
Page 529
19—
Atherosclerotic Cardiovascular Diseases
Atherosclerosis is the pathological process in the coronary arteries, cerebral arteries, iliac and femoral arteries, and aorta that is responsible for coronary heart disease (CHD), stroke, and peripheral arterial disease (PAD). It begins during childhood in the intima of the large elastic and muscular arteries with deposits of lipids, principally cholesterol and its esters, in macrophages and smooth muscle cells (Figure 19-1). The lesions, called fatty streaks, produce only minimal intimal thickening and cause no disturbances in blood flow during early childhood, but they rapidly become more extensive during adolescence. In young adults, more lipid is deposited at some sites, and a core of lipid and necrotic debris becomes covered by a cap of smooth muscle and fibrous tissue. These changes produce elevated lesions called fibrous plaques that project into the lumen and begin to disturb blood flow.
The relationship between fatty streaks and fibrous plaques has been one of the most controversial aspects of the pathogenesis of atherosclerosis. The coronary arteries differ from most other arteries by having a prominent intimal layer of longitudinal smooth muscle and fibrous tissue that is apparent even in childhood. By the age of 20, the thickness of this layer is about equal to that of the media, even when it does not contain abnormal lipid (Stary, 1987a,b). This fibromuscular intimal layer occurs in all populations, even in those not predisposed to coronary atherosclerosis in adulthood (Geer et al., 1968) and is considered to be a normal anatomic structure rather than an atherosclerotic lesion.
Some evidence suggests that fibrous plaques are created by cellular proliferation and subsequent fatty degeneration without prior lipid deposition (Benditt, 1974), and some observations are not consistent with the progression of fatty streaks to fibrous plaques. For example, fatty streaks are more extensive in the thoracic aortas of children, but fibrous plaques are more extensive in the abdominal aortas of adults. Young women have more extensive fatty streaks in their coronary arteries and aortas than do young men, but among adults this pattern is reversed. (McGill, 1968).
Overall, however, evidence supports the association of fatty streaks with fibrous plaques. Lesions in the arteries of young adults have many histological and chemical characteristics of fatty streaks as well as fibrous plaques—an observation suggesting a continuous progression from one type of lesion to the other (Geer et al., 1968; Katz, 1981; Stary, 1987a,b). Furthermore, in contrast to the differences in location of fatty streaks and fibrous plaques in the aorta, the sites of fatty streaks in the coronary arteries of children are the most common sites of fibrous plaques in adults (Montenegro and Eggen, 1968). The major risk factors, hypercholesterolemia and hypertension, are closely associ-
Page 530
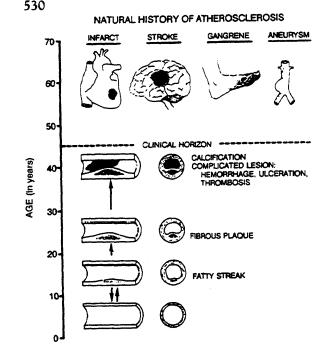
FIGURE 19-1
Natural history of atherosclerosis, showing progressive arterial
occlusion and resultant health effects. From McGill et al. (1963).
ated with the extent of fibrous plaques in adults (Solberg and Strong, 1983). The few relevant data indicate that there is an association between serum cholesterol and low-density lipoprotein (LDL) cholesterol concentrations with fatty streaks in childhood (Freedman et al., 1988; Newman et al., 1986). Furthermore, it seems most likely that fatty streaks in children are labile, i.e., some may regress or remain as fatty streaks whereas others progress and evolve into fibrous plaques. This later process occurs particularly in the coronary arteries and abdominal aorta, where some fatty streaks are gradually converted to fibrous plaques by continued lipid deposition and reactive chronic inflammation and repair. For a review of this subject, see McGill (1988).
Regardless of their origin, fibrous plaques undergo a variety of qualitative changes in early middle age in the U.S. population, as illustrated in Figure 19-1. These changes result in fibrous plaques that vary in their content of lipids, smooth muscle cells, connective tissue, calcium, and vessels. The most serious complication is ulceration of the connective tissue and smooth muscle cap of fibrous plaque, a change that exposes blood to the lipid-rich necrotic debris of the core and is likely to precipitate thrombosis. Another serious complication is hemorrhage into the plaque. This causes sudden swelling of the plaque and may precipitate ulceration and thrombosis.
Thrombosis overlying an advanced atherosclerotic fibrous plaque is the most common event that occludes the lumen of the coronary artery and causes ischemia. At a point, determined by such factors as blood pressure, collateral circulation, and tissue oxygen demand, the blood supply is reduced below a critical level and ischemic necrosis occurs in the tissue supplied by the affected artery.
Lesions in the coronary arteries lead to CHD, which is the most common and most serious manifestation of atherosclerotic cardiovascular diseases in middle-aged adults. The atherosclerotic process that occurs in the cerebral and peripheral arteries is similar to that which occurs in the coronary arteries, but the lesions usually develop a decade or two later than those in the coronary arteries.
In approximately one-third of all CHD cases, coronary artery occlusion causes a fatal arrhythmia within a few minutes or hours (sudden cardiac death). If the patient survives the first few hours, ischemic necrosis of the myocardium occurs (myocardial infarction). Afterward, the necrotic tissue is removed and replaced by connective tissue. The subsequent clinical outcome is determined, for the most part, by the amount and location of cardiac muscle that is lost. A few days after infarction, and before much connective tissue has formed, the heart may rupture at the site of infarction (cardiac tamponade). The patient surviving this stage may recover cardiac function as the remaining heart hypertrophies to compensate for myocardium lost by infarction. At any stage, the patient may die from failure of the heart to pump sufficient blood (congestive heart failure) or from a disturbance in the conduction system controlling the distribution of the contractile impulse (arrhythmia). Stenosis of the coronary arteries sometimes is sufficient to cause ischemic pain, but not infarction, especially on exertion (angina pectoris). This condition indicates the presence of severe lesions and high risk of myocardial infarction. All these syndromes (angina pectoris, myocardial infarction, sudden cardiac death) are included in the term coronary heart disease.
If thrombosis forms over an atherosclerotic plaque in a cerebral artery, ischemic necrosis occurs in the brain (cerebral infarct). Cerebral infarction (one type of stroke) typically causes paralysis on the contralateral side due to lack of upper motor neuron function, and disturbances of speech, vision, hearing, and memory, depending on the anatomic location of the infarct. Death may occur due to involvement of the brain centers
Page 531
controlling respiration or to cerebral edema. The necrotic tissue is converted to a liquid-filled cavity. Function is usually recovered to some degree as edema subsides, but neurons do not regenerate. Neural control of muscles and sensory organs may be regained in part as other pathways are developed. If the arterial occlusion is partial or temporary, temporary functional cerebral impairment may occur for a few minutes to a few hours (transient ischemic attacks). These episodes, which are analogous to angina pectoris, indicate that the patient has a high risk of developing cerebral infarction.
Another type of stroke is cerebral hemorrhage, which includes intracerebral hemorrhage (bleeding into the brain) and subarachnoid hemorrhage (bleeding into the space between the arachnoid membrane and the surface of the brain). In an intracerebral hemorrhage, an artery within the brain ruptures and causes a large area of tissue destruction. Its clinical manifestations are similar to those of cerebral infarction, except that it is more rapid in onset and more likely to be fatal. This type of stroke is almost always associated with severe hypertension. Since hypertension augments cerebral atherosclerosis, it is a major risk factor for both cerebral infarction and intracerebral hemorrhage.
The rupture of an artery into the subarachnoid space is usually at the site of a developmental defect in the artery wall. Either the defect, or its rupture, or both may be enhanced by hypertension. The clinical manifestations of a subarachnoid hemorrhage are similar to those of other types of stroke.
Peripheral arterial disease (PAD) occurs when atherosclerosis and its complications in the abdominal aorta, iliac arteries, and femoral arteries produce temporary arterial insufficiency in the lower extremities upon exertion (intermittent claudication) or ischemic necrosis of the extremities (gangrene). In the abdominal aorta, weakening of the media underlying the atherosclerotic plaque leads to an aneurysm, which may become filled with a thrombus or rupture into the abdominal cavity.
The major risk factors associated with clinically manifest atherosclerotic diseases also are associated with the severity of atherosclerosis. In particular, LDL cholesterol levels are positively correlated with fibrous plaques and other advanced lesions, and high-density lipoprotein (HDL) cholesterol levels are inversely associated with advanced lesions (Solberg and Strong, 1983). Hypertension is more closely associated with advanced atherosclerosis in the cerebral arteries than in other arteries, a selective effect consistent with the identification of hypertension as the dominant risk factor for stroke. Cigarette smoking is associated with advanced atherosclerosis of the abdominal aorta and iliac-femoral arteries, and consequently with PAD (DHHS, 1983). Smoking also is associated with advanced coronary atherosclerosis, but the increased coronary atherosclerosis in smokers is not sufficient to account for their much greater risk of CHD; other mechanisms, particularly thrombosis, are probably involved. Diabetes mellitus also is associated with severity of atherosclerosis in all arteries. Men have more severe coronary atherosclerosis than women, just as they have a higher frequency of CHD, but there is no sex difference in the severity of atherosclerosis of the aorta or cerebral arteries.
In populations with low serum cholesterol levels, atherosclerosis is less severe in those without hypertension and diabetes. However, among the latter, the severity of the disease is less than in populations where hyperlipidemia is prevalent (Robertson and Strong, 1968). Thus, hyperlipidemia, hypertension, and diabetes are additive in their effect on atherosclerosis, just as they are additive in their effect on risk of clinical disease. There is less information about the effects of cigarette smoking among different populations, but the evidence (Keys, 1980; Robertson et al., 1977) suggests that a similar relationship exists.
CHD risk factors for which no associations with severity of atherosclerosis have been found include physical activity and obesity (Solberg and Strong, 1983). The relationship of other putative risk factors to the severity of atherosclerosis has not been determined.
Results of animal experiments are consistent with observations in humans. LDL cholesterol and HDL cholesterol levels, and the ratio of the two lipoprotein cholesterol concentrations to one another are highly predictive of lesions in laboratory animals. High blood pressure combined with hyperlipidemia accelerates experimentally induced atherosclerosis. Despite several attempts, no effect of cigarette smoking on experimentally induced atherosclerosis has been demonstrated (Rogers et al., 1988).
Coronary Heart Disease
Occurrence in the U.S. Population
By 1920, CHD had become a major cause of death and a public health issue in the United States. CHD mortality rates increased thereafter by 1 to 2% per year until the mid-1960s and reached a peak of greater than 300 deaths per year
Page 532
per 100,000 population (Stallones, 1980; Woolsey and Moriyama, 1948). In the 1960s, a healthy 40-year-old man had a 20% chance of developing CHD before age 60 (DHHS, 1987), and 30 to 35% of first heart attacks were fatal within the first 3 weeks (Kuller, 1969; Stamler, 1967). U.S. mortality from CHD began to decline about 1967. The decline began earlier on the West Coast (Rosenberg and Klebba, 1979). By 1983, the most recent year for which official statistics have been published, age-adjusted risk of CHD death had declined by 28% from 328.1 to 236.1 per 100,000 population (DHHS, 1987). Unofficial reports indicate that the decline has continued at least through 1986 at a slightly slower rate. The decline has also occurred in men and women, in blacks and other nonwhite people, and in the young and the elderly. The decline was proportionately greater in the younger compared to the older groups (Pell and Fayerweather, 1985) and in salaried workers compared to wage earners (Thorn et al., 1985) (for further details, see Chapter 5). The decline has also occurred in out-of-hospital deaths, in sudden and unexpected deaths, in hospital case-fatality rates, and in acute nonfatal myocardial infarctions (Anastasiou-Nana et al., 1982; Folsom et al., 1987; Gillum et al., 1983; Gomez-Marin et al., 1987; Inter-Society Commission for Heart Disease Resources, 1984). Thus, improved medical care, better availability of medical care, and preventive measures in the population are probably all responsible for the decline. Despite the downward trend in mortality, CHD continues to be the leading cause of death in the United States. Of 2,091,200 deaths in the United States in 1983, 27% were attributed to CHD, 22% to malignant neoplasms, 21% to cardiovascular diseases other than CHD, 5% to accidents, and 25% to all other causes combined (DHHS, 1987).
Risk of CHD death is low during early adulthood but increases rapidly with age. The risk is higher for men than for women. Among men, risk is higher for whites than for blacks, but the reverse is found among women. Risk of CHD is inversely related to socioeconomic status now, but this association may have been positive earlier (Marmot et al., 1978).
Evidence Associating Dietary Factors with CHD
Animal Studies
The first animal model of atherosclerosis was discovered early in the twentieth century by Ignatowski (1909) while investigating the effects of animal protein fed to rabbits. A few years later in 1913, Anitschkow (1967) demonstrated that cholesterol was the dietary component responsible for experimentally induced hypercholesterolemia and atherosclerosis in rabbits. Subsequently, a variety of animal species, including guinea pigs, swine, fowl, and nonhuman primates, were also found to be susceptible to the serum cholesterol raising effects of dietary cholesterol. The excess serum cholesterol was carried primarily in the LDL fraction (see Chapter 7).
Similar effects were found in laboratory animals after the effects of dietary saturated fatty acids (SFAs) on serum lipids were discovered in 1952. Dietary SFAs elevated both LDL and HDL cholesterol, as in humans, and augmented experimentally induced atherosclerosis (see Chapter 7). In a few studies, prolonged feeding of cholesterol- and fat-enriched diets to laboratory animals produced severe obstructive atherosclerotic lesions and myocardial infarction or PAD.
The responses of serum lipids and lipoproteins to dietary cholesterol and SFAs vary among animal species, but experimentally induced atherosclerosis is strongly and consistently associated with elevated serum cholesterol levels. In particular, LDL cholesterol concentrations are directly associated, and HDL cholesterol is inversely associated, with experimentally induced atherosclerosis. Rabbits, guinea pigs, swine, and rhesus and cynomolgus monkeys are among the most susceptible species, more so than humans, whereas rats and dogs are more resistant than humans. Baboons and vervet monkeys are moderately susceptible, within the same range of susceptability as humans. Within species, there is interindividual variation in responsiveness to dietary fat and cholesterol. Variability among and within species is due in part to genetically determined differences in lipid metabolism. Heritability of this characteristic has been demonstrated in several species (see Chapter 7).
Proteins, carbohydrates, fibers, metals, trace elements, and vitamins have been examined for their effects on serum lipid levels and experimentally induced atherosclerosis. Evidence regarding these components is reviewed in Chapters 8, 9, 10, 11, 12, and 14 of this report. Although there have been some reports that these dietary components affect serum lipids, lipoproteins, and atherosclerosis in one or more animal species, none of the components has emerged as a consistent and strong determinant of either the biochemical intervening variables associated with atherosclerosis or of ath-
Page 533
erosclerosis itself. Thus, the evidence in laboratory animals is consistent with the evidence in humans and indicates that cholesterol and fats are the major dietary determinants of atherosclerotic cardiovascular diseases.
Human Studies
More than three decades of experiments in humans have demonstrated that dietary saturated fatty acids and cholesterol are major determinants of serum cholesterol and lipoprotein concentrations (see Chapter 7). The average response of adults to changes in dietary fat and cholesterol intake was expressed in the following equation by Keys (1965):
DChol = 1.35 (2S - P) + 1.52 Z,
where DChol is the change in serum cholesterol concentration in mg/dl; S and P are changes in percent of calories derived from saturated (S) and polyunsaturated (P) fatty acids; and Z is the difference between the square roots of the old and new cholesterol intakes expressed as mg/1,000 kcal.
This equation has been useful in predicting the effects of changes in dietary fat and cholesterol intakes, and its validity has been confirmed by subsequent experiments (Keys, 1984). However, not all SFAs raise serum cholesterol levels. The major cholesterol-raising SFAs are palmitic (C16) and myristic (C12) acids (see Chapter 7). In addition, individuals vary in their responses to SFAs (Grundy and Vega, 1988) and to cholesterol (Katan et al., 1986). Much of this variability is due to genetically controlled differences in lipid metabolism (see Chapters 4 and 7).
The Keys equation deals only with serum cholesterol concentrations. Dietary cholesterol leads predominantly to elevation of LDL cholesterol concentrations, whereas SFAs elevate both LDL and HDL cholesterol. When substituted for SFAs, polyunsaturated fatty acids (PUFAs) lead to both lower LDL and HDL cholesterol concentrations. Monounsaturated fatty acids (MUFAs) substituted for SFAs lead to decreased LDL cholesterol but have little or no effect on HDL cholesterol. Stearic acid has no effect on serum or lipoprotein cholesterol concentrations (see Chapter 7).
Most studies of PUFAs and plasma lipoproteins have used the more common w-6 PUFAs, which are abundant in vegetable oils. w-3 PUFAs derived from marine animals lower plasma triglyceride levels, but their effects on LDL cholesterol levels are not consistent. Their effects on lipid and lipoprotein metabolism are under investigation (see Chapter 7).
The effects of many other dietary components on serum lipids and lipoproteins are reviewed in detail in Chapters 6, 7, 8, 9, 10, 11, 12, 14, 16, and 17 of this report. Substitution of vegetable protein for animal protein reduces the serum cholesterol concentration slightly, although it probably is not an important factor within the usual ranges of intake (see Chapter 8). Dietary carbohydrates affect serum lipids and lipoproteins only when substituted for fats (see Chapter 9). Water-soluble, but not insoluble, dietary fiber lowers serum cholesterol levels (see Chapter 10). Alcohol consumption can elevate serum lipid levels, primarily by its elevating effect on serum triglyceride levels (see Chapter 16). Coffee consumption has been associated with slight elevations in serum cholesterol in some epidemiologic studies, but there is no consistent evidence that tea or other nonnutritive dietary components affect serum lipid concentrations (see Chapter 17).
The percentage of calories derived from SFAs in the food supply is strongly associated with mean population cholesterol levels and CHD rates (Keys, 1970, 1980), whereas the percentage of calories derived from PUFAs and MUFAs is not strongly related. Mean per capita intakes of other dietary components, such as starches and fiber, and habitual physical activity, mean body mass index, cigarette smoking, and blood pressure have little or no independent association with CHD rates for populations (Keys, 1980).
Until recently, cross-sectional studies detected little or no association between the fatty acid composition of the diet and serum cholesterol concentrations (see Chapter 7). In at least two studies, inverse associations were observed between the intake of SFAs and serum cholesterol concentrations (Shekelle et al., 1982). However, methodological problems in cross-sectional studies include unreliability in assessment of dietary intake (Beaton et al., 1979; Keys, 1965; Liu et al., 1978) and in measurement of serum cholesterol concentrations (Jacobs et al., 1979; Keys, 1965) and dietary changes by people after learning of their high serum cholesterol levels (Shekelle et al.,
Page 534
1982). Even after adjustment for these variables, cross-sectional studies have indicated that only a small proportion of the variation among individuals within a population can be attributed to the lipid composition of the diet.
In within-population studies carefully designed to reduce variation and in which the Keys equation was used, both SFAs alone and SFAs combined with PUFAs and cholesterol have been associated with serum cholesterol levels or CHD risk in individuals. Until recently, epidemiologic studies failed to show any association between a person's dietary cholesterol intake and serum cholesterol levels or CHD risk (see Chapter 7). However, these early studies did not adequately adjust for the confounding effect of caloric intake (Willet and Stampfer, 1986), were not accurate in measuring intake of dietary cholesterol (Beaton et al., 1979; Keys, 1965; Liu et al., 1978), or were biased due to systematic errors dietary change in people after learning of their hypercholesterolemia (Shekelle et al., 1981).
Since 1981, four prospective epidemiologic investigations—the Western Electric Study (Shekelle et al., 1981), the Ireland-Boston Diet-Heart Study (Kushi et al., 1985), the Zutphen Study (Kromhout and de Lezenne Coulander, 1984), and the Honolulu Heart Program (McGee et al., 1984)—have dealt adequately with these problems, and all found a positive association between the intake of dietary cholesterol and subsequent risk of CHD after adjustment for potentially confounding factors such as age, blood pressure, serum cholesterol concentration, and cigarette smoking.
Many studies have found that total energy intake is inversely associated with risk of CHD (see Chapter 6). This inverse association is probably due to a protective effect of physical activity on susceptibility to CHD and the positive correlation between physical activity and intake of energy.
Certain vegetable proteins (e.g., soy protein) and water-soluble dietary fiber can lower serum cholesterol in people with high cholesterol levels. The evidence for a protective role of these dietary constituents in CHD risk is, however, inconclusive (see Chapters 8 and 10).
Alcohol consumption in most cross-sectional epidemiologic studies is associated with an increased concentration of HDL cholesterol (Gordon et al., 1981), primarily of the HDL subclass, HDL3 (William et al., 1985), as well as of apolipoproteins AI (apo AI) and AII (apo All) (Camargo et al., 1985). Small intakes of alcohol (a range of 50-100 g distributed throughout the week) are associated in several studies with lower risk of CHD. A causal protective role for alcohol has not been established, however, and consumption of larger quantities of alcohol (e.g., >500 g/ week) is associated with increased risk of CHD, stroke, and other diseases (see Chapter 16).
The association between coffee consumption and CHD risk is weak and inconsistent. Some cohort studies have found that habitual consumption of five to six cups or more per day is associated with increased risk (La Croix et al., 1986; LeGrady et al., 1987). No association between consumption of tea and risk of CHD has been observed (see Chapter 16).
Early experiments in humans to prevent CHD by modification of diet had promising results (Dayton et al., 1968; Miettinen et al., 1972). These findings were not conclusive, however, either because of the small number of subjects or the absence of appropriate control groups. Although the National Heart, Lung, and Blood Institute Task Force on Arteriosclerosis (NHLBI, 1971) concluded that a large-scale diet-heart trial was not feasible, it recommended an intervention trial on multiple coronary risk factors. The results of that trial (MRFIT Research Group, 1982) showed no effect of intervention on risk of coronary death after 7 years of follow-up, but this finding has been questioned because of the inadequate power of the study.
In contrast, a primary prevention trial conducted in Oslo, Norway, with normotensive hypercholesterolemic men achieved a reduction in CHD risk following dietary changes to lower serum cholesterol and a reduction in cigarette smoking (Hjermann et al., 1981). Other clinical trials (e.g., the Lipid Research Clinics Program, 1984), as summarized in Chapter 7, demonstrated that CHD incidence in middle-aged hypercholesterolemic men decreased in proportion to the reduction in plasma LDL cholesterol concentrations.
Clinical trials to lower serum cholesterol and CHD are reviewed in detail in Chapter 7. The combined results of all such trials in which diet or drugs were used indicate that a reduction in CHD risk is proportional to the degree and duration of serum cholesterol lowering achieved. Although a statistically nonsignificant but consistent excess of non-CHD deaths occurred in the aggregate experimental groups, this excess was not directly attrib-
Page 535
utable either to the change in diet or to the lowering of total cholesterol; it was probably due to a combination of chance and the occurrence of deaths from causes that were previously masked by earlier death from CHD (Mann and Marr, 1981; Tyroler, 1985).
Evidence Associating Nondietary Factors with CHD
Cigarette Smoking
Cigarette smoking is an important risk factor for CHD in the United States and in other countries where the average diet is high in SFAs (i.e., >10% of total calories) and cholesterol and where serum cholesterol levels are high (Doll et al., 1980; Pooling Project Research Group, 1978). CHD risk within populations increases with the number of cigarettes smoked per day. A report by the Pooling Project Research Group (1978) indicated that the relative risk for men smoking more than one pack a day in comparison to nonsmokers was 3.2. In contrast, the association is weak in countries where intakes of SFAs and cholesterol and serum cholesterol concentrations are low. For example, cigarette smoking was not associated with the incidence of CHD in Japan, but was associated among men of Japanese descent living in Hawaii (Robertson et al., 1977). In the Seven Countries Study, cigarette smoking was strongly associated with risk of CHD death among U.S. and northern European men, but only weakly associated with risk in men from southern Europe where overall CHD risk was low (Keys, 1980).
Many observational studies have noted that former cigarette smokers have a substantially lower risk of CHD than current smokers (Doll and Hill, 1964; Friedman et al., 1981; Gordon et al., 1974). In high-risk men, randomized trials have shown a possible benefit of smoking cessation, even though the power of these studies to demonstrate an effect was only borderline (Holme, 1982; MRFIT Research Group, 1982; Rose et al., 1982).
High Blood Pressure
High blood pressure is a well-established major risk factor for CHD and stroke in the United States (Pooling Project Research Group, 1978) and other countries (Keys, 1980) (see Chapters 5 and 20). However, differences in prevalence of hypertension are less important than differences in serum cholesterol levels in accounting for population rates of CHD (Keys, 1980; Winkelstein et al., 1975).
Obesity
The precise role of obesity in the etiology of atherosclerosis and CHD is unclear. Cigarette smokers tend to be leaner than nonsmokers, and smokers who quit gain several pounds of body fat (Blitzer et al., 1977; Brozek and Keys, 1957; Goldbourt and Medalie, 1977; Gordon et al., 1975), but ex-smokers have lower risk of CHD. Adiposity is inversely correlated with HDL cholesterol concentration in some populations. Weight gain is associated with a decrease in HDL cholesterol levels (Garrison et al., 1980; Rhoads et al., 1976) and weight loss associated with moderate exercise with increased HDL cholesterol (Brownell et al., 1982). Adiposity is positively associated with the prevalence and incidence of hypertension (Kannel et al., 1967) and with the prevalence of glucose intolerance and hyperinsulinemia (Cahill, 1977) (see Chapter 21). Autopsy studies (Amad et al., 1965; Montenegro and Solberg, 1968) and angiographic studies (Anderson et al., 1978; Cramer et al., 1966) failed to show an association between obesity and severity of coronary artery disease.
Age-standardized 10-year incidence of nonfatal myocardial infarction and coronary death among 15 cohorts in the Seven Countries Study was not associated with mean body mass index or mean skinfold thickness (Keys, 1980). Within populations, however, prospective epidemiologic studies have generally shown an association between obesity and risk of coronary death at the upper range of body weight—e.g., ³ 140% of ideal body weight or a body mass index ³ 30 (see Chapter 21). For example, CHD incidence in the Honolulu Heart Study was positively associated with body mass index as well as subscapular skinfold thickness, even after controlling for other CHD risk factors (Donahue et al., 1987). Other studies (Lapidus et al., 1986; Larsson et al., 1984) support the hypothesis that the pattern of fat distribution (e.g., waist-to-hip ratio) may be an important risk factor for CHD; people with central fat distribution (typical in males) may be at increased risk of CHD in comparison to people with peripheral fat distribution (typical in females).
The evidence on body weight and CHD risk suggests that certain lifestyle variables, including diet, contribute to overweight and that obesity, especially severe obesity, increases CHD risk. Thus, maintaining moderate body weight is an important preventive measure to lower CHD risk in populations where obesity and high serum cholesterol levels are widespread.
Page 536
Genetic Factors
CHD has long been known to cluster in families. Osler (1897) described a family in which three generations were affected. In a pioneering study of young men with myocardial infarction, Gertler and White (1954) found that a family history of CHD was 2.5 times as frequent among the affected cases as among the controls. First-degree relatives of people with CHD had a higher than average risk of the disease (Rose, 1964; Shanoff et al., 1961; Slack and Evans, 1966; Thomas, 1958). There was a high degree of concordance between twins of the same sex in incidence of CHD (Harvald and Hauge, 1970). Thus, a family history of premature CHD was established as a risk factor. This genetic predisposition is likely to involve multiple mechanisms, but attention has been focused on the genetic control of serum cholesterol and lipoprotein concentrations, knowledge of which accumulated rapidly after 1970.
Familial hypercholesterolemia first described by Müller (1938) is the most extreme example of genetic dyslipoproteinemia predisposing to premature CHD. It results from an autosomal dominant trait that occurs in the heterozygous form in about 1 in 500 people and in a severe homozygous form in about 1 in 1 million people. Affected people are deficient in the LDL receptor, which is essential for the cellular uptake and internalization of LDL. Heterozygous people have about half the normal LDL receptor activity and approximately double the normal LDL concentrations from birth and often develop CHD in their forties. Homozygous people have very little or no LDL receptor activity, LDL concentrations 6 to 10 times the normal, and CHD in childhood. The molecular defect involved in familial hypercholesterolemia and the structure of the LDL receptor and its gene were determined by Brown and Goldstein (1986). Animal models of this defect have been found in rabbits (Goldstein et al., 1983; Tanzawa et al., 1980; Watanabe et al., 1985) and rhesus monkeys (Scanu et al., 1988).
LDL cholesterol concentrations of people with familial hypercholesterolemia are not reduced to desirable levels when dietary cholesterol and SFA intakes are reduced, and treatment depends on pharmacological intervention with cholesterol-lowering drugs. Since people with familial hypercholesterolemia make up less than 1% of the population and only a small percentage of all people with hyperlipidemia, this genetic dyslipoproteinemia is not a major consideration in making dietary recommendations for a population.
Another genetic dyslipoproteinemia associated with increased incidence of CHD is familial combined hyperlipidemia, characterized by elevated fasting plasma cholesterol and triglyceride concentrations (Goldstein et al., 1972, 1973a,b). The metabolic disorders involved in this condition are not well delineated and are under investigation.
The wide variation in responsiveness within a population to the serum cholesterol raising effects of dietary cholesterol and SFAs suggests genetic control, which has been demonstrated in nonhuman primates (Clarkson et al., 1971, 1985; Flow et al., 1981; La Ville et al., 1987; McGill et al., 1988). There is less direct evidence in humans, because it is not possible to undertake comparable dietary intervention in families.
Apolipoprotein E (Apo E) isoforms (see Chapter 7 for definitions) are associated with variations in serum cholesterol levels and in CHD risk, and there is considerable evidence regarding the molecular mechanism by which these effects are produced (Davignon et al., 1988; Mahley, 1988). Apo B variants are associated with serum cholesterol concentrations (Kwiterovich et al., 1987; Young et al., 1987a,b, 1988). Apo AI variants are associated with abnormalities in HDL cholesterol levels (Franceschini et al., 1987). Genetic control of lipoprotein metabolism is an active topic of investigation. Investigators hope to find many new associations between genetic variants and serum lipoprotein levels as well as genetic markers for susceptibility to the serum cholesterol raising effect of dietary fat and cholesterol.
A family history of CHD is also a risk factor for the disease, even after adjusting for other known CHD risk factors (Barrett-Connor and Khaw, 1984; Colditz et al., 1986; Hammond et al., 1971; Sholtz et al., 1975; ten Kate et al., 1982; and reviewed by Goldbourt and Neufeld, 1986). These studies suggest that increased risk in men with a family history of CHD is about 1.5 to 2 times greater than in men without such a history, but is less in women. The physiological mechanism by which this independent effect is produced is not known. An intervening variable could theoretically be a genetically modulated risk factor such as dyslipoproteinemia, but evidence for this is still lacking.
Physical Activity
Physical inactivity (measured directly, or indirectly by caloric intake) is associated with an
Page 537
increased CHD risk and mortality in some cohort studies (Ekelund et al., 1988; Morris et al., 1977; Paffenbarger et al., 1978), but not in all. For example, variation in CHD mortality among cohorts in the Seven Countries Study was unrelated to habitual occupational activity; the two most active populations resided in areas with the highest (East Finland) and lowest (Japan) CHD rates (Keys, 1970, 1980).
Sustained physical activity is associated with an increased concentration of HDL cholesterol in humans (Hartung et al., 1980; Huttunen et al., 1979; Wood and Haskell, 1979) and with retardation in the development of coronary atherosclerosis in monkeys (Macaca fascicularis) receiving an atherogenic diet (Kramsch et al., 1981).
The evidence suggests an indirect and possibly direct role of physical activity in reducing risk of fatal CHD. Evidence on the level of physical activity required to reduce risk is conflicting.
Psychosocial Factors
Research completed in the 1970s supported the hypothesis that the Type A behavior pattern—a pattern composed primarily of competitiveness, excessive drive, and an enhanced sense of time urgency—was associated with increased risk of CHD independently of other known risk factors (Review Panel on Coronary-Prone Behavior and Coronary Heart Disease, 1981). Although some later studies in healthy populations also indicated an association between Type A behavior and increased risk of CHD (French-Belgium Collaborative Group, 1982; Haynes et al., 1983), most subsequent studies in the United States, particularly in high-risk groups, have been unable to repeat these early findings (Case et al., 1985; Ragland and Brand, 1988a,b; Shekelle et al., 1985a,b). Inconsistent findings have also been reported in studies on the association of Type A behavior and the presence and degree of angiographically determined coronary atherosclerosis (Williams et al., 1988). These inconsistent findings do not support a conclusion that Type A behavior is an established risk factor for CHD. Evidence regarding other aspects of emotional stress or other psychosocial factors is insufficient to justify firm conclusions about their role in the etiology of CHD in humans (Shepard and Weiss, 1987). Furthermore, Type A behavior did not modify the association of such other established risk factors as serum cholesterol concentration, cigarette smoking, or blood pressure with CHD (Rosenman et al., 1976), and there is no substantial evidence to indicate that other psychosocial factors might do so.
Unstable social conditions were associated with coronary atherosclerosis in highly competitive male cynomolgus monkeys consuming an atherogenic diet (Kaplan et al., 1982), but there are no other laboratory data on this issue.
Summary
In summary, CHD is the most common clinical manifestation of atherosclerosis and it is the major cause of deaths among adults in the United States and many industrial societies. A large body of evidence indicates that the incidence of CHD is associated with three major risk factors: high serum cholesterol and low-density lipoprotein concentrations, high blood pressure, and cigarette smoking. Men are at much higher risk than are women. Other major risk factors include a low HDL-cholesterol concentration, diabetes mellitus, and a positive family history of early (premature) CHD. Other conditions (e.g., obesity, physical inactivity, personality type) also are associated with increased risk, but the associations are weaker and the evidence is less complete. There is a strong genetic component in risk of CHD in individuals and most of it is mediated through genetic/environmental interactions that determine the major risk factors. The predominant determinant of the average serum cholesterol and lipoprotein concentrations for populations is habitual diet, mainly the dietary intakes of cholesterol-raising SFAs and cholesterol. Individual CHD risk within populations is determined by genetic-environmental interactions affecting serum lipoproteins and blood pressure and by cigarette smoking. The observation that the established CHD risk factors account for about half the observed variation in CHD in multivariate analyses despite the lack of precision in measuring lifetime exposure to these variables underscores their importance as etiologic factors.
Peripheral Arterial Disease
Occurrence in the U.S. Population
PAD includes several clinical syndromes of arterial insufficiency in the extremities, characterized by pain, inflammation, and ischemic damage to soft tissues from partial or complete occlusion of major arteries. The most characteristic symptom of PAD is intermittent claudication, described as
Page 538
cramping, aching, and numbness of the extremities induced by exercise and resolved promptly by stopping the exercise. An advanced form of PAD is an aneurysm of the abdominal aorta, leading to occlusion of major aortic branches or to rupture and massive hemorrhage. PAD is caused by the obliteration of the arterial lumen due to thrombi overlying atherosclerotic plaques.
PAD is diagnosed by the clinical history of intermittent claudication, decreased arterial pulsations or pressure, and decreased blood flow. New ultrasound techniques promise effective noninvasive diagnosis, but x-ray angiography remains the definitive diagnostic method (Criqui et al., 1985c).
There are few systematic data pertaining to the frequency of PAD among or within populations. Mortality rates for PAD are unreliable because the disease occurs in a variety of syndromes and is not frequently a direct cause of death (Criqui et al., 1985a). Furthermore, there is no reliable information on trends in prevalence or deaths from PAD. In one U.S. study in which standardized diagnostic procedures were used, large-vessel PAD was found in 11.7% of the subjects, predominately whites with an average age of 66 years (Criqui et al., 1985b).
Evidence Associating Dietary Factors with PAD
Diet affects PAD through its effects on serum lipids and lipoproteins. The relationship of the serum cholesterol concentration to PAD risk or prevalence varies from strong in some studies to none in others. Where positive relationships occur, serum triglyceride and very-low-density lipoprotein (VLDL) cholesterol levels are more closely related to PAD than are HDL and LDL cholesterol levels (see Chapters 5 and 7).
No systematic studies have related dietary intakes to PAD risk; however, two diet-related conditions—diabetes and hypertension—are important PAD risk factors. Intermittent claudication (IC) is more common in diabetics than in nondiabetics in the Framingham study, and a substantial part of the risk for IC in that population was attributable to diabetes mellitus (Kannel and McGee, 1985). Risk ratios of 4 to 1 for IC were found with impaired glucose tolerance. PAD is a common late complication of diabetes; it was found in 45% of the diabetics studied in Rochester, Minnesota, for 20 years after diagnosis (Melton et al., 1980). Somewhat lower risk ratios are seen for hypertension. In the Framingham study, a multiple factor (age, blood cholesterol, electrocardiogram reading, systolic blood pressure, relative weight, hemoglobin, and cigarette smoking) coronary risk index was strongly related to the incidence of IC; IC risk doubled in the upper quintile of the coronary risk score. The clustering of IC with other cardiovascular diseases was also pronounced in the Framingham study (Kannel and McGee, 1985).
Evidence Associating Nondietary Factors with PAD
PAD increases steadily and dramatically with age, rising more steeply after age 55. The clinical onset of PAD may be delayed beyond that of other atherosclerotic manifestations, because an extreme degree of obstructive disease is required to impair blood flow through the large arteries serving the lower extremities (Kannel and McGee, 1985).
Clinical and population-based studies suggest that PAD, expressed as intermittent claudication, is more frequent in men than in women up to age 65—occurring in 11.6% of men and 5.3% of women over 26 years of follow-up (Kannel and McGee, 1985)—after which incidence rates in both sexes are similar. There are no data on ethnic and racial differences or on associations with socioeconomic and psychosocial factors. There is no strong familial or genetic clustering of PAD other than the association with diabetes. Older clinical studies suggested that most patients with PAD were cigarette smokers. In more systematic followup studies, the frequency of intermittent claudication rose steadily and steeply according to number of cigarettes smoked, even beyond age 65 (see Chapter 5). This observation was confirmed by studies showing more severe atherosclerosis in the aortas of smokers than in those of nonsmokers (Solberg and Strong, 1983).
Multiple Risk Factors
Age, smoking, diabetes and fasting plasma glucose level, and systolic blood pressure were associated with PAD, whereas obesity and levels of LDL and HDL cholesterol were only marginally related to large-vessel PAD (Criqui et al., 1980). More important is the difference emerging in risk-factor configurations associated with each major atherosclerotic end point, i.e., CHD, stroke, and PAD. For example, diabetes, cigarette smoking, serum
Page 539
triglyceride level, and glucose tolerance predict PAD better than do serum lipid concentrations and blood pressure, whereas LDL and HDL cholesterol levels more strongly predict CHD, and blood pressure more strongly predicts stroke.
Summary
In summary, PAD is a large but poorly documented public health problem. There is little information on its prevalence or incidence or on population differences or trends. Evidence suggests a different combination of risk factors for PAD than for other atherosclerotic manifestations, with an emphasis on diabetes, glucose intolerance, smoking, and plasma triglyceride concentrations. Because CHD risk factors strongly predict PAD risk in the U.S. population, measures that would control CHD would also be expected to control PAD.
Stroke
Occurrence in the U.S. Population
Stroke is a clinical syndrome of neurological disabilities due to infarction of the brain by thrombosis over an atherosclerotic plaque or to destruction of brain tissue by hemorrhage from a ruptured artery. Transient ischemic attacks (TIAs) are episodes of temporary neurological disability due to insufficient arterial blood supply to the brain.
Most strokes are due to cerebral infarction. The next most frequent causes are the two major forms of cerebral hemorrhage: intracerebral and subarachnoid. Stroke has been recognized since antiquity and remains a major cause of death in adults worldwide. In many industrial countries, it is third among causes of death, following heart diseases and cancer. In the United States, strokes of all types were responsible for the deaths of approximately 182,000 people in 1977 (DHHS, 1987). The American Heart Association estimated that the 1981 prevalence of stroke was 1.87 million compared to 4.6 million cases of CHD (AHA, 1983). In the United States, the short-term case fatality from stroke is about 15%. Another 16% of the cases require institutional care, and 50% of survivors are permanently disabled (Kannel and Wolf, 1983).
Prevalence rates for stroke differ greatly among populations. For example, age-adjusted prevalence rates for stroke in 1970 were approximately 556 per 100,000 people in Rochester, Minnesota, compared to 363 in the United Kingdom (Kurtzke, 1976). Among countries, the distribution of and mortality rates from the different types of stroke, particularly cerebral infarct versus hemorrhage, also vary widely (Omae et al., 1976). Generally, the frequency of cerebral infarct deaths parallels that of CHD, whereas the frequency of intracerebral hemorrhage parallels that of hypertension. For example, stroke is the leading cause of death among adults in Japan, where hypertension is prevalent but CHD is uncommon (Komachi, 1977). Hypertension selectively augments atherosclerosis of the large cerebral arteries and thereby contributes to cerebral infarction. It also damages smaller cerebral arteries and contributes to cerebral hemorrhage.
Stroke deaths in the United States have declined for several decades, and the decline has accelerated since 1972 (Levy, 1979). Between 1968 and 1981, the age-adjusted stroke death rate fell by 46% (Inter-Society Commission for Heart Disease Resources, 1984). The incidence of cerebral infarction and intracerebral hemorrhage is also declining in Rochester, Minnesota, and the latter began to decline well before the wide availability of computerized brain tomography for diagnosis (Garraway and Whisnant, 1987).
In contrast, short-term fatality and survival rates for hospitalized stroke patients have not changed appreciably. This observation suggests that stroke incidence and deaths have declined as a result of preventive measures in the population, including hypertension control (Gillum et al., 1985), rather than as a result of improved treatment.
Evidence Associating Dietary Factors with Stroke
As reviewed in Chapter 7, a U-shaped relationship has been found in several studies between serum cholesterol level and incidence of stroke or cerebral infarct (i.e., the highest stroke rates are associated with both low and high levels of serum cholesterol, and the lowest rates with moderate levels) (Kannel and Wolf, 1983; Reed et al., 1986). Improved discrimination between hemorrhage and thrombosis suggests that the left side of the U-shaped curve is related to a higher frequency of cerebral hemorrhage in persons with hypertension at low cholesterol levels; the right side reflects a positive relationship between serum cholesterol level and cerebral infarct. Similarly, there is an inverse relationship between HDL cholesterol lev-
Page 540
els and stroke rates in subjects in the Honolulu Heart Program (Reed et al., 1986).
Hypertension is consistently, strongly, and independently related to individual risk of stroke (Dyken et al., 1984). Relative body weight was positively related to stroke incidence in the Framingham population under age 65, and inversely related at older ages (Kannel and Wolf, 1983). Abdominal obesity was positively related to stroke in Göteborg men (Welin et al., 1987). Age-adjusted stroke incidence rises steadily with alcohol intake in several populations (Gill et al., 1986) (see Chapters 5 and 16).
The paradoxical relationship of certain dietary components to stroke is discussed in Chapters 7 and 28. Animal fat, SFAs, and total fat were positively related to the risk of cerebral infarct (Reed et al., 1986). However, where hemorrhage occurs in the major proportion of stroke cases, as in Japan, animal protein and saturated fatty acid intake were inversely related to the incidence of cerebral hemorrhage and, therefore, to overall stroke incidence (Tanaka et al., 1982, 1985). Among omnivorous Seventh-Day Adventists in the United States, consumption of meats, eggs, milk, or cheese was unrelated to stroke risk (Snowdon, 1988).
Evidence Associating Nondietary Factors with Stroke
Fatal and nonfatal stroke are uncommon under age 45, after which stroke rates climb dramatically with age—rising from about 1/1,000 at ages 45 to 54, to 3.5/1,000 at ages 55 to 64, 9.0/1,000 at ages 65 to 74, 20/1,000 at ages 75 to 84, and 40/1,000 at ages 85 and over. Before age 65, rates are higher for men. After that, the rates are approximately the same for both sexes.
Downward trends in stroke deaths in the United States started earlier and have been greater in women, partly because of more effective hypertension control in women (Garraway and Whisnant, 1987). Stroke incidence and death are higher in blacks than in whites. However, migrant studies suggest that environment has a greater influence on stroke incidence than race or ethnicity. For example, stroke rates were substantially greater in people of Japanese extraction living in Japan than in those who migrated to Hawaii and California (Kagan et al., 1980), and greater in New Zealand migrants than in those who remained in their island homeland (Bonita and Beaglehole, 1982). Close relatives of stroke patients are at slightly greater risk of stroke than are unrelated people, and maternal history of stroke confers a slightly greater risk than does paternal history (Heyden et al., 1969; Welin et al., 1987).
Smoking was associated with cerebral infarct below age 65 in the Honolulu and Framingham studies (Abbott et al., 1986) but not in the Chicago Stroke Study (Ostfeld et al., 1974). Other cardiovascular diseases were strongly associated with the risk of stroke, particularly CHD, PAD, and hypertensive heart disease (Ostfeld et al., 1974).
Summary
In summary, stroke is the third most frequent cause of death among adults in many industrial countries. Risk rises steeply with age, and there are proportionately more deaths among blacks than among whites in the United States. The proportion of strokes due to infarction compared to strokes due to hemorrhage varies among countries. The frequency of cerebral infarction parallels that of CHD, and the frequency of hemorrhage parallels that of hypertension. Despite little change in hospital case-fatality and survival rates, stroke deaths in the United States have been declining for several decades. Blood pressure is the most consistent characteristic associated with the risk of stroke in populations and in individuals, but all the major risk factors for atherosclerosis, including diet, serum lipids, and smoking, contribute to the risk of stroke. Alcohol is also a strong risk factor for cerebral hemorrhage.
Overall Summary
Atherosclerotic cardiovascular diseases make up the largest group of vascular diseases in the United States and have the greatest effect on morbidity and mortality. Comparisons of populations show large differences in incidence and mortality from the atherosclerotic cardiovascular diseases and in the underlying pathological process—atherosclerosis. The differences in population rates are strongly associated with average levels and distributions of the blood lipoproteins (for CHD) and with blood pressure (for stroke). The age-adjusted death rate from CHD in the United States declined 2 to 3% per year from 1968 to 1982 and has continued to fall since 1982 but at a slower rate. CHD deaths have increased at similar rates in some other countries. These trends in mortality and the rapid changes in risk factors and CHD risk observed among
Page 541
migrant populations indicate the potential for prevention. Despite the downward trend, CHD remains the major cause of deaths among U.S. adults.
CHD rates and population risk are most strongly related to the average serum cholesterol level (or, more specifically, to the average LDL cholesterol level). The mean cholesterol level in turn is strongly influenced by the composition of a population's habitual diet, chiefly its intake of saturated fatty acids and cholesterol. Within high-risk populations, the effect of diet on individual risk is strongly influenced by genetic differences in blood lipoprotein levels and by other factors such as arterial blood pressure and cigarette smoking. Within populations, the major risk factors—serum cholesterol concentration, blood pressure, and cigarette smoking—individually and combined are related to an individual's risk of clinical events in a continuously graded fashion.
Change in dietary composition, particularly in fatty acids (type and amount) and cholesterol, influences lipoprotein levels in small-group experiments. In randomized clinical trials, lowering of serum and LDL cholesterol, and possibly elevation of HDL cholesterol, has consistently demonstrated a reduction in CHD risk proportionate to the degree and duration of the reduced exposure. An excess of traumatic and other noncardiovascular deaths in the treated groups in these trials is not statistically significant and is not directly attributable to the lowering of serum cholesterol levels.
Hypertension, cigarette smoking, and diabetes mellitus are powerful influences on individual risk in populations with high CHD rates, but the role of overweight (except for severe obesity) and weight gain is variable, apparently determined more by dietary composition and lifestyle than by overweight itself. Genetic influences strongly affect an individual's susceptibility within high risk populations, but probably explain little of the large differences in rates among populations. Habitual physical activity has not been unequivocally related to population rates of CHD but may reduce individual risk as well as case-fatality rates in myocardial infarction. The evidence is inconclusive on the role of psychosocial factors.
PAD and cerebral infarction are influenced by the same risk factors as is CHD, but the relative importance of these risk factors is different. Smoking and diabetes are the most important risk factors for PAD, and hypertension is the most important risk factor for cerebral infarction and cerebral hemorrhage.
Congruence of evidence from laboratory, clinical, and population studies concerning the etiology and potential for prevention of atherosclerotic cardiovascular diseases provides a strong basis for public health recommendations. Furthermore, from the epidemiologic data, one can estimate the potential public health benefit of dietary modifications.
Directions for Research
Animal Studies
· The physiological and molecular mechanisms by which hyperlipidemia causes atherosclerosis; identification and characterization of the specific lipoprotein subclasses or the modified lipoproteins that are directly involved in lipid deposition in the arterial wall and in progression of atherosclerosis.
· The mechanisms that control the responses to dietary fat and cholesterol or to other dietary components affecting serum lipoproteins, experimental atherosclerosis, or both.
· The basis for the differences in susceptibility to atherosclerosis among species and among different arterial beds within a species.
Human Studies
· The nature and the regulation (including the dietary regulation) of heterogeneity within each major class of lipoproteins and the roles of different lipoprotein subclasses in atherosclerosis and CHD, PAD, and stroke.
· The role of postprandial lipoproteins in atherosclerosis and in CHD, PAD, and stroke; the effects of diet on postprandial lipoproteins.
· The major dietary determinants of plasma HDL and the role of HDL and the mechanism whereby it protects against CHD.
· The extent and mechanism of individual variability in response to dietary saturated fatty acid intake.
· The extent and mechanism of individual variability in response to dietary cholesterol.
· The U-shaped relationship between serum cholesterol level and cerebral hemorrhage and atherothrombotic brain infarction and possible mechanisms of action.
· The effects of monounsaturated fatty acids and stearic acid on plasma lipid and lipoprotein levels and on atherosclerotic cardiovascular disease risk.
· The long-term consequences of the ingestion of different levels of w-6 polyunsaturated vegetable oils on lipoproteins and atherosclerotic cardiovascular disease risk.
Page 542
· The effects of w-3 PUFAs (fish oils) on serum lipids and lipoproteins.
· The effects of total energy intake and expenditure on atherosclerotic cardiovascular disease risk factors and risk of atherosclerotic cardiovascular diseases, especially CHD.
· The effects of different types of protein (animal and vegetable) on serum lipid and lipoprotein levels and atherosclerotic cardiovascular disease risk.
· The long-term effects of increasing the proportion of dietary complex carbohydrates (starches and fibers) on serum lipid and lipoprotein levels and atherosclerotic cardiovascular disease risk.
· The metabolic mechanisms by which alcohol intake influences serum lipid and lipoprotein levels and risk of CHD and stroke.
· The influence of specific dietary components and dietary patterns compared to that of other environmental and genetic factors (and their interactions) on atherosclerotic cardiovascular disease risk.
References
Abbott, R.D., Y. Yin, D.M. Reed, and K. Yano. 1986. Risk of stroke in male cigarette smokers. N. Engl. J. Med. 315: 717-720.
AHA (American Heart Association). 1983. 1984 Heart Facts. Office of Communications, Dallas. 25 pp.
Amad, K.H., J.C. Brennan, and .K. Alexander. 1965. The cardiac pathology of chronic exogenous obesity. Circulation 32:740-745.
Anastasiou-Nana, M., S. Nanas, J. Stamler, J. Marquardt, R. Stamler, H.A. Lindberg, D.M. Berkson, K. Liu, E. Stevens, M. Mansour, and T. Kokich. 1982. Changes in rates of sudden CHD death with first vs. recurrent events, Chicago Peoples Gas Company Study, 1960-1980. Circulation, Suppl. 66:11-236.
Anderson, A.J., J.J. Barboriak, and A.A. Rimm. 1978. Risk factors and angiographically determined coronary occlusion. Am. J. Epidemiol. 107:8-14.
Anitschkow, N.N. 1967. A history of experimentation on arterial atherosclerosis in animals. Pp. 21-44 in H.T. Blumenthal, ed. Cowdry's Arteriosclerosis: a Survey of the Problem, 2nd ed. C.C. Thomas, Springfield, Ill.
Barrett-Connor, E., and K.T. Khaw. 1984. Family history of heart attack as an independent predictor of death due to cardiovascular disease. Circulation 69:1065-1069.
Beaton, G.H., J. Milner, P. Corey, V. McGuire, M. Cousins, E. Stewart, M. de Ramos, D. Hewitt, P.V. Grambsch, N. Kassim, and J.A. Little. 1979. Sources of variance in 24-hour dietary recall data: implications for nutrition study design and interpretation. Am. J. Clin. Nutr. 32:2546-2549.
Benditt, E.P. 1974. Evidence for a monoclonal origin of human atherosclerotic plaques and some implications. Circulation 50:650-652.
Blitzer, P.H., A.A. Rimm, and E.E. Giefer. 1977. The effect of cessation of smoking on body weight in 57,032 women: cross-sectional and longitudinal analyses. J. Chron. Dis. 30: 415-429.
Bonita, R., and R. Beaglehole. 1982. Trends in cerebrovascular disease mortality in New Zealand. N.Z. Med. J. 95:411-414.
Brown, M.S., and J.L. Goldstein. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science 232:34-47.
Brownell, K.D., P.S. Bachorik, and R.S. Ayerle. 1982. Changes in plasma lipid and lipoprotein levels in men and women after a program of moderate exercise. Circulation 65: 477-484.
Brozek, J., and A. Keys. 1957. Changes of body weight in normal men who stop smoking cigarettes. Science 125:1203.
Cahill, G.H., Jr. 1977. Obesity and diabetes. Pp. 101-110 in G. Bray, ed. Recent Advances in Obesity Research, Vol. II. John Libbey and Co., London.
Camargo, C.A., Jr., P.T. Williams, K.M. Vranizan, J.J. Albers, and P.D. Wood. 1985. The effect of moderate alcohol intake on serum apolipoproteins A-I and A-II. J. Am. Med. Assoc. 253:2854:2857.
Case, R.B., S.S. Heller, N.B. Case, A.J. Moss, and the Multicenter Post-Infarction Research Group. 1985. Type A behavior and survival after acute myocardial infarction. N. Engl. J. Med. 312:737-741.
Clarkson, T.B., H.B. Lofland, Jr., B.C. Bullock, and H.O. Goodman. 1971. Genetic control of plasma cholesterol: studies on squirrel monkeys. Arch. Pathol. 92:37-45.
Clarkson, T.B., J.R. Kaplan, and M.R. Adams. 1985. The role of individual differences in lipoprotein, artery wall, gender, and behavioral responses in the development of atherosclerosis. Ann. N.Y. Acad. Sci. 454:28-45.
Colditz, G.A., M.J. Stampfer, W.C. Willett, B. Rosner, F.E. Speizer, and C.H. Hennekens. 1986. A prospective study of parental history of myocardial infarction and coronary heart disease in women. Am. J. Epidemiol. 123:48-58
Cramér, K., S. Paulin, and L. Werkö. 1966. Coronary angiographic findings in correlation with age, body weight, blood pressure, serum lipids, and smoking habits. Circulation 33:888-900.
Criqui, M.H., E. Barrett-Connor, M.J. Holdbrook, M. Austin, and J.D. Turner. 1980. Clustering of cardiovascular disease risk factors. Prev. Med. 9:525-533.
Criqui, M.H., S.S. Coughlin, and A. Fronek. 1985a. Noninvasively diagnosed peripheral arterial disease as a predictor of mortality: results from a prospective study. Circulation 72:768-773.
Criqui, M.H., A. Fronek, E. Barrett-Connor, M.R. Klauber, S. Gabriel, and D. Goodman. 1985b. The prevalence of peripheral arterial disease in a defined population. Circulation 71:510-515.
Criqui, M.H., A. Fronek, M.R. Klauber, E. Barrett-Connor, and S. Gabriel. 1985c. The sensitivity, specificity, and predictive value of traditional clinical evaluation of peripheral arterial disease: results from noninvasive testing in a defined population. Circulation 71:516-522.
Davignon, J., R.E. Gregg, and C.F. Sing. 1988. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis 8: 1-21.
Dayton, S., M.L. Pearce, H. Goldman, A. Harnish, D. Plotkin, M. Shickman, M. Winfield, A. Zager, and W. Dixon. 1968. Controlled trial of a diet high in unsaturated fat for prevention of atherosclerotic complications. Lancet 2:1060-1062.
DHHS (U.S. Department of Health and Human Services).
Page 543
1983. Arteriosclerosis. Pp. 13-62 in The Health Consequences of Smoking: Cardiovascular Disease. DHHS (PHS) Publ. No. 84-50204. A Report of the Surgeon General, Office on Smoking and Health, Public Health Service, U.S. Department of Health and Human Services, Rockville, Md.
DHHS (U.S. Department of Health and Human Services). 1987. Monthly Vital Statistics Report, Vol. 36: The Advance Report of Final Mortality Statistics, 1985. DHHS Publ. No. (PHS) 87-1120. National Center for Health Statistics, Public Health Service, U.S. Department of Health and Human Services, Hyattsville, Md. 48 pp.
Doll, R., and A.B. Hill. 1964. Mortality in relation to smoking: ten years' observations of British doctors. Br. Med. J. 1:1399-1410.
Doll, R., R. Gray, B. Hafner, and R. Peto. 1980. Mortality in relation to smoking: 22 years' observations on female British doctors. Br. Med. J. 280:967-971.
Donahue, R.P., R.D. Abbott, E. Bloom, D.M. Reed, and K. Yano. 1987. Central obesity and coronary heart disease in men. Lancet 1:821-824.
Dyken, M.L., P.A. Wolf, H.J.M. Barnett, J.J. Bergan, W.K. Hass, W.B. Kannel, L. Kuller, J.F. Kurtzke, and T.M. Sundt. 1984. Risk factors in stroke: a statement for physicians by the Subcommittee on Risk Factors and Stroke of the Stroke Council. Stroke 15:1105-1111.
Ekelund, L.G., W.L. Haskell, J.L. Johnson, F.S. Whaley, M.H. Criqui, and D.S. Sheps. 1988. Physical fitness as a predictor of cardiovascular mortality in asymptomatic North American men: the Lipid Research Clinics Mortality Follow-up Study. N. Engl. J. Med. 319:1379-1384.
Flow, B.L., T.C. Cartwright, T.J. Kuehl, G.E. Mott, D.C. Kraemer, A.W. Kruski, J.D. Williams, and H.C. McGill, Jr. 1981. Genetic effects on serum cholesterol concentrations in baboons. J. Hered. 72:97-103.
Folsom, A.R., O. Gomez-Marin, R.F. Gillum, T.E. Kottke, W. Lohman, and D.R. Jacobs, Jr. 1987. Out-of-hospital coronary death in an urban population—validation of death certificate diagnosis: the Minnesota Heart Survey. Am. J. Epidemiol. 125:1012-1018.
Franceschini, G., L. Calabresi, C. Tosi, C.R. Sirtori, C. Fragiacomo, G. Noseda, E. Gong, P. Blanche, and A.V. Nichols. 1987. Apolipoprotein A-IMilano. Correlation between high density lipoprotein subclass distribution and triglyceridemia. Arteriosclerosis 7:426-435.
Freedman, D.S., W.P. Newman III, R.E. Tracy, A.E. Voors, S.R. Srinivasan, L.S. Webber, C. Restrepo, J.P. Strong, and G.S. Berenson. 1988. Black-white differences in aortic fatty streaks in adolescence and early adulthood: the Bogalusa Heart Study. Circulation 77:856-864.
French-Belgian Collaborative Group. 1982. Ischemic heart disease and psychological patterns: prevalence and incidence studies in Belgium and France. Adv. Cardiol. 29:25-31.
Friedman, G.D., D.B. Petitti, RD. Bawol, and A.B. Siegelaub. 1981. Mortality in cigarette smokers and quitters: effect of base-line differences. N. Engl. J. Med. 304:1407-1410.
Garraway, W.M., and J.P. Whisnant. 1987. The changing pattern of hypertension and the declining incidence of stroke. J. Am. Med. Assoc. 258:214-217.
Garrison, R.J., P.W. Wilson, W.P. Castelli, M. Feinleib, W.B. Kannel, and P.M. McNamara. 1980. Obesity and lipoprotein cholesterol in the Framingham Offspring Study. Metabolism 29:1053-1060.
Geer, J.C., H.C. McGill, Jr., W.B. Robertson, and J.P. Strong. 1968. Histologic characteristics of coronary artery fatty streaks. Lab. Invest. 18:565-570.
Gertler, M.M., and P.D. White. 1954. Coronary Heart Disease in Young Adults; a Multidisciplinary Study. Harvard University Press, Cambridge, Mass. 218 pp.
Gill, J.S., A.V. Zezulka, M.J. Shipley, S.K. Gill, and D.G. Beevers. 1986. Stroke and alcohol consumption. N. Engl. J. Med. 315:1041-1046.
Gillum, R.F., A. Folsom, R.V. Luepker, D.R. Jacobs, Jr., T.E. Kottke, O. Gomez-Marin, R.J. Prineas, H.L. Taylor, and H. Blackburn. 1983. Sudden death and acute myocardial infarction in a metropolitan area, 1970-1980: the Minnesota Heart Survey. N. Engl. J. Med. 309:1353-1358.
Gillum, R.F., O. Gomez-Marin, T.E. Kottke, D.R. Jacobs, Jr., R.J. Prineas, A.R. Folsom, R.V. Luepker, and H. Blackburn. 1985. Acute stroke in a metropolitan area, 1970 and 1980: the Minnesota Heart Survey. J. Chron. Dis. 38: 891-898.
Goldbourt, U., and J.H. Medalie. 1977. Characteristics of smokers, non-smokers and ex-smokers among 10,000 adult males in Israel. Am. J. Epidemiol. 105:75-86.
Goldbourt, U., and H.N. Neufeld. 1986. Genetic aspects of arteriosclerosis. Arteriosclerosis 6:357-377.
Goldstein, J.L., W.R. Hazzard, H.G. Schrott, E.L. Bierman, and A.G. Motulsky. 1972. Genetics of hyperlipidemia in coronary heart disease. Trans. Assoc. Am. Phys. 85:120-138.
Goldstein, J.L., W.R. Hazzard, H.G. Schrott, E.L Bierman, and A.G. Motulsky. 1973a. Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. J. Clin. Invest. 52:1533-1543.
Goldstein, J.L, H.G. Schrott, W.R. Hazzard, E.L. Bierman, and A.G. Motulsky. 1973b. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Invest. 52:1544-1568.
Goldstein, J.L., T. Kita, and M.S. Brown. 1983. Defective lipoprotein receptors and atherosclerosis: lessons from an animal counterpart of familial hypercholesterolemia. N. Engl. J. Med. 309:288-296.
Gomez-Marin, O., A.R. Folsom, T.E. Kottke, S.C. Wu, D.R. Jacobs Jr., R.F. Gillum, S.A. Edlavitch, and H. Blackburn. 1987. Improvement in long-term survival among patients hospitalized with acute myocardial infarction, 1970 to 1980: the Minnesota Heart Survey. N. Engl. J. Med. 316:1353-1359.
Gordon, T., W.B. Kannel, D. McGee, and T.R. Dawber. 1974. Death and coronary attacks in men after giving up cigarette smoking: a report from the Framingham Study. Lancet 2:1345-1348.
Gordon, T., W.B. Kannel, T.R. Dawber, and D. McGee. 1975. Changes associated with quitting cigarette smoking: the Framingham Study. Am. Heart. J. 90:322-328.
Gordon, T., N. Ernst, M. Fisher, and B.M. Rifkind. 1981. Alcohol and high-density lipoprotein cholesterol. Circulation, Suppl. 64:III63-III67.
Grundy, S.M., and G.L. Vega. 1988. Plasma cholesterol responsiveness to saturated fatty acids. Am. J. Clin. Nutr. 47:822-824.
Hammond, E.C., L. Garfinkel, and H. Seidmann. 1971. Longevity of parents and grandparents in relation to coronary heart disease and associated variables. Circulation 43: 31-44.
Page 544
Hartung, G.H., J.P. Foreyt, R.E. Mitchell, I. Vlasek, and A.M. Gotto, Jr. 1980. Relation of diet to high-density-lipoprotein cholesterol in middle-aged marathon runners, joggers, and inactive men. N. Engl. J. Med. 302:357-361.
Harvald, B., and M. Hauge. 1970. Coronary occlusion in twins. Acta Genet. Med. Gemellol. 19:248-250.
Haynes, S.G., M. Feinleib, and E.D. Eaker. 1983. Type A behaviour and the 10-year incidence of coronary heart disease in the Framingham Heart Study. Pp. 80-92 in R.H. Rosenman, ed. Psychosomatic Risk Factors and Coronary Heart Disease: Indications for Specific Preventive Therapy. Hans Huber Publishers, Bern, Switzerland.
Heyden, S., A. Heyman, and L. Camplong. 1969. Mortality patterns among parents of patients with atherosclerotic cerebrovascular disease. J. Chron. Dis. 22:105-110.
Hjermann, I., K.V. Byre, I. Holme, and P. Leren. 1981. Effect of diet and smoking intervention on the incidence of coronary heart disease. Lancet 2:1303-1310.
Holme, I. 1982. On the separation of the intervention effects of diet and antismoking advice on the incidence of major coronary events in coronary high risk men. The Oslo Study. J. Oslo City Hosp. 32:31-54.
Huttunen, J.K., E. Länsimies, E. Voutilainen, C. Ehnholm, E. Hietanen, I. Pentillä, O. Siitonen, and R. Rauramaa. 1979. Effect of moderate physical exercise on serum lipoproteins: a controlled clinical trial with special reference to serum high-density lipoproteins. Circulation 60:1220-1229.
Ignatowski, A. 1909. Über die Wirkung des tierischen Eiweisses auf die Aorta und die parenchymatösen Organe der Kaninchen. Virchows Arch. Pathol. 198:248-270.
Inter-Society Commission for Heart Disease Resources. 1984. Optimal resources for primary prevention of atherosclerotic diseases. Circulation 70:157A-205A.
Jacobs, D.R., Jr., J.T. Anderson, and H. Blackburn. 1979. Diet and serum cholesterol: do zero correlations negate the relationship? Am. J. Epidemiol. 110:77-87.
Kagan, A., J.S. Popper, and G.C. Rhoads. 1980. Factors related to stroke incidence in Hawaii Japanese men: the Honolulu Heart Study. Stroke 11:14-21.
Kannel, W.B., and D.L. McGee. 1985. Update on some epidemiologic features of intermittent claudication: the Framingham Study. J. Am. Geriatr. Soc. 33:13-18.
Kannel, W.B., and P.A. Wolf. 1983. Epidemiology of cerebrovascular disease. Pp. 1-24 in R.W.R. Russell, ed. Vascular Disease of the Central Nervous System. Churchill Livingstone, Edinburgh.
Kannel, W.B., N. Brand, J.J. Skinner, Jr., T.R. Dawber, and P.M. McNamara. 1967. The relation of adiposity to blood pressure and development of hypertension: the Framingham Study. Ann. Intern. Med. 67:48-59.
Kaplan, J.R., S.B. Manuck, T.B. Clarkson, F.M. Lusso, and D.M. Taub. 1982. Social status, environment, and atherosclerosis in cynomolgus monkeys. Arteriosclerosis 2:359-368.
Katan, M.B., A.C. Beynen, J.H.M. De Vries, and A. Nobels. 1986. Existence of consistent hypo- and hyperesponders to dietary cholesterol in man. Am. J. Epidemiol. 123:221-234.
Katz, S.S. 1981. The lipids of grossly normal human aortic intima from birth to old age. J. Biol. Chem. 256:12275-12280.
Keys, A. 1965. Dietary survey methods in studies on cardiovascular epidemiology. Voeding 26:464-483.
Keys, A. 1970. Coronary heart disease in seven countries. Circulation, Suppl. 41:11-1211.
Keys, A. 1980. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Disease. Harvard University Press, Cambridge, Mass. 381 pp.
Keys, A. 1984. Serum cholesterol response to dietary cholesterol. Am. J. Clin. Nutr. 40:351-359.
Komachi, Y. 1977. Recent problems in cerebrovascular accidents: characteristics of stroke in Japan. Nippon Ronen Igakkai Zasshi 14:359-364.
Kramsch, D.M., A.J. Aspen, B.M. Abramowitz, T. Kreimendahl, and W.B. Hood, Jr. 1981. Reduction of coronary atherosclerosis by moderate conditioning exercise in monkeys on an atherogenic diet. N. Engl. J. Med. 305:1483-1489.
Kromhout, D., and C. de Lezenne Coulander. 1984. Diet, prevalence and 10-year mortality from coronary heart disease in 871 middle-aged men: the Zutphen Study. Am. J. Epidemiol. 119:733-741.
Kuller, L. 1969. Sudden death in arteriosclerotic heart disease: the case for preventive medicine. Am. J. Cardiol. 24:617-628.
Kurtzke, J.F. 1976. An introduction to the epidemiology of cerebrovascular disease. Pp. 239-253 in P. Scheinberg, ed. Cerebrovascular Diseases: Tenth Princeton Conference. Raven Press, New York.
Kushi, L.H., R.A. Lew, F.J. Stare, C.R. Ellison, M. el Lozy, G. Bourke, L. Daly, I. Graham, N. Hickey, R. Mulcahy, and J. Kevaney. 1985. Diet and 20-year mortality from coronary heart disease: the Ireland-Boston Diet-Heart Study. N. Engl. J. Med. 312:811-818.
Kwiterovich, P.O., Jr., S. White, T. Forte, P.S. Bachorik, H. Smith, and A. Sniderman. 1987. Hyperapobetalipoproteinemia in a kindred with familial combined hyperlipidemia and familial hypercholesterolemia. Arteriosclerosis 7: 211-225.
La Croix, A.Z., L.A. Mead, K.Y. Liang, C.B. Thomas, and T.A. Pearson. 1986. Coffee consumption and the incidence of coronary heart disease. N. Engl. J. Med. 315:977-982.
Lapidus, L., H. Andersson, C. Bengtsson, and I. Bosaeus. 1986. Dietary habits in relation to incidence of cardiovascular disease and death in women: a 12-year follow-up of participants and the population study of women in Gothenburg, Sweden. Am. J. Clin. Nutr. 44:444-448.
Larsson, B., K. Svärdsudd, L. Welin, L. Wilhelmsen, P. Björntorp, and G. Tibblin. 1984. Abdominal adipose tissue distribution, obesity, and risk of cardiovascular disease and death: 13 year follow up of participants in the study of men born in 1913. Br. Med. J. 288:1401-1404.
La Ville, A., P.R. Turner, R.M. Pittilo, S. Martini, C.B. Marenah, P.M. Rowles, G. Morris, G.A. Thomson, N. Woolf, and B. Lewis. 1987. Hereditary hyperlipidemia in the rabbit due to overproduction of lipoproteins. I. Biochemical studies. Arteriosclerosis 7:105-112.
LeGrady, D., A.R. Dyer, R.B. Shekelle, J. Stamler, K. Liu, O. Paul, M. Lepper, and A.M. Shryock. 1987. Coffee consumption and mortality in the Chicago Western Electric Company Study. Am. J. Epidemiol. 126:803-812.
Levy, R.I. 1979. Stroke decline: implications and prospects. N. Engl. J. Med. 300:490-491.
Lipid Research Clinics Program. 1984. The Lipid Research Clinics Coronary Primary Prevention Trial Results. II. The relationship of reduction in incidence of coronary heart disease to cholesterol lowering. J. Am. Med. Assoc. 251: 365-374.
Liu, K., J. Stamler, A. Dyer, J. McKeever, and P. McKeever.
Page 545
1978. Statistical methods to assess and minimize the role of intra-individual variability in obscuring the relationship between dietary lipids and serum cholesterol. J. Chron. Dis. 31:399-418.
Mahley, R.W. 1988. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240: 622-630.
Mann, J.I., and J.W. Marr. 1981. Coronary heart disease prevention: trials of diets to control hyperlipidemia. Pp. 197-210 in N.E. Miller and B. Lewis, eds. Lipoproteins, Atherosclerosis and Coronary Heart Disease: Metabolic Aspects of Cardiovascular Disease, Vol. 1. Elsevier/North-Holland, Amsterdam.
Marmot, M.G., A.M. Adelstein, N. Robinson, and G.A. Rose. 1978. Changing social-class distribution of heart disease. Br. Med. J. 2:1109-1112.
McGee, D.L., D.M. Reed, K. Yano, A. Kagan, and J. Tillotson. 1984. Ten-year incidence of coronary heart disease in the Honolulu Heart Program: relationship to nutrient intake. Am. J. Epidemiol. 119:667-676.
McGill, H.C., Jr. 1968. Fatty streaks in the coronary arteries and aorta. Lab. Invest. 18:560-564.
McGill, H.C., Jr. 1988. The pathogenesis of atherosclerosis. Clin. Chem. 34:B33-B39.
McGill, H.C., Jr., J.C. Geer, and J.P. Strong. 1963. Natural history of human atherosclerotic lesions. Pp. 39-65 in M. Sandler and G.H. Bourne, eds. Atherosclerosis and Its Origin. Academic Press, New York.
McGill, H.C., Jr., C.A. McMahan, G.E. Mott, Y.N. Marinez, and T.J. Kuehl. 1988. Effects of selective breeding on the cholesterolemic responses to dietary saturated fat and cholesterol in baboons. Arteriosclerosis 8:33-39.
Melton, L.J., III, K.M. Macken, P.J. Palumbo, and LR. Elveback. 1980. Incidence and prevalence of clinical peripheral vascular disease in a population-based cohort of diabetic patients. Diabetes Care 3:650-654.
Miettinen, M., O. Turpeinen, M. Karvonen, R. Elosuo, and E Paavilainen. 1972. Effect of cholesterol-lowering diet on mortality from coronary heart-disease and other causes. Lancet 2:835-838.
Montenegro, M.R., and D.A. Eggen. 1968. Topography of atherosclerosis in the coronary arteries. Lab. Invest. 18:586-593.
Montenegro, M.R., and LA. Solberg. 1968. Obesity, body weight, body length, and atherosclerosis. Lab. Invest. 18: 594-603.
Morris, J.N., J.W. Marr, and D.G. Clayton. 1977. Diet and heart: a postscript. Br. Med. J. 2:1307-1314.
MRFIT (Multiple Risk Factor Intervention Trial) Research Group. 1982. Multiple Risk Factor Intervention Trial: risk factor changes and mortality results. J. Am. Med. Assoc. 248:1465-1477.
Müller, C. 1938. Xanthomata, hypercholesterolemia, angina pectoris. Acta Med. Scand. Suppl. 89:75-84.
Newman, W.P., III, D.S. Freedman, A.W. Voors, P.D. Gard, S.R. Srinivasan, J.L. Cresanta, G.D. Williamson, LS. Webber, and G.S. Berenson. 1986. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N. Engl. J. Med. 314: 138-144.
NHLBI (National Heart, Lung, and Blood Institute). 1971. Arteriosclerosis: A Report by the National Heart, Lung and Blood Task Force on Arteriosclerosis, Vol. 1. DHEW Publ. No. (NIH) 72-219. National Institutes of Health, Public Health Service, U.S. Department of Health, Education, and Welfare, Bethesda, Md. 365 pp.
Omae, T., M. Takeshita, and Y. Hirota. 1976. The Hisayama Study and Joint Study on cerebrovascular diseases in Japan. Pp. 255-265 in P. Scheinberg, ed. Cerebrovascular Diseases: Tenth Princeton Conference. Raven Press, New York.
Osler, W. 1897. Lectures on Angina Pectoris and Allied States. Appleton, New York. 160 pp.
Ostfeld, A.M., R.B. Shekelle, H. Klawans, and H.M. Tufo. 1974. Epidemiology of stroke in an elderly welfare population. Am. J. Public Health 64:450-458.
Paffenbarger, R.S., Jr., A.L. Wing, and R.T. Hyde. 1978. Physical activity as an index of heart attack risk in college alumni. Am. J. Epidemiol. 108:161-175.
Pell, S., and W.E. Fayerweather. 1985. Trends in the incidence of myocardial infarction and in associated mortality and morbidity in a large employed population, 1957-1983. N. Engl. J. Med. 312:1005-1011.
Pooling Project Research Group. 1978. Relationship of blood pressure, serum cholesterol, smoking habit, relative weight and ECG abnormalities to incidence of major coronary events: final report of the Pooling Project. J. Chron. Dis. 31: 201-306.
Ragland, D.R., and R.J. Brand. 1988a. Coronary heart disease mortality in the Western Collaborative Group Study: follow-up experience of 22 years. Am. J. Epidemiol. 127:462-475.
Ragland, D.R., and R.J. Brand. 1988b. Type A behavior and mortality from coronary heart disease. N. Engl. J. Med. 318: 65-69.
Reed, D., K. Yano, and A. Kagan. 1986. Lipids and lipoproteins as predictors of coronary heart disease, stroke, and cancer in the Honolulu Heart Program. Am. J. Med. 80: 871-878.
Review Panel on Coronary-Prone Behavior and Coronary Heart Disease. 1981. Coronary-prone behavior and coronary heart disease: a critical review. Circulation 63:1199-1215.
Rhoads, G.G., C.L. Gulbrandsen, and A. Kagan. 1976. Serum lipoproteins and coronary heart disease in a population study of Hawaii Japanese men. N. Engl. J. Med. 294: 293-298.
Robertson, W.B., and J.P. Strong. 1968. Atherosclerosis in persons with hypertension and diabetes mellitus. Lab. Invest. 18:538-551.
Robertson, T.L., H. Kato, T. Gordon, A. Kagan, G.G. Rhoads, C.E. Land, R.M. Worth, J.L. Belsky, D.S. Dock, M. Miyanishi, and S. Kawamoto. 1977. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California. Am. J. Cardiol. 39: 244-249.
Rogers, W.R., K.D. Cary, C.A. McMahan, M.M. Montiel, G.E. Mott, H.S. Wigodsky, and H.C. McGill, Jr. 1988. Cigarette smoking, dietary hyperlipidemia, and experimental atherosclerosis in the baboon. Exp. Mol. Pathol. 48:135-151.
Rose, G. 1964. Familial patterns in ischaemic heart disease. Br. J. Prev. Soc. Med. 18:75-80.
Rose, G., P.J.S. Hamilton, L. Colwell, and M.J. Shipley. 1982. A randomised controlled trial of anti-smoking advice: 10-year results. J. Epidemiol. Community Health 36:102-108.
Rosenberg, H.M., and A.J. Klebba. 1979. Trends in cardio-
Page 546
vascular mortality with a focus on ischemic heart disease: United States, 1950-1976. Pp. 11-41 in R.J. Havlik, M. Feinlieb, T. Thom, B. Krames, A.R. Sharrett, and R. Garrison, eds. Proceedings of the Conference on the Decline in Coronary Heart Disease Mortality. NIH Publ. No. 79-1610. National Heart, Lung, and Blood Institute, National Institutes of Health, Public Health Service, U.S. Department of Health, Education, and Welfare, Bethesda, Md.
Rosenman, R.H., R.J. Brand, R.I. Sholtz, and M. Friedman. 1976. Multivariate prediction of coronary heart disease during 8.5 year follow-up in the Western Collaborative Group Study. Am. J. Cardiol. 37:903-910.
Scanu, A.M., A. Khalil, L. Neven, M. Tidore, G. Dawson, D. Pfaffinger, E. Jackson, K.D. Carey, H.C. McGill, and G.M Fless. 1988. Genetically determined hypercholesterolemia in a rhesus monkey family due to a deficiency of the LDL receptor. J. Lipid Res. 29:1671-1681.
Shanoff, H.M., A. Little, E.A. Murphy, and H.E. Rykert. 1961. Studies of male survivors of myocardial infarction due to ''essential" atherosclerosis. I. Characteristics of the patients. Can. Med. Assoc. J. 84:519-530.
Shekelle, R.B., A.M. Shryock, O. Paul, M. Lepper, J. Stamler, S. Liu, and W.J. Raynor, Jr. 1981. Diet, serum cholesterol, and death from coronary heart disease: the Western Electric Study. N. Engl. J. Med. 304:65-70.
Shekelle, R.B., J. Stamler, O. Paul, A.M. Shryock, S. Liu, and M. Lepper. 1982. Dietary lipids and serum cholesterol level: change in diet confounds the cross-sectional association. Am. J. Epidemiol. 115:506-514.
Shekelle, R.B., S.B. Hulley, J.D. Neaton, J.H. Billings, N.O. Borhani, T.A. Gerace, D.R. Jacobs, N.L. Lasser, M.B. Mittlemark, and J. Stamler for the Multiple Risk Factor Intervention Trial Research Group. 1985a. The MRFIT Behavior Pattern Study. II. Type A behavior and incidence of coronary heart disease. Am. J. Epidemiol. 122:559-570.
Shekelle, R.B., M. Gale, and M. Norusis. 1985b. Type A score (Jenkins Activity Survey) and risk of recurrent coronary heart disease in the Aspirin Myocardial Infarction Study. Am. J. Cardiol. 56:221-225.
Shepard, J.T., and S.M. Weiss, eds. 1987. Conference on behavioral medicine and cardiovascular disease. Circulation, Suppl. 76:I1-I227.
Sholtz, R.I., R.H. Rosenman, and R.J. Brand. 1975. The relationship of reported parental history to the incidence of coronary heart disease in the Western Collaborative Group Study. Am. J. Epidemiol. 102:350-356.
Slack, J., and K.A. Evans. 1966. The increased risk of death from ischaemic heart disease in first degree relatives of 121 men and 96 women with ischaemic heart disease. J. Med. Genet. 3:239-257.
Snowdon, D.A. 1988. Animal product consumption and mortality because of all causes combined, coronary heart disease, stroke, diabetes, and cancer in Seventh-Day Adventists. Am. J. Clin. Nutr. 48:739-748.
Solberg, L.A., and J.P. Strong. 1983. Risk factors and atherosclerotic lesions. A review of autopsy studies. Arteriosclerosis 3:187-198.
Stallones, R.A. 1980. The rise and fall of ischemic heart disease. Sci. Am. 243:53-59.
Stamler, J. 1967. Lectures on Preventive Cardiology. Grune & Stratton, New York. 434 pp.
Stary, H.C. 1987a. Evolution and progression of atherosclerosis in the coronary arteries of children and adults. Pp 20-36 in S.R. Bates and E.C. Gangloff, eds. Atherogenesis and Aging. Springer-Verlag, New York.
Stary, H.C. 1987b. Macrophages, macrophage foam cells, and eccentric intimal thickening in the coronary arteries of young children. Atherosclerosis 64:91-108.
Tanaka, H., Y. Ueda, M. Hayashi, C. Date, T. Baba, H. Yamashita, H. Shoji, Y. Tanaka, K. Owada, and R. Detels. 1982. Risk factors for cerebral hemorrhage and cerebral infarction in a Japanese rural community. Stroke 13:62-73.
Tanaka, H., M. Hayashi, C. Date, K. Imai, M. Asada, H. Shoji, K. Okazaki, H. Yamamoto, K. Yoshikawa, T. Shimada, and S.I. Lee. 1985. Epidemiologic studies of stroke in Shibata, a Japanese provincial city: preliminary report on risk factors for cerebral infarction. Stroke 16:773-780.
Tanzawa, K., Y. Shimada, M. Kuroda, Y. Tsujita, M. Arai, and H. Watanabe. 1980. WHHL-rabbit: a low density lipoprotein receptor-deficient animal model for familial hypercholesterolemia. FEBS Lett. 118:81-84.
ten Kate, LP., H. Boman, S.P. Daiger, and A.G. Motulsky. 1982. Familial aggregation of coronary heart disease and its relation to known genetic risk factors. Am. J. Cardiol. 50: 945-953.
Thom, T.J., F.H. Epstein, J.J. Feldman, and P.E. Leaverton. 1985. Trends in total mortality and mortality from heart disease in 26 countries from 1950 to 1978. Int. J. Epidemiol. 14:510-520.
Thomas, C.B. 1958. Familial and epidemiologic aspects of coronary disease and hypertension. J. Chron. Dis. 7:198-208.
Tyroler, H.A. 1985. Total serum cholesterol and ischemic heart disease risk in clinical trials and observation studies. Am. J. Prev. Med. 1:18-24.
Watanabe, Y., T. Ito, and M. Shiomi. 1985. The effect of selective breeding on the development of coronary atherosclerosis in WHHL rabbits: an animal model for familial hypercholesterolemia. Atherosclerosis 56:71-79.
Welin, L, K. Svärdsudd, L. Wilhelmsen, B. Larsson, and G. Tibblin. 1987. Analysis of risk factors for stroke in a cohort of men born in 1913. N. Engl. J. Med. 317:521-526.
Willett, W., and M.J. Stampfer. 1986. Total energy intake: implications for epidemiologic analyses. Am. J. Epidemiol. 124:17-27.
William, P.T., R.M. Kraus, P.D. Wood, J.J. Albers, D. Dreon, and N. Ellsworth. 1985. Associations of diet and alcohol intake with high-density lipoprotein subclasses. Metab. Clin. Exp. 34:524-530.
Williams, R.B., Jr., J.C. Barefoot, T.L. Haney, F.E. Harrell, Jr., J.A. Blumenthal, D.B. Pryor, and B. Peterson. 1988. Type A behavior and angiographically documented coronary atherosclerosis in a sample of 2,289 patients. Psychosom. Med. 50:139-152.
Winkelstein, W., Jr., A. Kagan, H. Kato, and S.T. Sacks. 1975. Epidemiologic studies of coronary heart disease and stroke in Japanese men living in Japan, Hawaii and California: blood pressure distributions. Am. J. Epidemiol. 102: 502-513.
Wood, P.D., and W.L. Haskell. 1979. The effect of exercise on plasma high density lipoproteins. Lipids 14:417-427.
Woolsey, T.D., and I.M. Moriyama. 1948. Statistical studies of heart diseases. II. Important factors in heart disease mortality trends. Public Health Rep. 63:1247-1273.
Young, S.G., S.J. Bertics, T.M. Scott, B.W. Dubois, W.F. Beltz, L.K. Curtiss, and J.L Witztum. 1987a. Apolipoprotein B allotypes MB19(1) and MB19(2) in subjects with
Page 547
coronary artery disease and hypercholesterolemia. Arteriosclerosis 7:61-65.
Young, S.G., S.J. Bertics, L.K. Curtiss, B.W. Dubois, and J.L. Witztum. 1987b. Genetic analysis of a kindred with familial hypobetalipoproteinemia. Evidence for two separate gene defects: one associated with an abnormal apolipoprotein B species, apolipoprotein B-37; and a second associated with low plasma concentrations of apolipoprotein B-100. J. Clin. Invest. 79:1842-1851.
Young, S.G., S.T. Northey, and B.J. McCarthy. 1988. Low plasma cholesterol levels caused by a short deletion in the apolipoprotein B gene. Science 241:591-593.




















