variables as host age, host genotype, host body site, diet, and chemical or other experimental exposures. The experiments are typically performed on laboratory or wild animals that are colonized by complex microbial communities. Second, animals can be used to study the effects of the presence or composition of a microbiota on host phenotypes. To test whether microbiome composition contributes to host phenotypes, animals with an intact microbiome can be treated with broad-spectrum antibiotics to reduce microbial abundance and alter community composition. That is a relatively inexpensive and rapid way to disrupt the microbiome, but its disadvantage is that it does not distinguish between the effects induced by loss of antibiotic-sensitive microorganisms, by the remaining antibiotic-resistant microorganisms, or directly by the antibiotics (Morgun et al. 2015). Third, another inexpensive and rapid approach for testing the effect of a particular microbial community or strain is to introduce it directly into conventionally reared animals that are already colonized with a microbiota (a probiotic or super-colonization approach). Introduction can also be achieved by co-housing animals that initially contain distinct microbial communities or strains. However, introduction of microorganisms to compete with the pre-existing microbiota and establish stable colonization has had a low success rate and has resulted in considerable variation in experimental outcomes.
The effect of the presence or composition of a microbiota on host phenotypes can be addressed with substantial experimental control by using gnotobiotic animal models. As noted in Chapter 1, the term gnotobiotic refers to an animal that has no microorganisms (a germ-free animal) or an animal whose composition of associated microorganisms is fully defined by experimental methods. Germ-free animals can be colonized with microbial communities or strains of interest and then evaluated to assess effects on the host. The donor microbial communities can be derived from various sources; “humanized” animal models that are more relevant to the human condition are produced when a human source is used (Ridaura et al. 2013). Although gnotobiotic animal models provide strong experimental control, they are accompanied by distinct challenges and caveats, such as the relatively high cost and labor needs of gnotobiotic-animal facilities; developmental, immunologic, and physiologic anomalies of gnotobiotic animals; and augmented nutritional requirements of gnotobiotic animals (Falk et al. 1998).
The different experimental approaches described above have been used in a broad array of animal species, including mice, zebrafish, fruit flies, and Caenorhabditis elegans. Each species has a unique set of characteristics related to its relative size; transparency; microbiome complexity, composition, and function; genetic variance; and evolutionary distance from humans (Leulier et al. 2017). For example, using mice offers some advantages, such as powerful genetic resources that include inbred lines to reduce the effect of genetic variability, an extensive array of knockout strains, and their relatively close evolutionary distance and physiologic similarity to humans. But the disadvantages of using mice include the difficulty of in vivo imaging and the relatively high cost and low scalability of gnotobiotic and conventional husbandry. In contrast, zebrafish have such advantages as facile in vivo imaging owing to their optical transparency, small size that permits genetic and chemical screens, and scalable and inexpensive husbandry requirements that are easily adjusted for gnotobiotic methods. But the disadvantages of using zebrafish instead of mice include greater evolutionary distance from humans and smaller overlap in bacterial taxa1 in their microbiomes (Rawls et al. 2006; Hacquard et al. 2015). For all those animal models, best practices are emerging to promote interpretability and reproducibility of experimental results, partly by accounting and controlling for interfacility and interindividual variation in microbiome composition (Macpherson and McCoy 2015; Stappenbeck and Virgin 2016).
Engineered Systems for Studying Host–Microbiome Interactions In Vitro and Ex Vivo
Using in vitro and ex vivo experimental systems for studying host–microbiome interactions allows greater manipulation of experimental conditions and increased ability to examine interactions that are too complex to study in vivo. As defined in Chapter 1, the terms in vitro and ex vivo differ mainly in the source of the samples being used in the assay. Both require the use of an artificial setting for conducting an experiment: in vitro systems typically rely on such samples as cell lines or laboratory microbial strains whereas ex vivo systems typically rely on samples that are directly isolated from a host organism. The main systems currently in use for in vitro and ex vivo cultures that examine host–microbiota interactions include co-culture of microorganisms with or without host primary epithelial cells, tissues, or cell lines; microfluidic co-culture of microorganisms with or without engineered tissue; and organoid2 culture. Those systems are used primarily to examine bidirectional signaling between microorganisms or between target host tissue or cell types and a body-site microbiome. Perhaps central among the challenges of using the systems in an artificial setting is the propensity of microbial cultures to become ecologically imbalanced, with components either
___________________
1 A taxon (plural, taxa) is a taxonomic group of organisms, such as a family, genus, or species.
2 An organoid is “an in vitro 3D cellular cluster derived exclusively from primary tissue, embryonic stem cells, or induced pluripotent stem cells, capable of self-renewal or self-organization, and exhibiting similar organ functionality as the tissue of origin” (Fatehullah et al. 2016).
dying or overgrowing and preventing the cell-culture system from reflecting the in vivo community accurately. Although the following discussion focuses on gut-centric applications, analogous systems exist for the lung and, to a lesser extent, for the skin.
In the context of studying the gut microbiome, polarized epithelial monolayers are grown from primary or immortalized small intestinal or colonic cells on transwell membranes3 (Kauffman et al. 2013; Moon et al. 2014) or three dimensional scaffolds (Chen et al. 2015), and microorganisms are seeded on the apical face. Changes in the quality of the epithelial layer can be measured by assessing permeability, transmembrane resistance (used to measure how tightly connected neighboring cells are), active transport, absorption, and excretion. Miniaturization of culture systems to microliter or nanoliter scales renders them amenable to microfluidic manipulations, such as isolation of single bacterial cells from complex microbial communities and their study with imaging, gene-expression profiling, or mass spectrometric readouts (Ma et al. 2014a,b). Limitations of those techniques include their lack of secondary epithelial structures, such as villi and crypts; the absence of additional epithelial-cell subtypes, such as goblet, endocrine, and immune cells; the lack of mucus layers between host and microbial cells; and the difficulty of incorporating realistic multiorganism microbial-community components.
Some limitations are overcome by building structured epithelial layers with microfluidic and tissue-engineering approaches. Gut-on-a-chip technology uses microfluidic platforms to grow intestinal epithelial cells and mimic the movement of fluids through the gut (Kim and Ingber 2013); this promotes the formation of intestinal-tissue structures with specialized cell types, such as absorptive, goblet, enteroendocrine, and Paneth cells. The structures exhibit barrier properties, including mucosal linings and peristaltic motion. Continuous movement of fluids can enable persistent microbial microcolonization as a continuous-culture system (Kim et al. 2012, 2016). Limitations include the need for customized chip fabrication, specialized equipment and technical expertise, and difficulties in introducing diverse microbial components. Furthermore, the technology has thus far been tested and used only with immortalized cell lines and does not account for varied host genetics.
Growth of intestinal organoids, spheroids, or “mini-guts” is relatively accessible compared with that of microfluidic approaches and allows personalized organoid lines from different clinical donors or animal models to be generated. Several protocols have been developed and generally introduce specialized factors into cell-culture media to differentiate embryonic or induced pluripotent stem cells into clusters of villous epithelia or equivalent differentiated cell clusters of other body sites, such as the lung (Wilson et al. 2015; Nigro et al. in press). However, studying microorganisms in organoids requires careful microinjection into each cluster. Furthermore, the enclosed structures and lack of physiologic flow can result in rapid disruption of injected microorganisms, and this limits experimentation to relatively short timescales.
Microfluidic and organoid culture systems reproduce epithelial structures and various differentiated cell subtypes but typically lack integrated immune, muscle, and neuronal cells that are important for many host-microorganism interactions. No in vitro system faithfully captures all those elements in a unified technology. However, ex vivo culture systems can enable careful control of microbial colonization, luminal perfusion, and chemical exposures (Roeselers et al. 2013). Intestinal tissues can be isolated from model organisms and maintained in ex vivo culture for short durations. Chemicals, microorganisms, or both can be introduced into the systems particularly in combination with perfusion methods; this approach yields physiologic or molecular readouts that in the best cases closely mimic their in vivo counterparts. However, they have not yet been extensively explored to support multimicrobial model communities. As a technical intermediate between animal and culture-based models, ex vivo systems
___________________
3 Transwell membranes are inserts that can be placed inside a standard tissue-culture dish that has a permeable membrane on which the cells sit; this arrangement allows separation of the area above the cells (the apical face) and the area below the cells (the basolateral face). When cells are growing under ideal conditions, the cells control the passage of solutes between the two areas.
trade controllability for model accuracy. Advances in the development of parallel ex vivo multiculture systems that have increased experimental control and prolonged culture times are being explored.
Analogous tools are available to study host–microbiome interactions in the respiratory tract. Primary airway epithelial cells and cell lines are well-established tools in respiratory-disease research, but their application to study microbiota interactions has been limited. Recently, microfluidic platforms and organoid culture models for studying respiratory biology have been developed (Dye et al. 2015; Benam et al. 2016a,b). The former include lung-on-a-chip and small-airway-on-a-chip technologies that parallel the gut-on-a-chip platform. Substantial advances have also been made in ex vivo lung-perfusion models (animal and human), which are being used to conduct translational research on lung diseases. The ex vivo perfusion techniques now available have been so successful that clinical studies are investigating their use as a preservation method for donor lungs in human lung transplantation (Nelson et al. 2014; Tane et al. 2017).
Synthetic models of the skin microbiome are likewise in early development. One recent medium-throughput model system of the human stratum corneum (outermost skin layer) that uses collected sloughed human cells was used to evaluate survival of skin pathogens and commensals (van der Krieken et al. 2016). A commercial three-dimensional in vitro skin model is also available and can be populated with human skin microbiota and used to evaluate the effects of chemical exposure on skin colonization (Bojar 2015). These systems do not yet cover the diversity of microbial biochemical environments on skin, nor has their microbial suitability or modeling accuracy been ascertained.
Overall, the in vitro and ex vivo systems for examining host–microbiota interactions vary in experimental throughput, physiologic relevance, and experimental control. Conventional co-culture with primary epithelial cells or cell lines enables moderate experimental throughput that can be precisely controlled and manipulated. Microfluidic and engineered tissue systems are relatively high-throughput with potentially moderate physiologic relevance but require more technical infrastructure and are harder to manipulate. Organoid cultures offer moderate experimental throughput, moderate to high physiologic relevance, and moderate experimental control, whereas ex vivo perfusion systems are low-throughput and highly physiologically relevant and therefore offer more moderate control.
Culture Systems for Characterizing the Human Microbiome
The longest-standing in vitro technique for studying host-associated microorganisms is microbial culture. In tandem with the rise of culture-independent profiling, culture-based techniques have been refined to capture a wider array of organisms from the human microbiome than previously possible, including anaerobes and nonbacterial members, under ever more accurately controlled conditions. Bioreactors that contain microbial cultures, for example, can be used to test specific hypotheses about microorganism–microorganism interactions, microbial production of metabolites, microorganism–chemical transformations and kinetics, and effects of chemicals on microbiome structure and function. Studying microorganisms without the host component has several advantages: the system has increased reproducibility, microorganism–microorganism interactions can be studied in a more defined way, environmental conditions that affect microbiome composition and interactions can be easily controlled, and microbial biotransformations and metabolites can be precisely identified.
Studies have used bioreactors to simulate gut microbial communities to learn more about fermentation processes (Miller and Wolin 1981), biofilm formation (McDonald et al. 2015), and microbial-community responses to perturbations resulting from exposure to antibiotics (McDonald et al. 2015), nanoparticles (Taylor et al. 2015), metabolites from polyphenol transformations (Gross et al. 2010), and polycyclic aromatic hydrocarbons and polybrominated diphenyl ethers (Cui et al. 2016). For any culturing technique to be successful, knowledge of optimal environmental con-
ditions for the desired microorganism is required. Important conditions include pH, oxidation–reduction potential, temperature, and nutrients (Browne et al. 2016; Lagier et al. 2016; Lau et al. 2016). Microorganisms cultured from the human gut have been used to test biotransformations of specific pollutants, such as Eubacterium limosum metabolism of the insecticides methoxychlor and DDT (Yim et al. 2008).
Although development of in vitro host-microbiome simulator devices or bioreactors is in its infancy, several devices have found their way into basic and translational research. First, the simulator of the human intestinal microbial ecosystem (SHIME) (Van den Abbeele et al. 2012) is a model of the small and large intestines that contains stable and functional microbial communities similar to those found in the human (Joly et al. 2013). It is one of the earliest types of linked continuous culture systems that mimic the human digestive tract microbiome by controlling compartmentalization, nutrient availability, pH, and other environmental conditions. Another version of the SHIME model is the mucosal SHIME (M-SHIME); it permits the study of mucosa-associated microorganisms (Van den Abbeele et al. 2012). A simpler model is the minibioreactor array, which, unlike the SHIME model, is amenable to high-throughput screening, although it does not model multiple regions of the gastrointestinal tract (Auchtung et al. 2015).
Recent advances in culturing techniques that have been enhanced by sequencing and metabolomics techniques have increased the percentage of host-associated cultivable microorganisms (Browne et al. 2016; Lagier et al. 2016; Lau et al. 2016). As noted, however, culture conditions are critical. And as expected, culture outcomes are affected by collection and storage procedures and such factors as oxygen exposure, potential microbial growth, and changes resulting from freezing and thawing (Lau et al. 2016). Using selective culture media and choosing appropriate environmental conditions are critical for success. For example, a combination of anaerobic and microaerobic4 conditions at the correct pH is needed to isolate gut microorganisms. Isolation of anaerobes requires oxygen depletion in the media and airspace of the culture chamber and defined growth requirements, such as specialized media and targeted nutrient supplementation. Other challenges are the existence of syntrophic (mutually dependent) relationships, and the presence of many microorganisms in the host as a biofilm that is difficult to replicate externally. Special culture methods—such as the roll tube method in which the culture medium is rolled inside a test tube until it forms a thin film around the internal wall of the tube and methods that use soft agar plates in which the culture medium has a lower concentration of gelatin, which allows the detection of mobile microorganisms—can be used to encourage the growth of difficult microorganisms further (Dickson et al. 2017). Microfluidic devices that allow droplet separation and sequencing in tandem have been developed and used to isolate gut microorganisms that were previously considered uncultivable (Leung et al. 2012; Brouzes et al. 2015), and a microfluidic streak plate platform has been developed to facilitate cultivation of dominant and rare species in a microbial community (Dickson et al. 2017). Such novel platforms will allow physiologic microbial characterization and help to decipher the important roles of individual microorganisms, including their possible biotransformation pathways.
As noted above, there are clear advantages of studying microbial cultures and isolates that use the systems described. However, there are also some disadvantages: the host is not considered, syntrophic interactions are difficult to replicate, cultures or isolates rarely capture the physical structure of biofilms or other structured communities, enrichment and isolation techniques are often lower-throughput than molecular techniques, ideal culture conditions are not always known for many microorganisms of interest, and they can require more diverse expertise or facilities than do molecular techniques.
___________________
4 A microaerobic environment is one in which the oxygen concentration is lower than that found under standard atmospheric conditions.
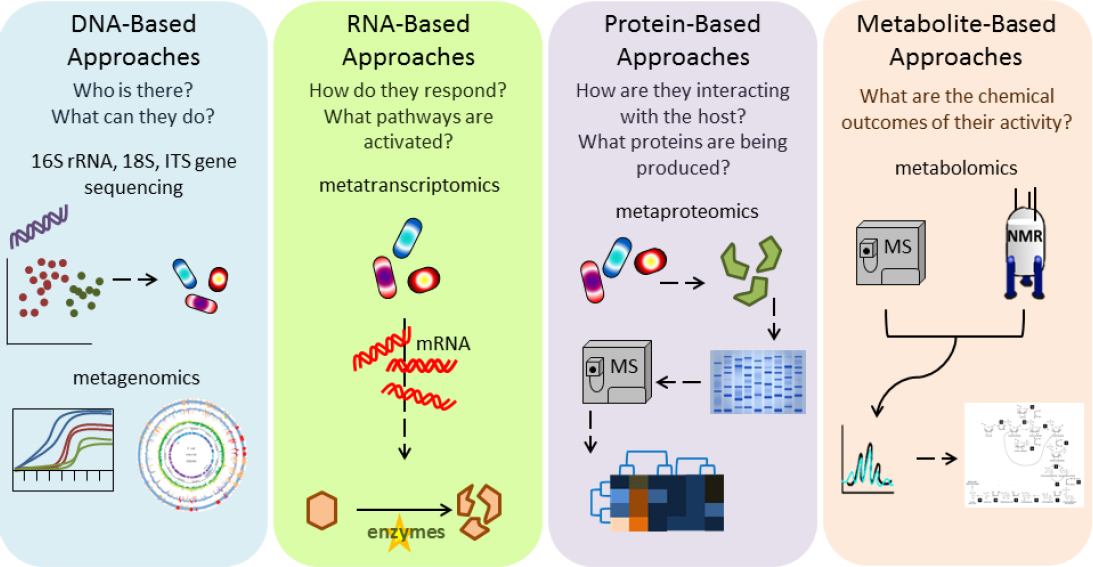
TECHNOLOGIES FOR ASSAYING THE MICROBIOME
Assaying the microbiome as described above can use various technologies as highlighted in Figure 4-1. The following sections describe nucleotide sequencing of DNA and RNA, other molecular profiling techniques, and methods for direct observation of the human microbiome.
Nucleotide Sequencing
The decreasing cost and increasing accessibility of nucleotide sequencing unquestionably boosted human-microbiome studies in population health, and it is still the primary tool used to study the microbiome (Franzosa et al. 2015). One of the earliest and most widespread techniques is amplicon5 sequencing, in which a single genomic locus is targeted for polymerase chain reaction (PCR) amplification; the chosen locus must be largely conserved throughout microorganisms of interest but contain sufficient variation to allow distinction of individual strains or species. Resulting PCR products are sequenced and compared with known reference sequences in a database. Amplicon sequencing most commonly targets the 16S rRNA gene (Hamady and Knight 2009), which is almost universal among bacteria, whereas the 18S rRNA gene and internal transcribed spacer (ITS) sequence variants are increasingly common for eukaryotic profiling6 (Findley et al. 2013). The meth-
___________________
5 An amplicon is a segment of DNA or RNA that is amplified during a replication event in the cell or during a polymerase chain reaction.
6 The 18S rRNA gene sequence variants are particularly well-suited for broad-spectrum assays, and the ITS sequence variants are particularly well suited for fungi.
ods rely on conserved targets of the PCR primers that are adjacent to sequences that are sufficiently variable to differentiate organisms of interest. As the price of sequencing technologies have decreased, whole-community metagenome sequencing of arbitrary short reads has become more common and today can provide billions of sequence reads (many gigabases) per community. Practical methods have also recently been developed to apply long-read metagenomic sequencing to RNA metatranscriptomes7 in the human microbiome (Franzosa et al. 2014), and protocols that use long-read high-throughput sequencing (Tsai et al. 2016) and single-cell sequencing (Gawad et al. 2016) are also emerging.
Amplicon sequencing, metagenome sequencing, and metatranscriptome sequencing have different strengths and weaknesses. All are sensitive to the specific protocols used for nucleotide extraction from samples, which requires care to avoid biasing experimental results. Microorganisms vary in their sensitivity to the reagents used for the extraction of genomic material, so researchers must be cautious to avoid destroying sensitive subsets of microorganisms while still extracting genomic material from more hardy or resistant organisms. If RNA is the desired genetic material, extra caution will be needed to avoid destroying the RNA during sample processing. Amplicon sequencing can be inexpensively carried out by using samples that have extremely low microorganism biomass or mixed samples that have, for example, substantial human or other nonmicrobial nucleotides (Hamady and Knight 2009). However, it provides information on only a relatively small region of a single gene. In most cases, that information is sufficient to generate taxonomic or phylogenetic profiles at about genus-level resolution. In some cases, more careful analysis makes it possible to get species-level or strain-level information. Amplicon sequencing can be highly sensitive to the details of amplification, primer composition, polymerase enzyme, and the PCR program (Gohl et al. 2016).
Shotgun metagenomics (a nontargeted sequencing process) can readily resolve species-level and strain-level classification and provide genome content, functional potential, and some genome assembly for organisms of even modest abundance. However, it remains more expensive than amplicon sequencing, it is less tolerant of low biomass or contaminated samples, and it requires substantially more complex and computationally expensive analytic approaches.
Metatranscriptomics is in its infancy. In addition to being more expensive because of challenging protocols and the scarcity of computational tools, it is not yet established in which environments or for which health-relevant phenotypes microbial community transcription will prove to be most informative (Franzosa et al. 2014).
Finally, most molecular techniques do not differentiate between current molecular activity (living microorganisms) and previously generated biomolecular pools (dead microorganisms), but those distinctions can be resolved better with culture-based or direct observation methods.
Other Culture-Independent Molecular Profiling Techniques
Metabolomic and metaproteomic techniques that use mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectrometry are among the most prevalent non-sequencing-based, culture-independent approaches to molecular profiling of the human microbiome. To date, MS-based and NMR-based profiling has been used to identify secreted and intracellular microbial products and metabolites, including fatty acids, vitamins, bile salts, and polyphenols. As a subset of the metabolome, lipids from microbiome samples have been profiled with MS-based detection methods after lipid extraction and separation. MS-based detection methods can be used after protein extraction and fractionation by two-dimensional electrophoresis or isotope tagging to profile metaproteomes from microbiome samples. Those approaches enable the quantification of cellular proteins from microbial cells and their post-translational modifica-
___________________
7 A metatranscriptome is the entirety of the RNA sequences expressed by the microbiome as identified by sequencing.
tions as the direct functional products of microbial metatranscriptomes and metagenomes (Kolmeder and de Vos 2014; Soufi and Soufi 2016). Emerging technologies for localized or in situ metabolomics profiling with such approaches as MS imaging, topographic mapping, and rapid evaporative ionization MS coupled with surgical diathermy devices enable spatial resolution of metabolic profiles within the microbial-community structures (Rath et al. 2012; Bouslimani et al. 2015; Golf et al. 2015).
Various platforms for targeted or untargeted metabolomic surveys and quantification of small molecules from biofluids include gas chromatography, liquid chromatography, capillary electrophoresis coupled with MS, Fourier transform infrared spectroscopy, and NMR spectroscopy (reviewed in Smirnov et al. 2016; Vernocchi et al. 2016). The methods differ in how specifically they can identify analytes, how well analytes can be distinguished, how sensitive the methods are to low molecular concentration, and their dynamic range of detectable molecules, data acquisition speed, and technical complexity of protocols.
MS-based profiling and NMR-based profiling are powerful tools for evaluating metaproteomic and metabolomic functional outputs of microbial activity and host–microorganism interactions. A primary advantage of those techniques over nucleic-acid–based microbiome profiling is the potential to identify microbial molecules that mediate microorganism–microorganism and host–microorganism signaling. However, methodologic limitations include the need to tailor sample preparation to target molecules and the inability to identify a wide array of molecule types simultaneously with a single sample-collection, handling, and preparation protocol. Furthermore, resources for determining accurate molecular identities and for differentiating between host-derived and microbially derived molecules are lacking. Further methodologic, technologic, and resource development is needed to create standardized protocols for metaproteomic and metabolomic profiling of microbiomes.
Direct Observation of the Human Microbiome
Most microbiome analyses have focused on DNA or RNA sequencing or metabolomic analyses, but useful insights into microbiome composition, function, and spatial organization can be gained by using a variety of imaging technologies. Transmission and scanning electron microscopy can be used to visualize microbial community organization in fixed samples but is not well suited to resolving individual taxa or traits in a complex community. Fluorescence in situ hybridization (FISH) can be used to evaluate the taxonomy, location, and organization of microbial community members in fixed microbiome samples. In the FISH method, fluorescently labeled DNA probes that recognize a gene sequence within targeted microbial taxa are hybridized to a fixed intact microbiome sample and imaged to visualize the location of the microbial cells that contain the corresponding DNA sequence with micrometer resolution. It can be performed with probes that recognize single taxa or multiplexed to target diverse taxa in a single sample (Earle et al. 2015; Mark Welch et al. 2016). Fluorescence-activated cell sorting can be used similarly to quantify and sort microbial cells that are dissociated from a microbiome sample and that display a phenotype that is detectable with a fluorescent marker, such as an exogenous fluorescent probe or genetically encoded fluorescent protein (Maurice et al. 2013; Ambriz-Aviña et al. 2014).
The above methods require fixation or dissociation of a microbial community, but other methods can be used to visualize microbial location and behavior in live animals. In mice, microbial taxa engineered to encode fluorescent reporter proteins can be visualized, although spatial resolution is low because of the opacity of host tissues (Wiles et al. 2006). In contrast, the optical transparency of the zebrafish permits high-resolution and longitudinal in vivo imaging of microbial cell location and behavior (Rawls et al. 2007; Jemielita et al. 2014) and location of nutrients (Semova et al. 2012).
When viable microbial community samples are available,8 their physiology can also be directly evaluated with enzymatic assays, which can measure growth (such as changes in optical density), colony (or microcolony) structure, or metabolic activity (such as pH or oxygen use). Direct enzymatic activity screens are more challenging to apply to microbiome samples but are practical in assessing the physiology of individual isolates from the microbiome that can be cultured (Tasse et al. 2010; Cohen et al. 2015; Koppel and Balscus 2016). There are high-throughput platforms for enzymatic assays (Jiang et al. 2015; Kaiko et al. 2016; Biggs et al. 2017), but they are not as well developed as high-throughput molecular profiling assays.
Finally, genetic screens and modifications can be used to observe microbial communities. Functional metagenomics (Lam et al. 2015) uses phenotypic screens that generally involve isolating large DNA fragments from a microbiome and generating a library of clones in a species, such as Escherichia coli, that lacks the function of interest. The library of clones can then be cultured under selective conditions, for example, with antibiotics. Assaying for a desired trait, such as antibiotic resistance or enzymatic activity, can identify the DNA sequence fragments that confer the trait and can potentially identify the microbiome member that encodes the given trait. Other single-organism genetic tools that can be extended to communities include transposon mutagenesis, forward and reverse genetics, and the introduction (or removal) of entire organisms (wild isolates or engineered organisms) to assess the resulting genetic or organismal effects on community phenotype. Recent advances in genetic manipulation, such as CRISPR-based editing and chemical mutagenesis, have begun to be applied to microbial communities (Mimee et al. 2015; Bae et al. 2016) and are expected to increase the ability to manipulate host-associated microbial interactions experimentally.
Direct observation of microbial communities can provide extremely precise, spatially detailed information regarding host-microbial interactions (Mark Welch et al. 2016). Likewise, microbial genetic manipulation has an extremely long and powerful history and allows precise molecular hypotheses to be tested in situ. Both techniques can be technically challenging in the human microbiome or associated models. Direct microscopy does not typically resolve more than tens of different organisms, for example, and taxa typically not higher than the genus. Likewise, genetic manipulation in whole microbial communities requires careful recolonization of a model by modified organisms, completely gnotobiotic manipulation in animal systems, or comprehensive transformation of community members in situ, all of which are technically challenging to conduct and verify. When they are appropriate, however, these systems offer among the most targeted mechanistic molecular tests in reductionist models of human microbial biotransformations.
ANALYZING MICROBIOME POPULATION AND EXPOSURE DATA
The Human Microbiome and Molecular-Epidemiology Analytic Approaches
As noted earlier, most current analytic methods for studying the human microbiome use techniques related to molecular epidemiology, which generally follow a strategy in which features of interest are bioinformatically quantified from culture-independent data and then statistically associated with environmental or health-related covariates and outcomes (Franzosa et al. 2015). Features used to describe the microbiome can include operational taxonomic unit9 counts or abundances derived from amplicon sequencing (Hamady and Knight 2009); species or strains detected with metagenome sequencing (Truong et al. 2015; Donati et al. 2016); functional profiles (gene or pathway quantifications) in metagenomes or metatranscriptomes (Abubucker et al. 2012); ecologic
___________________
8 Viability is surprisingly difficult to assess in a culture-independent manner, but sequencing has now been successfully coupled with a variety of DNA-intercalating dyes, such as propidium monoazide, for determining whole-community viability (Emerson et al. 2017).
9 Operational taxonomic units are used to cluster sequences on the basis of similarity (Nguyen et al. 2016).
summary statistics, such as species distributions or diversity (Hamady and Knight 2009); or partial to near-complete genome assemblies and annotations (Sangwan et al. 2016). Ultimately, any feature can be quantitatively modeled as a matrix of abundances or presence-or-absences, and samples can be additionally annotated with metadata, including outcome measures (health status or clinical phenotypes); host demographics or biometrics; population structure, such as ethnicity or genetic background; covariates, such as medications and diet; other molecular measures, such as microbial metabolites or gene expression; or environmental exposures.
Multivariate statistical modeling techniques—such as generalized linear modeling, factor analysis, variations on ordination, correspondence analysis, partial least-squares analysis, or nonparametric analysis of variance—are then applied. Such statistical or machine-learning methods are not unique to microbial-community epidemiology but are shared with other high-dimensional population analyses. For example, linear modeling is typically adapted to associate multiple population variables—such as health outcomes, demographics, biometrics, and chemical exposures—with microbial variables (Morgan et al. 2012, 2015), taking into account the mathematical properties of typical microbial measurements (sparse, zero-inflated, count-based, or proportional data). Nonparametric tests originally developed for quantitative ecology (Excoffier et al. 1992; Zapala and Schork 2006) are appropriate for determining whether overall variance in microbial community structure, as opposed to individual microbial features, is explained by covariates. Predictive models, such as random forests or support vector machines (Pasolli et al. 2016), can also be used to link microbial features to health outcomes or covariates. All the tests essentially detect microbial feature associations with covariates, including chemical exposures or exposure-related health outcomes, that occur more strongly than would be expected by chance (Paulson et al. 2013; Foxman and Martin 2015); these associations are similar to ones that can be observed and studied for gene expression or human genetic variation in other statistical -omics settings. The methods are typically well suited to large population studies that can indicate associations and can contribute to the generation of hypotheses that need to be probed in more detail with other methods to gain insight about causality and mechanisms.
Ecologic and Systems-Biology Analyses of the Human Microbiome
Other common analyses of the human microbiome use a systems-biology approach with the goal of identifying functional relationships among microorganisms, cells, or molecules. They might target molecular-interaction networks or ecologic structures in microbial communities directly (Faust et al. 2012; Friedman and Alm 2012; Kurtz et al. 2015) or in association with human immune-cell subsets (Amit et al. 2011). Molecular-network reconstruction techniques include identifying functionally related gene products by using co-expression data; this has been particularly successful in recovering human molecular regulatory programs during microbial exposure in immune-cell subsets (Haberman et al. 2014; Morgan et al. 2015; O’Connell et al. 2016). Similar data and techniques can be used to reconstruct regulatory and metabolic networks within microbial communities themselves, typically relying more on genomic potential (metagenome annotations) than on transcriptional profiling (Carr et al. 2013; Nielsen et al. 2014). The co-variation approach or other types of guilt-by-association approaches to identifying related molecules within a network can be extended to include phylogenetic information or profiling (Eisen 1998; Carr et al. 2013; Lan et al. 2014) or inferred metabolic capabilities by flux balance analysis (Zengler and Palsson 2012; Khandelwal et al. 2013; Hanemaaijer et al. 2015; Zelezniak et al. 2015). However, all the methods can be challenging to carry out in the microbiome, where, in contrast to the human genome, most microbial gene products are not annotated with well-characterized molecular or biochemical roles.
Analyses intended to characterize ecologic structure include models of microbial dispersion (such as entry of microorganisms into a communi-
ty) (Costello et al. 2012), transmission (movement of microorganisms between communities) (Blaser and Falkow 2009; Funkhouser and Bordenstein 2013; Milani et al. 2015), and co-occurrence (ecologic relationships, such as symbiosis or competition between microorganisms) (Faust et al. 2012; Friedman and Alm 2012; Kurtz et al. 2015). Because nearly all molecular assays measure relative abundance (compositions) rather than absolute cell counts, spurious correlations make it difficult to infer truly functional co-occurrence patterns (Tsilimigras and Fodor 2016). Dynamic systems models capture relationships in abundance patterns among organisms over time and have also been used to describe microbial interaction patterns. Examples of dynamic systems models include differential equations—for example, modified Lotka-Volterra systems (Stein et al. 2013; Marino et al. 2014; Bucci et al. 2016)—and probabilistic graphical models, for example, Gaussian processes (Tonner et al. 2017). Again, the level of detail can be difficult to reach with current data and modeling techniques because of the lack of taxonomically precise (strain-level) profiles sampled sufficiently densely over time to construct models outside simplified, in vitro systems.
STRENGTHS, WEAKNESSES, AND GAPS IN TECHNOLOGIES FOR STUDYING RELATIONSHIPS BETWEEN THE MICROBIOME AND CHEMICAL EXPOSURE
Systems
The microbiome field has available a diverse spectrum of experimental-animal systems that offer rigorous experimental control and provide distinct opportunities to define causality within host-microbiome–chemical interactions. However, as in all fields, researchers need to understand the strengths and weaknesses of each system and choose from among them appropriately. A persistent challenge in the use of nonhuman experimental systems to study the microbiome is to define which aspects of human-microbiome–chemical interactions can be effectively modeled and examined in each setting. To address that challenge, researchers need to improve their understanding of which aspects of each model system are reflective of humans, which ones are not, and which ones are likely to be relevant to host-microbiome–chemical interactions. Because the field relies heavily on microbiome transplant studies in animal models, experiments that include chemical treatment and microbiome transplantation will need to determine how to account and control for potential carryover of a chemical from the chemically exposed donor to the unexposed recipient via the transplanted microbiome. Finally, inasmuch as understanding of the field is based largely on cross-sectional sequence-based data, increased efforts need to complement the data with information on additional molecular activities and the spatial or temporal dynamics of microbial communities.
In vitro microbial-community model systems share many of the strengths and weaknesses of animal models but to a greater degree. For example, they are easier to manipulate and control, but they are less physiologically similar to a human, particularly because they lack host cellular and immune responses. Attention must be paid to how a chemical is introduced into the experimental systems and how the resulting exposure is measured and characterized. Specifically, in vitro systems that use static or flow-through technology present challenges in delivering specific, known amounts of chemical to the target organelles, cells, or tissues. In culture-based systems, genomic methods are well established for bacteria and their communities but less established for fungi, archaea, and viruses. Gaps for the other microorganisms include a lack of reference genomes, culture conditions for isolates, and adaptability for genetic manipulation. However, in vitro systems are often extremely cost-effective and scalable, and they are particularly well suited to screening assays, such as microorganism–microorganism or microorganism–chemical interaction testing. In vitro systems allow, for example, the introduction of potentially bioactive (positively or negatively) chemical exposures into a controlled microbial (typically not host-associated) setting with accompanying readout of microbial metabolism.
It is important in all such model systems to consider and integrate information from systems at various levels of reductionist scale, that is, from single microbial isolate cultures through human population measurements. A striking challenge in integrating results from systems across all scales is the small extent to which microbial gene products have been characterized. The lack of knowledge limits interpretation in vivo and manipulation in vitro.
Analyses
Analysis of human microbiome data, regardless of their source or assay method, can benefit from the approaches that have been developed over the last 2 decades in other fields that use molecular -omics approaches. Specifically, many associative studies share designs and methods with those in molecular epidemiology, such as genome-wide association studies or cancer-biomarker discovery that analyzes gene expression. With small statistical changes, computational methods and lessons learned from those other fields can be directly applied in microbiome research. The availability of individual microbial isolate reference data (primarily genome sequences) to contextualize microbial-community data is both a strength and a weakness: tens of thousands of reference genomes are available and constitute a powerful resource with which to interpret the microbiome, but these reference genomes are primarily bacterial, and there is a major gap if one wants to study viruses, fungi, and other microorganisms. Another major gap in the field is that most sequenced microbial genes and microorganism-associated chemicals that have been detected are not functionally or biochemically characterized; it is not even clear what fraction of them has been detected. That situation leads to a pool of biochemically functional “dark matter” with as-yet-unknown effects on microbial ecology or human health. Finally, as in most fields of molecular -omics, new computational methods will continue to be needed for integrating many types of microbial-community data; new methods will lead to increasingly accurate methods for identifying associations between molecular activities in the assays and human health outcomes.
FINDINGS
- Various animal models that have extensive conserved molecular and immunologic mechanisms provide appropriate experimental environments for controlled manipulation of host-associated microbial communities, although none mimics humans perfectly.
- Gnotobiotic animal models are particularly amenable to studies of the effect of microbial-community composition on host phenotype. Their use would benefit from more study of which aspects are shared (or not) with humans under different manipulations at each body site.
- Animal experiments that include chemical treatment and microbiome transplantation will need to determine how to differentiate carryover of a chemical from an exposed donor to an unexposed recipient via the transplanted microbiome.
- In vitro and ex vivo techniques can be usefully adapted to characterize diverse human-microbiome members and representative communities, but identifying appropriate culture conditions and models poses technical challenges.
- Human-microbiome experimental systems remain less developed outside the gut.
- As microbiome research is a young field, diversity in experimental protocols can make comparability of results among human-microbiome studies difficult.
- Computational methods and quantitative best practices of other -omics technologies can generally be applied to microbiome data with appropriate adaptations of statistical techniques.
- Most microbial genes and microbially associated chemicals in the microbiome are not functionally or biochemically characterized, and it is not even clear what fraction of them has been detected.
REFERENCES
Aagaard, K., J. Petrosino, W. Keital, M. Watson, J. Katancik, N. Garcia, S. Patel, M. Cutting, T. Madden, H. Hamilton, E. Harris, D. Gevers, G. Simone, P. McInnes, and J. Versalovic. 2013. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 27(3):1012-1022.
Abubucker, S., N. Segata, J. Goll, A.M. Schubert, J. Izard, B.L. Cantarel, B. Rodriguez-Mueller, J. Zucker, M. Thiagarajan, B. Henrissat, O. White, S.T. Kelley, B. Methé. P.D. Schloss, D. Gevers, M. Mitreva, and C. Huttenhower. 2012. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8(6):e1002358.
Ambriz-Aviña, V., J.A. Contreras-Garduño and M. PedrazaReyes. 2014. Applications of flow cytometry to characterize bacterial physiological responses. Biomed. Res. Int. 2014:461941.
Amit, I., A. Regev, and N. Hacohen. 2011. Strategies to discover regulatory circuits of the mammalian immune system. Nat. Rev. Immunol. 11(12):873-880.
Auchtung, J.M., C.D. Robinson, and R.A. Britton. 2015. Cultivation of stable, reproducible microbial communities from different fecal donors using minibioreactor arrays (MBRAs). Microbiome 3:42.
Bäckhed, F., C.M. Fraser, Y. Ringel, M.E. Sanders, R.B. Sartor, P.M. Sherman, J. Versalovic, V. Young, and B.B. Finlay. 2012. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 12(5):611-622.
Bae, S., O. Mueller, S. Wong, J.F. Rawls, and R.H. Valdivia. 2016. Genomic sequencing-based mutational enrichment analysis identifies motility genes in a genetically intractable gut microbe. Proc. Natl. Acad. Sci. USA 113(49):14127-14132.
Belkaid, Y., and J.A. Segre. 2014. Dialogue between skin microbiota and immunity. Science 346(6212):954-959.
Benam, K.H., R. Villenave, C. Lucchesi, A. Varone, C. Hubeau, H.H. Lee, S.E. Alves, M. Salmon, T.C. Ferrante, J.C. Weaver, A. Bahinski, G.A. Hamilton, and D.E. Ingber. 2016a. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 13(2):151-157.
Benam, K.H., R. Novak, J. Nawroth, M. Hirano-Kobayashi, T.C. Ferrante, Y. Choe, R. Prantil-Baun, J.C. Weaver, A. Bahinski, K.K. Parker, and D.E. Ingber. 2016b. Matched-comparative modeling of normal and diseased human airway responses using a microengineered breathing lung chip. Cell Syst. 3(5):456-466.e4.
Biggs, M.B., G.L. Medlock, T.J. Moutinho, H.J. Lees, J.R. Swann, G.L. Kolling, and J.A. Papin. 2017. Systems-level metabolism of the altered Schaedler flora, a complete gut microbiota. ISME J. 11(2):426-438.
Blaser, M.J., and S. Falkow. 2009. What are the consequences of the disappearing human microbiota? Nat. Rev. Microbiol. 7(12):887-894.
Bojar, R.A. 2015. Studying the human skin microbiome using 3D in vitro skin models. Appl. In Vitro Toxicol. 1(2):165-171.
Bouslimani, A., C. Porto, C.M. Rath, M. Wang, Y. Guo, A. Gonzalez, D. Berg-Lyon, G. Ackermann, G.J. Moeller Christensen, T. Nakatsuji, L. Zhang, A.W. Borkowski, M.J. Meehan, K. Dorrestein, R.L. Gallo, N. Bandeira, R. Knight, T. Alexandrov, and P.C. Dorrestein. 2015. Molecular cartography of the human skin surface in 3D. Proc. Natl. Acad. Sci. USA 112(17):E2120-E2129.
Brouzes, E., T. Kruse, R. Kimmerling, and H.H. Strey. 2015. Rapid and continuous magnetic separation in droplet microfluidic devices. Lab Chip 15(3):908-909.
Browne, H.P., S.C. Forster, B.O. Anonye, N. Kumar, B.A. Neville, M.D. Stares, D. Goulding, and T.D. Lawley. 2016. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533(7604):543-546.
Bucci, V., B. Tzen, N. Li, M. Simmons, T. Tanoue, E. Bogart, L. Deng, V. Yeliseyev, M.L. Delaney, Q. Liu, B. Olle, R.R. Stein, K. Honda, L. Bry, and G.K. Gerber. 2016. MDSINE: Microbial Dynamical Systems INference Engine for microbiome time-series analyses. Genome Biol. 17(1):121.
Carr, R., S.S. Shen-Orr, and E. Borenstein. 2013. Reconstructing the genomic content of microbiome taxa through shotgun metagenomic deconvolution. PLoS Comput. Biol. 9(10):e1003292.
Charlson, E.S., K. Bittinger, J. Chen, J.M. Diamond, H. Li, R.G. Collman, and F.D. Bushman. 2012. Assessing bacterial populations in the lung by replicate analysis of samples from the upper and lower respiratory tracts. PloS ONE 7(9): e42786.
Chen, Y., Y. Lin, K.M. Davis, Q. Wang, J. Rnjak-Kovacina, C. Li, R.R. Isberg, C.A. Kumamoto, J. Mecsas, and D.L. Kaplan. 2015. Robust bioengineered 3D functional human intestinal epithelium. Sci. Rep. 5:13708.
Chung, H., S.J. Pamp, J.A. Hill, N.K. Surana, S.M. Edelman, E.B. Troy, N.C. Reading, E.J. Villablanca, S. Wang, J.R. Mora, Y. Umesaki, D. Mathis, C. Benoist, D.A. Relman, and D.L. Kasper. 2012. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149(7):157-1593.
Cohen, L.J., H.S. Kang, J. Chu, Y.H. Huang, E.A. Gordon, B.V. Reddy, M.A. Ternei, J.W. Craig, and S.F. Brady. 2015. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc. Natl. Acad. Sci. U.S.A. 112(35):E4825-E4834.
Costello, E.K., K. Stagaman, L. Dethlefsen, B. J. Bohannan, and D.A. Relman. 2012. The application of ecological theory toward an understanding of the human microbiome. Science 336(6086):1255-1262.
Cui, X.Y., P. Xiang, R.W. He, A. Juhasz, and L.Q. Ma. 2016. Advances in in vitro methods to evaluate oral bioaccessibility of PAHs and PBDEs in environmental matrices. Chemosphere 150:378-389.
Dickson, R.P., J.R. Erb-Downward, C.M. Freeman, L. McCloskey, N.R. Falkowski, G.B. Huffnagle, and J.L. Curtis. 2017. Bacterial topography of the healthy human lower respiratory tract. MBio 8(1): e02287-16.
Donati, C., M. Zolfo, D. Albanese, D. Tin Truong, F. Asnicar, V. Iebba, D. Cavalieri, O. Jousson, C. De Filippo, C. Huttenhower, and N. Segata. 2016. Uncovering oral Neisseria tropism and persistence using metagenomic sequencing. Nat. Microbiol. 1(7):16070.
Dye, B.R., D.R. Hill, M.A. Ferguson, Y.H. Tsai, M.S. Nagy, R. Dyal, J.M. Wells, C.N. Mayhew, R. Nattiv, O.D. Klein, E.S. White, G.H. Deutsch, and J.R. Spence. 2015. In vitro generation of human pluripotent stem cell derived lung organoids. eLife 4:e05098.
Earle, K.A., G. Billings, M. Sigal, J.S. Lichtman, G.C. Hans-son, J.E. Elias, M.R. Amieva, K.C. Huang, and J.L. Sonnenburg. 2015. Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe 18(4):478-488.
Eisen, J.A. 1998. Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 8(3):163-167.
Emerson, J.B., R.I. Adams, C.M.B. Román, B. Brooks, D.A. Coil, K. Dahlhausen, H.H. Ganz, E.M. Hartmann, T. Hsu, N.B. Justice, I.G. Paulino-Lima, J.C. Luongo, D.S. Lymperopoulou, C. Gomez-Silvan, B. Rothschild-Mancinelli, M. Balk, C. Huttenhower, A. Nocker, P. Vaishampayan, and L.J. Rothschild. 2017. Schrödinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbe 5(1):86.
Excoffier, L., P.E. Smouse, and J.M. Quattro. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131(2):479-491.
Faith, J.J., F.E. Rey, D. O’Donnell, M. Karlsson, N.P. McNulty, G. Kallstrom, A.L. Goodman, and J.I. Gordon. 2010. Creating and characterizing communities of human gut microbes in gnotobiotic mice. ISME J. 4(9):1094-1098.
Falk, P.G., L.V. Hooper, T. Midtvedt, and J.I. Gordon. 1998. Creating and maintaining the gastrointestinal ecosystem: What we know and need to know from gnotobiology. Microbiol. Mol. Rev. 62(4):1157-1170.
Fatehullah, A., S.H. Tan, and N. Barker. 2016. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 18(3):246-254.
Faust, K., J.F. Sathirapongsasuti, J. Izard, N. Segata, D. Gevers, J. Raes, and C. Huttenhower. 2012. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 8(7):e1002606.
Findley, K., J. Oh, J. Yang, S. Conlan, C. Deming, J.A. Meyer, D. Schoenfeld, E. Nomicos, M. Park, NIH Intramural Sequencing Center Comparative Sequencing Program, H.H. Kong, and J.A. Segre. 2013. Topographic diversity of fungal and bacterial communities in human skin. Nature 498(7454):367-370.
Foxman, B., and E.T. Martin. 2015. Use of microbiome in the practice of epidemiology: A primer on -omic technologies. Am. J. Epidemiol. 182(1):1-8.
Franzosa, E.A., X.C. Morgan, N. Segata, L. Waldron, J. Reyes, A.M. Earl, G. Giannoukos, M.R. Boylan, D. CIulla, D. Gevers, J. Izard, W.S. Garrett, A.T. Chan, and C. Huttenhower. 2014. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 111(22):E2329-E2338.
Franzosa, E.A., T. Hsu, A. Sirota-Madi, A. Shafquat, G. Abu-Ali, X.C. Morgan, and C. Huttenhower. 2015. Sequencing and beyond: Integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 13(6):360-372.
Friedman, J., and E.J. Alm. 2012. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8(9):e1002687.
Fritz, J.V., M.S. Desai, P. Shah, J.G. Schneider, and P. Wilmes. 2013. From meta-omics to causality: Experimental models for human microbiome research. Microbiome 1(1):14.
Funkhouser, L.J., and S.R. Bordenstein. 2013. Mom knows best: The universality of maternal microbial transmission. PLoS Biol. 11(8):e1001631.
Garrett. W.S. 2015. Cancer and the microbiota. Science 348(6230):80-86.
Gawad, C., W. Koh, and S.R. Quake. 2016. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 17(3):175-188.
Gohl, D.M., P. Vangay, J. Garbe, A. MacLean A. Hauge, A. Becker, T.J. Gould, J.B. Clayton, T.J. Johnson, R. Hunter, D. Knights, and K.B. Beckman. 2016. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiomes studies. Nat. Biotehnol. 34(9):942-949.
Golf, O., N. Strittmatter, T. Karancsi, S.D. Pringle, A.V. Speller, A. Mroz, J.M. Kinross, N. Abbassi-Ghadi, E.A. Jones, and Z. Takats. 2015. Rapid evaporative ionization mass spectrometry imaging platform for direct mapping from bulk tissue and bacterial growth media. Anal. Chem. 87(5):2527-2534.
Grice, E.A., and J.A. Segre. 2011. The skin microbiome. Nat. Rev. Microbiol. 9(4):244-253.
Gross, G., D.M. Jacobs, S. Peters, S. Possemiers, J. van Duynhoven, E.E. Vaughan, and T. van de Wiele. 2010. In vitro bioconversion of polyphenols from black tea and red wine/grape juice by human intestinal microbiota displays strong interindividual variability. J. Agric. Food Chem. 58(18):10236-10246.
Haberman, Y., T.L. Tickle, P.J. Dexheimer, M.O. Kim, D. Tang, R. Karns, R.N. Baldassano, J.D. Noe, J. Rosh, J. Markowitz, M.B. Heyman, A.M. Griffiths, W.V. Crandall, D.R. Mack, S.S. Baker, C. Huttenhower, D.J. Keljo, J.S. Hyams, S. Kugathasan, T.D. Walters, B. Aronow, R.J. Xavier, D. Gevers, and L.A. Denson. 2014. Pedriatic Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 124(8):3617-3633.
Hacquard, S., R. Garrido-Oter, A. González, S. Spaepen, G. Ackermann, S. Lebeis, A.C. McHardy, J.L. Dangl, R. Knight, R. Ley, and P. Schulze-Lefert. 2015. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17(5):603-616.
Hamady, M., and R. Knight. 2009. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 19(7):1141-1152.
Hanemaajier, M., W.F. Röling, B.G. Olivier, R.A. Khandelwal, B. Teusink, and F.J. Bruggeman. 2015. Systems modeling approaches for microbial community studies from metagenomics to inference of the community structure. Front. Microbiol. 6:213.
HMP (Human Microbiome Project) Consortium. 2012a. Structure, function and diversity of the healthy human microbiome. Nature 486(7402):207-214.
HMP Consortium. 2012b. A framework for human microbiome research. Nature 486(7402):215-221.
Honda, K., and D.R. Littman. 2016. The microbiota in adaptive immune homeostasis and disease. Nature 535(7610):75-84.
Hsiao, E.Y., S.W. McBride, S. Hsien, G. Sharon, E.R. Hyde, T. McCue, J.A. Codelli, J. Chow, S.E. Reisman, J.F. Petrosino, P.H. Patterson, and S.K. Mazmanian. 2013. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155(7):1451-1463.
Ilhan, Z.E. 2016. Microbiome After Bariatric Surgery and Microbial Insights into Surgical Weight Loss. Ph. D. Disertation, Arizona State University. August 2016.
Jemielita, M., M.J. Taormina, A.R. Burns, J.S. Hampton, A.S. Rolig, K. Guillemin, and R. Parthasarathy. 2014. Spatial and temporal features of the growth of a bacterial species colonizing the zebrafish gut. MBio 5(6):e01751-14.
Jiang, C., C. Xie, Y. Lv, J. Li, K.W. Krausz, J. Shi, C.N. Brocker, D. Desai, S.G. Amin, W.H. Bisson, Y. Liu, O. Gavrilova, A.D. Patterson, and F.J. Gonzalez. 2015. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 6:10166.
Joly, C., J. Gay-Quéheillard, A. Léké, K. Chardon, S. Delanaud, V. Bach, and H. Khorsi-Cauet. 2013. Impact of chronic exposure to low doses of chlorpyrifos on the in-
testinal microbiota in the Simulator of the Human Intestical Microbial Ecosystem (SHME) and in the rat. Environ. Sci. Pollut. Res. Int. 20(5):2726-2734.
Kaiko, G.E., S.H. Ryu, O.I. Koues. P.L. Collins, L. Solnica-Krezel, E.J. Pearce, E.L. Pearce, E.M. Oltz, and T.S. Stappenbeck. 2016. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165(7):1708-1720.
Kauffman, A.L., A.V. Gyurdieva, J.R. Mabus, C. Ferguson, Z. Yan, and P.J. Hornby. 2013. Alternative functional in vitro models of human intestinal epithelia. Front. Pharmacol. 4:79.
Khandelwal, R.A., B.G. Olivier, W.F. Röling, B. Teusink, and F.J. Bruggeman. 2013. Community flux balance analysis for microbial consortia at balanced growth. PLoS ONE 8(5):e64567.
Kim, H.J., and D.E. Ingber. 2013. Gut-on-a-chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. (Camb.) 5(9):1130-1140.
Kim, H.J., D. Huh, G. Hamilton, and D.E. Ingber. 2012. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12(12):2165-2174.
Kim, H.J., H. Li, J.J. Collins, and D.E. Ingber. 2016. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA. 113(1):E7-E15.
Kolmeder, C.A., and W.M. de Vos. 2014. Metaproteomics of our microbiome- developing insight in function and activity in man and model systems. J. Proteomics. 97:3-16.
Koppel, N., and E.P. Balscus. 2016. Exploring and understanding the biochemical diversity of the human microbiota. Cell Chem. Biol. 23(1):18-30.
Kurtz, Z.D., C.L. Müller, E.R. Miraldi, D.R. Littman, M.J. Blaser, and R.A. Bonneau. 2015. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 11(5):e1004226.
Lagier, J.C., S. Khelaifia, and D. Raoult. 2016. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 1:16203.
Lam, K.N., J. Cheng, K. Engel, J.D. Neufeld, and T.C. Charles. 2015. Current and future resources for functional metagenomics. Front. Microbiol. 6:1196.
Lan, Y., J.C. Morrison, R. Hershberg, and G.L. Rosen. 2014. POGO-DB- a database of pairwise-comparisons of genomes and conserved orthologous genes. Nucleic Acids Res. 42(Database issue):D625-D632.
Lau, J.T., F.J. Whelan, I. Herath, C.H. Lee, S.M. Collins, P. Bercik, and M.G. Surette. 2016. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 8:72.
Lauder, A.P., A.M. Roche, S. Sherrill-Mix, A. Bailey, A.L. Laughlin, K. Bittinger, R. Leite, M.A. Elovitz, S. Parry, and F.D. Bushman. 2016. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4(1):29.
Leulier, F., L.T. MacNeil, W.J. Lee, J.F. Rawls, P.D. Cani, M. Schwarzer, L. Zhao, and S.J. Simpson. 2017. Integrative physiology: At the crossroads of nutrition, microbiota, animal physiology, and human health. Cell Metab. 25(3):522-534.
Leung, K., H. Zahn, T. Leaver, K.M. Konwar, N.W. Hanson, A.P. Pagé, C.C. Lo, P.S. Chain, S.J. Hallam, and C.L. Hansen. 2012. A programmable droplet-based microfluidic device applied to multiparameter analysis of single microbes and microbial communities. Proc. Natl. Acad. Sci. USA 109(20):7665-7670.
Ley, R.E., M. Hamady, C. Lozupone, P.J. Turnbaugh, R.R. Ramey, J.S. Bircher, M.L. Schlegel, T. A. Tucker, M.D. Schrenzel, R. Knight, and J.I. Gordon. 2008. Evolution of mammals and their gut microbes. Science 320(5883):1647-1651.
Lloyd-Price, J., G. Abu-Ali, and C. Huttenhower. 2016. The healthy human microbiome. Genome Med. 8(1):51.
Ma, L., J. Kim, R. Hatzenpichler, M.A. Karymov, N. Hubert, I.M. Hanan, E.B. Chang, and R.F. Ismagilov. 2014a. Gene-targeted microfluidic cultivation validated by isolation of a gut bacterium listed in Human Microbiome Project’s Most Wanted taxa. Proc. Natl. Acad. Sci. USA. 111(27):9768-9773.
Ma, L., S.S. Datta, M.A. Karymov, Q. Pan, S. Begolo, and R.F. Ismagilov. 2014b. Individually addressable arrays of replica microbial cultures enabled by splitting SlipChips. Integr. Biol. (Camb.) 6(8):796-805.
Macpherson, A.J., and K.D. McCoy. 2015. Standardised animal models of host microbial mutualism. Mucosal. Immunol. 8(3):476-486.
Marino, S., N.T. Baxter, G.B. Huffnagle, J.F. Petrosino, and P.D. Schloss. 2014. Mathematical modeling of primary succession of murine intestinal microbiota. Proc. Natl. Acad. Sci. USA 111(1):439-444.
Mark Welch, J.L., B.J. Rossetti, C.W. Rieken, F.E. Dewhirst, and G.G. Borisy. 2016. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. U.S.A. 113(6):E791-E800.
Maurice, C.F., H.J. Haiser, and P.J. Turnbaugh. 2013. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152(1-2):39-50.
McDonald, J.A., S. Fuentes, K. Schroeter, I. HeikampdeJong, C.M. Khursigara, W.M. de Vos, and E. AllenVercoe. 2015. Simulating distal gut mucosal and luminal communities using packed-column biofilm reactors and an in vitro chemostat model. J. Microbiol. Methods 108:36-44.
Milani, C., L. Mancabelli, G.A. Lugli, S. Duranti, F. Turroni, C. Ferrario, M. Mangifesta, A. Viappiani, P. Ferretti, V. Gorfer, A. Tett, N. Segata, D. van Sinderen, and M. Ventura. 2015. Exploring vertical transmission of bifidobacteria from mother to child. Appl. Environ. Microbiol. 81(20):7078-7087.
Miller, T.L., and M.J. Wolin. 1981. Fermentation by the human large intestine microbial community in an in vitro semicontinuous culture system. Appl. Environ. Microbiol. 42(3):400-407.
Mimee, M., A.C. Tucker, C.A. Voight, and T.K. Lu. 2015. Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 1(1):62-71.
Moon, C., K.L. VanDussen, H. Miyoshi, and T.S. Stappenbeck. 2014. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal. Immunol. 7(4):818-828.
Morgan, X.C., T.L. Tickle, H. Sokol, D. Gevers, K.L. Devaney, D.V. Ward, J.A. Reyes, S.A. Shah, N. LeLeiko, S.B. Snapper, A. Bousvaroa, J. Korzenik, B.E. Sands, R.J. Xavier, and C. Huttenhower. 2012. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13(9):R79.
Morgan, X.C., B. Kabakchiev, L. Waldron, A.D. Tyler, T.L. Tickle, R. Milgrom, J.M. Stempak, D. Gevers, R.J. Xavier, M.S. Silverberg, and C. Huttenhower. 2015. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. 16:67.
Morgun, A., A. Dzutsev, X. Dong, R.L. Greer, D.J. Sexton, J. Ravel, M. Schuster, W. Hsiao, P. Matzinger, and N. Shulzhenko. 2015. Uncovering effects of antibiotics on the host and microbiota using transkingdom gene networks. Gut. 64(11):1732-1743.
Nelson, K., C. Bobba, S. Ghadiali, D. Hayes, Jr, S.M. Black, and B.A. Whitson. 2014. Animal models of ex vivo lung perfusion as a platform for transplantation research. World J. Exp. Med. 4(2):7-15.
Nguyen, N.P., T. Warnow, M. Pop, and B. White. 2016. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2:16004.
Nielsen, H.B., M. Almeida, A.S. Juncker, S. Rasmussen, J. Li, S. Sunagawa, D.R. Plchta, L. Gautier, A.G. Pedersen, E. Le Chatelier, E. Pelletier, I. Bonde, T. Nielsen, C. Manichanh, M. Arumugam, J.M. Batto, M.B. Quintanilha Dos Santos, N. Blom, N. Borruel, K.S. Burgdorf, F. Boumezbeur, F. Casellas, J. Doré, P. Dworzynski, F. Guarner, T. Hansen, F. Hildebrand, R.S. Kaas, S. Kennedy, K. Kristiansen, J.R. Kultima, P. Léonard, F. Levenez, O. Lund, B. Moumen, D. Le Paslier, N. Pons, O. Pedersen, E. Prifti, J. Qin, J. Raes, S. Sørensen, J. Tap, S. Tims, D.W. Ussery, T. Yamada, MetaHIT Consortium, P. Renault, T. Sicheritz-Ponten, P. Bork, J. Wang, S. Brunak, and S.D. Ehrlich. 2014. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32(8):822-828.
Nigro, G., M. Hanson, C. Fevre, M. Lecuit, and P.J. Sansonetti. In Press. Intestinal organoids as a novel tool to study microbes-epithelium interactions. Methods Mol. Biol.
O’Connell, D.J., R. Kolde, M. Sooknah, D.B. Graham, T.B. Sundberg, I.J. Latorre, T.S. Mikkelsen, and R.J. Xavier. 2016. Simultaneous pathway activity inference and gene expression analysis using RNA sequencing. Cell Syst. 2(5):323-334.
Oh, J., A.L. Byrd, C. Deming, S. Conlan, NISC Comparative Sequencing Program, H.H. Kong, and J.A. Segre. 2014. Biogeography and individuality shape function in the human skin metagenome. Nature 514(7520):59-64.
Pasolli, E., D.T. Truong, F. Malik, L. Waldron, and N. Segata. 2016. Machine learning meta-analysis of large metagenomics datasets: Tools and biological insights. PLoS Comput. Viol. 12(7):e1004977.
Paulson, J.N., O.C. Stine, H.C. Bravo, and M. Pop. 2013. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10(12):1200-1202.
Pérez-Losada, M., K.A. Crandall, and R.J. Freishtat. 2016. Two sampling methods yield distinct microbial signatures in the nasopharynges of asthmatic children. Microbiome 4(1):25.
Petersen, C., and J.L. Round. 2014. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 16(7):1024-1033.
Rath, C.M., T. Alexandrov, S.K. Higginbottom, J. Song, M.E. Milla, M.A. Fischbach, J.L. Sonnernburg, and P.C. Dorrestein. 2012. Molecular analysis of model gut microbiotas by imaging mass spectrometry and nanodescription electrospray ionization reveals dietary metabolite transformations. Anal. Chem. 84(21):9259-9267.
Rawls, J.F., M.A. Mahowald, R.E. Ley, and J.I. Gordon. 2006. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127(2):423-433.
Rawls, J.F., M.A. Mahowald, A.L. Goodman, C.M. Trent, and J.I. Gordon. 2007. In vivo imaging and genetic analysis link bacterial motility and symbiosis in the zebrafish gut. Proc. Natl. Acad. Sci. USA 104(18):7622-7627.
Ridaura, V.K., J.J. Faith, F.E. Rey, J. Cheng, A.E. Duncan, A.L. Kau, N.W. Griffin, V. Lombard, B. Henrissat, J.R. Bain, M.J. Muehlbauer, O. Ilkayeva, C.F. Semenkovich, K. Funai, D.K. Hayashi, B.J. Lyle, M.C. Martini, L.K. Ursell, J.C. Clemente, W. Van Treuren, W.A. Walters, R. Knight, C.B. Newgard, A.C. Health, and J.I. Gordon. 2013. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341(6150):1241214.
Roeselers, G., M. Ponomarenko, S. Lukovac, and H.M. Wortelboer. 2013. Ex vivo systems to study host-microbiota interactions in the gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 27(1):101-113.
Salter, S.J., M.J. Cox, E.M. Turek, S.T. Calus, W.O. Cook-son, M.F. Moffatt, P. Turner, J. Parkhill, N.J. Loman, and A.W. Walker. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87.
Sangwan, N., F. Xia, and J.A. Gilbert. 2016. Recovering complete and draft population genomes from metagenome datasets. Microbiome 4:8.
Segata, N., D. Boernigen, T.L. Tickle, X.C. Morgan, W.S. Garrett, and C. Huttenhower. 2013. Computational meta’omics for microbial community studies. Mol. Syst. Biol. 9:666.
Semova, I., J.D. Carten, J. Stombaugh, L.C. Mackey, R. Knight, S.A. Farber, and J.F. Rawls. 2012. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12(3):277-288.
Sinha, R., C.C. Abnet, O. White, R. Knight, and C. Huttenhower. 2015. The microbiome quality control project: Baseline study design and future directions. Genome Biol. 16:276.
Sinha, R., G. Abu-Ali, E. Vogtmann, A.A. Fodor, B. Ren, A. Amir, E. Schwager, J. Crabtree, S. Ma, Microbiome Quality Control Project Consortium, C.C. Abnet, R. Knight, O. White, and C. Huttenhower. 2017. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 35(11):1077-1086.
Smirnov, K.S., T.V. Maier, A. Walker, S.S. Heinzmann, S. Forcisi, I. Martinex, J. Walter, and P. Schmitt-Kopplin. 2016. Challenges of metabolommics in human gut microbiota research. Int. J. Med. Microbiol. 306(5):266-279.
Song, S.J., A. Amir, J.L. Metcalf, K.R. Amato, Z.Z. Xu, G. Humphrey, and R. Knight. 2016. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems 1(3):00021-16.
Soufi, Y., and B. Soufi. 2016. Mass spectrometry-based bacterial proteomics: Focus on dermatologic microbial pathogens. Front. Microbiol. 7:181.
Stappenbeck, T.S., and H.W. Virgin. 2016. Accounting for reciprocal host-microbiome interactions in experimental science. Nature 534(7606):191-199.
Stein, R.R., V. Bucci, N.C. Toussaint, C.G. Buffie, G. Rätsch, E.G. Pamer, C. Sander, and J.B. Xavier. 2013. Ecological modeling from time-series inference: Insight into dynamics and stability of intestinal microbiota. PLoS Comput. Biol. 9(12):e1003388.
Tane, S., K. Noda, and N. Shigemura. 2017. Ex vivo lung perfusion a key tool for translational science in the lungs. Chest 151(6):1220-1228.
Tasse, L., J. Bercovici, S. Pizzut-Serin, P. Robe, J. Tap, C. Klopp, B.L. Cantarel, P.M. Coutinho, B. Henrissat, M. Leclerc, J. Doré, P. Monsan, M. Remaud-Simeon, and G. Potocki-Veronese. 2010. Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 20(11):1605-1612.
Taylor, A.A., I.M. Marcus, R.L. Guysi, and S.L. Walker. 2015. Metal oxide nanoparticles induce minimal phenotypic changes in a model colon gut microbiota. Environ. Eng. Sci. 32(7):602-612.
Tong, M., J.P. Jacobs, I.H. McHardy, and J. Braun. 2014. Sampling of intestinal microbiota and targeted amplification of bacterial 16S rRNA genes for microbial ecologic analysis. Curr. Protoc. Immunol. 107:7.41.
Tonner, P.D., C.L. Darnell, B.E. Engelhardt, and A.K. Schmid. 2017. Detecting differential growth of microbial populations with Gaussian process regression. Genome Res. 27(2):320-333.
Trompette, A., E.S. Gollwitzer, K. Yadava, A.K. Sichelstiel, N. Sprenger, C. Ngom-Bru, C. Blanchard, T. Junt, L.P. Nicod, N.L. Harris, and B.J. Marsland. 2014. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 20(2):159-166.
Truong, D.T., E.A. Franzosa, T.L. Tickle, M. Scholz, G. Weingart, E. Pasolli, A. Tett, C. Huttenhower and N. Segata. 2015. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12(10):902-903.
Tsai, Y.C., S. Conlan, C. Deming, NISC Comparative Sequencing Program, J.A. Segre, H.H. Kong, J. Korlach and J. Oh. 2016. Resolving the complexity of human skin metagenomes using single-molecule sequencing. MBio 7(1):e01948-15.
Tsilimigras, M.C., and A.A Fodor. 2016. Compositional data analysis of the microbiome: Fundamentals, tools, and challenges. Ann. Epidemiol. 26(5):330-335.
Van den Abbeele, P., S. Roos, V. Eeckhaut, D.A. MacKenzie, M. Derde, W. Verstraete, M. Marzorati, S. Possemiers, B. Vanhoecke, F. Van Immerseel, and T. Van de Wiele. 2012. Incorporating a mucosal environment in dynamic gut model results in a more representative colonization by lactobacilli. Microb. Biotechnol. 5(1):106-115.
van der Krieken, D.A., T.H. Ederveen, S.A. van Hijum, P.A. Jansen, W.J. Melchers, P.T. Scheepers, J. Schalkwijk, and P.L. Zeeuwen. 2016. An in vitro model for bacterial growth on human stratum corneum. Acta. Derm. Venereol. 96(7):873-879.
Vernocchi, P., F. Del Chierico, and L. Putignani. 2016. Gut microbiota profiling: Metabolomics based approach to unravel compounds affecting human health. Front. Microbiol. 7:1144.
Wiles, S., K.M. Pickard, K. Peng, T.T. MacDonald, and G. Frankel. 2006. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect. Immun. 74(9):5391-5396.
Wilson, S.S., A. Tocchi, M.K. Holly, W.C. Parks, and J.G. Smith. 2015. A small intestinal organoid model of noninvasive enteric pathogen-epithelial cell interactions. Mucosal. Immunol. 8(2):352-361.
Wolff, R.K. 1986. Effects of airborne pollutants on mucociliary clearance. Environ. Health Perspect. 66:223-237.
Yasuda, K., K. Oh, B. Ren, T.L. Tickle, E.A. Franzosa, L.M. Wachtman, A.D. Miller, S.V. Westmoreland, K.G. Mansfield, E.J. Vallender, G.M. Miller, J.K. Rowlett, D. Gevers, C. Huttenhowers, and X.C. Morgan. 2015. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe 17(3):385-391.
Yim, Y.J., J. Seo, S. Kang, J.H. Ahn, and H.G. Hur. 2008. Reductive dechlorination of methoxychlor and DDT by human intestinal bacterium Eubacterium limosum under anaerobic conditions. Arc. Environ. Contam. Toxicol. 54(3):406-411.
Zapala, M.A., and N.J. Schork. 2006. Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc. Natl. Acad. Sci. USA 103(51):19430-19435.
Zelezniak, A., S. Andrejev, O. Ponomarova, D.R. Mende, P. Bork, and K.R. Patil. 2015. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 112(20):6449-6454.
Zengler, K., and B.O. Palsson. 2012. A road map for the development of community systems (CoSy) biology. Nat. Rev. Microbiol. 10(5):366-372.






















