
As discussed by the first panel, transparent reporting of preclinical biomedical research is one element in the complex ecosystem of scientific research. Panelists discussed how the barriers to transparent reporting are rooted in the current culture of science and current incentive structures, and emphasized the importance of coordination across all stakeholders in fostering a culture of greater transparency. The second panel considered lessons learned and best practices for increased transparency from the field clinical research that could be applied to improving transparent reporting of preclinical studies (see Box 4-1 for corresponding workshop session objectives).
Veronique Kiermer, executive editor at the Public Library of Science and session moderator, mentioned the Consolidated Standards of Reporting Trials (CONSORT) guidelines as an example of an initiative to facilitate more complete reporting of randomized controlled trials.1 The development of the CONSORT guidelines began in the mid-1990s, leading to the release of the first CONSORT Statement in 1996. Revised CONSORT Statements were released in 2001 and 2010. The International Committee of
___________________
1 Further information on CONSORT is available at http://www.consort-statement.org (accessed December 14, 2019).
Medical Journal Editors (ICMJE) has endorsed the CONSORT Statement and encourages journals to adopt the CONSORT guidelines for manuscripts describing clinical studies. Kiermer also mentioned the Enhancing the QUAlity and Transparency Of health Research (EQUATOR) Network, which brings together different stakeholder working groups and other collaborators and collates and promotes guidelines and resources for health research reporting.2
Reporting the results of studies in journals is the end of the process, after the research has already been designed and conducted, Kiermer said. She pointed out that it can be difficult for researchers to comply with reporting guidelines if they have not built the elements needed to meet those guidelines into their studies early on in the process. Improving transparency needs to begin earlier in the process, and she said that funders and institutions have a role in coordinating with journals to improve transparency in reporting.
Three panelists working in different settings shared their perspectives on lessons and best practices from clinical research that could be applied broadly across biomedical research. An-Wen Chan, Phelan Scientist at Women’s College Research Institute and associate professor at the University of Toronto, described the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) initiative as an example of how guidelines can improve the reporting of clinical trial protocols and drive quality and efficiencies downstream. Geeta Swamy, vice dean for scientific integrity and associate vice president for research at Duke University, offered her perspective on developing a culture of research integrity and accountability at an institution through education, best practices, and scientific and analytical excellence. Magali Haas, chief executive officer (CEO) and president of Cohen Veterans Bioscience, described working with strategic partners to build enabling platforms that can accelerate rigorous, reproducible, and translatable preclinical science, and discussed the various ways that funders can influence and enable reproducibility.
LESSONS FROM THE SPIRIT INITIATIVE
An-Wen Chan, Phelan Scientist, Women’s College Research Institute, and Associate Professor, University of Toronto
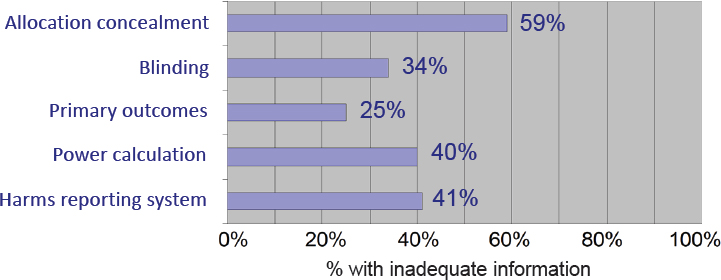
Analyses by Chan and others have found that many clinical trial protocols lack important information on key methodological elements such
___________________
2 Further information on the EQUATOR Network is available at https://www.equatornetwork.org (accessed December 14, 2019).
as primary outcomes, power calculations, or blinding, for example (see Figure 4-1). To address this, Chan and colleagues launched the SPIRIT initiative. In 2013, SPIRIT released a checklist of recommended information that should be included in a clinical trial protocol and a related paper explaining and elaborating on the supporting evidence and rationale for each item on the checklist (Chan et al., 2013a,b,c).
The SPIRIT checklist has been broadly endorsed and adopted as a standard for clinical trial protocols by more than 120 biomedical journals, Chan said. In addition, there are national ethics regulators that require submitted protocols to adhere to SPIRIT (e.g., the UK Health Research Authority), as well as funders and pharmaceutical companies that have adopted SPIRIT as the standard for their protocols. SPIRIT has been translated into six languages, and the SPIRIT website has 50,000 unique users per year.3 Chan noted that, although most protocols are not published, more than 600 published protocols are now based on SPIRIT.
Incentives
Efforts to promote quality and transparency in research are often perceived as creating additional administrative burdens, Chan observed. To encourage adoption, efforts are being made to promote the role of SPIRIT and other guidelines in improving clinical trial efficiency.
As an example, Chan said many of the delays in Institutional Review Board (IRB) or ethics approval of clinical studies are the result of protocols
SOURCES: Chan presentation, September 25, 2019; citing Chan et al., 2004, 2008, 2017; Hróbjartsson et al., 2009; Mhaskar et al., 2012; Pildal et al., 2005; and Scharf and Colevas, 2006.
___________________
3 For the SPIRIT guidelines, related publications, and additional information, see https://www.spirit-statement.org (accessed November 20, 2019).
being returned for revisions due to inadequate information (Russ et al., 2009). He suggested that IRB approval time could be reduced by using the SPIRIT checklist to submit more complete protocols. Once approved, trials are often amended (three amendments per trial on average). One study of more than 3,400 industry protocols found that one third of those amendments could have been avoided if more complete information had been incorporated in the protocol at the start. The study found that each amendment delays a trial by more than 2 months, delays trial registration by about 1 month, and adds significantly to the cost of the trial (Getz et al., 2011). Adhering to reporting guidelines upfront can lead to downstream efficiencies by helping to ensure that clinical trial protocols are complete and of high quality, Chan summarized.
Enforcement
As discussed by Brian Nosek and others, rewards and incentives can inspire behavior, but enforcement is also needed. Journal editors can play a key role in adopting and enforcing policies requiring adherence to reporting guidelines or transparency initiatives, Chan said. As an example, he said clinical trial registration was proposed in the 1980s to improve transparency and quality, but was not broadly practiced until after ICMJE instituted trial registration as a requirement for publication in 2005. As noted in the first panel discussion, it is important to recognize that these policies do not come without a cost. For example, the journal Trials requires that every protocol submit a SPIRIT checklist. While this has increased the implementation of SPIRIT, manual checking of the protocol against the checklist is very time consuming for reviewers and editors. Simply adopting a policy is insufficient, Chan said, and enforcement requires investment of significant resources.
Funders also have enforcement power, Chan said. The National Institute for Health Research in the United Kingdom, for example, withholds a portion of a grantee’s funding until a final report is published. In this way the Institute has achieved a 96 percent publication rate for their funded research (nearly twice the average rate across all types of funding agencies). Regulators have enforcement power through the implementation of legislation, although Chan noted that their authority only extends to products they regulate. IRBs can also require adherence to guidelines such as SPIRIT.
SPIRIT Electronic Protocol Tool and Resource
While the SPIRIT explanation and elaboration paper provides examples of good protocol reporting, Chan said the initiative believed it was
important to build the capacity for adherence by developing a software tool and to incentivize use by providing time-saving tools. The SPIRIT Electronic Protocol Tool and Resource (SEPTRE) “aims to help investigators author their protocols more efficiently while adhering to the SPIRIT guidance,” Chan said. He briefly showed participants how SEPTRE would be used to create and manage a clinical trial protocol. Drop-down menus facilitate entry of the protocol information in accordance with the SPIRIT checklist, and additional information and model examples are available. When all checklist information is entered, SEPTRE generates a formatted protocol document. SEPTRE also includes time-saving features, such as the ability to easily upload protocol information to ClinicalTrials.gov, which Chan said “reduces the registration time from hours to minutes.” Another feature is the ability to automatically track protocol amendments and easily carry them through to the next protocol version.
THE INSTITUTION’S ROLE IN IMPROVING REPRODUCIBILITY
Geeta Swamy, Vice Dean for Scientific Integrity and Associate Vice President for Research, Duke University
Although it is always hoped that people know what is right and will ultimately do what is right, things can still go wrong, Swamy said, and the stakes can be high for individuals, researchers, and institutions. Regardless of whether problematic scientific research is the result of mistakes or misconduct, the consequences can include harm to research participants, loss of research funding, data loss and publication retraction, inability to recruit top faculty and students, loss of sponsor confidence, and loss of public trust, Swamy said (see Figure 4-2).
Swamy mentioned two recent cases at Duke that demonstrate the impact of research misconduct. The omics case was covered widely in scientific journals and the news media.4 The case spanned 2006 through 2015, led to 11 retractions, and cost $10 million. Swamy said Duke runs a workshop to educate trainees and early career faculty about the omics case as many have not heard of it. The second example, a whistleblower case involving pulmonary medicine research, occurred simultaneously, but has been in the news more recently due to the time line of the investigation. This case spans 2005 to 2019 and led to 17 retractions, and the settlement cost the university $112 million.
___________________
4 A detailed case history is presented in Appendix D of the National Academies consensus study report Fostering Integrity in Research (NASEM, 2017).

SOURCE: Swamy presentation, September 25, 2019.
The Duke Research Integrity Culture
Swamy observed that a common theme in the workshop discussions thus far is the need for cultural change. As discussed, providing tools and resources for investigators, faculty, and trainees is important, she said, but for systematic change to occur across sectors and geographies, people must buy in to the need for change. To promote this cultural change, Duke has outlined key principles of a culture of research integrity. Swamy summarized that the research culture should be
- Inclusive, engaging all stakeholders in the process;
- Comprehensive, providing education, oversight, and accountability;
- Multifaceted, taking a holistic approach across all dimensions of research integrity;
- Pragmatic, providing the tools and resources needed to make it easier to “do the right thing”; and
- Empowering, enabling the research community and all stakeholders to speak up about concerns.
In its effort to support a culture of research integrity, the Duke Office of Scientific Integrity is focused on five main areas: (1) education and resources to translate the principles of integrity into routine practice; (2) standardized data management practices; (3) responsible conduct of research training for the more than 7,000 people at Duke who are engaged in research; (4) quality management strategies for both clinical and preclinical research; and (5) incident response and issue resolution. Although
there are cases of misconduct, she said most incidents can be attributed to missteps, a lack of awareness, or a lack of resources.
Foundational Principles and Initiatives for Rigor, Reproducibility, and Transparency
Drawing from the work of Arturo Casadevall, Duke developed its initiatives for rigor, reproducibility, and transparency in scientific research around four foundational principles: education and training, best practice, culture and accountability, and scientific and analytical excellence (see Casadevall and Fang, 2016). Swamy described some of the initiatives in each area.
Education and Training
Education and training initiatives include responsible conduct of research (RCR) and rigor, quality, and reproducibility (RQR) training for all faculty, staff, and administrators. There are monthly town hall meetings and interactive workshops to support and promote open dialogue on integrity. Despite initial hesitation, Swamy said these are well attended and provide an opportunity for discussion of ideas and concerns. Educational resources are also provided, including an interactive board game to engage graduate students and postdoctoral fellows, and an RCR/RQR toolbox with materials for journal club-style activities that investigators can do with their laboratory members. Swamy said the intent was to empower investigators with interactive, hands-on learning resources for their trainees rather than “one more electronic module to check a box” for compliance.
Best Practice
Among the best practices implemented are electronic research notebooks for preclinical research. Swamy explained that this is a centralized, auditable system that allows for preservation and tracking of data. Data management strategies have been implemented that require all laboratories to have a data management plan, including provisions for auditing. To ensure that trainees have adequate statistical support for their analyses, they are required to use the Duke statistics core.
Culture and Accountability
As mentioned, a key principle of establishing a culture of integrity and accountability is empowering the community to be able to voice concerns, Swamy reiterated. Duke also requires every department, center,
and institute to develop a science culture and accountability plan that establishes expectations of professionalism for all personnel. Compliance and oversight activities are coordinated, and there are regular communications among the units so that these activities are perceived as useful services and not as burdensome. Duke has also established a review and resolution mechanism for situations that are not already addressed by policies or regulations.
Thomas Curran asked how researchers could get more information to be able to implement the strategies discussed by Swamy at their own institutions. Swamy referred participants to a recent open-access publication describing the Duke RCR program5 as well as her presentation from the May 2019 Research Integrity Conference.6 She said that RCR is itself a field of research and RCR initiatives should be systematically evaluated and published so that institutions can share and implement successful strategies.
Scientific and Analytical Excellence
Swamy listed several of the Duke initiatives to promote scientific and analytical excellence. There is central review of the shared resources and core laboratories; systematic review of high-risk, high-profile investigator-initiated studies (e.g., first in human, rare disease); and external review of research programs with potential conflicts of interest (e.g., intellectual property, equity). Investigator-initiated clinical research is also subject to quality monitoring and risk-based monitoring, she said.
FUNDER/FOUNDATION ROLE IN INFLUENCING AND ENABLING REPRODUCIBILITY
Magali Haas, Chief Executive Officer and President, Cohen Veterans Bioscience
The mission of Cohen Veterans Bioscience is to accelerate the development of diagnostics and therapeutics for posttraumatic stress disorder (PTSD) and traumatic brain injury, Haas said.7 There are few U.S. Food and Drug Administration–approved diagnostics and therapeutics for PTSD and mild traumatic brain injury. Although there are products in
___________________
5 Available at https://www.tandfonline.com/doi/full/10.1080/08989621.2019.1621755 (accessed November 20, 2019).
6 Agenda available at https://www.researchintegrity.northwestern.edu/2019conference (accessed January 13, 2020).
7 For more information, see https://www.cohenveteransbioscience.org (accessed November 20, 2019).
the development pipeline, progress has been slow. There is much that is still not understood about the biology of these disorders, Haas said, but at the root of the slow progress are issues of reproducibility, robustness, and rigor. As a research funder, Cohen Veterans Bioscience requires quality data to make investment decisions.
Drawing on her background in pharmaceutical research and development and translational medicine, Haas recognized that the fundamental platforms and infrastructure needed to advance the field of traumatic brain injury and PTSD research were lacking. The approach of Cohen Veterans Bioscience is to work with strategic partners across industry, academia, and foundations to build those enabling platforms and foster a team science approach to accelerating product development (see Figure 4-3). “Reproducibility, robustness, and rigor are at the core of everything we do,” she said.
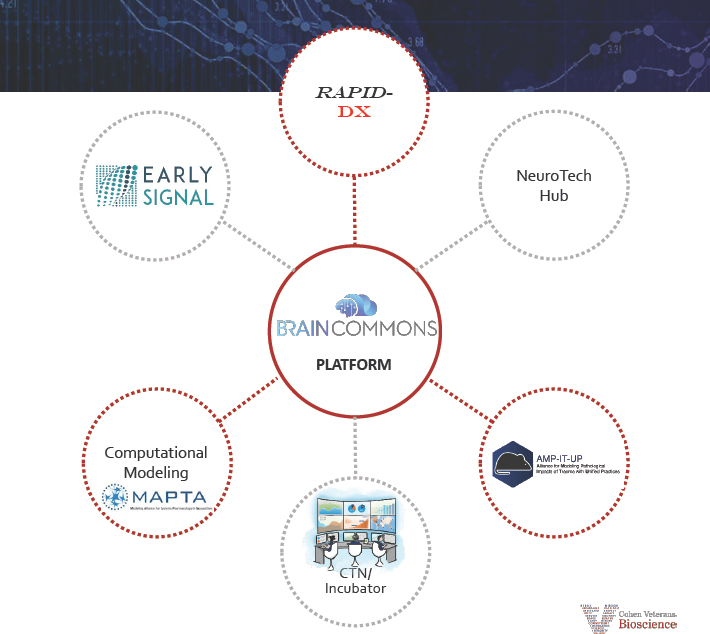
SOURCE: Haas presentation, September 25, 2019.
Enabling Solutions
In 2005, Haas said, researcher John Ioannidis drew attention to the potential lack of replicability in published scientific studies (Ioannidis, 2005). Subsequent reports from industry indicated that pharmaceutical scientists were often unable to reproduce the data from studies in the literature (Prinz et al., 2011). In 2014, the National Institutes of Health (NIH) committed to taking action to enhance reproducibility (Collins and Tabak, 2014), and a range of initiatives were then launched by scientific societies, foundations, and other stakeholders. Haas described several initiatives that Cohen Veterans Bioscience has established or helped support as examples of approaches that can be adopted to influence and/or enable reproducibility.
Preclinical Data Forum
Cohen Veterans Bioscience is a grant sponsor and founding member of the Preclinical Data Forum, a network of the European College of Neuropsychopharmacology (ECNP) with more than 50 academic, industry, publishing, and research member organizations.8 The Preclinical Data Forum focuses on enhancing reproducibility in preclinical neuroscience research. Early work sought to identify the problems and root causes, Haas said, and the focus is now on developing initiatives to address these issues.
One initiative resulting from Preclinical Data Forum efforts and described by Haas is the European Quality in Preclinical Data (EQIPD) project, which is funded by the Innovative Medicines Initiative and is focused on improving the quality of preclinical research data.9 Another activity is the sponsorship of workshops and training programs for early career investigators on reproducibility and rigor in research. The Preclinical Data Forum has also published guidelines and checklists such as a Consensus Preclinical Checklist for information related to the use of rodents in research. Cohen Veterans Bioscience also funded a $10,000 prize, awarded through the Preclinical Data Forum, for the best negative data publication. The intent of the prize, Haas explained, is to incentivize the sharing of negative results. “The literature is extraordinarily biased toward the positive findings,” she said, and as a funder investing in research, Cohen Veterans Bioscience believed it was important to incentivize a more balanced view of the data.
___________________
8 Further information on the ECNP Preclinical Data Forum network is available at https://www.ecnp.eu/research-innovation/ECNP-networks/List-ECNP-Networks/Preclinical-Data-Forum (accessed December 14, 2019).
9 Further information on the EQIPD project is available at https://quality-preclinical-data.eu (accessed December 14, 2019).
AMP-IT-UP Consortium
The AMP-IT-UP Consortium, launched by Cohen Veterans Bioscience, brings together academic and clinical researchers to address the robustness, reproducibility, and translational validity of preclinical models of brain trauma. One activity of the Consortium that Haas described is a partnership with Psychogenics, a clinical research organization (CRO), to “industrialize” animal models for PTSD and traumatic brain injury: Models developed and used in an academic setting are generally not validated for translational applications by industry, she explained, and the CRO can conduct preclinical modeling at scale. Thus far, however, efforts to reproduce some of the 16 available animal models of PTSD have been unsuccessful due to inconsistent reporting and non-standardized methods. Haas also raised the importance of funding enabling technologies that can be mass produced for use by researchers, as is being done in a consortium with the Wellcome Trust, IMEC, and the Howard Hughes Medical Institute to support a multi-channel “preclinical nanoprobe” to improve quality of the data being collected.
BRAIN Commons
Cohen Veterans Bioscience is also focused on improving data sharing and openness by sponsoring the BRAIN Commons, a cloud-based platform for data sharing of preclinical, clinical, omics, imaging, neuroimaging, and other data, Haas said. This platform, based on an open source code model, houses data in a secured environment and enables data integration and analysis across datasets and cohorts. BRAIN Commons team members are also working with NIH on incorporating preclinical common data elements. BRAIN Commons provides the infrastructure for Cohen Veterans Bioscience to require its grantees to share their results with the research community, Haas said.
Funder’s Levers
In closing, Haas shared her advice regarding the levers that funders can use to influence and enable rigor, robustness, and reproducibility:
- Use the Request for Applications process as an opportunity to give feedback about the robustness of the proposed methods. Haas added that blinding grant reviewers resulted in funding decisions based on the quality of the proposal, not the investigator.
- Have intramural experts work collaboratively with the grantees and partner institutions to improve the translational potential of their research. Haas said the experts at Cohen Veterans Bioscience
-
have broad industry experience in areas such as data science, methodology, diagnostics development, and preclinical modeling.
- Ensure that a statistical analysis plan is developed at the outset and is executed. Haas noted that Cohen Veterans Bioscience provides statistical support for grantees because many do not have access to statistical expertise.
- Provide the tools and training needed for conducting reproducible research (e.g., the Preclinical Checklist).
- Require and provide a platform for real-time data transfer of all results (e.g., the BRAIN Commons), Haas said, and require data sharing as part of the grant agreement and enforce it by linking to milestone payments.
- Ensure sufficient funding is awarded to cover costs such as data curation and management and open publication fees.
- Invest in consortia, education, training, technologies, and platforms.
DISCUSSION
Priorities
Kiermer prompted participants to identify priorities for preclinical research. Swamy said a preproject registration process would be helpful. Haas emphasized the need for adequate funding to conduct appropriately powered experiments. There is a variety of reasons for reducing the number of animals used in a study, she said, but it is also important to remember that underpowered studies are often uninterpretable. Chan said the current practice of registration of clinical trials demonstrates the role journals can play in leading change. He suggested that journals could require preclinical protocols to be preregistered and protocols to be submitted with the manuscript reporting the final results. Journals could also develop guidance for drafting a well-defined preclinical protocol. He added that there needs to be appropriate funding and infrastructure to support researchers in meeting these requirements. Steven Goodman suggested that the highest priority for preclinical research is design. He noted that many preclinical scientists were not taught proper study design, resulting in problems from the start. He added that a survey of doctoral students found that many were taking high-level statistics analysis courses (e.g., neural networks), but had a limited statistical foundation. Chan agreed that training in study design is essential for both preclinical and clinical research, but that currently there are limited training requirements in study design for someone to conduct research studies. A participant noted the importance of recording metadata for use in data analysis. Swamy agreed and reiterated that Duke trainees are required to consult
with the Duke statistics core to ensure they have the statistical capability for analysis of their data.
Protocol Development and Review
Panelists expanded on the role of IRBs in supporting transparency. Chan reiterated that IRBs can influence the design, conduct, and reporting of protocols by setting conditions such as trial registration or adherence to reporting guidelines. He noted, however, that the regulation of IRBs is highly variable across countries. In the United Kingdom, for example, research ethics committees are accountable to a national regulatory body that sets policy. In the United States, IRBs are independent and there is no central governing body. Chan observed that some IRBs require trial registration as a condition for review, and he encouraged IRBs to also recommend adherence to reporting guidelines, such as SPIRIT, to improve the content of submissions. He reiterated that is important to promote reporting guidelines to researchers as beneficial and not burdensome, in that submitting high-quality protocols facilitates more rapid review and leads to fewer queries and requests for revisions.
Swamy agreed and shared that a recent, in-depth review of the Duke Health IRB found delays were associated with the content and quality of the submissions. She suggested, however, that the primary role of an IRB is to be experts in research participant protections. IRBs are not experts in contractual publication requirements or managing conflicts of interest, nor should they be, and she raised concern about holding IRB approval “hostage” to these other interests. Duke has implemented an online tracking process for its IRB approval that allows researchers to see when each part of their submission is approved, and which sections are causing delays. The most common cause of delays is contractual agreements, she said, and she encouraged the use of standard contractual agreements, such as the Accelerated Clinical Trial Agreement that was developed by a group of Clinical and Translational Science Awards (CTSA) program investigators.10
Chan agreed that IRBs are overburdened and underresourced, and suggested that a separate institutional arm could take on the review of protocols for quality and transparency elements. He noted that some investigators question why the quality of the science in their submissions is being examined in an ethics review. Chan said quality and ethics are closely related, and a poorly designed study can be unethical.
Swamy pointed out that the investigator is not necessarily the author of the protocol. In industry, for example, the protocol might be written
___________________
10 For model agreements, see https://www.ara4us.org (accessed November 20, 2019).
by someone with specialized expertise in the regulatory requirements, working in collaboration with the investigators. She said protocols written with the assistance of a regulatory coordinator tend to progress more quickly through IRB review than an early career investigator-initiated and -written protocol. Haas agreed and added that many early career faculty have no background or experience in topics such as informed consent.
Chan described a study comparing the information in IRB-approved clinical trial protocols to information for the same trials in the clinical trial registry. For more than one-fifth of the studies evaluated, the primary outcomes in the protocol did not match those entered in the trial registry. He suggested that concordance of this information should be checked prospectively or made public so that those interested could conduct their own assessments. In addition, he said that using an electronic tool as the database for protocol information, such as SEPTRE, could help to ensure that information is consistent because protocol information can be exported from the tool to the IRB and the trial registry.
Haas raised the possibility of linking final publications back to the original protocol. Chan referred to the work of Altman on the concept of threaded publications, linking all publications that stem from a given protocol/registered clinical trial. Chan and Swamy mentioned the concept of a common identifier or “universal object identifier” for protocols that could be preserved across the different platforms used by journals, IRBs, and others.
Goodman asked how protocol review could be made a routine part of funding decisions, and how adherence to proposed protocols might be monitored after grants are awarded. Swamy noted that there are new requirements for the Human Subjects and Clinical Trials Information Form to be submitted to NIH with proposal and grant applications. Although a full protocol is not required, she said that a synopsis, primary outcome, and eligibility criteria must be included. She did not know the extent to which adherence to the proposed plans would continue to be evaluated over the course of the grant, but suggested that this information could be part of the annual progress report. Swamy noted that the recent update to the Common Rule removed the requirement to determine concordance between the grant and the protocol. This was done, in part, because protocols can change between when the grant application is submitted and when IRB approval is received. She suggested there is a need to find other ways to ensure consistency.
Funding for Development and Sustainability
Leslie McIntosh, co-founder and CEO of Ripeta, asked about securing funding to implement automated solutions. Chan said the develop-
ment of the SEPTRE tool was funded by interested government entities, including the Canadian Institutes of Health Research, the National Cancer Institute of Canada, and the Canadian Agency for Drugs and Technologies in Health. He noted that securing grants for the development of tools is challenging because these types of projects are not answering a research question. Once developed, it is difficult to fund the maintenance of the tools. One approach under consideration is a subscription model, Chan said, adding that researchers from lower income countries could access the tool for free. He noted that one potential source of support is investment by institutions that recognize the importance of promoting transparency and improving completeness of protocols.
Swamy said Duke leverages CTSA funds for new initiatives, but noted that CTSA funds are for developing solutions, not maintaining them once they are operational. Once a new initiative is operational, she said, a business proposal is presented to the leadership of the school of medicine and the university. All programs are assessed for impact, effectiveness, and acceptability, and that information is used to support the case for internal funding to support these programs.
Investigating Misconduct
Yvette Carter, health scientist administrator in the Office of Research Integrity (ORI) at NIH, said that like the journals, ORI does not conduct investigations. They are conducted by the awardee’s institution. She suggested that instead of contacting authors with concerns of misconduct, journal editors could contact the research integrity officer at the author’s institutions, or ORI at NIH, as appropriate. She urged contact “earlier rather than later.” She added that ORI can provide image analysis expertise to assist in investigations. Carter asked Swamy what lessons from the recent misconduct investigations at Duke could the ORI Division of Education and Integrity share to improve research integrity. The most recent case involved the “overwhelming fabrication of data,” Swamy said. While it is always exciting for investigators to get good results, she emphasized the importance of looking at raw data, test results, and data outputs, not just the final tables or figures or summary data for inclusion in a manuscript, to confirm that the findings make sense.
Common Data Elements
Stuart Hoffman, scientific program manager for the Department of Veterans Affairs’ Office of Research and Development, shared his experience with the development of common data elements for traumatic brain injury and noted the challenges of coming to consensus on those ele-
ments. As common data elements are now being considered for preclinical research, he raised the issue of novel methods that do not conform to common data elements, and the possibility that research could be steered toward techniques for which common data elements do exist.
Haas said common data elements are essential to enable the sharing and analysis of data across systems. Data platforms, such as the BRAIN Commons, rely on common data elements and standards, she said. She agreed that development of common data elements for preclinical research will be challenging, and noted that NIH has assembled a preclinical common data elements working group. The extent to which the common data elements are adopted depends on incentives and enforcement (“carrots and sticks”). She noted the need to educate the research community on the benefits of using common data elements for reproducibility and independent validation of studies.
This page intentionally left blank.


















