
Page 159
7—
Fats and Other Lipids
Lipids are compounds that are insoluble in water but are soluble in organic solvents such as ether and chloroform. Lipids that are important to our discussion include fats and oils (triglycerides or triacyglycerols), fatty acids, phospholipids, and cholesterol.
Fats and oils are esters of glycerol and three fatty acids. They are important in the diet as energy sources and as sources of essential fatty acids and fat-soluble vitamins, which tend to associate with fats. They also contribute satiety, flavor, and palatability to the diet.
Fatty acids generally consist of a straight alkyl chain, terminating with a carboxyl group. The number of carbons in the chain varies, and the compound may be saturated (containing no double bonds) or unsaturated (containing one or more double bonds). Short- and medium-chain saturated fatty acids (SFAs) (4 to 12 carbons in length) are found in milk fat, palm oil, and coconut oil. Other animal and vegetable fats contain predominantly longer-chain SFAs (more than 14 carbons in length) and are found chiefly in meats, butterfat, and some vegetable oils. Monounsaturated fatty acids (MUFAs), such as oleic acid, contain one double bond per molecule, whereas polyunsaturated fatty acids (PUFAs), such as linoleic acid, contain more than one. Linoleic acid is classified as an essential nutrient, since the body requires it but cannot synthesize it. Arachidonic acid is also required by the body but can be synthesized from linoleic acid, which is abundant in oils from corn, soybeans, and safflower seeds.
Linoleic acid (18 carbons with 2 double bonds) and arachidonic acid (20 carbons with 4 double bonds) belong to the omega(w)-6 group of fatty acids, since the first double bond, counting from the methyl end of the molecule, occurs at carbon number 6. Since linoleic acid has 18 carbon atoms and 2 double bonds, it is usually represented in shorthand as C18:2, w-6. Under this classification system, oleic acid (C18:1, w-9) belongs to the w-9 group, and the PUFAs in fish oils currently receiving much attention belong to the w-3 group. Chief among these w-3 fatty acids are eicosapentaenoic acid (EPA), which has 20 carbons and 5 double bonds (C20:5, w-3), and docosahexaenoic acid (DHA), which has 22 carbons and 6 double bonds (C22:6, w-3).
A growing body of evidence from studies in animals, including nonhuman primates, indicates that a-linolenic acid, or its longer-chain derivates EPA and DHA, are essential in the diet. These fatty acids appear to play distinctive roles in the structure and function of biologic membranes in the retina and central nervous system (Neuringer and Connor, 1986).
Unsaturated fatty acids form geometric isomers, i.e., the carbon chains are on the same side of the double bond in a cis isomer and on opposite sides of
Page 160
the bond in a trans isomer. Naturally occurring geometric isomers in food are mainly cis isomers, but hydrogenation of oils in the manufacture of margarine and shortening results in formation of some trans isomers. This latter process occurs naturally in the rumen of ruminants.
Phospholipids contain glycerol, fatty acids, phosphate, and, with such exceptions as phosphatidylglycerol and phosphatidylinositol, a nitrogenous component. Lecithin, for example, is made up of glycerol, two fatty acids (one saturated, usually), phosphate, and choline. Phospholipids are important structural components of brain and nervous tissue, of membranes throughout body tissues, and of lipoproteins—the carriers of cholesterol and fats in the blood.
Cholesterol and plant sterols, such as sitosterol, are high-molecular-weight alcohols with a characteristic cyclic nucleus and are unrelated to the structure of fats or phospholipids. Cholesterol frequently exists in foods and body tissues esterified to one fatty acid per molecule. It is a component of membranes in body cells and is required for normal development of the brain and nervous tissue. Furthermore, it is the precursor to bile acids, steroid hormones, and 7-dehydrocholesterol in the skin, which in turn is the precursor to vitamin D.
Cholesterol occurs naturally only in foods of animal origin. The highest concentrations are found in liver and egg yolk, but red meats, poultry (especially the skin), whole milk, and cheese make significant contributions to the diet.
Trends in the Food Supply
Trends in the quantities of lipids present in the food supply have been recorded by the U.S. Department of Agriculture (USDA) since 1909. These data represent amounts of lipids that "disappear" into wholesale and retail markets. No account is taken of amounts wasted, and no effort is made to measure intakes by individuals. Thus, food supply data do not represent amounts of lipids actually consumed and are referred to here as amounts available for consumption (see Chapters 2 and 3). Total amounts available are divided by the U.S. population to obtain amounts per capita. The following data on time trends were obtained from Marston and Raper (1987).
Fat available in the food supply increased from an average of 124 g/day per capita in 1909 to 172 g/day in 1985. Although the chief sources of fat during that time have been fats and oils; meat, poultry, and fish; and dairy products, great changes within each of these groups have occurred. The proportion of animal fat declined from 83 to 58% as butter and lard use declined, whereas the proportion of vegetable fat (in margarines and in salad and cooking oils) rose from 17 to 42%. Pork and beef have been major sources of fat in the food supply since 1909, but supplies of beef have declined somewhat recently from 90.7 lb/year per capita in 1975 to 79 lb/year in 1985. The supply of poultry has increased spectacularly since 1940 and continues to increase. In 1985, 70 lb per capita were available compared with 46 lb per capita from 1967 to 1969. The per-capita availability of whole milk dropped from 232 lb during 1967-1969 to 122 lb in 1985, whereas skim and low-fat milks increased from 44 to 112 lb (see Chapter 3).
Fatty acids available in the food supply have all increased since 1909, but the relative contributions of specific fatty acids have changed. The percentage of calories contributed by linoleic acid to total fat intake increased from 7% during 1909-1913 to about 16% in 1985, whereas the corresponding percentage from SFAs declined from approximately 42 to 34% (Figure 3-5). In 1985, linoleic acid was available at 7% of total calories, SFAs at 15%, and oleic acid at 17%.
Cholesterol availability reached its lowest levels of 464 mg/day per capita in 1917 and 1935, and its highest level of 596 mg/day in 1945. The supply declined to 480 mg/day per capita during 1977-1979, when it plateaued; the decline was due to diminished use of whole milk, butter, eggs, and lard. Food sources of cholesterol have changed somewhat over the century. In 1909, meat, poultry, and fish furnished 28% of the cholesterol in the food supply; in 1985, they supplied 40%. Fats and oils supplied 12% of the total cholesterol in 1909, but only 5% in 1985. Egg use has declined, but in 1985 still supplied 40% of the cholesterol in the food supply (see Chapter 3).
Lipid Intake: National Surveys
Actual intakes of various lipids have been estimated in national surveys, but the different surveys fail to agree on trends in actual consumption of fat. Data from the USDA's Nationwide Food Consumption Surveys (NFCS) of 1955, 1965, and 1977-1978 show little change in fat levels used by households, but mean individual intakes were lower during 1977-1978 than in 1965 (USDA, 1984). Furthermore, compared with 1977-1978, a decline in fat intake was indicated in the 1985 and 1986 USDA Continuing Survey of Food Intakes of
Page 161
Individuals (CSFII) (USDA 1986, 1987). On the other hand, data from the National Health and Nutrition Examination Surveys (NHANES) do not support a decline in fat intake. For example, data from the first and second NHANES (19711974 and 1976-1980, respectively) indicate that for women 19 to 50 years of age, mean fat intakes remained stable during the 1970s and early 1980s (see Table 3-4). Systematic biases due to methods used in the surveys appear to explain these differences in estimates of fat intake. For example, in the 1985 and 1986 CSFIIs, interviewers tried to determine whether or not fat was trimmed from meat and the skin removed from poultry before these foods were consumed, but this was not done in the 1977-1978 CSFII.
The 1985 and 1986 CSFIIs indicated that fat provided an average of 36 to 37% of total calories for men and women and 34% for children. The 1977-1978 NFCS reported an average of 41% of total calories as fat for women in this age group, but as noted above, the results of this survey were higher than those of other surveys.
On the basis of 4 nonconsecutive days of intake by women and their children, and on 1 day of intake by men, men and women 19 to 50 years of age consumed a mean of 13% of total calories from SFAs, 14% from MUFAs, and 7% from PUFAs. Children 1 to 5 years old consumed a mean of 14% of calories as SFAs, 13% as MUFAs, and 6% as PUFAs (USDA, 1986, 1987).
The daily intake of cholesterol averaged 280 mg for women 19 to 50 years old (187 mg/1,000 kcal) and 223 mg for children 1 to 5 years old (156 mg/ 1,000 kcal) (USDA, 1987). Intakes for men 19 to 50 years old averaged 439 mg/day (180 mg/1,000 kcal) (USDA, 1986). Cholesterol intake was higher in low-income groups than in high-income groups; black women had higher intakes than white women, but white men had higher intakes than other men.
In the 1977-1978 NFCS, people from infancy to 75 years of age and older averaged 385 mg of cholesterol per day (USDA, 1984). Dietary cholesterol levels, in absolute amounts and in mg/ 1,000 kcal, were higher for blacks, for those below the poverty level, for those living in the South and West, and for those living in inner cities.
In the 1985 CSFII, dietary cholesterol came chiefly from meat (48% for men and 45% for women). Eggs provided 18% of the cholesterol intake for men and 15% for women, and grain products furnished 17% of the cholesterol intake for women and 14% for men, but these figures are somewhat misleading in that grain products furnished cholesterol only because they contained milk, butter, and eggs. The milk group provided 14% of the cholesterol intake for men and 16% for women.
Evidence Associating Dietary Fats and Other Lipids with Chronic Diseases
Atherosclerosis and Cardiovascular Disease
Arterial lesions characterized by intimal thickening, lipid accumulation, and calcification in humans were identified and described at least as early as the seventeenth century. The lesions were named arteriosclerosis in 1829, and the distinctive form associated with lipid deposition was named atherosclerosis in 1904. However, atherosclerosis was not considered a common cause of death until decades after Herrick (1912) linked coronary atherosclerosis to thrombosis and myocardial infarction. Coronary heart disease (CHD) reached epidemic proportions in the United States before dietary fats were seriously suspected of being causative agents around 1950.
The first recorded evidence that diet had any association with atherosclerosis was the observation by Ignatovski (1908) that rabbits fed meat, milk, and eggs developed arterial lesions resembling atherosclerosis in humans. Anitschkow and Chalatow (1913) then identified cholesterol as the dietary component responsible for hypercholesterolemia and atherosclerosis in rabbits. In subsequent years, investigators demonstrated that many animal species were susceptible to dietary cholesterol, but this phenomenon was considered a laboratory curiosity that had no relevance to human nutrition nor to the rising incidence of CHD and related diseases in the Western world during the first half of the twentieth century.
De Langen, a Dutch physician working in Java, reported in 1916 that native Indonesians had lower levels of plasma cholesterol than did colonists from the Netherlands and associated this observation with a much lower frequency of CHD in the natives. He also observed that Javanese stewards on Dutch passenger ships who ate typical Dutch food had high plasma cholesterol levels and precocious CHD. These observations, published in Dutch in an obscure journal (De Langen, 1916, 1922), lay unnoticed for more than 40 years. In 1934, Rosenthal noted that the distribution of
Page 162
atherosclerosis and atherosclerotic diseases in many parts of the world corresponded to the consumption of fats and cholesterol (Rosenthal, 1934a,b,c).
The next published association of dietary fats with atherosclerosis and cardiovascular disease was the comment by Snapper (1941), based on his experience at Peking Union Medical College, that the high unsaturated fatty acid and low cholesterol content of the Chinese diet might be responsible for the remarkable scarcity of arteriosclerosis in China. This observation also lay unnoticed until after World War II, when reports that deaths from cardiovascular disease, especially those due to arteriosclerosis, declined dramatically in Scandinavia during the war when meat, eggs, and dairy products were scarce (Biörk, 1956; Malmros, 1950; Strøm and Jensen, 1951; Vartiainen, 1946; Vartiainen and Kanerva, 1947).
During the same period, a number of case-control studies of patients with myocardial infarction showed that affected people had higher serum cholesterol levels than did controls (Davis et al., 1937; Gertler et al., 1950b; Lerman and White, 1946; Morrison et al., 1948; Poindexter and Bruger, 1938; Steiner, 1948). The association of serum cholesterol concentrations with atherosclerosis and myocardial infarction was widely recognized by 1946 (Dock, 1946). The predictive value of serum cholesterol concentration for CHD was firmly established by the Framingham Study in 1957 (Dawber et al., 1957) and was confirmed by many similar longitudinal studies in the 1950s and 1960s (Pooling Project Research Group, 1978).
Nevertheless, appreciation of the relationship of diet to serum cholesterol levels, and thereby to CHD, developed more slowly. Although there had been an accumulation of epidemiologic evidence (mainly ecological correlations) supporting the concept that diet, especially dietary fat, was associated with elevated serum cholesterol concentrations and with CHD (Keys, 1957; Keys and Anderson, 1954), there also was much skepticism, as illustrated by the comments of Yudkin (1957), Yerushalmy and Hilleboe (1957), and Mann (1957).
Early in the 1950s, the serum-cholesterol-lowering effects of PUFAs were discovered, and epidemiologic and human experimental studies were focused on this issue. The role of dietary cholesterol remained uncertain until the 1960s, when several careful experiments in humans showed that it had a modest but definite effect. The early development of these concepts, along with the controversies, are found in reviews of the topic by Keys (1957, 1975) and Ahrens (1957). Subsequent sections of this chapter review in detail the epidemiologic and experimental evidence on the relationship between serum cholesterol and CHD, between diet and serum cholesterol, and between diet and CHD.
Plasma Lipids and Lipoproteins
Lipids are insoluble in water and circulate in plasma in association with certain specific proteins called apolipoproteins. The lipoproteins are large, macromolecular complexes of apolipoproteins and lipids in varying proportions. The four classes of specific lipoproteins that circulate in plasma are called chylomicrons, very-low-density lipoproteins (VLDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL).
The primary function of plasma lipoproteins is lipid transport. The major lipid transported in lipoproteins—triglyceride—is only slightly soluble in water, yet up to several hundred grams must be transported through the blood daily. Hence, transport mechanisms have evolved to permit the packaging of thousands of triglyceride molecules in individual lipoprotein particles, which deliver the transported lipid to specific cells. Fatty acids esterified to glycerol constitute approximately 90% of the mass and about 95% of the potential energy of the triglyceride molecule. Free fatty acids are transported in noncovalent linkage as albumin-fatty acid complexes. This latter mode of transport does not permit the high degree of selective targeting of fatty acids to specific sites that is permitted by transport in lipoproteins, but these two modes of fatty acid transport together provide a more versatile system for bulk movement of a major substrate for energy metabolism.
Cholesterol is the other major lipid transported in lipoproteins. It is not used for energy; it is the precursor of steroid hormones and bile acids and is a structural component of cellular membranes. In higher animals, including all mammals, it is transported mainly in the form of cholesteryl esters, which are synthesized in cells or in the plasma compartment itself. As with triglycerides, the transport of cholesteryl esters in lipoproteins permits specific targeting of cholesterol to tissues that require it for structural purposes or for making its metabolic products.
Two of the lipoprotein classes, chylomicrons and VLDL, are composed primarily of triglyceride.
Page 163
Chylomicrons transport exogenous (dietary) triglyceride, and VLDLs transport endogenous triglyceride. Chylomicrons are not normally present in postabsorptive plasma after an overnight fast. The VLDLs normally contain 10 to 15% of the plasma total cholesterol. LDLs contain cholesterol as their major component and normally contain most (60 to 70%) of the plasma cholesterol. HDLs are approximately half protein and half lipid and usually contain 20 to 30% of the total plasma cholesterol. Lipoproteins are lighter than the other plasma proteins because of their high lipid content. This characteristic permits both the operational classification and the ultracentrifugal separation of the different classes of lipoproteins.
Each lipoprotein class is heterogeneous in its protein constituents. Nine distinct apolipoproteins have been separated and described. Most investigators group the apolipoproteins into five families (designated apo A, apo B, apo C, apo D, and apo E) on the basis of their chemical, immunologic, and metabolic characteristics. Apo A refers to the apolipoproteins (apo A-I and apo A-II) that are primarily, but not exclusively, found in HDL. A third member of the apo A family, apo A-IV, is a minor component of chylomicrons. Apo B is the major apoprotein of LDL but also comprises about 35% of VLDL protein. There are two forms of apo B: a large form called apo B-100 and found in LDL and a smaller form called apo B-48 and produced mainly in the intestine. Apo C represents a group of apoproteins (apo C-I, apo C-II, apo C-III) that were originally described as major components of VLDL but that are also present as minor components in HDL. Apo D is a minor component of HDL. Apo E is a major component of VLDL and a minor one of HDL. The apolipoproteins serve both structural and functional roles. Some apoproteins are ligands for specific cell surface receptors, e.g., apo B-100 and apo E for the LDL (or apo B/E) receptor (Brown and Goldstein, 1986); others are cofactors for enzymes, e.g., apo C-II is a necessary activating cofactor for lipoprotein lipase. For reviews of lipoprotein structure and metabolism, see Havel (1987) and Stanbury et al. (1983).
Relationship of Plasma Lipid and Lipoprotein Levels to Atherosclerotic Cardiovascular Diseases
Most major epidemiologic studies have focused on white men, but a few have provided information about women and nonwhites of both sexes.
Total Cholesterol (Tc)
TC is used in this chapter as an abbreviation for the total cholesterol in either serum or plasma. TC concentration is usually expressed as milligrams of cholesterol per 100 ml of serum or plasma (mg/dl). TC concentrations in serum are about 2% higher than those measured in corresponding plasma (Folsom et al., 1983). Although this difference should be considered in comparing the results of studies with one another when numbers of subjects are large and small systematic biases might affect the comparison, it does not affect the major results or conclusions of studies discussed in this report in which serum or plasma is used in analyses of cholesterol. Thus, TC is used interchangeably for both serum and plasma total cholesterol.
Until the past decade, TC, rather than lipoprotein cholesterol, was measured in most epidemiologic studies because reliable methods for measuring lipoprotein cholesterol in large numbers of people were not available. Therefore, most data on disease risk are based on TC level.
Variation in Mean TC Among Populations
Meanlevels of TC vary widely among populations. In the Seven Countries Study, investigators studied 16 populations of middle-aged men residing in seven countries: Finland, the Netherlands, Italy, Yugoslavia, Greece, the United States, and Japan (Keys, 1970, 1980b). Examination methods, laboratory procedures, and quality control procedures were standardized. Mean TC for men ages 50 to 54 years varied from 157 mg/dl in a Japanese population to 262 mg/dl in eastern Finland (Keys, 1980b). In the Ni-Hon-San Study, three population-based samples of men of Japanese ancestry were compared. Mean TC levels for men ages 50 to 54 were 182, 219, and 228 mg/dl in Japan, Hawaii, and California, respectively (Nichaman et al., 1975). In the Israel Ischemic Heart Disease Study, the age-adjusted mean TC level in male civil servants age 40 and older varied from 195 mg/dl for those born in Africa to 219 mg/dl for those born in Central Europe (Kahn et al., 1969). Similar differences were found some 15 years later for male and female adolescents and adults in the Jerusalem Lipid Research Clinics Prevalence Study (Halfon et al., 1982a,b).
Other differences among populations have been observed for men in Puerto Rico, Hawaii, and Framingham, Massachusetts (Gordon et al., 1974), and for men and women in London, Naples, Uppsala, and Geneva (Lewis et al., 1978). Some of this evidence is reviewed in the report of
Page 164
a Conference on Health Effects of Blood Lipoproteins (1979). The results of these various studies, particularly the studies of migrants, indicate that the differences in mean TC levels among populations are due largely to environmental factors, principally diet, rather than to constitutional factors.
Large population differences in mean TC levels have also been observed among children and adolescents; the pattern of variation in these means closely parallels that of the adult values, but at lower absolute values (Conference on Blood Lipids in Children, 1983).
Variation in CHD Rates Among Populations
Large differences also exist among populations in the incidence of and mortality from CHD and in the prevalence and severity of atherosclerosis. For example, in the Seven Countries Study, age-standardized, 10-year incidence of first major CHD events (myocardial infarction and coronary death) among men free of CHD at entry varied from 3 in 1,000 on Crete to 107 in 1,000 in eastern Finland (Keys, 1980b). Corresponding figures for 10-year CHD mortality were 0 and 68 in 1,000, respectively. In the Ni-Hon-San Study, relative risks of first major CHD event were 0.46, 1.00, and 1.54 for the cohorts in Japan, Hawaii, and California, respectively (Kagan et al., 1981; Marmot et al., 1975; Robertson et al., 1977). Incidence of first major CHD events among middle-aged men in Framingham was twice that in Puerto Rico and Hawaii (Gordon et al., 1974).
Variation in Atherosclerosis Among Populations
Inthe International Atherosclerosis Project, the extent of atherosclerosis in the coronary arteries and aortas was measured in 23,207 autopsied people from 19 populations in 14 countries (McGill, 1968b). The mean percentage of intimal surface with raised lesions varied from 6% in Durban Bantu to 18% in New Orleans whites. Differences among populations were noticeable at ages 15 to 24 and marked at ages 25 to 34. With few exceptions, ranking the populations according to extent of raised lesions corresponded closely to ranking them by CHD mortality rate.
Correlations Between Mean TC and CHD Rates Among Populations
Variation in mean TC levels among populations is highly correlated with variation in CHD incidence and extent of atherosclerosis. The correlation coefficient for median level of TC with age-standardized, 10-year CHD death rates for 16 cohorts in the Seven Countries Study was .82 (Keys, 1980b). The correlations between median TC and national CHD death rates for these seven countries at 0, 5, 10, and 15 years after TC was measured were .86, .90, .93, and .96, respectively (Rose, 1982). In the International Atherosclerosis Project, there was a correlation of .76 between the extent of atherosclerosis and mean TC concentration in 19 populations (Scrimshaw and Guzman, 1968). Populations with mean TC levels less than 180 mg/dl are largely free of atherosclerosis and CHD, whereas those with mean TC levels above 220 mg/dl are characterized by high rates of CHD (Conference on Health Effects of Blood Lipoproteins, 1979). These results support the conclusion that variation in CHD rates among populations is determined predominantly by differences in levels of TC.
CHD Incidence and Mortality Among Individuals Within Populations
In prospective studies of middle-aged men, TC levels above 200-220 mg/dl are positively associated with risk of CHD in the United States (Pooling Project Research Group, 1978; Stamler et al., 1986) as well as in Norway (Holme et al., 1981), France (Ducimetiere et al., 1980), Japan (Johnson et al., 1968), Israel (Goldbourt et al., 1985), England (Rose and Shipley, 1980), Italy (Italian National Research Council, 1982), Finland, the Netherlands, Greece, and Serbia (Keys, 1980b). The association may be weak or absent in some populations with low mean levels of TC and low absolute risk of CHD, e.g., in rural areas of Bosnia and Croatia (Keys, 1980b; Kozarevic et al., 1976, 1981).
Results have been less consistent regarding the association of TC levels below 200 mg/dl with the risk of CHD. In fact, questions have been raised as to whether the association of serum TC with CHD risk is continuous or whether there is some level of serum TC below which it is not related to risk of CHD (e.g., Goldbourt, 1987). In four of the eight studies in the U.S. Pooling Project, age-standardized incidence of CHD for men ages 40 to 64 years was lower in the second quintile of serum TC (194 to 218 mg/dl) than in the first quintile (<194 mg/ dl) (Pooling Project Research Group, 1978). In the Israel Ischemic Heart Disease Study of 9,902 male civil servants 40 or more years old, age-standardized 15-year CHD mortality rates according to quintile of serum TC (<176, 176 to 197, 198 to 216, 217 to 241, and >241 mg/dl) were 4.5, 4.9, 4.4, 6.7, and 10.2 per 100, respectively (Goldbourt et al., 1985). In the same study, how-
Page 165
ever, the corresponding 7-year CHD mortality rates previously showed a steadily increasing pattern: 10, 12, 16, 17, and 30 per 1,000, respectively (Yaari et al., 1981). Also, in that study, the 5-year incidence rates for myocardial infarction were 29, 39, and 60 per 1,000 for men in the serum TC tertiles of 77 to 189, 190 to 219, and 220 to 500 mg/dl, respectively (Medalie et al., 1973).
Many other large prospective studies have also shown a clear monotonic association of CHD with TC levels below 200 mg/dl. In the Hiroshima Adult Health Study of 4,256 men age 40 and older, 6-year age-standardized CHD morbidity ratios associated with three levels of TC (<180, 180 to 219, and >220 mg/dl) were 72, 162, and 333, respectively (100 representing risk for the whole group) (Johnson et al., 1968). Ten-year CHD mortality rates among 17,718 British civil servants ages 40 to 64 according to quintile of TC (<159, 159 to 183, 184 to 203, 204 to 233, and >233) were 28, 34, 36, 44, and 54 per 1,000, respectively (Rose and Shipley, 1980). In the Framingham Study, the 20-year CHD incidence for men ages 33 to 49 according to level of TC (114 to 193, 194 to 213, 214 to 230, 231 to 255, and 256 to 514 mg/ dl) was 86, 153, 220, 268, and 306 per 1,000, respectively (Kannel and Gordon, 1982). Among 356,222 men ages 35 to 57 who were initially screened in the Multiple Risk Factor Intervention Trial, age-standardized 6-year CHD mortality increased steadily according to decile of TC from 3 per 1,000 for TC <168 mg/dl to 13 per 1,000 for TC >263 mg/dl (Stamler et al., 1986). The data for that trial are shown in Figure 7-1.
In that trial the 6-year mortality rate doubled between 153 and 226 mg/dl (3.16 to 6.94 per 1,000) and doubled again between 226 and 290 mg/dl (6.94 to 13.05 per 1,000). The weight of evidence supports the idea that TC level, at least from 150 mg/dl upward, is positively associated in a continuous fashion with CHD risk. Because the incidence of CHD is low at TC levels under 200 mg/dl, occasional exceptions to this rule are more likely due to statistical artifact than to biologic diversity. Results from the observations of screenees in the Multiple Risk Factor Intervention Trial also indicated that the association between TC and 5-year risk of CHD death for 23,490 black men was similar to that for 325,384 white men (Neaton et al., 1984).
The association between TC and the relative risk of CHD declines with age. In the pooled results of five U.S. studies, the relative risk of CHD in the highest quintile of TC (>256 mg/dl) compared to
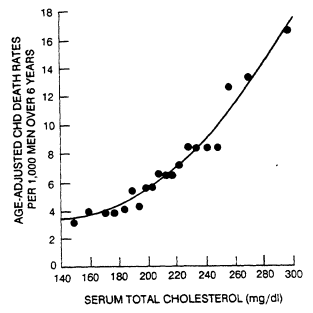
FIGURE 7-1
Relationship of serum cholesterol to CHD death in 361,662 men ages
35 to 57 during an average followup of 6 years. From Martin et al. (1986).
Each point represents the median value for 5% of the population.
the lowest quintile (<194 mg/dl) (defined as the ratio of the risks in these two groups) was 3.6, 1.9, 1.8, and 1.5 for men ages 45 to 49, 50 to 54, 55 to 59, and 60 to 64, respectively (Pooling Project Research Group, 1978). This observation has sometimes been misinterpreted to mean that the level of TC is relatively unimportant in elderly people. The committee believes that this misinterpretation may arise from failure to distinguish between the concepts of relative risk (the ratio between two risks) and attributable risk (the difference between two risks). The former is commonly used to evaluate the magnitude of an epidemiologic association; the latter is commonly used to evaluate its public health importance. In fact, in the set of studies cited above, the attributable risk (calculated as the difference in risk between the highest and lowest quintiles) did not vary consistently with age. Thus, the corresponding attributable risks were 46, 28, 40, and 42 per 1,000 in 8 years for men ages 45 to 49, 50 to 54, 55 to 59, and 60 to 64, respectively.
Atherosclerosis at Autopsy
TC measured by standardized procedures in apparently healthy men was strongly associated with extent of atherosclerosis at autopsy in the Hiroshima Adult Health Study (Rickert et al., 1968), Honolulu Heart Program (Reed et al., 1987; Rhoads et al., 1978),
Page 166
the Framingham Study (Feinleib et al., 1979), the Oslo Study (Holme et al., 1981), and the Puerto Rico Heart Study (Sorlie et al., 1981). There are few data on women.
In the Framingham Study, the extent of coronary atherosclerosis in men was positively correlated with TC measured 1, 5, and 9 years before death; in women, only TC measured 9 years before death but not TC measured 5 and 1 years before death correlated significantly (Feinleib et al., 1979). In the Bogalusa Heart Study, TC measured at 3 to 18 years of age was correlated (R = .67) with percentage of aortic intimal surface involved with fatty streaks as measured during the autopsies of 35 people who died between 7 and 24 years of age (Newman et al., 1986; Strong et al., 1986). Correlations with raised lesions and with lesions in the coronary arteries could not be reliably determined in this series because only 6 people had fibrous plaques and the extent of coronary intimal surface involved in fatty streaks was small, varying only from 0 to 6% (mean, 1%). In a follow-up of the initial report, Freedman et al. (1988) increased the number of cases to 44 and examined the relationship within races. As anticipated, blacks had more extensive fatty streaks than did whites, but there also was a strong positive association between the extent of aortic fatty streaks and the LDL cholesterol concentration within each race group.
In summary, epidemiologic findings among populations and for individuals within populations consistently indicate a strong, continuous, and positive relationship between TC levels and the prevalence and incidence of, as well as mortality from, atherosclerotic CHD. This relationship has been confirmed in autopsy studies.
LDL and HDL Cholesterol
There is less epidemiologic evidence on lipoprotein cholesterol than on TC. Few early studies included measurements of lipoprotein cholesterol because of technical difficulties and cost.
Differences Among Populations
Lipoprotein cholesterol was not measured in the Seven Countries Study; thus, variation among populations is less well characterized for LDL cholesterol (LDL-C) and HDL cholesterol (HDL-C) than for TC. Apparently, average values of LDL-C vary widely among populations, but the evidence is less clear for HDL-C. In the Four European Communities Study, mean LDL-C varied from 116 to 168 mg/dl for men and from 114 to 146 mg/dl for women; in contrast, mean HDL-C varied only from 52 to 55 mg/dl for men and from 60 to 64 mg/dl for women (Lewis et al., 1978). In the 1950s, studies of three groups of sedentary Japanese men matched for fatness showed that mean b-lipoprotein cholesterol (LDL-C) was 120, 183, and 213 mg/dl for those residing in Shime, Honolulu, and Los Angeles, respectively; corresponding mean values for a-lipoprotein cholesterol (HDL-C) were 40, 40, and 35 mg/dl (Keys et al., 1958b). Results from the Jerusalem Lipid Research Clinic Study showed that differences in mean TC according to place of birth (Asia, North Africa, Europe, and Israel) were due mainly to differences in LDL-C (Harlap et al., 1982). On the other hand, a preliminary report of a survey of men in 13 countries indicated that mean HDL-C varied from 27 mg/dl in an African population to 58 mg/dl in eastern Finland (Knuiman and West, 1981). In a study of schoolboys 6 to 7 years old selected from 26 rural and urban populations in 16 countries (Knuiman et al., 1980), mean HDL-C varied from 31 mg/dl in rural Nigeria to 65 mg/dl in rural Finland and correlated (.90) with mean TC. The basis for these different results is not clear.
There is no firm evidence regarding the ecological association of mean HDL-C to risk of CHD. In one study, a correlation of -.57 was found between mean HDL-C and CHD mortality for 19 countries (Simons, 1986), but these results were based on nonstandardized measurements of undefined samples of people. Knuiman and West (1981) obtained standardized measurements of HDL-C in a survey of small samples of men in 13 countries and found a correlation of .57 with national CHD mortality in those countries. One of the lowest population mean values for HDL-C was found in Mexico in Tarahumara Indians, who also had very low values of LDL-C and TC (Connor et al., 1978). Thus, the extent to which variation among populations in CHD rates may be affected by (associated with) variations in mean HDL-C levels is uncertain. Clearly, the association is substantially less than that between population CHD rates and mean LDL-C levels.
Differences Among Individuals
Baseline levels of LDL-C were positively associated with risk of CHD in men and women ages 50 to 79 years in the Framingham Study (Gordon et al., 1977), in Israeli male civil servants age 40 and older (Medalie et al., 1973), and in men ages 35 to 57, black and white, separately identified, who were assigned to the Usual-Care Group in the Multiple Risk
Page 167
Factor Intervention Trial (Watkins et al., 1986). Change in LDL-C level has also been related directly to change in CHD risk; among hypercholesterolemic men treated with cholestyramine, reduction of 11% in LDL-C was associated with a decrease of 19% in risk of CHD (Lipid Research Clinics Program, 1984b).
In the Framingham Study, HDL-C was inversely related to incidence of CHD in men and women ages 49 to 82 years (Castelli et al., 1986; Gordon et al., 1977); an increment in HDL-C of 25 mg/dl was associated with nearly a 50% decrement in risk of CHD for men and women after adjustment for age, body mass index (see Chapter 21 for definition), cigarette smoking, systolic blood pressure, and TC (Castelli et al., 1986). Among the hypercholesterolemic men treated with cholestyramine in the Lipid Research Clinics Coronary Primary Prevention Trial, 1 mg/dl increments in HDL-C at baseline and afterward were associated with decrements of 5.5 and 4.4% in risk of CHD, respectively (Gordon et al., 1986). Baseline level of HDL-C was inversely associated with risk of CHD among men assigned to the Usual-Care Group in the Multiple Risk Factor Intervention Trial (Watkins et al., 1986), but the association was weaker than that observed in the Framingham Study or the Lipid Research Clinics Program. These findings of an inverse relationship between HDL-C and CHD have been supported by observations in the Israel Ischemic Heart Disease Study (Goldbourt et al., 1985; Medalie et al., 1973) and in prospective case-comparison studies of frozen serum samples in Oslo (Enger et al., 1979) and in Tromsö, Norway (Miller et al., 1977).
HDL-C was not significantly associated with the 24-year CHD death rate in a cohort of 526 Finnish men ages 36 to 61 (Keys et al., 1984) and was only weakly associated with CHD death rate in a cohort of 284 Minneapolis business and professional men ages 45 to 55 at entry into the study (Keys, 1980a; Keys et al., 1963). HDL-C was weakly and not significantly (inversely) associated with the incidence of CHD after adjustment for age, body mass index, blood pressure, cigarette smoking, and non-HDL cholesterol in the British Regional Heart Study (Pocock et al., 1986). A study of men in the USSR also failed to find an inverse association between HDL-C and CHD (Levy and Klimov, 1987).
In the Oslo Study of men ages 40 to 49 years at entry in 1972 and 1973, antemortem measurements of plasma lipids and other characteristics were available for 129 men for whom there were also postmortem measurements of the extent of atherosclerosis. The percentage of the coronary intimal surface involved with raised lesions was positively correlated with TC (.32) and negatively correlated with HDL-C (-.25) (Holme et al., 1981).
Although changes in HDL-C are related to changes in CHD risk in most populations, the benefit that can be expected from raising HDL as a preventive strategy in itself is not entirely clear. Added benefit, over that derived from lowering LDL, is suggested, however, by the long-term results of the Coronary Drug Project (Canner et al., 1986), by the results of the Cholesterol Lowering Atherosclerosis Study (Blankenhorn et al., 1987), and by those of the Helsinki Heart Study (Frick et al., 1987; Manninen at al., 1988), in which HDL-C was raised and TC, LDL-C, and VLDL-C were lowered.
Among people ages 50 to 79 years in the Framingham Study, the ratio of TC to HDL-C was strongly associated with risk of CHD in men and women. The strength of the association was not significantly improved by adding TC or HDL-C to the equation containing this ratio (Castelli et al., 1983). A similar result was obtained with the ratio of LDL-C to HDL-C for women, but information about the level of LDL-C did add to the strength of the association in men. The authors concluded that ratios can be useful predictors of risk, but warned that they may not always be as informative as the joint use of the two individual figures used to calculate the ratio. It seems reasonable to expect that increasing knowledge about the various classes and subclasses of lipoproteins will lead to improved ability to predict risk of atherosclerotic diseases. Whether these prediction formulas will take the form of ratios or more detailed specifications of lipoprotein levels is uncertain.
Although the ratio of TC or LDL-C to HDL-C may be a very good predictor of CHD risk in the U.S. population, it is probably not the best target for clinical management or therapy. The recent report of the National Cholesterol Education Program (1988) identified the absolute level of LDL-C as the key index for clinical decision-making about cholesterol-lowering therapy and as the specific target for therapy. The authors of the report stated, ''Reliance on a ratio of either total or LDL-cholesterol to HDL-cholesterol as a key factor in decisions regarding treatment is not a practice recommended in this report. Blood pressure and smoking are not combined into a single number because the clinician needs to know both facts
Page 168
separately in order to recommend an intervention. Similarly, HDL-cholesterol and LDL-cholesterol are independent risk factors with different determinants, and combining them into a single number conceals information that may be useful to the clinician" (National Cholesterol Education Program, 1988).
In summary, of the lipoprotein fractions, LDL has the strongest and most consistent relationship to individual and population risk of CHD. HDL has generally been found to be inversely associated with risk of CHD in individuals within a population, but in at least three long-term population studies, this inverse association was not seen (Keys et al., 1984; Levy and Klimov, 1987; Pocock et al., 1986). Population rates of CHD are much more strongly related to average TC and LDL-C values than to HDL-C.
These findings (together with the results of animal experiments and clinical research reviewed later in this chapter) strongly support the conclusions that LDL-C is centrally and causally important in the pathogenic chain leading to atherosclerosis and CHD. Variation in LDL-C levels explains a large part of individual risk within high-risk cultures and explains almost all the differences in CHD rates among populations. HDL-C is associated inversely with individual susceptibility to CHD within populations where there is widespread elevation of LDL-C and TC. Data are inadequate to characterize ratios of LDL to HDL as a major determinant of the atherosclerotic disease burden among populations; however, the ratio provides improved individual prediction, again within high-risk, high-LDL cultures.
Apolipoproteins
Apolipoproteins play key roles in both the structure and function of plasma lipoproteins. Research on the molecular structure, genetic variability, and metabolism of plasma apolipoproteins has progressed rapidly in recent years, particularly with the application of the new techniques of molecular biology. Knowledge about apolipoproteins has added greatly to our understanding of lipoprotein metabolism and how it is related to atherosclerosis. In a number of instances, genetically controlled variations in apolipoproteins affect lipoprotein structure, composition, and metabolism. For example, polymorphic forms of apolipoprotein E (apo E) interact with dietary fats to influence plasma lipoprotein concentrations, and assessment of apo E phenotypes is an essential procedure in the diagnosis of familial dysbetalipoproteinemia. However, the evidence currently available does not clearly show that plasma apolipoprotein levels are better predictors of CHD than are the plasma levels of cholesterol in the major lipoprotein classes.
Apo E Polymorphism and Hyperlipidemia
In 1977, Utermann et al. (1977, 1979) demonstrated that genetic polymorphisms in apo E were associated with different plasma cholesterol, LDL-C, and b-VLDL concentrations. The apo E phenotypes were shown to be due to segregation of three alleles at a single locus (Zannis and Breslow, 1981) and the major isoforms to be determined by substitution of the amino acid cysteine for arginine (Weisgraber et al., 1981). The three major isoforms are called apo E2, E3, and E4. If the most common phenotype, designated E3/3, is considered to be normal, women who are heterozygous, with phenotype E3/2, have more than 40% higher plasma VLDL-C and intermediate-density-lipoprotein (IDL)-C levels, and 12% lower LDL-C levels (Robertson and Cumming, 1985). Men with the E3/2 phenotype have an approximately 15% increase in plasma VLDL-C and IDL-C concentrations and 20% lower LDL-C concentrations. The E2/2 phenotype is uncommon but is associated with extreme variations in plasma lipoprotein levels. Patients with familial dysbetalipoproteinemia (type 3 hyperlipoproteinemia) generally have the E2/2 phenotype. Most people with this phenotype do not have familial dysbetalipoproteinemia, however, and in fact have lower plasma cholesterol levels than the general population (see Davignon et al., 1988). In contrast, the presence of the E4 allele (and the E4/3 or 4/4 phenotype) appears to be associated with an increased level of LDL and an increase in coronary risk. Thus, a single amino acid substitution in one apolipoprotein can have a substantial effect on plasma cholesterol concentrations and on the plasma lipoprotein profile. This topic has been reviewed thoroughly by Davignon et al. (1988).
The Apolipoproteins and Atherosclerosis
In the 1980s, the availability of better methods for fractionating lipoproteins, for measuring serum apolipoproteins, and for detecting apolipoprotein variants made possible a new series of studies that sought relationships among lipoproteins, apolipoproteins, and atherosclerosis. This topic was reviewed by Brunzell et al. (1984) and by Wallace and Anderson (1987).
Ishikawa et al. (1978) were among the first to examine the relationship of plasma apolipoprotein concentrations to CHD. In the Tromsö Heart
Page 169
Study, apo A-I levels were associated inversely with CHD, but were less powerful predictors than HDL-C levels. A number of investigators subsequently compared CHD patients with controls and generally found apo B levels higher and apo A-I and apo A-II levels lower in diseased subjects (DeBacker et al., 1982; Fager et al., 1980, 1981; Franceschini et al., 1982; Maciejko et al., 1983; Naito, 1985; Onitiri and Jover, 1980; Riesin et al., 1980; Sniderman et al., 1980; Whayne et al., 1981). However, these investigators were not in agreement about whether apolipoprotein levels or lipoprotein cholesterol levels were better indicators of CHD. Pilger et al. (1983) used stepwise discriminant analysis to determine which measures best differentiated subjects with and without peripheral vascular disease. The best model included 14 variables, among which, in addition to HDL-C and LDL-C, were apo B, apo A-I, and apo A-II concentrations.
Menzel et al. (1983) examined the relationship of six different apo E phenotypes to coronary lesions assessed by angiography in 1,000 patients. Heterozygotes (E3/2) were more frequent in patients with little or no coronary atherosclerosis, but E2/2 homozygosity, despite the presence of b-VLDL in the plasma, was not associated with more severe coronary lesions.
Wallace and Anderson (1987) concluded their review by stating that apolipoprotein levels appeared promising as predictors of CHD, but that large cohort studies would be required to determine whether they are better predictors of atherosclerotic disease than plasma lipoprotein lipid levels and whether they are useful clinically.
Lp(a) and Human Atherosclerosis
The role of lipoprotein(a) [Lp(a)] in atherosclerosis and atherosclerotic heart disease has been controversial ever since Lp(a) was discovered by Berg (1963). The apoprotein of Lp(a) has been shown to consist of two peptides—apo(a) and apo B—linked by one or more disulfide bonds (Gaubatz et al., 1983). The recent sequencing of a cloned apo(a) complementary DNA (cDNA) showed that apo(a) is very similar to plasminogen (McLean et al., 1987).
Several studies have shown a relationship between serum Lp(a) levels and CHD (Berg et al., 1974; Dahlén et al., 1975). High scores for coronary arteriosclerosis on angiography were correlated with serum Lp(a) levels (Dahlén et al., 1976). Walton et al. (1974) demonstrated Lp(a) in arterial lesions by immunofluorescence. Overall, the evidence concerning the association of Lp(a) with atherosclerosis is rather scanty, but highly suggestive. Lp(a) levels in plasma are generally believed not to be affected by diet.
Lipoproteins and Apolipoproteins in Atherosclerotic Lesions
Kao and Wissler (1965) demonstrated the presence of LDL in human lesions by immunofluorescence with rabbit antisera to LDL. Hoff and associates have demonstrated the presence of apo B, apo A-I, and apo C in the arteries of humans and several animal species (Hoff and Gaubatz, 1975; Hoff et al., 1974, 1975a,b,c; Yomantas et al., 1984).
The same investigators quantified apo B in lesions (Hoff et al., 1977a,b, 1978, 1979b; reviewed by Hoff and Morton, 1985) and showed that LDL in lesions differed from plasma LDL (Hoff and Gaubatz, 1982; Hoff et al., 1979a). LDL in lesions stimulated the production of cholesteryl esters by macrophages (Clevidence et al., 1984; Goldstein et al., 1981). Apo A-I-containing lipoprotein extracted from arteries also differed in composition from plasma HDL (Heideman and Hoff, 1982).
These studies clearly show that apo B-containing lipoproteins accumulated in atherosclerotic lesions. LDL from atherosclerotic lesions contained particles larger than those found in plasma LDL, was more electronegative, and stimulated cholesterol esterification in mouse peritoneal macrophages. The extent to which these alterations in LDL occurred in the arterial intima, after the LDL had entered the vessel wall, is not clear. This combined evidence implicates apo B-containing lipoproteins in the pathogenesis of atherosclerosis.
Apolipoproteins and Dietary Responsiveness
In view of the critical roles of apolipoproteins in lipoprotein metabolism, genetic variants in addition to those of apo E probably influence the lipemic responses of individuals to dietary fats and thereby affect the risk of CHD. This research area is quite active, and new findings are likely to be available in the near future.
Summary
Apolipoproteins show promise of helping us to understand the mechanisms linking diet to atherosclerosis and cardiovascular disease, but as yet they have not provided predictive power for atherosclerotic diseases beyond that provided by plasma lipid and lipoprotein cholesterol concentrations. They are not now useful in assessing the relationship of dietary fats to atherogenesis or
Page 170
atherosclerotic cardiovascular diseases on a population-wide basis.
Differences Among Populations
Fasting plasma triglyceride or VLDL-C levels have not been systematically ascertained in standardized, population-based surveys designed to compare cultures. Nonfasting triglyceride levels were measured in the Framingham, Honolulu, and Puerto Rico studies. Results indicated that relatively high mean nonfasting triglyceride levels are compatible with low-population CHD rates when the mean TC is also low (Gordon et al., 1974). However, in most populations found to have low average TC levels, triglyceride levels are also low (Conference on Health Effects of Blood Lipoproteins, 1979). The correlation was small (R = .26) and not statistically significant between 1980 CHD death rates in 19 countries and mean triglyceride levels measured in undefined population samples in those countries during the 1970s (Simons, 1986).
Differences Among Individuals
A fairly large number of epidemiologic studies have explored relationships between plasma triglyceride levels and the risk of CHD. The studies conducted before 1980 were reviewed in full by Hulley et al. (1980). In almost all reported studies, a significant positive association between triglyceride levels and CHD was found upon univariate analysis. However, triglyceride has not held up as an independent risk factor in most of the studies that have provided multivariate adjustment for a full set of major risk variables. It is well established that a reciprocal relationship generally exists, both within populations and within individuals, between plasma triglyceride (and, therefore, VLDL) and HDL levels. Thus, individuals with high triglyceride levels tend to have lower HDL levels, and an increase or decrease in triglyceride level in a patient is generally accompanied by an opposite change in HDL-C level. On the other hand, our understanding of the association of VLDL and VLDL remnants to atherosclerosis is still incomplete, and there is some evidence that the latter particles may have an important role in atherogenesis. Hence, the answer to the question of whether or not elevated triglyceride (and VLDL) levels are causally linked to atherosclerotic disease in some patients, or groups of people, is not clear. What is clear is that, as a group, people with high triglyceride levels have elevated coronary risk. Overall, however, the potential atherogenicity of the triglyceride-rich lipoproteins has not been determined. (For reviews and reports of recent studies, see Aberg et al., 1985; Carlson and Bottiger, 1981; Hulley et al., 1980; NIH, 1984; Wallace and Anderson, 1987.)
In the Framingham Heart Study, the plasma triglyceride level was found to be an independent predictor of CHD in women (Castelli, 1986). Several studies have observed that triglycerides (and VLDL) may have independent predictive power for CHD in women and a greater association with peripheral arterial disease than with CHD, but none of the associations found was strong (Aberg et al., 1985; Gordon et al., 1977, 1981b).
The National Institutes of Health (NIH) Consensus Conference on Treatment of Hypertriglyceridemia concluded, "Careful evaluation of existing data indicates that in the presence of normal cholesterol levels mild elevations of plasma triglycerides do not necessarily increase the risk of CVD. When triglycerides are below 250 mg/dl, risk generally does not exceed that of other Americans, and changes in lifestyle are unnecessary beyond those recommended for the general public. The same can be said for many normocholesterolemic individuals with borderline hypertriglyceridemia (defined as triglyceride levels ranging from 250 to 500 mg/dl) who have no risk factors or family history for cardiovascular disease. However, triglyceride levels in the range of 250 to 500 mg/dl can be a marker for secondary disorders or for a subset of patients with genetic forms of hyperlipoproteinemia who are at increased risk and who need specific therapy. Dietary intervention is the primary approach to therapy in these patients, but drugs have a role in selected individuals not responding to dietary management. Finally, the danger of pancreatitis is present in frank hypertriglyceridemia (triglycerides >500 mg/dl), and triglyceride lowering by diet and, if necessary, by drugs is indicated" (NIH, 1984). The Committee on Diet and Health endorses these conclusions.
Summary
The relationship between plasma triglyceride levels and cardiovascular diseases is somewhat controversial and unclear. In most within-population studies, plasma triglyceride levels were positively associated with increased risk for cardiovascular diseases but they were not independently predictive for CHD after statistical adjustment for closely associated -attributes such as TC, HDL-C, hypertension, cigarette smoking, and obesity. As indicated above, however, in the Framingham Heart Study the plasma triglyceride level was found to be an independent predictor of
Page 171
CHD in women. Even so, the plasma total triglyceride level, rather than being a direct cause of atherosclerotic disease, probably reflects the presence of certain atherogenic lipoproteins. It is well known that VLDLs are highly heterogeneous; a number of different kinds of lipoprotein particles are contained within this density class. Many disease entities that elevate triglyceride levels, such as diabetes mellitus, nephrotic syndrome, chronic renal disease, and certain primary hyperlipidemias, carry an increased risk of CHD. In these situations, the high triglyceride level may be a clue to the presence of other lipoprotein abnormalities that are more directly associated with CHD, such as low HDL-C, elevated apo B or LDL-C, or atherogenic remnant triglyceride-rich lipoprotein particles that have not been well defined. Thus, whether or not VLDL and triglycerides are directly involved in the atherogenic process, elevated levels can be helpful in identifying people at increased risk of cardiovascular diseases.
A number of genetic disorders of lipoprotein metabolism have been identified and characterized (Brown and Ginsberg, 1987; Stanbury et al., 1983). The study of these disorders has provided many insights into the structure, metabolism, and regulation of the plasma lipoproteins and apolipoproteins. Several of these disorders are characterized by severe hypercholesterolemia and premature atherosclerosis and CHD. Such disorders include familial hypercholesterolemia (FH), familial combined hyperlipidemia, and familial dysbetalipoproteinemia (type 3 hyperlipoproteinemia).
FH provides strong evidence that a high LDL level per se is a cause of accelerated atherosclerosis and premature CHD. FH is caused by a defect in the gene encoding for the LDL receptor—the cell surface receptor that normally removes LDL from the circulation (Brown and Goldstein, 1986). The defective gene results in the receptor being either absent or nonfunctional. One gene for the LDL receptor normally is inherited from each parent. In the heterozygous form of FH, the patient has one normal and one abnormal gene for LDL receptors, and LDL-C levels are approximately doubled to levels exceeding 200 mg/dl.
Heterozygous FH occurs in the general population at a frequency of about 1 in 500, making FH one of the most common single-gene-determined diseases in humans. FH heterozygotes have TC levels that usually exceed 300 mg/dl and frequently have tendon xanthomas, corneal arcus, premature CHD, and a strong family history of hypercholesterolemia. Approximately 5% of patients with myocardial infarction before age 60 will have heterozygous FH. Affected men often develop CHD in their 30s and 40s; in women with FH, CHD often occurs in their 50s and 60s.
Rarely, about once per million people, individuals inherit two abnormal genes for LDL receptors and hence are homozygous for FH. Homozygous FH patients have TC levels that range from 600 to 1,000 mg/dl, and they usually have planar and tuberous xanthomas as well as tendon xanthomas. Severe and often fatal coronary disease frequently develops while these people are in their teens. One homozygote had an acute myocardial infarction as early as 18 months of age; another died of an acute myocardial infarction at 3 years of age. Very few FH homozygotes survive past age 30 (Goldstein and Brown, 1983).
An animal counterpart of FH has been identified in a strain of rabbits called the Watanabe heritable hyperlipidemic (WHHL) rabbit (Goldstein et al., 1983). These rabbits have very high TC and LDL-C levels and develop severe atherosclerosis that is similar to that seen in humans. This genetic animal model provides further strong evidence of the causal relationship between high LDL-C levels and atherosclerosis. Elevated LDL-C levels due to a defect in the LDL receptor have also been observed in a rhesus monkey family by Scanu et al. (1988).
The research on FH and on the LDL receptor has provided insights into the mechanisms that might be involved in the more common problem of the mild to moderately elevated blood cholesterol levels that are seen in the general population. Potential relationships between LDL receptors, diet, and atherosclerosis have been discussed by Brown and Goldstein (1986). It is likely that the dietary components that raise plasma TC and LDL-C levels (specifically, dietary SFAs and cholesterol, as discussed in detail later in this chapter) act, at least in part, by suppressing hepatic LDL receptor activity. Recent studies (Spady and Dietschy, 1988) in hamsters powerfully demonstrate the role of dietary saturated triglycerides in augmenting the effect of dietary cholesterol in suppressing hepatic LDL receptor activity and elevating plasma LDL-C levels.
Small-Scale Trials (Angiographic End Points)
Evidence has been accumulating and a number of studies are under way to determine the effect of
Page 172
lowering TC by diet, drugs, or ileal bypass surgery on the progression or regression of atherosclerotic lesions of coronary and large peripheral arteries as determined by angiography and ultrasound. As shown angiographically, in the earliest reported randomized controlled trial, patients with intermittent claudication had less angiographic progression and more regression of plaques in the diet- and drug-treated group than did a control group over a 19-month period. This was directly related to change in plasma LDL-C concentration (Duffield et al., 1983). In a recently completed randomized controlled trial in which angiographic techniques were used, patients treated with diet and several TC-lowering drugs had less progression and more actual regression of plaques in natural and bypass graft coronary vessels compared to controls over a 2-year period (Blankenhorn et al., 1987). These results and those of other systematic studies cited below are generally consistent with the conclusion that reducing LDL-C in hyperlipidemic patients reduces the rate of progression (and can induce regression) of atherosclerosis according to the degree of blood lipid reduction (Arntzenius et al., 1985; Brensike et al., 1984; Cohn et al., 1975; Kuo et al., 1979; Nash et al., 1982; Nikkila et al., 1984).
Large-Scale Preventive Trials (CHD End Points)
Nine randomized controlled trials on the primary prevention of CHD have been conducted. Key elements of their designs are summarized in Table 7-1. In five trials, various methods were used to change the plasma lipid concentrations as the only intervention; in two, diet was used; and in three, drugs. The other four studies, in which diet was also used, were multifactorial trials in which effects of change in plasma lipids were confounded with effects of change in cigarette smoking and blood pressure. Only one of the six trials involving change in diet—the Los Angeles Veterans Administration Domiciliary Study—was conducted under double-blind conditions (Dayton et al., 1968). In two studies—the World Health Organization Multifactor Trial (WHO European Collaborative Group, 1983) and the Finnish Mental Hospital Study (Hjermann et al., 1981)—groups of participants were randomized. In the others, individuals were randomized. The number of participants in these studies varied from approximately 800 to nearly 50,000.
Results of the trials summarized in Table 7-2 have been listed according to the percentage difference in mean serum cholesterol that was created between the treatment and comparison groups—from 0% in the Göteborg Multifactor Trial (Wilhelmsen et al., 1986) to -15% in the Finnish Mental Hospital Study (Hjermann et al., 1981; Miettinen et al., 1972). (A negative sign here indicates that the experimental group had a lower mean level than the control group.) These results indicate that the magnitude of the difference in risk of CHD varied directly with the magnitude of the difference in mean TC. Studies that achieved little or no difference in mean TC had little or no difference in rate of CHD, whereas studies that achieved larger difference in mean TC experienced larger differences in CHD rates.
As indicated in Table 7-2, differences in CHD rates were statistically significant in five of the seven studies that achieved a difference of 8% or more in mean TC. The number of cardiac events in the other two studies—in the entire cohort of men in the Los Angeles Veterans Administration Domiciliary Study and in the sample of women in the Finnish Mental Hospital Study—were relatively small, and the percentage differences in CHD rates, although comparable in magnitude to those that were statistically significant, did not reach the 5% criterion for statistical significance.
Combined with the evidence from epidemiologic, clinical, and laboratory animal studies, this body of evidence establishes unequivocally that lowering serum TC, particularly LDL-C, reduces the incidence of CHD in middle-aged, hypercholesterolemic men. High-risk populations were used in most of these studies for quite practical reasons; the cost and problems of conducting such a trial in the general population would be very large and the results might well be inconclusive. However, in light of the total body of evidence, it seems reasonable to extend this conclusion to the general population of men and women with serum TC of 200 mg/dl or higher. Results of the Los Angeles Veterans Administration Domiciliary Trial support the inference that the conclusion may extend to people over 65 years of age as well, perhaps with a somewhat smaller effect, but the evidence bearing on this important question is still weak.
Most of the trials listed in Tables 7-1 and 7-2 showed an excess of non-CHD deaths in the intervention group as compared to the comparison group. How much of this can be attributed to chance and whether any of it reflects adverse effects of intervention are complex questions that may never be completely resolved. An analysis of 20 randomized controlled clinical trials that involved change in serum TC as the only systematic intervention indicates no statistically significant
Page 173
| TABLE 7-1 Design of Randomized Controlled Trials on Primary Prevention of Coronary Heart Diseasea | |||
| Study | Study Population | Double | Interventions and |
| Göteborg Multifactor Trialb | 20,015 men ages 47 to 55 | No | Diet, cigarette smoking, high blood pressure |
| WHO Multifactor Trialc | 66 employed groups totaling | No | Diet, cigarette smoking, high blood pressure |
| Multiple Risk Factor | 12,886 high-risk men ages | No | Diet, cigarette |
| Lipid Research Clinics Coronary | 3,806 hypercholesterolemic | Yes | Cholestyramine |
| WHO Clofibrate Trialf | 11,627 hypercholesterolemic | Yes | Clofibrate |
| Helsinki Heart Studyg | 4,081 hypercholesterolemic | Yes | Gemfibrozil |
| Los Angeles Veterans | 846 men ages 55 to 89 | Yes | Diet only |
| Oslo Studyi | 1,232 hypercholesterolemic normotensive men ages 40 to 49 | No | Diet, cigarette |
| Finnish Mental Hospital Studyj | 2 mental hospitals totaling | No | Diet only |
| a Unpublished table compiled by R. Peto, University of Oxford, 1988. Not included in this table are data from a paper by the WHO European Collaborative Group (1983), which included populations in Warsaw, because it did not present results on percentage reduction in serum cholesterol, and a paper on the Finnish Mental Hospital Study (Turpeinen et al., 1979), because it lacked results for total mortality and for women. | |||
effect on risk of non-CHD death (Richard Peto, University of Oxford, personal communication, 1987). In the trials shown in Table 7-2, none of the percentage differences in risk of non-CHD death or total mortality was statistically significant except for the WHO Clofibrate Trial. In the final mortality follow-up, however, the Committee of Principal Investigators (1978) of that study found that the excess mortality in the clofibrate-treated group did not continue after the end of treatment, and they could not find a reasonable explanation for the excess mortality that did occur during the period of active treatment. The excess deaths were due to a variety of non-CHD causes and had no apparent association either with the extent of reduction in TC or with duration of treatment with clofibrate. These results do not rule out the possibility of a toxic effect, but seem more consistent with the hypothesis that the excess of deaths in the treated group was the result of random sampling variation.
The possible adverse effects of the drugs used in these trials are not strictly relevant to a report on diet and health and are not discussed further. The safety of dietary changes to lower serum TC is discussed in Chapter 28 of this report. There is no
Page 174
| TABLE 7-2 Results of Randomized Controlled Trials on Primary Prevention of Coronary Heart Diseasea | |||
| Percentage Differences Between | |||
| Treated and Control Groupsc | |||
| Serum Cholesterol at Entry (mg/dl) | Serum Cholesterol | CHDd | |
| Studyb | |||
| Göteborg Multifactor Trial | 250 | 0 | 0 |
| WHO Multifactor Trial | 216 | -1 | -7 |
| Multiple Risk Factor Intervention Trial | 254 | -2 | -7 |
| Lipid Research Clinics Coronary Primary Prevention Trial | 292 | -8 | -19e |
| WHO Clofibrate Trial | 242 | -9 | - 20e |
| Helsinki Heart Study | 270 | -9 | -34e |
| Finnish Mental Hospital Study (women) | 275 | -12 | -34 |
| Los Angeles Veterans Administration Domiciliary Study | 233 | -13 | -24 |
| Oslo Study | 329 | -13 | -47e |
| Finnish Mental Hospital Study (men) | 267 | -15 | -53e |
| a From R. Peto, University of Oxford, unpublished data, 1988. | |||
firm evidence to indicate that any of the dietary interventions used in the trials described above increased the risk of non-CHD deaths. In the Los Angeles Veterans Administration Domiciliary Trial, an excess of cancer deaths was observed in the experimental group that ate a diet high in PUFAs (on average, 15% of calories as PUFAs), but the difference was larger in the subgroup that adhered poorly to the prescribed diets than in the subgroup that adhered well (Pearce and Dayton, 1971). Also, results from other clinical trials did not indicate an excess of cancer deaths in groups assigned to a high-PUFA diet (Ederer et al., 1971). These considerations support the conclusion that the excess of cancer deaths observed in the Los Angeles study did not result from the high-PUFA diet.
Rose et al. (1974) analyzed the pooled data from six prospective studies of CHD and found that the mean serum TC of men who subsequently developed colon cancer was significantly lower than expected. The extensive literature that has since developed on this topic has been reviewed by McMichael et al. (1984). In some studies, an inverse association was found over a protracted period; in others, an inverse association was observed during the first few years of follow-up but disappeared during continued follow-up; in yet others, no association was observed between initial serum TC level and subsequent risk of cancer. Some of the results may have reflected a preclinical effect of cancer on serum TC and some may have been due to metabolic characteristics (e.g., increased secretion of bile) of people who maintain low serum TC despite a diet high in SFAs and cholesterol. There is no evidence from studies in humans that low consumption of SFAs and cholesterol are positively correlated with rates of mortality from colon cancer (Liu et al., 1979). This evidence is discussed in more detail later in this chapter and in Chapter 28.
Richard Peto (Oxford University, personal communication, 1987) contends that the primary information to be obtained from the randomized clinical trials is the time required for changes in TC to have an effect on CHD rates. He has obtained information on 18 published and 2 unpublished randomized trials that involved changes in TC as the only systematic intervention, regardless of the mode of intervention (diet or drugs) and regardless of whether they were trials of disease prevention or disease treatment. Analysis of all
Page 175
data from these trials indicates that a 10% reduction in TC was associated with an average reduction of 16% in CHD risk (95% confidence interval of 10.6 to 20.4%) in trials lasting 4 years. Peto also noted that the reduction in risk varied according to the duration of treatment: 11% (±5%) in 13 shorter trials and 21% (±4%) in 7 longer trials, for a 10% reduction in TC. There have been no trials lasting a decade or longer, but results from the observational studies suggest that a reduction of 10% in TC over decades would be associated with a 33% reduction in rate of fatal CHD.
Peripheral Vascular Diseases (PVD)
There are few systematic data on the relationship of blood lipids and lipoproteins to the prevalence or risk of PVD or large-vessel peripheral arterial disease (PAD). In the Framingham Study, the 26-year cumulative incidence of intermittent claudication showed a concentration of cases in the upper part of the distribution for TC values in men but not in women (i.e., 11.6% in the upper quintile versus 5.3% in the lower) (Kannel and McGee, 1985). The results are, however, inconsistent among studies. For example, serum cholesterol was strongly related in some studies (Bothig et al., 1976; Reunanen et al., 1982) but only weakly or not related in others (Cavallo-Perin et al., 1984; Davignon et al., 1977; Greenhalgh et al., 1971; Hughson et al., 1978; Isacsson, 1972; Sirtori et al., 1974).
Triglycerides (and VLDL) may be more highly related to PAD than either HDL or LDL. In a British in-patient case-control study, there was a suggestion that prebetalipoprotein (i.e., VLDL) and serum triglyceride levels were disproportionately elevated in PVD patients, leading the authors to speculate about differences in the time course or severity of atherosclerosis with this blood lipid pattern, compared to that in coronary disease (Greenhalgh et al., 1971). At any rate, the overlap was considerable, in that a third of their PVD patients also had CHD. Clustering of intermittent claudication with other atherosclerotic diseases was also very strong in the Framingham Study. Overt coronary disease, cerebrovascular disease, or congestive heart failure were found in one out of three cases of intermittent claudication at the time of diagnosis, indicating the strong likelihood of common risk characteristics (Kannel and McGee, 1985).
The linkage in the basic processes leading to manifestations of PVD and CHD is further suggested by the finding of a strong curvilinear relationship between a Framingham risk index involving multiple risk characteristics for coronary disease and the incidence over time of manifest intermittent claudication, primarily due to an excess risk of intermittent claudication in the upper quintile of the coronary index (Kannel and McGee, 1985). A similar predominance of hypertriglyceridemia was observed in older men and in women among a series of Swedish patients with PVD (Leren and Haabrekke, 1971).
Because the underlying pathogenesis of PVD and CHD is atherothrombotic disease, it is likely, but not established, that the same evidence outlined for the relationship of diet to CHD holds for PVD as well. This is also suggested by the epidemiologic evidence about PVD, i.e., the positive relationship of intermittent claudication to the coronary disease risk index in the Framingham population. Finally, although the pathogenesis of atherosclerosis is similar in the different arterial beds, the relative importance of different risk factors in causing the disease process clearly differs among different anatomic sites. This issue is discussed further in Chapter 19.
Stroke
There are few systematic data relating blood lipids and lipoproteins to stroke incidence. A U-shaped relationship between lipids and stroke was suggested in the Chicago Stroke Study, in which a lower stroke risk was found in the central part of the TC distribution (Ostfeld et al., 1974). This was more striking in the 20-year Framingham follow-up of the older population, ages 65 to 74, in which the highest risk of stroke and acute brain infarction was observed at TC levels below 190 and above 295 mg/dl. This U-shaped relationship was stronger for acute brain infarction than for all strokes. In the same series, there was a negative relationship between LDL-C and overall stroke as well as for HDL-C and overall stroke (Kannel and Wolf, 1983). A similar U-shaped relationship was also observed in the Honolulu Heart Program, both for TC and for LDL-C, whereas HDL-C was negatively related and VLDL was positively but not significantly related to stroke risk (D. Reed et al., 1986). The left side of the U-shaped relationship to stroke in the Honolulu Study was due primarily to the inverse relationship between TC and hemorrhagic stroke (Kagan et al., 1980).
In later studies involving improved ability to discriminate between intracerebral hemorrhage and thrombosis by the use of computed tomography (CT) scanning, a clearer epidemiologic pic-
Page 176
ture emerges. For example, in recent data from the Multiple Risk Factor Intervention Trial on more than 360,000 screenees observed for 10 years, there is a clear, positive, monotonic, linear relationship between TC level and death ascribed to cerebral thrombosis (Iso et al., 1988). This explains the right side of the U-shaped distribution of TC to overall stroke risk. A strong linear negative relationship was found between TC level and deaths coded to cerebral hemorrhage, the major explanation for the left side of the U-shaped TC-stroke risk curve. However, the negative relationship between TC and cerebral hemorrhage was limited to people with TC levels under 160 mg/dl and diastolic pressures greater than 90 mm Hg (Iso et al., 1988).
Hypercholesterolemia has been the common denominator of experimental atherosclerosis in a variety of animal species since shortly after the turn of the century (reviewed by Anitschkow, 1967; Duff, 1935; Jokinen et al., 1985; Katz and Stamler, 1953; Roberts and Straus, 1965; Strong, 1976; Wissler and Vesselinovitch, 1987a,b). In most species, hypercholesterolemia is induced by a diet enriched in cholesterol, SFAs, or both. Some animal species, such as the rat and the dog, are resistant to diet-induced hypercholesterolemia and develop atherosclerotic lesions when hormonal or other manipulations are used in conjunction with diet to induce hypercholesterolemia. When hypercholesterolemia is prolonged over several years, these animals develop all the complications seen in advanced atherosclerosis in humans and, eventually, myocardial infarction and other clinical manifestations of atherosclerosis in humans (Taylor et al., 1963).
Not only has hypercholesterolemia been the sine qua non of experimental atherosclerosis, but the relationship of the specific plasma lipoproteins to atherosclerosis in animals is similar to that in humans. For example, in humans and nonhuman primates, LDL is positively, and HDL is negatively, associated with atherosclerosis. In multiple regression analyses, plasma LDL and HDL concentrations account for as much as 50% of the variance in the extent of atherosclerosis (McGill et al., 1981a, 1985; Rudel, 1980). In some experiments, physical characteristics of LDL, such as molecular size, are also associated with atherosclerosis (Rudel et al., 1985). When diet-induced hypercholesterolemia is combined with other conditions known to augment atherosclerosis in humans, such as hypertension, the experimental lesions also are increased in extent and severity (McGill et al., 1985). Thus, the association of altered plasma lipoprotein concentrations in laboratory animals is not only strong and consistent, but the pattern of the association is similar to that in humans.
Furthermore, many studies have demonstrated that reduction of plasma cholesterol concentrations by withdrawal of cholesterol or fat, or both, from the diet, or by administration of drugs, leads to regression of experimental atherosclerosis (reviewed by Malinow, 1983). These regression studies, which were conducted in nonhuman primates, gave considerable support to the concept that the progression of atherosclerosis in humans can be retarded and possibly that lesions can be reversed by treating hyperlipidemia with diet and drugs.
The strong relationship between high levels of plasma LDL and atherosclerosis has provided an important background for in vitro studies dealing with the cellular and molecular mechanisms of atherogenesis. These in vitro studies have sought to determine how LDL and other atherogenic lipoproteins could lead to the formation of cholesteryl ester-filled macrophages (foam cells) and other elements of the atherosclerotic lesion. The results of such studies have, in turn, supported data on humans and animals that suggest a primary etiologic role for LDL and related lipoproteins in the development of atherosclerosis.
A prominent and early feature of atherosclerotic lesions is the foam cell. In order to understand the mechanisms of foam cell formation, investigators have studied the interaction of plasma lipoproteins with various types of cultured macrophages. Native LDL does not lead to the accumulation of cholesteryl ester in many types of macrophages (reviewed in Brown and Goldstein, 1983). This lack of native LDL-induced cholesteryl ester accumulation is due, at least in part, to down-regulation of the LDL receptor on these cells by small amounts of excess cellular cholesterol, thus preventing a large influx of LDL-C. However, modified forms of LDL, such as acetyl-LDL, oxidized LDL, and malondialdehyde-LDL, do lead to massive accumulations of cholesteryl ester in cultured macrophages. These modified forms of LDL enter the cell by a receptor (called the scavenger receptor) that is distinct from the LDL receptor and that is not subject to down-regulation (Brown and Goldstein, 1983, 1986). Thus, cellular influx of these modi-
Page 177
fled forms of LDL continues at a high rate, leading to significant accumulation of cholesteryl ester.
These in vitro observations have spawned a widespread search for evidence of LDL modification in vivo. Several recent studies have shed light on this topic. Pitas et al. (1983) demonstrated that foam cell macrophages from explants of rabbit aorta atheroma have scavenger receptors capable of binding modified LDL. Kita et al. (1987) and Carew et al. (1987) took advantage of the in vitro finding that the drug probucol, which is an antioxidant transported in LDL, prevents oxidative modification of LDL. These investigators demonstrated that probucol treatment of LDL receptor-deficient WHHL rabbits decreased LDL uptake in foam cell lesions and reduced the rate of development of these lesions independently of plasma cholesterol levels. These results are consistent with the hypothesis that oxidative modification of LDL may be important in the development of foam cell lesions.
Haberland et al. (1988) found immunochemical evidence for the existence of malondialdehyde-LDL in atherosclerotic aortas. Malondialdehyde is a byproduct of arachidonic acid metabolism, which is an active process in the arterial wall. Whether malondialdehyde-LDL is involved in foam cell formation in vivo has yet to be determined.
A potential role for native LDL in foam cell formation has recently been suggested by findings that another cultured macrophage, the J774 macrophage, accumulates large amounts of cholesteryl ester in the presence of native LDL (Tabas et al., 1985). In this case, the LDL receptor is poorly down-regulated by LDL-C because of intracellular shunting of cholesterol away from the regulatory pathway into a hyperactive cholesterol esterification pathway. Since LDL receptors are abundant on foam cells in vivo (see below), indicative of poor LDL receptor down-regulation, this mechanism of foam cell formation may help explain the atherogenic effects of native LDL.
Foam cell formation has also been demonstrated in vitro by lipoproteins other than native and modified forms of LDL. One such lipoprotein, b-VLDL, is a cholesterol-enriched lipoprotein in the plasma of cholesterol-fed animals and in the plasma of humans with the genetic disease familial dysbetalipoproteinemia. b-VLDL causes massive deposition of cholesteryl ester in macrophages in vitro (Brown and Goldstein, 1983; Koo et al., 1986) and enters the macrophage by binding to the same receptor as does native LDL, and this receptor demonstrates poor down-regulation (Koo et al., 1986). Receptors for b-VLDL (and thus for native LDL) are located on foam cells from rabbit aorta atheroma (Pitas et al., 1983). These observations, together with the strong correlation between b-VLDL plasma levels and atherosclerosis in cholesterol-fed animals and dysbetalipoproteinemic humans, indicate that b-VLDL is atherogenic.
b-VLDL has been found only in humans with familial dysbetalipoproteinemia. However, chylomicron remnants, which occur commonly in humans, interact with macrophages as does b-VLDL and cause the accumulation of cholesteryl ester as well as a large amount of triglyceride (Van Lentern et al., 1985). These and other findings have led some investigators to speculate that postprandial lipoproteins, particularly cholesterol-enriched particles such as chylomicron remnants, may be atherogenic in humans.
Foam cell research is an active area of in vitro investigation exploring the relationship between LDL and atherogenesis. However, other areas of study have also focused on this relationship (reviewed in Steinberg, 1983). For instance, LDL, either native or oxidized, causes endothelial cell injury in vitro. Endothelial injury, in turn, has been hypothesized to initiate atherogenesis by causing platelet adherence to the vessel wall and release of growth factors. In addition, LDL has been reported to directly augment platelet aggregation and to stimulate the growth of smooth muscle cells—two important features of the atherosclerotic lesion. It is clear, moreover, that other components of cells and vessel walls, not yet well defined, are also important factors in atherogenesis. The potential roles of growth factors, endothelial injury, and the different cells in arterial walls in the pathogenesis of atherosclerosis have been reviewed by Ross (1986).
In summary, numerous in vitro studies have demonstrated the ability of LDL and related lipoproteins to cause the formation of foam cells and other elements of atherosclerosis. Although there may be other important metabolic, cellular, and vessel wall factors in atherogenesis, in vitro evidence strongly suggests an important causative role for LDL and related lipoproteins.
There are large interpopulation differences in mean plasma lipid and lipoprotein levels and CHD rates. Average population TC levels also differ between adults and children; levels in children parallel those in adults at lower absolute values. Population correlations between CHD risk and mean TC level are strong but are weak for VLDL,
Page 178
triglyceride, and HDL-C. TC means and CHD rates among migrants change toward those of the adopted country, whether higher or lower than the country of origin.
Individual correlations of TC and LDL-C levels with CHD risk within populations are strong, positive, and continuous. Individual HDL-C level is inversely related to CHD risk in studies in the United States, Norway, and Israel, but not in Finland and the USSR. HDL-C population means are weakly correlated, if at all, with population TC levels and CHD rates.
Randomized clinical trials show a consistent relationship between decreases in TC and LDL-C levels resulting from altered diets and drugs and decreases in CHD incidence; the magnitude of the difference in risk varies directly with the magnitude of the difference in mean TC between the experimental and control groups. Differences in CHD rates between the experimental and control groups were statistically significant in five of the seven trials that achieved a difference of 8% or more in mean TC. The magnitude of the benefit that can be expected from lowering TC and LDL levels can be estimated from observational epidemiologic studies and from clinical trials. The estimates from these different kinds of studies are consistent in projecting that for individuals with TC levels initially in the 250- to 300-mg/dl range, each 1% reduction in TC level reduces CHD rates by approximately 2%. Thus, observational studies and clinical trials indicate that relatively small differences or changes in average TC level have a relatively large population effect or public health impact on disease. In addition, the trials indicate that risk can be reduced in individuals at moderate to high risk and that much of the effect of a lower TC on risk is seen after only a very few years.
Effects of Dietary Fats on Plasma Lipids and Lipoproteins
Evidence concerning the effects of dietary fats on plasma lipid and lipoprotein levels is reviewed in this section. Evidence specifically about the effects of dietary sterols is reviewed later in the section on the Relationship Between Dietary Sterols and Cardiovascular Disease. However, it is often difficult to separate clearly the effects of SFAs from those of dietary cholesterol, especially in observational studies. Therefore, terms such as ''SFAs and cholesterol" are sometimes used in order to avoid giving the impression that a statement applies only to the fatty acid intake.
In many early studies, only serum TC was measured. When reviewing that body of evidence, variation in TC has been interpreted as a surrogate for variation in LDL-C. Some of the early studies assessed only the percentage of calories from total fat and did not assess the fatty acid composition of the diets. Where it seemed appropriate, differences in percentage of calories from total fat have been interpreted as reflecting differences in percentage of calories from SFAs.
Studies of Populations
Before World War II, reports from many parts of the world indicated that the prevalence of atherosclerotic cardiovascular diseases varied according to intake of fat and cholesterol (Rosenthal, 1934a,b,c). In the decade following World War II, further studies provided evidence that populations with low mean intakes of SFAs and cholesterol had low mean levels of serum TC in comparison to populations with diets high in SFAs and cholesterol (Bronte-Stewart et al., 1955; Keys and Anderson, 1954; Scrimshaw et al., 1957). [Only representative publications have been cited here. This large body of literature was reviewed by Katz et al. (1958) and by Keys (1963).] Although consistent with the hypothesis that dietary factors play an important role in determining the distribution of TC in populations, these studies often suffered from inadequate sampling and lack of standardized procedures.
In the Seven Countries Study (Keys, 1970, 1980b), standardized procedures (including 7-day weighed food records, a central laboratory, and chemical analysis of duplicate meals) were used. Median TC levels varied from 156 to 264 mg/dl. The mean percentage of calories from fat varied from 9 to 40% of total calories, with 3 to 22% from SFAs, 3 to 29% from MUFAs, and 3 to 7% from PUFAs. The mean concentration of TC correlated with mean percentages of calories from total fats (R = .67) and from SFAs (R = .87), and with estimated mean serum TC (R = .90) calculated from the formula, 164 + 1.35 (2S - P), where S and P represent the percentage of calories from SFAs and PUFAs, respectively (Keys, 1980b). Neither mean intake of PUFAs nor mean relative body weight was significantly correlated with mean serum TC. Dietary cholesterol was not measured in this study, but mean intake of SFAs probably had a strong correlation with mean intake of cholesterol, and results for SFAs probably apply to cholesterol as well.
The strong correlation between mean serum TC and mean percentage of calories from SFAs has been observed in other studies in which groups
Page 179
have formed the unit of analysis. As noted by Keys (1980b), the correlation coefficient was .94 for participants in the Israeli Ischemic Heart Disease Study (Kahn et al., 1969) when categorized according to region of birth: Israel, Asia, Africa, and eastern, central, and southern Europe. Stallones (1983) reviewed a large array of data from population surveys and found that the correlations between mean percentage calories from fats and mean serum TC was .86 for 76 groups of men and .79 for 27 groups of women. Stallones found the magnitude of these correlations to be remarkable since the studies were conducted with different procedures at different times; measurements of diet and of serum TC were not always on the same people; dietary data were not always presented separately by gender; and serum TC and fat intake were subject to appreciable errors of measurement and are surrogates for the variables of real interest—serum LDL-C, dietary SFAs, and cholesterol.
These observations indicate that 60 to 80% of the variance among populations in mean serum TC can be accounted for on a statistical basis by variation in mean percentage of calories from SFAs. (Dietary cholesterol probably contributes to this result as well, but the evidence from cross-population studies on dietary cholesterol is much less than the evidence on SFAs). Many feeding experiments (reviewed below) have established that isocaloric changes in the lipid composition of the diet—i.e., in the percentage of calories from SFAs with 12 to 16 carbon atoms, the percentage of calories from PUFAs, and the amount of dietary cholesterol—affects the concentration of serum TC. However, these studies focused on the assessment of short-term effects; they did not attempt to determine whether long-term changes in the lipid composition of the diet would change the mean serum TC of populations characterized by a high incidence of CHD to levels (<200 mg/dl) commonly seen in populations with a low incidence of CHD. Evidence relating to this important question must come from a variety of sources, e.g., surveys of migrants, comparisons of socioeconomic classes that differ in access to meat and dairy products, analyses of secular trends, and feeding experiments. Some of this literature is reviewed below. Other reviews of this extensive literature have been done by Katz and Stamler (1953), Katz et al. (1958), Keys (1963), and Stamler (1967, 1979).
South Africa
In a survey of employed, middle-aged men in Cape Town, South Africa, mean serum TC and mean consumption of fats were found to be lowest in Bantus, intermediate in "Cape Coloureds," and highest in "European Whites" (Bronte-Stewart et al., 1955). Mean TC levels were 166, 204, and 234 mg/dl, respectively; means for b-lipoprotein cholesterol were 122, 160, and 194 mg/dl; and means for fat intake were approximately 42, 62, and 88 g/day. (Given the differences among these groups in the sources of fats, it seems reasonable to infer that the groups differed substantially in intake of SFAs and cholesterol.) When each racial group was stratified according to income, differences in mean serum TC were closely paralleled by corresponding differences in mean consumption of fats, and racial groups similar in economic status also had similar intakes of fat and mean serum TC. The authors interpreted this evidence as supporting the hypothesis that the observed differences in serum TC could be accounted for by the observed differences in diet and that invoking genetic factors was unnecessary. This interpretation was supported by the results of feeding experiments in humans (Antonis and Bersohn, 1962b; Bronte-Stewart et al., 1955).
Southern Italy
In the early 1950s, working-class inhabitants of Naples consumed 20% of their calories from fats (Keys et al., 1955). The great bulk of their diet consisted of bread and pasta. Cheese and meat were consumed in small amounts, olive oil was used sparingly, and butter was not used at all. Mean serum TC was 172 mg/ dl in a sample of 230 clinically healthy male steel workers, firemen, and clerks 35 to 60 years of age (Keys et al., 1955).
In Boston, a survey was conducted on 189 clinically healthy male factory workers ages 20 to 50 who had lived all their adult lives in the United States but whose parents had been born near Naples. Investigators found that 43% of their calories came from fats (approximately 32% from animal fats) and that their mean serum TC was 239 mg/dl (Miller et al., 1958). It seems unlikely that the difference in mean serum TC between Naples and Boston was due to differences in body fatness; when the Boston sample was stratified according to relative weight, mean serum TC was 223, 243, and 232 mg/dl for men with relative weights less than 100, between 100 and 119, and 120% of standard or greater.
Ferro-Luzzi et al. (1984) partially substituted butter and other foods rich in SFAs and cholesterol for the customary olive oil in the diets of healthy middle-aged men and women who resided in a
Page 180
rural area south of Naples and who habitually consumed the traditional diet. Mean serum TC changed from 214 to 245 mg/dl in men and from 179 to 203 mg/dl in women. These differences were due almost entirely to changes in LDL-C, which were 148 to 182 mg/dl for men and 119 to 142 mg/dl for women.
The Japanese
A survey of Japanese men in Japan, Hawaii, and Los Angeles indicated that mean serum TC was directly correlated with mean percentage of calories from fats, and that the mean serum TC of Japanese men in Los Angeles (about 240 mg/dl) was similar to that of American men generally (Keys et al., 1958b). A feeding experiment conducted in Japanese men indicated that the change in mean serum TC associated with change in lipid composition of the diet was comparable in magnitude to that of white subjects in Minnesota (Keys et al., 1957b).
In the Ni-Hon-San Study, standardized methods were used beginning in 1965 to investigate the three cohorts of middle-aged men of Japanese ancestry in Hiroshima and Nagasaki, in Honolulu, and in San Francisco (Kagan et al., 1974; Kato et al., 1973; Tillotson et al., 1973). In the latter two areas, more than 85% of the participants were Nisei who had been born in the United States to parents who had emigrated from Japan; the remainder were Issei who had migrated to the United States before the Exclusion Act of 1924. Sources of migration for more than 70% of the Issei and Nisei were the areas around Hiroshima and Nagasaki in the southwestern region of Japan. All three cohorts had similar distributions of the A, B, and O blood groups.
Data on diet were obtained by a 24-hour diet recall interview and a self-administered dietary acculturation questionnaire designed to ascertain adherence to a traditional Japanese diet (Tillotson et al., 1973). Information on fatty acid composition of foods in Japan was not available, and a food grouping system was used in order to categorize fats as primarily saturated or primarily unsaturated.
Mean values for serum TC for all cohorts in Japan, Honolulu, and San Francisco were 181, 218, and 228 mg/dl, respectively. Corresponding mean values for total energy intake were 2,164, 2,275, and 2,262 kcal/day; for percentage of calories from fats, 15, 33, and 38; for percentage of calories from saturated fats, 7, 23, and 26; for intake of cholesterol, 464, 545, and 533 mg/day; and for subscapular skinfold thickness, 10, 16, and 16 mm.
Although data on fatty acid composition of the diet were not available for the cohort in Japan, such data have been published for the cohorts in Hawaii and California (Tillotson et al., 1973). These data indicate that SFAs accounted for 54% of saturated fats in both Hawaii and California and that PUFAs accounted for 58% of unsaturated fats in Hawaii and 52% in California. The mean percentages of calories from SFAs and PUFAs in Japan were estimated, based on the assumption that the proportions were the same as those in Hawaii. The age-adjusted data for men ages 45 to 59 in Japan, Hawaii, and California are as follows: mean percentage of calories from SFAs was 3.5, 12.4, and 14.1% and from PUFAS, 5.3, 6.1, and 6.1%; mean intake of cholesterol was 217, 237, and 243 mg/1,000 kcal; and serum TC was 175.9, 219.2, and 226.1 mg/dl, respectively.
The effect of the differences in lipid composition of these diets on mean serum TC can be estimated by applying the equations of Keys et al. (1965c) and of Hegsted et al. (1965) (discussed in the next section of this chapter)—and subsequently modified by Hegsted (1986) for diets with 0 to 400 mg of dietary cholesterol per 1,000 kcal of intake. For men ages 45 to 59, the Keys equation accounts for more than 50% of the observed differences between the three groups. The Hegsted equation yields slightly lower values: 47% of the difference between Hawaii and Japan, 54% of the difference between California and Japan, and 57% of the difference between California and Hawaii. The predictive equations were based on short-term feeding experiments lasting for several weeks, and it is reasonable to expect that effects of long-term exposure to different diets would be greater. Therefore, these results support the concept that the major portion of the differences in mean serum TC between the cohorts in Japan, Hawaii, and California can be accounted for by differences in lipid composition of the diet.
The Yemenite Jews
Toor et al. (1960) studied three groups of people who migrated from Yemen to Israel and had lived in Israel for approximately 5 years, 9 to 10 years, and more than 20 years, respectively. The economic status of the most recent migrants was very poor during 1953 and 1954. A survey showed that their diet was low in calories (mean, 1,750 kcal/day) and contained 12 g of animal protein and 30 g of total fat per day—almost all from vegetable sources. The economic status of the earliest migrants was better. Their mean intakes were: energy, 2,500 kcal/day;
Page 181
animal protein, 29 g/day; and total fat, 65 g/day (28% from animal sources). For men ages 45 to 54, mean values for serum TC from the most recent to the earliest migrants were 158, 180, and 197 mg/ dl, respectively. Corresponding values for women of the same age group were 172, 189, and 206 mg/ dl. These differences in mean serum TC were due primarily to differences in b-lipoprotein (LDL) cholesterol; differences in the a-lipoprotein (HDL) fraction were small and not statistically significant. Mean weight-to-height ratios [weight in kg x 100/ (height in cm - 100)] were 92, 98, and 96 for men and 95, 94, and 108 for women.
The Irish
Results from the Ireland-Boston Diet-Heart Study (Kushi et al., 1985) indicate that migration without substantial change in lipid composition of the diet is not associated with substantial change in mean serum TC. Comparison of middle-aged Irishmen living in Ireland with their brothers and a sample of unrelated Irishmen living in Boston showed that the age-adjusted means for serum TC were similar—216, 218, and 215 mg/dl, respectively. Corresponding mean values for energy intake were 4,033, 3,099, and 2,946 kcal/day. The proportions of calories from SFAs were 18, 17, and 16% and from PUFAs, 2, 3, and 3%; the intakes of cholesterol were 233, 273, and 240 mg/1,000 kcal. Mean values for a diet score intended to estimate the effect of dietary lipids on serum TC were 60, 59, and 54 when calculated according to the equation of Keys et al. (1965c) and 89, 96, and 84 when calculated according to a modification of the equation of Hegsted et al. (1965). Mean relative weights were 100, 107, and 106% of standard.
These results indicate that the mean serum TC level of populations is strongly correlated with mean intake of SFAs (and probably of cholesterol as well, although data on this point are not adequate). Correlations of TC level are weaker with mean intake of PUFAs and mean relative weight. In short-term feeding experiments in small groups of people, mean TC changes predictably when intakes of SFAs, PUFAs, and cholesterol are changed under controlled conditions. Most of the differences among populations in mean TC can be accounted for by these short-term effects, and it seems reasonable to believe that long-term effects would be greater. Although populations differ in genetic factors, in body fatness, and in other factors affecting TC, it is not necessary to invoke such differences in order to account for the major portion of the differences among populations in mean TC levels. In view of the evidence, it seems reasonable to infer that variation among populations in the lipid composition of the diet, principally in mean intake of SFAs (and cholesterol), is an important factor, perhaps the major factor, in determining differences in mean serum TC.
Studies of Individuals
Cross-sectional studies of individuals within populations have generally shown little or no association between the fatty acid composition of the diet and serum TC. [See reviews of the extensive literature by Stallones (1983) and Stamler (1979).] Initially, this observation was difficult to reconcile with the clear experimental evidence for groups of people that changes in the lipid composition of the diet predictably affect mean LDL-C and TC in the blood and with the evidence from observational studies that mean SFA intake is strongly correlated with mean TC across populations. However, subsequent investigations have shown that these results are not inconsistent and, in fact, are expected.
Levels of LDL-C (and consequently of TC) are determined by a variety of intrinsic and extrinsic factors—the lipid composition of the diet being a key extrinsic factor (Hegsted et al., 1965; Keys et al., 1965a). However, people differ substantially in their TC levels, even when maintained on the same diet under controlled conditions. Jacobs et al. (1979) showed that weak cross-sectional correlations between the individual intake of SFAs and level of TC are a consequence of the large variation in intrinsic TC levels among individuals. Moreover, random error in determining the usual intake of nutrients by individuals biases these already weak correlations further toward zero (Beaton et al., 1979; Keys, 1965; Liu et al., 1978; Rush and Kristal, 1982). Knowledge of hypercholesterolemia can lead people to change their diets, thereby producing a negative cross-sectional correlation between SFA intake and level of TC (Shekelle et al., 1981b). Although the correlations were weak, after adjustment for intraindividual variability Shekelle et al. (1981b) showed that the magnitude of the regression of TC on lipid composition of the diet was consistent with the effect demonstrated in controlled feeding experiments.
In summary, the epidemiologic evidence obtained between and within populations indicates that increased SFA intake is associated with increased plasma TC and that SFA intake is a major extrinsic determinant of TC levels.
Page 182
Effects Of Saturation Of Dietary Fatty Acids On Plasma Cholesterol And Lipoprotein Concentrations
In 1952, Kinsell et al. (1952) studied the effects of formula diets on the responses of patients to various hormones and found that diets high in vegetable fat dramatically lowered TC concentrations. In the same year, Groen et al. (1952) observed that a diet rich in vegetable fat fed to volunteers resulted in much lower TC levels than a diet containing an equal amount of animal fat, regardless of cholesterol intake. Despite the skepticism typical of new findings, these initial observations were soon confirmed (Ahrens et al., 1954, 1957; Beveridge et al., 1955, 1957; Bronte-Stewart et al., 1955; Keys et al., 1957a,b; Kinsell et al., 1953; Malmros and Wigand, 1957). It was established that the serum cholesterol concentration was more responsive to the saturation of dietary fatty acids than to the total amount of fats or to the amount of dietary cholesterol. After many comparisons of different fats and oils, several investigators (Ahrens et al., 1957; Malmros and Wigand, 1957; Keys et al., 1957a) proposed that the presence of PUFAs in vegetable oils was responsible for the hypocholesterolemic effect. The effect was observed principally or exclusively on LDL-C.
Using the results of a series of experiments, Keys et al. (1957b, 1959) developed a formula predicting the change in serum cholesterol concentration from the change in the amounts of SFAs and PUFAs in the diet:
D Chol = 2.74 S - 1.31 P,
where DChol is the change in serum cholesterol in mg/dl and S and P are the changes in the percentage of calories derived from saturated (S) and polyunsaturated (P) fatty acids. At that time, the prevailing opinion was that dietary cholesterol did not affect the serum cholesterol concentration. Therefore, it was not part of the equation.
After additional experiments with dietary fats and cholesterol, Keys et al. (1965c) modified the equation and incorporated the effect of dietary cholesterol as follows:
DChol = 1.35 (2 S - P) + 1.5 Z,
where Z is the difference between the square root of the initial intake of cholesterol and the square root of the subsequent intake of cholesterol, both expressed as mg per 1,000 kcal.
Hegsted et al. (1965) conducted a comprehensive experiment in which they examined the effects of different types of fats (safflower, olive, and coconut oils), amounts of fat (22 or 40% of calories), and amounts of cholesterol. The amounts of dietary fats within these limits did not influence serum cholesterol concentration; the proportion of saturated and unsaturated fatty acids was a major determinant. Dietary cholesterol produced a small but statistically significant effect independently of the amount or type of fat. These investigators also developed regression equations that predicted the responses of plasma cholesterol concentrations to changes in dietary fats. The most practical equation was the following:
DChol = 2.16 S - 1.65 P + 6.77 C - 0.53,
where C is a change of 100 mg dietary cholesterol per day (or 100 mg per 2,600 kcal, or approximately 38 mg per 1,000 kcal) and S and P have the same meaning as above. This formula was subsequently modified by Hegsted (1986) to:
DChol = 2.16 S - 1.65 P + 0.097 C,
where C is the difference in cholesterol intake between the two diets in mg/1,000 kcal. In these studies, the SFAs alone accounted for 72% of the variation in serum cholesterol concentrations.
Keys and Parlin (1966) compared the predictive power of the equation of Hegsted et al. (1965) with those he and his associates had proposed previously (Keys et al., 1957b, 1959, 1965c) and found them to be quite similar. The principal difference resided in the computation of the dietary cholesterol effect; the Keys equation treated it as a square-root function, whereas the Hegsted equation treated it as a linear function. Both equations predicted approximately 90% of the effect of the dietary fat changes on the observed serum cholesterol concentrations.
Equations of the type developed by Keys, Hegsted, and their associates in the publications cited above, and similar ones devised by other investigators, were intended to indicate an average effect of dietary modifications on plasma cholesterol concentrations in a group of individuals—not to represent precise physiological or metabolic mathematical models. The equations were useful in subsequent studies to compare the results of experiments that varied in proportions of dietary fatty acids and cholesterol. Most such experiments have
Page 183
confirmed the validity of these equations as predictors of group averages of responses to dietary manipulations, but at the same time many investigators have commented on the large individual variability in responses (Keys et al., 1959, 1965b). This individual variability was more prominent in experiments involving dietary cholesterol, but it also appeared in experiments involving types of dietary fats. For example, Grundy and Vega (1988) reviewed the results of four experiments on 46 subjects to determine the effects of PUFAs and SFAs on plasma cholesterol and LDL-C. Six (about 13%) of the 46 subjects did not respond to a change in dietary fatty acid saturation with any substantial change in plasma cholesterol concentration.
Katan et al. (1988) characterized the responsiveness of people to dietary cholesterol by subjecting them to repeated dietary challenges and then compared the responses of these same people to high-SFA and high-PUFA diets. Average egg eaters (who consumed an average of approximately 500 mg of cholesterol per day) showed a high degree of congruence between responsiveness to dietary cholesterol and responsiveness to dietary SFAs (R = .62, p <.01), whereas eaters of large amounts of eggs (approximately 1,130 mg of cholesterol per day) showed a low degree of congruence (R = .15, not significant). There was no ready explanation for the difference between the two groups. It will be important in future research to determine the extent to which the responsiveness to dietary fatty acids is a genetically determined characteristic, whether it is influenced by previous diets, whether it is dependent on an interaction between genetics and diet, and whether it involves the same metabolic processes as does responsiveness to dietary cholesterol.
One of the most convincing demonstrations of the effects of dietary fatty acid saturation was the Lipid Research Clinics Coronary Primary Prevention Trial, in which more than 6,000 hypercholesterolemic men were instructed to adopt a diet lower in cholesterol and higher in the relative proportion of PUFAs to SFAs. The change in dietary SFAs was correlated directly, and the change in PUFA intake was correlated inversely, with lowered plasma cholesterol concentration—mainly LDL-C (Gordon et al., 1982).
Within a few years of the initial observations, the weight of the evidence led to the clear conclusion that the effect was real and consistent, and research efforts thereafter were directed toward identifying the specific fatty acids responsible for the effect, the mechanisms of the effect, and the role of SFAs relative to that of cholesterol in causing hypercholesterolemia in humans. These topics have been reviewed by Ahrens (1957), Goodnight et al. (1982), Grundy (1979, 1987), Jackson et al. (1978), McNamara (1987), and Nestel (1987).
Saturated Fatty Acids
In early experiments concerned with the effects of fatty acid saturation on serum cholesterol concentrations, cocoa butter, which is approximately 35% stearic acid (C18:0), did not elevate serum cholesterol as did most other natural foods rich in SFAs (Ahrens et al., 1957; Erickson et al., 1964). Keys et al. (1965c) found that the predictive equations were more accurate when stearic acid was eliminated from the computation of the percentage of calories derived from SFAs. The SFAs with chain lengths shorter than 12 carbon atoms likewise appeared not to influence serum cholesterol levels, and the equations were more predictive when they were eliminated from consideration. Grande et al. (1970) demonstrated that stearic acid did not affect serum cholesterol concentrations. This finding was reconfirmed by Reiser et al. (1985), who compared the effects of beef fat, which is about 20% stearic acid, with those of coconut oil and safflower oil on serum cholesterol concentrations, and more recently by Bonanome and Grundy (1988), who compared the effects of formula diets high in palmitic, stearic, and oleic acids.
In early experiments, butter and coconut oil were strongly hypercholesterolemic. Because these fats have a high proportion of saturated medium- and short-chain fatty acids (C6 to C12), it was suspected that their effects might be due to these fatty acids. Beveridge et al. (1959) and Hashim et al. (1960) demonstrated that medium-chain triglycerides, which were 99% SFAs with chain lengths from C6 to C12, produced little or no elevations in serum cholesterol concentrations over those produced by corn oil diets. Keys et al. (1965c) found that SFAs with fewer than 12 carbon atoms did not contribute to the predictive accuracy of their equation for estimating serum cholesterol changes due to fatty acid intake, but included lauric acid (C12) in their equation. Hegsted et al. (1965) found that C10 and C12 SFAs did not contribute significantly to the predictive accuracy of their equation for estimating the cholesterol-raising effects of dietary fats. There has been little or no investigation of the metabolic effects of these fatty acids since 1965, and the role of lauric acid (C12) remains uncertain.
Page 184
The average effects of the various fatty acids commonly encountered in human diets are summarized graphically in Figure 7-2 (Grundy, 1981). This figure compares the predictions of the Keys and Hegsted equations for serum cholesterol concentration changes when the fatty acid content of the diet is changed.
In conclusion, palmitic (C16) and myristic (C14) acids are the SFAs most effective in elevating serum cholesterol concentrations. Stearic acid (C18:0) and SFAs with chain lengths of 10 or less have no effect. Lauric acid (C12) also raises the serum cholesterol level but the quantitative extent of its effects, compared to those of C14 and C16 acids, requires further investigation.
Monounsaturated Fatty Acids
Early investigators noted that olive oil, which is rich in oleic acid (C 18:1), lowered serum cholesterol concentrations when substituted for SFAs, as did the PUFAs (Ahrens et al., 1957; Bronte-Stewart et al., 1955; Keys 1957, 1958a). Keys et al. (1958a) found that substituting oleic acid for carbohydrates in isocaloric quantities did not affect the serum cholesterol level. Both Keys et al. (1965a) and Hegsted et al. (1965) reported that including MUFAs in regression equations did not increase the predictive value of the equation.
The effects of dietary MUFAs on serum lipoprotein levels were reexamined by Baggio et al. (1988), Becker et al. (1983), Grundy (1986), Grundy et al. (1986, 1988), Mattson and Grundy (1985), and Mensink and Katan (1987), all of whom confirmed that MUFAs did not elevate serum cholesterol concentrations as did SFAs. Furthermore, they found that high-MUFA diets did not lower HDL-C concentrations as did replacement of SFAs by carbohydrates.
The effects of MUFAs on serum lipoprotein levels are important, because olive oil or other oils rich in these fatty acids could be used, along with or as an alternative to carbohydrates, as replacement for SFAs in diets designed to lower serum cholesterol and, particularly, LDL-C levels. If the early and recent findings regarding the effects of MUFAs on LDL-C and HDL-C levels are consistently confirmed, the use of oils rich in these fatty acids makes possible the design of diets that would lower LDL-C levels, maintain HDL-C levels, and be more palatable than very-low-fat diets.
For patients with high serum cholesterol levels, it remains to be determined whether attempts to minimize HDL-C lowering (in the course of dietary treatment to lower LDL-C) by partial substitution
FIGURE 7-2
Responses to changes in dietary fatty acids predicted from
the studies of Keys et al. (1965c) and Hegsted et al. (1965).
Figure adapted from Grundy (1981).
of SFAs with MUFAs might be more beneficial than substitution with carbohydrates.
w-6 PUFAs
The major dietary PUFA with the double bond in the w-6 position is linoleic acid (C18:2). The proportion of linoleic acid in plant oils, expressed as percent of all fatty acids, ranges from 1 to 2% in coconut and palm kernel oils to 50 to 60% in corn, cottonseed, and soybean oils and to about 75% in safflower oil. Corn oil, cottonseed oil, and safflower oil are the natural sources most frequently used in the dietary experiments in humans cited in this report. The other important PUFA found in plants is linolenic acid (C18:3), which has a double bond in the w-3 position.
Keys et al. (1957b, 1965a) and Hegsted et al. (1965) showed that isocaloric exchange of PUFAs for carbohydrates or MUFAs lowered the serum cholesterol concentration in men consuming mixed-food diets that provided 10 to 40% of calories from fat. Predictive equations developed by linear regression analysis of data from those experiments, described in the previous section, indicated that the isocaloric exchange of PUFAs for 10% of calories derived from MUFAs or from carbohydrates decreased serum cholesterol by 13 mg/dl (Keys et al., 1965c) or by 16.5 mg/dl (Hegsted et al., 1965) when other factors were held constant. This effect was observed directly in experiments in which PUFAs were exchanged for
Page 185
carbohydrates or for MUFAs with little change in intake of other nutrients.
Although Keys et al. (1957b, 1965a, 1965c) and Hegsted et al. (1965) published results only for serum cholesterol, both measured b-lipoprotein (the term then used for LDL) and stated that the changes observed in that fraction paralleled the changes in serum cholesterol. The observed differences in serum cholesterol were larger than would be accounted for by changes in HDL-C. These results suggested that PUFAs had a specific serum-cholesterol- (particularly, an LDL-C-) lowering effect when substituted isocalorically for carbohydrates as well as when substituted for SFAs.
Between 1959 and 1986, several studies in humans addressed the effects of PUFAs substituted for carbohydrates and suggested that dietary PUFAs did not lower serum TC or LDL-C levels when substituted for carbohydrates (Brussaard et al., 1980, 1982; Grundy et al., 1986; Horlick, 1959, 1960; Kuusi et al., 1985). Some of these experiments involved women, who may not have responded as did men, and the observations were not controlled for possible effects of the menstrual cycle on serum lipids and lipoproteins (Oliver and Boyd, 1953); some involved small numbers of subjects and had limited power; some were confounded by variation in other dietary components, particularly cholesterol; and in one, formula diets were used. These reports have, however, raised the question of whether there is a specific LDL-C-lowering effect of PUFAs when substituted for carbohydrates.
There have been no recent reports of experiments designed to test this question directly and rigorously in people eating mixed-food diets. The early experiments conducted by Keys, Hegsted, and their associates (described above) indicated that there was such an effect in men consuming mixed-food diets. Furthermore, the analysis of effects of dietary changes in the Lipid Research Clinics Coronary Primary Prevention Trial showed an association of increased intake of PUFAs with decreased serum cholesterol concentration (mainly in LDL-C). This association was independent of changes in body weight and intake of SFAs and cholesterol (Gordon et al., 1982).
This issue is not important in the design of practical diets for lowering serum cholesterol levels, because for other reasons, diets containing more than approximately 7% of calories as PUFAs are not recommended. The issue is of interest as part of the overall question of the mechanism by which dietary PUFAs lower LDL-C concentrations. The evidence bearing on this mechanism is discussed below.
A consistent finding in all comparisons of isocaloric diets in which fats of any type were substituted for carbohydrates is that serum triglyceride levels were lowered. These studies have been of short duration. The effect may well be temporary, since populations that habitually consume a low-fat, high-carbohydrate diet are not characterized by a high prevalence of hypertriglyceridemia.The comparative effects of carbohydrates versus fats in both short- and long-term studies are discussed in more detail in Chapter 9.
w-3 PUFAs
Soon after the hypocholesterolemic effects of PUFAs were discovered in 1952, some investigators found that oils derived from fish and marine mammals were as effective as vegetable oils in lowering serum cholesterol levels in humans (Ahrens et al., 1959; Bronte-Stewart et al., 1955; Keys et al., 1957a). This observation received little attention for nearly 25 years until interest was stimulated by fragmented and poorly documented reports of low rates of CHD among fish-eating Greenland Eskimos. The recent literature on the effects of fish oils on plasma lipids, plasma lipoproteins, and lipoprotein metabolism has been reviewed by Carroll (1986a), Goodnight et al. (1982), Herold and Kinsella (1986), Klasing and Pilch (1986), Leaf and Weber (1988), Nestel (1987), von Schacky (1987), and Carroll and Woodward (1989).
Fish oils consistently reduce serum triglyceride concentrations, particularly in hypertriglyceridemic subjects (Harris et al., 1983, 1984; Illingworth et al., 1984; Nestel et al., 1984; Phillipson et al., 1985; Sanders et al., 1985; Sullivan et al., 1986). This effect appears to result from their inhibition of the synthesis of VLDL-triglycerides, leading to a reduced total secretion of VLDL-triglycerides (Connor, 1986; Nestel et al., 1984). Substitution of w-3 PUFAs for SFAs in the diet results in a lowering of LDL-C. This response appears to be the result of removing SFAs from the diet. Thus, supplementation of the diet with w-3 PUFAs without altering SFA intake has been reported not to cause a lowering of LDL-C levels (Rogers et al., 1986). In hypertriglyceridemic patients who are treated with w-3 PUFAs, the lowering of plasma triglycerides is often associated with an increase in LDL-C levels (Connor, 1986). This response is typical of other triglyceride-lowering agents as well.
Fish oils also affect the hemostatic system (de-
Page 186
scribed below in the section on Effects of Dietary Fats and Other Lipids on Hemostasis and Eicosanoid Metabolism and reviewed in depth by Herold and Kinsella, 1986). The effects of the w-3 PUFAs are currently being investigated intensively, and a considerable amount of new information regarding their effects on lipoprotein metabolism may be anticipated in the near future.
Hydrogenated Fatty Acids
The hydrogenation of vegetable oils with high proportions of unsaturated fatty acids was developed as a process to make the oils more palatable and useful as substitutes for animal fats. This process not only reduces the degree of unsaturation of the oils, but also generates geometric isomers—the trans fatty acids (see section on Trans Fatty Acids below for a discussion of the effects of these isomers). In early experiments shortly after the discovery of the serum-cholesterol-lowering effects of the PUFAs, hydrogenation was found to raise serum cholesterol levels. Malmros and Wigand (1957) found that hydrogenated coconut oil elevated serum cholesterol more than other plant oils rich in PUFAs, but they did not test the effects of hydrogenation alone.
Anderson et al. (1961) reported that hydrogenated corn and safflower oils, compared to their native (naturally occurring) forms, consistently raised serum cholesterol concentrations, but the elevation was less than that predicted by the equations developed by Keys et al. (1957b). They attributed the less-than-predicted effect to the presence of trans fatty acids, which they tested in another experiment. Horlick (1960) observed that hydrogenated corn oil, as compared to a low-fat diet, elevated serum cholesterol in four subjects but not to the same degree as did butter. McOsker et al. (1962) found that the effects of hydrogenated cottonseed and soybean oils did not differ from effects of the native oils on serum cholesterol concentrations.
There is a widespread belief that hydrogenated vegetable oils, as consumed in margarines, shortenings, and salad oils, elevate serum cholesterol levels because of a reduction in the number of double bonds, but no controlled experiments have been conducted since 1962 to test this. The experiments with hydrogenated oils have focused on the trans fatty acids produced in the hydrogenation process and have attempted to eliminate saturation of fatty acids as a variable by using fat blends as controls.
Hydrogenated coconut oil was frequently used in the early experiments cited above. This oil was already rich in the cholesterol-raising SFAs (C12 to C16) and thus may not have been representative of the other vegetable oils. When oils rich in oleic and linoleic acids are hydrogenated, stearic and oleic acids, both of which do not elevate serum cholesterol, are produced; almost no myristic or palmitic acids (the main fatty acids responsible for raising serum cholesterol) are produced because the oils contain very few C14 or C16 unsaturated fatty acids. Furthermore, some PUFAs may remain. For example, analyses of more than 80 different margarines prepared by hydrogenation of vegetable oils showed that the palmitic acid content ranged from 8.5 to 13.2% of all fatty acids, with a mean of approximately 10% (Slover et al., 1985). Palmitic acid constituted 24% of one margarine made from lard. In the vegetable oil-based margarines, the linoleic acid content ranged from 6 to 46% (average, about 27%), and the linolenic acid content ranged from 0.2 to 3.6% (average, about 2%). Each hydrogenated oil preparation must be examined with regard to the spectrum of fatty acids it contains, and hydrogenated fats cannot be classified in general as cholesterol raising. This conclusion, based principally on the application of the predictive equations described above, needs verification by experimentation in humans.
The formation of trans fatty acids during hydrogenation is another matter that confounded many of the experiments with hydrogenated oils. This issue is discussed in the following section.
Trans Fatty Acids
The trans fatty acids are geometric isomers of the unsaturated fatty acids (mostly oleic, linoleic, and linolenic acids) occurring naturally in the fats of ruminants and formed in vegetable oils during hydrogenation. From 2 to 7% of beef fat and butterfat, and from 10 to 30% of margarines, shortenings, and salad oils, are trans fatty acids. Between 1965 and 1983, the daily per-capita consumption was about 8 g/day or 6% of the total fat consumed in the United States. The estimates of daily consumption have been confirmed by analyses of trans fatty acids in the adipose tissue of humans (reviewed by Senti, 1985). The biologic properties of the trans fatty acids have been studied extensively, and the original reports have been reviewed by Applewhite (1981), Beare-Rogers (1983), Emken (1984), Hunter and Applewhite (1986), Kinsella et al. (1981), and Senti (1985). A recent survey of more than 80 samples of commercial margarines produced and sold in the United States showed that trans-unsaturated octa-
Page 187
decanoic acids ranged from 10.7 to 30.1% of all fatty acids and that small amounts of tras, trans, trans, cis, and cis, trans isomers of linoleate were present (Slover et al., 1985).
Anderson et al. (1961) compared the effects of native and hydrogenated oils similar in their proportions of SFAs, MUFAS, and PUFAS, but varying in their proportions of trans isomers. The oils with high proportions of trans isomers produced higher serum levels of cholesterol and triglycerides than did the native oils. However, the hydrogenated corn oil contained half as much of the essential fatty acids as did the native corn oil. Beveridge and Connell (1962) compared the effects of margarines, corn oil, and butter in formula diets and found that margarines produced higher serum TC levels than did corn oil diets, but not as high as did butter. Grasso et al. (1962) found that the trans fatty acid composition of a hydrogenated oil lowered serum cholesterol levels as did an oil with the same distribution of SFAs, MUFAs, and PUFAs.
McOsker et al. (1962) reported no difference in serum cholesterol concentrations in six subjects fed hydrogenated oils containing 15 to 20% trans fatty acids. Erickson et al. (1964) compared the effects of partially hydrogenated oils with blends of native oils having a similar distribution of SFAs, MUFAs, and PUFAs, but up to 10% trans isomers, and found no difference in serum cholesterol levels and no interaction with dietary cholesterol. After a series of experiments, de Iongh et al. (1965) also concluded that trans fatty acid isomers had no effect on serum cholesterol concentrations; the levels attained followed the predictive equation of Keys et al. (1965c) and were not influenced by the trans fatty acid content. Mishkel and Spritz (1969) tested the effects of high levels of trans isomers of linoleic acid in six patients and found elevations of serum triglycerides but inconsistent changes in serum cholesterol concentrations.
Mattson et al. (1975) conducted the most thorough and controlled test of trans fatty acid effects in 33 men who consumed formula diets containing similar distributions of SFAs, MUFAs, and PUFAs, but differing to an extreme in trans fatty acid isomers. More than 60% of the MUFAs and approximately 50% of the PUFAs in the contrasting diet were in the trans configuration. There were no differences in serum cholesterol or triglyceride levels after 4 weeks on the diets.
In conclusion, some effects of the trans fatty acid isomers on lipid or lipoprotein metabolism may remain undetected. However, most evidence indicates that these isomers, in the quantities usually consumed in the U.S. diet, do not influence serum cholesterol concentrations. There may be other effects unrelated to lipid and lipoprotein metabolism. These possibilities deserve careful attention and additional investigation.
Peanut Oil
Peanut oil, which is rich in MUFAs and PUFAs, was found to be unusually atherogenic in some experiments in animals (see following section on Animal Studies). Very few tests of the effects of peanut oil on serum lipoproteins have been conducted in humans. In a few experiments in humans (Ahrens et al., 1957; Baudet et al., 1984; Bronte-Stewart et al., 1955), peanut oil resulted in serum cholesterol and VLDL-C, LDL-C, and HDL-C concentrations similar to those produced by other unsaturated vegetable oils.
Fatty Acid Effects on Plasma Lipoproteins
There have been many attempts to determine the mechanisms of action of the SFAs in raising serum cholesterol concentrations or of the PUFAs in lowering them. The unsaturated dietary fatty acids were incorporated into the lipoprotein lipids within 14 days (Spritz and Mishkel, 1969). In studies of humans fed high-PUFA diets, Shepherd et al. (1980a) found decreases in the cholesterol-to-protein ratio in LDL, and Pownall et al. (1980) observed a number of differences in the thermal properties of plasma LDL related to the fatty acid composition of the diet. Kuksis et al. (1982) found many changes in lipoprotein composition and decreased numbers of LDL particles, but no changes in the sizes of the particles, in people consuming a high-PUFA diet. Zanni et al. (1987) also found decreased numbers of LDL and HDL particles related to PUFA intake.
PUFA increased the fractional catabolic rate of LDL apo B (Shepherd et al., 1980a) and in some studies decreased the production of LDL (Cortese et al., 1983; Turner et al., 1981). However, these changes were small and did not occur consistently.
As yet, the only evidence regarding the type of effects exerted by fats on LDL receptor activity are the result of experiments in animals. Spady and Dietschy (1985) found that receptor-mediated uptake of LDL in hamsters was suppressed about 30% by dietary cholesterol when the animals were consuming PUFAs (safflower oil) and was suppressed about 90% when they were consuming SFAs (coconut oil). In more recent studies in hamsters, Spady and Dietschy (1988) observed that dietary cholesterol suppressed hepatic LDL receptor activity and raised plasma LDL levels in a
Page 188
dose-dependent manner. At each level of cholesterol intake, polyunsaturated triglycerides diminished and saturated triglycerides accentuated the effects of dietary cholesterol. In baboons, hepatic LDL receptor messenger RNA (mRNA) was decreased when the animals were fed SFAs with cholesterol as compared to those fed either MUFAs or PUFAs (Fox et al., 1987). In all these studies, the effects on LDL receptor activity were consistent with the observed fat-induced changes in plasma lipoprotein concentrations.
Plasma triglyceride levels, representing VLDL and other triglyceride-rich lipoproteins, were lower in humans on PUFA diets than on SFA diets. The difference was usually proportionally greater than for plasma cholesterol (Chait et al., 1974).
Initially, plasma HDL levels were believed to be insensitive to dietary factors (Keys et al., 1959). Between 1958 and 1980, 6 of 15 observational studies and experiments suggested an inverse relationship between HDL-C concentration and intake of PUFAs (reviewed by McGill et al., 1981c). PUFAs also decreased the synthesis of apo A-I but did not change its fractional catabolic rate (Shepherd et al., 1978).
Since 1981, a number of experiments in humans have been conducted to examine the effects of PUFAs on HDL. Brussaard et al. (1982), Ehnholm et al. (1984), Jackson et al. (1984), Kuusi et al. (1985), Mattson and Grundy (1985), Schonfeld et al. (1982), Shepherd et al. (1980b), and Vega et al. (1982) found that dietary PUFAs decreased HDL-C, but Baudet et al. (1984), Becker et al. (1983), Kraemer et al. (1982), McNamara et al. (1987), Oh and Miller (1985), and Weisweiler et al. (1985, 1986) reported no change in HDL-C in subjects on similar diets. Both Brussaard et al. (1982) and Kuusi et al. (1985) observed that the decrease in HDL was due primarily to a drop in HDL2-C, whereas HDL3-C was unchanged (HDL2 and HDL3 are the two major subfractions of HDL). Jones et al. (1987b) found that the reduction in HDL-C associated with an increased intake of PUFAs occurred only when total fat intake was low (20% of calories).
In conclusion, dietary PUFAs probably lower plasma HDL-C concentrations, particularly HDL2 levels, to some degree, but individual variability in the response probably leads to varying results among experiments involving small numbers of subjects.
Effects of Fat on Cholesterol Absorption, Synthesis, and Excretion
There is little evidence that dietary fat saturation influences cholesterol absorption in humans (Grundy and Ahrens, 1970; Nestel et al., 1975). However, there are several reports of enhanced cholesterol absorption by PUFAs in squirrel monkeys (Tanaka and Portman, 1977). Measurements by sterol balance methods in humans (Grundy and Ahrens, 1970) and by in vitro methods in rat liver (Wilson and Siperstein, 1959) indicated that cholesterol synthesis was not influenced by saturation of dietary fat.
Although there is little support for the concept that fatty acid saturation influences cholesterol absorption or synthesis in humans, some results have shown a transient enhanced sterol excretion when PUFAs were substituted for SFAs in the diet (Connor et al., 1969; Moore et al., 1968; Nestel et al., 1973, 1975; Wood et al., 1966). The initial increase in sterol excretion may account for the reduction in plasma cholesterol resulting from dietary PUFAs. However, if cholesterol absorption and synthesis were not increased, increased sterol excretion would not be expected upon attainment of a new steady state. This may explain the failure to detect consistently enhanced sterol excretion in humans fed PUFAs as reported by Avigan and Steinberg (1965), Shepherd et al. (1980a), and Spritz et al. (1965) and in patients with hypertriglyceridemia (Grundy, 1975) or hypercholesterolemia (Grundy and Ahrens, 1970).
Grundy and Ahrens (1970) suggested that PUFAs cause a shift in cholesterol from plasma to liver. Hepatic cholesterol accumulation upon PUFA feeding has been reported in several animal experiments (Avigan and Steinberg, 1958; Bloomfield, 1964; Tidwell et al., 1962). However, in a group of 12 hypercholesterolemic patients, polyunsaturated fats, compared to saturated fats, decreased the liver cholesterol content (Frantz and Carey, 1961).
In summary, transient increased sterol excretion may explain the decrement in plasma cholesterol among normolipidemic subjects, but not among those with hypercholesterolemia. A plausible, but not firmly established, mechanism is that PUFAs accelerate the transport of lipoprotein cholesterol to the liver and promote higher sterol excretion until a new steady state is achieved. Precisely how PUFAs mediate the decrease in LDL production and LDL plasma concentration is not known. However, effects on LDL receptor activity in the liver are likely to play an important role in the mechanism of the effects of dietary SFAs and PUFAs. SFA intake may decrease LDL receptor activity, thereby leading to an increase in LDL and TC levels.
Page 189
Summary
Some dietary SFAs (palmitic, myristic, and lauric acids) elevate serum LDL-C and HDL-C concentrations. w-6 PUFAs lower serum TC and LDL-C levels when substituted for SFAs and are also likely to do so to a small extent when isocalorically exchanged for MUFAs or carbohydrates in mixed-food diets that provide 10 to 40% of calories from fats. w-6 PUFAs also lower HDL-C levels. MUFAs do not affect serum TC or LDL-C levels when isocalorically substituted for carbohydrates in mixed-food diets, but they do lower serum TC and LDL-C levels, without lowering HDL-C, when substituted for SFAs. The average effect on TC levels brought about by variations in the proportions of the various dietary fatty acids can be predicted largely from equations based on experimental observations. However, the mechanism of the effect is not clearly defined, despite many proposed hypothetical mechanisms. Recent results from experiments in animals suggest that the effects of dietary SFAs and PUFAs on hepatic LDL receptor activity play a role in this phenomenon. There is considerable interindividual variability in response of plasma lipoproteins to dietary fatty acid saturation. This is probably controlled genetically but has not been studied as intensively as has the response to dietary cholesterol.
After the initial discovery that dietary cholesterol produced hypercholesterolemia and atherosclerosis in rabbits, a variety of species were found also to be sensitive to dietary cholesterol. Fat in the experimental diet was considered simply as a vehicle for the added cholesterol until the effects of SFAs and PUFAs on serum cholesterol levels were discovered in humans in 1952. Most studies of the effects of various types of fats on plasma lipid and lipoprotein levels and on lipoprotein metabolism have been conducted in humans, but a few investigators have examined these effects in animal models.
SFAs Compared To PUFAs
In 1954, Kritchevsky et al. (1954) demonstrated that dietary cholesterol elevated serum cholesterol to a greater extent when fed to rabbits with hydrogenated vegetable oil than when fed with corn oil. This effect was associated with saturation of dietary fatty acids (Kritchevsky et al., 1956).
Similar observations were made in nonhuman primates. SFAs usually produced higher serum cholesterol levels than did PUFAs when fed with low or moderate levels of dietary cholesterol to squirrel monkeys (Corey et al., 1974; Lofland et al., 1970; Robison et al., 1971), cebus monkeys (Corey et al., 1974; Portman and Andrus, 1965; Wissler et al., 1962), vervet monkeys (Mendelsohn et al., 1980), and rhesus monkeys, but not when fed to baboons (Strong and McGill, 1967). In some experiments, two or more types of fats differing in origin and in fatty acid chain length were compared. For example, in cebus monkeys (Wissler et al., 1962), coconut oil produced higher serum cholesterol levels than did butterfat. However, only a few such experiments were conducted, and it is difficult to reach a firm conclusion other than the generalization that SFAs elevate serum cholesterol and cause more severe atherosclerosis in most nonhuman primates than do PUFAs.
More recent experiments have been conducted to examine the effects of various types of fatty acids on the major serum lipoprotein classes. In baboons, for example, SFAs, as compared to unsaturated fatty acids, elevated serum LDL-C and HDL-C concentrations to approximately the same extent. (McGill et al., 1981b; Mott et al., 1982).
In rhesus monkeys, coconut oil, unlike corn oil, elevated plasma LDL-C proportionately more than it did HDL-C in a short-term experiment (Chong et al., 1987), but coconut oil elevated LDL-C only when fed with cholesterol in a long-term experiment (Ershow et al., 1981). Coconut oil also elevated serum cholesterol in cebus and squirrel monkeys (Corey et al., 1974). Although there are some variations among the results in nonhuman primates, the preponderance of evidence indicates that these animals respond to saturated and unsaturated fatty acids as do humans, i.e., with an elevation in the plasma concentrations of both LDL-C and HDL-C in response to SFAs.
A number of studies in animals have been conducted in an attempt to identify the mechanisms responsible for these effects of saturated and unsaturated fatty acids on plasma lipoproteins (Chong et al., 1987; Ershow et al., 1981; Johnson et al., 1985; Parks and Rudel, 1982; Rudel et al., 1981, 1983, 1986; Spady and Dietschy, 1985). The observation promising the most insight into the process (and discussed above) is that dietary SFAs fed with cholesterol suppress LDL receptor activity in the hamster liver to a much greater extent than does dietary cholesterol alone (Spady and Dietschy, 1985, 1988)—a change that would be expected to elevate plasma LDL-C levels.
Page 190
There is no conclusive evidence that hemostatic variables such as platelet function and blood coagulation as measured in vitro are influenced by dietary intake of SFAs, MUFAs, or w-6 PUFAs, such as linoleic, dihomogamma linolenic, or arachidonic acids (von Schacky, 1987). However, some reports have suggested that long-term differences in the intake of SFAs or of w-6 PUFAs may influence platelet function or thrombogenesis (Renaud et al., 1970, 1985). Additional definitive studies would be useful in this area.
More definite evidence about such effects is available for the w-3 PUFAs, such as eicosapentaenoic acid (EPA, C20:5, w-3) and docosahexaenoic acid (DHA; C22:6, w-3). These w-3 PUFAs appear to have inhibitory effects on platelet function and prolong bleeding time—phenomena somewhat similar to those elicited by aspirin (Knapp et al., 1986; Leaf and Weber, 1988). However, no studies have as yet determined whether w-3 PUFAs reduce morbidity or mortality in cardiovascular disease (for full reviews and references, see Leaf and Weber, 1988, and von Schacky, 1987).
Shortened coagulation times measurable in hyperlipemic plasma were originally believed to represent hypercoagulability, but this was later shown to be an artifact induced by platelets trapped in the lipemic plasma (Marcus, 1966). With the possible exception of w-3 PUFAs, dietary lipids do not influence eicosanoid metabolism as presently understood (Mahadevappa and Holub, 1987).
Of greatest interest concerning effects of dietary fats and other lipids on hemostasis and eicosanoid metabolism are the w-3 PUFAs of marine origin (von Schacky et al., 1985b). Some investigators have reported that Eskimo populations in Greenland appear to have reduced mortality from occlusive vascular disease, putatively related to consumption of fish and fish oils containing w-3 fatty acids (Bang and Dyerberg, 1980). However, the epidemiologic evidence on this assertion is quite incomplete. Furthermore, there is no evidence that dietary supplementation with w-3 fish oils will reduce the risk of cardiovascular disease (Carroll, 1986a).
w-3 Polyunsaturated Fatty Acids
The w-3 group of PUFAs includes not only EPA and DHA, but also a-linolenic acid (C18:3, w-3). These PUFAs are cyclooxygenated and lipoxygenated in a manner similar to w-6 fatty acids, but the end products may differ functionally from those of the w-6 group.
The w-6 PUFAs include linoleic acid (C18:2, w-6), dihomogamma linolenic acid (C20:3, w-6), and arachidonic acid (C20:4, w-6). Arachidonic acid is cyclooxygenated to prostaglandins E2, D2, F2a, prostacyclin (PGI2, measured as 6-keto-PGF1a), thromboxane (TX) A2, HHT, and malondialdehyde (Marcus, 1978, 1984), whereas EPA is metabolized to PGE3, PGF3a, PGI3, and TXA3. EPA is also lipoxygenated in stimulated leukocytes to the leukotrienes LTB5, LTC5, LTD5, and LTE5 and to 5-hydroxyeicosapentaenoic acid (5-HEPE). This is in contrast to leukocyte arachidonate, which is lipoxygenated to LTB4, LTC4, LTD4, LTE4, and 5-hydroxyeicosatetraenoic acid (5-HETE). In activated platelets, arachidonate is lipoxygenated to 12-HETE, and EPA to 12-HEPE (von Schacky, 1987).
Some studies indicate that TXA3 does not possess the same proaggregatory or vasoconstrictory properties as TXA2, whereas PGI3 is reported to retain the vasodilatory and platelet inhibitory properties of PGI2. More definitive studies are necessary to confirm and examine further the spectrum of biologic properties of PGI3 and TXA3.
Terminology
The widespread interdisciplinary interest in dietary lipids as possible modulators of hemostasis, eicosanoid metabolism, and cardiovascular disease has led to the inadvertent conceptualization of these different processes as synonymous events (Marcus, 1988b). Thrombogenesis, atherosclerosis, hypercoagulability, and platelet hyperreactivity are frequently referred to as phenomena occurring in parallel. Atherosclerosis, however, is not clearly related to blood coagulation or to platelet function, until it reaches the advanced occlusive stages in which thrombosis occurs.
Patients with clinically manifest coronary artery disease may have circulating hyperactive platelets, but these could result from increasing cell-to-cell encounters between platelets and atherosclerotic plaques. Platelet hyperreactivity may represent a secondary event such as a response to injury (Marcus, 1988a).
A direct relationship between diet and hemostasis or eicosanoid metabolism has not been established. The only possible exception may be an association with w-3 PUFAs (Mahadevappa and Holub, 1987).
Page 191
Effects Of w-3 Fatty Acids On Platelet Function
Studies have been conducted in volunteers and patients with vascular disease who have ingested cold water marine fish such as mackerel or salmon or fish oils per se. Cod liver oil, which contains 10% EPA and 15% DHA, has been administered in doses ranging from 10 to 40 ml daily for varying periods as has the commercial preparation known as MaxEPA—a blend of fish body oil containing 20% EPA and 10% DHA. Purified EPA and DHA also have been administered separately in a short-term study. Bleeding time, a major indicator of platelet function, was prolonged to some degree in almost all study participants (von Schacky, 1987). In Greenland Eskimos, bleeding time averaged 8 minutes; in a Danish control population, it was 5 minutes. Bleeding time in patients with atherosclerosis extended from a control of 3.3 minutes to 5.6 minutes during the third week in which their diet was supplemented with MaxEPA. Although these findings are suggestive, it is difficult to know with certainty whether such lengthening of the bleeding time indeed signifies protection against vascular disease. Nevertheless, the prolongation is consistent and is comparable to changes in bleeding time observed following the ingestion of aspirin (Knapp et al., 1986).
In addition to lengthening bleeding time, the ingestion of cod liver oil reduces ex vivo platelet aggregation in response to adenosine diphosphate (ADP) and collagen. Responsiveness to agonists would be more adequately interpretable if each stimulus were studied over a concentration curve, because high doses of agonists such as thrombin and collagen may mask mild inhibitory effects of fish oils.
When EPA and DHA were administered separately for 6 days at 6 g/day, EPA reduced platelet responsiveness to collagen. In contrast, DHA reduced platelet aggregation to both ADP and collagen. Thus, DHA may be an important antithrombotic agent in its own right (von Schacky et al., 1985b).
A Comparison Of Arachidonate And Epa Metabolism In Humans
Early in vitro studies on human or animal platelets demonstrated that added EPA was incorporated into all phospholipid subclasses whether or not arachidonate was also present. In contrast, EPA ingested by volunteers was incorporated mainly into platelet phosphatidylcholine and phosphatidylethanolamine, but not phosphatidylinositol or phosphatidylserine. Upon stimulation of washed platelets obtained from these volunteers, EPA was released by way of the phospholipase A2 mechanism, acting on phosphatidylcholine and phosphatidylethanolamine. Moreover, the dietary EPA released was demonstrably converted to TXA3 (von Schacky et al., 1985a).
When arachidonic acid is added to platelets in vitro, it is rapidly metabolized to TXA2, HHT, and 12-HETE, and platelet aggregation ensues via release of ADP (Marcus, 1987). In contrast, the addition of EPA does not induce platelet aggregation. EPA will compete with free arachidonate for platelet cyclooxygenase; however, compared to arachidonate, EPA is a poor platelet agonist. More detailed research on the biologic action of TXA3 in vitro is desirable (von Schacky et al., 1985b).
DHA is incorporated into platelets in vivo in a manner similar to EPA. At high concentrations (150 µM in vitro), DHA appears to inhibit platelet aggregation, but at low concentrations (2 to 10 µM) platelet aggregation is not inhibited. Following dietary supplementation, DHA is not released from stimulated human platelets or neutrophils. In contrast, dietary DHA is partially retroconverted to EPA (C20:5, w-3). Further studies on the effects of DHA on platelets ex vivo should be of great interest (Mori et al., 1987; von Schacky et al., 1985b).
During w-3 PUFA dietary supplementation of healthy volunteers or patients with atherosclerosis, the quantity of TXA2 formed by stimulated platelets ex vivo is reduced. This could be due to preferential incorporation and may lead to less arachidonate being available for cyclooxygenation. There may also be competition for cyclooxygenase between arachidonate and EPA (which is oxygenated less efficiently than arachidonate), resulting in less TXA2 formation. This may represent part of the suggested protective action of dietary w-3 PUFAs.
Metabolism Of EPA In Endothelial Cells
EPA added to endothelial cells of human umbilical cord origin in the presence of arachidonate results in formation of PGI3, whose biologic activity has been reported to be similar to that of PGI2. During dietary supplementation with EPA, the urinary metabolite of PGI3 is detectable. Metabolism of w-3 PUFAs acids in various types of cultured cells should be a fruitful subject for future investigation. Results obtained thus far suggest that dietary lipid modification or supplementation may benefit blood vessel function (von Schacky, 1987).
Page 192
Summary
There are many compelling reasons for pursuing basic and applied research on the effects of dietary w-3 PUFAs in vascular, inflammatory, and allergy diseases. Among these are the broad spectrum of possible beneficial effects of two natural compounds (EPA and DHA), avoidance of pharmacologic intervention with its inherent side effects and problems of compliance, and the long-term economic advantages of successful prophylaxis in coronary artery disease and cerebrovascular disease. The effects of these w-3 PUFAs can include reductions in platelet responsiveness, as well as favorable changes in blood lipids, blood pressure, blood rheology, and eicosanoid biosynthesis. More detailed information on the effects of w-3 PUFAs on platelet function, coagulation, and eicosanoid metabolism is desirable. In studies of each disease entity possibly amenable to improvement by the ingestion of w-3 PUFAs, problems of patient selection, precise dosages, criteria for evaluation of results, and definition of end points remain to be resolved.
Consumption of w-3 PUFAs should be differentiated from that of fish. Epidemiologic data suggest that consumption of fish is associated with a reduced risk of CHD (Kromhout et al., 1985; Shekelle et al., 1981a). In contrast, it has not been established that intake of w-3 fish oils per se will reduce the risk of CHD. Furthermore, it is not known whether long-term ingestion of these PUFAs will lead to undesirable side effects. The information available does not support a recommendation to use fish oil supplements to reduce the risk of CHD.
Associations of Dietary Fats and Fatty Acids with Cardiovascular Disease
Self-reported survey data from longitudinal population studies indicate that specific dietary fatty acids are related to CHD risk. Data from national sources on food consumption and vital statistics on causes of death also suggest an association.
Studies Of Differences Among Populations
In the Seven Countries Study, correlations between mean intake of SFAs and 10-year CHD mortality and incidence were .84 and .73, respectively (Keys, 1980b). CHD was not significantly associated with mean intake of PUFAs, which varied little among the cohorts. Another salient finding from these comparisons was the observation on Crete that a high intake of total fat (40% of daily calories) was compatible with very low risk of CHD, along with a very low total death rate, when the mean intake of SFA was low (8% of daily calories, while monounsaturates accounted for 29%).
Data from national food balance sheets and vital statistics show positive correlations of CHD deaths with eggs, meat, milk, sugar, and coffee, and negative correlations for flour (Armstrong et al., 1975; Stamler, 1979). However, such data have severe limitations, and the results should be interpreted with great caution, even when appropriate adjustments have been made for changes in the International Classification of Diseases and when data are used only from countries having the more reliable mortality statistics. These correlations require assumptions about lag time between measurement of diet and manifestation of disease. Moreover, confounders cannot usually be measured or accounted for in analysis. The diet and disease data bases are usually different, and the dietary data may not be representative for the populations of interest (e.g., middle-aged men). Nevertheless, these data suggest that more systematically acquired data, as from the Seven Countries Study, can be generalized.
Greenland Eskimos have been cited as an example of a population with a high intake of fats and oils from marine sources and very low rates of CHD (Bang and Dyerberg, 1980), but the epidemiologic data are insufficient to confirm this assertion. In fact, the risk of CHD is unknown in this small population, whose members live under harsh conditions and usually die before middle age.
The ecological associations between composition of the diet and CHD rates, based on systematic and standardized survey comparisons, indicate that low CHD rates may indeed occur in the presence of moderately high total (and monounsaturated) fatty acid intake—when intake of dietary SFAs and cholesterol is low. A low SFA intake, i.e., <10% of daily energy intake, along with a low intake of cholesterol, is common to all populations found to have truly low CHD rates.
Studies Of Individuals Within Populations
Studies in which intake of nutrients was not adjusted for energy intake and dietary information was not obtained at the initial examination (when participants were unaware of their CHD risk status) have not been considered here. The committee considers their results to be uninterpretable with regard to associations between diet and CHD risk (Shekelle et al., 1981b; Willett and Stampfer, 1986).
Page 193
Even studies that met these conditions have produced inconsistent results. For instance, the percentage of calories from SFAs was positively associated with risk of CHD in the rural sample of the Puerto Rico Study (Garcia-Palmieri et al., 1980), the Honolulu Heart Program (McGee et al., 1984), and the Ireland-Boston Diet-Heart Study (Kushi et al, 1985), but was not significantly associated in the Israel Ischemic Heart Disease Study (Medalie et al., 1973), the urban sample in Puerto Rico (Garcia-Palmieri et al., 1980), the Western Electric Study (Shekelle et al., 1981a), or the Zutphen Study (Kromhout and de Lezenne Coulander, 1984). The percentage of calories from PUFAs was inversely associated with risk of CHD in the Western Electric Study (Shekelle et al., 1981a), but not in the other studies; in fact, it was positively associated with CHD in the urban sample in Puerto Rico (Garcia-Palmieri et al., 1980). The ratio of PUFAs to SFAs (the P:S ratio) was inversely associated with risk of CHD in the small cohort of British men studied by Morris et al. (1977), but not in Israeli civil servants (Medalie et al., 1973).
In two studies, the Western Electric Study and the Ireland-Boston Diet-Heart Study, the lipid composition of the diet was characterized by scores based on the formulas developed by Keys et al. (1965c) and by Hegsted et al. (1965) to summarize the effect of change in dietary SFAs, PUFAs, and cholesterol on change in TC. In both studies, the scores were significantly associated with risk of CHD.
Animal foods in general were related to CHD risk and total deaths in Seventh-Day Adventists (Phillips et al., 1978). Fish-eating habits of men in the Zutphen Study and in the Western Electric Study were related to subsequent individual CHD risk. In these studies, men who rarely or never ate fish had a higher risk of CHD in comparison to men who usually consumed fish one or more times per week (Kromhout et al., 1985; Shekelle et al., 1985). In other population studies, however, fish intake was not significantly related to risk of death from CHD (Curb and Reed, 1985; Snowdon, 1981; Vollset et al., 1985).
The results of studies of individuals within populations have in general been somewhat inconsistent and inconclusive. Investigation of the basis for these inconsistencies has led to greater awareness of the problems inherent in using cohort and case-comparison studies to investigate a diet-disease hypothesis. These problems include the limited variation in characteristic intake of dietary lipids among individuals within populations; the large misclassification of persons according to characteristic intake of dietary lipids; and the substantial misclassification of individuals according to severity of coronary atherosclerosis (the dependent variable of real interest) when classification is based on signs and symptoms of CHD. These factors will produce weak and inconsistent associations in case-comparison and cohort studies of dietary lipids and CHD, even when important causal associations exist. When combined with results from metabolic ward studies, data from carefully conducted ecological studies, such as the Seven Countries Study (Keys, 1980b), provide a more accurate estimate of the role of dietary lipids in producing widespread atherosclerosis and CHD.
Trans Fatty Acids
Thomas et al. (1981, 1983a,b) compared the trans fatty acid content of adipose tissues from men dying of CHD and from other causes. CHD subjects had a higher proportion of shorter chain trans fatty acids than did non-CHD subjects. The investigators concluded that the CHD subjects had consumed more hydrogenated fats than had the controls; however, they did not adjust their findings for other risk factors. In contrast, Heckers et al. (1977) found no differences in tissue trans fatty acids related to atherosclerosis in a few cases. No other systematic epidemiologic data regarding the possible association of trans fatty acid consumption and atherosclerotic disease are available.
Atherosclerosis at Autopsy
In an autopsy study of 253 men in New Orleans (Moore et al., 1976), data on diet were obtained retrospectively from surviving spouses. The extent of raised lesions in the coronary arteries was positively associated with reported intake (expressed as percentage of total calories) of fat and animal protein and was inversely associated with intake of vegetable protein and starch. In contrast, however, antemortem data on diet in the prospective Honolulu Heart Program (Reed et al., 1987) were not significantly associated with the extent of coronary atherosclerosis. Results from the prospective Puerto Rico Heart Health Program (Sorlie et al., 1981) showed small negative correlations between extent of coronary atherosclerosis and reported intake of calories, starch, and fat, but the results are difficult to interpret because the data on intake of nutrients were not adjusted for intake of calories.
Diet and Other Cardiovascular Disease Manifestations
Even though data may be collected sys-
Page 194
tematically, they are insufficient for comparing habitual diet and rates of brain infarction and peripheral vascular disease among populations. This is partly because of the difficulty of obtaining accurate diagnoses and because rates of stroke and peripheral vascular disease rates are much lower than those for CHD, thus giving less power for analysis.
Animal fat, saturated fat, and total fat have been positively related to the risk of thrombotic brain infarction (D. Reed et al., 1986). However, where hemorrhage accounts for most cases of stroke, as in Japan, animal protein and saturated fat are inversely related to cerebral hemorrhage and overall incidence (Kagan et al., 1980, 1985; Kato et al., 1973; Takeya et al., 1984). The components of diets consumed by omnivorous Seventh-Day Adventists, including meat, eggs, milk, and cheese, were shown to be unrelated to risk of stroke (Snowden, 1988).
In studies of Japanese populations, investigators have hypothesized that a Western-type diet (i.e., with a fat content higher than the usual Japanese diet), which is associated with higher TC levels, is protective against stroke in Japan (Komachi 1977). This idea arose not only from the findings of the negative association of TC level to cerebral hemorrhage, but also from the decline in overall stroke rates in Japan with acculturation to Western ways in the last 20 years (Shimamoto et al., 1989). This hypothesis has been supported in Western countries by the negative association of TC with cerebral hemorrhage deaths among hypertensives in the Multiple Risk Factor Intervention Trial screening population and by the positive association of TC with cerebral thrombotic deaths cited above. However, it is still questionable whether the Western diet with its higher saturated and total fat intake and resultant higher TC levels is causally related to decreased risk of cerebral hemorrhage, because obvious confounders, such as the traditional Japanese diet of high salt and relatively low fat and protein intake, have not been sufficiently considered. Another potential confounder, alcohol and alcohol-related bleeding diseases, has also not been excluded.
The randomized controlled trials on primary prevention of CHD are summarized above in Tables 7-1 and 7-2. Results indicated that the percentage difference in risk of CHD between the experimental and control groups varied directly with the percentage difference in mean serum TC: the larger the difference in mean TC, the larger the difference in risk of CHD.
Only two small studies included changes in diet as the only intervention. The double-blind Los Angeles Veterans Administration Domiciliary Study (Dayton et al., 1968) of 846 men ages 55 to 89 years was not strictly focused on primary prevention, since 20% of the participants had evidence of old myocardial infarction at entry. In this study, investigators compared the effects of two diets, each providing approximately 40% of calories as fat, but with a much higher proportion of PUFAs and a lower proportion of SFAs in the experimental diet than in the control diet. The experimental diet (which contained linoleic and oleic acid each at 35 to 40% of total fatty acids) induced a prompt and sustained difference in mean TC level between the experimental and control groups, amounting to 12.7% of the starting level. Incidence of the primary end point, sudden death and definite myocardial infarction, was 24% lower in the experimental group than in the control group (54 compared to 71 cases, respectively), but the difference was not statistically significant. However, when these end points were pooled with cerebral infarction and other secondary end points, the totals were 66 in the experimental group and 96 in the control group (31% reduction; p = .01). Fatal atherosclerotic events numbered 48 in the experimental group and 70 in the control group (31% reduction; p <.05).
Initially, there was some concern that the experimental diet might have increased the risk of cancer, since 31 of 174 deaths in the experimental group were due to cancer as compared to 17 of 178 deaths in the control group (p = .06) (Pearce and Dayton, 1971). However, analysis of results from four other clinical trials in which high-PUFA diets were used to lower serum TC (Ederer et al., 1971) showed that the incidence of and mortality from all cancers were lower in the experimental group than in the control group; the relative risks were .75 for incidence and .62 for death. None of the relative risks were significantly different from unity, and the combined results from all five studies were consistent with the conclusion that the cholesterol-lowering diets did not influence risk of cancer. However, these studies could not rule out the possibility of small effects in either direction (increasing or decreasing the risk of cancer).
Further examination of the results of the Los Angeles Veterans Administration Domiciliary
Page 195
Study showed an association between the experimental diet and cholelithiasis (Sturdevant et al., 1973). Autopsied men who had eaten more than 33% of the experimental meals served from entry into the trial to death were more likely to have gallstones than were control subjects; the prevalence rates were 34 and 14%, respectively (p < .01). The prevalence of gallstones at autopsy was positively correlated with number of trial meals eaten by men in the experimental group (p <.05) but not by men in the control group.
High-PUFA diets (i.e., more than 10% of total calories from PUFAs) are no longer recommended in the United States for lowering serum TC, in part because of concerns about the long-term safety of such diets.
In the Finnish Mental Hospital Study (Turpenien et al., 1979), participants were 450 men ages 34 to 64 who were hospitalized in one of two mental hospitals near Helsinki. At the beginning of the study, one hospital received a diet low in SFA and cholesterol and relatively high in PUFA, while the other hospital continued with the usual hospital diet. Six years later, the diets were reversed, and the study was continued for another 6 years. In both hospitals, the special (high PUFA) diet was associated with markedly lower mean TC. The difference in one hospital was -50.9 mg/dl (-19%) and in the other, -32.0 mg/dl (-12%). The mean difference between the special-diet and usual-diet periods in the two hospitals was -41.4 mg/dl (-15%). The incidence of coronary events in the special-diet and usual-diet periods for both hospitals combined was 2.3 and 4.3 per 1,000 person-years, respectively, for coronary death (p = .10); 4.2 and 12.7 per 1,000 person-years for coronary death plus major electrocardiogram (ECG) change (p = .001); and 13.5 and 24.3 per 1,000 person-years for coronary death plus intermediate or major ECG changes (p = .008).
A large-scale trial on primary prevention of CHD with change in diet as the only intervention was never conducted because of concerns about feasibility (NHLBI, 1971). Thus, the remaining diet-heart trials are either 2-factor or 3-factor trials in which any effect of dietary change was confounded with effects of changes in the other factors. Three of those trials (the Göteborg Multifactor Trial, the WHO Multifactor Trial, and Multiple Risk Factor Intervention Trial) achieved differences in mean TC between the intervention and comparison groups of 0, -1, and -2%, respectively; the differences in coronary rates were correspondingly small (0, -4, and -7%, respectively).
The Oslo Study (Hjermann et al., 1981), which began with 1,232 hypercholesterolemic men ages 40 to 49, achieved a 13% reduction in mean TC levels in the experimental group as compared to the control group. At the end of the follow-up period, 7.5 years later, the incidence of myocardial infarction (fatal and nonfatal) plus sudden death was 47% lower in the experimental group than in the control group (p = .028). Total mortality in the experimental and control groups was 26/1,000 and 38/1,000, respectively (p = .25). From their own analysis of the data in this study, the authors concluded that changes in cigarette smoking accounted for at most approximately one-fourth of the observed difference in risk of CHD and that most of the difference in CHD was apparently due to diet-induced changes in serum cholesterol. Although this quantitative conclusion is subject to some question, it is clear that the diet-induced lowering of TC played a major role in the observed significant reduction in CHD.
Summary
Epidemiologic evidence shows that serum TC levels are positively associated with risk of CHD, experimental studies in animals and clinical research have elucidated in large part the mechanisms for this association, and results of clinical trials strongly support the concept that lowering TC levels reduces incidence of CHD. Experimental studies in humans have demonstrated that intake of dietary SFAs, PUFAs, and cholesterol affects the level of serum TC. The mechanisms of these effects are now being elucidated. Epidemiologic studies support the concept that variation among populations in mean intake of SFAs (and probably cholesterol) is a major factor accounting for variation in mean TC levels. Results of dietary intervention clinical trials are consistent with the conclusion that changing the lipid composition of the diet in ways expected to lower the TC levels (i.e., decreasing SFA and cholesterol intake, and increasing PUFA intake) in fact does lower mean levels of serum TC and reduces incidence of CHD.
SFAs and PUFAs
The relationship of dietary fatty acid saturation to experimental atherosclerosis parallels closely the fatty acid effects on levels of serum cholesterol, particularly LDL-C. Cholesterol fed with hydrogenated vegetable oil (source unspecified) produced more severe atherosclerosis
Page 196
in rabbits than cholesterol fed with corn oil (Kritchevsky et al., 1954). Subsequently, SFAs were found to be more atherogenic than unsaturated fatty acids in several species of monkeys (Lofland et al., 1970; Mendelsohn et al., 1980; Robison et al., 1971; Wissler et al., 1962).
Effects Of Dietary Fats With No Cholesterol
Hypercholesterolemia and atherosclerosis were produced in rabbits by feeding them semisynthetic diets enriched with hydrogenated coconut oil, butterfat, or margarines and no added cholesterol (Carroll, 1971; Funch et al., 1960, 1962; Lambert et al., 1958; Malmros and Wigand, 1959; Wigand, 1959). It was later shown, however, that these diets were deficient in essential fatty acids, and a small amount of PUFAs prevented hypercholesterolemia (Diller et al., 1961). Finally, an improved semisynthetic diet eliminated the essential fatty acid deficiency, and it was shown that hypercholesterolemia and atherosclerosis were related to fatty acid saturation when fed with a low dose of cholesterol (Wilson et al., 1973). These results were confirmed by other investigators (Kritchevsky and Tepper, 1965a,b; McMillan et al., 1960). Cholesterol-free semisynthetic diets also produced hypercholesterolemia and atherosclerosis in vervet monkeys and in baboons (Kritchevsky et al., 1974a,b).
Peanut Oil
Gresham and Howard (1960) reported that peanut oil, which has a high proportion of PUFAs, had the unanticipated effect of being more atherogenic than other fats when fed with cholesterol to rats. This finding was confirmed in rats (Scott et al., 1964) and extended to rabbits (Imai et al., 1966; Kritchevsky et al., 1971) and rhesus monkeys (Scott et al., 1967a,b; Vesselinovitch et al., 1974, 1980). Later experiments suggest that the unusual atherogenicity may be related to triglyceride structure (Kritchevsky et al., 1973, 1981; Myher et al., 1977), but there are no studies that explain this effect in terms of an effect on lipoprotein metabolism.
In cynomolgus monkeys, peanut oil fed with a concentration of dietary cholesterol equivalent to that consumed by humans affected serum lipoproteins and atherosclerosis as did other unsaturated vegetable oils (Alderson et al., 1986).
The contradictory results have not been explained. Kritchevsky (1988) suggested that because positional randomization of the triglyceride fatty acids of peanut oil reduced its atherogenicity, the unusual effect might be due to its triglyceride structure or that a lectin in peanut oil might be responsible. Further investigation is needed to resolve these discrepant results.
Trans Fatty Acids
Effects of trans fatty acid isomers have been studied in laboratory animals (reviewed by Kritchevsky, 1982; Senti, 1985). These isomers are not metabolized in the same way as the cis isomers. However, at dietary intakes similar to those consumed by humans, and with a small proportion of linoleic acid in the diets, the trans fatty acid isomers appear to have no deleterious health effects (Jackson et al., 1977; Kritchevsky, 1983; Royce et al., 1984).
Summary And Conclusions
In most animal species, SFA intake is associated with more severe atherosclerosis than that achieved with similar diets without (or with little) SFA. Some types of SFAs, especially coconut oil, are more atherogenic than indicated by their effects on plasma lipoproteins alone. The observations in animals support the putative role of SFAs in causing hypercholesterolemia and atherosclerosis in humans.
Relationship Between Dietary Sterols and Cardiovascular Disease
The first evidence that dietary cholesterol may be involved in the etiology of atherosclerosis and its sequelae (CHD, stroke, or peripheral vascular disease) came from the demonstration by Anitschkow and Chalatow in 1913 that cholesterol fed to rabbits produced arterial lesions similar to those of human atherosclerosis (Anitschkow, 1967; Anitschkow and Chalatow, 1913). Over the next 50 years, it was shown that almost all animal species were to some degree susceptible to dietary cholesterol-induced hyperlipidemia and experimentally induced atherosclerosis.
In the late 1940s, interest in the possible dietary etiology of atherosclerosis in humans was stimulated by the observations on CHD in Europe during World War II. A large number of epidemiologic, clinical, and animal studies were conducted from 1950 to the present to examine the roles of dietary fats, including cholesterol, in causing hyperlipidemia, atherosclerosis, and clinical atherosclerotic disease. Dietary SFAs became the major focus, because their effects on serum cholesterol levels in humans were more consistent and their associations with atherosclerotic diseases were stronger than those of cholesterol. Despite the high susceptibility of many test animals to dietary cholesterol, evidence regarding
Page 197
dietary cholesterol in humans was for many years variable and often conflicting. In the past decade, a number of studies have shed light on this difference in findings by indicating that there is considerable variability in the plasma lipid responses to dietary cholesterol among individuals and that a substantial proportion of the population is highly susceptible to the cholesterol-raising effects of dietary cholesterol. This variability probably accounts for much of the initial uncertainty about the role of dietary cholesterol and for many of the apparently conflicting results (reviewed by McGill, 1979).
Studies Of Populations
Stamler et al. (1970, 1972) examined the relationship of mean per-capita consumption of dietary cholesterol to arteriosclerotic disease mortality rates in 20 developed countries. The correlation coefficient for daily cholesterol intake and mortality from CHD in 1964 was .617 (p <.01) for 45- to 54-year-old men and .685 (p <.01) for 55- to 64-year-old men. These mortality rates were also correlated with national per-capita income, cigarette smoking, total protein consumption, total fat consumption, and other environmental variables.
Armstrong et al. (1975) compared the consumption of various commodities with mortality from ischemic heart disease in nine regions of the United Kingdom and in 30 countries. They found strong correlations between egg consumption and ischemic heart disease. These results are consistent with the hypothesis that dietary cholesterol is causally associated with incidence of CHD, but evidence from other types of studies must be considered in order to assess alternative hypotheses and to reach a conclusion.
The Effect of Dietary Cholesterol on Plasma Lipids
With one exception (Connor et al., 1978), cross-sectional studies of individuals within populations have shown little or no association between dietary cholesterol and concentration of serum TC (Frank et al., 1978; Garcia-Palmieri et al., 1977; Harlan et al., 1983; Kahn et al., 1969; Nichols et al., 1976; Shekelle et al., 1981a; U.S.-U.S.S.R. Steering Committee, 1984; Yano et al., 1986). Jacobs et al. (1979) reported that correlations close to zero are a necessary result of cross-sectional studies because of the large differences in intrinsic TC levels among individuals. They concluded that cross-sectional studies are inappropriate for studying the effects of dietary cholesterol on serum TC.
In contrast, longitudinal observational studies have generally shown a positive association between changes in dietary cholesterol and changes in TC concentrations. For example, in the Lipid Research Clinics Coronary Primary Prevention Trial, 6,000 hypercholesterolemic men were advised to lower their intake of cholesterol and SFAs (Gordon et al., 1982). Multiple regression analysis showed that the reduction in dietary cholesterol was independently associated with reduction in serum TC concentrations. The reduction in TC was due primarily to a reduction in LDL-C. A similar association of changes in dietary cholesterol with changes in serum TC was found in the Zutphen Study (Kromhout, 1983) and in the Western Electric Study (Shekelle et al., 1981a). The magnitude of the association found in observational studies has been less than that found in experimental studies due to the well-known effects of day-to-day intraindividual variation in intake of nutrients and in serum TC levels (Beaton et al., 1979; Keys, 1965; Liu et al., 1978).
Dietary Cholesterol and Coronary Heart Disease
Before 1981, epidemiologic studies of individuals within populations showed little or no association between dietary cholesterol and risk of CHD (Garcia-Palmieri et al., 1980; Gertler et al., 1950a; Gordon et al., 1981a; Paul et al., 1963; Yano et al., 1978). However, in these studies, investigators did not adequately consider one or more of three important methodological issues: adjustment for the confounding effect of caloric intake (Willett and Stampfer, 1986); limitations in the reliability of dietary measurements (Beaton et al., 1979; Keys, 1965; Liu et al., 1978); and bias resulting from systematic dietary changes made by hypercholesterolemic persons after learning of their condition (Shekelle et al., 1981b). Since then, four long-term cohort studies—the Western Electric Study (Shekelle et al., 1981a), the Zutphen Study (Kromhout and de Lezenne Coulander, 1984; Kromhout et al., 1985), the Ireland-Boston Diet-Heart Study (Kushi et al., 1985), and the Honolulu Heart Program (McGee et al., 1984)—have shown that dietary cholesterol, assessed reliably at entry and expressed as mg/1,000 kcal, was positively associated with risk of CHD after adjustment for age, blood pressure, cigarette smoking, and serum cholesterol level. Combining the results of these studies, Stamler and Shekelle (1988) calculated that a dietary cholesterol incre-
Page 198
ment of 200 mg/1,000 kcal daily was associated with a 30% greater risk of CHD.
Considering the difficulties in measuring cholesterol intake, the many etiologic factors involved in CHD, the long natural history of the underlying atherosclerosis, and the variability among individuals in response to diet, the committee interprets these observations as supporting an etiologic role of dietary cholesterol in CHD. Since the association between dietary cholesterol and CHD persisted after adjustment for serum TC, the results suggest that dietary cholesterol may affect atherogenesis and CHD by other mechanisms in addition to its effect on LDL-C concentration (Zilversmit, 1979). The possible contribution of dietary cholesterol to cerebral vascular disease and peripheral vascular disease was not addressed in these studies.
Response Of Serum Total Cholesterol
Many experiments have been conducted to measure the response of serum or lipoprotein cholesterol concentrations in humans to different amounts of dietary cholesterol. Cholesterol intake was usually modulated by adding or removing eggs from the diet. These experiments have varied widely in the degree of control of dietary intake among the subjects. They range from experiments in which the subjects were confined to a metabolic ward and fed laboratory-prepared formula diets to those in which noninstitutionalized subjects were advised to add eggs to or remove eggs from their habitual diets.
Examples of rigorously controlled experiments include those reported by Connor et al. (1961a,b, 1964). In each of three experiments, six men were placed on cholesterol-free and high-cholesterol diets that were either formula diets or carefully selected natural foods. Cholesterol was added as egg yolk. In all three experiments, substantial increases in serum TC resulted from the addition of egg yolk to the diet.
Beveridge et al. (1960) fed formula diets containing varying amounts of added crystalline cholesterol for 8 days to 67 noninstitutionalized university students. There was a linear increase in serum TC with increases in daily intake up to about 600 mg. A similar study was conducted by Mattson et al. (1972) in 56 healthy prison inmates who consumed formula diets with varying amounts of added egg yolk. There was an approximately linear relationship between cholesterol intake and elevation of serum TC levels.
An unusual opportunity to examine the effects of changes in habitual diet on plasma lipoprotein cholesterol levels was provided by the Lipid Research Clinics Coronary Primary Prevention Trial (Gordon et al., 1982). As in other cross-sectional studies, there was no association of any dietary component with either LDL-C or HDL-C before subjects were instructed to change their diets. However, the change in cholesterol intake (of about 27 mg/1,000 kcal per day) was associated with a change in plasma cholesterol concentration in a multiple regression model. The association was independent of the effects of changes in saturated and unsaturated fat and of weight loss, and there was no interaction between the type of fat and cholesterol intake. The change in plasma cholesterol was almost entirely due to a change in LDL-C.
In a follow-up analysis of data from the same trial after it was terminated, serum TC and LDL-C levels were less strongly and less consistently correlated with decreases in cholesterol intake up to 7 years after the initial dietary instruction period (Glueck et al., 1986). The partial regression coefficients were statistically significant at 4 years, but not at 0.8 and 7 years. In contrast, the coefficients for SFA intake were significant at all times.
In another cross-sectional dietary study, the investigators focused on subfractions of the lower-density lipoproteins measured by analytic ultracentrifugation (Williams et al., 1986). Daily cholesterol intake averaged 190 mg/1,000 kcal. Dietary cholesterol was not significantly correlated with serum TC or LDL-C, but was significantly correlated with the mass of small LDL in the Sf0-7 range (Rs = .30, p <.01). Although there is considerable interest in the relative atherogenicity of lipoprotein subclasses, we do not know whether Sf0-7 LDL has any special significance.
Predictive Equations Relating Dietary Cholesterol To Serum Cholesterol
The first report of an equation to describe the effect of dietary cholesterol or serum cholesterol was made by Grande et al. (1965) in 1965. Keys (1984) reviewed this equation in light of 39 subsequent reports and concluded that the same equation, shown below, remained the best description:
DChol = 1.5 (C2½ - C1½),
where DChol is the change in serum TC concentration in mg/dl, C2 is the new intake of cholesterol in mg/1,000 kcal per day, and C1 is the initial intake.
Page 199
Hegsted (1986) reviewed the same 39 reports plus several others and concluded that the association was essentially linear from 0 to 400 mg per 1,000 kcal and could be described by the following equation:
DChol = 0.0974 C,
where DChol is the change in serum cholesterol in mg/dl and C is the difference in cholesterol intake between the two diets in mg/1,000 kcal.
Variability In Response To Dietary Cholesterol
Most reports of experiments on dietary cholesterol in humans mention the high variability of responses of serum cholesterol and LDL-C among individuals. Some people do not respond at all; many have small responses; and a few have large responses. Indeed, as discussed above, the variability among individuals is so great that it obscures correlations in cross-sectional studies.
Reports after 1980 began to focus more attention on this variability. A group of investigators in the Netherlands conducted an intensive study of individual variability in responsiveness to dietary cholesterol. Katan et al. (1986) tested 94 people on diets providing 121 and 625 mg of cholesterol per day in egg yolk. The highest quartile and the lowest quintile of responders were challenged 1 month and again 7 months after the initial challenge. The high and low responders were significantly different from one another in the repeated challenges. Standardized correlation coefficients of the responses to the three challenges, taken in pairs, ranged from .34 to .53 (all p <.05). The mean of each subject's responses during the three challenges, taken as the best estimate of the response, showed a normal unimodal distribution from -12 to +46 mg/dl. Approximately 90% of the change in serum TC was accounted for by changes in LDL-C. McNamara et al. (1987) found that people who responded to dietary cholesterol with an increase in TC were also those in whom endogenous cholesterol synthesis was not suppressed.
There now appears to be convincing evidence that consistent differences in responsiveness of serum cholesterol to dietary cholesterol do exist in humans. When these responses are measured in terms of specific lipoprotein classes, the differences among individuals become even more prominent. No simple marker for sensitivity is available. A test for responsiveness involves feeding a controlled diet for 6 weeks or longer and analyzing several blood samples. The procedure is laborious and cannot be performed on large numbers of people. The definition of a responder, or a high responder, is arbitrary, but is the basis for determining the proportion affected, since the distribution of responses is continuous. However, the results indicate that approximately 20% of adults are likely to have a response more than 10% higher than that predicted by the Hegsted (1986) or Keys (1984) equations.
Effects Of Dietary Cholesterol On Lipoprotein Metabolism
In most studies up to about 1980, the effects of dietary cholesterol were measured only as changes in serum TC. As new analytical methods became available, investigators began to measure the effects of dietary cholesterol on the major lipoprotein classes or the apolipoproteins or on variables such as lipoprotein secretion or degradation. Only in the late 1970s did reliable techniques become available for measuring cholesterol in major lipoprotein fractions and apolipoproteins in a large number of plasma samples. Many cholesterol feeding experiments were repeated to take advantage of these new techniques. For example, Applebaum-Bowden et al. (1979) found that elevations in serum cholesterol were due principally to an elevation of LDL-C and that plasma apolipoprotein B concentrations were also increased. Similar results were obtained by Mistry et al. (1981), Packard et al. (1983), and Schonfeld et al. (1982).
Beynen and Katan (1985) and Schonfeld et al. (1982) reported that most of the change in serum cholesterol was accounted for by a change in LDL-C. Zanni et al. (1987) demonstrated that dietary cholesterol affected HDL-C and found considerable heterogeneity in response among individuals. Applebaum-Bowden et al. (1984) observed that LDL receptor activity in mononuclear cells, as measured by ability to degrade LDL, was decreased by dietary cholesterol proportionally to the increase in serum LDL-C levels. Lin and Connor (1980) found that the percentage of dietary cholesterol absorbed in two subjects did not change within the range of intake from 45 mg to 1,000 mg per day. Ginsberg et al. (1981) reported no change in the rates of production or clearance of VLDL or LDL with changes in dietary cholesterol in five subjects. In all the studies in which the characteristics of the elevated serum LDL were examined, the LDL particles were essentially normal in size and other characteristics; there were simply more particles.
Katan and Beynen (1987) looked for differences in metabolic characteristics between high
Page 200
and low responders and found that only habitual cholesterol intake and HDL2 cholesterol levels were associated with responsiveness. McNamara et al. (1987) observed that two-thirds of a group of 75 men responded to a 600 mg/day increase in dietary cholesterol with no change in plasma cholesterol level, whereas approximately one-third of the subjects responded with an increase greater than 5%. The nonresponders suppressed cholesterol biosynthesis in blood mononuclear cells, but endogenous biosynthesis was not decreased in the responders.
Zilversmit (1979) suggested that absorbed dietary cholesterol is concentrated in the postprandial lipoproteins, which are thereby made more atherogenic. Investigation of the postprandial lipoproteins on a larger scale has been hampered by limitations in the methods for separating and quantitating these lipoproteins. Therefore, this mechanism remains an attractive but speculative possibility.
In summary, the effect of dietary cholesterol on serum lipoprotein concentrations is principally an increase in the concentration of normal serum LDL particles. There may be other effects on the characteristics of VLDL or HDL that are related to the increased LDL, but these have not been clearly identified and linked to the change in LDL.
Interactions Of Dietary Cholesterol With Other Nutrients
Many investigators have looked for an interaction between dietary cholesterol and type of fat with regard to their effects on serum lipoproteins. A positive interaction with SFAs was found by the National Diet-Heart Study Research Group in the Faribault Study (1968). Bronsgeest-Schoute et al. (1979a) observed no significant interaction in one experiment, but did find one in a later experiment (1979b). Neither Chenoweth et al. (1981) nor Oh and Monaco (1985) reported an interaction with any type of fat.
In summary, despite the widespread belief that there is a positive interaction between dietary cholesterol and SFAs in their effects on serum lipid or lipoprotein concentrations, the evidence from studies in humans is inconsistent, perhaps due to varying proportions of responders and nonresponders in the different studies. Further research is needed to resolve this question.
Since the initial demonstration of dietary cholesterol-induced atherosclerosis in rabbits in 1913 (Anitschkow, 1967; Anitschkow and Chalatow, 1913), a variety of animal species have been shown to be sensitive to cholesterol. Guinea pigs (Bailey, 1916), mice (Löwenthal, 1925), goats (Chalatow, 1929), swine (Bragdon et al., 1957), chickens (Dauber and Katz, 1942), and most species of nonhuman primates (Clarkson et al., 1976a,b) are quite sensitive; dogs (Steiner and Kendall, 1946) and rats (Wissler et al., 1954) are relatively resistant. Among the nonhuman primates, rhesus, cynomolgus, squirrel, and vervet monkeys are moderately sensitive, whereas baboons are only slightly sensitive (Corey et al., 1974; Eggen, 1974; McGill et al., 1976; Portman and Andrus, 1965; Srinivasan et al., 1976). In each species, as in humans, there is considerable interindividual variability, and it has been shown in several species that much of this variability is under genetic control (Clarkson et al., 1971, 1985; Flow et al., 1981). The metabolic basis of the genetic variability has not been identified.
In animal models, as in humans, the elevated serum cholesterol resulting from dietary cholesterol is carried primarily in LDL (McGill et al., 1981b). Effects on HDL vary considerably from one species to another. In some, dietary cholesterol increases HDL; in others, it decreases HDL.
The increases in serum lipids and lipoproteins caused by dietary cholesterol (usually accompanied by a high level of dietary SFAs) result in arterial lesions similar to those found in humans with atherosclerosis. A few experiments with prolonged feeding of a cholesterol-supplemented diet showed that arterial lesions in animals can undergo complications typical of those seen in advanced atherosclerosis in humans and occasionally result in thrombotic occlusion of the coronary artery and myocardial infarction or occlusion of the iliac or femoral arteries and peripheral vascular disease (Taylor et al., 1963).
In summary, dietary cholesterol is the dominant feature of experimental hyperlipidemia and atherosclerosis in animal models. Humans resemble the more resistant species of nonhuman primates in their sensitivity to dietary cholesterol. The results of studies in which cholesterol is fed to laboratory animals are of particular value in examining physiological mechanisms. The role of dietary cholesterol as an etiologic agent in atherosclerosis in humans should be interpreted with caution, because the relative amount of cholesterol fed to animals usually is much greater than that consumed by human populations. Experimental diets often contain 1,000 to 2,000 mg of cholesterol per 1,000 kcal, whereas a high intake for humans is
Page 201
300 to 500 mg per 1,000 kcal. These very high cholesterol diets are used to produce high serum cholesterol levels and atherosclerotic lesions within a limited time. Lower dietary intakes, well within the range of human intakes, fed over longer periods to susceptible species have produced atherosclerotic lesions. Thus, the observations in laboratory animals are consistent with the causative role of dietary cholesterol in human disease.
There has been much speculation about the possible toxicity of cholesterol derivatives. Several reports, all from one laboratory (Imai et al., 1976, 1980; Janakidevi et al., 1984; K.T. Lee et al., 1985; Murray et al., 1981; Peng et al., 1978, 1979; Taylor et al., 1979) indicate that 25-hydroxycholesterol and cholestane triol cause necrosis of arterial smooth muscle cells and influence the growth of these cells in culture. None of these effects has been confirmed by other investigators. There is no epidemiologic or clinical evidence suggesting that these derivatives cause hyperlipidemia or atherosclerosis in humans.
The intake of plant sterol by humans ranges from 150 to 400 mg/day—about the same daily intake as cholesterol. Most plant sterols are poorly absorbed by the intestinal tract, and those that are absorbed are rapidly excreted. Sitosterol is the most abundant plant sterol; others include stigmasterol, campesterol, brassicasterol, and 24-methylene cholesterol. The amounts that are absorbed are inversely proportional to the molecular weight of the sterols.
Long ago, plant sterols (b-sitosterol and related compounds) were found to prevent the absorption of dietary cholesterol (Best et al., 1955; Farquhar and Sokolow, 1958; Farquhar et al., 1956; Lees et al., 1977; Peterson et al., 1959), apparently by blocking absorption of cholesterol in the intestine (Davis, 1955; Grundy and Mok, 1977; Jandacek et al., 1977; Mattson et al., 1977). More recent reports indicate that these compounds may be more effective in small doses than previously believed (Mattson et al., 1982). No deleterious side effects have been identified, but b-sitosterol has not been widely used in a lipid-lowering diet.
A rare genetic disorder causes people to absorb a large proportion of dietary plant sterols (Bhattacharyya and Connor, 1974; Lin et al., 1983; McArthur et al., 1986; Miettinen, 1980; Shulman et al., 1976; Tilvis and Miettinen, 1986). In this condition, known as phytosterolemia, the affected subjects have high plasma levels of plant sterols and xanthomas. The patients appear to be at an increased risk for atherosclerotic cardiovascular disease and should therefore greatly limit their intake of plant sterols.
Dietary cholesterol causes hypercholesterolemia and atherosclerosis in a variety of animal species. In humans, within the usual range of intakes (300 to 600 mg/day, or about 150 to 300 mg/1,000 kcal), dietary cholesterol influences the plasma cholesterol concentration, primarily LDL-C. On average, 100 mg of dietary cholesterol per 1,000 kcal elevates LDL-C by 8 to 10 mg/dl. Individual responses vary considerably on both sides of this average response in a continuous distribution. Some people are resistant to the cholesterol-raising effects of dietary cholesterol; others are quite sensitive. For those in the upper range of this distribution—at least one-third of the population—dietary cholesterol is a substantial contributor to serum TC and LDL-C concentration and thus to the progression of atherosclerosis and to the risk of clinically manifest atherosclerotic disease. The only way to identify a susceptible person is by repeated dietary challenges—a procedure that is not feasible on a large scale. For the overall population, the evidence indicates that dietary cholesterol contributes to the development of atherosclerosis and the risk of CHD.
Dietary Lipids/Fats and Hypertension
Human Studies
Epidemiologic studies suggest that people who subsist on complete vegetarian diets have lower mean blood pressure levels than those consuming omnivorous diets (Sacks et al., 1974) (see Chapters 8 and 10). The specific dietary components involved have not been identified, but they probably do not include sodium since salt intake is similar in many vegetarian and omnivorous groups (Armstrong et al., 1974). The fact that complete vegetarians tend to eat more PUFAs and less total fat, SFAs, and cholesterol (Rouse et al., 1981) has led researchers to design a number of observational, clinical, and community intervention studies to examine the relationship of dietary fatty acids to blood pressure (Weinsier and Norris, 1985).
In general, observational studies of blood pressure and of indirect indices of high blood pressure
Page 202
such as stroke in noninstitutionalized populations offer little support for an effect of total fat, but suggest a modest and more consistent effect for specific fatty acids. Tuomilhehto et al. (1984) compared random samples of the Finnish population, ages 30 to 59, living in two counties in eastern Finland in 1972, 1977, and 1982, and reported that intake of total fat was positively associated with blood pressure in each study period and with change in blood pressure over the three study periods. In two other studies, no association with total fat was found. McCarron et al. (1983) used cross-sectional data from the 1971-1974 NHANES I to evaluate representative subgroups of the U.S. population for whom intake of 17 nutrients was determined and a single blood pressure measurement was taken. They found no difference in total fat intake between hypertensive (>160/95 mm Hg) and normotensive (<160/95 mm Hg) individuals. The lack of association was found in both men and women and across the three age categories examined: 20 to 34, 35 to 54, and 55 to 74 years. Thulin et al. (1980) compared the nutrient intakes of 36 untreated hypertensive women to that of an equal number of normotensive age-matched women and found no difference in total fat intake between the two groups. The authors did not, however, control for other dietary variables.
A lack of association of hypertension with total fat has also been observed in studies using stroke as a surrogate end point. In a 10-year follow-up study of 7,895 men of Japanese ancestry ages 45 to 68 living in Hawaii with no history of stroke at baseline, Kagan et al. (1985) observed an inverse association of total dietary fat with incidence of thromboembolic stroke and total stroke in univariate analysis. This association disappeared when the investigators controlled other factors associated with stroke (e.g., age, blood pressure, serum glucose, and cigarette smoking). No association of total fat with hemorrhagic stroke was seen in either univariate or multivariate analyses.
In a pilot cross-sectional study comparing blood pressure level to dietary variables in a small number of people living in rural Finland, Italy, and the United States, no association with total dietary fat was found (Carvalho et al., 1981; Iacono et al., 1982). The authors did report, however, that the Finnish sample, which had the highest mean blood pressures of all three groups, also had a higher intake of SFAs and a lower intake of PUFAs than did the two other national samples. Groen et al. (1962) compared dietary intakes by Benedictine and Trappist monks who had a prevalence of hypertension (diastolic blood pressure >95 mm Hg) of 51 and 12%, respectively, and found no difference in total fat consumption between the two groups. Trappist monks, however, consumed a greater percentage of their fat as vegetable fat.
The types and amounts of dietary lipids and their relationships to blood pressure have been examined in clinical studies of both normotensive and hypertensive people but have produced inconsistent results. In a study at the USDA Lipid Nutrition Laboratory, Judd et al. (1981) compared blood pressures among four groups of normal and mildly hypertensive male subjects fed natural diets of either high fat (43% of calories from fat) or low fat (25% of calories from fat) with a high P:S ratio (1.0) or low P:S ratio (0.3) for 6 weeks. Salt intake was slightly decreased (from 8.6 to 8.1 g/day) and potassium intake was slightly increased (from @4.1 to @5.1 g/day) in the groups consuming low compared to high fat. Body weights changed only slightly. On average, systolic and diastolic blood pressures were lower in persons on the high P:S diets than in those on the low P:S diets, regardless of total fat intake, although the magnitude of the difference between the groups (<4 mm Hg for both diastolic and systolic blood pressure) was modest (Iacono et al., 1982).
Vergroesen et al. (1978) assessed blood pressure changes in eight normotensive people (five men and three women) who were fed a diet with a moderately increased level of linoleic acid and no other dietary changes. A decrease in diastolic but not systolic blood pressure was noted after approximately 4 weeks on the diet. The diastolic pressure returned to baseline values after the subjects resumed their normal dietary regimen. In a similar but larger study, Fleischman et al. (1979) reported decreases in both systolic and diastolic blood pressures in 28 mildly hypertensive subjects fed a diet substituting linoleic-rich lipids for SFAs. Further evidence for an effect of linoleic acid was provided by Rao et al. (1981), who randomized 24 hypertensive Indians into one of three treatment groups receiving capsules containing groundnut oil (21% PUFA), safflower oil (12% PUFA), or starch. The authors calculated that the groundnut and safflower oil supplements increased linoleic acid intake by 7 to 10%. All three groups received a standard diet containing 2,000 kcal and 15 to 18% energy from fat. After 1 month of treatment, diastolic blood pressure decreased approximately 6.5 and 9.0% in the groups consuming supplements of groundnut and safflower oil, respectively;
Page 203
no change was observed in the group consuming starch.
Not all clinical studies have demonstrated that a change in dietary fatty acids affects blood pressure. Sacks et al. (1984) reduced SFAs from 21 g/day to 10 g/day (maintaining a constant level of PUFA, carbohydrate, and total fiber) in the diets of 19 normotensive volunteers and observed no change in blood pressure. Margetts et al. (1984), in a randomized double-blind study of 54 normotensive volunteers, reported no change in blood pressure in people allocated to either a low or high P:S ratio diet. In a subsequent randomized, double-blind crossover study, 21 mildly hypertensive subjects were fed dietary supplements of cream, safflower oil, and carbohydrate for 6 weeks in random sequence (Sacks et al., 1987). The investigators noted no difference in blood pressure among the three study groups.
Three metabolic epidemiologic studies comparing blood pressure levels to fatty acid concentrations in tissue and erythrocytes in noninstitutionalized populations offer conflicting data. Oster et al. (1979) studied 650 German males and observed a small inverse correlation (R = -.16 to -.12) between the concentration of linoleic acid in adipose tissue and both systolic and diastolic blood pressure. These small correlations were reduced even further when the investigators controlled for age and body weight. Berry and Hirsch (1986) found no association between linoleic acid concentration in adipose tissue and blood pressure in 399 males in the United States, but they reported that there was an inverse association between linolenic acid concentrations in adipose tissue and systolic and diastolic blood pressure, both of which decreased approximately 5 mm Hg for every 1% increase in adipose linolenic acid content. The authors concluded that the level of dietary linolenic acid has a disproportionate effect on blood pressure since it constituted only one-eighth as much of the fatty acid content of the adipose tissue as did linoleic acid. Ciocca et al. (1987) compared the fatty acid composition of erythrocytes to arterial blood pressure in a large population of urban Italian females and noted no differences in the relative amounts of SFAs, MUFAs, and PUFAs or in the mean P:S ratio and linoleic:oleic acid ratio in erythrocytes across quartiles of systolic and diastolic blood pressure.
Several population intervention studies on dietary lipids and blood pressure have been conducted. In Finland and Italy, 30 normotensive couples were given an experimental diet in which total fat was decreased and the P:S ratio and vegetable intake were increased. The authors observed a reduction in blood pressure that was reversed when the couples resumed their usual diets (Ehnholm et al., 1982; Puska et al., 1982).
Using a different approach, Strazzullo et al. (1986) investigated whether or not adding SFAs to the typical Mediterranean diet of rural southern Italians would raise blood pressure. Fifty-seven normotensive people (29 males and 28 females) ages 30 to 50 were followed for 2 weeks on their customary diet (P:S ratio of 0.44), then switched to a 6-week isocaloric diet with a 70% increase in SFAs and a corresponding decrease in carbohydrate and MUFAs (P:S ratio of 0.23). By the end of 6 weeks, systolic blood pressure had increased by 2.6 mm Hg in men and 4.8 mm Hg in women; no change in diastolic blood pressure was observed in either sex.
In an intervention study on 57 adult normotensive spouse pairs (diastolic blood pressure <90 mm Hg) and untreated hypertensive people (diastolic blood pressure >90 mm Hg) living in two communities in North Karelia, Finland, Puska et al. (1983) randomly allocated couples to one of three study groups: Group 1 followed a low-fat diet (23% of energy intake) with a high P:S ratio (1.0); Group II reduced daily salt intake from 192 mmol to 77 mmol; and Group III continued their usual diet. After a 6-week intervention period, systolic blood pressure in Group I declined from 138.4 to 129.5 mm Hg, while diastolic pressure decreased from 88.9 to 81.3 mm Hg. The mean decrease in blood pressure was greater in hypertensive than in normotensive subjects. Both diastolic and systolic blood pressures returned to baseline values once couples in Group I resumed their usual diets. No blood pressure differences were observed over the course of the study in Groups II or III.
In a subsequent study of 84 middle-aged people from the same population, Puska et al. (1985) randomly allocated subjects to one of two study groups. Total fat intake was reduced from 38 to 24% in both groups. The dietary P:S ratio was increased from 0.2 to 0.9 in Group I and from 0.2 to 0.4 in Group II. During the 12-week intervention period, systolic and diastolic blood pressure in Group I decreased 4 mm Hg and 5 mm Hg, respectively. The corresponding decrease in Group II was 3 mm Hg and 4 mm Hg, respectively. Again, blood pressure reduction was reversed when subjects assumed their usual diets. The findings suggest that in people on a low fat diet, a moderate
Page 204
increase in the dietary P:S ratio is as effective in lowering blood pressure as is a greater increase.
In a study in 36 children ages 8 to 18 in the same two communities in North Karelia, Vartiainen et al. (1986) assessed the effect of total fat reduction (from 35 to 24% of total energy) and an increase in P:S ratio (from 0.18 to 0.61) on blood pressure. Neither systolic nor diastolic blood pressure changed significantly over the 12-week study period.
A few reports indicate that MUFAs, like PUFAs, may reduce blood pressure. In a cross-sectional survey of 76 middle-aged persons, Williams et al. (1987) found a strong inverse correlation between MUFA intake and both systolic and diastolic blood pressures that persisted after controlling for other potentially confounding variables, including age, cigarette smoking, stress, Quetelet Index, percentage of body fat, and waist-to-hip ratio. Rao et al. (1981) reported an inverse association between supplemental dietary MUFAs and blood pressure in hypertensive Indians on a traditional low-fat diet. Further studies, especially controlled clinical studies, are needed to replicate these findings.
In summary, studies of dietary lipids and blood pressure in humans suggest that a diet with a high P:S ratio and low total fat can produce modest reductions in blood pressure in normotensive and hypertensive people. Reduced total fat intake by itself appears to have little influence on blood pressure.
The inconsistency among study findings may reflect, in part, the inadequacy of the P:S ratio as a measure of the fatty acid composition of the diet, imprecise measurements of the dietary PUFAs and SFAs consumed by study participants, and inadequate control of other dietary and nondietary variables that may confound the association between dietary lipids and blood pressure.
Animal Studies
Studies in animals provide additional evidence for an association between intake of specific fatty acids (e.g., PUFAs) and blood pressure. In many animal studies, however, hypertension is induced by extremely high levels of dietary salt, which can confound interpretation of results. The following section distinguishes such studies from those restricting dietary salt or providing basal salt levels.
Lipid Studies On Hypertension Not Induced By Salt
Smith-Barbaro et al. (1981) investigated the relative effects of diets high in PUFAs (20% corn oil), high in SFAs (5% corn oil and 15% coconut oil), and low in total fat (5% corn oil) in Sprague Dawley rats. Salt was maintained at normal levels in all three diets. After 9 weeks, blood pressure was highest in animals fed the high SFA diets, intermediate in animals fed the high PUFA diets, and lowest in animals fed the low-fat diet.
Dusing et al. (1983) reported that the addition of even small amounts (5%) of linoleic acid to the diets of rats was sufficient to blunt the increase in blood pressure associated with diets deficient in linoleic acid. The moderating effect was evident even when animals were fed varying amounts of SFAs. The authors concluded that reduced linoleic acid intake was a more important factor in elevating blood pressure than increased SFAs. MacDonald et al. (1981) noted that the effect of linoleic acid intake on blood pressure is not continuous but, rather, exhibits a threshold (between 4.2 and 9.4% of total calories) above which little effect on blood pressure is observed.
Total dietary fat appears to modify the effect of dietary linoleate on blood pressure. Moritz et al. (1985) reported that low dietary fat (15% of energy from fat) can moderate the development of hypertension in susceptible rats and that high PUFA diets (15% sunflower or linseed oil by weight) can lower blood pressure in both hypertensive and normotensive animals.
Like studies in humans, studies in animals have not produced consistent results. Murray et al. (1986) reported higher blood pressures in Wistar-Kyoto spontaneously hypertensive rats (SHRs) fed high fat (40% of energy from fat) maize-oil diets compared to SHRs fed high-fat coconut-oil diets. Wexler (1981) observed that the onset of hypertension was blocked in Okamoto-Aoki SHRs fed a high SFA (20% suet) diet but not a low-fat (4% fat) diet.
In one study in rabbits (Bursztyn and King, 1986) and one in the SHR and normotensive Wistar-Kyoto and Wistar-Schonwalde rat strains (Singer et al., 1986), no effect on blood pressure was observed from specific fatty acids with diets that differed in their proportions of linoleic, g-linolenic, a-linolenic, and eicosapentaenoic acids.
Lipid Studies On Salt-Induced Hypertension
Studies on salt-induced hypertension in sodium-sensitive Wistar (Ten Hoor and Van de Graaf, 1978) and Dahl rats (Tobian et al., 1985) indicate that the addition of linoleic acid can blunt the hypertensive
Page 205
response of dietary salt and result in lower blood pressures. Hoffman et al. (1978) compared blood pressures in Wistar rats fed low saturated fat (7% hydrogenated palm kernel oil) or unsaturated fat (7% sunflower seed oil) diets with 1.5% added salt. They concluded that when chain length is similar, degree of unsaturation is inversely associated with systolic blood pressure.
Smith-Barbaro et al. (1981) compared blood pressure responses in Sprague Dawley rats on low total fat (5% corn oil), high PUFA (20% corn oil), and high SFA (5% corn oil and 15% coconut oil) diets who were also randomized to two levels of salt intake (8 and 15% sodium chloride). The authors reported that high salt intake produced hypertension when consumed with the low-fat diet. However, substitution of a high-SFA diet appeared to minimize the hypertensive effect of salt.
Codde and colleagues (1987a,b) conducted two studies comparing the effects of w-3 PUFA (fish oil), ¬-6 PUFA (safflower oil), and SFAs on blood pressure in rats challenged with desoxycorticosterone and salt. In the first study (Codde et al., 1987a), blood pressure in all three groups increased markedly by the end of the study (8 weeks), but rats fed the w-3 and w-6 PUFA diets had significantly lower blood pressures than those fed the SFA diet. In the second study (Codde et al., 1987b), SHRs and normotensive Wistar-Kyoto rats fed either an w-3-rich diet of fish oil or an SFA-rich diet exhibited no differences in blood pressure before the addition of 1.5% saline to the diet. With salt loading, however, the blood pressure of SHRs on the SFA diet increased 21 mm Hg over that of the SHRs given the fish oil diets. No change was seen in the Wistar-Kyoto group.
The effect of dietary lipids on blood pressure has also been investigated by Kennedy et al. (1978), who reported that blood pressure in New Zealand white rabbits fed palm oil diets (high in SFAs) increased more than in rabbits fed safflower oil diets high in w-6 PUFAs. Elevations in blood pressure have also been observed in rabbits fed a high SFA diet of coconut oil (Gardey et al., 1978).
In summary, animal studies indicate that diets deficient in linoleic acid increase blood pressure, and they provide weaker and less consistent evidence that diets with high levels of PUFAs lower blood pressure whereas those high in SFAs increase blood pressure. A few studies suggest that the hypertensive effect of high salt intake is moderated by high-fat diets.
The mechanism by which unsaturated fatty acids, especially linoleic acid, influence blood pressure has not been identified. Postulated mechanisms include alterations in prostaglandin production or effects on the renin-angiotensin system (Comberg et al., 1978; Frohlich, 1977; Weinsier and Norris, 1985).
Summary
Observational, clinical, and community intervention studies in humans suggest that although reduction of total dietary fat alone has little to no effect on reducing blood pressure in normotensive and mildly hypertensive individuals, increasing the dietary P:S ratio, with or without decreasing total intake of fat, results in a modest decrease in the blood pressure of hypertensives. The inconsistency of findings in humans, however, may reflect the inadequacy of the P:S ratio as an accurate measure of the dietary composition of fatty acids, as well as inadequate control of other dietary factors (e.g., total calories, salt, potassium, calcium, and alcohol) that might confound the association between lipids and blood pressure.
Findings in animal studies support an association of dietary lipids with blood pressure. The strongest and most consistent evidence links linoleic acid restriction to elevated blood pressure. Weaker evidence suggests that increasing the proportion of MUFAs or PUFAs relative to SFAs reduces blood pressure but that increasing dietary SFAs alone increases blood pressure. The exact nature of the response of blood pressure to graded increases in the intake of linoleic acid beyond the amounts necessary to prevent deficiency is unclear.
Cancer
The National Research Council's Committee on Diet, Nutrition, and Cancer (NRC, 1982) concluded ''that of all the dietary components it studied, the combined epidemiological and experimental evidence is most suggestive for a causal relationship between fat intake and the occurrence of cancer. Both epidemiological studies and experiments in animals provide convincing evidence that increasing the intake of total fat increases the incidence of cancer at certain sites, particularly the breast and colon, and, conversely, that the risk is lower with lower intakes of fat. Data from studies in animals suggest that when fat intake is low, polyunsaturated fats are more effective than saturated fats in enhancing tumorigenesis, whereas the data on humans do not permit a clear distinction to be made between the effects of different components of fat. In general, however, the evidence
Page 206
from epidemiological and laboratory studies is consistent."
The committee further concluded that "the relationship between dietary cholesterol and cancer is not clear. Many studies of serum cholesterol levels and cancer mortality in human populations have demonstrated an inverse correlation with colon cancer among men, but the evidence is not conclusive. Data on cholesterol and cancer risk from studies in animals are too limited to permit any inferences to be drawn" (NRC, 1982, pp. 1-4 and 1-5).
The conclusion on fat was based largely on the consistency between epidemiologic and animal evidence. International correlation studies show direct associations between per-capita availability of dietary fat and the incidence of mortality from cancer at such sites as the breast, prostate, and gastrointestinal tract. Further evidence is provided by observational epidemiologic studies and by experimental results showing that animals fed high-fat diets develop cancers of the mammary gland, pancreas, and intestinal tract more readily than do animals on low-fat diets (NRC, 1982).
Rather than repeat discussions of the evidence on dietary fat, cholesterol, and cancer reviewed by the Committee on Diet, Nutrition, and Cancer (NRC, 1982), this section is concerned primarily with data that have accumulated since that time. Much work continues to be focused on breast cancer and colon cancer. Other studies have focused on the effects of dietary fat on pancreatic cancer and on the development of better models of prostate cancer that can be used to study effects of dietary fat. Interest has been concentrated primarily on effects of different levels and types of dietary fat. The mechanisms by which dietary fat may influence carcinogenesis have also been the subject of much speculation and some studies.
Epidemiologic data from different countries show that cancer incidence and mortality correlate more positively with total dietary fat than with any one type of dietary fat. On the other hand, results of experiments in animals demonstrate that vegetable oils containing w-6 PUFAs promote carcinogenesis more effectively than SFAs, whereas fish oils containing w-3 PUFAs tend to inhibit carcinogenesis. These findings are not necessarily in conflict with the epidemiologic data, since the studies were conducted primarily with individual fats and oils with fatty acid compositions that are quite different from those of the mixtures of fats and oils present in the diets of humans.
Recent studies on dietary fat and carcinogenesis are summarized in books, conference proceedings, and review articles (e.g., ACS, 1983, 1986; Carroll, 1985; CAST, 1982; ECP/IUNS, 1986; Finley and Schwass, 1985; Hayashi et al., 1986; Holman et al., 1986; Hopkins and Carroll, 1985; ILSI/NF, 1987; Ip et al., 1986; loosens et al., 1985; Knudsen, 1986; Milner and Perkins, 1984; Perkins and Wisek, 1983; Poirier et al., 1987; Reddy and Cohen, 1986a,b; Roe, 1983; Stich, 1982; Walker, 1986; Wynder et al., 1983).
Epidemiologic Evidence
The National Research Council's report Diet, Nutrition, and Cancer (NRC, 1982) indicated that fat has probably been studied more thoroughly and has been associated with various cancers more frequently than any other dietary factor. Since that report was published, studies have focused on the relationship between dietary fat and risk of colorectal, breast, and some other cancers, including cancers of the prostate and ovary. In this section, the committee considers the evidence first on dietary fat and then on dietary and serum cholesterol.
The evidence from epidemiologic studies has been based in part on geographical comparisons correlating differences in incidence of and mortality from various cancers with estimated fat intake by populations. Strong associations were found for cancer of the breast, colorectum, endometrium, and prostate, and, to a lesser extent, kidney and testicles (Armstrong and Doll, 1975; Carroll, 1975; Hems, 1980; Knox, 1977). In studies based on data from personal interviews in Hawaii, Kolonel et al. (1981) found significant associations of total fat, animal fat, and saturated and unsaturated fat intakes with ethnic patterns of breast, prostate, and corpus uteri cancer incidence but not of colon cancer.
Other studies have suggested an association of dietary fat with breast cancer (Lubin et al., 1981; Miller et al., 1978; Nomura et al., 1978; Phillips, 1975), prostate cancer (Hirayama, 1977; Rotkin, 1977; Schuman et al., 1982), and colorectal cancer (Dales et al., 1979; Jain et al., 1980; Martinez et al. 1979; Phillips, 1975). Negative studies were also reported, especially for colorectal cancer (Bjelke, 1978; Graham et al., 1978; Haenzel et al., 1980). As noted by the Committee on Diet, Nutrition, and Cancer (NRC, 1982), however, some of these studies were conducted on groups of relatively homogeneous populations, and some
Page 207
were not specifically designed to test the hypothesis that fat consumption is associated with colorectal cancer. The studies specifically designed to test the dietary fat hypothesis (e.g., Dales et al., 1979; Jain et al., 1980) tend to show the most striking direct associations.
The remainder of this section presents the evidence by cancer site, the way most of it is reported.
Breast Cancer
Since the 1982 report of the Committee on Diet, Nutrition, and Cancer (NRC, 1982), data from an earlier case-control study in Canada (Miller et al., 1978) have been reanalyzed with more modem methods and in light of the contribution made by different dietary factors (Howe, 1985). As a result of this analysis, the strongest association was found for SFA intake, and the dose-response relationship was particularly significant in premenopausal women. In this group, the risk for those in the highest SFA consumption tertile (>35.4 g of SFAs per day) relative to the lowest (<23.4 g of SFAs per day) was 5.9. Hirohata et al. (1987) studied 183 case-control sets of Japanese and 161 case-control sets of Caucasian subjects in Hawaii. Each case was matched to one hospital and one neighborhood control, and all were between the ages of 45 and 74. No statistically significant differences were found between cases and controls in their mean intake of total fat, SFAs, oleic acid, linoleic acid, animal protein, and cholesterol. However, there was a suggestion that cases consumed more SFAs and oleic acid than did neighborhood controls. As the authors noted, one difficulty with this investigation may have been the use of a relatively limited dietary questionnaire, which was designed to collect information on a selected list of 43 food items believed to represent the major sources of total and saturated fat, cholesterol, and animal protein.
Lubin et al. (1986) reported the findings of a large case-control study in Israel in which neighborhood controls were also used. When consumption levels were considered in quartiles, an increased risk of breast cancer was found among high fat, high animal protein, and low fiber consumers compared to those with the lowest consumption of fat and animal protein and highest consumption of fiber. For women under the age of 50, the relative risk was 1.6; for women age 50 or more, it was 2.0. A significant trend (p <.02) was found for the latter group.
In British Columbia, Canada, Hislop et al. (1986) administered a food frequency questionnaire on current diet and dietary practices in childhood. Although unable to determine nutrient intake, they found positive associations of breast cancer with current intake of fat-associated foods, especially whole milk and beef, and an increased risk among those who consumed visible fat on meat, both recently and in childhood. In northern Italy, risk estimates for indices of fat intake based on selected food items with a high fat content were found to be moderately elevated (Talamini et al., 1984). In a large case-control study in France involving 1,010 breast cancer cases and 1,950 controls with nonmalignant diseases, increased risk was found for consumption of dairy products but not for butter or yogurt; for the latter, there was a suggestion of a protective effect (Lê et al., 1986). For daily cheese consumption relative to never, the risk was 1.5, and for full cream milk consumption relative to none, the risk was 1.8.
At least three recent studies, however, were negative. In a case-control study in Greece involving 120 patients with breast cancer and 120 controls from an orthopedic hospital, Katsouyanni et al. (1986) found no association with consumption of fats and oils, but a protective effect for the consumption of vegetables. However, the authors pointed out that animal fat consumption in Greece is less than one-third of that in the United States or Canada and all study subjects may have therefore been at low risk of breast cancer, whereas vegetable consumption in Greece is twice as high. In a case-control study in Australia involving 451 cases and 451 controls, Rohan et al. (1988) found no association between dietary fat, protein, or caloric intake, but a protective effect of b-carotene.
A cohort study of nurses who completed a self-administered questionnaire also showed no association between dietary fat and risk of breast cancer (Willett et al., 1987). Among the possible reasons for discrepancies between this large cohort study (89,538 nurses ages 34 to 59 who had 601 breast cancers during 4 years' follow-up) and most case-control studies are recall bias in the case-control studies, and in the cohort study, less than complete estimates of fat intake in the self-administered questionnaire compared to a detailed dietary history (Jain et al., 1982), a short follow-up period (4 years), a narrow range of fat intakes in the study sample, and measurement error combined with a weak association. Another cohort study based on NHANES I data showed an inverse association between dietary fat and breast cancer occurrence that was no longer significant when adjusted for total energy intake. However, the find-
Page 208
ings may have been influenced by methodologic problems with the dietary assessment; indeed, the design of the NHANES study has been considered unsuitable for analyses based on individual intakes (Jones et al., 1987a) (see Chapter 2).
In addition to the limitations noted above, part of the inconsistencies in the epidemiologic studies could be that fat intake early in life may have a greater influence on breast cancer risk than intake later in life. This could account for the slow change in breast cancer rates in some migrant groups and could be reflected in the failure to observe reduced breast cancer incidence in population groups who as adults have made major dietary changes resulting in lower total fat intake. This applies to groups of nuns who adopted a low-fat diet in adult life (Kinlen, 1982). Furthermore, measurement errors in dietary recall in case-control and cohort studies can account for weak or absent associations of dietary fat with breast cancer, even if the relationship is real (Freudenheim and Marshall, 1988; Prentice et al., 1988).
In summary, the overall evidence is inconsistent. Nevertheless, it tends to support a weak association between dietary fat and breast cancer risk in humans.
Colorectal Cancer
Miller et al. (1983) compared the intake of food groups and various nutrients, particularly SFAs, in relation to risk of colorectal cancer. This was a further analysis of the case-control study first reported by Jain et al. (1980). The risk associated with a higher consumption of SFAs relative to lower consumption, considered in tertiles, for colon and rectal cancer cases combined relative to the combined neighborhood and surgical control series was 2.4 for males and 2.6 for females. In females, increased intake of SFAs consistently increased risk for colon cancer and also had an important independent effect over and above an apparent effect of some meats in increasing risk of rectal cancers. In males, different meat products appeared to increase risk and intake of SFAs did not appear to have an independent effect. Nevertheless, it seems possible that imprecision in consumption estimates, particularly for men, may have contributed to this result.
Willett and Stampfer (1986), in a commentary on differences in total energy intake in dietary studies, recalculated nutrient densities for SFA intake (SFAs divided by total calories) from the study of Jain et al. (1980) and found that the association with colorectal cancer largely disappeared. They noted, however, that inverse associations (as observed for crude fiber intake) can be produced by this process even when such nutrients are not independently associated with disease. This caused Howe et al. (1986) to reanalyze the data from the Jain et al. (1980) study once more, constructing logistic regression models that included variables representing the effect of SFAs and calories from sources other than SFAs. In this approach, the risk for highest, relative to low, consumption levels for SFAs was found to be 2.0 for males and 2.3 for females. No effects were found for calories or for fiber.
Manousos et al. (1983) in a case-control study conducted in Greece based on a food frequency questionnaire observed a positive association between intake of beef and lamb and colorectal cancer risk and a negative association with intake of cabbage, lettuce, spinach, and beets.
Stemmermann et al. (1984) reported the results of a 14- to 17-year follow-up of 8,006 men of Japanese ancestry whose intake of total fat was assessed by a 24-hour recall administered at baseline. The estimated mean intake of fat by the 99 cases was 69 g/day. For the 378 controls, it was 77 g/day. Thus this study did not support the dietary fat hypothesis.
In the Marseilles region of southern France, 399 cases and a corresponding number of surgical controls were studied by Macquart-Moulin et al. (1986), who used a food frequency dietary questionaire and estimated consumption of fat. There was no evidence of any difference in the consumption of total fat between cases and controls. Indeed, a reduced risk appeared to be associated with increasing consumption of oil, largely monounsaturated oil. Vegetables, particularly vegetables with low fiber content, appeared to be protective in this study.
In Belgium, a study conducted with a similar questionnaire showed no effect of fat intake, but there was a clear dose-response effect for intake of mono- and disaccharides. A protection effect was found for linoleic acid and dietary fiber (Tuyns et al., 1987).
In a case-control study conducted in Adelaide, Australia, Potter and McMichael (1986) found evidence of increasing risk with increasing consumption of a number of nutrients and a pattern of food intake involving high dietary fat and possibly SFAs. Although risk associated with SFAs and various components of fat was increased, it was the impression of the authors that the major contribution to increased risk came from dietary protein.
Page 209
A case-control study of colorectal cancer in Melbourne, Australia, included 715 cases and 727 age- and sex-matched community controls. Fat intake was found to be a risk factor for both males and females (Kune et al., 1987); however, the relative risk associated with high fat intake was not large (1.4) and became apparent only after adjustment for meat and vegetable intake. The effect was similar, however, for colon and rectal cancer.
In summary, the weight of the evidence points to an association between colorectal cancer and a pattern of food intake involving high dietary fat. The negative outcome of some studies may be partly attributable to methodological difficulties, but it is possible that different cultures differ in the extent to which factors operate either in increasing risk or in providing protection against colorectal cancer.
Prostate Cancer
Results from at least four case-control studies have been published since the 1982 report Diet, Nutrition, and Cancer (NRC, 1982). Heshmat et al. (1985) reported a case-control study of 181 blacks with histologically confirmed prostate cancer and 181 controls from Washington, D.C. Risk enhancement was associated with increased intake of proteins, total fat, SFAs, and oleic acid. These items were related to intake by people ages 30 to 49 years, but not for those age 50 or more.
Graham et al. (1983) compared 262 patients from Buffalo, New York, with two different control series. Risk of prostate cancer increased with increasing ingestion of animal fats. Risk ratios of approximately 2.0 were found for fat consumption greater than 2,100 g/month relative to fat consumption of less than 1,200 g/month.
In a study of 452 prostate cancer cases and 899 age-matched population controls in Hawaii, Kolonel et al. (1988) found a dose-response gradient in risk of prostate cancer associated with intake of SFAs among men 70 years of age and older. The risk ratio was 1.7 for the highest relative to the lowest quartile of SFA intake.
In a follow-up study of 6,763 Seventh-Day Adventists who completed a dietary questionnaire in 1960, Snowdon et al. (1984) found positive associations between fatal prostate cancer and the consumption of milk, cheese, eggs, and meat. There was a graded dose-response between each of the four animal products and risk. The predicted relative risk of fatal prostate cancer was 3.6 for those who consumed all four animal products heavily. Although the dietary instrument used could not provide an estimate of fat consumption, it does seem possible that dietary fat at least contributed to the increased risk of fatal prostate cancer in this study.
In summary, though not entirely consistent, there is evidence of an association of dietary fat, especially from animal sources, with prostate cancer. In one study, there was an association with incidence of prostate cancer, particularly in younger men, and in another, with fatal prostate cancer.
Ovarian Cancer
Two case-control studies on dietary fat and risk of ovarian cancer have produced contradictory results. In one, based on data collected between 1957 and 1965, 274 white women ages 30 to 79 with epithelial carcinoma of the ovary were compared with 1,034 hospital controls (Byers et al., 1983). The investigators found no risk associated with consumption of fat at any age. However, Cramer et al. (1984), who studied 215 white women with epithelial ovarian cancer and 215 control women matched by age, race, and residence, found that the women with ovarian cancer favored, and consumed greater amounts of, foods high in animal fats and significantly less vegetable fat compared with control subjects. There was a significant trend toward an increased risk of ovarian cancer with increasing animal fat consumption. The risk controlling for Quetelet's Index for those with a high intake of animal fat (intake score of 225 or more) was 1.83 compared to those with a low intake (intake score of less than 125).
Details of the methods of dietary assessment used by Cramer et al. (1984) were not given. It is known that in the method used by Byers et al. (1983), relatively limited information was collected on food items contributing to dietary fat.
The geographic correlations for dietary fat and ovarian cancer are somewhat lower than those found for breast, colorectal, and prostate cancer (Armstrong and Doll, 1975). The positive results of Cramer et al. (1984) suggest that further study of this reported association is warranted.
Conclusions
In summary, evidence suggests that the risk of breast cancer and, to a possibly greater extent, of colon, prostate, and ovarian cancers, is associated with dietary fat. Although studies on diet and endometrial cancer have not been reported since 1982, the incidence of endometrial cancer has also been associated geographically with high total fat intake and with the incidence of these other cancers. The data suggest
Page 210
that the endocrine-related cancers of the breast, prostate, ovary, and endometrium, and also colorectal cancer, which itself has some features of an endocrine-related disease, are all associated with diet and possibly with total fat or SFA intake. This evidence supports the evidence from animals in studies reviewed below in the section on Animal Studies.
Cholesterol intake was greater in premenopausal cases than in controls in a case-control study of diet and breast cancer (Miller et al., 1978). However, this association was not statistically significant and was quantitatively weaker than an association with dietary fat. In a reanalysis of the same data, dietary cholesterol did not have an effect independent of SFA intake (Howe, 1985). Cholesterol intake by cases was greater than that by controls in a case-control study of colorectal cancer, but the risk ratios were lower than for SFA intake (Jain et al., 1980). Risk ratios were also elevated for dietary cholesterol and colon cancer in a second case-control study, but they were lower for rectal cancer where risk was more strongly associated with dietary protein (Potter and McMichael, 1986). In a study comparing cholesterol intake by Seventh-Day Adventist lacto-ovovegetarians and nonvegetarians, differences were not "striking" (Turjman et al., 1984b). However, Liu et al. (1979), using food disappearance data from 20 countries, found that when they controlled for dietary cholesterol, the associations of dietary fat and fiber with colon cancer mortality that they previously observed were no longer significant.
Dietary cholesterol was associated with an increased risk of lung cancer in a case-control study by Hinds et al. (1983). The association was found in cigarette smokers and in males but not in females. Dietary cholesterol has also been associated with an increased risk of bladder cancer in a case-control study by Risch et al. (1988). In these studies, however, relatively restricted dietary questionnaires were used and it was not possible to determine whether the association with dietary cholesterol was due to other dietary factors associated with cholesterol intake.
No significant association was found between dietary cholesterol and cancer mortality in a 10-year follow-up of the Honolulu Heart Program (McGee et al., 1985), but data for individual cancer sites were not reported. In a cohort study of 89,538 U.S. nurses, no association between dietary cholesterol and breast cancer was found (Willett et al., 1987).
Some studies suggest that low serum TC is associated with an increased risk of cancer. These are discussed in the following paragraphs. The topic of competing risks is discussed further in Chapter 28.
Data on TC and cancer collected up to 1983 were reviewed by McMichael et al. (1984), who concluded that observational studies provide substantial evidence that preclinical cancer causes a lowering of TC and that limited but biologically plausible evidence suggests that males with low TC despite a diet high in SFAs and cholesterol are at increased risk of colon cancer. Since the studies reviewed by McMichael and colleagues (i.e., from 1983 on), there have been reports of seven studies primarily related to follow-up of cohorts established for the study of cardiovascular disease. In a study of 4,035 residents of California ages 40 to 89, Wingard et al. (1984) found no association between serum TC and cancer morbidity or mortality over a 7-year period for either men or women for any cancer site.
In a 5-year follow-up of 10,940 participants in the Hypertension Detection and Follow-up Program, a small but statistically significant inverse relationship was found between baseline TC and cancer incidence (Morris et al., 1983). When cases diagnosed during the first 2 years were excluded, the association was similar in magnitude but no longer statistically significant. The number of cases did not permit analysis by cancer site. In up to 6 years of follow-up (mean, 3 years) of 10,000 middle-aged men in the Malmo Prevention Program (Peterson and Trell, 1983), TC was inversely related to cancer mortality (44 deaths)—a relationship also observed for the 25 cancer deaths that occurred more than 2.5 years after screening. Serum urate levels at screening in this study were correlated with early but not late (more than 2.5 years after screening) cancer mortality (Peterson and Trell, 1983). As urate levels might indicate proliferation of cancer cells, the association of elevated TC with the late deaths may be due to a factor other than cancer operating at the time of screening.
In the Busselton Community Study in western Australia, 1,564 subjects ages 40 to 74 were followed for 13 years (Cullen et al., 1983). In men ages 60 to 74, but not in men ages 40 to 59 or in women, a negative association was found between serum TC and cancer mortality. In this study, however, it was not indicated whether the association persisted if early cancer deaths were ex-
Page 211
cluded. In New Zealand, 630 Maoris ages 25 to 74 were followed for more than 17 years (Salmond et al., 1985). A significant inverse relationship between cancer mortality and TC was found for men and women considered together. The relative risk derived by comparing cancer mortality in men and women in the 10th and 90th percentiles of TC concentration decreased from 3.0 to 2.4 after excluding deaths during the first 5 years.
In the Seven Countries Study, 11,325 healthy men ages 40 to 59 were followed for 15 years (Keys et al., 1985). Among 477 cancer deaths 5 or more years after TC measurement, there was a significant excess of deaths from lung cancer in the lowest 20% of the TC distribution. Nevertheless, regional comparisons of cancer mortality showed that the highest cancer rates occurred in northern Europe where the TC levels were highest. In contrast, in a cohort study of 92,000 subjects less than 75 years old examined during 1963-1965 and followed by linkage to the Swedish Cancer Registry to 1975, there was a positive association between TC levels and risk of rectal cancer in men (Tornberg et al., 1986). The risk for men with elevated levels of both serum TC (³250 mg per deciliter) and b-lipoprotein (³2.2 g/liter), relative to those with lower levels of both, was 1.62 for colon cancer (95% confidence interval [CI], 1.18-2.22) and 1.70 for rectal cancer (95% CI, 1.18-2.44).
Cancer incidence was determined in 160,135 men and women who were members of a prepaid health plan and whose TC was determined as part of a multiphasic health examination (Hiatt and Fireman, 1986). These subjects were followed from 8 to 16 years. No consistent association of low TC with cancer incidence was found, but cancer incidence was highest among those in the lowest quintile of TC in the first 2 years after the measurement. In the largest study so far reported, a follow-up of the 361,662 men screened for the MRFIT study, there was a significant excess of cancer in those in the lowest decile of TC (<168 mg/dl), which attenuated over time (Sherwin et al., 1987).
Serum TC was assessed in five case-control studies. In one, elevated levels of TC were found in 37 cases of primary brain tumors compared to 74 controls (Abramson and Kark, 1985). The difference was not reduced by controlling for potential confounders (including weight). In another case-control study, TC was measured in 244 patients with adenomatous polyps of the colon, 182 patients with Dukes' A or B colon cancer, and 688 hospital controls (Neugat et al., 1986). The mean TC levels were lower in the cancer cases than in the controls, the Dukes' B cases accounting for most of the difference. There was no difference between the mean levels for those with adenomatous polyps and the controls. After adjustment for nutritional status based on serum albumin level, however, there were no differences among any of the groups. In a nested case-control study within a cohort of members of a prepaid health care plan, 245 newly diagnosed cases of large bowel cancer and 5 matched controls for each case were compared on the basis of TC levels measured during a multiphasic health examination before diagnosis of the cases (Sidney et al., 1986). No direct or inverse relationship between TC and large bowel cancer was found. A fourth case-control study was based on a cohort of 18,995 people examined at a health center between 1970 and 1973 (Gerhardsson et al., 1986). The investigators found medical records for 100 of 176 cancer cases who died by 1979, for 393 of 900 controls still alive in 1979, and for 69 of 153 people who died of cardiovascular disease by 1979. Serum TC levels in the cancer cases were significantly lower than those of controls only in the 2-year period before death.
In a fifth study, a positive association was found between TC levels and the prevalence of adenomatous polyps found during colonoscopies of 842 patients (Mannes et al., 1986). For large bowel adenoma, the odds ratio between the highest and lowest quintiles of serum cholesterol was 1.9 (95% CI, 1.1-3.5) after adjustment for age and was 2.0 (95% CI, 1.1-3.6) after adjustment for body mass index. In a study by Tartter et al. (1984), serum TC was assessed in relation to disease-free survival of 279 colon cancer patients. There was an 11% but nonsignificant lower cumulative disease-free survival at 5 years in those whose TC levels were below the median than in those with levels above the median. In another study, a family history of cancer was found to be positively associated with TC levels in young adults (T. Reed et al., 1986).
Thus, although some epidemiologic studies of cohorts assembled to study cardiovascular disease risk show associations of low serum TC with cancer incidence and mortality, the studies designed specifically to assess the relationship have not in general found an association. When site-specific data are available, they are not consistent.
Clinical and Metabolic Studies
Metabolic studies have explored variables possibly related to the question of whether or not diet
Page 212
plays a role in the etiology of colon cancer. Several studies of fecal steroids have been undertaken in a number of different populations. Antonis and Bersohn (1962a) found that an increase in calories from dietary fat from 15 to 40% of the diet consumed by 43 white and Bantu prisoners resulted in an increased fecal excretion of neutral sterols and bile acids. The effect was greater for unsaturated fat. Cummings et al. (1978) observed that healthy volunteers placed on controlled diets containing first high and then low levels of animal fat had significant increases in fecal steroid excretion when on the high-fat diet. In a similar study, Reddy et al. (1980a) observed an increase in the enzymic activity of fecal microflora in addition to increases in excretion of fecal steroids.
Studies in various populations have shown an association of fecal excretion of steroids and cholesterol metabolites with the incidence of colorectal cancer in volunteers from England, Scotland, the United States, Uganda, Japan, India, and Hong Kong (Crowther et al. 1976; Hill et al., 1971). Finegold et al. (1974, 1975) were unable to confirm the findings reported by the previous investigators. Hill (1981) pointed out the limitations of traditional bacteriological techniques to investigate the microflora of the colonic lumen.
Reddy et al. (1978) found that volunteers in the United States and from rural Kuopio in Finland excreted the same amount of secondary bile acids daily, but the concentration of fecal bile acids in the U.S. subjects was 2.5 times greater than that of the Finns due to the greater stool output of the Finns. Similar studies conducted in New York and Sweden produced comparable results (Reddy et al., 1983). In another international study, dietary intake and fecal characteristics of random population samples from Kuopio, Finland, and Copenhagen, Denmark, were compared (Maclennan and Jensen, 1977). This study failed to confirm the correlation between the incidence of large bowel cancer and fecal steroid output or concentration. No difference was found in fat intake, but there were higher intakes of dietary fiber and milk in low-incidence Kuopio.
Another study conducted by the same group included areas with intermediate colon cancer risks in Finland and Denmark (Jensen et al., 1982). Daily fecal loss of bile acids was found to be identical among high-, medium-, and low-risk groups, but fecal bile acid concentrations among these groups were positively correlated with large bowel cancer risk, a finding probably related to the negative association between fecal bulk—which increases with increased fiber intake—and fecal bile acid concentration. Comparisons between Japanese living in Hawaii and those in Japan showed rather similar findings (Nomura et al., 1983).
Turjman et al. (1984a) found different concentrations of total bile acids among pure vegetarians, lacto-ovovegetarians, nonvegetarian Seventh-Day Adventists, and a demographically comparable group from the general population—groups that have different risks for colon cancer. Fecal cholesterol and its metabolites were lower among vegetarians; the ratio of secondary to primary bile acids also differed among the groups, suggesting a positive correlation between the formation of secondary bile acids and risk for developing colon cancer (Nair et al., 1984).
Several investigators have studied the excretion of bile acids in patients with colon cancer and in controls (Hill et al., 1977; Murray et al., 1980; Reddy and Wynder, 1977). These studies have generally produced inconsistent findings, possibly because of sampling limitations and the difficulties in sorting out causal factors from metabolic side effects of the disease or its treatment.
A completely different line of investigation has been followed by some investigators who studied the relationship between fecal mutagens and potential colon cancer. Ehrich et al. (1979) compared three South African populations with different colon cancer risks and observed that 19% of the fecal samples from urban white South Africans who are at higher risk of colon cancer were mutagenic compared to only 2% for urban blacks and 0% for rural blacks who are at lower risk. Reddy et al. (1980b) studied fecal mutagen activity among omnivorous volunteers and vegetarian Seventh-Day Adventists from New York, and rural Finns from Kuopio. A significantly higher percentage of omnivorous volunteers had fecal mutagenic activity compared to subjects in Finland. None of the samples from Seventh-Day Adventists had mutagenic activity. Similar results were obtained by Kuhnlein et al. (1981), but two small case-control studies failed to show a significant association of fecal mutagens with adenomatous polyps in autopsy specimens (Correa et al., 1981) or with colon cancer in newly diagnosed patients (Bruce and Dion, 1980). Bruce and Dion (1980) also found no differences in fecal mutagenic activity among people with different fat intakes.
In summary, clinical and metabolic studies focusing on colon cancer have explored three postulated intermediate mechanisms in colonic carcino-
Page 213
genesis, involving fecal bacteria, steroids and bile acids, and mutagens. Some consistency in research findings has been attained only for fecal steroids and bile acids, possibly because of the complexity of metabolic pathways and, in some instances, inadequate attention to appropriate sampling procedures. These findings have suggested that it is the concentrations of fecal steroids and bile acids that may enhance carcinogenesis rather than the total amounts excreted.
Animal Studies
Recent studies on dietary fat and carcinogenesis in laboratory animals have generally confirmed earlier evidence that animals fed high-fat diets develop tumors of the mammary gland, intestinal tract, and pancreas more readily that those fed low-fat diets ad libitum (NRC, 1982). The results can be modified, however, by the type of dietary fat.
Dietary fats increase the yield of mammary tumors only when they contain adequate amounts of w-6 PUFAs, which are normally present as linoleate in fats derived from plants and land animals. This probably explains why fats such as butter, coconut oil, and beef tallow have little effect on mammary carcinogenesis (Carroll et al., 1981). The requirements for w-6 PUFAs in mammary tumor promotion have been explored systematically by Ip et al. (1985), who reported 4 to 5% of total calories as the threshold at which the yield of mammary tumors increased. Increasing the amount of linoleate above this level appeared to have little additional effect on tumor promotion (Ip, 1987).
The amount of linoleate required to promote pancreatic tumors in rats appears to be somewhat higher (up to 8% of calories) (Birt and Roebuck, 1986; Roebuck et al., 1985). Much smaller amounts are evidently required for colon cancer (Bull et al., 1988), but in general, PUFAs appear to promote tumorigenesis more effectively than SFAs or MUFAs (Reddy, 1986a; Reddy and Maeura, 1984). However, some investigators have failed to observe the promotion of colon cancer by either SFAs or PUFAs (Nauss et al., 1987; Newberne and Nauss, 1986).
There is some evidence that tumor initiation can be influenced by the type of dietary fat. For example, lard has been observed to increase the yield of mammary tumors (Rogers and Lee, 1986; Rogers et al., 1986; Sylvester et al., 1986a) and colon tumors (Reddy, 1986a) when fed before or during the initiation.
The effects of dietary PUFAs on tumor metastasis have been investigated in two animal studies. Katz and Boylan (1987) found that a high-fat diet containing 23% corn oil stimulated metastasis of a mammary adenocarcinoma in retired female breeder rats compared to a diet containing 5% corn oil. Hubbard and Erickson (1987) reported that metastasis from transplanted mammary tumors in mice was enhanced by diets containing 8 to 12% linoleic acid as compared to diets with lower levels of linoleic acid.
In contrast to fats containing w-6 PUFAs, fish oils, which contain mainly w-3 PUFAs, fail to promote tumorigenesis when fed at high levels in the diet and may even have an inhibitory effect. This has been observed for tumors of the mammary gland (Braden and Carroll, 1986; Jurkowski and Cave, 1985; Karmali et al., 1984), intestine (Reddy, 1986a), and pancreas (O'Connor et al., 1985).
Diets used in these experiments may have been deficient in w-6 PUFAs, because fish oils contain only small amounts of these PUFAs and because w-3 PUFAs act as competitive inhibitors of reactions involving w-6 PUFAs. This may have affected the results of the experiments on tumorigenesis (Karmali, 1987). In more recent studies, vegetable oils have been added to fish oil diets to provide a source of w-6 PUFAs. In one such study, Kort et al. (1987a) reported that dietary menhaden oil inhibited the growth of transplantable mammary adenocarcinomas in rats but had no effect on metastases. In another, the tumor growth inhibition was not observed when the fish oil diet was fed only after transplantation of the tumor (Kort et al., 1987b). Other studies on experimental models of mammary cancer (Cave and Jurkowski, 1987), colon cancer (Reddy, 1987), and pancreatic cancer (O'Connor et al., 1987) indicate that a relatively high ratio of dietary fish oil to polyunsaturated vegetable oil is required to counteract the promoting effects of the vegetable oil.
Studies on animal models of mammary cancer and colon cancer indicate that trans fatty acids behave like SFAs with respect to their effects on tumorigenesis and do not promote tumors to a greater extent than do the cis isomers (Ip et al., 1986; Reddy et al., 1985; Selenskas et al., 1984). The evidence regarding dietary MUFAs, such as that derived from studies with olive oil, whose principal MUFA is oleic acid, is somewhat variable. In some experiments, olive oil promoted tumorigenesis about as well as PUFAs containing a high proportion of linoleic acid (Carroll and Khor,
Page 214
1971), but more recent reports suggest that olive oil is not a good promoter (L.A. Cohen et al., 1986; Reddy, 1986a). The amount of linoleic acid in olive oil is insufficient for maximum promoting activity (Ip et al., 1985), and variations in linoleic acid content may account for some of the difference among results.
Although experiments in animals indicate that the promoting effect of dietary fat is influenced by its fatty acid composition, cancer mortality in human populations seems to correlate with total dietary fat about as well as with animal fat, and there is little or no correlation with vegetable fat or PUFAs (Carroll, 1983; Carroll et al., 1986). This does not necessarily conflict with the results of animal studies that show a tumor-promoting effect of PUFAs. In fact, the experiments in animals suggest a twofold requirement in promotion of carcinogenesis by dietary fat (Carroll, 1986b). The dietary fat must provide a certain amount of w-6 PUFA. When this requirement is satisfied, the promoting effect of additional dietary fat appears to be unrelated to the type of fat (Ip, 1987). This promotion related to total fat intake may correspond to the positive correlations between total dietary fat and cancer incidence and mortality seen in epidemiologic data.
Dietary cholesterol has been studied primarily with respect to intestinal cancer. Although the data are inconsistent, Broitman (1986) in his review of the literature concluded that most evidence points to a possible relationship between dietary cholesterol and colon tumor development.
In animals treated with a single dose of a chemical carcinogen to induce mammary tumors, the predominant effect of dietary fat is exerted after exposure to the carcinogen, is positively correlated with the duration of feeding the high-fat diet, and is inversely related to the time between exposure to carcinogen and introduction of the high-fat diet (Dao and Chan, 1983; Rogers and Lee, 1986). This indicates that dietary fat is acting primarily as a promoter (Carroll and Khor, 1975).
As indicated above, some dietary fats, e.g., lard and beef tallow (Rogers et al., 1986; Sylvester et al., 1986a), enhance mammary tumorigenesis when fed only at or before exposure to a carcinogen (Cohen, 1986; Rogers and Lee, 1986; Welsch, 1986). The mechanism of action for this is not known (Rogers and Lee, 1986) but could involve alteration of the rates of formation and inactivation of carcinogens or an influence on their binding to DNA (Hopkins and West, 1976; Wade and Dhardwadkar, 1986).
Many of the hypotheses advanced to account for the promoting action of dietary fat have been concerned specifically with mammary cancer (Carroll, 1986a; Cohen, 1986; Kidwell, 1986; Rogers et al., 1986; Welsch, 1986; Welsch and Aylsworth, 1983). One of the first and most intensively studied hypotheses is that dietary fat perturbs the endocrine system, especially prolactin (Cohen, 1986). After reviewing this literature, Welsch (1986) concluded that there is no consistent support for the concept that dietary fat enhances mammary tumorigenesis by increasing blood levels of pituitary or ovarian hormones. This concept is also weakened by evidence that high-fat diets promote the development of tumors that both are and are not dependent on hormones (Sylvester et al., 1986b: Welsch, 1986). Other studies have shown little difference between target tissue receptor levels and responsiveness to estrogen and prolactin in animals fed moderate and high levels of fat. Animals fed low levels of fat have reduced hormone receptor levels, which may be related to essential fatty acid deficiency (Welsch, 1986).
Other proposed mechanisms are related to the observation that dietary PUFAs promote mammary carcinogenesis in animals more effectively than SFAs (Carroll et al., 1986). As indicated above, this difference appears to be related to a requirement for w-6 PUFAs, which are precursors of a variety of biologically active eicosanoids, including prostaglandins, thromboxanes, and leucotrienes. Evidence that the promoting effect of dietary fat can be prevented by inhibitors of prostaglandin synthesis (Carter et al., 1983; Hillyard and Abraham, 1979) suggests that the effect of fat may be mediated by eicosanoids. Further support for this idea is provided by the failure of dietary fish oils to promote mammary carcinogenesis (see the section on Breast Cancer). The w-3 PUFAs in fish oils competitively inhibit the formation of eicosanoids from w-6 PUFAs, and eicosanoids formed from w-3 PUFAs differ in structure and biologic properties from those derived from w-6 PUFAs. However, the prostaglandin synthesis inhibitor carprofen marginally enhanced the induction of mammary tumors induced by dimethylbenzanthracene in rats fed diets containing 5 or 20% corn oil, although the average weight of tumors was reduced significantly (Carter et al., 1987).
Although this is an attractive hypothesis, it is based mainly on speculation. The highly unsaturated fatty acids in fish oils are very susceptible to
Page 215
oxidation. Furthermore, while products of lipoxidation may initiate or promote carcinogenesis (Carroll, 1986a), there is also the alternative possibility that such compounds may inhibit carcinogenesis by exerting a toxic effect on tumor cells.
PUFAs of the w-6 and w-3 families can be incorporated into cellular membrane lipids. Since these fatty acids cannot be synthesized by animals, the amounts in tissues are influenced by the dietary supply (Holman, 1986). A model in which dietary unsaturated fatty acids are first taken up by adipose tissue and then transferred to epithelial tissue of the mammary gland has been described by Kidwell (1986).
The effects of dietary fat on the composition and properties of cellular membranes could influence carcinogenesis in a number of different ways. One possibility is an alteration in immune responses, which are known to be affected by PUFAs. These effects may be mediated by alterations in structure and function of cellular membranes or by modulation of prostaglandin biosynthesis (Erickson, 1986). Alterations in membrane structure may affect gap junction-mediated intercellular communication and thus serve as a mechanism by which dietary fat may promote carcinogenesis (Aylsworth, 1986).
These and other studies have led to an interest in diglyceride and protein kinase C as potential mediators of the effects of dietary fat on carcinogenesis. Protein kinase C is activated by diglyceride and also by phorbol ester—a known tumor promoter. Activation of protein kinase C has in turn been associated with cellular proliferation. Since unsaturated diglycerides activate protein kinase C more effectively than saturated diglycerides, it is possible that these compounds could mediate effects of dietary fat on carcinogenesis (Welsch, 1986).
Most proposed mechanisms involve the possible effects of dietary fat on cellular proliferation. Evidence presented by Hillyard and Abraham (1979) suggests, however, that dietary PUFAs may enhance tumor growth by prolonging the life of tumor cells and thus altering the balance between accretion and loss of cells by the tumor.
In epidemiologic studies, total dietary fat shows the strongest correlation with breast cancer incidence and mortality, whereas dietary PUFAs show little or no correlation (Carroll et al., 1986). This has been explained on the basis that human diets normally contain enough PUFAs to satisfy the threshold requirement for tumor promotion observed in animal studies. The positive correlations observed in epidemiologic studies are consistent with the promotional effect of dietary fat observed in animal models (Carroll, 1986a). Therefore, mechanisms by which high-fat diets per se may promote carcinogenesis should be considered.
There is a strong positive correlation between caloric intake and level of dietary fat (Carroll and Khor, 1975), and it is well known that carcinogenesis can be inhibited in animals by caloric restriction (Beth et al., 1987; Cohen et al., 1988; Klurfeld et al., 1987; Kritchevsky, 1986; Reddy et al., 1987; Sylvester, 1986; Tannenbaum, 1959). It is therefore possible that promotion of tumorigenesis by high-fat diets is due to excess caloric intake, but this has not been demonstrated experimentally. The metabolism of animals on high-fat diets is substantially different from that of animals on low-fat diets (Clarke, 1986); some of these differences may favor carcinogenesis.
The mechanisms by which dietary fat affects mammary carcinogenesis are not necessarily the same as those by which it affects other sites. In colon cancer, excretion of secondary bile acids has been positively correlated with the promotion of colon cancer by different types of dietary fat (Reddy, 1986a,b). No specific mechanisms have yet been proposed for effects of dietary fat on pancreatic cancer (Birt and Roebuck, 1986).
Summary
Together, the epidemologic and animal data generally support an association between dietary fat and risk of several cancers. The data are most consistent for colorectal cancer and less so for prostate and breast cancer. Overall, the association with total fat is the strongest and most consistent. However, some evidence obtained in studies on humans suggests that high SFA intake is associated with an increased risk of and mortality from cancers of the colon, prostate, and breast. In animals, w-6 PUFAs increase risk to the greatest extent, but high SFA intake also increases risk, provided that the minimum requirement for w-6 PUFAs is satisfied. There is some evidence from animal experiments that diets with a high content of w-3 PUFAs may decrease risk of cancers of the mammary gland, intestine, and pancreas. In some studies, MUFAs have been shown either to have no effect on cancer risk or to be protective. Dietary cholesterol is highly correlated with SFA intake in humans. There is no consistent evidence that low levels of serum cholesterol increase the risk of cancer in any site, including the colon and rectum.
Page 216
Dietary Phospholipids
Phospholipids are major constituents of biologic membranes and as such are normal constituents of the diet. Phosphatidylcholine (PtdCho), also called lecithin, makes up approximately 40 to 70% of mammalian membrane phospholipids. It is also added to foods as an emulsifying agent. Lipid emulsions used for parenteral nutrition contain about 1% PtdCho. In the United States, the intake of PtdCho from natural sources is 3 to 6 g per day (Wurtman, 1979; Zeisel et al., 1980). Some people also take nutritional supplements containing PtdCho.
Two phospholipids, PtdCho and phosphatidylserine, are undergoing testing for their use as drugs for treatment or prophylaxis for various diseases. This section focuses mainly on PtdCho and on the nutritional aspects of dietary phospholipids.
Choline as an Essential Nutrient
Phospholipids and their constituents can be synthesized in the body and are thus not normally required in the diet. Choline, a constituent of PtdCho, may, however, be an essential dietary ingredient under certain circumstances (Zeisel, 1981).
Chronic ingestion of a diet deficient in choline has been associated with memory impairment and with hepatic, renal, and growth disorders. In the rat, hamster, pig, dog, chicken, and baboon, choline deficiency resulted in fatty infiltration of the liver (Zeisel, 1981), probably by disturbing the synthesis of PtdCho needed to secrete triglycerides as part of the lipoproteins. Renal function was also compromised. Infertility, growth impairment, bony abnormalities, decreased hematopoiesis, and hypertension have also been associated with consumption of diets low in choline (Zeisel, 1981). Deficiency of choline and methionine produces a high incidence of liver tumors in rats in the absence of any known carcinogen (Ghoshal and Farber, 1984; Mikol et al., 1983; Newberne and Rogers, 1986). A relatively short exposure to severe methyl group deficiency also enhanced development of carcinogen-induced liver tumors (Hoover et al., 1984). In mice, choline deficiency was associated with memory impairment (Bartus et al., 1980).
Decreased serum choline concentrations have been observed in humans fed by total parenteral nutrition with commercial solutions low in choline. Liver disease associated with parenteral nutrition may be related to choline deficiency (Sheard et al., 1986). Marked depressions of plasma choline levels were noted among trained athletes running the Boston Marathon, and this might have affected performance (Conlay et al., 1986).
Despite this evidence of pathology associated with choline-deficient diets, it is not possible to state definitely that humans require dietary choline, because there is uncertainty about the extent to which methionine and other labile methyl donors can be substituted for choline. In rats, biosynthesis of PtdCho by methylation is decreased by dietary deficiencies of methionine and vitamin B12 (Akesson et al., 1978; Hoffman et al., 1980).
PtdCho and Other Choline-Containing Compounds in the Treatment of Human Diseases
Administration of choline or PtdCho can benefit patients with tardive dyskinesia (Gelenberg et al., 1979; Zeisel, 1981). This disorder is a side effect of virtually all antipsychotic drugs marketed in the United States and presumably results from an imbalance between dopaminergic and cholinergic neurotransmission.
Cholinergic neurons are essential for memory and other cognitive functions in rats and humans. An age-related diminution of memory in mice was exacerbated by a diet low in choline and was alleviated by a choline-rich diet (Bartus et al., 1980). Administration of choline reportedly improved memory in serial-learning and selective-reminding tasks among humans who were poor initial learners (Sitaram et al., 1978; Zeisel, 1981).
Supplemental choline (usually as PtdCho) has also been tested in the prophylaxis or treatment of memory disorders associated with Alzheimer's disease in which central cholinergic neurotransmission is known to be deficient (Zeisel, 1981). Preliminary nonblinded studies in Alzheimer's patients yielded positive findings, but most well-controlled, double-blind studies have failed to show clear-cut improvement (Corkin et al., 1982). In a single long-term, double-blind study, approximately 20 g of highly purified PtdCho or placebo given daily for 6 months led to a significant improvement in the oldest third of 50 patients (Little et al., 1985).
Supplemental choline has also been used to treat mania, which may reflect excessive activation of adrenergic receptors in the brain. When PtdCho
Page 217
was given to manic patients with lithium, the improvement in behavior was marked but temporary (Cohen et al., 1980).
PtdCho produced mild improvement in a few patients with Friedreich's ataxia, but no improvement in patients with another ataxic disorder, spinocerebellar degeneration (Barbeau, 1978). Administration of choline only occasionally resulted in reproducible improvement in other ataxic disorders (Lawrence et al., 1980). Some improvement was observed in one patient with the myasthenic syndrome (Kranz et al., 1980). Supplemental choline does not appear to be beneficial in myasthenia gravis, but this difference has not been explored extensively.
Choline increases release of acetylcholine by the heart in vitro (Loffelholz et al., 1979) but has not been found to affect cardiac function in healthy humans. Rats exposed to transient periods of choline deficiency develop irreversible hypertension (Kratzing and Perry, 1971; Michael et al., 1975). This may be related to renal as well as cardiovascular function.
In a study on rabbits fed an atherogenic diet, soya lecithin (1 or 5 g daily) was found to decrease serum cholesterol and atherosclerosis (Kesten and Silbowitz, 1942). Other investigators reported reductions in serum cholesterol in hypercholesterolemic patients treated with soya lecithin (Morrison, 1958; Steiner and Domanski, 1944), but subsequent studies had less encouraging results (Cobb et al., 1980; Davies and Murdoch, 1959; ter Welle et al., 1974). Childs et al. (1981) observed small but significant reductions in LDL-C and an increase in HDL-C in normolipidemic and familial hypercholesterolemic subjects given 36 g of an oral soya lecithin daily. Since soy lecithin preparations used for such studies generally contained less than 30% PtdCho, observed effects could have been due to other components.
High-PUFA PtdCho has been given in some studies. Svanberg et al. (1974) reported a decrease in serum triglycerides and VLDL-C and an increase in HDL-C in hypertriglyceridemic subjects treated with lecithin high in linoleate. Greten et al. (1980) observed no change in serum lipid or lipoprotein concentrations in patients with familial hypercholesterolemia or normal controls treated with lecithin with high linoleate content, but fecal stool excretion was increased. PtdCho has been found to interfere with intestinal absorption of cholesterol both in humans and in laboratory animals (Beil and Grudy, 1980; Rampone and Machida, 1981). In some of these studies, care was taken to distinguish between effects of PtdCho and its constituent PUFAs. Disaturated PtdChos adsorb to surfaces more readily than those with unsaturated fatty acids. Elevated levels of disaturated PtdChos have been observed in some patients with atherosclerotic heart disease and may constitute a risk factor (Gershfeld, 1979).
Inositol and Phosphoinositides
Although phosphoinositides are minor components of the lipids of cellular membranes, they have recently attracted much attention as precursors of second messenger molecules (Majerus et al., 1986). In this capacity, they can exert profound effects on cellular function and metabolism.
Like most other phospholipids, phosphoinositides and their component parts can normally be synthesized in adequate amounts in the body. There are reports, however, of deficiency symptoms associated with inositol-deficient diets in animals (Holub, 1982, 1986). Liver triglycerides accumulate in inositol-deficient animals, possibly because of inadequate release of lipoproteins (Hoover et al., 1978) or increased mobilization of fatty acids from adipose depots to the liver due to activation of hormone-sensitive lipase in adipose tissue (Hayashi et al., 1978). Further research on the role of inositol in cardiovascular disease may therefore be warranted.
Gallbladder Disease
Pathophysiology of Gallbladder Disease
Gallstones are the major cause of gallbladder disease in the United States, affecting approximately 10% of the adult population. Cholecystectomy for gallstones is one of the most common surgical procedures. Gallstones are composed primarily of cholesterol and bilirubin. The pathogenesis of cholesterol gallstones has been reviewed by Admirand and Small (1968), Bennion and Grundy (1978a,b), Carey and Small (1978), Grundy (1979), Schoenfield (1980), and Small (1976).
Cholesterol, bile acids, and phospholipids are secreted by the liver in varying proportions. Cholesterol is solubilized in bile by the detergent properties of the bile salts and phospholipids. When cholesterol exceeds a certain level relative to the concentrations of other lipids, the bile
Page 218
becomes supersaturated, increasing the probability that cholesterol crystals will precipitate and begin to form stones. Thus, supersaturated bile predisposes to cholesterol gallstones, and conditions that increase the proportion of cholesterol in bile relative to that of bile salts are considered to be possible etiologic factors. Gallstones are frequently asymptomatic, but they predispose to cholecystitis and obstruction of the biliary duct—complications requiring surgical intervention.
Epidemiology of Gallstones
As with most chronic diseases, the wide geographic and ethnic variations in the frequency of cholesterol and pigment gallstones (Brett and Barker, 1976; reviewed by Heaton, 1973; Yamase and McNamara, 1972) suggest that both environmental and genetic factors are involved in their causation. Cholesterol gallstones are much more common among American Indians (Comess et al., 1967; Sampliner et al., 1970) and Hispanics (Arevalo et al., 1987; Weiss et al., 1984) than among black or white Americans. They are the predominant type of gallstones in North and South America and in Europe, but are rare in the Orient (Sutor and Wooley, 1971; Trotman and Soloway, 1975). Pigment gallstones are most frequent in the Orient, but cholesterol stones are becoming more frequent in Japan (Nagase et al., 1978).
Within the U.S. white population, the major conditions associated with a greater probability of developing cholesterol gallstones are obesity, female sex, number of pregnancies, and age (Friedman et al., 1966). Despite widespread suspicion in the medical community and the general population that diet contributes to cholesterol gallstone formation, no convincing epidemiologic evidence except for that pertaining to obesity supports this concept (see review by Low-Beer, 1985).
Clinical and Metabolic Studies
The major body of evidence linking dietary fats and sterols to cholesterol gallstones and thereby to chronic gallbladder disease derives from many observational and experimental studies of humans indicating that dietary cholesterol and PUFAs may increase the cholesterol saturation of bile, thereby increasing the probability of stone formation.
Case-control studies have shown no association between cholelithiasis and consumption of eggs (Sarles et al., 1969) or cholesterol (Scragg et al., 1984). In a cohort study, cholesterol intake did not predict the occurrence of cholelithiasis (Friedman et al., 1966). The lithogenicity of bile (bile saturation index) was increased modestly by large intakes of cholesterol as egg yolk in one experiment (DenBesten et al., 1973) and was increased substantially in another experiment (D.W. Lee et al., 1985), especially in patients who already had gallstones. However, dietary cholesterol did not increase the bile saturation index in three other experiments (Andersen and Hellstrom, 1979; Dam et al., 1971; Sarles et al., 1970). Thus, despite the obvious logic relating cholesterol intake to cholesterol gallstones, little evidence from studies in humans supports the relationship. The variability in results from different experiments, each involving small numbers of subjects, suggests that a few persons may be sensitive to the lithogenic effects of dietary cholesterol but that most persons are not.
A number of experiments in humans between 1958 and 1967 showed no effect of dietary PUFAs on the lithogenicity of bile (Dam et al., 1967; Hellstrom and Lindstedt, 1966; Lewis, 1958; Lindstedt et al., 1965; Sodhi et al., 1967; Watanabe et al., 1962). In 1973, attention was redirected to this issue by an observation from a dietary trial to prevent coronary heart disease. The prevalence of cholelithiasis (as detected at autopsy) was about twice as high in men who had consumed a low-cholesterol, high-PUFA diet as in men who had consumed a high-cholesterol, high-SFA diet (Sturdevant et al., 1973). However, no difference related to PUFAs was observed in another, similar dietary trial (Miettinen et al., 1976).
Grundy (1975) identified 4 of 11 patients with hypertriglyceridemia in whom bile cholesterol saturation was increased by PUFA feeding. However, this difference could not be distinguished from random or individual variability, and it could not be extrapolated to the general population because the subjects were highly selected. More recently, Kohlmeier et al. (1985) found extreme variability in the response of bile cholesterol saturation to a high-PUFA, low-cholesterol diet fed to 12 women, 2 of whom developed much higher cholesterol saturation indices, while the rest experienced no significant change.
In summary, the evidence suggests that a few people may be susceptible to the lithogenic effects of PUFAs when the cholesterol saturation index of bile is increased, but that most humans do not
Page 219
seem to be susceptible. It would be important to identify such subjects if they exist.
Numerous epidemiologic studies have shown that gallstones are more frequent among obese persons than among persons of appropriate weight for height (for example, Friedman et al., 1966; Scragg et al., 1984). Obese people have higher bile saturation indices than do nonobese persons, and the condition is reversible (Bennion and Grundy, 1975). This is caused by increased hepatic secretion of cholesterol into the bile (Leijd, 1980) (see also Chapter 21, Obesity and Digestive Diseases). A high-calorie diet also increases the bile cholesterol saturation index (Sarles et al., 1971; Scragg et al., 1984).
Obesity is at least one factor involved in the predisposition of Pima Indians to gallstones (Howard, 1986). Thus, it is well established that obesity possibly resulting from high caloric intake is associated with increased risk of cholelithiasis through greater secretion of cholesterol by the liver and higher bile cholesterol saturation.
Recent research indicates that the lithogenicity of bile may be much more complex than the interrelationships among cholesterol, bile salts, and phospholipids (reviewed by Somjen and Gilat, 1986). Vesicles composed mainly of phospholipids and cholesterol have been observed in the bile of humans, and some proteins may be involved in the processes that influence stone formation. Thus, the cholesterol saturation index, which does not take into account other carriers and other mechanisms stabilizing cholesterol in bile, may be limited in its ability to detect the conditions that favor the formation of gallstones. There is no information concerning the effects of dietary cholesterol, fat, or other nutrients on these newly identified properties of bile that are likely to influence gallstone formation. Such information should lead to new insights into the genetic-environmental interactions involved in cholelithiasis.
Animal Studies
Scattered reports between 1912 and 1950 describe gallstones induced in rabbits by feeding them excess cholesterol, but the results were inconsistent and many cholesterol-fed rabbits did not develop gallstones (reviewed by Freston and Bouchier, 1968). Interest in animal models was stimulated by the discovery of diet-induced gallstones in hamsters in 1952. Subsequently, cholesterol gallstones were observed in mice, prairie dogs, and several species of monkeys. These animal models appear to involve different mechanisms. Dam (1971), Freston and Bouchier (1968), Gurll and DenBesten (1978), and Holzbach (1984) reviewed animal models of gallstones.
While examining the relationship of nutrition to muscular dystrophy in hamsters, Dam and Christensen (1952) found that a high-carbohydrate, low-fat diet produced gallstones. These investigators carried out a number of experiments (reviewed by Dam, 1971) concerned with the etiology and pathogenesis of this phenomenon. It was eventually determined that the nutritional factor was an essential fatty acid deficiency that did not alter the secretion of bile salts but did increase biliary cholesterol secretion several-fold (Robins and Fasulo, 1973). The enhanced biliary cholesterol secretion was not accompanied by increased tissue cholesterol levels. The addition of cholesterol to the diet yielded varying results, but most investigators have found that increased lithogenicity of bile was associated with dietary cholesterol, e.g., as reported by Singhal et al. (1983). The hamster, which appears to be genetically more susceptible than other rodents to the lithogenic effects of dietary cholesterol (Ho, 1976), continues to be the subject of studies to investigate the pathogenesis and prevention of lithogenic bile and gallstones, but this animal model is of little help in identifying nutritional factors contributing to gallstones in humans.
A high-cholesterol (1.2% by weight, derived from egg yolk), high-fat (41% of calories from egg yolk) diet for 6 months invariably produced cholesterol gallstones in prairie dogs (Cynomys ludovicianus) (Brenneman et al., 1972). The bile became lithogenic primarily through the increased secretion of cholesterol. The mechanism was interpreted as a massive overloading of the hepatic system for metabolism of cholesterol, apparently because the prairie dog absorbed cholesterol efficiently from the intestine. The overload occurred quite rapidly after beginning the diet, since bile cholesterol was increased within 36 hours, cholesterol crystals were found in the gallbladder within 5 days, and most animals had gallstones within 14 days (DenBesten et al., 1974). A semisynthetic
Page 220
diet with cholesterol was even more effective in inducing gallstones (B.I. Cohen et al., 1986). Susceptibility was not due to lack of responsiveness of 7a-hydroxylase in the liver (Howell et al., 1983). Thus, this animal model also may be useful in studying the pathogenesis of gallstones.
MacPherson et al. (1987) produced cholesterol gallstones rapidly in ground squirrels (Spermphilus richardsonii) by giving them a cholesterol-enriched (2%) rat chow diet. The features of this model appear to be similar to those of the cholesterol-fed prairie dog.
Tepperman et al. (1964) produced cholesterol gallstones in mice by feeding them a diet containing 1% cholesterol, 0.5% cholic acid, and 31% lard. There has been no subsequent exploration of this phenomenon in inbred strains of mice.
Osuga and Portman (1971, 1972) and Osuga et al. (1974) produced lithogenic bile and cholesterol gallstones in squirrel monkeys (Saimiri sciureus) with a diet containing 0.9 mg of cholesterol per kcal and 45% of total calories from butter. They concluded that dietary cholesterol was the major factor in the production of these effects (Osuga et al., 1976). Armstrong et al. (1982) found that dietary cholesterol increased the cholesterol bile saturation index of squirrel monkeys, whereas dietary fat saturation (corn vs. coconut oil) had no independent effect. The effect of cholesterol was greater when it was fed with PUFAs (Melchior et al., 1978; Tanaka et al., 1976). African green monkeys (Cercopithecus aethiops) also developed a more lithogenic bile when fed cholesterol with PUFAs (St. Clair et al., 1980). In contrast, the bile of Cebus monkeys, which do not develop gallstones from dietary cholesterol, was much less influenced by diet.
Baboons (Papio sp.) occasionally develop cholesterol gallstones spontaneously (Glenn and McSherry, 1970), and their bile is frequently supersaturated with cholesterol (McSherry et al., 1971a). The bile of these animals has a cyclic variation that reflects the enterohepatic circulation of bile salts (McSherry et al., 1971b).
The animal models of cholelithiasis present a mixed picture. Several species of rodents (but not the rat) are susceptible to the lithogenic effects of dietary cholesterol, but they require large doses of cholesterol and are accompanied by high levels of serum cholesterol. The common denominator of these rodent models is enhanced secretion of cholesterol into the bile, as in humans with cholelithiasis, but the underlying genetically determined metabolic characteristic that makes them susceptible to dietary cholesterol is not known. Among nonhuman primates, the squirrel monkey is similar to humans in its sensitivity to dietary cholesterol. The baboon appears to simulate more closely the human pathophysiology. The extreme susceptibility of some species to dietary cholesterol does not seem to have any parallel in humans with cholelithiasis.
Summary
Cholesterol gallstones are the major cause of chronic gallbladder disease in the United States and are responsible for substantial morbidity (see Chapter 5). Excessive secretion of cholesterol into the bile relative to the secretion of bile salts and phospholipids is the common denominator in both human and test animals with cholelithiasis. The only nutritional factor clearly identified in humans is obesity possibly due to excessive caloric intake. The major nutritional factor identified from animal models is dietary cholesterol, although other conditions, such as essential fatty acid deficiency, also may produce gallstones. Remarkably few intensive epidemiologic studies have been undertaken to determine whether dietary fats and sterols are causative agents in cholelithiasis. And despite the evidence from studies in animals, no conclusive statements about their role in the development of cholelithiasis in humans can be made. It seems likely that, as with hyperlipidemia and atherosclerosis, lithogenic bile and cholelithiasis result from an interaction between dietary factors (such as dietary fats and sterols) and unidentified, genetically programmed metabolic characteristics. Research to identify both dietary and metabolic factors responsible for cholelithiasis, following the pattern of investigation used in hyperlipidemia and atherosclerosis, might lead to dietary recommendations to prevent cholelithiasis and chronic gallbladder disease in susceptible persons.
Dietary Fat and Obesity
Epidemiologic Studies
As noted in Chapter 6, epidemiologic studies indicate that average caloric intake has been de-
Page 221
creasing while mean body weight and body mass index have increased. There is no epidemiologic evidence indicating that total fat intake per se, independent of total caloric intake, is associated with increased adiposity in the overall population. Although some diseases commonly associated with obesity (e.g., cardiovascular disease and some cancers) are associated either with an increase in type of fat in the diet or with the percentage of fat in the diet (see earlier sections in this chapter), no clear association of either percentage or type of fat (independent of total caloric intake) has been associated with human obesity in epidemiologic studies. Thus, although the type or amount of dietary fat may be associated with diseases common in obese individuals, obesity itself has not been found to be associated with dietary fat in either between- or within-population studies.
Clinical Studies
Most knowledge of the effect of dietary fat on obesity in humans comes from studies in which diets differing in percent or type of fat have been fed to individuals for short periods. This was most frequently done through isocaloric substitution of fat for carbohydrate in isonitrogenous diets. In the typical study design, weight gain on a high-fat diet can be compared to gains on a low-fat or high-carbohydrate diet. Thus, the evidence does not permit an effective analysis of changes in weight or adiposity over a range of dietary fat intakes. In some studies, weight gain on a high-fat diet, or weight loss on a low-fat diet, have been been observed (e.g., Duncan et al., 1983). However, when dietary fat was replaced with sucrose polyester (Glueck et al., 1982) or carbohydrate (Porikos et al., 1977), the subjects compensated partially or completely for the calories deleted from the diet as fat.
In one study (Lissner et al., 1987), caloric compensation was studied in 24 women who consumed in sequence three 2-week diets with low (15-20%), medium (30-35%), and high (4045%) fat contents, similar in appearance and apparent palatability. Subjects on the low-fat diet spontaneously consumed 11.3% fewer calories, and those on the high-fat diet consumed 15.4% more calories than did those on the medium-fat diet. Thus these diets, differing in fat content and consumed spontaneously, resulted in significant short-term changes in body weight due to changes in caloric intake.
Some data suggest that obese people and formerly obese people prefer high-fat stimuli (Drewnowski et al., 1985) and may overconsume high-fat foods (Gates et al., 1975). A few studies suggest that a high SFA intake may be associated with obesity. For example, Berry et al. (1986) reported a positive association between body mass index and SFAs as a marker of animal fat intake. In addition, an analysis of the Nurses' Health Study (Romieu et al., 1988) suggests that obese women report higher intakes of both total fat and SFAs. Some people spontaneously reduce total caloric consumption when regularly given a low-fat diet; others spontaneously increase caloric intake when a high-fat diet is easily available.
Hence, the short-term clinical studies suggest that low-fat diets may be beneficial to obese people or those at risk for obesity. Nonetheless, it is difficult to implicate excess dietary fat ingestion per se (independent of total caloric intake) as a mechanism for the cause of obesity and the development of obesity. It is more likely that a combination of increased total caloric intake and decreased physical activity are the variables directly associated with the onset of human obesity.
Animal Studies
Studies in animals have provided evidence that both the type and the amount of dietary fat are significant in the production of obesity in rodents and other animal species. Several spontaneously obese rodent models show preferences for dietary fat at least under some experimental conditions (Maggio et al., 1983). When rats are allowed to spontaneously select macronutrient diets, they will consume approximately 30% of their calories as fat (Kanarek et al., 1981) and sometimes higher percentages during weight cycling (i.e., repeated episodes of weight loss and regain) (Reed, 1988). When medium-chain triglycerides are substituted isocalorically for long-chain triglycerides, both obese and lean rats gain less weight (Turkenkopf, 1982).
Some effects of high-fat intake have been attributed to the fact that the energy cost of converting dietary fat to stored triglycerides is lower than the cost of converting dietary carbohydrates to stored triglyceride (Donato and Hegsted, 1985). Furthermore, early exposure to a high-fat diet may predispose to future weight gain on high-fat diets (Peckham et al., 1962).
There is considerable genetic heterogeneity in animal populations, even among inbred strains of normally lean rats. Some rats, normally lean when fed standard chow, will gain body fat and develop hyperphagia when fed isonitrogenous diets con-
Page 222
raining increasingly higher percentages of fat (Schemmel et al., 1982).
Responses to high-fat diets are different between the sexes. Female rats spontaneously overingest high-fat diets compared to male rats and become fatter than males when fed isocaloric high-fat and high-carbohydrate diets (Hoyenga and Hoyenga, 1982; Sclafani and Gorman, 1977).
Summary
Animal data suggest that dietary fat, independent of total caloric intake, may contribute to obesity, possibly through more efficient metabolism of this nutrient relative to other nutrients. This effect is particularly prominent in female rats of some inbred strains (e.g., Wistar and Sprague Dawley rats) and certain genetic backgrounds (e.g., Osborne-Mendel). Unfortunately, there are no equivalent data on humans. Short-term studies in humans, primarily clinical studies, indicate that reduced fat content may be accompanied by weight loss. Reductions in caloric intake were noted in some of these reports and although not always specifically noted, may have occurred in others. This in itself is significant and may indicate that substantial reductions in the percentages of fat in the diet of humans may result in beneficial total caloric reductions as well, perhaps because diets with reduced dietary fat are generally regarded as less palatable. From a public health perspective, this phenomenon may be important, regardless of whether obesity is decreased by reduced percentages of dietary fat per se or is secondary only to concomitant reductions in caloric intake.
Effects of Dietary Fats and Other Lipids on Health and Disease in Children
Cardiovascular Diseases
The Origin of Atherosclerosis in Childhood
Atherosclerosis and its clinical sequelae are found primarily in adults. In rare cases (about 1 in a million), however, children with homozygous familial hypercholesterolemia develop severe atherosclerosis and clinical CHD. Holman—a pioneer in the exploration of atherosclerosis in childhood (Holman et al., 1958)—in 1959 presented a paper posing the question, ''Atherosclerosis—a pediatric nutrition problem?" at the Ninth International Congress of Pediatrics (Holman, 1961). At that time, the question could not be answered with certainty, because there was no evidence supporting the relationship in children between diet and subsequent atherosclerosis, in contrast to the considerable amount of data supporting such an association in adults. Abnormal lipid deposits (fatty streaks), believed by many to be the precursors of raised atherosclerotic lesions, were found in the aortas of all children by 3 years of age in all populations around the world, regardless of dietary fat and cholesterol intake (Strong et al., 1958; Tejada et al., 1968). Furthermore, these simple lipid deposits had not been firmly linked to the fibrous plaques that lead directly to arterial occlusion (see Chapter 19).
Long before there was any concern about hyperlipidemia or atherosclerosis in children, Enos et al. (1953) described advanced coronary artery lesions in young American adults in a landmark study of Korean battlefield casualties. In this study, 77% of men averaging 22 years of age had grossly detectable coronary artery lesions, and 15% had coronary artery lesions that obstructed the lumen by 50% or more. This remarkable prevalence of coronary atherosclerosis in young men was confirmed in other studies of young people who died from accidental causes in the United States (Strong and McGill, 1962).
Subsequent intensive examination of large numbers of children and young adults showed that a continuous progression of lesions could be traced from fatty streaks to fibrous plaques (Geer et al., 1968; McGill, 1984; Robertson et al., 1963; Stary, 1987a,b; Strong and McGill, 1969; reviewed by McGill et al., 1963, 1980). These observations and many more left little doubt that many fatty streaks in the coronary arteries and some fatty streaks in the aortas of children are converted to fibrous plaques in adolescents and young adults.
Effects of Dietary Fats and Cholesterol in Infants
Preweaning (suckling) infants are quite sensitive to the plasma cholesterol-raising (or lowering) effects of dietary fat and cholesterol. Approximately 50% of the calories in human breast milk is supplied by fat, half of which is SFA (Lo, 1985; reviewed by Jensen et al., 1978). Its cholesterol content averages 11 mg/dl (range, 3 to 29 mg/dl)
Page 223
(Picciano et al., 1978; reviewed by Jensen et al., 1978). In comparison to human milk, cow's milk has more SFA (65.6 versus 48.2% of total fatty acids), less MUFA (30.3 versus 39.8%), and much less PUFA (4.1 versus 12.0%) (Lo, 1985). In contrast, commercially prepared infant formulas are made with soy, corn, safflower, and coconut oils. Although these formulas provide the same proportion of calories as fat as does breast milk, they contain a higher proportion of PUFAs (up to 50% as linoleic acid) and virtually no cholesterol (Benson, 1981; Jensen et al., 1978). In several studies, as might be anticipated from their higher intake of cholesterol and SFAs, breast-fed infants had higher serum cholesterol concentrations than did formula-fed infants (Carlson et al., 1982; Pomeranze, 1961; reviewed by Wissler and McGill, 1983).
In a study of the nutrient composition of diets given to infants over the age of 6 months, Montaldo and coworkers (1985) compared solid food diets supplemented with whole milk from cows or with formula. Using data from NHANES II, they found that infants fed the cow's milk received less iron and linoleic acid. Manipulation of the fat and cholesterol content of infant formulas showed the anticipated effects (Sweeney et al., 1961). Similar observations have been made in animals, particularly nonhuman primates, whose breast milk is similar in composition to that of humans (Mott et al., 1978). The effect of plasma cholesterol concentrations in the suckling child on the subsequent development of atherosclerosis is totally unknown. It has generally been believed not to be significant because of the limited duration of exposure and the overriding importance of other nutritional considerations during the first year of life.
In 1972, Reiser proposed that a high cholesterol intake before weaning, such as that provided by breast milk, would induce a higher level of tolerance for dietary cholesterol in adulthood and presented results from studies in swine and rats to support this hypothesis (Reiser and Sidelman, 1972; Reiser et al., 1979). Tests of this hypothesis in other animal species, including nonhuman primates, have generally been negative (Mott et al., 1982; reviewed by Innis, 1985). However, Mott et al. (1982) found that breast-fed infant baboons had lower plasma HDL-C and apo AI concentrations when they reached adolescence than did formula-fed baboons, regardless of the cholesterol content of the formula. In another experiment, Lewis et al. (1988) also found that breast-fed baboon infants reared on an atherogenic diet had lower HDL-C levels and more extensive atherosclerosis at age 5 than baboons fed a formula as infants and reared on an identical diet. No similar effects have been detected in humans.
Plasma lipid and lipoprotein concentrations in infants and children under 2 years of age are quite sensitive to dietary fat and cholesterol, but there is no evidence that elevated plasma lipoproteins in this age group are factors in the etiology of atherosclerosis. Other nutritional considerations are more important during this period. Results from experiments in animals indicate that breast feeding, as compared to formula feeding, exerts a long-term deferred effect on lipoprotein metabolism that continues into adulthood, but no such effect has yet been detected in humans. Therefore, the remaining concerns regarding dietary fat and cholesterol during childhood are focused on children over the age of 2 years.
Relationship of Childhood Lesions to Plasma Lipids and Lipoproteins
It has been difficult to obtain information relating the arterial lesions observed in children and young adults to plasma lipid and lipoprotein levels. Many early observations seemed to indicate that there was no relationship when population means were compared (McGill, 1968; Tejada et al., 1968).
Fatty streaks were found to be ubiquitous among children throughout the world. Little or no excess was observed in populations such as that of the United States where TC levels were high. Indeed, aortic fatty streaks were equally prevalent in poorly nourished children in post-World War I Europe (Zinserling, 1925). Holman et al. (1959) observed that children dying from cystic fibrosis had difficulty in absorbing fat, had very low TC concentrations, and had one-fourth the fatty streaks as did the children without the disease.
An unusual opportunity to study the relationship of childhood lesions to plasma lipid or lipoprotein concentrations was provided by examinations of the arteries of young accidental death victims (average age, 18 years) whose plasma lipids and lipoproteins had been measured in the Bogalusa Heart Study (Berenson, 1980). In this study, both aortic and coronary fatty streaks were correlated strongly with antemortem LDL-C levels (Newman et al., 1986). The relationship contin-
Page 224
ued to be strong, even when the confounding effect of race was taken into account in a larger number of cases (Freedman et al., 1988). These findings from a small number of cases are the only observations that relate atherosclerotic lesions directly to plasma lipid or lipoprotein concentrations in children.
Plasma Lipids and Lipoproteins in Children
Until about 1970, descriptive studies of the distribution of serum cholesterol and lipoprotein cholesterol concentrations focused on adults. In the early 1970s, several research groups began to survey these variables in U.S. children (Berenson et al., 1979; Frerichs et al., 1976, 1978a,b; Friedman and Goldberg, 1973; Lauer et al., 1975; Morrison et al., 1979a,b; NHLBI, 1978; Wilmore and McNamara, 1974). Results of these studies indicate that TC concentrations at birth were approximately 70 mg/dl (half of it as HDL-C) and rose rapidly to approximately double that value at 1 year and increased slowly thereafter to about 160 mg/dl (roughly two-thirds LDL-C and one-third HDL-C) at 10 years of age. Between 12 and 18 years of age, TC in boys declined slightly to about 150 mg/dl. The 10 mg/dl decrease was accounted for primarily by HDL-C and was associated with an increase in plasma testosterone levels (Kirkland et al., 1987). Serum TC concentrations and lipoprotein cholesterol distributions in girls remained at about the same level. At any age, the frequency distribution of TC levels was skewed to the high end similar to that of the adult population. In the 10- to 14-year age group, the fifth percentile levels were approximately 120 mg/dl, and the 95th percentile levels were about 200 mg/dl (Lipid Research Clinics Population Studies Data Book, 1980).
In the Bogalusa Heart Study, which periodically screens a pediatric population of approximately 5,000 children from a semirural community, Berenson et al. (1988) found that the schoolchildren's daily diet contained an average of 38% fat and 300 mg of cholesterol; the P:S ratio was 0.4. These data are similar to the findings of the National Health Survey (Carroll et al., 1983).
Emmons (1986) studied the nutritional adequacy of diets consumed by low-income people participating in the Special Supplemental Food Program for Women, Infants, and Children (WIC Program) and found that both white and black children consumed approximately 35% of their calories as fat. However, black children consumed more animal fat than vegetable fat and thus had a diet with a lower P:S ratio than that of white children. On average, therefore, it appears that children in the United States, especially those in lower socioeconomic groups, consume diets that tend to increase their LDL-C.
The H.J. Heinz Company of Canada conducted a longitudinal survey of the eating habits of infants and children (Leung et al., 1984). The food intake of more than 400 4-year-old children was obtained from data collected from a 4-day dietary record. This study documented that preschool Canadian children obtained 34% of their calories as fat. Their snacks, unlike typical processed food snacks in the United States, had the lowest caloric density of any of the meals, suggesting that they were composed mainly of carbohydrates. These Canadian data contrast strongly with the diet of U.S. children (Carroll et al., 1983).
The Lipid Research Clinics Program Prevalence Study presented the dietary data for white American males and females between the ages of 6 and 25 years. Enrolled within that study were more than 1,100 schoolchildren. The percentage of calories as fat was 39% for the boys and 38% for the girls. The P:S ratio was 0.4 for both sexes (Beaglehole et al., 1980).
Serum cholesterol levels in U.S. children were considerably higher than those in children of other regional groups or countries where CHD was much less frequent (Knuiman et al., 1980; Savage et al., 1976; Sullivan et al., 1987). The families of children with high TC levels had a greater incidence of deaths from CHD than did the families of children with low TC levels (Schrott et al., 1979). Families of children with low HDL-C levels also had an excess of deaths from CHD (Bodurtha et al., 1987). Conversely, approximately one-third of the children selected because their parents had precocious CHD had serum TC or triglyceride levels in the upper percentiles for their age (Chase et al., 1974; Glueck et al., 1974; Tamir et al., 1972).
Of importance in studies of childhood serum lipid levels is tracking, that is, whether children maintain similar levels (relative to one another) of serum total and lipoprotein cholesterol as they
Page 225
grow. Tracking is important in determining whether a child in a high percentile of serum cholesterol concentration would remain in the same high percentile several years later when he or she entered adulthood and therefore would be at high risk of CHD. Most studies have demonstrated that serum TC concentrations track to a moderate degree during childhood (Clarke et al., 1978; Frerichs et al., 1979; Laskarzewski et al., 1979; Webber et al., 1983, 1986) and from childhood into young adulthood (Orchard et al., 1983). For example, in the study by Orchard and colleagues, 49% of children with a TC level in the top quintile at 12 years of age were in the top quintile when 21 years old, and the correlation coefficient between the 12- and 21-year values was .52. Children who were obese had both higher TC and blood pressure and increased left ventricular mass (Frerichs et al., 1978a; Schieken et al., 1981).
Walter and Hofman (1987) studied the differences in the distribution of risk factors in children stratified by race and socioeconomic status in the "Know Your Body" program. They found that white children in the lower socioeconomic strata had the highest level of risk as defined by blood pressure, physical conditioning, and serum lipoprotein levels. These children also reportedly consumed the most atherogenic diets of the groups in the study. Thus, the evidence indicates that children with high TC values will be likely to become adults with high serum cholesterol values.
Effects of Diet on Serum Lipids and Lipoproteins in Children
Observational cross-sectional studies of diet and serum lipids in children have yielded results similar to those obtained from adults—correlations that are either zero or weak (Frank et al., 1978; McGandy, 1971; Morrison et al., 1980; Rasanen et al., 1978; Vartiainen et al., 1982). For example, Frank et al. (1978) studied 185 10-year-old children and found a positive but statistically insignificant association of TC concentration with total fat and SFA intake and no association with dietary cholesterol. Some statistically significant correlations emerged from other surveys, but they were low in magnitude (Frank et al., 1986).
Like studies in adults, early experiments on the effects of dietary cholesterol on serum TC levels in children were negative (Heymann and Rack, 1943). In later studies, positive results were obtained. In one study conducted in a boys' boarding school, meals were modified to reduce total fat intake from 38 to 33% of calories, SFAs from 16 to 10% of calories, and cholesterol from 720 mg/day to 380 mg/day; PUFA intake was increased from 3 to 13% of calories (Ford et al., 1972; McGandy et al., 1972). The average TC concentration of the 63 13- to 18-year-old boys fell from approximately 190 mg/dl, equivalent to that of other age-matched boys in similar schools (McGandy et al., 1972), to about 170 mg/dl—a reduction of 15%. The boys with the highest TC levels experienced the greatest decreases. Several other dietary interventions in children subsequently showed a similar responsiveness of TC concentrations to reductions in dietary SFAs and cholesterol (Nestel et al., 1979; Puska et al., 1985; Stein et al., 1982; Vartiainen et al., 1986). Thus, adolescent children are as responsive to dietary fat modification as are adults.
Safety of Fat-Modified Diets for Children
When it was first recommended in 1970 that the diets of children as well as those of adults be reduced in fat and cholesterol content, the question of safety was immediately raised. This concern stemmed from the natural conservatism of physicians combined with the great improvements in growth and health of children in the United States associated with the conventional diets they consumed. Many major childhood infectious diseases and conditions related to nutritional deficiency, e.g., rheumatic fever, tuberculosis, rickets, scurvy, and others, had virtually disappeared, and their disappearance was attributed in large part to improved nutrition. Therefore, physicians were reluctant to tamper with success.
Since cholesterol is an essential component of mammalian tissues, and since it is a precursor of steroid hormones, dietary cholesterol has been hypothesized to be necessary for optimum growth and development, particularly of the central nervous system. However, there is no experimental or observational evidence to support the concept that cholesterol is an essential nutrient.
The well-known capacity of mammalian cells to synthesize cholesterol suggests that dietary cholesterol is not required for normal growth. However, there have been no experiments testing the effects of cholesterol deprivation on children or on growing animals, probably because
Page 226
the hypothesis is considered to be very unlikely. As in other situations, proving the lack of a requirement is more difficult than proving that a requirement exists.
Evidence indicates that there is no need for a fat intake of more than 10% of calories, provided the diet contains an adequate amount of essential fatty acids. The requirement for essential fatty acids in infants is met if they ingest 3% of their total calories as linoleic acid. This requirement drops to between 1 and 2% of total calories as linoleic acid in adults (NRC, 1980)—an amount well below the usually recommended 30% of calories from fat or even the 20% of calories from fat sometimes recommended. Energy requirements can readily be met by an increased intake of carbohydrate.
Friedman and Goldberg (1976) found no differences associated with a low-fat, low-cholesterol diet for a number of developmental and biochemical variables of children. Dwyer (1980) discussed the preparation of diets for children that would fulfill the recommendations for lowering fat and cholesterol intake, but would also meet the Recommended Dietary Allowances for other nutrients. If care is not taken, she pointed out, deficiencies in the intakes of some vitamins and minerals might result. These and other aspects of safety have been reviewed by Kwiterovich (1980, 1986) and by Voller and Strong (1981). It is clear that caloric intake needs to be monitored to ensure its adequacy, if fat intake is decreased, in order to maintain optimal growth and development.
Recommendations of Other Expert Groups Regarding Dietary Modifications for Children
The American Heart Association recommended in 1983 that all children over 2 years of age should reduce total fat intake to no more than 30% of calories, 10% or less being provided by SFAs and not more than 10% by PUFAs, and should reduce cholesterol intake to 100 mg/1,000 kcal (Weidman et al., 1983). That report did not discuss safety concerns, which had been the basis for earlier reluctance to extend to children recommendations made for adults. About the same time, the Committee on Nutrition of the American Academy of Pediatrics stated, "Current dietary trends in the United States toward a reduced consumption of saturated fats, cholesterol, and salt and an increased intake of polyunsaturated fats should be followed with caution. Diets that avoid extremes are safe for children" (AAP, 1983). However, the committee did not recommend specific targets.
The recommendations of these two committees are rather similar. Dietary fat and cholesterol intakes of children have declined along with those of adults, and continued reductions would bring actual intakes very close to those recommended by the American Heart Association. Thus, the only real difference between the two recommendations is the absence of quantitative targets in the statement issued by the American Academy of Pediatrics.
The most recent recommendation regarding fat-modified diets for children was made by the National Institutes of Health Consensus Conference on Cholesterol (NIH, 1985). This recommendation corresponds closely to that of the American Heart Association for children over 2 years of age. Subsequent published comments have both supported and opposed these recommendations. In a detailed and comprehensive review, Kwiterovich (1986) concluded that the recommendation by the American Heart Association was safe and beneficial. Barness (1986) approved the recommendations to limit total fat to 30% of calories, but believed that PUFA intake should be less than 10% of calories and saw no reason to limit cholesterol intake.
Conclusions and Recommendations of the Committee on Diet and Health
After reviewing the evidence, the Committee on Diet and Health concluded that recommendations concerning dietary fats and other lipids for children over 2 years of age should be generally similar to those for adults. In general, children over 2 years of age should eat a diet that includes a total fat intake of no more than 30% of calories, with less than 10% of calories from SFAs, and PUFAs at approximately current levels—6 to 8% of calories and not to exceed 10% of calories in any individual and a cholesterol intake of 100 mg or less per 1,000 calories—not to exceed 300 mg per day. A diet supplying macronutrients from the major food groups, with an increase in fruits and vegetables rather than refined sugar, should provide ample amounts of essential fatty acids, protein for growth, and an abundance of trace minerals and vitamins.
Between the ages of 2 and 4, children are often fed diets rather different from those consumed by older members of their family. Thus, it would not be
Page 227
unreasonable to gradually phase in the recommended diet by slowly limiting the intake of fat and cholesterol, so that by age 5 the child is eating the recommended diet (similar to the rest of the family).
Diets similar to this recommended diet are consumed by children in Mediterranean countries where the prevalence of CHD is much lower than in the United States. No abnormalities of growth and development have been observed in these children. The evidence indicates that the recommended diet is both safe and hygienically advisable.
Overall Summary
A substantial body of evidence supports the conclusion that there is a direct relationship between dietary fat and the risk of both CHD and certain kinds of cancer, and possibly also obesity. The evidence is extremely powerful for CHD and is somewhat more limited for cancer. Fortunately, the major public health conclusions and recommendations that flow from the evidence, namely, that Americans should eat less total fat, especially less SFAs, are congruent with respect to both CHD and cancer, and possibly obesity.
Cardiovascular Disease
Many lines of evidence—from epidemiologic, clinical, genetic, and laboratory animal studies—indicate that high plasma levels of total and LDL cholesterol are causally related to atherosclerosis and increased risk of CHD. The epidemiologic evidence includes comparisons among various populations and prospective studies of individuals within populations. The predictive association between serum TC levels and the future occurrence of CHD is a continuous one throughout the range of values seen in the U.S. population and meets all the criteria for an etiological relationship. Clinically, premature CHD can result from high LDL-C levels, even in the absence of any other contributing risk factors. This is most clearly demonstrated in patients with familial hypercholesterolemia.
Many animal species (including monkeys and baboons) develop atherosclerosis when fed diets that raise their TC levels. Severe atherosclerotic lesions in monkeys regress when the TC is lowered substantially for an extended period by diet or by drugs.
Clinical trials have shown that lowering TC and LDL-C levels by diet or drugs decreases the subsequent incidence of CHD. The direct evidence from such trials is strongest in middle-aged men with highest initial cholesterol levels. However, the complete set of evidence, including that from epidemiologic studies and experiments in animals, strongly suggests that reducing TC and LDL-C levels is also likely to decrease CHD incidence in younger and older men, in women, and in individuals with more moderate levels of TC. Moreover, the epidemiologic studies and clinical trials are remarkably consistent in their quantitative conclusions. The data suggest that for people with TC levels in the upper range, each 1% of reduction in serum TC levels leads to an approximately 2% reduction in CHD rates after 5 to 7 years and an even greater reduction in CHD rates after decades.
Consumption of specific fatty acids strongly influences plasma levels of TC and LDL-C in individuals and in populations. Many kinds of studies of relationships between diet, serum cholesterol, and CHD, including clinical-metabolic, epidemiologic, and animal studies, have implicated both the amount and nature of dietary fats as important determinants of serum cholesterol levels and CHD risk.
Clinical and controlled metabolic ward studies have clearly shown that increased intakes of some SFAs (palmitic, myristic, and lauric acids) raises TC and LDL-C levels in humans and, conversely, that reduced intake of these SFAs has the opposite effect. Although there is much interindividual variability in quantitative response to changes in SFA intake, the average response in the population can be quantitatively described by formulas developed in the laboratories of Keys and of Hegsted.
There are two kinds of PUFAs: w-6 and w-3. In general, w-6 PUFAs lower TC, including reductions in both LDL-C and HDL-C levels. Substitution of linoleic acid for dietary SFAs results in a significant drop in TC levels. Although high intakes of linoleic acid were once advocated as a means of lowering TC, lack of information about the consequences of long-term ingestion of large amounts of linoleic acid, including concern about effects on cancer risk (see below), has led most public health authorities to recommend a ceiling of 10% or less of total calories from such fatty acids.
The major sources of w-3 PUFAs are fish oils. Whether w-3 PUFAs are useful in the prevention of CHD has not been determined. They have been found to lower serum triglyceride levels when given in relatively high doses; however, there is little evidence that they lead to reductions in concentrations of LDL-C beyond those obtained by removal of SFAs from the diet. It is not known
Page 228
whether long-term ingestion of these oils will lead to undesirable side effects, but there is no evidence that such side effects occur when intake of fish oils is through ingestion of fresh fish.
MUFAs have long been considered to be neutral in their effects on serum TC levels. However, recent evidence indicates that replacing SFAs with oleic acid may lead to a significant decrease in LDL-C levels without the concomitant decrease in HDL-C observed when linoleic acid is substituted for SFAs in the diet.
A variety of epidemiologic studies have focused on the relationships between dietary fat (both the amount and kind), serum cholesterol levels, and CHD. Several types of international and population studies have explored population (group) differences in diet, TC distributions in the population, and CHD rates. These studies have included comparisons of populations in different countries, of migrant groups from a single origin, and of special population groups within a country. These studies have generally demonstrated that mean intake of SFAs is strongly correlated with mean TC and LDL-C levels and with CHD rates in populations.
In contrast to the strong correlations observed between dietary fat and serum TC between populations, only weak (albeit significant) associations have been found between diet and TC in cross-sectional studies of individuals within a given population. There are methodological difficulties, however, suggesting that this might be a false-negative result. Thus, a number of factors identified would make it difficult to demonstrate such relationships in individuals within a fairly homogeneous population.
Studies in animals have shown that SFAs increase TC levels in a variety of species. In most species, SFA intake also is associated with severe atherosclerosis. These observations further support the conclusion that SFAs are important contributors to high TC levels and to atherosclerosis in humans.
Many studies have explored relationships between dietary cholesterol intake, serum cholesterol, and CHD. The evidence includes results from epidemiologic, clinical-metabolic, and experimental animal studies. Epidemiologic studies have shown high correlations between populations (both within and between countries) with respect to average dietary cholesterol intake, average TC levels, and CHD rates. In contrast to these high ecological correlations, however, individual correlations of dietary cholesterol with TC levels and CHD rates within a population have generally been found to be weak. These two methods of assessing correlations measure different kinds of correlations, and several factors could weaken the ability to demonstrate individual correlations within a given group. Moreover, since 1980, a small but definite power of dietary cholesterol to predict subsequent CHD within given populations has emerged from several longitudinal epidemiologic studies.
Dietary cholesterol causes hypercholesterolemia and atherosclerosis in a variety of animal species, including nonhuman primates. In clinical and controlled metabolic ward studies in humans, variable effects of dietary cholesterol (from large to negligibly small) have been reported. In general, however, within the range of intakes ordinarily consumed (200 to 600 mg/day, or approximately 150 to 400 mg/1,000 kcal), dietary cholesterol influences serum TC concentration, primarily LDL-C. On the average, in a large group of people, 100 mg of dietary cholesterol per 1,000 kcal elevates LDL-C by approximately 8 mg/dl. There is, however, much individual variability in the response to dietary cholesterol. Some people are resistant to the TC-elevating effect of dietary cholesterol, and others are quite sensitive. For the one-third or more of the population in the upper range of the distribution of response, dietary cholesterol is a meaningful contributor to TC and LDL-C concentrations and thereby to CHD risk. Dietary cholesterol may also have an effect on atherogenesis independent of its effect on fasting plasma lipid concentrations. Thus, for the overall population, it is clear that dietary cholesterol definitely contributes to the development of atherosclerosis and risk of CHD, although there is extensive interindividual variability in response.
Cancer
The National Research Council's Committee on Diet, Nutrition, and Cancer (NRC, 1982) concluded "that of all the dietary components [it] studied, the combined epidemiological and experimental evidence is most suggestive for a causal relationship between fat intake and the occurrence of cancer." This conclusion was based largely on the consistency between the epidemiologic and experimental evidence.
Epidemiologic and laboratory animal evidence obtained since 1981 bears on the possible relationship between dietary fat and the risk of certain kinds of cancer. The epidemiologic evidence in-
Page 229
cludes comparisons of populations in different geographical areas as well as case-control studies of cancer of the colon and rectum, prostate, ovary, and breast. In general, these studies support conclusions from the earlier studies, namely, that there are significant associations between dietary fat and risk of these cancers; however, such associations have not been found in all studies.
In the epidemiologic studies, cancer incidence and mortality have generally had a stronger positive correlation with total dietary fat than with any particular type of dietary fat. Some studies of breast and prostate cancer have shown especially strong associations of cancer risk with the intake of SFAs and with dietary fat from animal sources, much of which is saturated. Overall, however, cancer mortality in human populations seems to correlate with total dietary fat about as well as with animal fat, and there is little or no correlation with vegetable fat or PUFAs.
Recent studies in animals have generally confirmed earlier evidence that animals fed high-fat diets develop tumors of the mammary gland, intestinal tract, and pancreas more readily than those fed low-fat diets. Dietary fat has its greatest effect during the promotion phase of carcinogenesis, but a lesser effect on initiation has been observed in some studies. The effect of dietary fat differs according to the type of fat. Vegetable oils containing w-6 PUFAs promote carcinogenesis in animals more effectively than SFAs. Fish oils containing w-3 PUFAs tend to have an inhibitory effect on carcinogenesis in animals. These findings are not necessarily in conflict with the epidemiologic data on humans that show the greatest effects from total and SFA intake, since the animals are usually fed large amounts of specific fats and oils, representing extremes of intake not found in the diets of human.
Epidemiologic studies have focused on potential relationships between low levels of serum cholesterol and cancer risk. In some studies of cohorts assembled to study cardiovascular disease risk, low serum cholesterol levels have been associated with increased cancer incidence and mortality. Such associations have not been found in some other studies and have been found inconsistently in subgroups within a given study population. The site-specific cancer data that are available are not consistent. In general, studies designed specifically to assess the relationship do not confirm the association. Thus, the complete set of evidence does not support the existence of a causally linked association between low levels of serum cholesterol and cancer risk.
Obesity and Gallbladder Disease
High-fat intake is associated with the development of obesity in animals and possibly in humans. In short-term clinical studies, a marked reduction in the percentage of calories derived from dietary fat has been associated with weight loss.
Obesity is the only nutritional factor clearly identified with cholesterol gallstones (gallbladder disease) in humans. In several animal models, increased dietary cholesterol causes cholelithiasis. However, few epidemiologic studies have been conducted to examine relationships between dietary fats and sterols and cholelithiasis. No conclusive evidence exists about the possible roles of dietary fat and cholesterol in the development of cholelithiasis.
Conclusions
The evidence concerning cardiovascular disease, summarized above, clearly indicates that dietary changes to lower the plasma levels of TC and LDL-C in North America would be desirable from a public health perspective. Two approaches can be used to accomplish this. First is a population (public health) strategy to shift the distribution of cholesterol levels in the entire population to a lower range. Second is a high-risk, patient-based approach that seeks to identify individuals at high risk and bring them into medical intervention. These two approaches are complementary and together represent a coordinated strategy to reduce cholesterol levels and coronary risk. Both approaches merit pursuit.
Although the evidence relating dietary fat to cancer is weaker than that relating dietary fat to CHD, the conclusions for both of these major chronic diseases are entirely congruent. Thus, recommendations to reduce the dietary intake of total fat and of SFAs will lead to reductions in coronary risk and perhaps in cancer risk.
Directions for Research
More information is needed on many issues concerning relationships between dietary fats and other lipids and health and disease. Thus, the committee believes that future research directed at the following issues would be of value.
Cardiovascular Disease
· The role of postprandial lipoproteins and their remnants in atherogenesis and in coronary risk;
Page 230
relationships between intake of specific fats and lipids and postprandial and remnant lipoproteins.
· The nature and regulation (including the dietary regulation) of heterogeneity within each major class of lipoproteins and the roles of different lipoprotein subclasses in atherosclerosis and CHD.
· The major dietary determinants of plasma HDL and the role and mechanism of HDL as protective against CHD.
· Characterization of the extent of interindividual variability in response to SFA intake. Are there human hyper- and hyporesponders to SFA intake? Can markers of such responsiveness be identified?
· Further characterization of the effects of MUFAs and stearic acid on serum lipid and lipoprotein profiles and levels.
· More systematic study of the effects of trans fatty acids on health.
· Is there a threshold for the unusually atherogenic effects of peanut oil observed in animals? Does peanut oil, consumed above a certain level, increase the risk of atherosclerotic disease in humans?
· Further characterization of the long-term consequences of the ingestion of different levels of w-6 PUFA vegetable oils (i.e., on lipoproteins, atherosclerosis, cancer, and gallstones). What is the desirable level of intake of w-6 PUFAs?
· The effects of w-3 PUFAs (fish oils) on serum lipids and lipoproteins and on other parameters (including platelet function, coagulation, and eicosanoid metabolism). What are the long-term effects of w-3 PUFA ingestion (e.g., potential benefits, untoward effects, possible regression of atherosclerosis)? More information is needed on the biologic actions of thromboxane A3.
· Characterization of the basis, particularly the genetic basis, of the variability in response to dietary cholesterol. Can we define human hyper- and hyporesponders?
· Effects of dietary cholesterol and dietary fats, and their interactions, on receptor-mediated clearance of IDL and LDL.
· Is there a minimum level of dietary cholesterol that affects plasma LDL level? Does dietary cholesterol lead to the formation of particularly atherogenic postprandial remnant lipoproteins?
· The associations of diet, and the levels and profiles of serum lipoproteins, with peripheral vascular and cerebrovascular diseases.
· Mechanisms and extent to which genetic factors control responses to dietary fats and other lipids, their interaction, and impact on specific chronic diseases, especially cardiovascular diseases, cancer, and gallbladder disease.
· The influence of dietary factors other than fats on serum lipids, the atherosclerotic process, and cardiovascular disease.
Cancer
· Further characterization of the association of dietary fat and caloric intake with increased risk of breast, colorectal, prostate, endometrial, and ovarian cancer.
· Further exploration of the possible mechanisms through which the amount and type of dietary fat (e.g., SFAs, w-6 and w-3 PUFAs) may influence different stages of carcinogenesis.
· Clarification of the relationship between dietary fat and other macronutrients (e.g., protein, carbohydrate) in colorectal cancer.
· Evaluation of the possible reciprocal relationship of dietary fat and dietary fiber intake to risk of colorectal cancer.
· Further assessment of the relationship between specific types of dietary fat (SFAs, MUFAs, w-6 and w-3 PUFAs) and risk of breast, colorectal, and prostate cancer.
· Evaluation of the possible relationship of dietary fat and elevated cholesterol intake to risk of lung and bladder cancer.
· Exploration of the quantitative relationship between reduced fat intake and decreased cancer rates, and thus the optimal range of intake of dietary fat and its component types.
· Consideration of the feasibility of intervention studies, preferably on defined populations, to determine the effects of reduced fat intake on the incidence of common cancers, especially colorectal and perhaps breast cancer. Large sample sizes and long-term follow-up will be required for such studies.
Obesity
· The proportion of fats in the diet may be a factor in the development of positive energy balance and possibly obesity, but the validity of this claim and the possible mechanisms of action require elucidation.
Gallstones
· Further exploration of the epidemiology of cholesterol gallstones. Identification of dietary and metabolic factors, and their interplay, in the etiology of cholelithiasis.
Page 231
· Dietary studies in humans to relate defined dietary components to changes in the lithogenicity of bile.
References
AAP (American Academy of Pediatrics). 1983. Committee on Nutrition: toward a prudent diet for children. Pediatrics 71: 78-80.
Aberg, H., H. Lithell, I. Selinus, and H. Hedstrand. 1985. Serum triglycerides are a risk factor for myocardial infarction but not for angina pectoris. Results from a 10-year followup of Uppsala Primary Preventive Study. Atherosclerosis 54:89-97.
Abramson, Z.H., and J.D. Kark. 1985. Serum cholesterol and primary brain tumours: a case-control study. Br. J. Cancer 52:93-98.
ACS (American Cancer Society). 1983. Workshop conference on nutrition in cancer causation and prevention. Fort Lauderdale, Florida, October 18-20, 1982. Cancer Res. 43: 2398S-2519S.
ACS (American Cancer Society). 1986. Second National Conference on Diet, Nutrition and Cancer. Houston, Texas, September 5-7, 1985. Cancer 58:1791-1962.
Admirand, W.H., and D.M. Small. 1968. The physicochemical basis of cholesterol gallstone formation in man. J. Clin. Invest. 47:1043-1052.
Ahrens, E.H., Jr. 1957. Nutritional factors and serum lipid levels. Am. J. Med. 23:928-952.
Ahrens, E.H., Jr., D.H. Blankenhorn, and T.T. Tsaltas. 1954. Effect on human serum lipids of substituting plant for animal fat in diet. Proc. Soc. Exp. Biol. Med. 86:872-878.
Ahrens, E.H., Jr., J. Hirsch, W. Insull, Jr., T.T. Tsaltas, R. Blomstrand, and M.L Peterson. 1957. The influence of dietary fats on serum-lipid levels in man. Lancet 1:943- 953.
Ahrens, E.H., Jr., W. Insull, Jr., J. Hirsch, W. Stoffel, M.L. Peterson, J.W. Farquher, T. Miller, and H.J. Thomasson . 1959. The effect on human serum-lipids of a dietary fat, highly unsaturated, but poor in essential fatty acids. Lancet 1:115-119.
Akesson, B., C. Fehling, and M. Jagerstad. 1978. Effect of vitamin B deficiency on phosphatidylethanolamine methylation in rat liver. Br. J. Nutr. 40:521-527.
Alderson, L.M., K.C. Hayes, and R.J. Nicholosi. 1986. Peanut oil reduces diet-induced atherosclerosis in cynomolgus monkeys. Arteriosclerosis 6:465-474.
American Health Foundation. 1979. Conference on the health effects of blood lipids: optimal distributions for populations. Prev. Med. 8:580-759.
Andersen, E., and K. Hellstrom. 1979. The effect of cholesterol feeding on bile acid kinetics and biliary lipids in normolipidemic and hypertriglyceridemic subjects. J. Lipid Res. 20:1020-1027.
Anderson, J.T., F. Grande, and A. Keys. 1961. Hydrogenated fats in the diet and lipids in the serum of man. J. Nutr. 75: 388-394.
Anitschkow, N.N. 1967. A history of experimentation on arterial atherosclerosis in animals. Pp. 21-44 in H.T. Blumenthal, ed. Cowdry's Arteriosclerosis: A Survey of the Problem, 2nd ed. C.C. Thomas, Springfield, Ill.
Anitschkow, N., and S. Chalatow. 1913. Ueber Experimentelle Cholesterinsteatose und ihre Bedeutung für die Entstehung einiger pathologischer Prozesse. Zentralbl. Allg. Pathol. Pathol. Anat. 24:1-9.
Antonis, A., and I. Bersohn. 1962a. The influence of diet on fecal lipids in South African White and Bantu prisoners. Am. J. Clin. Nutr. 11:142-155.
Antonis, A., and 1. Bersohn. 1962b. The influence of diet on serum lipids in South African White and Bantu prisoners. Am. J. Clin. Nutr. 10:484-499.
Applebaum-Bowden, D., W.R. Hazzard, J. Cain, M.C. Cheung, R.S. Kushwaha, and J.J. Albers. 1979. Short-term egg yolk feeding in humans. Increase in apolipoprotein B and low density lipoprotein cholesterol. Atherosclerosis 33:385-396.
Applebaum-Bowden. D., S.M. Haffner, E. Hartsook, K.H. Luk, J.J. Albers, and W.R. Hazzard. 1984. Down-regulation of the low-density lipoprotein receptor by dietary cholesterol. Am. J. Clin. Nutr. 39:360-367.
Applewhite, T.H. 1981. Nutritional effects of hydrogenated soya oil. J. Am. Oil Chem. Soc. 58:260-269.
Arevalo, J.A., A.O. Wollitzer, M.B. Corporon, M. Larios, D. Huante, and M.T. Ortiz. 1987. Ethnic variability in cholelithiasis—an autopsy study. West. J. Med. 147:44-47.
Armstrong, B., and R. Doll. 1975. Environmental factors and cancer incidence and mortality in different countries, with special reference to dietary practices. Int. J. Cancer 15:617-631.
Armstrong, B.K., J.I. Mann, A.M. Adelstein, and F. Eskin. 1975. Commodity consumption and ischemic heart disease mortality, with special reference to dietary practices. J. Chronic Dis. 28:455-469.
Armstrong, M.J., Z. Stephan, and K.C. Hayes. 1982. Biliary lipids in New World monkeys: dietary cholesterol, fat, and species interactions. Am. J. Clin. Nutr. 36:592-601.
Arntzenius, A.C., D. Kromhout, J.D. Barth, J.H. Reiber, A.V. Bruschke, B. Buis, C.M. van Gent, N. Kempen-Voogd, S. Strikwerda, and E.A. van der Bruschke. 1985. Diet, lipoproteins, and the progression of coronary atherosclerosis: the Leiden Intervention Trial. N. Engl. J. Med. 312:805-811.
Avigan, J., and D. Steinberg. 1958. Effects of saturated and unsaturated fat on cholesterol metabolism in the rat. Proc. Soc. Exp. Biol. Med. 97:814-816.
Avigan, J., and D. Steinberg. 1965. Sterol and bile acid excretion in man and the effects of dietary fat. J. Clin. Invest. 44:1845-1856.
Aylsworth, C.F. 1986. Effects of lipids on gap junctionally-mediated intercellular communication: possible role of the promotion of tumorigenesis by dietary fat. Prog. Clin. Biol. Res. 222:607-622.
Baggio, G., A. Pagnan, M. Muraca, S. Martini, A. Opportuno, A. Bonanome, G.B. Ambrosio, S. Ferrari, P. Guarini, D. Piccolo, E. Manzato, R. Corrocher, and G. Crepaldi. 1988. Olive-oil-enriched diet: effect on serum lipoprotein levels and biliary cholesterol saturation. Am. J. Clin. Nutr. 47:960-964.
Bailey, C.H. 1916. Observations on cholesterol-fed guinea pigs. Proc. Soc. Exp. Biol. Med. 13:60-62.
Bang, H.O., and J. Dyerberg. 1980. Lipid metabolism and ischemic heart disease in Greenland Eskimos. Pp. 1-22 in H.H. Draper, ed. Advances in Nutritional Research, Vol. 3. Plenum Press, New York.
Barbeau, A. 1978. Lecithin in neurologic disorders. N. Engl. J. Med. 299:200-201.
Barness, L.A. 1986. Cholesterol and children. J. Am. Med. Assoc. 256:2871.
Bartus, R.T., R.L. Dean, J.A. Goas, and A.S. Lippa. 1980.
Page 232
Age-related changes in passive avoidance retention: modulation with dietary choline. Science 209:301-303.
Baudet, M.F., C. Dachet, M. Lasserre, O. Esteva, and B. Jacotot. 1984. Modification in the composition and metabolic properties of human low density and high density lipoproteins by different dietary fats. J. Lipid. Res. 25:456-468.
Beaglehole, R., D.C. Trost, I. Tamir, P. Kwiterovich, C.J. Glueck, W. Insull, and B. Christensen. 1980. Plasma high-density lipoprotein cholesterol in children and young adults. The Lipid Research Clinics Program Prevalence Study. Circulation 62:IV83-IV92.
Beare-Rogers, J.L. 1983. Trans- and positional isomers of common fatty acids. Adv. Nutr. Res. 5:171-200.
Beaton, G.H., J. Milner, P. Corey, V. McGuire, M. Cousins, E. Stewart, M. de Ramos, D. Hewitt, P.V. Grambsch, N. Kassim, and J.A. Little. 1979. Sources of variance in 24-hour dietary recall data: implications for nutrition study design and interpretation. Am. J. Clin. Nutr 32:2546-2549.
Becker, N., D.R. Illingworth, P. Alaupovic, W.E. Connor, and E.E. Sundberg. 1983. Effects of saturated, monounsaturated, and omega-6 polyunsaturated fatty acids on plasma lipids, lipoproteins, and apoproteins in humans. Am. J. Clin. Nurr. 37:355-360.
Beil, F.U., and S.M. Grundy. 1980. Studies on plasma lipoproteins during absorption of exogenous lecithin in man. J. Lipid Res. 21:525-536.
Bennion, L.J., and S.M. Grundy. 1975. Effects of obesity and caloric intake on biliary lipid metabolism in man. J. Clin. Invest. 56:996-1011.
Bennion, L.J., and S.M. Grundy. 1978a. Risk factors for the development of cholelithiasis in man (first of two parts). N. Engl. J. Med. 299:1161-1167.
Bennion, L.J., and S.M. Grundy. 1978b. Risk factors for the development of cholelithiasis in man (second of two parts). N. Engl. J. Med. 299:1221-1227.
Benson, J. 1981. Fats of human milk and infant formulas. Pp. 553-560 in E. Lebenthal, ed. Textbook of Gastroenterology and Nutrition in Infancy, Vol. 1. Gastrointestinal Development in Perinatal Nutrtion. Raven Press, New York.
Berenson, G.A. 1980. Cardiovascular Risk Factors in Children: The Early Natural History of Atherosclerosis and Essential Hypertension. Oxford University Press, New York. 453 pp.
Berenson, G.S., and F.H. Epstein. 1983. Conference on blood lipids in children: optimal levels for early prevention of coronary artery disease. Workshop report: epidemiological section. April 18 and 19, 1983, American Health Foundation. Prev. Med. 12:741-797.
Berenson, G.S., S.R. Srinivasan, R.R. Frerichs, and L.S. Webber. 1979. Serum high density lipoprotein and its relationship to cardiovascular disease risk factor variables in children—the Bogalusa Heart Study. Lipids 14:91-98.
Berenson, G.S., S.R. Srinivasan, T.A. Nicklas, and LS. Webber. 1988. Cardiovascular risk factors in children and early prevention of heart disease. Clin. Chem. 34:B115-B122.
Berg, K. 1963. A new serum type system in man—the Lp system. Acta Pathol. Microbiol. Scand. 59:369-382.
Berg, K., G. Dahlen, and M.H. Frick. 1974. Lp(a) lipoprotein and pre-b1-lipoprotein in patients with coronary heart disease. Clin. Genet. 6:230-235.
Berry, E.M., and J. Hirsch. 1986. Does dietary linoleic acid influence blood pressure? Am. J. Clin. Nutr. 44:336-340.
Berry, E.M., J. Hirsch, J. Most, D.J. McNamara, and J. Thorton. 1986. The relationship of dietary fat to plasma lipid levels as studied by factor analysis of adipose tissue fatty acid composition in a free living population of middle aged American men. Am. J. Clin. Nutr. 44:220-231.
Best, M.M., C.H. Duncan, E.J. Van Loon, and J.D. Wathen. 1955. The effects of sitosterol on serum lipids. Am. J. Med. 19:61-70.
Beth, M., M.R. Berger, M. Aksoy, and D. Schmahl. 1987. Comparison between the effects of dietary fat level and of calorie intake on methylnitrosourea-induced mammary carcinogenesis in female SD rats. Int. J. Cancer 39:737-744.
Beveridge, J.M.R., and W.F. Connell. 1962. The effect of commercial margarines on plasma cholesterol levels in man. Am. J. Clin. Nutr. 10:391-397.
Beveridge, J.M.R., W.F. Connell, G.A. Mayer, J.B. Firstbrook, and M.S. DeWolfe. 1955. The effects of certain vegetable and animal fats on the plasma lipids of humans. J. Nutr. 56:311-320.
Beveridge, J.M.R., W.F. Connell, and G.A. Mayer. 1957. The nature of the substances in dietary fat affecting the level of plasma cholesterol in humans. Can. J. Biochem. Physiol. 35:257-270.
Beveridge, J.M.R., W.F. Connell, H.L. Haust, and G.A. Mayer. 1959. Dietary cholesterol and plasma cholesterol levels in man. Can. J. Biochem. Physiol. 37:575-582.
Beveridge, J.M.R., W.F. Connell, G.A. Mayer, and H.L. Haust. 1960. The response of man to dietary cholesterol. J. Nutr. 71:61-65.
Beynen, A.C., and M.B. Katan. 1985. Effect of egg yolk feeding on the concentration and composition of serum lipoprotein in man. Atherosclerosis 54:157-166.
Bhattacharyya, A.K., and W.E. Connor. 1974. Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J. Clin. Invest. 53:1033-1043.
Biörck, G. 1956. Wartime lessons on arteriosclerotic heart disease from northern Europe. Pp. 8-21 in A. Keys and P. D. White, eds. Cardiovascular Epidemiology. Second World Congress of Cardiology and Twenty-Seventh Annual Scientific Sessions of the American Heart Association. Hoeber-Haper, New York.
Birt, D.F., and B.D. Roebuck. 1986. Enhancement of pancreatic carcinogenesis by dietary fat in the hamster and rat models. Prog. Clin. Biol. Res. 222:331-335.
Bjelke, E. 1978. Dietary factors and the epidemiology of cancer of the stomach and large bowel. Aktuel. Ernaehrungsmed. Klin. Prax. Suppl. 2:10-17.
Blankenhorn, D.H., S.A. Nessim, R.L Johnson, M.E. Sanmarko, S.P. Azen, and L Cashin-Hemphill. 1987. Beneficial effects of combined colestipol-niacin therapy on coronary atherosclerosis and coronary venous bypass grafts. J. Am. Med. Assoc. 257:3233-3240.
Bloomfield, D.K. 1964. Cholesterol metabolism. Ill. Enhancement of cholesterol absorption and accumulation in safflower oil-fed rats. J. Lab. Clin. Med. 64:613-623.
Bodurtha, J.N., R.M. Schieken, J. Segrest, and W.E. Nance. 1987. High-density lipoprotein-cholesterol subfractions in adolescent twins. Pediatrics 79:181-189.
Bonanome, A., and S.M. Grundy. 1988. Effect of dietary stearic acid on plasma cholesterol and lipoprotein levels. N. Engl. J. Med. 318:1244-1248.
Bothig, S., V.I. Metelitsa, W. Barth, A.A. Aleksandrov, 1. Schneider, T.P. Ostrovskaya, E.V. Kokurina, I.I. Saposhinkov, I.P. Iliushina, and L.S. Gurevich. 1976. Prev-
Page 233
alence of ischaemic heart disease, arterial hypertension and intermittent claudication, and distribution of risk factors among middle-aged men in Moscow and Berlin. Cor Vasa 18:104-118.
Braden, L.M., and K.K. Carroll. 1986. Dietary polyunsaturated fat in relation to mammary carcinogenesis in rats. Lipids 21:285-288.
Bragdon, J.H., J.H. Zeller, and J.W. Stevenson. 1957. Swine and experimental atherosclerosis. Proc. Soc. Exp. Biol. Med. 95:282-284.
Brenneman, D.E., W.E. Connor, EL. Forker, and L. DenBesten. 1972. The formation of abnormal bile and cholesterol gallstones from dietary cholesterol in the prairie dog. J. Clin. Invest. 51:1495-1503.
Brensike, J.F., R.I. Levy, S.F. Kelsey, E.R. Passamani, J.M. Richardson, I.K. Loh, N.J. Stone, R.F. Aldrich, J.W. Battaglini, D.J. Moriarity, M.R. Fisher, L. Friedman, W. Friedewald, K.M. Detre, and S.E. Epstein. 1984. Effects of therapy with cholestyramine on progression of coronary arteriosclerosis: results of the NHLBI Type II Coronary Intervention Study. Circulation 69:313-324.
Brett, M., and D.J. Barker. 1976. The world distribution of gallstones. Int. J. Epidemiol. 5:355-341.
Broitman, S.A. 1986. Cholesterol conundrums: the relationship between dietary and serum cholesterol in colon cancer. Prog. Clin. Biol. Res. 222:435-459.
Bronsgeest-Schoute, D.C., J.G. Hautvast, and R.J. Hermus. 1979a. Dependence of the effects of dietary cholesterol and experimental conditions on serum lipids in man. I. Effects of dietary cholesterol in a linoleic acid-rich diet. Am. J. Clin. Nutr. 32:2183-2187.
Bronsgeest-Schoute, D.C., R.J. Hermus, G.M. Dallinga-Thie, and J.G. Hautvast. 1979b. Dependence of the effects of dietary cholesterol and experimental conditions on serum lipids in man. II. Effects of dietary cholesterol in a linoleic acid-poor diet. Am. J. Clin. Nutr. 32:2188-2192.
Bronte-Stewart, B., A. Keys, J.F. Brock, A.D. Moodie, M.H. Keys, and A. Antonis. 1955. Serum-cholesterol, diet, and coronary heart-disease: an inter-racial survey in the Cape Peninsula. Lancet 269:1103-1108.
Brown, M.S., and J.L. Goldstein. 1983. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 52:223-261.
Brown, M.S., and J.L. Goldstein. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science 232:34-47.
Brown, W.V., and H. Ginsberg. 1987. Classification and diagnosis of the hyperlipidemias. Pp. 143-168 in D. Steinberg and J.M. Olefsky, eds. Hypercholesterolemia and Atherosclerosis: Pathogenesis and Prevention. Churchill Livingstone, New York.
Bruce, W.R., and P.W. Dion. 1980. Studies relative to a fecal mutagen. Am. J. Clin. Nutr. 33:2511-2512.
Brunzell, J.D., A.D. Sniderman, J.J. Albers, and P.O. Kwiterovich, Jr. 1984. Apoproteins B and A-I and coronary artery disease in humans. Arteriosclerosis 4:79-83.
Brussaard, J.H., F. Dallinga-Thie, P.H. Groot, and M.B. Katan. 1980. Effects of amount and type of dietary fat on serum lipids, lipoproteins and apolipoproteins in man. A controlled 8-week trial. Atherosclerosis 36:515-527.
Brussaard, J.H., M.B. Katan, P.H. Groot, L.M. Havekes, and J.G. Hautvast. 1982. Serum lipoproteins of healthy persons fed a low-fat diet or a polyunsaturated fat diet for three months. A comparison of two cholesterol-lowering diets. Atherosclerosis 42:205-219.
Bull, A.W., J.C. Bronstein, and N.D. Nigro. 1988. Role of essential fatty acids in azoxymethane-induced colon carcinogenesis. Proc. Am. Assoc. Cancer Res. 29:149.
Bursztyn, P.G., and M.H. King. 1986. Fat-induced hypertension in rabbits: the effects of dietary linoleic and linolenic acid. J. Hypertens. 4:699-702.
Byers, T., J. Marshall, S. Graham, C. Mettlin, and M. Swanson. 1983. A case-control study of dietary and nondietary factors in ovarian cancer. J. Natl. Cancer Inst. 71: 681-686.
Canner, P.L., K.G. Berge, N.K. Wenger, J. Stamler, L. Friedman, R.J. Prineas, and W. Friedewald. 1986. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J. Am. Coll. Cardiol. 8:1245-1255.
Carew, T.E., D.C. Schwenke, and D. Steinberg. 1987. Antiatherogenic effect of probucol unrelated to its hypocholesterolemic effect: evidence that antioxidants in vivo can selectively inhibit low density lipoprotein degradation in macrophage-rich fatty streaks and slow the progression of atherosclerosis in the Watanabe heritable hyperlipidemic rabbit. Proc. Natl. Acad. Sci. U.S.A. 84:7725-7729.
Carey, M.C., and D.M. Small. 1978. The physical chemistry of cholesterol solubility in bile. Relationship to gallstone formation and dissolution in man. J. Clin. Invest. 61: 998-1026.
Carlson, L.A., and L.E. Bottiger. 1981. Serum triglycerides, to be or not to be a risk factor for ischaemic heart disease? Atherosclerosis 39:287-291.
Carlson, S.E., P.W. DeVoe, and LA. Barness. 1982. Effect of infant diets with different polyunsaturated to saturated fat ratios on circulating high-density lipoproteins. J. Pediatr. Gastroenterol. Nutr. 1:303-309.
Carroll, K.K. 1971. Plasma cholesterol levels and liver cholesterol biosynthesis in rabbits fed commercial or semisynthetic diets with and without added fats or oils. Atherosclerosis 13:67-76.
Carroll, K.K. 1975. Experimental evidence of dietary factors and hormone-dependent cancers. Cancer Res. 35:3374-3383.
Carroll, K.K. 1983. Diet and carcinogenesis. Pp. 223-227 in F.G. Schettler, A.M. Gotto, G. Middelhoff, A.J.R. Habenicht, and K.R. Jurutka, eds. Atherosclerosis: Proceedings of the Sixth International Symposium. Springer-Verlag, Berlin.
Carroll, K.K. 1985. Dietary fat and breast cancer. Pp. 29-47 in J. Weininger, ed. Nutrition Update, Vol. 2. Wiley, New York.
Carroll, K.K. 1986a. Biological effects of fish oils in relation to chronic diseases. Lipids 21:731-732.
Carroll, K.K. 1986b. Experimental studies on dietary fat and cancer in relation to epidemiological data. Prog. Clin. Biol. Res. 222:231-248.
Carroll, K.K., and H.T. Khor. 1971. Effects of level and type of dietary fat on incidence of mammary tumors induced in female Sprague-Dawley rats by 7,12-dimethylbenz(a)anthracene. Lipids 6:415-420.
Carroll, K.K., and H.T. Khor. 1975. Dietary fat in relation to tumorigenesis. Prog. Biochem. Pharmacol. 10:308-353.
Carroll, K.K., and C.J.H. Woodward. 1989. Nutrition and human health aspects of marine oils and lipids. Pp. 435-456 in R.G. Ackman, ed. Marine Biogenic Lipids, Fats, and Oils, Vol. 2. CRC Press, Boca Raton, Fla.
Carroll, K.K., G.J. Hopkins, T.G. Kennedy, and M.B. Davidson. 1981. Essential fatty acids in relation to mam-
Page 234
mary carcinogenesis. Prog. Lipid. Res. 20:685-690.
Carroll, K.K., L.M. Braden, J.A. Bell, and R. Kalameghan. 1986. Fat and cancer. Cancer 58:1818-1825.
Carroll, M.D., S. Abraham, and C.M. Dresser. 1983. Dietary Intake Source Data: United States, 1976-80. Vital and Health Statistics, Ser. 11, No. 231. DHHS Publ. No. (PHS) 83-1681. National Center for Health Statistics, Public Health Service, U.S. Department of Health and Human Services, Hyattsville, Md. 483 pp.
Carter, C.A., R.J. Milholland, W. Shea, and M.M. Ip. 1983. Effect of the prostaglandin synthetase inhibitor indomethacin on 7,12-dimethylbenz(a)anthracene-induced mammary tumorigenesis in rats fed different levels of fat. Cancer Res. 43:3559-3562.
Carter, C.A., M.M. Ip, and C. Ip. 1987. Response of mammary carcinogenesis to dietary linoleate and fat levels and its modulation by prostaglandin synthesis inhibitors. Pp. 253-260 in W.E.M. Lands, ed. Proceedings of the AOCS Short Course on Polyunsaturated Fatty Acids and Eicosanoids. American Oil Chemists' Society, Champaign, Ill.
Carvalho, A.C.A., C. Galli, R. Paoletti, J.M. Iacono, and A. Keys. 1981. Platelets, thrombosis and dietary fats. International pilot epidemiological study in thrombosis. Pp. 125-142 in N.G. Basán, R. Paoletti, and J.M. Iacono, eds. New Trends in Nutrition, Lipid Research, and Cardiovascular Diseases. Current Topics in Nutrition and Disease, Vol. 5. Alan R. Liss, New York.
CAST (Council for Agricultural Science and Technology). 1982. Diet, Nutrition, and Cancer. A Critique. Report of the Task Force on Diet, Nutrition, and Cancer. Special Publ. No. 13. CAST, Ames, Iowa. 80 pp.
Castelli, W.P. 1986. The triglyceride issue: a view from Framingham. Am. Heart J. 112:432-437.
Castelli, W.P., RD. Abbott, P.M. McNamara. 1983. Summary estimates of cholesterol used to predict coronary heart disease. Circulation 67:730-734.
Castelli, W.P., R.J. Garrison, P.W. Wilson, R.D. Abbott, S. Kalousdian, and W.B. Kannel. 1986. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. J. Am. Med. Assoc. 256:2835-2838.
Cavallo-Perin, P., C. Barile, A. Ozello, M. La Rosa, G. Pagano, and G. Lenti. 1984. Peripheral vascular disease and risk factors of atherosclerosis: an epidemiologic study. Panminerva Med. 26:139-143.
Cave, W.T., Jr., and J.]. Jurkowski. 1987. Comparative effects of omega-3 and omega-6 dietary lipids on rat mammary tumor development. Pp. 261-266 in W.E.M. Lands, ed. Proceedings of the AOCS Short Course on Polyunsaturated Fatty Acids and Eicosanoids. American Oil Chemists' Society, Champaign, Ill.
Chait, A., A. Onitiri, A. Nicoll, E. Rabaya, J. Davies, and B. Lewis. 1974. Reduction of serum triglyceride levels by polyunsaturated fat. Studies on the mode of action and on very low density lipoprotein composition. Atherosclerosis 20:347-364.
Chalatow, S.S. 1929. Bemerkungen zu den Arbeiten über die sogenannte. Experimentelle Cholesterinsteatose oder Experimentelle Cholesterinkrankheit des Kaninchens und anderer Tiere. Virchows. Arch. f. Pathol. Anat. 272:691-708.
Chase, H.P., R.J. O'Quin, and D. O'Brien. 1974. Screening for hyperlipidemia in childhood. J. Am. Med. Assoc. 230: 1535-1537.
Chenoweth, W., M. Ullmann, R. Simpson, and G. Leveille. 1981. Influence of dietary cholesterol and fat on serum lipids in men. J. Nutr. 111:2069-2080.
Childs, M.T., J.A. Bowlin, J.T. Ogilvie, W.R. Hazard, and J.J. Albers. 1981. The contrasting effects of a dietary soya lecithin product and corn oil on lipoprotein lipids in normolipidemic and familial hypercholesterolemic subjects. Atherosclerosis 38:217-228.
Chong, K.S., R.J. Nicolosi, R.F. Rodger, D.A. Arrigo, R.W. Yuan, J.J. MacKey, S. Georas, and P.N. Herbert. 1987. Effect of dietary fat saturation on plasma lipoproteins and high density lipoprotein metabolism of the rhesus monkey. J. Clin. Invest. 79:675-683.
Ciocca, S., M. Arca, A. Montali, S. Fazio, A. Bucci, and F. Angelico. 1987. Lack of association between arterial blood pressure and erythrocyte fatty acid composition in an Italian population sample. Scand. J. Clin. Lab. Invest. 47:105-110.
Clarke, S.D. 1986. Metabolic adaptations to dietary fats. Prog. Clin. Biol. Res. 222:531-553.
Clarke, W.R., H.G. Schrott, P.E. Leaverton, W.E. Connor, and R.M. Lauer. 1978. Tracking of blood lipids and blood pressures in school age children: the Muscatine Study. Circulation 58:626-634.
Clarkson, T.B., H.B. Lofland, Jr., B.C. Bullock, and H.O. Goodman. 1971. Genetic control of plasma cholesterol. Studies on squirrel monkeys. Arch. Pathol. 92:37-45.
Clarkson, T.B., N.D. Lehner, B.C. Bullock, H.B. Lofland, and W.D. Wagner. 1976a. Atherosclerosis in new world monkeys. Primates Med. 9:90-144.
Clarkson, T.B., T.E. Hamm, B.C. Bullock, and N.D. Lehner. 1976b. Atherosclerosis in old world monkeys. Primates Med. 9:66-89.
Clarkson, T.B., J.R. Kaplan, and M.R. Adams. 1985. The role of individual differences in lipoprotein, artery wall, gender, and behavioral responses in the development of atherosclerosis. Ann. N.Y. Acad. Sci. 454:28-45.
Clevidence, B.A., R.E. Morton, G. West, D.M. Dusek, and H.F. Hoff. 1984. Cholesterol esterification in macrophages. Stimulation by lipoproteins containing apo B isolated from human aortas. Arteriosclerosis 4:196-207.
Cobb, M., P. Turkki, W. Linscheer, and K. Raheja. 1980. Lechithin supplementation in healthy volunteers: effect on cholesterol esterification and plasma, and bile lipids. Nutr. Metab. 24:228-237.
Codde, J.P., K.D. Croft, and L.J. Beilin. 1987a. Dietary suppression of prostaglandin synthesis does not accelerate DOCA/salt hypertension in rats. Clin. Exp. Pharmacol. Physiol. 14:513-523.
Codde, J.P., L.J. Beilin, K.D. Croft, and R. Vandongen. 1987b. The effect of dietary fish oil and salt on blood pressure and eicosanoid metabolism of spontaneously hypertensive rats. J. Hypertens. 5:137-142.
Cohen, B.I., E.H. Mosbach, C.K. McSherry, R.J. Stenger, S. Kuroki, and B. Rzigalinski. 1986. Gallstone prevention in prairie dogs: comparison of chow vs semisynthetic diets. Hepatology 6:874-880.
Cohen, B.M., A.L. Miller, J.F. Lipinski, and H.G. Pope. 1980. Lecithin in mania: a preliminary report. Am. J. Psychiatry 137:242-243.
Cohen, L.A. 1986. Dietary fat and mammary cancer. Pp. 77-100 in B.S. Reddy and L.A. Cohen, eds. Diet, Nutrition and Cancer: A Critical Evaluation, Vol. 1. Macronutrients and Cancer. CRC Press, Boca Raton, Fla.
Cohen, L.A., D.O. Thompson, V. Maeura, K. Choi, M.E.
Page 235
Blank, and D.P. Rose. 1986. Dietary fat and mammary cancer. 1. Promoting effects of different dietary fats on N-nitrosomethylurea-induced rat mammary tumorigenesis. J. Natl. Cancer Inst. 77:33-42.
Cohen, LA., K.W. Choi, and C.X. Wang. 1988. Influence of dietary far, caloric restriction, and voluntary exercise on N-nitrosomethylurea-induced mammary tumorigenesis in rats. Cancer Res. 48:4276-4283.
Cohn, K., F.J. Sakai, and M.F. Langston, Jr. 1975. Effect of clofibrate on progression of coronary disease: a prospective angiographic study in man. Am. Heart J. 89:591-598.
Comberg, H.U., S. Heyden, C.G. Hames, A.J. Vergroesen, and A.I. Fleischman. 1978. Hypotensive effect of dietary prostaglandin precursor in hypertensive man. Prostaglandins 15:193-197.
Comess, L.J., P.H. Bennett, and T.A. Burch. 1967. Clinical gallbladder disease in Pima Indians. Its high prevalence in contrast to Framington, Massachusetts. N. Engl. J. Med. 277:894-898.
Committee of Principal Investigators. 1978. A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. Br. Heart J. 40:1069-1118.
Conlay, L.A., R.J. Wurtman, K. Blusztajn, I.L Coviella, T.J. Maher, and G.E. Evoniuk. 1986. Decreased plasma choline concentrations in marathon runners. N. Engl. J. Med. 315: 892.
Connor, W.E. 1986. Hypolipidemic effects of dietary omega-3 fatty acids in normal and hyperlipidemic humans: effectiveness and mechanisms. Pp. 173-210 in A.P. Simopoulos, R.R. Kifer, and R.E Martin, eds. Health Effects of Polyunsaturated Fatty Acids in Seafoods. Academic Press, New York.
Connor, W.E., R.E Hodges, and R.E. Bleiler. 1961a. Effect of dietary cholesterol upon serum lipids in man. J. Lab. Clin. Med. 57:331-342.
Connor, W.E., R.E. Hodges, and R.E. Bleiler. 1961b. The serum lipids in men receiving high cholesterol and cholesterol-free diets. J. Clin. Invest. 40:894-901.
Connor, W.E., D.B. Stone, and R.E. Hodges. 1964. The interrelated effects of dietary cholesterol and fat upon human serum lipid levels. J. Clin. Invest. 43:1691-1696.
Connor, W.E., D.T. Witiak, D.B. Stone, and M.L. Armstrong. 1969. Cholesterol balance and fecal neutral steroid and bile acid excretion in normal men fed dietary fats of different fatty acid composition. J. Clin. Invest. 48:1363-1375.
Connor, W.E., M.T. Cerqueira, R.W. Connor, R.B. Wallace, M.R. Malinow, and H.R. Casdorph. 1978. The plasma lipids, lipoproteins, and diet of the Tarahumara Indians of Mexico. Am. J. Clin. Nutr. 31:1131-1142.
Corey, J.E., K.C. Hayes, B. Dorr, and D.M. Hegsted. 1974. Comparative lipid response of four primate species to dietary changes in fat and carbohydrate. Atherosclerosis 19:119-134.
Corkin, S., K.L. Davis, J.H. Growdon, E. Usdin, and R.J. Wurtman, eds. 1982. Aging, Vol. 19. Alzheimer's Diseases: A Report of Progress in Research. Raven Press, New York. 525 pp.
Correa, P., J. Paschal, P. Pizzolato, W. Pelon, and D.E. Lesley. 1981. Fecal mutagens and colorectal polyps: preliminary report of an autopsy study. Pp. 119-127 in W.R. Bruce, P. Correa, M. Lipkin, S.R. Tannenbaum, and T.D. Wilkins, eds. Banbury Report 7. Gastrointestinal Cancer: Endogenous Factors. Cold Spring Harbor Laboratory, New York.
Cortese, C., Y. Levy, E.D. Janus, P.R. Turner, S.N. Rao, N.E. Miller, and B. Lewis. 1983. Modes of action of lipid-lowering diets in man: studies of apolipoprotein B kinetics in relation to fat consumption and dietary fatty acid composition. Eur. J. Clin. Invest. 13:79-85.
Cramer, D.W., W.R. Welch, G.B. Hutchinson, W. Willett, and R.E. Scully. 1984. Dietary animal fat in relation to ovarian cancer risk. Obstet. Gynecol. 63:833-838.
Crowther, J.S., B.S. Drasar, M.J. Hill, B. Maclennen, D. Magnin, S. Peach, and C.H. Teoh-Chan. 1976. Faecal steroids and bacteria and large bowel cancer in Hong Kong by socio-economic groups. Br. J. Cancer 34:191-198.
Cullen, K., N.S. Stenhouse, K.L. Wearne, and T.A. Welborn. 1983. Multiple regression analysis of risk factors for cardiovascular disease and cancer mortality in Busselton, Western Australia—13-year study. J. Chronic Dis. 36:371-377.
Cummings, J.H., H.S. Wiggins, D.J. Jenkins, H. Houston, T. Jivraj, B.S. Drasar, and M.J. Hill. 1978. Influence of diets high and low in animal fat on bowel habit, gastrointestinal transit time, fecal microflora, bile acid, and fat excretion. J. Clin. Invest. 61:953-963.
Curb, J.D., and D.M. Reed. 1985. Fish consumption and mortality from coronary heart disease. N. Engl. J. Med. 313: 821.
Dahlén, G., K. Berg, T. Gillnäs, and C. Ericson. 1975. Lp(a) lipoprotein/pre-b1-lipoprotein in Swedish middle-aged males and in patients with coronary heart disease. Clin. Genet. 7:334-341.
Dahlén, G., K. Berg, and M.H. Frick. 1976. Lp(a) lipoprotein/pre-beta1-lipoprotein, serum lipids and atherosclerotic disease. Clin. Genet. 9:558-566.
Dales, LG., G.D. Friedman, H.K. Ury, S. Grossman, and S.R. Williams. 1979. A case-control study of relationships of diet and other traits to colorectal cancer in American blacks. Am. J. Epidemiol. 109:132-144.
Dam, H. 1971. Determinants of cholesterol cholelithiasis in man and animals. Am. J. Med. 51:596613.
Dam, H., and F. Christensen. 1952. Alimentary production of gallstones in hamsters. Acta Path. Microbiol. Scand. 30: 236-242.
Dam, H., I. Kruse, M.K. Jensen, and H.E. Kallehauge. 1967. Studies on human bile. II. Influence of two different fats on the composition of human bile. Scand. J. Clin. Lab. Invest. 19:367-378.
Dam, H., I. Prange, M.K. Hensen, H.E. Kallehauge, and H.J. Fenger. 1971. Studies on human bile. IV. Influence of ingestion of cholesterol in the form of eggs on the composition of bile in healthy subjects. Z. Ernaehrungswiss. 10:178-187.
Dao, T.L., and P.C. Chan. 1983. Effect of duration of high fat intake on enhancement of mammary carcinogenesis in rats. J. Natl. Cancer Inst. 71:201-205.
Dauber, D.V., and LN. Katz. 1942. Experimental cholesterol atheromatosis in an omnivorous animal, the chick. Arch. Pathol. 34:937-950.
Davies, LG.G., and L Murdoch. 1959. ''Lipostabil": A pilot study. Br. Med. J. 2:619-620.
Davignon, J., S. Lussier-Cacan, M. Ortin-George, M. Lelievre, D. Bertagna, A. Gattereau, and A. Fontaine. 1977. Plasma lipids and lipoprotein patterns in angiographically graded atherosclerosis of the legs and in coronary heart disease. Can. Med. Assoc. J. 116:1245-1250.
Davignon, J., R.E. Gregg, and C.F. Sing. 1988. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis 8: 1-21.
Page 236
Davis, D., B. Stem, and G. Lesnick. 1937. The lipid and cholesterol content of blood of patients with angina pectoris and arteriosclerosis. Ann. Int. Med. 11:354-369.
Davis, W.W., III. 1955. Symposium on sitosterol. III. The physical chemistery of cholesterol and b-sitosterol related to the intestinal absorption of cholesterol. Trans. N.Y. Acad. Sci. 18:123-128.
Dawber, T.R., F.E. Moore, and G.V. Mann. 1957. Coronary heart disease in the Framingham Study. Am. J. Public Health 47 Suppl. April:4-24.
Dayton, S., M.L. Pearce, H. Goldman, A. Harnish, D. Plotkin, M. Schickman, M. Winfield, A. Zager, and W. Dixon. 1968. Controlled trial of a diet high in unsaturated fat for prevention of atherosclerotic complications. Lancet 2:1060-1062.
DeBacker, G., M. Rosseneu, and J.P. Deslypere. 1982. Discriminative value of lipids and apoproteins in coronary heart diease. Atherosclerosis 42:197-203.
de Iongh, H., R.K. Beerthuis, C. den Hartog, L.M. Dalderup, and P.A. van der Spek. 1965. The influence of some dietary fats on serum lipids in man. Bibl. Nutr. Dieta. 7:137-152.
de Langen, C.D. 1916. Cholesterine-Stofwisseling en Rassenpathologie. Geneeskd. Tijdschr. Ned. Indie 56:1-34.
de Langen, C.D. 1922. Her cholesterinegehalte van her bloed in Indie. Geneeskd. Tijdschr. Ned. Indie 62:1-4.
DenBesten, L., W.E. Connor, and S. Bell. 1973. The effect of dietary cholesterol on the composition of human bile. Surgery 73:266-273.
DenBesten, L., S. Safaie-Shirazi, W.E. Connor, and S. Bell. 1974. Early changes in bile composition and gallstone formation induced by a high cholesterol diet in prairie dogs. Gastroenterology 66:1036-1045.
Diller, E.R., M. Korzenovsky, and O.A. Harvey. 1961. Endogenous hypercholesterosis in rabbits fed a fat-free purified diet and the effect of unsaturated lipid. J. Nutr. 73:14-16.
Dock, W. 1946. The predilection of atherosclerosis for the coronary arteries. J. Am. Med. Assoc. 131:875-878.
Donato, K., and D.M. Hegsted. 1985. Efficiency of utilization of various sources of energy for growth. Proc. Natl. Acad. Sci. U.S.A. 82:4866-4870.
Drewnowski, A., J.D. Brunzell, K. Sande, P.H. Iverius, and M.R. Greenwood. 1985. Sweet tooth reconsidered: taste responsiveness in human obesity. Physiol. Behav. 35:617-622.
Ducimetiere, P., J.L. Richard, F. Cambien, R. Rakotovao, and J.R. Claude. 1980. Coronary heart disease in middle-aged Frenchmen: comparisons between Paris Prospective Study, Seven Countries Study, and Pooling Project. Lancet 1:1346-1350.
Duff, G.L. 1935. Experimental cholesterol arteriosclerosis and its relationship to human arteriosclerosis. Arch. Pathol. 20: 81-124, 259-304.
Duffield, R.G., B. Lewis, N.E. Miller, C.W. Jamieson, J.N. Brunt, and A.C. Colchester. 1983. Treatment of hyperlipidemia retards progression of symptomatic femoral atherosclerosis. A randomized control trial. Lancet 2:639-642.
Duncan, K.H., J.A. Bacon, and R.L. Weinsier. 1983. The effects of high and low energy density diets on satiety, energy intake, and eating time of obese and nonobese subjects. Am. J. Clin. Nutr. 37:763-767.
Dusing, R., R. Scherhag, K. Glanzer, U. Budde, and H.J. Kramer. 1983. Dietary linoleic acid deprivation: effects on blood pressure and PGI2 synthesis. Am. J. Physiol. 244: H228-H233.
Dwyer, J. 1980. Diets for children and adolescents that meet the dietary goals. Am. J. Dis. Child. 134:1073-1080.
ECP/IUNS (European Organization for Cooperation on Cancer Prevention Studies/International Union for Nutritional Sciences). 1986. Proceedings of a joint ECP-IUNS workshop on diet and human carcinogenesis (Arhus, Denmark; June 1985). Nutr. Cancer 8:1-40.
Ederer, F., P. Leren, O. Turpeinen, and I.D. Frantz, Jr. 1971. Cancer among men on cholesterol-lowering diets. Experience from five clinical trials. Lancet 2:203-206.
Eggen, D.A. 1974. Cholesterol metabolism in rhesus monkey, squirrel monkey, and baboon. J. Lipid Res. 15:139-145.
Ehnholm, C., J. Huttunen, P. Pietinen, U. Leino, M. Mutanen, E. Kostiainen, J Pikkarainen, R. Dougherty, J.M. Iacono, and P. Puska. 1982. Effect of diet on serum lipoproteins in a population with a high risk of coronary heart disease. N. Engl. J. Med. 307:850-855.
Ehnholm, C., J.K. Huttunen, P. Pietinen, U. Leino, M. Mutanen, E. Kostiainen, J.M. Iacono, R. Dougherty, and P. Puska. 1984. Effect of a diet low in saturated fatty acids on plasma lipids, lipoproteins, and HDL subfractions. Arteriosclerosis 4:265-269.
Ehrich, M., J.E. Aswell, R.L. Van Tassell, T.D. Wilkins, A.R. Walker, and N.J. Richardson. 1979. Mutagens in the feces of 3 South-African populations at different levels of risk for colon cancer. Mutagen. Res. 64:231-240.
Emken, E.A. 1984. Nutrition and biochemistry of trans and positional fatty acid isomers in hydrogenated oils. Annu. Rev. Nutr. 4:339-376.
Emmons, L. 1986. Food procurement and the nutritional adequacy of diets in low-income families. J. Am. Diet. Assoc. 86:1684-1693.
Enger, S.C., I. Hjermann, O.P. Foss, A. Helgeland, J. Holme, P. Leren, and K.R. Norum. 1979. High density lipoprotein cholesterol and myocardial infarction or sudden coronary death: a prospective case-control study in middle-aged men of the Oslo Study. Artery 5:170-181.
Enos, W.F., R.H. Holmes, and J. Beyer. 1953. Coronary disease among United States soldiers killed in action in Korea. J. Am. Med. Assoc. 152:1090-1093.
Erickson, B.A., R.H. Coots, F.H. Mattson, and A.M. Kligman. 1964. The effect of partial hydrogenation of dietary fats, of the ratio of polyunsaturated to saturated fatty acids, and of dietary cholesterol upon plasma lipids in man. J. Clin. Invest. 43:2017-2025.
Erickson, K.L. 1986. Mechanisms of dietary fat modulation of tumorigenesis: changes in immune response. Prog. Clin. Biol. Res. 222:555-586.
Ershow, A.G., R.J. Nocolosi, and K.C. Hayes. 1981. Separation of the dietary fat and cholesterol influences on plasma lipoproteins of rhesus monkeys. Am. J. Clin. Nutr. 34:830-840.
Fager, G., O. Wiklund, S.O. Olofsson, C. Wihelmsson, and G. Bondjers. 1980. Serum apolipoprotein levels in relation to acute myocardial infarction and its risk factors. Apolipoprotein A-I levels in male survivors of myocardial infarction. Atherosclerosis 36:67-74.
Fager, G., O. Wiklund, S.O. Olofsson, L. Wilhelmsen, and G. Bondjers. 1981. Multivariate analyses of serum apolipoproteins and risk factors in relation to acute myocardial infarction. Arteriosclerosis 1:273-279.
Farquhar, J.W., and M. Sokolow. 1958. Response of serum lipids and lipoproteins of man to beta-sitosterol and safflower
Page 237
oil: a long-term study. Circulation 17:890-899.
Farquhar, J.W., R.E. Smith, and M.E. Dempsey. 1956. The effect of beta-sirosterol on the serum lipids of young men with arteriosclerotic heart disease. Circulation 14:77-82.
Feinleib, M., W.B. Kannel, C.G. Tedeschi, T.K. Landau, and R.J. Garrison. 1979. The relation of antemortem characteristics to cardiovascular findings at necropsy—The Framingham Study. Atherosclerosis 34:145-157.
Ferro-Luzzi, A., P. Strazzullo, C. Scaccini, A. Siani, S. Sette, M.A. Mariani, P. Mastranzo, R.M. Dougherty, J.M. Iacono, and M. Mancini. 1984. Changing the Mediterranean diet: effects on blood lipids. Am. J. Clin. Nutr. 40:10271037.
Finegold, S.M., H.R. Attebery, and V.L. Sutter. 1974. Effect of diet on human fecal flora: comparison of Japanese and American diets. Am. J. Clin. Nutr. 27:1456-1469.
Finegold, S.M., D.J. Flora. H.R. Attebery, and V.L. Sutter. 1975. Fecal bacteriology of colonic polyp patients and control patients. Cancer Res. 35:3407-3417.
Finley, J.W., and D.E. Schwass. 1985. Xenobiotic Metabolism: Nutritional Effects. American Chemical Society Symposium Series 277. American Chemical Society, Washington, D.C. 382 pp.
Fleischman, A.T., M.L. Bierenbaum, A. Stier, H. Somol, P. Watson, and A.M. Naso. 1979. Hypotensive effect of increased dietary linoleic acid in mildly hypertensive humans. J. Med. Soc. N.J. 76:181-183.
Flow, B.L., T.C. Cartwright, T.J. Keuhl, G.E. Mott, D.C. Kraemer, A.W. Kruski, J.D. Williams, and H.C. McGill, Jr. 1981. Genetic effects on serum cholesterol concentrations in baboons. J. Hered. 72:97-103.
Folsom, A.R., K. Kuba, R.V. Leupker, D.R. Jacobs, and I.D. Frantz, Jr. 1983. Lipid concentrations in serum and EDTA-treated plasma from fasting and nonfasting normal persons, with particular regard to high-density lipoprotein cholesterol. Clin. Chem. 29:505-508.
Ford, C.H., R.B. McGandy, and F.J. Stare. 1972. An institutional approach to the dietary regulation of blood cholesterol in adolescent males. Prev. Med. 1:426-445.
Fox, J.C., H.C. McGill, Jr., K.D. Carey, and G.S. Getz. 1987. In vivo regulation of hepatic LDL receptor mRNA in the baboon. J. Biol. Chem. 262:7014-7020.
Franceschini, G., A. Bondioli, M. Mantero, M. Sirtori, G. Tattoni, G. Biasi, and C.R. Sirtori. 1982. Increased apoprotein B in very low density lipoproteins of patients with peripheral vascular disease. Arteriosclerosis 2:74-80.
Frank, G.C., G.S. Berenson, and L.S. Webber. 1978. Dietary studies and the relationship of diet to cardiovascular disease risk factor variables in 10-year-old children—The Bogalusa Heart Study. Am. J. Clin. Nutr. 31:328-340.
Frank, G.C., R.P. Farris, J.L. Cresanta, and T.A. Nicklas. 1986. Dietary intake as a determinant of cardiovascular risk factor variables. Part A: observations in a pediatric population. Pp. 254-291 in G.S. Berenson, ed. Causation of Cardiovascular Risk Factors in Children: Perspectives on Cardiovascular Risk in Early Life. Raven Press, New York.
Frantz, I.D., Jr., and J.B. Carey, Jr. 1961. Cholesterol content of human liver after feeding of corn oil and hydrogenated coconut oil. Proc. Soc. Exp. Biol. Med. 106:800-801.
Freedman, D.S., W.P. Newman III, R.E. Tracy, A.E. Voors, S.R. Srinivasan, L.S. Webber, C. Restrepo, J.P. Strong, and G.S. Berenson. 1988. Black-white differences in aortic fatty streaks in adolescence and early adulthood: the Bogalusa Heart Study. Circulation 77:856-864.
Frerichs, R.R., S.R. Srinivasan, L.S. Webber, and G.S. Berenson. 1976. Serum cholesterol and triglyceride levels in 3,446 children from a biracial community: the Bogalusa Heart Study. Circulation 54:302-309.
Frerichs, R.R., L.S. Webber, S.R. Srinivasan, and G.S. Berenson. 1978a. Relation of serum lipids and lipoproteins to obesity and sexual maturity in white and black children. Am. J. Epidemiol. 108:486-496.
Frerichs, R.R., S.R. Srinivasan, L.S. Webber, M.C. Rieth, and G.S. Berenson. 1978b. Serum lipids and lipoproteins at birth in a biracial population: the Bogalusa Heart Study. Pediatr. Res. 12:858-863.
Frerichs, R.R., L.S. Webber, A.W. Voors, S.R. Srinivasan, and G.S. Berenson. 1979. Cardiovascular disease risk factor variables in children at two successive years—the Bogalusa Heart Study. J. Chronic Dis. 32:251-262.
Freston, J.W., and I.A. Bouchier. 1968. Experimental cholelithiasis. Gut 9:2-4.
Freudenheim, J.L., and J.R. Marshall. 1988. The problem of profound mismeasurement and the power of epidemiological studies of diet and cancer. Nutr. Cancer 11:243-250.
Frick, M.H., O. Elo, K. Haapa, O.P. Heinonen, P. Heinsalmi, P. Helo, J.K. Huttunen, P. Kaitaniemi, P. Koskinen, V. Manninen, H. Maenpaa, M. Malkonen, M. Manttari, S. Norola, A. Pasternack, J. Pikkarainen, M. Romo, T. Sjoblom, and E.A. Nikkila. 1987. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N. Engl. J. Med. 317:1237-1245.
Friedman, G., and S.J. Goldberg. 1973. Normal serum cholesterol values. Percentile ranking in a middle-class pediatric population. J. Am. Med. Assoc. 225:610-612.
Friedman, G., and S.J. Goldberg. 1976. An evaluation of the safety of a low-saturated-fat, low-cholesterol diet beginning in infancy. Pediatrics 58:655-657.
Friedman, G.D., W.B. Kannel, and T.R. Dawber. 1966. The epidemiology of gallbladder disease: observations in the Framingham Study. J. Chronic Dis. 19:273-292.
Frohlich, E.D. 1977. Hemodynamics of hypertension. Pp. 1549 in J. Genest, E. Koiw, and O. Kuchel, eds. Hypertension: Physiopathology and Treatment. McGraw-Hill, New York.
Funch, J.P., B. Krogh, and H. Dam. 1960. Effects of butter, some margarines and arachis oil in purified diets on serum lipids and atherosclerosis in rabbits. Br. J. Nutr. 14:355360.
Funch, J.P., G. Kristensen, and H. Dam. 1962. Effects of various dietary fats on serum cholesterol, liver lipids and tissue pathology in rabbits. Br. J. Nutr. 16:497-506.
Garcia-Palmieri, M.R., J. Tillotson, E. Cordero, R. Costas, Jr., P. Sorlie, T. Gordon, W.B. Kannel, and A.A. Colon. 1977. Nutrient intake and serum lipids in urban and rural Puerto Rican men. Am. J. Clin. Nutr. 30:2092-2100.
Garcia-Palmieri, M.R., P. Sorlie, J. Tillotson, R. Costas, Jr., E. Cordero, and M. Rodriguez. 1980. Relationship of dietary intake to subsequent coronary heart disease incidence: the Puerto Rico Heart Health Program. Am. J. Clin. Nutr. 33:1818-1827.
Gardey, T., P.G. Burstyn, and T.G. Taylor. 1978. Fat induced hypertension in rabbits. I. The effects of fibre on the blood pressure increase induced by coconut oil. Proc. Nutr. Soc. 37:97A.
Gates, J.C., R.L. Huenemann, and R.J. Brand. 1975. Food
Page 238
choices of obese and non-obese persons. J. Am. Diet. Assoc. 67:339-343.
Gaubatz, J.W., C. Heideman, A.M. Gotto, Jr., J.D. Morrisett, and G.H. Dahlen. 1983. Human plasma lipoprotein [a]. Structural properties. J. Biol. Chem. 258:4582-4589.
Geer, J.C., H.C. McGill, Jr., W.B. Robertson, and J.P. Strong. 1968. Histologic characteristics of coronary artery fatty streaks. Lab. Invest. 18:565-570.
Gelenberg, A.J., J.D. Wojcik, and J.H. Growdon. 1979. Lecithin for the treatment of tardive dyskinesia. Pp. 285303 in A. Barbeau, J.H. Growdon, and R.J. Wurtman, eds. Nutrition and the Brain, Vol. 5. Choline and Lecithin in Brain Disorders. Raven Press, New York.
Gerhardsson, M., U. Rosenqvist, A. Ahlbom, and L.A. Carlson. 1986. Serum cholesterol and cancer—a retrospective case-control study. Int. J. Epidemiol. 15:155-159.
Gershfeld, N.L. 1979. Selective phospholipid adsorption and atherosclerosis. Science 204:506-508.
Gertler, M.M., S.M. Garn, and P.D. White. 1950a. Diet, serum cholesterol and coronary artery disease. Circulation 2: 696-704.
Gertler, M.M., S.M. Garn, and J. Lerman. 1950b. The interrelationships of serum cholesterol, cholesterol esters and phospholipids in health and in coronary artery disease. Circulation 2:205-214.
Ghoshal, A.K., and E. Farber. 1984. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis 5:1367-1370.
Ginsberg, J., N.A. Le, C. Mays, J. Gibson, and W.V. Brown. 1981. Lipoprotein metabolism in nonresponders to increased dietary cholesterol. Arteriosclerosis 1:463-470.
Glenn, F., and C.K. McSherry. 1970. The baboon and experimental cholelithiasis. Arch. Surg. 100:105-108.
Glueck, C.J., R.W. Fallat, R. Tsang, and C.R. Buncher. 1974. Hyperlipidemia in progeny of parents with myocardial infarction before age 50. Am. J. Dis. Child. 127:70-75.
Glueck, C.J., M.M. Hastings, C. Allen, E. Hogg, L. Baehler, P.S. Gartside, D. Phillips, M. Jones, E.J. Hollenbach, B. Braun, and J.B. Anastasia. 1982. Sucrose polyester and covert caloric dilution. Am. J. Clin. Nutr. 35:1352-1359.
Glueck, C.J., D.J. Gordon, J.J. Nelson, C.E. Davis, and H.A. Tyroler. 1986. Dietary and other correlates of changes in total and low density lipoprotein cholesterol in hypercholesterolemic men: the Lipid Research Clinics Coronary Primary Prevention Trial. Am. J. Clin. Nutr. 44:489-500.
Goldbourt, U. 1987. High risk versus public health strategies in primary prevention of coronary heart disease. Am. J. Clin. Nutr. 45 Suppl. 5:1185-1192.
Goldbourt, U., E. Holtzman, and H.N. Neufeld. 1985. Total and high density lipoprotein cholesterol in the serum and risk of mortality: evidence of a threshold effect. Br. Med. J. 290:1239-1243.
Goldstein, J.L., and M.S. Brown. 1983. Familial hypercholesterolemia. Pp. 672-712 in J.B. Stanbury, J.B. Wyngaarden, D.S. Fredrickson, J.L Goldstein, and M.S. Brown, eds. The Metabolic Basis of Inherited Disease, 5th ed. McGraw-Hill, New York.
Goldstein, J.L., H.F. Hoff, Y.K. Ho, S.K. Basu, and M.S. Brown. 1981. Stimulation of cholesteryl ester synthesis in macrophages by extracts of atherosclerotic human aortas and complexes of albumin/cholesteryl esters. Arteriosclerosis 1: 210-226.
Goldstein, J.L., T. Kita, and M.S. Brown. 1983. Defective lipoprotein receptors and atherosclerosis. Lessons from an animal counterpart of familial hypercholesterolemia. N. Engl. J. Med. 309:288-296.
Goodnight, S.H., Jr., W.S. Harris, W.E. Connor, and D.R. Illingworth. 1982. Polyunsaturated fatty acids, hyperlipidemia, and thrombosis. Arteriosclerosis 2:87-113.
Gordon, D.J., K.M. Salz, K.J. Roggenkamp, and F.A. Franklin, Jr. 1982. Dietary determinants of plasma cholesterol change in the recruitment phase of the Lipid Research Clinics Coronary Primary Prevention Trial. Arteriosclerosis 2:537-548.
Gordon, D.J., J. Knoke, J.L. Probstfield, R. Superko, and H.A. Tyroler. 1986. High-density lipoprotein cholesterol and coronary heart disease in hypercholesterolemic men: the Lipid Research Clinics Coronary Primary Prevention Trial. Circulation 74:1217-1225.
Gordon, T., M.R. Garcia-Palmieri, A. Kagan, W.B. Kannel, and J. Schiffman. 1974. Differences in coronary heart disease mortality in Framingham, Honolulu and Puerto Rico. J. Chronic Dis. 27:329-344.
Gordon, T., W.P. Castelli, M.C. Hjortland, W.B. Kannel, and T.R. Dawber. 1977. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 62:707-714.
Gordon, T., A. Kagan, M. Garcia-Palmieri, W.B. Kannel, W.J. Zukel, J. Tillotson, P. Sorlie, and M. Hjortland. 1981a. Diet and its relation to coronary heart disease and death in three populations. Circulation 63:500-515.
Gordon, T., W.B. Kannel, W.P. Castelli, and T.R. Dawber. 1981b. Lipoproteins, cardiovascular disease, and death: the Framingham Study. Arch. Intern. Med. 141:1128-1131.
Graham, S., H. Dayal, M. Swanson, A. Mittelman, and G. Wilkinson. 1978. Diet in the epidemiology of cancer of the colon and rectum. J. Natl. Cancer Inst. 61:709-714.
Graham, S., B. Humphey, J. Marshall, R. Priore, T. Byers, T. Rzepka, C. Mettlin, and J.E. Pontes. 1983. Diet in the epidemiology of carcinoma of the prostate gland. J. Natl. Cancer Inst. 70:687-692.
Grande, F., J.T. Anderson, C. Chlouverakis, M. Proja, and A. Keys. 1965. Effect of dietary cholesterol on man's serum lipids. J. Nutr. 87:52-62.
Grande, F., J.T. Anderson, and A. Keys. 1970. Comparison of effects of palmitic and stearic acids in the diet on serum cholesterol in man. Am. J. Clin. Nutr. 23:1184-1193.
Grasso, S., B. Gunning, K. Imaichi, G. Michaels, and L. Kinsell. 1962. Effects of natural and hydrogenated fats of approximately equal dienoic acid content upon plasma lipids. Metabolism 11:920-924.
Greenhalgh, R.M., D.S. Rosengarten, I. Mervart, J.S. Calnan, B. Lewis, and P. Martin. 1971. Serum lipids and lipoproteins in peripheral vascular disease. Lancet 2:947-950.
Gresham, G.A., and A.N. Howard. 1960. The independent production of atherosclerosis and thrombosis in the rat. Br. J. Exp. Pathol. 41:395-402.
Greten, H., H. Raetzer, A. Stiehl, and G. Schettler. 1980. The effect of polyunsaturated phosphatidylcholine on plasma lipids and fecal sterol excretion. Atherosclerosis 36: 81-88.
Groen, J., B.K. Tjiong, C.E. Kamminga, and A.F. Willebrands. 1952. The influence of nutrition, individuality and some other factors, including various forms of stress, on the serum cholesterol; an experiment of nine months' duration in 60 normal human volunteers. Voeding 13:556-587.
Groen, J.J., K.B. Tijong, M. Koster, A.F. Willebrands, G. Verdonck, and M. Pierloot. 1962. The influence of nutri-
Page 239
tion and ways of life on blood cholesterol and the prevalence of hypertension and coronary heart disease among Trappist and Benedictine Monks. Am. J. Clin. Nutr. 10:456-470.
Grundy, S.M. 1975. Effects of polyunsaturated fats on lipid metabolism in patients with hypertriglyceridemia. j. Clin. Invest. 55:269-282.
Grundy, S.M. 1979. Dietary fats and sterols. Pp. 89-118 in R.I. Levy, B.M. Rifkind, B.H. Dennis, and N. Ernst, eds. Nutrition, Lipids, and Coronary Heart Disease: A Global View. Nutrition in Health and Disease, Vol. 1. Raven Press, New York.
Grundy, S.M. 1981. Saturated fats and coronary heart disease. Pp. 57-78 in M. Winick, ed. Nutrition and the Killer Diseases. Wiley, New York.
Grundy, S.M. 1986. Comparison of monounsaturated fatty acids and carbohydrates for lowering plasma cholesterol. N. Engl. J. Med. 314:745-748.
Grundy, S.M. 1987. Monounsaturated fatty acids, plasma cholesterol, and coronary heart disease. Am. J. Clin. Nutr. 45:1168-1175.
Grundy, S.M., and E.H. Ahrens, Jr. 1970. The effects of unsaturated dietary fats on absorption, excretion, synthesis, and distribution of cholesterol in man. J. Clin. Invest. 49: 1135-1152.
Grundy, S.M., and H.Y. Mok. 1977. Determination of cholesterol absorption in man by intestinal perfusion. J. Lipid Res. 18:263-271.
Grundy, S.M., and G.L. Vega. 1988. Plasma cholesterol responsiveness to saturated fatty acids. Am. J. Clin. Nutr. 47:822-824.
Grundy, S.M., D. Nix, M.F. Whelan, and L Franklin. 1986. Comparison of three cholesterol-lowering diets in normolipidemic men. J. Am. Med. Assoc. 256:2351-2355.
Grundy, S.M., L. Florentin, D. Nix, and M.F. Whelan. 1988. Comparison of monounsaturated fatty acids and carbohydrates for reducing raised levels of plasma cholesterol in man. Am. J. Clin. Nutr. 47:965-969.
Gurll, N., and L. DenBesten. 1978. Animal models of human cholesterol gallstone disease: a review. Lab. Animal Sci. 28: 428-432.
Haberland, M.E., D. Fong, and L. Cheng. 1988. Malondialdehyde-altered protein occurs in atheroma of Watanabe heritable hyperlipidemic rabbits. Science 241:215-218.
Haenzel, W., F.B. Locke, and M. Segi. 1980. A case-control study of large bowel cancer in Japan. J. Natl. Cancer Inst. 64:17-22.
Halfon, S.T., S. Eisenberg, M. Baras, A.M. Davies, G. Halperin, and Y. Stein. 1982a. Plasma cholesterol, triglyceride and high-density lipoprotein-cholesterol levels in 17-year-old Jerusalem offspring of Jews from 19 countries of birth. Isr. J. Med. Sci. 18:1121-1130.
Halfon, S.T., B.M. Rifkind, S. Harlap, N.A. Kaufmann, M. Baras, P.E. Slater, G. Halperin, S. Eisenberg, A.M. Davies, and Y. Stein. 1982b. Plasma lipids and lipoproteins in adult Jews of different origin: the Jerusalem Lipid Research Clinic prevalence study. Isr. J. Med. Sci. 18:1113-1120.
Harlan, W.R., A.L Hull, R.P. Schmouder, F.E. Thompson, F.A. Larkin, and J.R. Landis. 1983. Dietary Intake and Cardiovascular Risk Factors, Part II. Serum Urate, Serum Cholesterol, and Correlates. Vital and Health Statistics, Ser. 11, No. 227. DHHS Publ. No. (PHS) 83-1677. National Center for Health Statistics, Public Health Service, U.S. Department of Health and Human Services, Hyattsville, Md. 94 pp.
Harlap, S., M. Baras, Y. Friedlander, N.A. Kaufmann, S. Eisenberg, A.M. Davies, and Y. Stein. 1982. Contributions of different lipoprotein fractions to variations in total cholesterol between Israeli origin groups and social classes. Isr. J. Med. Sci. 18:1131-1136.
Harris, W.S., W.E. Connon, and M.P. McMurry. 1983. The comparative reductions of the plasma lipids and lipoproteins by dietary polyunsaturated fats: salmon oil versus vegetable oils. Metabolism 32:179-184.
Harris, W.S., W.E. Connor, S.B. Inkeles, and D.R. Illingworth. 1984. Dietary omega-3 fatty acids prevent carbohydrate-induced hypertriglyceridemia. Metabolism 33:1016-1019.
Hashim, S.A., A. Argeaga, and T.B. Van Itallie. 1960. Effect of a saturated medium-chain triglyceride on serum-lipids in man. Lancet 1:1105-1108.
Havel, R.J. 1987. Origin, metabolic fate, and metabolic function of plasma lipoproteins. Pp. 117-141 in D. Steinberg and J.M. Olefsky, eds. Hypercholesterolemia and Atherosclerosis: Pathogenesis and Prevention. Churchill Livingstone, New York.
Hayashi, E, T. Maeda, R. Hasegawa, and T. Tomita. 1978. The effect of myo-inositol deficiency on lipid metabolism in rats. III. The mechanism of an enhancement in lipolysis due to myo-inositol deficiency in rats. Biochem. Biophys. Acta 531:197-205.
Hayashi, Y., M. Nagao, T. Sugimura, S. Takayama, L Tomatis, L.W. Wattenberg, and G.N. Wogan. 1986. Proceedings of the 16th International Symposium of the Princess Takamatsu Cancer Research Fund, Tokyo, 1985. Diet, Nutrition and Cancer. Japan Scientific Society Press, Tokyo. 345 pp.
Heaton, K.W. 1973. The epidemiology of gallstones and suggested aetiology. Clin. Gastroenterol. 2:67-83.
Heckers, H., M. Korner, T.W.L. Tuschen, and F.M. Melcher. 1977. Occurrence of individual trans-isomeric fatty acids in human myocardium, jejunum and aorta in relation to different degrees of atherosclerosis. Atherosclerosis 28:389-398.
Hegsted, D.M. 1986. Serum-cholesterol reponse to dietary cholesterol: a re-evaluation. Am. J. Clin. Nutr. 44:299-305.
Hegsted, D.M., R.B. McGandy, M.L Myers, and F.J. Stare. 1965. Quantitative effects of dietary fat on serum cholesterol in man. Am. J. Clin. Nutr. 17:281-295.
Heideman, C.L, and H.F. Hoff. 1982. Lipoproteins containing apolipoprotein A-I extracted from human aortas. Biochem. Biophys. Acta 711:431-444.
Hellstrom, K., and S. Lindstedt. 1966. Studies on the formation of cholic acid in subjects given standardized diet with butter or corn oil as dietary fat. Am. J. Clin. Nutr. 18:46-59.
Hems, G. 1980. Associations between breast-cancer and mortality rates, childbearing and diet in the United Kingdom. Br. J. Cancer 41:429-437.
Herold, P.M., and J.E. Kinsella. 1986. Fish oil consumption and decreased risk of cardiovascular disease: a comparison of findings from animal and human feeding trials. Am. J. Clin. Nutr. 43:566-598.
Herrick, J.B. 1912. Clinical features of sudden obstruction of the coronary arteries. J. Am. Med. Assoc. 59:2015-2020.
Heshmat, M.Y., L. Kaul, J. Kovi, M.A. Jackson, A.G. Jackson, G.W. Jones, M. Edson, J.P. Enterline, R.G. Worrell, and S.L. Perry. 1985. Nutrition and prostate cancer: a case-control study. Prostate 6:7-17.
Page 240
Heymann, W., and F. Rack. 1943. Independence of serum cholesterol from exogenous cholesterol in infants and in children. Am. J. Dis. Child. 65:235-246.
Hiatt, R.A., and B.H. Fireman. 1986. Serum cholesterol and the incidence of cancer in a large cohort. J. Chronic Dis. 39: 861-870.
Hill, M.J. 1981. Diet and the human intestinal flora. Cancer Res. 41:3778-3780.
Hill, M.J., B.S. Drasar, G. Hawksworth, V. Aries, J.S. Crowther, and R.E. Williams. 1971. Bacteria and aetiology of cancer of the large bowel. Lancet 1:95-100.
Hill, M.J., B.S. Drasar, R.E. Williams, T.W. Meade, A.G. Cox, J.E. Simpson, and B.C. Morson. 1977. Faecal bile-acids and clostridia in patients with cancer of the large bowel. Lancet 1:535-539.
Hillyard, L.A., and S. Abraham. 1979. Effect of dietary polyunsaturated fatty acids on growth of mammary adenocarcinomas in mice and rats. Cancer Res. 39:4430-4437.
Hinds, M.W., L.N. Kolonel, J.H. Hankin, and J. Lee. 1983. Dietary cholesterol and lung cancer risk in a multiethnic population in Hawaii. Int. J. Cancer 32:727-732.
Hirayama, T. 1977. Changing patterns of cancer in Japan with special reference to the decrease in stomach cancer mortality. Pp. 55-75 in H.H. Hiatt, J.D. Watson, and J.A. Winston, eds. Origins of Human Cancer, Book A. Incidence of Cancer in Humans. Cold Spring Harbor Laboratory, New York.
Hirohata, T., A.G. Nomura, J.H. Hankin, L.N. Kolonel, and J. Lee. 1987. An epidemiologic study on the association between diet and breast cancer. J. Natl. Cancer Inst. 78: 595-600.
Hislop, T.G., A.J. Coldman, J.M. Elwood, G. Brauer, and L. Kan. 1986. Childhood and recent eating patterns and risk of breast cancer. Cancer Detect. Prev. 9:47-58.
Hjermann, I., K. Velve Byre, I. Holme, and P. Leren. 1981. Effect of diet and smoking intervention on the incidence of coronary heart disease. Report from the Oslo Study Group of a randomised trial in healthy men. Lancet 2:1303-1310.
Ho, K.J. 1976. Comparative studies on the effect of cholesterol feeding on biliary composition. Am. J. Clin. Nutr. 29:698-704.
Hoff, H.F., and J.W. Gaubatz. 1975. Ultrastructural localization of plasma lipoproteins in human intracranial arteries. Virchows Arch. 369:111-121.
Hoff, H.F., and J.W. Gaubatz. 1982. Isolation, purification, and characterization of a lipoprotein containing Apo B from the human aorta. Atherosclerosis 42:273-297.
Hoff, H.F., and R.E. Morton. 1985. Lipoproteins containing apo B extracted from human aortas. Structure and function. Ann. N.Y. Acad. Sci. 454:183-194.
Hoff, H.F., R.L. Jackson, S.J. Mao, and A.M. Gotto, Jr. 1974. Localization of low-density lipoproteins in atherosclerotic lesions from human normolipemics employing a purified fluorescent-labeled antibody. Biochem. Biophys. Acta 351:407-415.
Hoff, H.F., J.L. Titus, R.J. Bajardo, R.L. Jackson, A.M. Gotto, M.E. DeBakey, and .T. Lie. 1975a. Lipoproteins in atherosclerotic lesions. Localization by immunofluorescence of apo-low-density lipoproteins in human atherosclerotic arteries from normal and hyperlipoproteinemics. Arch. Pathol. 99:253-258.
Hoff, H.F., C.L. Heideman, G.P. Noon, and J.S. Meyer. 1975b. Localization of apo-lipoproteins in human carotid artery plaques. Stroke 6:531-534.
Hoff, H.F., C.L Heideman, R.L. Jackson, R.J. Bayardo, H.S. Kim, and A.M. Gotto, Jr. 1975c. Localization patterns of plasma apolipoproteins in human atherosclerotic lesions. Circ. Res. 37:72-79.
Hoff, H.F., C.L Heideman, A.M. Gotto, Jr., and J.W. Gaubatz. 1977a. Apolipoprotein B retention in the grossly normal and atherosclerotic human aorta. Circ. Res. 41:684-690.
Hoff, H.F., C.L. Heideman, J.W. Gaubatz, A.M. Gotto, Jr., E.E. Erickson, and R.L. Jackson. 1977b. Quantification of apolipoprotein B in grossly normal human aorta. Circ. Res. 40:56-4.
Hoff, H.F., J.W. Gaubatz, and A.M. Gotto, Jr. 1978. Apo B concentration in the normal human aorta. Biochem. Biophys. Res. Commun. 85:1424-1430.
Hoff, H.F., W.A. Bradley, C.L. Heideman, J.W. Gaubatz, M.D. Karagas, and A.M. Gotto, Jr. 1979a. Characterization of low density lipoprotein-like particle in the human aorta from grossly normal and atherosclerotic regions. Biochem. Biophys. Acta 573:361-374.
Hoff, H.F., M. Karagas, C.L. Heideman, J.W. Gaubatz, and A.M. Gotto, Jr. 1979b. Correlation in the human aorta of apo B fractions with tissue cholesterol and collagen content. Atherosclerosis 32:259-268.
Hoffman, D.R., E.O. Uthus, and W.E. Cornatzer. 1980. Effect of diet on choline phosphotransferase, phosphatidylethanolamine methyltransferase and phosphatidyldimethylethanolamine methyltransferase in liver microsomes. Lipids 15:439-446.
Hoffmann, P., C. Taube, K. Pönicke, W. Forster, L. Somova, V. Orbetova, and F. Davidova. 1978. Influence of linoleic acid content on arterial blood pressure of salt loaded rats. I. Effects on prostaglandin metabolism and sympathetic nervous system. Acta Bio. Med. Ger. 37:863-867.
Holman, R.L. 1961. Atherosclerosis-a pediatric nutrition problem? Am. J. Clin. Nutr. 9:565-569.
Holman, R.L., H.C. McGill, Jr., J.P. Strong, and J.C. Geer. 1958. The natural history of atherosclerosis. The early aortic lesions as seen in New Orleans in the middle of the 20th century. Am. J. Pathol. 34:209-235.
Holman, R.L., W.A. Blanc, and D. Andersen. 1959. Decreased aortic atherosclerosis in cystic fibrosis of the pancreas. Pediatrics 24:34-39.
Holman, R.T. 1986. Nutrition and functional requirements for essential fatty acids. Prog. Clin. Biol. Res. 222:211-228.
Holman, R.T., W.W. Christie, H. Sprecher, M. Crawford, B. Lewis, K.K. Carroll, and K.K. Wahle, eds. 1986. Dietary fats and cancer. Pp. 527-553 in Progress in Lipid Research, Vol. 25. Essential Fatty Acids, Prostaglandins and Leukotrienes. Pergamon Press, Oxford.
Holme, I., S.C. Enger, A. Helgeland, I. Hjermann, P. Leren, P.G. Lund-Larsen, L.A. Solberg, and J.P. Strong. 1981. Risk factors and raised atherosclerotic lesions in coronary and cerebral arteries. Statistical analysis from the Oslo study. Arteriosclerosis 1:250-256.
Holub, B.J. 1982. The nutritional significance, metabolism, and function of myo-inositol and phosphatidylinositol in health and disease. Adv. Nutr. Res. 4:107-141.
Holub, B.J. 1986. Metabolism and function of myo-inositol and inositol phospholipids. Annu. Rev. Nutr. 6:563-597.
Holzbach, R.T. 1984. Animal models of cholesterol gallstone disease. Hepatology 4:191S-198S.
Hoover, G.A., R.J. Nocolosi, J.E. Corey, M. el Lozy, and K.C. Hayes. 1978. Inositol deficiency in the gerbil: altered
Page 241
hepatic lipid metabolism and triglyceride secretion. J. Nutr. 108:1588-1594.
Hoover, K.L., P.H. Lynch, and L.A. Poirier. 1984. Profound postinitiation enhancement by short-term severe methionine, choline, vitamin B12 and folate deficiency of hepatocarcinogenesis in F344 rats given a single low-dose diethylnitrosamine injection. J. Natl. Cancer Inst. 73:1327-1336.
Hopkins, G.J., and K.K. Carroll. 1985. Role of diet in cancer prevention. J. Environ. Pathol. Toxicol. Oncol. 5:279-298.
Hopkins, G.J., and C.E. West. 1976. Possible roles of dietary fats in carcinogenesis. Life Sci. 19:1103-1116.
Horlick, L. 1959. Studies on the regulation of serum-cholesterol levels in man; the effects of corn oil, ethyl stearate, hydrogenated soybean oil, and nicotinic acid when added to a very low-fat basal diet. Lab. Invest. 8:723-735.
Horlick, L. 1960. The effect of artificial modification of food on the serum cholesterol level. Can. Med. Assoc. J. 83: 1186-1192.
Howard, B.V. 1986. Obesity, cholelithiasis, and lipoprotein metabolism in man. Atherosclerosis Rev. 15:169-186.
Howe, G.R. 1985. The use of polytomous dual response data to increase power in case-control studies: an application to the association between dietary fat and breast cancer. J. Chronic Dis. 38:663-670.
Howe, G.R., A.B. Miller, and M. Jain. 1986. Re: "Total energy intake": implications for epidemiologic analyses. Am. J. Epidemiol. 124:157-159.
Howell, J.H., M.E. Marsh, E.S. Thiel, and R.T. Holzbach. 1983. Hepatic microsomal activities of cholesterol 7 alphahydroxylase and 3-hydroxy-3-methylglutaryl-CoA reductase in the prairie dog. An animal model for cholesterol gallstone disease. Biochim. Biophys. Acta 753:32-39.
Hoyenga, K.B., and K.T. Hoyenga. 1982. Gender and energy balance: sex differences in adaptations for feast and famine. Physiol. Behav. 28:545-563.
Hubbard, N.E., and K.L. Erickson. 1987. Enhancement of metastasis from a transplantable mouse mammary tumor by dietary linoleic acid. Cancer Res. 47:6171-6175.
Hughson, W.G., J.I. Mann, and A. Garrod. 1978. Intermittent claudication: prevalence and risk factors. Br. Med. J. 1: 1379-1381.
Hulley, S.B., R.H. Rosenman, R.D. Bawol, and R.J. Brand. 1980. Epidemiology as a guide to clinical decisions. The association between triglycerides and coronary heart disease. N. Engl. J. Med. 302:1383-1389.
Hunter, J.E., and T.H. Applewhite. 1986. Isometric fatty acids in the U.S. diet: levels and health perspectives. Am. J. Clin. Nutr. 44:707-717.
Iacono, J.M., R.M. Dougherty, and P. Puska. 1982. Reduction of blood pressure associated with dietary polyunsaturated fat. Hypertension 4:III34-III42.
Ignatovski, A.I. 1908. Influence of animal food on the organism of rabbits. Izv. Imp. Voyenno-Med. Akad. Peter. 16:154-176.
Illingworth, D.R., W.S. Harris, and W.E. Connor. 1984. Inhibition of low density lipoprotein synthesis by dietary omega-3 fatty acids in humans. Arteriosclerosis 4:270-275.
ILSI/NF (International Life Sciences Institute/Nutrition Foundation). 1987. Calories and energy expenditure in carcinogenesis. Proceedings of a symposium, Washington, D.C., February 24 and 25, 1986. Am. J. Clin. Nutr. 45:149-372.
Imai, H., K.T. Lee, S. Pastori, E. Panlilio, R. Florentin, and W.A. Thomas. 1966. Arteriosclerosis in rabbits. Architectural and subcellular alterations of smooth muscle cells of aortas in response to hyperlipemia. Exp. Mol. Pathol. 5: 273-310.
Imai, H., N.T. Werthessen, C.B. Taylor, and K.T. Lee. 1976. Angiotoxicity and atherosclerosis due to contaminants to USP-grade cholesterol. Arch. Pathol. Lab. Med. 100:565-572.
Imai, H., N.T. Werthessen, V. Subramanyam, P.W. LeQuesne, A.H. Soloway, and M. Kanisawa. 1980. Angiotoxicity of oxygenated sterols and possible precursors. Science 207:651-653.
Innis, S.M. 1985. The role of diet during development on the regulation of adult cholesterol homeostasis. Can. J. Physiol. Pharmacol. 63:557-564.
Ip, C. 1987. Fat and essential fatty acid in mammary carcinogenesis. Am. J. Clin. Nutr. 45:218-224.
Ip, C., C.A. Carter, and M.M. Ip. 1985. Requirement of essential fatty acid for mammary tumorigenesis in the rat. Cancer Res. 45:1997-2001.
Ip, C., M.M. Ip, and P. Sylvester. 1986. Relevance of trans fatty acids and fish oil on animal tumorigenesis studies. Prog. Clin. Biol. Res. 222:283-294.
Isacsson, S.O. 1972. Venous occlusion plethysmography in 55-year old men. A population study in Malmo, Sweden. Acta Med. Scand., Suppl. 537:1-62.
Ishikawa, T., N. Fidge, D.S. Thelle, O.H. Forde, and N.E. Miller. 1978. The Tromso Heart Study: serum apolipoprotein Al concentration in relation to future coronary heart disease. Eur. J. Clin. Invest. 8:179-182.
Iso, H., D.R. Jacobs, Jr., D. Wentworth, J.D. Neaton, and J. Cohen. 1988. Relationship of serum cholesterol to risk of different types of stroke: 28th conference on cardiovascular disease epidemiology. CVD Epidemiol. Newsletter (AHA) 43:40.
Italian National Research Council. 1982. Incidence and prediction of coronary heart disease in two Italian rural population samples followed-up for 20 years. Acta Cardiol. 37: 129-145.
Jackson, R.L., J.D. Morrisett, H.J. Pownall, A.M. Gotto, Jr., A. Kamio, H. Imai, R. Tracy, and F.A. Kummerow. 1977. Influence of dietary trans-fatty acids on swine lipoprotein composition and structure. J. Lipid Res. 18:182-190.
Jackson, R.L., O.D. Taunton, J.D. Morrisett, and A.M. Gotto, Jr. 1978. The role of dietary polyunsaturated fat in lowering blood cholesterol in man. Circ. Res. 42:447-453.
Jackson, R.L, M.L Kashyap, R.L Barnhart, C Allen, E. Hogg, and C.J. Glueck. 1984. Influence of polyunsaturated and saturated fats on plasma lipids and lipoproteins in man. Am. J. Clin. Nutr. 39:589-597.
Jacobs, D.R., Jr., J.T. Anderson, and H. Blackburn. 1979. Diet and serum cholesterol: do zero correlations negate the relationship? Am. J. Epidemiol. 110:77-87.
Jain, M., G.M. Cook, F.G. Davis, M.G. Grace, G.H. Howe, and A.B. Miller. 1980. A case-control study of diet and colo-rectal cancer. Int. J. Cancer 26:757-768.
Jain, M.G., L. Harrison, G.R. Howe, and A.B. Miller. 1982. Evaluation of a self-administered dietary questionnaire for use in a cohort study. Am. J. Clin. Nutr. 36:931-935.
Janakidevi, K., K.T. Lee, M. Kroms, H. Imai, and W.A. Thomas. 1984. Mosaicism in female hybrid hares heterozygous for glucose-6-phospate dehydrogenase. VI. Production of monotypism in the aortas of 4 of 10 mosaic hares fed cholesterol oxidation products. Exp. Mol. Pathol. 41:354-362.
Jandacek, R.J., M.R. Webb, and F.H. Mattson. 1977. Effect
Page 242
of an aqueous phase on the solubility of cholesterol in an oil phase. J. Lipid Res. 18:203-210.
Jensen, O.M., R. MacLennan, and J. Wahrendorf. 1982. Diet, bowel function, fecal characteristics, and large bowel cancer in Denmark and Finland. Nutr. Cancer 4:5-19.
Jensen, R.G., M.M. Hagerty, and K.E. McMahon. 1978. Lipids of human milk and infant formulas: a review. Am. J. Clin. Nutr. 31:990-1016.
Johnson, F.L., R.W. St. Clair, and L.L. Rudel. 1985. Effects of the degree of saturation of dietary fat on the hepatic production of lipoproteins in the African green monkey. J. Lipid Res. 26:403-417.
Johnson, K.G., K. Yano, and H. Kato. 1968. Coronary heart disease in Hiroshima, Japan: a report of a six-year period of surveillance, 1958-1964. Am. J. Public Health 58:1355-1367.
Jokinen, M.P., T.B. Clarkson, and R.W. Prichard. 1985. Animal models in atherosclerosis research. Exp. Mol. Pathol. 42:1-28.
Jones, D.Y., A. Schatzkin, S.B. Green, G. Block, L.A. Brinton, R.G. Ziegler, R. Hoover, and P.R. Taylor. 1987a. Dietary fat and breast cancer in the National Health and Nutrition Examination Survey. 1. Epidemiological Followup Study. J. Natl. Cancer Inst. 79:465-471.
Jones, D.Y., J.T. Judd, P.R. Taylor, W.S. Campbell, and P.P. Nair. 1987b. Influence of caloric contribution and saturation of dietary fat on plasma lipids in premenopausal women. Am. J. Clin. Nutr. 45:1451-1456.
Joossens, J.V., M.J. Hill, and J. Geboers, eds. 1985. Diet and Human Carcinogenesis: Proceeding of the 3rd Annual Symposium of the European Organization for Cooperation in Cancer Prevention Studies (ECP). International Congress Series No. 685. Excerpta Medica, Amsterdam. 343 pp.
Judd, J.T., M.W. Marshall, and J.J. Canary. 1981. Changes in blood pressure and blood lipids of adult men consuming modified fat dies. Pp. 129-141 in G.R. Beecher, ed. Beltsville Symposia in Agricultural Research, IV. Human Nutrition Research. Allanheld, Osmun, & Co., Totowa, N.J.
Jurkowski, J.J., and W.T. Cave, Jr. 1985. Dietary effects of menhaden oil on the growth and membrane lipid composition of rat mammary tumors. J. Natl. Cancer Inst. 74:1145-1150.
Kagan, A., B.R. Harris, W. Winkerstein, Jr., K.G. Johnson, H. Kato, S.L. Syme, G.G. Rhoads, M.L Gay, M.Z. Nichaman, H.B. Hamilton, and J. Tillotson. 1974. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: Demographic, physical, dietary and biochemical characteristics. J. Chronic Dis. 27:345-364.
Kagan, A.K, J.S. Popper, and G.G. Rhoads. 1980. Factors related to stroke incidence in Hawaii Japanese men. The Honolulu Heart Study. Stroke 11:14-21.
Kagan, A., D.L. McGee, K. Yano, G.G. Rhoads, and A. Nomura. 1981. Serum cholesterol and mortality in a Japanese-American population: the Honolulu Heart Program. Am. J. Epidemiol. 114:11-20.
Kagan, A., J.S. Popper, G.G. Rhoads, and K. Yano. 1985. Dietary and other risk factors for stroke in Hawaiian Japanese men. Stroke 16:390-396.
Kahn, H.A., J.H. Medalie, H.N. Neufeld, E. Riss, M. Balogh, and J.J. Groen. 1969. Serum cholesterol: its distribution and association with dietary and other variables in a survey of 10,000 men. Isr. J. Med. Sci. 5:1117-1127.
Kanarek, R.B., T.G. Feldman, and C. Hanes. 1981 Patterns of dietary self selection in VMH lesioned rats. Physiol. Behav. 29:337-343.
Kannel, W.B., and T. Gordon. 1982. The search for an optimum serum cholesterol. Lancet 2:374-375.
Kannel, W.B., and D.L. McGee. 1985. Update on some epidemiologic features of intermittent claudication: the Framingham Study. J. Am. Geriatr. Soc. 33:13-18.
Kannel, W.B., and P.A. Wolf. 1983. Epidemiology of cerebrovascular disease. Pp. 1-24 in R.W.R. Russel, ed. Vascular Disease of the Central Nervous System, 2nd ed. Churchill Livingstone, Edinburgh.
Kao, V.C., and R.W. Wissler. 1965. A study of the immunohistochemical localization of serum lipoproteins and other plasma proteins in human atherosclerotic lesions. Exp. Mol. Pathol. 4:465-479.
Karmali, R.A. 1987. Omega-3 fatty acids and cancer: a review. Pp. 222-232 in W.E.M. Lands, ed. Proceedings of the AOCS Short Course on Polyunsaturated Fatty Acids and Eicosanoids. American Oil Chemists' Society, Champaign, Ill.
Karmali, R.A., J. Marsh, and C. Fuchs. 1984. Effect of omega-3 fatty acids on growth of a rat tumor. J. Natl. Cancer Inst. 73:457-461.
Katan, M.B., and A.C. Beynen. 1987. Characteristics of human hypo- and hyperresponders to dietary cholesterol. Am. J. Epidemiol. 125:387-399.
Katan, M.B., A.C. Beynen, J.H. de Vries, and A. Nobels. 1986. Existence of consistent hypo- and hyperresponders to dietary cholesterol in man. Am. J. Epidemiol. 123:221-234.
Katan, M.B., M.A. Berns, J.F. Glatz, J.T. Knuiman, A. Nobels, and J.H. de Vries. 1988. Congruence of individual responsiveness to dietary cholesterol and to saturated fat in humans. J. Lipid Res. 29:883-892.
Kato, H., J. Tillotson, M.Z. Nichaman, G.G. Rhoads, and H.B. Hamilton. 1973. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California. Serum lipids and diet. Am. J. Epidemiol. 97:372-385.
Katsouyanni, K., D. Trichopoulos, P. Boyle, E. Xirouchaki, A. Trichopoula, B. Lisseos, S. Vasilaros, and B. MacMahon. 1986. Diet and breast cancer: a case-control study in Greece. Int. J. Cancer 38:815-820.
Katz, E.B., and E.S. Boylan. 1987. Stimulating effect of high polyunsaturated fat diet on lung metastasis from the 13762 mammary adenocarcinoma in female retired breeder rats. J. Natl. Cancer Inst. 79:351-358.
Katz, L.N., and J. Stamler. 1953. Experimental atherosclerosis. Pp. 120-290 in Experimental Atherosclerosis. C.C. Thomas, Springfield, Ill.
Katz, L.N., J. Stamler, and R. Pick. 1958. Nutrition and Atherosclerosis. Lea & Febiger, Philadelphia. 146 pp.
Kennedy, M., P.G. Burstyn, and D.R. Husbands. 1978. Fat induced hypertension in rabbits. 2. The effect of feeding diets containing high concentrations of safflower oil and palm oil. Proc. Nutr. Soc. 37:98A.
Kesten, A.D., and R. Silbowitz. 1942. Experimental atherosclerosis and soya lecithin. Proc. Soc. Exp. Biol. Med. 49:71-73.
Keys, A. 1957. Diet and the epidemiology of coronary heart disease. J. Am. Med. Assoc. 164:1912-1919.
Keys, A. 1963. The role of the diet in human atherosclerosis and its complications. Pp. 263-299 in M. Sandler and G.H. Bourne, eds. Atherosclerosis and Its Origin. Academic Press, New York.
Page 243
Keys, A. 1965. Dietary survey methods in studies on cardiovascular epidemiology. Voeding 26:464-483.
Keys, A. 1970. Coronary heart disease in seven countries. Circulation Suppl. 41:11-1211.
Keys, A. 1975. Coronary heart disease—the global picture. Atherosclerosis 22:149-192.
Keys, A. 1980a. Alpha lipoprotein (HDL) cholesterol in the serum and the risk of coronary heart disease and death. Lancet 2:603-606.
Keys, A. 1980b. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Harvard University Press, Cambridge, Mass. 381 pp.
Keys, A. 1984. Serum cholesterol response to dietary cholesterol. Am. J. Clin. Nutr. 40:351-359.
Keys, A., and J.T. Anderson. 1954. The relationship of the diet to the development of atherosclerosis in man. Pp. 181-197 in Symposium on Atherosclerosis. Publ. No. 338. National Research Council, National Academy of Sciences, Washington, D.C.
Keys, A., and R.W. Parlin. 1966. Serum cholesterol response to changes in dietary lipids. Am. J. Clin. Nutr. 19:175-181.
Keys, A., F. Fidanza, and M.H. Keys. 1955. Further studies on serum cholesterol of clinically healthy men in Italy. Voeding 16:492-498.
Keys, A., J.T. Anderson, and F. Grande. 1957a. "Essential" fatty acids, degree of unsaturation, and effect of corn (maize) oil on the serum-cholesterol level in man. Lancet 1:66-68.
Keys, A., J.T. Anderson, and F. Grande. 1957b. Prediction of serum-cholesterol responses of man to changes in fats in the diet. Lancet 2:959-966.
Keys, A., J.T. Anderson, and F. Grande. 1958a. Effect on serum cholesterol in man of mono-ene fatty acid (oleic acid) in the diet. Proc. Soc. Exp. Biol. Med. 98:387-391.
Keys, A., N. Kimura, A. Kusukawa, B. Bronte-Stewart, N. Larsen, and M.H. Keys. 1958b. Lessons from serum cholesterol studies in Japan, Hawaii and Los Angeles. Ann. Intern. Med. 48:83-94. .
Keys, A., J.T. Anderson, and F. Grande. 1959. Serum cholesterol in man: diet fat and intrinsic responsiveness. Circulation 19:201-214.
Keys, A., H.L. Taylor, H. Blackburn, J. Brozek, J.T. Anderson, and E. Simonson. 1963. Coronary heart disease among Minnesota business and professional men followed fifteen years. Circulation 28:381-395.
Keys, A., J.T. Anderson, and F. Grande. 1965a. Serum cholesterol response to changes in the diet. I. Iodine value of dietary fat versus 2S-P. Metabolism 14:747-758.
Keys, A., J.T. Anderson, and F. Grande. 1965b. Serum cholesterol response to changes in the diet. III. Differences among individuals. Metabolism 14:766-775.
Keys, A., J.T. Anderson, and F. Grande. 1965c. Serum cholesterol response to changes in the diet. IV. Particular saturated fatty acids in the diet. Metabolism 14:776-787.
Keys, A., M.J. Karvonen, S. Punsar, A. Menotti, F. Fidanza, and G. Farchi. 1984. HDL serum cholesterol and 24-year mortality of men in Finland. Int. J. Epidemiol. 13:428-435.
Keys, A., C. Aravanis, H. Blackburn, R. Buzina, A.S. Dontas, F. Fidanza, M.J. Karvonen, A. Menotti, S. Nedeljkovic, S. Punsar, and H. Toshima. 1985. Serum cholesterol and cancer mortality in the Seven Countries Study. Am. J. Epidemiol. 121:870-883.
Kidwell, W.R. 1986. Fatty acid growth requirements of normal and neoplastic mammary epithelium. Prog. Clin. Biol. Res. 222:699-706.
Kinlen, L.J. 1982. Meat and fat consumption and cancer mortality: a study of strict religious orders in Britain. Lancet 1:946-949.
Kinsell, L.W., J. Partridge, L. Boling, S. Margen, and G. Michaels. 1952. Dietary modification of serum cholesterol and phospholipid levels. J. Clin. Endocrinol. 12:909-913.
Kinsell, L.W., G.D. Michaels, J.W. Partridge, L.A. Boling, H.E. Balch, and G.C. Cochrane. 1953. Effect upon serum cholesterol and phospholipids of diets containing large amounts of vegetable fat. J. Clin. Nutr. 1:224-231.
Kinsella, J.E., G. Bruckner, J. Mai, and J. Shimp. 1981. Metabolism of trans fatty acids with emphasis on the effects of trans, trans-octadecadienoate on lipid composition, essential fatty acid, and prostaglandins: an overview. Am. J. Clin. Nutr. 34:2307-2318.
Kirkland, R.T., B.S. Keenan, J.L. Probstfield, W. Patsch, T.L. Lin, G.W. Clayton, and W. Insull, Jr. 1987. Decrease in plasma high-density lipoprotein cholesterol levels at puberty in boys with delayed adolescence. Correlation with plasma testosterone levels. J. Am. Med. Assoc. 257:502-507.
Kita, T., Y. Nagano, M. Yokode, K. Ishii, N. Kume, A. Ooshima, H. Yoshida, and C. Kawai. 1987. Proc. Natl. Acad. Sci. U.S.A. 84:5928-5931.
Klasing, S.A., and S.M. Pilch. 1986. Review of Epidemiological and Clinical Evidence on the Role of Omega-3 Fatty Acids in Health and Disease. Quick Response Report #3. Life Sciences Research Office, Federation of American Societies for Experimental Biology, Bethesda, Md. 66 pp.
Klurfeld, D.M., M.M. Weber, and D. Kritchevsky. 1987. Inhibition of chemically induced mammary and colon tumor promotion by caloric restriction in rats fed increased dietary fat. Cancer Res. 47:2759-2762.
Knapp, H.R., I.A. Reilly, P. Alessandrini, and G.A. Fitz-Gerald. 1986. In vivo indexes of platelet and vascular function during fish-oil administration in patients with atherosclerosis. N. Engl. J. Med. 314:937-942.
Knox, E.G. 1977. Foods and diseases. Br. J. Prev. Soc. Med. 31:71-80.
Knudsen, I., ed. 1986. Genetic Toxicology of the Diet. Progress in Clinical and Biological Research, Vol. 206. Alan R. Liss, New York. 351 pp.
Knuiman, J.T., and C.E. West. 1981. HDL-cholesterol in men from thirteen countries. Lancet 2:367-368.
Knuiman, J.T., R.J. Hermus, and J.G. Hautvast. 1980. Serum total and high density lipoprotein (HDL) cholesterol concentrations in rural and urban boys from 16 countries. Atherosclerosis 36:529-537.
Kohlmeier, M., G. Stricker, and G. Schlierf. 1985. Influences of "normal" and "prudent" diets on biliary and serum lipids in healthy women. Am. J. Clin. Nutr. 42:1201-1205.
Kolonel, L.N., J. H. Hankin, J. Lee, S.Y. Chu, A.M. Nomura, and M.W. Hinds. 1981. Nutrient intakes in relation to cancer incidence in Hawaii. Br. J. Cancer 44: 332-339.
Kolonel, LN., C.N. Yoshizawa, and J.H. Hankin. 1988. Diet and prostatic cancer: a case-control study in Hawaii. Am. J. Epidemiol. 127:999-1012.
Komachi, Y. 1977. Recent problems in cerebrovascular accidents: characteristics of stroke in Japan. Nippon Ronen Igakkai Zasshi 14:359-364.
Koo, C., M.E. Wernette-Hammond, and T.L. Innerarity. 1986. Uptake of canine beta-very low density lipoproteins by mouse peritoneal macrophages is mediated by a low
Page 244
density lipoprotein receptor. J. Biol. Chem. 261:11194-11201.
Kort, W.J., I.M. Weijma, A.J. Vergroesen, and D.L. Westbroek. 1987a. Conversion of diets at tumor induction shows the pattern of tumor growth and metastasis of the first given diet. Carcinogenesis 8:611-614.
Kort, W.J., I.M. Weijma, A.M. Bijma, W.P. van Schalkwijk, A.J. Vergroesen, and D.L. Westbroek. 1987b. Omega-3 fatty acids inhibiting the growth of a transplantable rat mammary adenocarcinoma. J. Natl. Cancer Inst. 79:593-599.
Kozarevic, D., B. Pirc, Z. Racic, T.R. Dawber, T. Gordon, and W.J. Zukel. 1976. The Yugoslavia cardiovascular disease study. II. Factors in the incidence of coronary heart disease. Am. J. Epidemiol. 104:133-140.
Kozarevic, D., D. McGee, N. Vojvodic, T. Gordon, Z. Racic, W. Zukel, and T. Dawber. 1981. Serum cholesterol and mortality: the Yugoslavia Cardiovascular Disease Study. Am. J. Epidemiol. 114:21-28.
Kraemer, F.B., M. Greenfield, T.A. Tobey, and G.M. Reaven. 1982. Effects of moderate increases in dietary polyunsaturated:saturated fat on plasma triglyceride and cholesterol levels in man. Br. J. Nutr. 47:259-266.
Kranz, H., D.J. Caddy, A.M. Williams, and W. Gay. 1980. Myasthenic syndrome: effect of choline, plasmapheresis and tests for circulating factor. J. Neurol. Neurosurg. Psychiatry 43:483-488.
Kratzing, C.C., and J.J. Perry. 1971. Hypertension in young rats following choline deficiency in maternal diets. J. Nutr. 101:1657-1661.
Kritchevsky, D. 1982. Trans fatty acid effects in experimental atherosclerosis. Fed. Proc. 41:2813-2817.
Kritchevsky, D. 1983. Influence of trans unsaturated fat on experimental atherosclerosis. Pp. 403-413 in E.G. Perkins and W.J. Visek, eds. Dietary Fats and Health. American Oil Chemists' Society, Champaign, Ill.
Kritchevsky, D. 1986. Fat, calories and fiber. Prog. Clin. Biol. Res. 222:495-515.
Kritchevsky, D. 1988. Cholesterol vehicle in experimental atherosclerosis. A brief review with special reference to peanut oil. Arch. Pathol. Lab. Med. 112:1041-1044.
Kritchevsky, D., and S.A. Tepper. 1965a. Cholesterol vehicle in experimental atherosclerosis. VII. Influence of naturally occurring saturated fats. Med. Pharmacol. Exp. 12:315-320.
Kritchevsky, D., and S.A. Tepper. 1965b. Cholesterol vehicle in experimental atherosclerosis. 8. Effect of a medium chain triglyceride (MCT). Exp. Mol. Pathol. 4:489-499.
Kritchevsky, D., A.W. Moyer, W.C. Tesar, J.B. Logan, R.A. Brown, M.C. Davies, and H.R. Cox. 1954. Effect of cholesterol vehicle in experimental atherosclerosis. Am. J. Physiol. 178:30-32.
Kritchevsky, D., A.W. Moyer, W.C. Tesar, R.F.J. McCandless, J.B. Logan, R.A Brown, and M.E. Englert. 1956. Cholesterol vehicle in experimental atherosclerosis. II. Influence of unsaturation. Am. J. Physiol. 185:279-280.
Kritchevsky, D., S.A. Tepper, D. Vesselinovitch, and R.W. Wissler. 1971. Cholesterol vehicle in experimental atherosclerosis. 11. Peanut oil. Atherosclerosis 14:53-64.
Kritchevsky, D., S.A. Tepper, D. Vesselinovitch, and R.W. Wissler. 1973. Cholesterol vehicle in experimental atherosclerosis. 13. Randomized peanut oil. Atherosclerosis 17: 225-243.
Kritchevsky, D., L.M. Davidson, J.J. van der Watt, P.A. Winter, and 1. Bersohn. 1974a. Hypercholesterolaemia and atherosclerosis induced in vervet monkeys by cholesterol-free, semisynthetic diets. S. Afr. Med. J. 48:2413-2414.
Kritchevsky, D., L.M. Davidson, I.L. Shapiro, H.K. Kim, M. Kitagawa, S. Malhotra, P.P. Nair, T.B. Clarkson, I. Bersohn, and P.A. Winter. 1974b. Lipid metabolism and experimental atherosclerosis in baboons: influence of cholesterol-free, semi-synthetic diets. Am. J. Clin. Nutr. 27: 29-50.
Kritchevsky, D., S.A. Tepper, D.A. Scott, D.M. Klurfeld, D. Vesselinovitch, and R.W. Wissler. 1981. Cholesterol vehicle in experimental atherosclerosis. 18. Comparison of North American, African and South American peanut oils. Atherosclerosis 38:291-299.
Kromhout, D. 1983. Body weight, diet, and serum cholesterol in 871 middle-aged men during 10 years of follow-up (the Zutphen Study). Am. J. Clin. Nutr. 38:591-598.
Kromhout, D., and C. de Lezenne Coulander. 1984. Diet, prevalence and 10-year mortality from coronary heart disease in 871 middle-aged men. The Zutphen Study. Am. J. Epidemiol. 119:733-741.
Kromhout, D., E.B. Bosschieter, and C. de Lezenne Coulander. 1985. The inverse relation between fish consumption and 20-year mortality from coronary heart disease. N. Engl. J. Med. 312:1205-1209.
Kuhnlein, U., D. Bergstrom, and H. Kuhnlein. 1981. Mutagens in feces from vegetarians and non-vegetarians. Mutat. Res. 85:1-12.
Kuksis, A., J.J. Myher, K. Geher, G.J. Jones, J. Shepherd, C.J. Packard, J.D. Morrisett, O.D. Taunton, and A.M. Gotto. 1982. Effect of saturated and unsaturated fat diets on lipid profiles of plasma lipoproteins. Atherosclerosis 41:221-240.
Kune, S., G.A. Kune, and LF. Watson. 1987. Case-control study of dietary etiological factors: the Melbourne Colorectal Cancer Study. Nutr. Cancer 9:21-42.
Kuo, P.T., K. Hayase, J.B. Kostis, and A.E. Moreyra. 1979. Use of combined diet and colestipol in long-term (7-7½ years) treatment of patients with type II hyperlipoproteinemia. Circulation 59:199-211.
Kushi, LH., R.A. Lew, F.J. Stare, C.R. Ellison, M. el Lozy, G. Bourke, L Daly, I. Graham, N. Hickey, R. Mulcahy, and J. Kevaney. 1985. Diet and 20-year mortality from coronary heart disease. The Ireland-Boston Diet-Heart Study. N. Engl. J. Med. 312:811-818.
Kuusi, T., C. Ehnholm, J.K. Huttunen, E. Kostiainen, P. Pietinen, U. Leino, U. Uusitalo, T. Nikkari, J.M. Iacono, and P. Puska. 1985. Concentration and composition of serum lipoproteins during a low-fat diet at two levels of polyunsaturated fat. J. Lipid. Res. 26:360-367.
Kwiterovich, P.O., Jr. 1980. Can an effective fat-modified diet be safely recommended after weaning for infants and children in general? a. Some theoretical and practical considerations of the use of a low-fat diet in childhood. Pp. 375-382 in R.M. Lauer and R.B. Shekelle, eds. Childhood Prevention of Atherosclerosis and Hypertension. Raven Press, New York.
Kwiterovich, P.O., Jr. 1986. Biochemical, clinical, epidemiologic, genetic, and pathologic data in the pediatric age group relevant to the cholesterol hypothesis. Pediatrics 78: 349-362.
Lambert, G.F., J.P. Miller, R.T. Olsen, and D.V. Frost. 1958. Hypercholesterolemia and atherosclerosis induced in rabbits by purified high fat rations devoid of cholesterol. Proc. Soc. Exp. Biol. 97:544-549.
Page 245
Laskarzewski, P., J.A. Morrison, I. deGroot, K.A. Kelly, M.J. Mellies, P. Khoury, and C.J. Glueck. 1979. Lipid and lipoprotein tracking in 108 children over a four-year period. Pediatrics 64:584-591.
Lauer, R.M., W.E. Connor, P.E. Leaverton, M.A. Reiter, and W.R. Clarke. 1975. Coronary heart disease risk factors in school children: the Muscatine Study. J. Pediatr. 86:697-706.
Lawrence, C.M., P. Millac, G.S. Stout, and J.W. Ward. 1980. The use of choline chloride in ataxic disorders. J. Neurol. Neurosurg. Psychiatry 43:452-454.
Lê, M.G., L.H. Moulton, C. Hill, and A. Kramer. 1986. Consumption of dairy produce and alcohol in a case-control study of breast cancer. J. Natl. Cancer Inst. 77:633-636.
Leaf, A., and P.C. Weber. 1988. Cardiovascular effects of n-3 fatty acids. N. Engl. J. Med 318:549-557.
Lee, D.W., C.J. Gilmore, G. Bonorris, H. Cohen, J.W. Marks, M. Cho-Sue, M.S. Meiselman, and L.J. Schoenfield. 1985. Effect of dietary cholesterol on biliary lipids in patients with gallstones and normal subjects. Am. J. Clin. Nutr. 42:414-420.
Lee, K.T., K. Janakidevi, M. Kroms, J. Schmee, and W.A. Thomas. 1985. Mosaicism in female hybrid hares heterozygous for glucose-6-phosphate dehydrogenase. VII. Evidence for selective advantage of one phenotype over the other in ditypic samples from aortas of hares fed cholesterol oxidation products. Exp. Mol. Pathol. 42:71-77.
Lees, A.M., H.Y. Mok, R.S. Lees, M.A. McCluskey, and S.M. Grundy. 1977. Plant sterols as cholesterol-lowering agents: clinical trials in patients with hypercholesterolemia and studies of sterol balance. Atherosclerosis 28:325-338.
Leijd, B. 1980. Cholesterol and bile acid metabolism in obesity. Clin. Sci. 59:203-206.
Leren, P., and O. Haabrekke. 1971. Blood lipids in patients with atherosclerosis obliterans of the lower limbs. Acta Med. Scand. 189:511-513.
Lerman, J., and P.D. White. 1946. Metabolic changes in young people with coronary heart disease. J. Clin. Invest. 25:914.
Leung, M., D.L. Yeung, M.D. Pennell, and J. Hall. 1984. Dietary intakes of preschoolers. J. Am. Diet. Assoc. 84: 551-554.
Levy, R.I., and A.N. Klimov. 1987. High density lipoprotein cholesterol (HDL-C) and mortality in U.S.S.R. and U.S. middle-aged men: Collaborative U.S.-U.S.S.R. Mortality Follow-up Study. Arteriosclerosis 7:508a.
Lewis, B. 1958. Effect of certain dietary oils on bile-acid secretion and serum-cholesterol. Lancet 1:1090-1092.
Lewis, B., A. Chait, and G. Sigurdsson. 1978. Serum lipoproteins in four European communities: a quantitative comparison. Eur. J. Clin. Invest. 8:165-173.
Lewis, D.S., G.E. Mott, C.A. McMahan, E.J. Masoro, K.D. Carey, and H.C. McGill, Jr. 1988. Deferred effects of preweaning diet on atherosclerosis in adolescent baboons. Arteriosclerosis 8:274-280.
Lin, D.S., and W.E. Connor. 1980. The long term effects of dietary cholesterol upon the plasma lipids, lipoproteins, cholesterol absorption, and the sterol balance in man: the demonstration of feedback inhibition of cholesterol biosynthesis and increased bile acid excretion. J. Lipid. Res. 21: 1042-1052.
Lin, H.J., C. Wang, G. Salen, K.C. Lam, and T.K. Chan. 1983. Sitosterol and cholesterol metabolism in a patient with coexisting phytosterolemia and cholestanolemia. Metabolism 32:126-133.
Lindstedt, S., J. Avigan, D.S. Goodman, J. Sjovall, and D. Steinberg. 1965. The effect of dietary fat on the turnover of cholic acid and on the composition of the biliary bile acids in man. J. Clin. Invest. 44:1754-1765.
Lipid Research Clinics Program. 1984a. The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. J. Am. Med. Assoc. 251:351-364.
Lipid Research Clinics Program. 1984b. The Lipid Research Clinics Coronary Primary Prevention Trial results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. J. Am. Med. Assoc. 251: 365-374.
Lissner, L., D.A. Levitsky, B.J. Strupp, H.J. Kalkwarf, and D.A. Roe. 1987. Dietary fat and the regulation of energy intake in human subjects. Am. J. Clin. Nutr. 46:886-892.
Little, A., R. Levy, P. Chuaqui-Kidd, and D. Hand. 1985. A double-blind, placebo controlled trial of high-dose lecithin in Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 48: 736-742.
Liu, K., J. Stamler, A. Dyer, J. McKeever, and P. McKeever. 1978. Statistical methods to assess and minimize the role of intra-individual variability in obscuring the relationship between dietary lipids and serum cholesterol. J. Chronic Dis. 31:399-418.
Liu, K., J. Stamler, D. Moss, D. Garside, V. Persky, and I. Soltero. 1979. Dietary cholesterol, fat, and fibre, and colon-cancer mortality. An analysis of international data. Lancet 2:782-785.
Lo, C.W. 1985. Human milk: nutritional properties. Pp. 797-818 in W.A. Walker and J.B. Watkins, eds. Nutrition in Pediatrics: Basic Science and Clinical Application. Little, Brown and Co., Boston.
Löffelholz, K., R. Lindmar, and W. Weide. 1979. The relationship between choline and acetylcholine release in the autonomic nervous system. Pp. 233-241 in A. Barbeau, J.H. Growdon, and R.J. Wurtman, eds. Nutrition and the Brain, Vol. 5. Choline and Lecithin in Brain Disorders. Raven Press, New York.
Lofland, H.B., Jr., T.B. Clarkson, and B.C. Bullock. 1970. Whole body sterol metabolism in squirrel monkeys (Saimiri sciureus). Exp. Mol. Pathol. 13:1-11.
Low-Beer, T.S. 1985. Nutrition and cholesterol gallstones. Proc. Nutr. Soc. 44:127-134.
Löwenthal, K. 1925. Cholesterinfütterung bei der Maus. Verh. Dtsch. Pathol. Ges. 20:137-139.
Lubin, F., Y. Wax, and B. Modan. 1986. Role of fat, animal protein, and dietary fiber in breast cancer etiology: a case-control study. J. Natl. Cancer Inst. 77:605-612.
Lubin, J.H., P.E. Burns, W.J. Blot, R.G. Ziegler, A.W. Lees, and J.F. Fraumeni, Jr. 1981. Dietary factors and breast cancer risk. Int. J. Cancer 28:685-689.
MacDonald, M.C., R.L. Kline, and G.J. Mogenson. 1981. Dietary linoleic acid and salt-induced hypertension. Can. J. Physiol. Pharmacol. 59:872-875.
Maciejko, J.J., D.R. Holmes, B.A. Kottke, A.R. Zinsmeister, D.M. Dinh, and S.J. Mao. 1983. Apolipoprotein A-I as a marker of angiographically assessed coronary-artery disease. N. Engl. J. Med. 309:385-389.
Maclennan, R., and O.M. Jensen. 1977. Dietary fibre, transit-time, faecal bacteria, steroids and colon cancer in two Scandinavian populations. Report from the International Agency for Research on Cancer Intestinal Microecology Group. Lancet 2:207-211.
Page 246
MacPherson, B.R., R.S. Pemsingh, and G.W. Scott. 1987. Experimental cholelithiasis in the ground squirrel. Lab. Invest. 56:138-145.
Macquart-Moulin, G., E. Riboli, J. Cornee, B. Charnay, P. Berthezene, and N. Day. 1986. Case-control study on colorectal cancer and diet in Marseilles. Int. J. Cancer. 38:183-191.
Maggio, C.A., M.R.C. Greenwood, and J.R. Visselli. 1983. The satiety effects of intragastric macronutrient infusions in fatty and lean Zucker rats. Physiol. Behav. 31:367-372.
Mahadevappa, V.G., and B.J. Holub. 1987. Quantitative loss of individual eicosapentaenoyl-relative to arachidonoyl-containing phospholipids in thrombin-stimulated human platelets. J. Lipid Res. 28:1275-1280.
Majerus, P.W., T.M. Connolly, H. Deckmyn, T.S. Ross, T.E. Bross, H. Ishii, V.S. Bansal, and D.B. Wilson. 1986. The metabolism of phosphoinositide-derived messenger molecules. Science 234:1519-1526.
Malinow, M.R. 1983. Experimental models of atherosclerosis regression. Atherosclerosis 48:105-118.
Malmros, H. 1950. The relation of nutrition to health: a statistical study of the effect of the war-time on arteriosclerosis, cardiosclerosis, tuberculosis and diabetes. Acta Med. Scand. Suppl. 246:137-153.
Malmros, H., and G. Wigand. 1957. The effect on serum-cholesterol of diets containing different fats. Lancet 2:1-7.
Malmros, H., and G. Wigand. 1959. Atherosclerosis and deficiency of essential fatty acids. Lancet 2:749-751.
Mann, G.V. 1957. The epidemiology of coronary heart disease. Am. J. Med. 23:463-480.
Mannes, G.A., A. Maier, C. Thieme, B. Wiebecke, and G. Paumgartner. 1986. Relation between the frequency of colorectal adenoma and the serum cholesterol level. N. Engl. J. Med. 315:1634-1638.
Manninen V., M.O. Elo, M.H. Frick, K. Haapa, O.P. Heinonen, P. Heinsalmi, P Helo, J.K. Huttunen, P. Kaitaniemi, P. Koskinen, H. Mäenpää, M. Mälkönen, M. Mänttäri, S. Norola, A. Pasternack, J. Pikkarainen, M. Romo, T. Sjöblom, and E.A. Nikkilä. 1988. Lipid alterations and decline in the incidence of coronary heart disease in the Helsinki Heart Study. J. Am. Med. Assoc. 260:641-651.
Manousos, O., N.E. Day, D. Trichopoulos, F. Gerovassilis, A. Tzonou, and A. Polychronopoulou. 1983. Diet and colorectal cancer: a case-control study in Greece. Int. J. Cancer. 32:1-5.
Marcus, A.J. 1966. The role of lipids in blood coagulation. Adv. Lipid Res. 4:1-37.
Marcus, A.J. 1978. The role of lipids in platelet function: with particular reference to the arachidonic acid pathway. J. Lipid Res. 19:793-826.
Marcus, A.J. 1984. The eicosanoids in biology and medicine. J. Lipid Res. 25:1511-1516.
Marcus, A.J. 1987. Platelet eicosanoid metabolism. Pp. 676-688 in R.W. Coleman, J. Hirsch, V.J. Marder, and E.W. Salzman, eds. Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 2nd ed. J.B Lippincott, Philadelphia.
Marcus, A.J. 1988a. Eicosanoids: transcellular metabolism. Pp. 129-137 in J.I. Gallin, I.M. Goldstein, and R. Snyderman, eds. Inflammation: Basic Principles and Clinical Correlates. Raven Press, New York.
Marcus, A.J. 1988b. Hemorrhagic disorders: abnormalities of platelet and vascular function. Pp. 1042-1060 in J.B. Wyngaarden and L.H. Smith, Jr., eds. Cecil Textbook of Medicine, 18th ed., Vol. 1. Saunders, Philadelphia.
Margetts, B.M., L.J. Beilin, B.K. Armstrong, R. Vandongen, and K.D. Croft. 1984. Dietary fat intake and blood pressure: a double blind controlled trial of changing polyunsaturated to saturated fat ratio. J. Hypertens. 2:S201-S203.
Marmot, S.G., S.L. Syme, A. Kagan, H. Kato, J.B. Cohen, and J. Belsky. 1975. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: prevalence of coronary and hypertensive heart disease and associated risk factors. Am. J. Epidemiol. 102:514-525.
Marston R., and N. Raper. 1987. Nutrient content of the U.S. food supply. National Food Review (Winter-Spring) 36:18-23.
Martin, M.J., S.B. Hulley, W.S. Browner, L.H. Kuller, and D. Wentworth. 1986. Serum cholesterol, blood pressure, and mortality: implications from a cohort of 361,662 men. Lancet 2:933-936.
Martinez, I., R. Torres, Z. Frias, J.R. Colón, and M. Fernàndez. 1979. Factors associated with adenocarcinomas of the large bowel in Puerto Rico. Pp. 45-52 in J.M. Birch, ed. Advances in Medical Oncology, Research and Education, Vol. III. Epidemiology. Pergamon Press, Oxford.
Mattson, F.H., and S.M. Grundy. 1985. Comparison of effects of dietary saturated, monunsaturated, and polyunsaturated fatty acids on plasma lipids and lipoproteins in man. J. Lipid Res. 26:194-202.
Mattson, F.H., B.A. Erickson, and A.M. Kligman. 1972. Effect of dietary cholesterol on serum cholesterol in man. Am. J. Clin. Nutr. 25:589-594.
Mattson, F.H., E.J. Hollenbach, and A.M. Kligman. 1975. Effect of hydrogenated fat on the plasma cholesterol and triglyceride levels of man. Am. J. Clin. Nutr. 28:726-731.
Mattson, F.H., R.A. Volpenhein, and B.A. Erikson. 1977. Effect of plant sterol esters on the absorption of dietary cholesterol. J. Nutr. 107:1139-1146.
Mattson, F.H., S.M. Grundy, and J.R. Crouse. 1982. Optimizing the effect of plant sterols on cholesterol absorption in man. Am. J. Clin. Nutr. 35:697-700.
McArthur, R.G., D.A. Roncari, J.A. Little, A. Kuksis, J.J. Myher, and L. Marai. 1986. Phytosterolemia and hypercholesterolemia in childhood. J. Pediatr. 108:254-256.
McCarron, D.A., J. Stanton, H. Henry, and C. Morns. 1983. Assessment of nutritional correlates of blood pressure. Ann. Intern. Med. 98:715-719.
McGandy, R.B. 1971. Adolescence and the onset of atherosclerosis. Bull. N.Y. Acad. Med. 47:590-600.
McGandy, R.B., B. Hall, C. Ford, and F.J. Stare. 1972. Dietary regulation of blood cholesterol in adolescent males: a pilot study. Am. J. Clin. Nutr. 25:61-66.
McGee, D.L., D.M. Reed, K. Yano, A. Kagan, and J. Tillotson. 1984. Ten-year incidence of coronary heart disease in the Honolulu Heart Program. Relationship to nutrient intake. Am. J. Epidemiol. 119:667-676.
McGee, D., D. Reed, G. Stemmerman, G. Rhoads, K. Yano, and M. Feinleib. 1985. The relationship of dietary fat and cholesterol to mortality in 10 years: the Honolulu Heart Program. Int. J. Epidemiol. 14:97-105.
McGill, H.C., Jr. 1968a. Fatty streaks in the coronary arteries and aorta. Lab. Invest. 18:560-564.
McGill, H.C., Jr., ed. 1968b. The Geographic Pathology of Atherosclerosis. Williams & Wilkins, Baltimore. 193 pp.
McGill, H.C., Jr. 1979. The relationship of dietary cholesterol to serum cholesterol concentration and to atherosclerosis in man. Am. J. Clin. Nutr. 32:2664-2702.
Page 247
McGill, H.C., Jr. 1980. Morphologic development of the atherosclerotic plaque. Pp. 41-49 in R.M. Lauer and R.B. Shekelle, eds. Childhood Prevention of Atherosclerosis and Hypertension. Raven Press, New York.
McGill, H.C., Jr. 1984. George Lyman Duff memorial lecture. Persistent problems in the pathogenesis of atherosclerosis. Arteriosclerosis 4:443-451.
McGill, H.C., Jr., J.C. Geer, and J.P. Strong. 1963. Natural history of human atherosclerotic lesions. Pp. 39-65 in M. Sandler and G.H. Bourne, eds. Atherosclerosis and Its Origin. Academic Press, New York.
McGill, H.C., Jr., G.E. Mott, and C.A. Bramblett. 1976. Experimental atherosclerosis in the baboon. Primates Med. 9:41-65.
McGill, H.C., Jr., C.A. McMahan, A.W. Kruski, and G.E. Mott. 1981a. Relationship of lipoprotein cholesterol concentrations to experimental atherosclerosis in baboons. Arteriosclerosis 1:3-12.
McGill, H.C., Jr., C.A. McMahan, A.W. Kruski, J.L. Kelley, and G.E. Mott. 1981b. Responses of serum lipoproteins to dietary cholesterol and type of fat in the baboon. Arteriosclerosis 5:337-344.
McGill, H.C., Jr., C.A. McMahan, and J.D. Wene. 1981c. Unresolved problems in the diet-heart issue. Arteriosclerosis 1:164-176.
McGill, H.C., Jr., K.D. Carey, C.A. McMahan, Y.N. Marinez, T.E. Cooper, G.E. Mott, and C.J. Schwartz. 1985. Effects of two forms of hypertension on atherosclerosis in the hyperlipidemic baboon. Arteriosclerosis 5:481-493.
McLean, J.W., J.E. Tomlinson, W.J. Kuang, D.L. Eaton, E.Y. Chen, G.M. Fless, A.M. Scanu, and R.M. Lawn. 1987. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 330:132-137.
McMichael, A.J., O.M. Jensen, D.M. Parkin, and D.G. Zaridze. 1984. Dietary and endogenous cholesterol and human cancer. Epidemiol. Rev. 6:192-216.
McMillan, G.C., B.I. Weigensberg, and A.C. Ritchie. 1960. Effects of dietary fats in rabbits fed cholesterol. Arch. Pathol. 70:220-225.
McNamara, D.J. 1987. Effects of fat-modified diets on cholesterol and lipoprotein metabolism. Annu. Rev. Nutr. 7:273-290.
McNamara, D.J., R. Kolb, T.S. Parker, H. Batwin, P. Samuel, C.D. Brown, and E.H. Ahrens, Jr. 1987. Heterogeneity of cholesterol homeostasis in man. Response to changes in dietary fat quality and cholesterol quantity. J. Clin. Invest. 79:1729-1739.
McOsker, D.E., F.H. Mattson, H.B. Sweringen, and A.M. Kligman. 1962. The influence of partially hydrogenated dietary fats on serum cholesterol levels. J. Am. Med. Assoc. 180:380-385.
McSherry, C.K., N.B. Javitt, J.M. De Carvalho, and F. Glenn. 1971a. Cholesterol gallstones and the chemical composition of bile in baboons. Ann. Surg. 173:569-577.
McSherry, C.K., F. Glenn, and N.B. Javitt. 1971b. Composition of basal and stimulated hepatic bile in baboons, and the formation of cholesterol gallstones. Proc. Natl. Acad. Sci. U.S.A. 68:1564-1568.
Medalie, J.H., H.A. Kahn, H.N. Neufeld, E. Riss, and U. Goldbourt. 1973. Five-year myocardial infarction incidence. II. Association of single variables to age and birthplace. J. Chronic Dis. 26:325-349.
Melchior, G.W., H.B. Lofland, and R.W. St. Clair. 1978. The effect of polyunsaturated fats on bile acid metabolism and cholelithiasis in squirrel monkeys. Metabolism 27: 1471-1484.
Mendelsohn, D., L. Mendelsohn, and D.G. Hamilton. 1980. Effect of polyunsaturated fat on atheroma formation in the non-human primate. S. Afr. J. Sci. 76:225-228.
Mensink, R.P., and M.B. Katan. 1987. Effect of monounsaturated fatty acids versus complex carbohydrates on high-density lipoproteins in healthy men and women. Lancet 1: 122-125.
Menzel, H.J., R.G. Kladetzky, and G. Assmann. 1983. Apolipoprotein E polymorphism and coronary artery disease. Arteriosclerosis 3:310-315.
Michael, U.F., S.L. Cookson, R. Chavez, and V. Pardo. 1975. Renal function in the choline deficient rat. Proc. Soc. Exp. Biol. Med. 150:672-676.
Miettinen, M., O. Turpeinen, M.J. Karvanon, R. Elosuo, and E. Paavilainen. 1972. Effect of cholesterol-lowering diet on mortality from coronary heart-disease and other causes. A twelve-year clinical trial in men and women. Lancet 2:835-838.
Miettinen, M., O. Turpeinen, M.J. Karvonen, E. Paavilainen, and R. Elosuo. 1976. Prevalence of cholelithiasis in men and women ingesting a serum-cholesterol-lowering diet. Ann. Clin. Res. 8:111-116.
Miettinen, T.A. 1980. Phytosterolaemia, xanthomatosis and premature atherosclerotic arterial disease: a case with high plant sterol absorption, impaired sterol elimination and low cholesterol synthesis. Eur. J. Clin. Invest. 10:27-35.
Mikol, Y.B., K.L. Hoover, D. Creasia, and LA. Poirier. 1983. Hepatocarcinogenesis in rats fed methyl-deficient, amino acid-defined diets. Carcinogenesis 4:1619-1629.
Miller, A.B., A. Kelly, N.W. Choi, V. Matthews, R.W. Morgan, L. Munan, J.D. Burch, J. Feather, G.R. Howe, and M. Jain. 1978. A study of diet and breast cancer. Am. J. Epidemiol. 107:499-509.
Miller, A.B., G.R. Howe, M. Jain, K.J. Craib, and L Harrison. 1983. Food items and food groups as risk factors in a case-control study of diet and colo-rectal cancer. Int. J. Cancer 32:155-161.
Miller, D.C., M.F. Trulson, M.B. McCann, P.D. White, and F.J. Stare. 1958. Diet, blood lipids and health of Italian men in Boston. Ann. Intern. Med. 49:1178-1200.
Miller, N.E., D.S. Thelle, O.H. Forde, and O.D. Mjos. 1977. The Tromso Heart-Study. High-density lipoprotein and coronary heart-disease: a prospective case-control study. Lancet 1:965-968.
Milner, J.A., and E.G. Perkins, chairmen. 1984. Papers from the symposium on cancer—a molecular event. J. Am. Oil. Chem. Soc. 61:1881-1918.
Mishkel, M.A., and N. Spritz. 1969. The effects of trans isomerized trilinolein on plasma lipids of man. Pp. 355-364 in W.L. Holmes, L.A. Carlson, and R. Paoletti, eds. Drugs Affecting Lipid Metabolism. Advances in Experimental Medicine and Biology, Vol. 4. Plenum Press, New York.
Mistry, P., N.E. Miller, M. Laker, W.R. Hazzard, and B. Lewis. 1981. Individual variation in the effects of dietary cholesterol on plasma lipoproteins and cellular cholesterol homeostasis in man. Studies of low density lipoprotein receptor activity and 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in blood mononuclear cells. J. Clin. Invest. 67:493-502.
Montaldo, M.B., J.D. Benson, and G.A. Martinez. 1985. Nutrient intakes of formula-fed infants and infants fed cow's milk. Pediatrics 75:343-351.
Page 248
Moore, M.C., M.A. Guzman, P.E. Schilling, and J.P. Strong. 1976. Dietary-atherosclerosis study on deceased persons. Relation of selected dietary components to raised coronary lesions. J. Am. Diet. Assoc. 68:216-223.
Moore, R.B., J.T. Anderson, H.L. Taylor, A. Keys, and I.D. Frantz, Jr. 1968. Effect of dietary fat on the fecal excretion of cholesterol and its degradation products in man. J. Clin. Invest. 47:1517-1534.
Mori, T.A., J.P. Codde, R. Vandongen, and L.J. Beilin. 1967. New findings in the fatty acid composition of individual platelet phospholipids in man after dietary fish oil supplementation. Lipids 22:744-750.
Moritz, V., P. Singer, D. Forster, I. Bergen, and S. Massow. 1985. Changes of blood pressure in spontaneously hypertensive rats dependent on the quantity and quality of fat intake. Biomed. Biochim. Acta 44:1491-1505.
Morris, D.L, N.O. Borhani, E. Fitzsimmons, R.J. Hardy, CM. Hawkins, J.F. Kraus, D.R. Labarthe, L. Mastbaum, and G.H. Payne. 1983. Serum cholesterol and cancer in the Hypertension Detection and Follow-up Program. Cancer 52: 1754-1759.
Morris, J.N., J.W. Marr, and D.G. Clayton. 1977. Diet and heart: a postscript. Br. Med. J. 2:1307-1314.
Morrison, J.A., I. deGroot, K.A. Kelly, B.K. Edwards, M.J. Mellies, S. Tillett, P. Khoury, and C.J. Glueck. 1979a. High and low density lipoprotein cholesterol levels in hypercholesterolemic school children. Lipids 14:99-104.
Morrison, J.A., P.M. Laskarzewski, J.L Rauh, R. Brookman, M. Mellies, M. Frazer, P. Khoury, 1. deGroot, K. Kelly, and C.J. Glueck. 1979b. Lipids, lipoproteins, and sexual maturation during adolescence: the Princeton maturation study. Metabolism 28:641-649.
Morrison, J.A., R. Larsen, L. Glatfelter, D. Boggs, K. Burton, C. Smith, K. Kelly, M.J. Mellies, P. Khoury, and C.J. Glueck. 1980. Interrelationships between nutrient intake and plasma lipids and lipoproteins in schoolchildren aged 6 to 19: the Princeton School District Study. Pediatrics 65: 727-734.
Morrison, LM. 1958. Serum cholesterol reduction with lecithin. Geriatrics 13:12-19.
Morrison, L.M., L Hall, and A.L. Chaney. 1948. Cholesterol metabolism: blood serum cholesterol and ester levels in 200 cases of acute coronary thrombosis. Am. J. Med. Sci. 216: 32-38.
Mott, G.E., C.A. McMahan, and H.C. McGill, Jr. 1978. Diet and sire effects on serum cholesterol and cholesterol absorption in infant baboons (Papio cynocephalus). Circ. Res. 43:364-371.
Mott, G.E., C.A. McMahan, J.L Kelly, C.M. Farley, and H.C. McGill, Jr. 1982. Influence of infant and juvenile diets on serum cholesterol, lipoprotein cholesterol, and apolipoprotein concentrations in juvenile baboons (Papio sp.). Atherosclerosis 45:191-202.
MRFIT (Multiple Risk Factor Intervention Trial) Research Group. 1982. Multiple Risk Factor Intervention Trial: risk factor changes and mortality results. J. Am. Med. Assoc. 248:1465-1477.
Murray, C.D., K.T. Lee, W.A. Thomas, J.M. Reiner, and K. Janakidevi. 1981. Mosaicism in female hybrid hares heterozygous for glucose-6-phosphate dehydrogenase (G-6-PD). III. Changes in the ratios of G-6-PD types in skin fibroblast cultures exposed to 25-hydroxycholesterol. Exp. Mol. Pathol. 34:209-215.
Murray, G.E., R. Nair, and J. Patrick. 1986. The effect of dietary polyunsaturated fat on cation transport and hypertension in the rat. Br. J. Nutr. 56:587-593.
Murray, W.R., A. Backwood, J.M. Trotter, K.C. Calman, and C. MacKay. 1980. Faecal bile acids and clostridia in the aetiology of colorectal cancer. Br. J. Cancer 41:923-928.
Myher, J.J., L. Marai, and A. Kuksis. 1977. Acylglycerol structure of peanut oils of different atherogenic potential. Lipids 12:775-785.
Nagase, M., H. Tanimura, M. Setoyama, and Y. Hikasa. 1978. Present features of gallstones in Japan. A collective review of 2,144 cases. Am. J. Surg. 135:788-790.
Nair, P.P., N. Turjman, G.T. Goodman, C. Guidry, and B.M. Calkins. 1984. Diet, nutrition intake, and metabolism in populations at high and low risk for colon cancer. Metabolism of neutral sterols. Am. J. Clin. Nutr. 40:931-936.
Naito, H.K. 1985. The association of serum lipids, lipoproteins, and apolipoproteins with coronary artery disease assessed by coronary arteriography. Ann. N.Y. Acad. Sci. 454:230-238.
Nash, D.T., G. Gensini, and P. Esente. 1982. Effect of lipid-lowering therapy on the progression of coronary atherosclerosis assessed by scheduled repetitive coronary arteriography. Int. J. Cardiol. 2:43-55.
National Cholesterol Education Program. 1988. Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. 1988. Arch. Intern. Med. 148:36-69.
National Diet-Heart Study Research Group. 1968. The National Diet-Heart Study Final Report. Chapter XVII: Faribault Second Study. Circulation 37:1260-1274.
Nauss, K.M., L.R. Jacobs, and P.M. Newberne. 1987. Dietary fat and fiber: relationship to caloric intake, body growth, and colon tumorigeneis. Am. J. Clin. Nutr. 45:243-251.
Neaton, J.D., LH. Kuller, D. Wentworth, and N.O. Borhani. 1984. Total and cardiovascular mortality in relation to cigarette smoking, serum cholesterol concentration, and diastolic blood pressure among black and white males followed up for five years. Am. Heart J. 108:759-769.
Nestel, P.J. 1987. Polyunsaturated fatty acids (n-3, n-6). Am. J. Clin. Nutr. 45:1161-1167.
Nestel, P.J., N. Havenstein, H.M. Whyte, T.J. Scott, and L.J. Cook. 1973. Lowering of plasma cholesterol and enhanced sterol excretion with the consumption of polyunsaturated ruminant fats. N. Engl. J. Med. 288:379-382.
Nestel, P.J., N. Havenstein, Y. Homma, T.W. Scott, and L.J. Cook. 1975. Increased sterol excretion with polyunsaturated-fat high-cholesterol diets. Metabolism 24:189-198.
Nestel, P.J., A. Poyser, and T.J. Boulton. 1979. Changes in cholesterol metabolism in infants in response to dietary cholesterol and fat. Am. J. Clin. Nutr. 32:2177-2182.
Nestel, P.J., W.E. Connor, M.F. Reardon, S. Connor, S. Wong, and R. Boston. 1984. Suppression by diets rich in fish oil of very low density lipoprotein production in man. J. Clin. Invest. 74:82-89.
Neugat, A.I., C.M. Johnsen, and D.J. Fink. 1986. Serum cholesterol levels in adenomatous polyps and cancer of the colon. A case-control study. J. Am. Med. Assoc. 255: 365-367.
Neuringer, M., and W.E. Connor. 1986. N-3 fatty acids in the brain and retina: evidence for essentiality. Nutr. Rev. 44: 285-294.
Newberne, P.M., and K.M. Nauss. 1986. Dietary fat and colon cancer. variable results in animal models. Prog. Clin. Biol. Res. 222:311-330.
Page 249
Newberne, P.M., and A.E. Rogers. 1986. Labile methyl groups and the promotion of cancer. Annu. Rev. Nutr. 6: 407-432.
Newman, W.P., III, D.S. Freedman, A.W. Voors, P.D. Gard, S.R. Srinivasan, J.L. Cresanta, G.D. Williamson, L.S. Webber, and G.S. Berenson. 1986. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N. Engl. J. Med. 314: 138-144.
NHLBI (National Heart, Lung, and Blood Institute). 1971. Arteriosclerosis: A Report by the National, Heart, Lung and Blood Task Force on Arteriosclerosis, Vol. 1. DHEW Publ. No. (NIH) 72-219. National Institutes of Health, Public Health Service, U.S. Department of Health, Education, and Welfare, Bethesda, Md. 365 pp.
NHLBI (National Heart, Lung, and Blood Institute). 1978. Cardiovascular Profile of 15,000 Children of School Age in Three Communities 1971-1975. DHEW Publ. No. (NIH) 78-1472. National Institutes of Health, Public Health Service, U.S. Department of Health, Education, and Welfare, Bethesda, Md. 806 pp.
NHLBI (National Heart, Lung, and Blood Institute). 1980. The Lipid Resarch Clinics Population Studies Data Book, Vol. 1. The Prevalence Study: Aggregate Distributions of Lipids, Lipoproteins and Selected Variables in 11 North American Populations. NIH Publ. No. 80-1527. National Institutes of Health, Public Health Service, U.S. Department of Health and Human Services, Bethesda, Md. 136 pp.
Nichaman, M.Z., H.B. Hamilton, A. Kagan, T. Grier, T. Sacks, and S.L. Syme. 1975. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: distribution of biochemical risk factors. Am. J. Epidemiol. 102:491-501.
Nichols, A.B., C. Ravenscroft, D.E. Lamphiear, and L.D. Ostrander, Jr. 1976. Daily nutritional intake and serum lipid levels. The Tecumseh Study. Am. J. Clin. Nutr. 29:1384-1392.
NIH (National Institutes of Health). 1984. Treatment of hypertriglyceridemia: consensus conference. J. Am. Med. Assoc. 251:1196-1200.
NIH (National Institutes of Health). 1985. Lowering blood cholesterol to prevent heart disease: consensus conference. J. Am. Med. Assoc. 253:2080-2086.
Nikkila, E.A., P. Viikinkoski, M. Valle, and M.H. Frick. 1984. Prevention of progression of coronary atherosclerosis by treatment of hyperlipidaemia: a seven year prospective angiographic study. Br. Med. J. 289:220-223.
Nomura, A., B.E. Henderson, and J. Lee. 1978. Breast cancer and diet among the Japanese in Hawaii. Am. J. Clin. Nutr. 31:2020-2025.
Nomura, A.M., T.D. Wilkins, S. Kamiyama, L.K. Heilbrun, A. Shimada, G.N. Stemmerman, and H.F. Mower. 1983. Fecal neutral steroids in two Japanese populations with different colon cancer risks. Cancer Res. 43:1910-1913.
NRC (National Research Council). 1980. Recommended Dietary Allowances, 9th ed. Report of the Committee on Dietary Allowances, Food and Nutrition Board, Division of Biological Sciences, Assembly of Life Sciences. National Academy Press, Washington, D.C. 185 pp.
NRC (National Research Council). 1982. Diet, Nutrition, and Cancer. Report of the Committee on Diet, Nutrition, and Cancer, Assembly of Life Sciences. National Academy Press, Washington, D.C. 478 pp.
O'Connor, T.P., B.D. Roebuck, F. Peterson, and T.C. Campbell. 1985. Effect of dietary intake of fish oil and fish protein on the development of L-azaserine-induced preneoplastic lesions in the rat pancreas. J. Natl. Cancer Inst. 75: 959-962.
O'Connor, T.P., B.D. Roebuck, and T.C. Campbell. 1987. Effect of varying dietary omega-3:omega-6 fatty acid ratio on L-azaserine induced preneoplastic development in rat pancreas. Pp. 238-240 in W.E.M. Lands, ed. Proceedings of the AOCS Short Course on Polyunsaturated Fatty Acids and Eicosanoids. American Oil Chemists' Society, Champaign, Ill.
Oh, S.Y., and L.T. Miller. 1985. Effect of dietary egg on variability of plasma cholesterol levels and lipoprotein cholesterol. Am. J. Clin. Nutr. 42:421-431.
Oh, S.Y., and P.A. Monaco. 1985. Effect of dietary cholesterol and degree of fat unsaturation on plasma lipid levels, lipoprotein composition, and fecal steroid excretion in normal young adult men. Am. J. Clin. Nutr. 42:399413.
Oliver, M.F., and G.S. Boyd. 1953. Changes in the plasma lipids during the menstrual cycle. Clin. Sci. 12:217-222.
Onitiri, A.C., and E. Jover. 1980. Comparative serum apolipoprotein studies in ischaemic heart disease and control subjects. Clin. Chim. Acta 108:25-30.
Orchard, T.J., R.P. Donahue, LH. Kuller, P.N. Hodge, and A.L. Drash. 1983. Cholesterol screening in childhood: does it predict adult hypercholesterolemia? The Beaver County experience. J. Pediatr. 103:687-691.
Oster, P., L. Arab, B. Schellenberg, C.C. Hueck, R. Mordasini, and G. Schlierf. 1979. Blood pressure and adipose tissue linoleic acid. Res. Exp. Med. 175:287-291.
Ostfeld, A.M., R.B. Shekelle, H. Klawans, and H.M. Tufo. 1974. Epidemiology of stroke in an elderly welfare population. Am. J. Public Health 64:450-458.
Osuga, T., and O.W. Portman. 1971. Experimental formation of gallstones in the squirrel monkey. Proc. Soc. Exp. Biol. Med. 136:722-726.
Osuga, T., and O.W. Portman. 1972. Relationship between bile composition and gallstone formation in squirrel monkeys. Gastroenterology 63:122-133.
Osuga, T., O.W. Portman, K. Matamura, and M. Alexander. 1974. A morphologic study of gallstone development in the squirrel monkey. Lab. Invest. 30:486-493.
Osuga, T., O.W. Portman, N. Tanaka, M. Alexander, and A.J. Ochsner III. 1976. The effect of diet on hepatic bile formation and bile acid metabolism in squirrel monkeys with and without cholesterol gallstones. J. Lab. Clin. Med. 88: 649-661.
Packard, C.J., L. McKinney, K. Carr, and . Shepherd. 1983. Cholesterol feeding increases low density lipoprotein synthesis. J. Clin. Invest. 72:45-51.
Parks, J.S., and L.L. Rudel. 1982. Different kinetic fates of apolipoproteins A-I and A-II from lymph chylomicra of nonhuman primates. Effect of saturated versus polyunsaturated dietary fat. J. Lipid Res. 23:410-421.
Paul, O., M.H. Lepper, W.H. Phelan, G.W. Dupertuis, A. MacMillan, H. McKean, and H. Park. 1963. A longitudinal study of coronary heart disease. Circulation 28:20-31.
Pearce, M.L., and S. Dayton. 1971. Incidence of cancer in men on a diet high in polyunsaturated fat. Lancet 1:464-467.
Peckham, S.C., C. Entenman, and H.W. Carroll. 1962. The influence of a hypercaloric diet on gross body and adipose tissue composition in the rat. J. Nutr. 77:187-197.
Page 250
Peng, S.K., C.B. Taylor, P. Tham, N.T. Werthessen, and B. Mikkelson. 1978. Effect of auto-oxidation products from cholesterol on aortic smooth muscle cells: an in vitro study. Arch. Pathol. Lab. Med. 102:57-61.
Peng, S.K., P. Tham, C.B. Taylor, and B. Mikkelson. 1979. Cytotoxicity of oxidation derivatives of cholesterol on cultured aortic smooth muscle cells and their effect on cholesterol biosynthesis. Am. J. Clin. Nutr. 32:1033-1042.
Perkins, E.G., and W.J. Wisek, eds. 1983. Dietary Fats and Health. American Oil Chemists' Society, Champaign, Ill. 978 pp.
Peterson, B., and E. Trell. 1983. Premature mortality in middle-aged men: Serum cholesterol as risk factor. Klin. Wochenschr. 61:795-801.
Peterson, D.W., C.W. Nichols, Jr., N.F. Peek, and I.L. Chaikoff. 1959. Depression of plasma cholesterol in human subjects consuming butter containing soy sterols. Fed. Proc. 15:569.
Petersson, B., and E. Trell. 1983. Raised serum urate concentration as risk factor for premature mortality in middle-aged men: relation to death from cancer. Br. Med. J. 287:7-9.
Phillips, R.L. 1975. Role of life-style and dietary habits in risk of cancer among Seventh-Day Adventists. Cancer. Res. 35: 3513-3522.
Phillips, R.L., F.R. Lemon, W.L. Beeson, and J.W. Kuzma. 1978. Coronary heart disease mortality among Seventh-Day Adventists with differing dietary habits: a preliminary report. Am. J. Clin. Nutr. 31:S191-S198.
Phillipson, B.E., D.W. Rothrock, W.E. Connor, W.S. Harris, and D.R. Illingworth. 1985. Reduction of plasma lipoproteins, and apoproteins by dietary fish oils in patients with hypertriglyceridemia. N. Engl. J. Med. 312:1210-1216.
Picciano, M.F., H.A. Guthrie, and D.M. Sheehe. 1978. The cholesterol content of human milk. A variable constituent among women and within the same woman. Clin. Pediatr. 17:359-362.
Pilger, E., H. Pristautz, K.P. Pfeiffer, and G. Kostner. 1983. Risk factors for peripheral atherosclerosis. Retrospective evaluation by stepwise discriminant analysis. Arteriosclerosis 3:57-63.
Pitas, R.E., T.L Innerarity, and R.W. Mahley. 1983. Foam cells in explants of atherosclerotic rabbit aortas have receptors for beta-very low density lipoproteins and modified low density lipoproteins. Arteriosclerosis 3:2-12.
Pocock, S.J., A.G. Shaper, A.N. Phillips, M. Walker, and T.P. Whitehead. 1986. HDL cholesterol is not a major risk factor for ischaemic heart disease in British men. Br. Med. J. 292:515-519.
Poindexter, C.A., and M. Bruger. 1938. Cholesterol content of the blood in heart disease. Arch. Intern. Med. 61:714-719.
Poirier, LA., P.M. Newberne, and M.W. Pariza. 1987. Essential Nutrients in Carcinogenesis. Advances in Experimental Medicine and Biology, Vol. 206. Plenum Publishing Company, New York. 562 pp.
Pomeranze, J. 1961. Serum cholesterol studies in infants. A comparison of infants fed breast milk, evaporated milk and prepared milk formulas. Am. J. Clin. Nutr. 9:570-572.
Pooling Project Research Group. 1978. Relationship of blood pressure, serum cholesterol, smoking habit, relative weight and ECG abnormalities to incidence of major coronary events: final report of the Pooling Project. J. Chronic Dis. 31:201-306.
Porikos, K.P., G. Booth, and T.B. Van Itallie. 1977. Effect of covert nutritive dilution on the spontaneous food intake of obese individuals: a pilot study. Am. J. Clin. Nutr. 30: 1638-1644.
Portman, O.W., and S.B. Andrus. 1965. Comparative evaluation of three species of new world monkeys for studies of dietary factors, tissue lipids, and atherogenesis. J. Nutr. 87: 429-438.
Potter, J.D., and A.J. McMichael. 1986. Diet and cancer of the colon and rectum: a case-control study. J. Natl. Cancer Inst. 76:557-569.
Pownall, H.J., J. Shepherd, W.W. Mantulin, L.A. Sklar, and A.M. Gotto, Jr. 1980. Effect of saturated and polyunsaturated fat diets on the composition and structure of human low density lipoproteins. Atherosclerosis 36:299-314.
Prentice, R.L., F. Kakar, S. Hursting, L. Sheppard, R. Klein, and LH. Kushi. 1988. Aspects of the rationale for the women's health trial. J. Natl. Cancer Inst. 80:802-814.
Puska, P., E. Vartiainen, U. Pallonen, J.T. Salonen, P. Poyhia, K. Koskela, and A. McAlister. 1982. The North Karelia Youth Project: evaluation of two years of intervention on health behavior and CVD risk factors among 13- to 15-year old children. Prev. Med. 11:550-570.
Puska, P., J.M. Iacono, A. Nissinen, H.J. Korhonen, E. Vartianinen, P. Pietinen, R. Dougherty, U. Leino, M. Mutanen, S. Moisio, and J. Huttunen. 1983. Controlled, randomized trial of the effect of dietary fat on blood pressure. Lancet 1:1-5.
Puska, P., J.M. Iacono, A. Nissinen, E. Vartiainen, R. Dougherty, P. Pietinen, U. Leino, U. Uusitalo, T. Kuusi, E. Kostiainen, T. Nikkari, E. Seppälä, H. Vapaatalo, and J.K. Huttunen. 1985. Dietary fat and blood pressure: an intervention study on the effects of a low-fat diet with two levels of polyunsaturated fat. Prev. Med. 14:573-584.
Rampone, A.J., and C.M. Machida. 1981. Mode of action of lecithin in suppressing cholesterol absorption. J. Lipid Res. 22:744-752.
Rao, R.H., U.B. Rao, and S.G. Srikantia. 1981. Effect of polyunsaturate-rich vegetable oils on blood pressure in essential hypertension. Clin. Exper. Hypertens. 3:27-38.
Rasanen, L., M. Wilska, R.L. Kantero, V. Nanto, A. Ahlstrom, and N. Hallman. 1978. Nutrition survey of Finnish rural children. IV. Serum cholesterol values in relation to dietary variables. Am. J. Clin. Nutr. 31:1050-1056.
Reddy, B.S. 1986a. Amount and type of dietary fat and colon cancer: animal model studies. Prog. Clin. Biol. Res. 222: 295-309.
Reddy, B.S. 1986b. Diet and colon cancer: evidence from human and animal model studies. Pp. 47-65 in B.S. Reddy and LA. Cohen, eds. Diet, Nutrition, and Cancer: A Critical Evaluation, Vol. I. Macronutrients and Cancer. CRC Press, Boca Raton, Fla.
Reddy, B.S. 1987. Dietary fat and colon cancer: effect of fish oil. Pp. 233-237 in W.E.M. Lands, ed. Proceedings of the AOCS Short Course on Polyunsaturated Fatty Acids and Eicosanoids. American Oil Chemists' Society. Champaign, Ill.
Reddy, B.S., and L.A. Cohen, eds. 1986a. Diet, Nutrition, and Cancer: A Critical Evaluation, Vol. I. Macronutrients and Cancer. CRC Press, Boca Raton, Fla. 175 pp.
Reddy, B.S., and L.A. Cohen, eds. 1986b. Diet, Nutrition, and Cancer: A Critical Evaluation, Vol. II. Micronutrients, Nonnutritive Dietary Factors, and Cancer. CRC Press, Boca Raton, Fla. 181 pp.
Reddy, B.S., and Y. Maeura. 1984. Tumor promotion by
Page 251
dietary fat in azoxymethane-induced colon carcinogenesis in female F344 rats: influence of amount and source of dietary fat. J. Natl. Cancer Inst. 72:745-750.
Reddy, B.S., and E.L. Wynder. 1977. Metabolic epidemiology of colon cancer. Fecal bile acids and neutral sterols in colon cancer patients and patients with adenomatous polyps. Cancer 39:2533-2539.
Reddy, B.S., A.R. Hedges, K. Laakso, and E.L. Wynder. 1978. Metabolic epidemiology of large bowel cancer: fecal bulk and constituents of high-risk North American and low-risk Finnish population. Cancer 42:2832-2838.
Reddy, B.S., B. Hanson, S. Mangat, L. Mathews, M. Sbaschnig, C. Sharma, and B. Simi. 1980a. Effect of high-fat, high-beef diet and of mode of cooking of beef in the diet on fecal bacterial enzymes and fecal bile acids and neutral sterols. J. Nutr. 110:1880-1887.
Reddy, B.S., C. Sharma, L. Darby, K. Laakso, and EL. Wynder. 1980b. Metabolic epidemiology of large bowel cancer. Fecal mutagens in high- and low-risk population for colon cancer. A preliminary report. Mutat. Res. 72:511-522.
Reddy, B.S., G. Ekelund, M. Bohe, A. Engle, and L. Domellof. 1983. Metabolic epidemiology of colon cancer: dietary pattern and fecal sterol concentrations of three populations. Nutr. Cancer 5:34-40.
Reddy, B.S., T. Tanaka, and B. Simi. 1985. Effect of different levels of dietary trans fat or corn oil on azoxymethane-induced colon carcinogenesis in F344 rats. J. Natl. Cancer Inst. 75:791-798.
Reddy, B.S., C.X. Wang, and H. Maruyama. 1987. Effect of restricted caloric intake on azoxymethane-induced colon tumor incidence in male F344 rats. Cancer Res. 47:1226-1228.
Reed, D., K. Yano, and A. Kagan. 1986. Lipids and lipoproteins as predictors of coronary heart disease, stroke, and cancer in the Honolulu Heart Program. Am. J. Med. 80: 871-878.
Reed, D.M., C.J. MacLean, and T. Hayashi. 1987. Predictors of atherosclerosis in the Honolulu Heart Program. I. Biologic, dietary, and lifestyle characteristics. Am. J. Epidemiol. 126:214-225.
Reed, D., R.J. Contreras, C. Maggio, M.R.C. Greenwood, and J. Rodin. 1988. Weight cycling in female rats increases dietary fat selection and adiposity. Physiol. Behav. 42:389-395.
Reed, T., D.K. Wagner, R.P. Donahue, and L.H. Kuller. 1986. Family history of cancer related to cholesterol level in young adults. Genet. Epidemiol. 3:63-71.
Reisen, W.F., R. Mordasini, C. Salzmann, A. Theler, and H.P. Gurtner. 1980. Apoproteins and lipids as discriminators of severity of coronary heart disease. Atherosclerosis 37: 157-162.
Reiser, R., and Z. Sidelman. 1972. Control of serum cholesterol homeostasis by cholesterol in the milk of the suckling rat. J. Nutr. 102:1009-1016.
Reiser, R., B.C. O'Brien, G.R. Henderson, and R.W. Moore. 1979. Studies on a possible function for cholesterol in milk. Nutr. Rep. Int. 19:835-849.
Reiser, R., J.L. Probstfield, A. Silvers, L.W. Scott, M.L. Shorney, R.D. Wood, B.C. O'Brien, A.M. Gotto, Jr., and W. Insull, Jr. 1985. Plasma lipid and lipoprotein response of humans to beef fat, coconut oil and safflower oil. Am. J. Clin. Nutr. 42:190-197.
Renaud, S., K. Kuba, C. Goulet, Y. Lemire, and C. Allard. 1970. Relationship between fatty acid composition of platelets and platelet aggregation in the rat and man. Relation to thrombosis. Circ. Res. 26:553-561.
Renaud, S., R. Morazain, F. Godsey, C. Thevenon, J. Martin, and F. Mendy. 1986. Nutrients, platelet function and composition in nine groups of French and British farmers. Atherosclerosis 60:37-48.
Reunanen, A., H. Takkunen, and A. Aromaa. 1982. Prevalence of intermittent claudication and its effect on mortality. Acta Med. Scand. 211:249-256.
Rhoads, G.G., W.C. Blackwelder, G.N. Stemmerman, T. Hayashi, and A. Kagan. 1978. Coronary risk factors and autopsy findings in Japanese-American men. Lab. Invest. 38:304-311.
Rickert, R.R., K.G. Johnson, H. Kato, T. Yamamoto, and K. Yano. 1968. Atherosclerosis in a defined Japanese population: a clinicopathologic appraisal. Am. J. Clin. Pathol. 49: 371-386.
Risch, H.A., J.D. Burch, A.B. Miller, G.B. Hill, R. Steele, and G.R. Howe. 1988. Dietary factors and the incidence of cancer of the urinary bladder. Am. J. Epidemiol. 127:1179-1191.
Roberts, J.C., Jr., and R. Straus, eds. 1965. Comparative Atherosclerosis; the Morphology of Spontaneous and Induced Atherosclerotic Lesions in Animals and Its Relation to Human Disease, by 56 Authors. Harper & Row, New York. 426 pp.
Robertson, F.W., and A.M. Cumming. 1985. Effects of apoprotein E polymorphism on serum lipoprotein concentration. Arteriosclerosis 5:283-292.
Robertson, T.L, H. Kato, G.G. Rhoads, A. Kagan, M. Marmot, S.L. Syme, T. Gordon, R.M. Worth, J.L Belsky, D.S. Dock, M. Miyanishi, and S. Kawamoto. 1977. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California. Am. J. Cardiol. 39:239-243.
Robertson, W.B., J.C. Geer, J.P. Strong, and H.C. McGill, Jr. 1963. The fate of the fatty streak. Exp. Mol. Pathol. 2 Suppl. 1:28-39.
Robins, S.J., and J. Fasulo. 1973. Mechanism of lithogenic bile production: studies in the hamster fed an essential fatty acid-deficient diet. Gastroenterology 65:104-114.
Robison, R.L, K.C. Hayes, H.L McCombs, and T.P. Faherty. 1971. Effect of dietary fat and cholesterol on circulating lipids and aortic ultrastructure of squirrel monkeys. Exp. Mol. Pathol. 15:281-303.
Roe, D.A., ed. 1983. Diet, Nutrition, and Cancer: From Basic Research to Policy Implications. Current Topics in Nutrition and Disease, Vol. 9. Alan R. Liss, New York. 294 pp.
Roebuck, B.D., D.S. Longnecker, K.J. Baumgartner, and C.D. Thron. 1985. Carcinogen-induced lesions in the rat pancreas: effects of varying levels of essential fatty acid. Cancer Res. 45:5252-5256.
Rogers, A.E, and S.Y. Lee. 1986. Chemically-induced mammary gland tumors in rats: modulation by dietary fat. Prog. Clin. Biol. Res. 222:255-282.
Rogers, A.E., B. Connor, C. Boulanger, and S. Lee. 1986. Mammary tumorigenesis in rats fed diets high in lard. Lipids 21:275-280.
Rohan, T.E., A.J. McMichael, and P.A. Baghurst. 1988. A population-based case-control study of diet and breast cancer in Australia. Am. J. Epidemiol. 128:478-489.
Romieu, I., W.C. Willett, M.J. Stampfer, G.A. Colditz, L. Sampson, B. Rosner, C.H. Hennekens, and F.E. Speizer.
Page 252
1988. Energy intake and other determinants of relative weight. Am. J. Clin. Nutr. 47:406-412.
Rose, G. 1982. Incubation period of coronary heart disease. Br. Med. J. 284:1600-1601.
Rose, G., and M.J. Shipley. 1980. Plasma lipids and mortality: a source of error. Lancet 1:523-526.
Rose, G., H. Blackburn, A. Keys, H.L. Taylor, W.B. Kannel, O. Paul, D.D. Reid, and J. Stamler. 1974. Colon cancer and blood-cholesterol. Lancet 1:181-183.
Rosenthal, S.R. 1934a. Studies in atherosclerosis: chemical, experimental and morphologic. I and II. Roles of cholesterol metabolism, blood pressure and structure of the aorta; the fat angle of the aorta (f.a.a.), and the infiltration-expression theory of lipoid deposit. Arch. Pathol. 18:473-506.
Rosenthal, S.R. 1934b. Studies in atherosclerosis: chemical, experimental and morphologic. III and IV. Roles of cholesterol metabolism, blood pressure and structure of the aorta; the fat angle of the aorta (f.a.a.), and the infiltration-expression theory of lipoid deposit. Arch. Pathol. 18:660-698.
Rosenthal, S.R. 1934c. Studies in atherosclerosis: chemical, experimental and morphologic. V. Possible dangers of iodine therapy in atherosclerosis of aorta seen from an experimental standpoint. Arch. Pathol. 18:827-842.
Ross, R. 1986. The pathogenesis of atherosclerosis—an update. N. Engl. J. Med. 314:488-500.
Rotkin, I.D. 1977. Studies in the epidemiology of prostatic cancers: expanded sampling. Cancer Treat. Rep. 61:173-180.
Rouse, I.L., B.K. Armstrong, B.M. Margetts, and L.J. Beilin. 1981. The dietary habits and nutrient intakes of Seventh-Day Adventists vegetarians and Mormon omnivores. Proc. Nutr. Soc. Aust. 6:117.
Royce, S.M., R.P. Holmes, T. Takagi, and F.A. Kummerow. 1984. The influence of dietary isomeric and saturated fatty acids on atherosclerosis and eicosanoid synthesis in swine. Am. J. Clin. Nutr. 39:215-222.
Rudel, L.L. 1980. Plasma lipoproteins in atherogenesis in nonhuman primates. Pp. 37-57 in S.S. Kalter, ed. The Use of Nonhuman Primates in Cardiovascular Diseases. University of Texas Press, Austin, Tex.
Rudel, L.L, J.A. Reynolds, and B.C. Bullock. 1981. Nutritional effects on blood lipid and HDL cholesterol concentrations in two subspecies of African green monkeys (Cercopithecus aethiops). J. Lipid Res. 22:278-286.
Rudel, L.L., J.S. Parks, and R.M. Carroll. 1983. Effects of polyunsaturated versus saturated dietary fat on nonhuman primate HDL. Pp. 649-666 in E.G. Perkins and W.J. Visek, eds. Dietary Fats and Health. American Oil Chemists' Society, Champaign, Ill.
Rudel, L.L., M.G. Bond, and B.C. Bullock. 1985. LDL heterogeneity and atherosclerosis in nonhuman primates. Ann. N.Y. Acad. Sci. 454:248-253.
Rudel, L.L., J.S. Parks, and M.G. Bond. 1986. Dietary polyunsaturated fat effects on atherosclerosis and plasma lipoproteins in African green monkeys. Pp. 501-523 in D.G. Scarpelli and G. Migaki, eds. Nutritional Diseases: Research Directions in Comparative Pathobiology. Current Topics in Nutrition and Disease, Vol. 15. Alan R. Liss, New York.
Rush, D., and A.R. Kristal. 1982. Methodologic studies during pregnancy: the reliability of the 24-hour dietary recall. Am. J. Clin. Nutt. 35:1259-1268.
Sacks, F.M., B. Rosner, and E.H. Kass. 1974. Blood pressure in vegetarians. Am. J. Epidemiol. 100:390-398.
Sacks, F.M., G.E. Marais, G. Handysides, J. Salazar, L. Miller, J.M. Foster, B. Rosner, and E.H. Kass. 1984. Lack of an effect of dietary saturated fat and cholesterol on blood pressure in normotensives. Hypertension 6:193-198.
Sacks, F.M., I.L. Rouse, M.J. Stampfer, L.M. Bishop, C.F. Lenherr, and R.J. Walther. 1987. Effect of dietary fats and carbohydrate on blood pressure of mildly hypertensive patients. Hypertension 10:452-460.
Salmond, C.E., R. Beaglehole, and I.A.M. Prior. 1985. Are low cholesterol values associated with excess mortality? Br. Med. J. 290:422-424.
Sampliner, R.E., P.H. Bennett, L.J. Comess, F.A. Rosé, and T.A. Burch. 1970. Gallbladder disease in Pima Indians. Demonstration of high prevalence and early onset by cholecystography. N. Engl. J. Med. 283:1358-1364.
Sanders, T.A., D.R. Sullivan, J. Reeve, and G.R. Thompson. 1985. Triglyceride-lowering effect of marine polyunsaturates in patients with hypertriglyceridemia. Arteriosclerosis 5: 459-465.
Sarles, H., C. Chabert, Y. Pommeau, E. Save, H. Mouret, and A. Gerolami. 1969. Diet and cholesterol gallstones. A study of 101 patients with cholelithiasis compared to 101 matched controls. Am. J. Dig. Dis. 14:531-537.
Sarles, H., J. Hauton, N.E. Planche, H. Lafont, and A. Gerolami. 1970. Diet, cholesterol gallstones, and composition of the bile. Am. J. Dig. Dis. 15:251-260.
Sarles, H., C. Crotte, A. Gerolami, A. Mule, N. Domingo, and J. Hauton. 1971. The influence of calorie intake and of dietary protein on the bile lipids. Scand. J. Gastroenterol. 6: 189-191.
Savage, P.J., R.F. Hamman, G. Bartha, S.E. Dippe, M. Miller, and P.H. Bennett. 1976. Serum cholesterol levels in American (Pima) Indian children and adolescents Pediatrics 58:274-282.
Scanu, A.M., A. Khalil, L Neven, M. Tidore, G. Dawson, D. Pfaffinger, E. Jackson, K.D. Carey, H.C. McGill, and G.M. Fless. 1988. Genetically determined hypercholesterolemia in a rhesus monkey family due to a deficiency of the LDL receptor. J. Lipid Res. 29:1671-1681.
Schemmel, R., O. Mickelsen, and K. Motawi. 1982. Conversion of dietary to body energy in rats as affected by strain, sex and ration. J. Nutr. 102:1187-1197.
Schieken, R.M., W.R. Clarke, and R.M. Lauer. 1981. Left ventricular hypertrophy in children with blood pressures in the upper quintile of the distribution. The Muscatine Study. Hypertension 3:669-675.
Schoenfield, L.J. 1980. Diseases of the gallbladder and bile ducts. Pp. 1489-1498 in K.J. Isselbacher, R.D. Adams, E. Braunwald, R.G. Petersdorf, and J.D. Wilson, eds. Harrison's Principles of Internal Medicine, 9th ed., Vol. 2. McGraw-Hill, New York.
Schonfeld, G., W. Patsch, LL Rudel, C. Nelson, M. Epstein, and R.E. Olson. 1982. Effects of dietary cholesterol and fatty acids on plasma lipoproteins. J. Clin. Invest. 69: 1072-1080.
Schrott, H.G., W.R. Clarke, D.A. Wiebe, W.E. Connor, and R.M. Lauer. 1979. Increased coronary mortality in relatives of hypercholesterolemic school children: the Muscatine Study. Circulation 59:320-326.
Schuman, LM., J.S. Mandel, A. Radke, U. Seal, and F. Halberg. 1982. Some selected features of the epidemiology of prostatic cancer. Minneapolis-St. Paul, Minnesota Case-Control Study, 1976-1979. Pp. 345-354 in K. Magnus, ed.
Page 253
Trends in Cancer Incidence: Causes and Practical Implications. Hemisphere Publishing Corp., Washington, D.C.
Sclafani, A., and A.N. Gorman. 1977. Effects of age, sex, and prior body weight on the development of dietary obesity in adult rats. Physiol. Behav. 18:1021-1026.
Scott, R.F., E.S. Morrison, W.A. Thomas, R. Jones, and S.C. Nam. 1964. Short-term feeding of unsaturated versus saturated fat in the production of atherosclerosis in the rat. Exp. Mol. Pathol. 3:421-443.
Scott, R.F., E.S. Morrison, J. Jarmolych, S.C. Nam, M. Kroms, and F. Coulston. 1967a. Experimental atherosclerosis in rhesus monkeys. I. Gross and light microscopy features and lipid values in serum and aorta. Exp. Mol. Pathol. 7:11-33.
Scott, R.F., R. Jones, A.S. Daoud, O. Zumbo, F. Coulston, and W.A. Thomas. 1967b. Experimental atherosclerosis in rhesus monkeys. II. Cellular elements of proliferative lesions and possible role of cytoplasmic degeneration in pathogenesis as studied by electron microscopy. Exp. Mol. Pathol. 7: 34-57.
Scragg, R.K.R, A.J. McMichael, and P.A. Baghurst. 1984. Diet, alcohol, and relative weight in gall stone disease: a case-control study. Br. Med. J. 288:1113-1119.
Scrimshaw, N.S., and M.A. Guzman. 1968. Diet and atherosclerosis. Lab. Invest. 18:623-628.
Scrimshaw, N.S., M. Trulson, C. Tejada, D.M. Hegsted, F.J. Stare. 1957. Serum lipoprotein and cholesterol concentrations; comparison of rural Costa Rican, Guatemalan, and United States populations. Circulation 15:805-813.
Selenskas, S.L., M.M. Ip, and C. Ip. 1984. Similarity between trans fat and saturated fat in the modification of rat mammary carcinogenesis. Cancer Res. 44:1321-1326.
Senti, F.R., ed. 1985. Health Aspects of Dietary trans Fatty Acids. Federation of American Societies for Experimental Biology, Bethesda, Md. 148 pp.
Sheard, N.F., J.A. Tayek, B.R. Bistrian, G.L. Blackburn, and S.H. Zeisel. 1986. Plasma choline concentration in humans fed parenterally. Am. J. Clin. Nutr. 43:219-224.
Shekelle, R.B., A.M. Shryock, O. Paul, M. Lepper, J. Stamler, S. Liu, and W.J. Raynor, Jr. 1981a. Diet, serum cholesterol, and death from coronary heart disease. The Western Electric Study. N. Engl. J. Med. 304:65-70.
Shekelle, R.B., J. Stamler, O. Paul, A.M. Shyrock, S. Liu, and M. Lepper. 1981b. Dietary lipids and serum cholesterol level: change in diet confounds the cross-sectional association. Am. J. Epidemiol. 115:506-514.
Shekelle, R.B., L. Missell, O. Paul, A.M. Shryock, and J. Stamler. 1985. Fish consumption and mortality from coronary heart disease. N. Engl. J. Med. 313:820.
Shepherd, J., C.J. Packard, J.R. Patsch, A.M. Gotto, Jr., and O.D. Taunton. 1978. Effects of dietary polyunsaturated and saturated fat on the properties of high density lipoproteins and the metabolism of apolipoprotein A-I. J. Clin. Invest. 61:1582-1592.
Shepherd, J., C.J. Packard, S.M. Grundy, D. Yeshurun, A.M. Gotto, Jr., and O.D. Taunton. 1980a. Effects of saturated and polyunsaturated fat diets on the chemical composition and metabolism of low density lipoproteins in man. J. Lipid Res. 21:91-99.
Shepherd, J., J.M. Stewart, J.G. Clark, and K. Cart. 1980b. Sequential changes in plasma lipoproteins and body fat composition during polyunsaturated fat feeding in man. Br. J. Nutr. 44:265-271.
Sherwin, R.W., D.N. Wentworth, J.A. Cutler, S.B. Hulley, L.H. Kuller, and J. Stamler. 1987. Serum cholesterol levels and cancer mortality in 361,662 men screened for the Multiple Risk Factor Intervention Trial. J. Am. Med. Assoc. 257:943-948.
Shimamoto, T., Y. Komachi, H. Inada, M. Doi, H. Iso, S. Sato, A. Kitamura, M. lida, M. Konishi, N. Nakanishi, A. Terao, Y. Naito, and S. Kojima. 1989. Trends for coronary heart disease and stroke and their risk factors in Japan. Circulation 79:503-516.
Shulman, R.S., A.K. Bhattacharyya, W.E. Connor, and D.S. Fredrickson. 1976. Beta-sitosterolemia and xanthomatosis. N. Engl. J. Med 294:482-483.
Sidney, S., G.D. Friedman, and R.A. Hiatt. 1986. Serum cholesterol and large bowel cancer. A case-control study. Am. J. Epidemiol. 124:33-38.
Simons, L.A. 1986. Interrelations of lipids and lipoproteins with coronary artery disease mortality in 19 countries. Am. J. Cardiol. 57:5G-10G.
Singer, P., U. Gerhard, V. Moritz, D. Förster, I. Berger, and H. Heine. 1986. Different changes of N-6 and N-3 fatty acids in adipose tissue from spontaneously hypertensive (SHR) and normotensive rats after diets supplemented with linolenic or eicosapentaenoic acids. Prost. Leuk. Med. 24: 163-172.
Singhal, A.K., J. Finver-Sadowsky, C.K. McSherry, and E.H. Mosbach. 1983. Effect of cholesterol and bile acids on the regulation of cholesterol metabolism in hamster. Biochim. Biophys. Acta 752:214-222.
Sirtori, C.R., G. Biasi, G. Vercellio, E. Agradi, and E. Malan. 1974. Diet, lipids and lipoproteins in patients with peripheral vascular disease. Am. J. Med. Sci. 268:325-332.
Sitaram, N., H. Weingartner, E.D. Caine, and J.C. Gillin. 1978. Choline: selective enhancement of serial learning and encoding of low imagery words in man. Life Sci. 22:1555-1560.
Slover, H.T., R.H. Thompson, Jr., C.S. Davis, and G.V. Merola. 1985. Lipids in margarines and margarine-like foods. J. Am. Oil Chem. Soc. 62:775-786.
Small, D.M. 1976. Part I. The etiology and pathogenesis of gallstones. Adv. Surg. 10:63-85.
Smith-Barbaro, P., M.R. Quinn, H. Fisher, and D.M. Hegstead. 1981. The effect of dietary fat and salt on blood pressure, renal and aortic prostaglandins. Nutr. Res. 1:277-287.
Snapper, I. 1941. Chinese Lessons to Western Medicine. Interscience Publishers, New York. 380 pp.
Sniderman, A., S. Shapiro, D. Marpole, B. Skinner, B. Teng, and P.O. Kwiterovich, Jr. 1980. Association of coronary atherosclerosis with hyperapobetalipoproteinemia [increased protein but normal cholesterol levels in human plasma low density (beta) lipoproteins]. Proc. Natl. Acad. Sci. U.S.A. 77:604-608.
Snowdon, D.A. 1981. Alcohol Use and Mortality from Cancer and Heart Disease Among Members of the Lutheran Brotherhood Cohort. University Microfilms, Ann Arbor, Mich.
Snowdon, D.A. 1988. Animal product consumption and mortality because of all causes combined, coronary heart disease, stroke, diabetes, and cancer in Seventh-Day Adventists. Am. J. Clin. Nutr. 48:739-748.
Snowdon, D.A., R.L. Phillips, and W. Choi. 1984. Diet, obesity, and risk of fatal prostate cancer. Am. J. Epidemiol. 120:244-250.
Sodhi, H.S., P.D. Wood, G. Schlierf, and L.W. Kinsell.
Page 254
1967. Plasma, bile and fecal sterols in relation to diet. Metabolism 16:334-344.
Somjen, G.I., and T. Gilat. 1986. Changing concepts of cholesterol solubility in bile. Gastroenterology 91:772-775.
Sorlie, P.D., M.R. Garcia-Palmieri, M.I. Castillo-Stabb, R. Costas, Jr., M.C. Oalmann, and R. Havlik. 1981. The relation of antemortem factors to atherosclerosis at autopsy. The Puerto Rico Heart Health Study. Am. J. Pathol. 103: 345-352.
Spady, D.K., and J.M. Dietschy. 1985. Dietary saturated triacylglycerols suppress hepatic low density lipoprotein receptor activity in the hamster. Proc. Natl. Acad. Sci. U.S.A. 82:4526-4530.
Spady, D.K., and J.M. Dietschy. 1988. Interaction of dietary cholesterol and triglycerides in the regulation of hepatic low density lipoprotein transport in the hamster. J. Clin. Invest. 81:300-309.
Spritz, N., and M.A. Mishkel. 1969. Effects of dietary fats on plasma lipids and lipoproteins: an hypothesis for the lipid-lowering effect of unsaturated fatty acids. J. Clin. Invest. 48: 78-86.
Spritz, N., E.H. Ahrens, Jr., and S. Grundy. 1965. Sterol balance in man as plasma cholesterol concentrations are altered by exchanges of dietary fats. J. Clin. Invest. 44: 1482-1493.
Srinivasan, S.R., B. Radhakrishnamurthy, C.C. Smith, R.H. Wolf, and G.S. Berenson. 1976. Serum lipid and lipoprotein responses of six nonhuman primate species to dietary changes in cholesterol levels. J. Nutr. 106:1757-1767.
St. Clair, R.W., G.R. Henderson, V. Heaster, W.D. Wagner, M.G. Bond, and M.R. McMahan. 1980. Influence of dietary fats and oral contraceptive on plasma lipids, high density lipoproteins, gallstones, and atherosclerosis in African green monkeys. Atherosclerosis 37:103-121.
Stallones, R.A. 1983. Ischemic heart disease and lipids in blood and diet. Annu. Rev. Nutr. 3:155-185.
Stamler, J. 1967. Lectures on Preventive Cardiology. Grune & Stratton, New York. 434 pp.
Stamler, J. 1979. Population studies. Pp. 25-88 in R.I. Levy, B.M. Rifkind, B.H. Dennis, and N. Ernst, eds. Nutrition, Lipids, and Coronary Heart Disease: A Global View. Nutrition in Health and Disease, Vol. 1. Raven Press, New York.
Stamler, J., and R. Shekelle. 1988. Dietary cholesterol and human coronary heart disease: the epidemiologic evidence. Arch. Pathol. Lab. Med. 112:1032-1040.
Stamler, J., R. Stamler, and R.B. Shekelle. 1970. Regional differences in prevalence, incidence and mortality from atherosclerotic coronary heart disease. Pp. 84-127 in J.H. de Haas, H.C. Hemker, and H.A. Snellen, eds. Ischaemic Heart Disease. Leiden University Press, Leiden, The Netherlands.
Stamler, J., D.M. Berkson, and H.A. Lindberg. 1972. Risk factors: their role in the etiology and pathogenesis of the atherosclerotic diseases. Pp. 41-119 in R.W. Wissler, J.C. Geer, and N. Kaufman, eds. The Pathogenesis of Atherosclerosis. Williams & Wilkins, Baltimore.
Stamler, J., D. Wentworth, and J.D. Neaton. 1986. Is the relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Findings in 356,222 primary screenees of the Multiple Risk Factor Intervention Trial (MRFIT). J. Am. Med. Assoc. 256:2823-2828.
Stanbury, J.B., J.B. Wyngaarden, D.S. Fredrickson, J.L. Goldstein, and M.S. Brown, eds. 1983. The Metabolic Basis of Inherited Disease, 5th ed., Part 4. McGraw-Hill, New York. 2032 pp.
Stary, H.C. 1987a. Evolution and progression of atherosclerosis in the coronary arteries of children and adults. Pp. 20-36 in S.R. Bates and E.C. Gangloff, eds. Atherogenesis and Aging. Springer-Verlag, New York.
Stary, H.C. 1987b. Macrophages, macrophage foam cells, and eccentric intimal thickening in the coronary arteries of young children. Atherosclerosis 64:91-108.
Stein, E.A., J. Shapero, C. McNerey, C.J. Glueck, T. Tracy, and P. Gartside. 1982. Changes in plasma lipid and lipoprotein fractions after alteration in dietary cholesterol, polyunsaturated, saturated, and total fat in free-living normal and hypercholesterolemic children. Am. J. Clin. Nutr. 35:1375-1390.
Steinberg, D. 1983. Lipoproteins and atherosclerosis. A look back and a look ahead. Arteriosclerosis 3:283-301.
Steiner, A. 1948. Significance of cholesterol in coronary arteriosclerosis. N.Y. State J. Med. 48:1814-1818.
Steiner, A., and B. Domanski. 1944. Effect of feeding of ''soya lecithin" on serum cholesterol level of man. Proc. Soc. Exp. Biol. Med. 55:236-238.
Steiner, A., and F.E. Kendall. 1946. Atherosclerosis and arteriosclerosis in dogs following ingestion of cholesterol and thiouracil. Arch. Pathol. 42:433-444.
Stemmerman, G.N., A.M. Nomura, and L.K. Heilbrun. 1984. Dietary fat and the risk of colorectal cancer. Cancer Res. 44:4633-4637.
Stich, H.F., ed. 1982. Carcinogens and Mutagens in the Environment, Vol. I. Food Products. CRC Press, Boca Raton, Fla. 310 pp.
Strazzullo, P., A. Ferro-Luzzi, A. Siani, C. Scaccini, S. Sette, G. Catasta, and M. Mancini. 1986. Changing the Mediterranean diet: effects on blood pressure. J. Hypertens. 4:407-412.
Strøm, A., and R.A. Jensen. 1951. Mortality from circulatory diseases in Norway 1940-1945. Lancet I:126-129.
Strong, J.P. 1976. Atherosclerosis in primates. Introduction and overview. Primates Med. 9:1-15.
Strong, J.P., and H.C. McGill, Jr. 1962. The natural history of coronary atherosclerosis. Am. J. Pathol. 40:37-49.
Strong, J.P., and H.C. McGill, Jr. 1967. Diet and experimental atherosclerosis in baboons. Am. J. Pathol. 50:669-690.
Strong, J.P., and H.C. McGill, Jr. 1969. The pediatric aspects of atherosclerosis. J. Atheroscler. Res. 9:251-265.
Strong, J.P., H.C. McGill, Jr., C. Tejada, and R.L. Holman. 1958. The natural history of atherosclerosis. Comparison of the early aortic lesions in New Orleans, Guatemala, and Costa Rica. Am. J. Pathol. 34:731-744.
Strong, J.P., W.P. Newman III, D.S. Freedman, P.D. Gard, R.E. Tracy, and L.A. Solberg. 1986. Atherosclerotic disease in children and young adults: relationship to cardiovascular risk factors. Pp. 27-41 in G.S. Berenson, ed. Causation of Cardiovascular Risk Factors in Children. Perspectives on Cardiovascular Risk in Early Life. Raven Press, New York.
Sturdevant, R.A., M.L. Pearce, and S. Dayton. 1973. Increased prevalence of cholelithiasis in men ingesting a serum-cholesterol-lowering diet. N. Engl. J. Med. 288:24-27.
Sullivan, D.R., T.A. Sanders, I.M. Trayner, and G.R. Thompson. 1986. Paradoxical elevation of LDL apoprotein B levels in hypertriglyceridaemic patients and normal sub-
Page 255
jects ingesting fish oil. Atherosclerosis 61:129-134.
Sullivan, D.R., C.E. West, M.B. Katan, I. Halferkamps, and H. van der Torre. 1987. Atherogenic and protective lipoproteins in boys from ten countries differing in dietary carbohydrate intake. Pp. 73-81 in B.S. Hetzel and G.S. Berenson, eds. Cardiovascular Risk Factors in Childhood: Epidemiology and Prevention. Elsevier, Amsterdam.
Sutor, D.J., and S.E. Wooley. 1971. A statistical survey of the composition of gallstones in eight countries. Gut 12:55-64.
Svanberg, U., A. Gustafson, and R. Ohlson. 1974. Polyunsaturated fatty acids in hyperlipoproteinemia. II. Administration of essential phospholipids in hypertriglyceridemia. Nutr. Metab. 17:338-346.
Sweeney, M.J., J.N. Etteldorf, W.T. Dobbins, B. Somervil, R. Fischer, and C. Ferrell. 1961. Dietary fat and concentrations of lipid in the serum during the first six to eight weeks of life. Pediatrics 27:765-771.
Sylvester, P.W. 1986. Role of acute caloric-restriction in murine tumorigenesis. Prog. Clin. Biol. Res. 222:517-528.
Sylvester, P.W., M. Russell, M.M. Ip., and C. Ip. 1986a. Comparative effects of different animal and vegetable fats fed before and during carcinogen administration on mammary tumorigenesis, sexual maturation, and endocrine function in rats. Cancer Res. 46:757-762.
Sylvester, P.W., C. Ip., and M.M. Ip. 1986b. Effects of high dietary fat on the growth and development of ovarian-independent carcinogen-induced mammary tumors in rats. Cancer Res. 46:763-769.
Tabas, I., D.A. Weiland, and A.R. Tall. 1985. Unmodified low density lipoprotein causes cholesteryl ester accumulation in J774 macrophages. Proc. Natl. Acad. Sci. U.S.A. 82:416-420.
Takeya, Y., J.S. Popper, Y. Shimizu, H. Kato, G.G. Rhoads, and A. Kagan. 1984. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: incidence of stroke in Japan and Hawaii. Stroke 15:15-23.
Talamini, R., C. La Veccia, A. Decarli, S. Franceschi, E Grattoni, E. Grigoletto, A. Liberati, and G. Tognoni. 1984. Social factors, diet and breast cancer in a northern Italian population. Br. J. Cancer 49:723-729.
Tamir, I., Y. Bojanower, O. Levtow, D. Heldenberg, Z. Dickerman, and B. Werbin. 1972. Serum lipids and lipoproteins in children from families with early coronary heart disease. Arch. Dis. Child. 47:808-810.
Tanaka, N., and O.W. Portman. 1977. Effect of type of dietary fat and cholesterol on cholesterol absorption rate in squirrel monkeys. J. Nutr. 107:814-821.
Tanaka, N., O.W. Portman, and T. Osuga. 1976. Effect of type of dietary fat, cholesterol and chenodeoxycholic acid on gallstone formation, bile acid kinetics and plasma lipids in squirrel monkeys. J. Nutr. 106:1123-1134.
Tannenbaum, A. 1959. Nutrition and cancer. Pp. 517-562 in F. Homburger, ed. The Physiopathology of Cancer by 32 Authors, 2nd ed. Hoeber-Harper, New York.
Tartter, P.I., G. Slater, A.E. Paptestas, and A.H. Aufses, Jr. 1984. Cholesterol, weight, height, Quetelet's index, and colon cancer recurrence. J. Surg. Oncol. 27:232-235.
Taylor, C.B., D.E. Patton, and G.E. Cox. 1963. Atherosclerosis in rhesus monkeys. VI. Fatal myocardial infarction in a monkey fed fat and cholesterol. Arch. Pathol. 76:404-423.
Taylor, C.B., S.K. Peng, N.T. Werthessen, P. Tham, and K.T. Lee. 1979. Spontaneously occurring angiotoxic derivatives of cholesterol. Am. J. Clin. Nutr. 32:40-57.
Tejada, C., J.P. Strong, M.R. Montenegro, C. Restrepo, and LA. Solberg. 1968. Distribution of coronary and aortic atherosclerosis by geographic location, race, and sex. Lab. Invest. 18:509-526.
ten Hoor, F., and H.M. van de Graaf. 1978. The influence of a linoleic acid-rich diet and of acetyl salicylic acid on NaCl induced hypertension, Na+—and H2O-balance and urinary prostaglandin excretion in rats. Acta Biol. Med. Ger. 37: 875-877.
Tepperman, J., F.T. Caldwell, and H.M. Tepperman. 1964. Induction of gallstones in mice by feeding a cholesterol-cholic acid containing diet. Am. J. Physiol. 206:628-634.
ter Welle, H.F., C.M. van Gent, W. Dekker, and A.F. Willebrands. 1974. The effect of soya lecithin on serum lipid values in type II hyperlipoproteinemia. Acta Med. Scand. 195:267-271.
Thomas, L.H., P.R. Jones, J.A. Winter, and H. Smith. 1981. Hydrogenated oils and fats: the presence of chemically-modified fatty acids in human adipose tissue. Am. J. Clin. Nutr. 34:877-886.
Thomas, L.H., J.A. Winter, and R.G. Scott. 1983a. Concentration of transunsaturated fatty acids in the adipose body tissue of decedents dying of ischaemic heart disease compared with controls. J. Epidemiol. Community Health 37: 22-24.
Thomas, L.H., J.A. Winter, and R.G. Scott. 1983b. Concentration of 18:1 and 16:1 transunsaturated fatty acids in the adipose body tissue of decedents dying of ischaemic heart disease compared with controls: analysis by gas liquid chromatography. J. Epidemiol. Community Health 37:16-21.
Thulin, T., M. Abdulla, I. Denker, M. Jagerstad, A. Melander, A. Norden, B. Schersten, and B. Akesson. 1980. Comparison of energy and nutrient intakes in women with high and low blood pressure levels. Acta Med. Scand. 208: 367-373.
Tidwell, H.C., J.C. McPherson, and W.W. Burr, Jr. 1962. Effect of the saturation of fats upon the disposition of ingested cholesterol. Am. J. Clin. Nutr. 11:108-114.
Tillotson, J.L, H. Kato, M.Z. Nichaman, D.C. Miller, M.L Gay, K.G. Johnson, and G.G. Rhoads. 1973. Epidemiology of coronary heart disease and stroke in Japanese men living in Japan, Hawaii, and California: methodology for comparison of diet. Am. J. Clin. Nutr. 26:177-184.
Tilvis, RS., and T.A. Miettinen. 1986. Serum plant sterols and their relation to cholesterol absorption. Am. J. Clin. Nutr. 43:92-97.
Tobian, L., M. Ganguli, M.A. Johnson, and J. Iwai. 1985. The influence of renal prostaglandins and dietary linoleate on hypertension of Dahl salt-sensitive rats. Pp. 349-359 in M.J. Horan, M. Blaustein, J.B. Dunbar, W. Kachadorian, N.M. Kaplan, and A.P. Simopoulos, eds. NIH Workshop on Nutrition and Hypertension: Proceedings from a Symposium. Biochemical Information Corp., New York.
Toor, M., A. Katchalsky, J. Agmon, and D. Allalouf. 1960. Atherosclerosis and related factors in immigrants to Israel. Circulation 22:265-279.
Tornberg, S.A., L.E. Holm, J.M. Carstensen, and G.A. Eklund. 1986. Risks of cancer of the colon and rectum in relation to serum cholesterol and beta-lipoprotein. N. Engl. J. Med. 315:1629-1633.
Trotman, B.W., and R.D. Soloway. 1975. Pigment vs cholesterol cholelithiasis: clinical and epidemiological aspects. Am. J. Dig. Dis. 20:735-740.
Tuomilhehto, J., P. Puska, A. Nissinen, J. Salonen, A.
Page 256
Tanskanen, P. Pietinen, and E. Wolf. 1984. Community-based prevention of hypertension in North Karelia, Finland. Ann. Clin. Res. 16 Suppl. 43:18-27.
Turjman, N., G.T. Goodman, B. Jaeger, and P.P. Nair. 1984a. Diet, nutrition intake, and metabolism in populations at high and low risk for colon cancer. Metabolism of bile acids. Am. J. Clin. Nutr. 40:937-941.
Turjman, N., B. Calkins, R. Phillips, G.T. Goodman, and P.P. Nair. 1984b. Dietary intake of plant sterols and cholesterol among population groups at different risks for colon cancer. Fed. Proc. 43:855.
Turkenkopf, I.J., C.A. Maggio, and M.R. Greenwood. 1982. Effect of high fat weanling diets containing either medium-chain triglycerides or long-chain triglycerides on the development of obesity in the Zucker rat. J. Nutr. 112:1254-1263.
Turner, J.D., N.A. Le, and W.V. Brown. 1981. Effect of changing dietary fat saturation on low-density lipoprotein metabolism in man. Am. J. Physiol. 241:E57-E63.
Turpeinen, O., M.J. Karvonen, M. Pekkarinen, M. Miettinen, R. Elosuo, and E. Paavilainen. 1979. Dietary prevention of coronary heart disease: the Finnish Mental Hospital Study. Int. J. Epidemiol. 8:99-118.
Tuyns, A.J., M. Haelterman, and R. Kaaks. 1987. Colorectal cancer and the intake of nutrients: oligosaccharides are a risk factor, fats are not. A case-control study in Belgium. Nutr. Cancer 10:181-196.
USDA (U.S. Department of Agriculture). 1984. Nationwide Food Consumption Survey. Nutrient Intakes: Individuals in 48 States, Year 1977-78. Report No. 1-2. Consumer Nutrition Division, Human Nutrition Information Service, Hyattsville, Md. 439 pp.
USDA (U.S. Department of Agriculture). 1986. Nationwide Food Consumption Survey. Continuing Survey of Food Intakes of Individuals. Men 19-50 Years, 1 Day, 1985. Report No. 85-3. Nutrition Monitoring Division, Human Nutrition Information Service, Hyattsville, Md. 94 pp.
USDA (U.S. Department of Agriculture). 1987. Nationwide Food Consumption Survey. Continuing Survey of Food Intakes of Individuals. Women 19-50 Years and Their Children 1-5 Years, 4 Days, 1985. Report No. 85-4. Nutrition Monitoring Division, Human Nutrition Information Service, Hyattsville, Md. 182 pp.
U.S.-U.S.S.R. Steering Committee. 1984. Nutrient intake and its association with high-density lipoprotein and low-density lipoprotein cholesterol in selected U.S. and U.S.S.R. subpopulations. The U.S.-U.S.S.R. Steering Committee for Problem Area 1: the pathogenesis of atherosclerosis. Am. J. Clin. Nutr. 39:942-952.
Utermann, G., M. Hees, and A. Steinmetz. 1977. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature 269:604-607.
Utermann, G., N. Pruin, and A. Steinmetz. 1979. Polymorphism of apolipoprotein E. III. Effect of a single polymorphic gene locus on plasma lipid levels in man. Clin. Genet. 15: 63-72.
Van Lenten, B.J., A.M. Fogelman, R.L. Jackson, S. Shapiro, M.E. Haberland, and P.A. Edwards. 1985. Receptor-mediated uptake of remnant lipoproteins by cholesterol-loaded human monocyte-macrophages. J. Biol. Chem. 260:8783-8788.
Vartiainen, E., P. Puska, and J.T. Salonen. 1982. Serum total cholesterol, HDL cholesterol and blood pressure levels in 13-year-old children in Eastern Finland. The North Karelia Youth Project. Acta Med. Scand. 211:95-103.
Vartiainen, E., P. Puska, P. Pietinen, A. Nissinen, U. Leino, and U. Uusitalo. 1986. Effects of dietary fat modifications on serum lipids and blood pressure in children. Acta Pediatr. Scand. 75:396-401.
Vartiainen, 1. 1946. War-time and mortality in certain diseases in Finland. Ann. Med. Int. Fenn. 35:234-240.
Vartiainen, I., and K. Kanerva. 1947. Arteriosclerosis and war-time. Ann. Med. Int. Fenn. 36:748-758.
Vega, G.L., E. Groszek, R. Wolf, and S.M. Grundy. 1982. Influence of polyunsaturated fats on composition of plasma lipoproteins and apolipoproteins. J. Lipid Res. 23:811-822.
Vergroesen, A.J., A.I. Fleischman, H.U. Comberg, S. Heyden, and C.G. Hames. 1978. The influence of dietary linoleate on essential hypertension in man. Acta Biol. Med. Ger. 37:879-883.
Vesselinovitch, D., G.S. Getz, R.H. Hughes, and R.W. Wissler. 1974. Atherosclerosis in the rhesus monkey fed three food fats. Atherosclerosis 20:303-321.
Vesselinovitch, D., R.W. Wissler, T.J. Schaffner, and J. Borensztajn. 1980. The effect of various diets on atherogenesis in rhesus monkeys. Atherosclerosis 35:189-207.
Voller, R.D., Jr., and W.B. Strong. 1981. Pediatric aspects of atherosclerosis. Am. Heart J. 101:815-836.
Vollset, S.E., I. Heuch, and E. Bjelke. 1985. Fish consumption and mortality from coronary heart disease. N. Engl. J. Med. 313:820-821.
von Schacky, C. 1987. Prophylaxis of atherosclerosis with marine omega-3 fatty acids. A comprehensive strategy. Ann. Intern. Med. 107:890-899.
von Schacky, C., W. Siess, S. Fischer, and P.C. Weber. 1985a. A comparative study of eicosapentaenoic acid metabolism by human platelets in vivo and in vitro. J. Lipid Res. 26:457-464.
von Schacky, C., S. Fischer, and P.C. Weber. 1985b. Long-term effects of dietary marine omega-3 fatty acids upon plasma and cellular lipids, platelet function, and eicosanoid formation in humans. J. Clin. Invest. 76:1626-1631.
Wade, A.E., and S. Dhardwadkar. 1986. Metabolic activation of carcinogens. Prog. Clin. Biol. Res. 222:587-606.
Walker, B.L. (Memorial Symposium). 1986. Lipids and cancer. Papers from the Brian L. Walker Memorial Symposium presented at the 76th AOCS Annual Meeting in Philadelphia, Pennsylvania, May 1985. Lipids 21:271-307.
Wallace, R.B., and R.A. Anderson. 1987. Blood lipids, lipid-related measures, and the risk of atherosclerotic cardiovascular disease. Epidemiol. Rev. 9:95-119.
Walter, H.J., and A. Hofman. 1987. Socioeconomic status, ethnic origin, and risk factors for coronary heart disease in children. Am. Heart. J. 113:812-818.
Walton, K.W., J. Hitchens, H.N. Magnani, and M. Khan. 1974. A study of methods of identification and estimation of LP(a) lipoprotein and of its significance in health, hyperlipidaemia and atherosclerosis. Atherosclerosis 20:323-346.
Watanabe, N., N.S. Gimbel, and C.G. Johnston. 1962. Effect of polyunsaturated and saturated fatty acids on the cholesterol holding capacity of human bile. Arch. Surg. 85:136-141.
Watkins, L.O., J.D. Neaton, and L.H. Kuller. 1986. Racial differences in high-density lipoprotein cholesterol and coronary heart disease incidence in the usual-care group of the Multiple Risk Factor Intervention Trial. Am. J. Cardiol. 57: 538-545.
Webber, L.S., J.L. Cresanta, A.W. Voors, and G.S. Beren-
Page 257
son. 1983. Tracking of cardiovascular disease risk factor variables in school-age children. J. Chronic Dis. 36:647-660.
Webber, L.S., D.S. Freedman, and J.L. Cresanta. 1986. Tracking of cardiovascular disease risk factor variables in school-age children. Pp. 42-64 in G.S. Berenson, ed. Causation of Cardiovascular Risk Factors in Children. Perspectives on Cardiovascular Risk in Early Life. Raven Press, New York.
Weidman, W., P. Kwiterovich, Jr., M.J. Jesse, and E. Nugent. 1983. Diet in the healthy child. Task Force Committee of the Nutrition Committee and the Cardiovascular Disease in the Young Council of the American Heart Association. Circulation 67:1411A-1414A.
Weinsier, R.L., and D. Norris. 1985. Recent developments in the etiology and treatment of hypertension: dietary calcium, fat, and magnesium. Am. J. Clin. Nutr. 42:1331-1338.
Weisgraber, K.H., S.C. Rall, Jr., and R.W. Mahley. 1981. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 256:9077-9083.
Weiss, K.M., R.E. Ferrell, C.L. Hanis, and P.N. Styne. 1984. Genetics and epidemiology of gallbladder disease in new world native peoples. Am. J. Hum. Genet. 36:1259-1278.
Weisweiler, P., P. Janetschek, and P. Schwandt. 1985. Influence of polyunsaturated fats and fat restriction on serum lipoproteins in humans. Metabolism 34:83-87.
Weisweiler, P., P. Janetschek, and P. Schwandt. 1986. Fat restriction alters the composition of apolipoprotein B-100 containing very low-density lipoproteins in humans. Am. J. Clin. Nutr. 43:903-909.
Welsch, C.W. 1986. Interrelationship between dietary fat and endocrine processes in mammary gland tumorigenesis. Prog. Clin. Biol. Res. 222:623-654.
Welsch, C.W., and C.F. Aylsworth. 1983. Enhancement of murine mammary tumorigenesis by feeding high levels of dietary fat: a hormonal mechanism? J. Natl. Cancer Inst. 70:215-221.
Wexler, B.C. 1981. Inhibition of the pathogenesis of spontaneous hypertension in spontaneously hypertensive rats by feeding a high fat diet. Endocrinology 108:981-989.
Whayne, T.F., P. Alaupovic, M.D. Curry, ET. Lee, P.S. Anderson, and E. Schechter. 1981. Plasma apolipoprotein B and VLDL-, LDL-, and HDL-cholesterol as risk factors in the development of coronary artery disease in male patients examined by angiography. Atherosclerosis 39:411-424.
WHO (World Health Organization). 1982. Prevention of Coronary Heart Disease. Technical Report Series No. 678. World Health Organization, Geneva. 53 pp.
WHO (World Health Organization) European Collaborative Group. 1983. Multifactorial trial in the prevention of coronary heart disease: 3. Incidence and mortality results. Eur. Heart J. 4:141-147.
Wigand, G. 1959. Production of hypercholesterolemia and atherosclerosis in rabbits by feeding different fats without supplementary cholesterol. Acta Med. Scand. 166 suppl. 351:1-91.
Wilhelmsen, L., G. Berglund, D. Elmfeldt, G. Tibblin, H. Wedel, K. Pennert, A. Vedin, C. Wilhelmsson, and L Werkö. 1986. The multifactor primary prevention trial in Göteborg, Sweden. Eur. Heart J. 7:279-288.
Willett, W., and M.J. Stampfer. 1986. Total energy intake: implications for epidemiologic analyses. Am. J. Epidemiol. 124:17-27.
Willett, W., M.J. Stampfer, G.A. Colditz, B.A. Rosner, C.H. Hennekeris, and F.E. Speizer. 1987. Dietary fat and the risk of breast cancer. N. Engl. J. Med. 316:22-28.
Williams, P.T., R.M. Krauss, S. Kindel-Joyce, D.M. Dreon, K.M. Vranizan, and P.D. Wood. 1986. Relationship of dietary fat, protein, cholesterol, and fiber intake to atherogenic lipoproteins in men. Am. J. Clin. Med. 44:788-797.
Williams, P.T., S.P. Fortmann, R.B. Terry, S.C. Garay, K.M. Vranizan, N. Ellsworth, and P.D. Wood. 1987. Associations of dietary fats, regional adiposity, and blood pressure in men. J. Am. Med. Assoc. 257:3251-3256.
Wilmore, J.H., and J.J. McNamara. 1974. Prevalence of coronary heart disease risk factors in boys, 8 to 12 years of age. J. Pediatr. 84:527-533.
Wilson, J.D., and M.D. Siperstein. 1959. Effect of saturated and unsaturated fats on hepatic synthesis and biliary excretion of cholesterol by the rat. Am. J. Physiol. 196:599-602.
Wilson, R.B., P.M. Newberne, and M.W. Conner. 1973. An improved semisynthetic atherogenic diet for rabbits. Dietary fat-carbohydrate interaction in atherogenesis. Arch. Pathol. 96:355-359.
Wingard, D.L., M.H. Criqui, M.J. Holbrook, and E Barrett-Connor. 1984. Plasma cholesterol and cancer morbidity and mortality in an adult community. J. Chronic Dis. 37:401- 406.
Wissler, R.W., and H.C. McGill, Jr. 1983. Conference on blood lipids in children: optimal levels for early prevention of coronary artery disease. Workshop report: experimental section. American Health Foundation, April 18 and 19, 1983. Prev. Med. 12:868-902.
Wissler, R.W., and D. Vesselinovitch. 1987a. Animal models for hyperlipidemia-induced atherosclerosis. Pp. 111-116 in R. Paoletti, D. Kritchevsky, and W.L. Holmes, eds. Drugs Affecting Lipid Metabolism. Springer-Verlag, Berlin.
Wissler, R.W., and D. Vesselinovitch. 1987b. The development and use of animal models in atherosclerosis research. Pp. 337-357 in L.L Gallo, ed. Cardiovascular Disease: Molecular and Cellular Mechanisms, Prevention and Treatment. Plenum Press, New York.
Wissler, R.W., M.L Eilert, M.A. Schroeder, and L. Cohen. 1954. Production of lipomatous and atheromatous arterial lesions in the albino rat. Arch. Pathol. 57:333-351.
Wissler, R.W., L.E. Frazier, R.H. Hughes, and RA. Rasmussen. 1962. Atherogenesis in the cebus monkey. I. A comparison of three food fats under controlled dietary conditions. Arch. Pathol. 74:312-322.
Wood, P.D.S., R. Shioda, and L.W. Kinsell. 1966. Dietary regulation of cholesterol metabolism. Lancet 2:604-607.
Wurtman, J.J. 1979. Sources of choline and lecithin in the diet. Pp. 73-81 in A. Barbeau, J.H. Growdon, and R.J. Wurtman, eds. Nutrition and the Brain, Vol. 5. Choline and Lecithin in Brain Disorders. Raven Press, New York.
Wynder, EL., G.A. Leveille, J.H. Weisburger, and G.E. Livingston, eds. 1983. Environmental Aspects of Cancer. The Role of Macro and Micro Components of Foods Proceedings of an International Conference Sponsored by the American Health Foundation, March 31-April 1, 1982, New York, New York. Food & Nutrition Press, Westport, Conn. 295 pp.
Yaari, S., U. Goldbourt, S. Even-Zohar, and H.N. Neufeld. 1981. Associations of serum high-density lipoprotein and total cholesterol with total, cardiovascular, and cancer mortality in a 7-year prospective study of 10,000 men. Lancet 1:1011-1015.
Yamase, H., and J.J. McNamara. 1972. Geographic differences in the incidence of gallbladder disease. Influence of
Page 258
environment and ethnic background. Am. J. Surg. 123: 667-670.
Yano, K., G.G. Rhoads, A. Kagan, and J. Tillotson. 1978. Dietary intake and the risk of coronary heart disease in Japanese men living in Hawaii. Am. J. Clin. Nutr. 31: 1270-1279.
Yano, K., D.M. Reed, J.D. Curb., J.H. Hankin, and J.J. Albers. 1986. Biological and dietary correlates of plasma lipids and lipoproteins among elderly Japanese men in Hawaii. Arteriosclerosis 6:422-433.
Yerushalmy, J., and H.E. Hilleboe. 1957. Fat in the diet and mortality from heart disease: a methodologic note. N.Y. State J. Med. 57:2343-2354.
Yomantas, S., V.M. Elner, T. Schaffner, and R.W. Wissler. 1984. Immunohistochemical localization and apolipoprotein B in human atherosclerotic lesions. Arch. Pathol. Lab. Med. 108:374-378.
Yudkin, J. 1957. Diet and coronary thrombosis: hypothesis and fact. Lancet 2:155-162.
Zanni, E.E., Zannis, V.I., C.B. Blum, P.N. Herbert, and J.L. Breslow. 1987. Effect of egg cholesterol and dietary fats on plasma lipids, lipoproteins, and apoproteins of normal women consuming natural diets. J. Lipid Res. 28: 518-527.
Zannis, V.I., and J.L. Breslow. 1981. Human very low density lipoprotein apolipoprotein E isoprotein polymorphism is explained by genetic variation and posttranslational modification. Biochemistry 20:1033-1041.
Zeisel, S.H. 1981. Dietary choline: biochemistry, physiology, and pharmacology. Annu. Rev. Nutr. 1:95-121.
Zeisel, S.H., J.H. Growdon, R.J. Wurtman, S.G. Magil, and M. Logue. 1980. Normal plasma choline responses to ingested lecithin. Neurology 30:1226-1229.
Zilversmit, D.B. 1979. Atherogenesis: a postprandial phenomenon. Circulation 60:473-485.
Zinserling, W.D. 1925. Untersuchungen über Atherosklerose. I. Über die Aortaverfettung bei Kindern. Virchows Arch. f. Pathol. Anat. 255:677-705.




































































































