
The Biology of Ovarian Cancers
An improved understanding of ovarian cancer biology can serve as a foundation for many other types of research and, as such, may ultimately underlie many improvements in the prevention, screening and early detection, diagnosis, and treatment of—and survival from—ovarian cancer. This chapter outlines the current state of the science in the biology of ovarian cancers along with the challenges and opportunities that exist in advancing research in that area. In particular, this chapter provides an overview of the most common types of ovarian cancer, including their origins, pathobiology, and molecular features, and highlights the research tools needed to address knowledge gaps in our understanding of the biology of this heterogeneous group of tumors.
FEATURES OF OVARIAN CARCINOMAS
The ovaries are composed of different cell types, including germ cells, specialized gonadal stromal cells, and epithelial cells. Ovarian cancer can arise from any of these cell types, but ovarian carcinomas (cancers with epithelial differentiation) make up the majority of ovarian cancers and are responsible for most ovarian cancer–related deaths (Kurman, 2013). The major subtypes of ovarian carcinomas include high-grade serous carcinoma (HGSC), endometrioid carcinoma (EC), clear cell carcinoma (CCC), low-grade serous carcinoma (LGSC), and mucinous carcinoma (MC). Pathologists classify ovarian carcinomas into these different subtypes based largely on their appearance under the microscope.
High-Grade Serous Carcinoma
HGSC is the most common type of ovarian cancer. HGSCs account for roughly 70 to 74 percent of ovarian carcinomas, and fewer than 5 percent of HGSCs are diagnosed at Stage I (when the disease is confined to the ovaries) (Braicu et al., 2011; Seidman et al., 2004). HGSC tends to present in women much later than LGSC, with the average age of presentation for HGSC—60 years—about 10 years more than the average age for LGSC. Pathologic examinations of HGSCs usually find papillary or solid growth with slit-like spaces, with the tumor cells characterized by abnormal cell nuclei, variability in size and shape, and abundant cell proliferation. HGSCs typically present with a widely disseminated disease that may include sizable masses in the ovaries, omentum, and other intra-abdominal locations, with or without ascites. HGSCs are genetically unstable, which may be due to frequent mutations in the TP53, BRCA1, and BRCA2 tumor suppressor genes. Because HGSCs usually present at an advanced stage, the earliest events of HGSC tumorigenesis have been elusive. However, data suggest that a substantial portion of HGSCs originate from the epithelium of the fallopian tube rather than of the ovary (discussed in detail later in this chapter).
Endometrioid Carcinoma
Almost half of ovarian ECs present at Stage I, and, of these, around 15 percent involve both ovaries (Gilks et al., 2008). The prognosis of patients with EC is generally favorable, largely because women with Stage I disease tend to have excellent outcomes (Storey et al., 2008). However, the overall 5-year survival of patients presenting with EC at a higher stage is poor (Storey et al., 2008). Along with CCC (described below), EC is one of the two major types of ovarian carcinoma with a well-defined association with endometriosis (ectopic endometrial tissue) (DePriest et al., 1992; Erzen et al., 2001; Yoshikawa et al., 2000). Around 5 percent of ovarian ECs are associated with synchronous uterine EC at the time of diagnosis (Soliman et al., 2004; Zaino et al., 2001). In these cases, it can be challenging for pathologists to determine, based solely on morphological criteria, whether the endometrial and ovarian tumors represent two independent primary carcinomas or a single primary cancer arising in one organ and metastasizing to the other. Whether it is one or the other distinction has significant implications for both therapy and prognosis, as the outcome is expected to be favorable with two early-stage primary cancers, while the prognosis of an EC that originated in the uterus and then metastasized to one or both ovaries is significantly worse (Soliman et al., 2004; Zaino et al., 2001). Several genes are characteristically mutated in ECs, including
CTNNB1, PIK3CA, KRAS, ARID1A, PTEN, and PPP2R1A (McConechy et al., 2014). CTNNB1 mutations are very common in ECs but rare in all of the other major ovarian carcinoma subtypes. In contrast, genes such as PIK3CA and ARID1A are frequently mutated in both EC and CCC.
Clear Cell Carcinoma
CCCs are so named because the tumor cells typically have abundant clear cytoplasm because of the presence of intracytoplasmic glycogen. Although nearly half of CCCs are diagnosed at Stage I, several studies have noted a relatively unfavorable prognosis for women with these tumors (Anglesio et al., 2011; Jenison et al., 1989; Tammela et al., 1998). As noted previously, CCC is the other major type of ovarian carcinoma that is associated with endometriosis. Researchers have observed several growth patterns for CCCs (e.g., solid, papillary, and tubulocystic), with many CCCs arising in association with cysts or with benign tumors known as clear cell adenofibromas. CCCs that arise from adenofibromas are less likely to be associated with endometriosis than cystic CCCs (Veras et al., 2009). Approximately 50 percent of CCCs contain mutations in ARID1A, and around 36 percent of CCCs harbor mutations of PIK3CA (Jones et al., 2010; Matsumoto et al., 2015). CCCs also have mutations in PPP2R1A, PTEN, KRAS, and TP53, but at a lower frequency (Cho and Shih, 2009; McConechy et al., 2011; I. M. Shih et al., 2011).
Low-Grade Serous Carcinoma
LGSCs are uncommon, and only 10 to 20 percent of them are diagnosed at Stage I (Bell, 2014; Bodurka et al., 2012; Malpica et al., 2004). In a recent series from a large single-institution registry, only 2 percent of LGSCs were Stage I at diagnosis (Gershenson et al., 2015). Although LGSCs generally display more indolent behavior than HGSCs, they are relatively more chemoresistant (Gourley et al., 2014), and the overall survival rate for women diagnosed with the advanced-stage disease remains poor (Gershenson et al., 2015). LGSCs frequently arise in association with serous borderline tumors (SBTs) and present as palpable masses in one or both ovaries. Histologically, LGSCs are characterized by papillary architecture and cells with low mitotic activity and relatively uniform, small nuclei. Metastases from LGSCs often manifest as small solid nests of tumor cells or as micropapillae surrounded by clear spaces or clefts that invade haphazardly into involved tissue. Despite the similarity in names, LGSCs infrequently progress to HGSCs, and, for the most part, the two types of ovarian carcinomas have non-overlapping mutational patterns (Vang et al., 2009). However, there have been several documented cases of LGSCs
transitioning to an “intermediate grade” and then transitioning to HGSCs (Dehari et al., 2007; Parker et al., 2004; Silva et al., 1997). Characteristic mutations in LGSCs include the mutually exclusive activating mutations of KRAS or BRAF and activating mutations of ERBB2 (Tone et al., 2014; Vang et al., 2009).
Mucinous Carcinoma
Although benign mucinous tumors of the ovary represent approximately 12 percent of all ovarian tumors in the Western world, MCs are the least common of the major types of ovarian carcinoma. The carcinoma’s stage at diagnosis is the most important prognostic factor for MCs, as patients with Stage I disease have an excellent prognosis, while outcomes for those with advanced-stage disease tend to be very poor (Ledermann et al., 2014; Zaino et al., 2011). Until the 1990s researchers believed that the relative frequency of primary MCs was significantly higher than it is now known to be; in the 1990s and 2000s several studies collectively showed that many ovarian MCs are actually metastases from MCs that arose in other sites, such as the gastrointestinal and biliary tracts or pancreas (Lee and Young, 2003; Riopel et al., 1999; Ronnett et al., 1997; Szych et al., 1999; Vang et al., 2006a,b). Histologically, MCs are characterized by glandular architecture and stratified columnar cells with basally located nuclei and pale-staining mucin in the apical cytoplasm. The cytoplasm tends to become mucin-depleted in high-grade MCs. Differentiating between primary and metastatic MC can be challenging because most MCs display evidence of intestinal-type differentiation. Therefore, it is likely that many previous clinical and molecular analyses of MCs were compromised by the inadvertent misclassification of metastatic adenocarcinoma to the ovaries as primary ovarian MCs (Hart, 2005). KRAS and TP53 mutations are found in roughly half of invasive MCs and frequently co-occur in the same tumors (Rechsteiner et al., 2013). ERBB2 amplification is found in 19 percent of MCs (Anglesio et al., 2013a). Both ERBB2 amplification and KRAS mutation may be associated with improved survival (Anglesio et al., 2013a). There are few good data on the frequency of other molecular alterations in MCs.
Ovarian Carcinoma Classification and Nomenclature
The classification and nomenclature of ovarian carcinomas has evolved over many decades and may continue to evolve as the understanding of the carcinomas’ origin and molecular features becomes more refined. Historically, more than one term has been used for some ovarian carcinoma types, which may have hindered progress in understanding their biology and clini-
cal behavior. For example, the terms “micropapillary serous carcinoma” and “psammocarcinoma” have been used for certain subgroups of LGSCs, and HGSCs have been variably referred to as “serous cystadenocarcinomas” or “papillary serous carcinomas” (or “serous papillary carcinomas”). As mentioned above, many tumors previously classified as MCs are actually ovarian metastases from non-ovarian primaries. Furthermore, the systems that pathologists use to grade ovarian carcinomas have evolved over the past several years and vary with subtype. Pathologists continue to use a three-grade system for ECs; CCCs are not currently assigned a grade (they are considered high grade by default); and the grading of serous carcinomas has changed from three grades to two grades (LGSC versus HGSC). Many tumors previously diagnosed as high-grade EC would be classified as HGSC today. A two-grade system for grading MCs is also gaining favor (Seidman et al., 2011). The changes in tumor classification and nomenclature, while necessary to reflect the evolving understanding of ovarian cancer heterogeneity, have undoubtedly contributed to confusion among pathologists, clinicians, and researchers alike.
TISSUE AND CELL OF ORIGIN OF OVARIAN CARCINOMAS
Despite the fact that most cancers involving the ovaries are called ovarian cancer, many of them may not actually originate in the ovaries (see Figure 2-1). Even cancers that originate in the ovaries may arise from cell types that are not considered intrinsic to normal ovaries (e.g., endometrial- or fallopian tube–type epithelium). The incomplete understanding of the origins of each type of ovarian cancer may impede the development of effective prevention, early detection, and treatment methods. In fact, detecting ovarian cancers early may require looking in locations other than the ovaries themselves, because a growing evidence base suggests that many ovarian carcinomas arise from sites outside the ovaries and spread to the ovaries secondarily.
The Origins of HGSCs
Historically, researchers and clinicians assumed that ovarian carcinomas develop from the ovarian surface epithelium (OSE), primarily because the dominant mass is often found in the ovaries. Precursor lesions were scarce and hard to identify, but periodically tubal carcinoma and dysplasia would be reported as having presented concomitantly with ovarian carcinoma (Woolas et al., 1994). Even so, attention did not shift away from the ovaries and toward the fallopian tubes until Dutch investigators studied the fallopian tubes prophylactically removed from women with a genetic predisposition to ovarian cancer and noted lesions, now called serous tubal
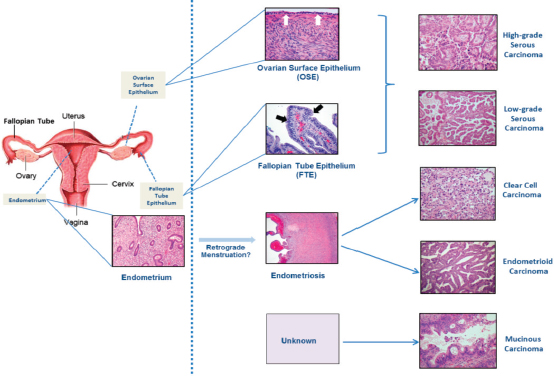
FIGURE 2-1 Potential cellular origins of ovarian carcinomas.
NOTES: Data suggest that many ovarian carcinomas may originate from outside the ovaries. However, questions still remain regarding their origins and progression. White arrows indicate OSE, black arrows indicate FTE.
SOURCE: Photographs of pathology slides reprinted with permission from Kathleen Cho (2016).
intraepithelial carcinomas (STICs), that closely resembled HGSC (Piek et al., 2001a). Subsequent studies identified STICs in about 6 to 10 percent of tubes that had been prophylactically removed from genetically predisposed women, but the studies failed to identify comparable HGSC precursor lesions in their ovaries (Callahan et al., 2007; Shaw et al., 2009). The development of a protocol in which the fimbriated end of the fallopian tube was sectioned and extensively examined (the SEE-FIM protocol) allowed for a more detailed analysis of the fallopian tube, and researchers subsequently identified STICs or small tubal HGSCs in 50 to 70 percent of women with HGSC, most of whom had advanced-stage disease (Kindelberger et al., 2007; Lee et al., 2006; Przybycin et al., 2010). STICs were also identified in women with HGSC who lacked germline BRCA1 or BRCA2 mutations, which indicated that these lesions can also form in women without a known hereditary mutation (Kindelberger et al., 2007). Moreover, STICs were frequently detected in the fimbria, finger-like projections at the end of the fallopian tube that are closely associated with the ovaries. Collectively, these findings suggest that many HGSCs may actually originate in fallopian tube epithelium (FTE). Cancer cells from STICs or early tubal HGSCs implanted in the ovary could give the false impression that the tumor originated in the ovary (Piek et al., 2001b, 2003). Various studies have supported a clonal relationship between STICs and concomitant HGSC by identifying identical TP53 and other mutations (Kindelberger et al., 2007; Lee et al., 2007; McDaniel et al., 2015). Furthermore, gene expression profiling has demonstrated that HGSCs are more closely related to FTE than to OSE (Kurman and Shih, 2010; Marquez et al., 2005).
During ovulation, breaches in the OSE may allow detached tubal epithelium to implant in the ovary. Indeed, benign tubal-type epithelium is often identified in the ovary, but it remains unclear whether this ectopic tubal epithelium, known as endosalpingiosis, is actually imported to the ovary or arises from cells intrinsic to the ovary that are capable of acquiring tubal-type differentiation through a process known as metaplasia. The OSE can invaginate into the underlying ovarian stroma, forming so-called inclusion glands or cysts. Some HGSCs may arise from the OSE, from inclusion glands and cysts, from ovarian endosalpingiosis, or from other cells in the ovary without first involving the fallopian tube (Auersperg, 2013). Recent work done in a mouse model suggests that ovarian carcinomas may arise from a susceptible population of cells with stem cell–like properties that are present where the OSE, FTE, and peritoneal mesothelium converge (Flesken-Nikitin et al., 2013; Ng and Barker, 2015).
Despite a high proportion of STIC lesions being identified in women with HGSCs, questions still remain regarding HGSC origin, particularly those carcinomas that show no involvement of the fallopian tube. It will be necessary to develop additional studies and models in order to better
understand which cells undergo neoplastic transformation to HGSC and whether these are cells with stem cell–like properties. Given the remaining uncertainties regarding the origin of HGSC, the current (revised in 2014) International Federation of Gynecology and Obstetrics staging criteria for ovarian cancer combine cancers of the ovary, fallopian tube, and peritoneum into a single unified staging system (Mutch and Prat, 2014).
Sources of Other Ovarian Carcinomas
HGSC is not the only ovarian carcinoma that has been suggested to arise from nonovarian tissues. Studies indicate that endometriosis is associated with 15 to 50 percent of CCCs and ECs and that women with endometriosis are two to three times more likely to develop ovarian cancer (Brinton et al., 2005; DePriest et al., 1992; Erzen et al., 2001; Forte et al., 2014; Pavone and Lyttle, 2015; Rossing et al., 2008; Sainz de la Cuesta et al., 1996; Yoshikawa et al., 2000). The mechanisms by which endometriosis develops remain poorly understood. More than 90 years ago it was suggested that endometrial tissue might implant outside of the endometrium via retrograde menstruation (Sampson, 1927). More recently, it has been suggested that endometriosis arises from endometrial stem cells or progenitor cells that are disseminated outside the endometrium around the time of birth and then stimulated to differentiate after menarche (Brosens and Benagiano, 2015). In some cases, a transition from endometriotic cysts to EC or CCC has been observed by morphologic and molecular genetic studies (Fukunaga et al., 1997; Sainz de la Cuesta et al., 1996; Veras et al., 2009). Around 10 percent of ovarian ECs are associated with synchronous uterine ECs at the time of diagnosis (Soliman et al., 2004; Zaino et al., 2001). The shared mutational spectrum of endometriosis-associated ovarian cancers and uterine ECs suggests a common origin, likely attributable to endometriosis.
LGSCs frequently arise in association with serous borderline tumors—ovarian tumors that have some features similar to carcinomas—but they usually behave in a benign fashion. However, the origin of serous borderline tumors is unclear. Some investigators have proposed that cells from a fallopian tube lesion, termed papillary tubal hyperplasia, detach and implant in the ovary and subsequently give rise to endosalpingiosis and serous borderline tumors (Kurman et al., 2011; Robey and Silva, 1989).
Finally, the cellular origin of MCs remains unclear. Most MCs and the mucinous cystadenomas and borderline tumors from which they often arise display intestinal-type glandular differentiation and express intestinal-type protein markers (e.g., cytokeratin 20 and CDX2). The reason for this is poorly understood, particularly because intestinal differentiation is not a feature of normal epithelia in the female genital tract (e.g., organs derived
from the Müllerian ducts during embryological development). Only a small subset of MCs display the sort of endocervical-type differentiation that suggests Müllerian origin. Recent studies suggest that at least some MCs may be derived from a type of ovarian germ cell tumor known as a mature teratoma (Fujii et al., 2014; Kerr et al., 2013). Mature teratomas are ovarian germ cell tumors that are typically composed of tissues derived from two or three germ layers in the developing embryo (i.e., ectoderm, mesoderm, and endoderm). Occasionally, teratomas may display tissue from only one germ layer (the so-called monodermal teratomas). If this is the case, the least common of the major types of ovarian cancer may prove to be the only one that arises from cells intrinsic to the ovaries. Recently, it has been suggested that some mucinous tumors may arise from another type of ovarian tumor known as a Brenner tumor, based on a clonal relationship between the Brenner tumor and associated mucinous tumor components (Wang et al., 2015).
Implications
In summary, only a small fraction of ovarian carcinomas may actually originate in the ovaries, and in response to that understanding, a paradigm shift is occurring that is moving the focus away from seeing the ovaries as the source of most ovarian carcinomas (Kuhn et al., 2012). This shift in thinking has significant clinical implications. For example, the performance of prophylactic salpingectomy alone may prove to be a strategy for reducing the incidence (and mortality) of ovarian carcinomas. (See Chapter 3 for more on prophylactic salpingectomy.) Furthermore, knowing where these early precursor lesions start can help direct the development of new imaging and other techniques to detect precursor lesions before they spread to the ovaries and elsewhere.
Although more work is required to determine the origins of ovarian carcinomas definitively, it is clear that the ovaries provide a highly receptive site from which malignant cells can spread and grow (Asotra et al., 2009). This is not a new insight. The ability for cancer cells (seeds) to target specific organs (soil) has been recognized since the late 18th century, when Paget described the “seed and soil hypothesis” (Fidler, 2003; Paget, 1889). However, as will be discussed later in this chapter, evaluating these theories, developing better methods of early detection and prevention, and designing new therapies is difficult without having the proper experimental systems with which to test their validity.
Advancing technologies and improved analytical methods are making it possible for researchers to explore the landscape of ovarian cancers in a variety of novel ways and, in particular, to uncover new information on the genomics, transcriptomics, epigenomics, proteomics, and metabolomics of ovarian cancers. These studies accrue large amounts of what is referred to as omics data, and it seems likely that the best way to analyze and study this information is to use a systems biology approach that explores the interactions of proteins, gene expression, and metabolism in order to gain useful insights into the biology of ovarian cancer (Gehlenborg et al., 2010). This section describes some of the highlights of research in ovarian cancer omics; because of the rapid development of these technologies, it was not possible to offer a completely comprehensive look at such research.
Genomics
All cancers develop as a consequence of an accumulation of genetic alterations or other molecular defects. The majority of these accumulated genetic alterations lead to abnormal cellular functions such as uncontrolled growth, angiogenesis, and immune evasion (Hanahan and Weinberg, 2011). Some cancers develop through a germline (inherited) mutation, but most advance through a series of steps that begin with somatic (acquired) mutations in the tissue of origin. Many of these mutations result in the inactivation or activation of genes known, respectively, as tumor suppressors and oncogenes. These genes encode proteins that can either inhibit (tumor suppressors) or promote (oncogenes) the growth of ovarian carcinomas. The Cancer Genome Atlas (TCGA),2 a National Cancer Institute–supported effort to molecularly characterize various cancers, developed a catalogue of molecular abnormalities identified from a pool of 489 HGSC tumor samples. However, many research groups have been identifying other molecular abnormalities in the other ovarian carcinoma subtypes (see Table 2-1).
Inherited Mutations
A strong risk factor for ovarian cancer is having a family history of ovarian cancer. Women in families with several cases of ovarian carcinomas usually have mutations in BRCA1 and BRCA2, which are good risk predictors. Together, BRCA1 and BRCA2 account for around 15 percent
________________
1Omics is a term encompassing multiple molecular disciplines that involve the characterization of global sets of biological molecules such as DNAs, RNAs, proteins, and metabolites (IOM, 2012).
2 For more information, see http://www.tcga.cancer.gov (accessed September 1, 2015).
TABLE 2-1
Characteristic Mutations in Ovarian Carcinomas
| Carcinoma Subtype | Gene Activation | Gene Inactivation |
| HGSC |
|
|
| LGSC |
|
|
| MC |
|
|
| EC |
|
|
| CCC |
|
|
NOTE: The descriptions describe the functions of the proteins encoded by the genes listed above.
SOURCES: Anglesio et al., 2013a; Belanger et al., 2015; Cancer Genome Atlas Research Network, 2011; Della Pepa et al., 2015; Garrett et al., 2001; Gemignani et al., 2003; Jones et al., 2010; McConechy et al., 2011, 2014; Merritt and Cramer, 2010; Ryland et al., 2015; I. M. Shih et al., 2011; Zhai et al., 2015.
of all ovarian cancers (Pal et al., 2005). In one study looking at Ashkenazi Jewish women, BRCA1 mutation carriers were found to have a 40 to 50 percent cumulative lifetime risk of ovarian cancer and BRCA2 mutation carriers to have a 20 to 30 percent cumulative lifetime risk (King et al., 2003). BRCA1 and BRCA2 mutations also account for about 5 to 10 percent of all breast cancers and 20 to 25 percent of hereditary breast cancers (Campeau et al., 2008; Easton, 1999). Additional high-risk ovarian cancer susceptibility genes include MLH1, MSH2, MSH6, PMS2, EPCAM, and TP53; other potential susceptibility genes continue to emerge through the use of genome-wide association studies (Kuchenbaecker et al., 2015), which have been able to identify a spectrum of susceptibility genes in ovarian tumor samples, ranging from common to rare variants (Pharoah et al., 2002). Many susceptibility genes are involved in DNA repair; mutations in such genes leave a cell unable to properly correct DNA damage, which helps explain why women with mutations in these genes have a higher risk of getting ovarian cancer in their lifetimes. Chapter 3 discusses the importance of identifying women with genetic susceptibility in order to improve prevention and early detection.
Acquired Mutations
HGSCs have frequent mutations in TP53 and, indeed, are likely the type of solid tumor with the greatest number of TP53 mutations, except for select inherited cancer syndromes (Belanger et al., 2015; Cancer Genome Atlas Research Network, 2011). TP53 mutations are found in more than approximately 95 percent of HGSCs (Ahmed et al., 2010; Cancer Genome Atlas Research Network, 2011). Although most serous tumors develop as de novo high-grade lesions with early alterations of TP53, it is thought that in some cases the tumors stem from previously established lower-grade tumors (Dehari et al., 2007). One reanalysis of molecularly characterized TP53 mutation-negative cases by specialty pathologists indicated that most of these cases were not truly HGSCs (Vang et al., 2015). Some of the previous genetic studies of serous carcinoma have been confounded by the inclusion of other, non-serous subtypes of ovarian carcinoma.
Somatic mutations in BRCA1 and BRCA2 are found to occur in almost one-third of HGSCs (Hennessy et al., 2010). A recent survey of putative homologous recombination gene mutations identified rare somatic mutations in BRIP1, CHEK2, and RAD51C among the advanced-stage HGSCs (Pennington et al., 2014). TCGA, which has comprehensively sequenced the exomes of more than 300 cases, not only found mutations in TP53, BRCA1, and BRCA2, but also identified NF1 and FAT3 as rare genes that were mutated in more than 3 percent of cases studied. Copy number alterations are extensive in HGSCs and spread throughout the genome, resulting
in HGSC being a very complex solid tumor. Recurrent amplifications found in CCNE1, PIK3CA, KRAS, and MYC may have prognostic or therapeutic significance (Pennington et al., 2014). Focal deletions identified in PTEN, RB1, NF1, and CDKN2A also have the potential to affect prognosis and treatment outcomes (Martins et al., 2014).
Transcriptomics
Transcriptomics is the study of all RNAs—including mRNA, microRNA (miR), long noncoding RNA, and small RNA transcripts from DNA—in a cell or tissue. Historically, the study of tumor-specific alterations to DNA represented the major focus in cancer research. It is now recognized, however, that RNA may also be a useful diagnostic and therapeutic target. The study of transcriptomics has progressed significantly in large part because RNA-sequencing has advanced to the point that it is possible to make genome-wide expression measurements. The comparison of RNA-seq data from normal and malignant tissues makes it possible to identify tumor-specific RNA. For instance, one recent analysis of mRNA in HGSC successfully identified tumor-specific mRNA (Barrett et al., 2015). Just as DNA in circulating tumor cells is being investigated as a non-invasive means to detect the presence of ovarian cancer, it is possible that RNA can serve a similar function (Kinde et al., 2013). RNA is also capable of regulating the expression of protein. For example, miR can be used to target mRNA that encodes specific genes and thus to control gene expression and biologic function by regulating mRNA turnover and translation (Eitan et al., 2009; K. K. Shih et al., 2011). Studies of ovarian cancer have noted differential expressions of miR that are associated with prognosis and platinum resistance (Eitan et al., 2009; K. K. Shih et al., 2011). Work continues to progress in this field and may eventually provide a successful avenue for tumor detection and therapy.
Epigenomics
Epigenomics is the study of reversible, inheritable modifications to DNA and chromatin that are independent of changes in DNA sequence. These modifications can alter how accessible DNA is to transcription factors and either promote or inhibit the expression of genes. The most common epigenetic modifications are DNA methylation and histone modification. Epigenetic studies in ovarian cancer identified hypermethylated genes in the hedgehog signaling pathway, which is known to promote the development of cancers and is associated with poor prognosis (Huang et al., 2013). Promoter methylation of BRCA1 is found in approximately 10 percent of HGSC cases and is correlated with decreased gene and protein expression
(Cancer Genome Atlas Research Network, 2011; Garg et al., 2013), but the clinical significance of this BRCA1 promoter methylation is unclear. Demethylating agents have been shown to have activity in platinum-resistant tumors (Matei et al., 2012). Treatment with demethylating agents has also been shown to have broad immune stimulatory effects and may work well in combination with immunotherapy (Li et al., 2014). MiR-mediated epigenetic regulation can also lead to changes in gene expression and cell phenotype (Mishra and Johnsen, 2014). This epigenetic regulation can allow cancer cells to better adapt to their environment, which can even promote drug resistance (Berry and Bapat, 2008). Research in this area may lead to the development of new targets and more specific ovarian cancer therapies.
Proteomics and Metabolomics
Proteomic and metabolomic profiling can provide new insights into ovarian cancer that are not apparent from genomic analysis. With the possibility of analyzing thousands of proteins, which can be simultaneously altered, comparative proteomics represents a promising model of biomarker discovery for ovarian cancer detection and monitoring (Orsini et al., 2013). Using proteomic analysis to map out signaling pathways in ovarian cancer cells analysis should make it possible to design novel drugs and to optimize the use of molecularly targeted agents against crucial biologically active pathways (Toss et al., 2013). Researchers are analyzing ovarian tumor tissue, cell lines, urine, ascites fluid, and blood samples in search of metabolites and proteins to serve as promising biomarkers (Emori and Drapkin, 2014; Toss et al., 2013). Several such markers have been identified (e.g., inter-alpha-trypsin inhibitor heavy chain H4 and transferrin), but many of them have turned out to not be specific to cancer (Ahmed et al., 2005; Zhang et al., 2004). The significance of and degree of specificity and sensitivity of these markers for ovarian cancer both remain to be explored, and to date no single test or modality has been able to provide an early diagnosis of ovarian cancer.
Ovarian cancers are heterogeneous tumors that can change in various ways as the disease progresses. Only a few small studies have investigated these variations in a comprehensive manner. One recent study of 14 patients indicated that most tumors undergo clonal expansion in a metastasis-to-metastasis pattern, suggesting a continued evolution throughout anatomic dissemination rather than multiple simultaneous expansions from a common precursor clone (Schwarz et al., 2015). Furthermore, there appeared to be limited clonal expansion between primary tumor specimens and tumors
after neoadjuvant chemotherapy. However, in two cases of recurrent disease there was marked clonal expansion from a subclone that was present at diagnosis (Schwarz et al., 2015). Array comparative-genomics hybridization studies have found low copy-number variations between pre- and post-neoadjuvant chemotherapy specimens, suggesting that a dominant clone is present at diagnosis (Cooke et al., 2010).
Regarding mutational burdens, a study of six patients found that approximately 50 percent of mutations were shared through disease progression, with TP53 mutations being the only genetic mutation found in all specimens—a state of affairs that reflects TP53’s early role in tumorigenesis (Bashashati et al., 2013). Both copy-number alterations and mutations were highly heterogeneous and independent of one another. The phylogenic evolution of each of the tumors was unique and complex, reflecting the varied mechanisms of drug resistance. Pre- and post-treatment magnetic resonance imaging parameters have also been shown to vary by anatomic site, with more changes in the ovary than in metastatic deposits, and these measurements correspond with treatment response (Sala et al., 2012). Taken together, the limited studies to date demonstrate that HGSCs are molecularly diverse and continually evolving and have broad subclonal structure. An improved understanding of clonal diversity and tumor heterogeneity is needed.
ADAPTATION AND DRUG RESISTANCE
Both acquired and de novo chemoresistance remain a significant clinical challenge in ovarian cancer (discussed further in Chapter 4). The biological underpinnings of resistance are not well understood. Identifying the genes involved in responding to chemotherapy and survival may contribute to a better understanding of prognosis and potentially guide the selection of treatment options to help circumvent chemoresistance. Drug resistance, especially platinum resistance, is not limited to ovarian cancer, and there is much still to learn regarding drug resistance in all cancers. However, most cancers other than ovarian cancer have a variety of treatment options available to overcome resistance when a specific therapeutic is no longer effective. Ovarian cancer is hindered by the lack of additional therapeutic options, and therefore an improved understanding of the molecular mechanisms underlying drug resistance in ovarian carcinomas could be useful for devising new targeted therapeutic approaches to overcome or bypass resistance.
To date, several mechanisms have been proposed to explain the development of chemoresistance. Recent genetic analyses suggest that resistant clones may be present in small populations at diagnosis and then undergo selection in the face of chemotherapy. Alternatively, adaptive responses
may exist that induce resistance mechanisms during active chemotherapy treatment (Cunnea and Stronach, 2014). Other mechanisms that have been well established include reversion mutations in BRCA1 or BRCA2 that restore homologous recombination DNA repair and decrease sensitivity to platinum and other chemotherapeutic agents (Edwards et al., 2008; Sakai et al., 2008). Targeting the drivers of BRCA1 and BRCA2 expression is one possible approach to inducing homologous recombination deficiency in tumors with intact BRCA1 and BRCA2 function. Potential therapeutic targets include the E26 transformation-specific family of transcription factors, the retinoblastoma tumor suppressor (RB) pathway, and other regulators of the DNA damage response (Wiedemeyer et al., 2014). CCNE1 is overexpressed in about 20 percent of HGSC tumors and appears to upregulate members of the BRCA/homologous recombination damage-repair complex (Etemadmoghadam et al., 2013). Therefore, the inhibition of the proteasome and homologous recombination is one potential approach to overcoming the platinum resistance seen in CCNE1-amplified tumors.
Among the mechanisms that have already been employed to overcome platinum resistance are dose-dense chemotherapy and varying dose intensity. Dose-dense treatment is often applied by giving paclitaxel on a weekly, rather than 3-weekly, schedule. This approach may have effects on growth kinetics, log-kill, and neovascularization (Pinato et al., 2013). Another potential approach to overcoming platinum resistance is using targeted inhibitors to regulate signaling pathways that affect various mechanisms, including drug uptake, efflux, and binding. The use of different drugs with non-overlapping mechanisms of action may also be an important approach to overcoming platinum and other types of drug resistance.
Cancers form because of a series of mutations in oncogenes or tumor suppressor genes, but the specific tumor microenvironment can shape the transcriptional and functional diversity of the resulting cancer cells (Abelson et al., 2013; Cancer Genome Atlas Research Network, 2011; Hanahan and Weinberg, 2011; Myers et al., 2006; Schwarz et al., 2015; Tlsty and Coussens, 2006; Touboul et al., 2014). Ovarian tumor cells exist in a complex, dynamic, and multifaceted microenvironment that includes blood vessels (e.g., endothelial cells) and lymphatic networks, immune cells (e.g., infiltrating myeloid- and lymphoid-lineage cells), mesenchymal stem cells, extracellular matrix (e.g., fibroblasts, collagen, and proteoglycans), and connective tissue (e.g., adipose cells) (Hanahan and Coussens, 2012; Kenny et al., 2007; Quail and Joyce, 2013; Tlsty and Coussens, 2006). Analyses of the sequence of histologic changes that occur between tumor cells and the surrounding stromal tissues demonstrate the importance of
the tumor microenvironment (Hanahan and Weinberg, 2011). In addition, clonally expanded cell lines from single ovarian cancer cells demonstrate a phenotypic heterogeneity (plasticity) within the individual tumors with the capacity to restore self-renewal markers that are dependent on the tumor microenvironment (Abelson et al., 2013).
The Role of the Tumor Microenvironment in Ovarian Cancers
The microenvironment plays a key role in a number of stages of cancer progression, including local evasion from immune surveillance, sustained growth, invasion, and metastasis (Chen et al., 2015). Tumor and host cells physically interact and also secrete cytokines, chemokines, growth factors, and proteases that cleave and modify the structure of the extracellular matrix. Infiltrating immune cells, cancer-associated fibroblastic cells, and angiogenic vascular cells all contribute to the ability of cancer cells to keep proliferating, evade growth suppressors, avoid immune destruction, activate invasion and metastasis, induce angiogenesis, and resist cell death (Hanahan and Coussens, 2012). For example, myeloid-derived suppressor cells are mobilized during tumorigenesis and infiltrate tumors in order to promote vascularization and disrupt immune surveillance (Quail and Joyce, 2013). Infiltrating immune cells can also bind directly to cancer cells in order to suppress the activation of cell death pathways. Furthermore, activated macrophages that secrete proteases are recruited to the neoplastic site (Hanahan and Weinberg, 2011; Junttila and de Sauvage, 2013; Mroue and Bissell, 2013).
Cancer cells and stromal cells also stimulate angiogenesis. Angiogenic vascular cells not only attenuate cell death by the vascularization of tumors, they also secrete growth-promoting trophic factors as well as inhibitors of cell death (Butler et al., 2010; Castells et al., 2013). Although heterotypic signaling through paracrine signaling loops of cytokines or growth factors and their receptors is a key means of intercellular communication within the tumor microenvironment, exosome shedding from both tumor and stromal cells has recently been suggested as another possible mode of cell–cell signaling in the tumor microenvironment (Barcellos-Hoff et al., 2013). Thus, the interactions between the tumor and the stromal cells accelerate disease progression by eliciting effects on the cellular growth and metabolism of both neoplastic and stromal cell types.
The Role of the Immune System in Ovarian Cancers
Cancer cells evade immunological elimination by inducing the expression of T cell inhibitory receptors on tumor cells and immune cells and by recruiting immunosuppressive cells such as regulatory T cells and tumor-
associated macrophages (Wefers et al., 2015). The presence of macrophages correlates with malignancy in both serous and mucinous ovarian carcinomas (Hagemann et al., 2006; Kawamura et al., 2009; Takaishi et al., 2010). Tumor-associated macrophages turn into immunosuppressive cells as the tumor progresses (Hagemann et al., 2006; Mantovani and Sica, 2010). The macrophages continue to recruit regulatory T cells, which further potentiates the suppressive activity of the macrophages through cytokine production (Wefers et al., 2015). Macrophage proteases also remodel the extracellular matrix, resulting in the disruption of tissue architecture, which in turn allows cancer cells to escape the constraints imposed by the microenvironment (Balkwill et al., 2005; Karin and Greten, 2005). The degraded extracellular matrix possesses fragments thought to exert potent effects on processes such as angiogenesis (Folkman, 2006). Angiogenesis is essential for supplying blood and oxygen. Conversely, inadequate vascular function can result in hypoxia around tumor vessels, which contributes to metastasis by regulating the expression of genes through hypoxia-inducible transcription factors that alter vascular integrity (Kashiwagi et al., 2005).
Mechanisms of Metastasis
The pelvic area provides a unique environment for ovarian cancer cells to grow and metastasize. Many of the organs in the pelvic region are in close proximity and lack the physical barriers among them that could hinder the spread of cancer. The epithelial–mesenchymal transition of cancer cells is an important step in tumor metastasis. Recent data indicate that proteins that regulate actin cross-linking and coordinate the assembly of cell junctions may be critical regulators of this transition (Zhu et al., 2015).
Peritoneal recurrences of ovarian cancer indicate that a niche exists where cells are protected. Diverse stromal cell types that enhance ectopic cell survival by stimulating cells to exit dormancy also infiltrate metastatic lesions (Catena et al., 2013; Granot et al., 2011; Quail and Joyce, 2013; Tlsty and Coussens, 2006). For example, metastatic ovarian carcinomas typically seed into the adipose tissue of the peritoneum, which results in the reprogramming of adipocytes to generate free fatty acids that are then used by the cancer cells to generate ATP. This protects the cancer cells from apoptotic cell death, thereby enhancing their colonization (Nieman et al., 2011). The metastatic cells continue to acquire genetic changes that can lead toward more aggressive clones (Cancer Genome Atlas Research Network, 2011; Myers et al., 2006).
Cancer stem cells are able to produce primary and recurrent disease (Ffrench et al., 2014). One of the properties of cancer stem cells is self-renewal, which underlies tumorigenesis and differentiation and contributes
to the heterogeneity of cancer cells. Cancer stem cells have been identified in ovarian cancers. These cells may mediate tumor metastasis and, by virtue of their relative resistance to chemotherapy and radiotherapy, may contribute to the treatment resistance commonly seen in ovarian cancers (Zhang et al., 2008). Cancer stem cells are typically resistant to chemotherapy because of their decreased oxidative stress response, increased genomic stability, and expression of multiple drug resistance transporters, and they are therefore a source for tumor relapse (Visvader and Lindeman, 2008). Cancer stem cells are believed to spread by direct surface contact as well as by migration to distant areas following the flow of peritoneal fluids (Ffrench et al., 2014). Furthermore, the successful seeding of cancer stem cells at secondary sites can occur when cells breach the endothelial basement membrane (Bissell and Hines, 2011). Using anatomically joined mice, Sood and colleagues demonstrated the metastasis of ovarian cancer cells through the blood circulation (Pradeep et al., 2014).
Abnormal regulation of miR expression has been documented in ovarian cancers (Miles et al., 2012). One study identified and characterized a microenvironment-induced downregulation of miR-193b in metastasizing ovarian cancer cells (Mitra et al., 2015). The reduction in miR-193b resulted in an increased expression of its target urokinase-type plasminogen activator, a known tumor-associated protease. These changes correlated with the invasion and proliferation of the cancer cells. Another study showed that a copy number gain of miR-569 led to a loss of TP53INP1, which contributed to the proliferation and survival of ovarian epithelial cancer cells (Chaluvally-Raghavan et al., 2014). A small number of long noncoding RNAs have also been associated with ovarian cancer (Ren et al., 2015). However, it is not clear whether these long noncoding RNAs have a function in the cancer phenotype or what their mechanism of action is.
Mesenchymal stem cells are multipotent adherent cells that incorporate into the stroma of solid tumors (Karnoub et al., 2007; Klopp et al., 2007; Studeny et al., 2004). These cells contribute to the proliferation, chemoresistance, infiltration, and metastasis of ovarian cancer cells (Bianco et al., 2008), and they are able to mobilize in the circulation of ovarian cancer patients (Roodhart et al., 2008). Mesenchymal stem cells also have an immunosuppressive effect on T lymphocytes, inhibit apoptosis, stimulate angiogenesis, recruit and regulate the proliferation of cancer stem cells, and attenuate oxidative stress (Le Blanc and Ringden, 2007; Strioga et al., 2012). Cytokine secretion by ovarian cancer cells results in the mesenchymal stem cell infiltration of the tumor stromal environment (Wels et al., 2008). Expression profiles change with an increase in secretion of paracrine factors that stimulates motility and metastatic abilities of tumor cells as well as pro-angiogenic molecules (Karnoub et al., 2007). In vitro co-culture
experiments have shown that mesenchymal stem cells trigger expression differences in ovarian cancer cells, leading to metastatic characteristics such as adherence, invasion, and migration (Lis et al., 2012).
DEVELOPMENT OF EXPERIMENTAL MODEL SYSTEMS
Because of the extreme heterogeneity of ovarian cancers, it is difficult to design a single system to replicate the myriad human manifestations of the disease. Furthermore, many experimental models may not accurately represent the disease pathogenesis (e.g., assuming origination in the ovaries). However, research efforts have led to the development of a number of newer in vitro and in vivo models that may assist in the development of new strategies for the prevention, early detection, and treatment of ovarian cancers.
Cell Culture Models
Cancer cell lines are the most commonly used models in cancer research, and their use has advanced the understanding of cancer biology. However, because of the heterogeneity and the distinct molecular features of different ovarian carcinomas, not all ovarian cancer cell lines are representative of all ovarian cancers (see Table 2-2). One study analyzed a panel of commonly used ovarian cancer cell lines and found significant genetic differences between them and HGSC tumor samples from women (Domcke et al., 2013). Furthermore, the genetics of several rarely used cell lines were found to more closely resemble the genetics of the primary tumors than of some of the more commonly used cell lines found in the literature. Another study found that several commonly used cell lines were incorrectly classified with respect to their tumor subtype (Anglesio et al., 2013b). Because cancer cell lines are often used to identify and test new biomarkers and drug targets, cell lines need to be properly validated (IOM, 2012). Recently, a collection of 25 rigorously validated ovarian cancer cell lines was reported to represent the tumor types from which they are derived (Ince et al., 2015).
Researchers study ovarian cancer by growing cancer cells obtained from ovarian cancer patients in culture dishes. Cancer cells are typically grown in a single layer and spread across the cell culture dish. However, newer three-dimensional cell culture systems are becoming more widely used because researchers can introduce various components (e.g., adipocytes, immune cells, and endothelial cells) in order to reconstruct the tumor microenvironment (White et al., 2014). Three-dimensional culture systems also produce a histologic morphology reminiscent of the tumor from which they are derived. In addition, as compared with monolayer cell cultures, three-dimensional culture systems more accurately depict cell prolifera-
TABLE 2-2
Representative Ovarian Carcinoma Cell Lines
| Histologic Subtype | Molecular Features | Cell Lines |
| HGSC | Near universal somatic TP53 mutations, high-frequency BRCA1 and BRCA2 alterations, extensive genomic instability, common MYC and CCNE1 amplifications | CAOV3*, CAOV4*, COV318*, COV362*, COV504*, JHOS2, JHOS4, Kuramochi*, OAW28*, OVCAR3*, OVCAR4*, OVCAR5, OVKATE, OVSAHO, PEA1, PEA2, PEO1, PEO4, PEO14, PEO23, SNU119 |
| EC | Few TP53, BRCA1, BRCA2 mutations, frequent ARID1A mutations, common PTEN, PIK3CA, and CTNNB1 mutations or loss | A2780*, SKOV3*, TOV112D* |
| CCC | Few TP53, BRCA1, BRCA2 mutations, frequent ARID1A, PIK3CA, and PTEN mutations or loss, high expression of HNF1B | 2008, JHOC-5, JHOC-7, JHOC-9, OVMANA*, OVTOKO*, RMG-2 |
| MC | Some TP53 mutations, few BRCA1, BRCA2 mutations, frequent KRAS mutations, some ERBB2 amplification | COV644, MCAS* |
| Other | Mixed features of more than one subtype, features precluding classification, or conflicting classification by referenced sources | ES2*, IGROV1*, OV90*, OVCAR8*, TOV21G* |
NOTE: Asterisk indicates classification supported by more than one reference.
SOURCES: Anglesio et al., 2013b; Beaufort et al., 2014; Domcke et al., 2013; Ince et al., 2015.
tion, drug response, phenotypic heterogeneity, and changes in gene expression and cell behavior. A human fallopian tube co-culture system of both secretory and ciliated cells was developed from primary human surgical specimens (Fotheringham et al., 2011; Levanon et al., 2010). This model mimics the properties of tubal epithelium in situ, including morphological and immune-phenotypic properties. A similar model was developed using oviduct epithelium (equivalent to human FTE) from pigs (Miessen et al., 2011). Recently, even more complex in vitro model systems called human tissue chips have been developed (Benam et al., 2015). These models incorporate the tissue structure and physiology to provide a more accurate setting in order to better evaluate new therapies and disease mechanisms. These robust models more accurately represent the human disease. However, more models are needed to incorporate other important factors in ovarian cancer, such as the endocrine system.
Animal Models3
Genetically engineered mouse models (GEMMs) that replicate the morphological and biological features of a particular histologic type of ovarian cancer are useful for studying tumor biology and for the preclinical testing of new strategies for prevention, early detection, and treatment of ovarian cancer. A number of GEMMs developed over the past several years are outlined in Table 2-3; no GEMM of mucinous ovarian carcinoma has yet been reported.
Developing an ovarian cancer GEMM requires being able to modify the genes of interest in the cells of interest. The genes of interest are usually selected because they are characteristically mutated in a particular subtype of ovarian cancer (e.g., activating mutations of Pik3ca in ECs or CCCs, and inactivating mutations of Trp53 and Brca1 and Brca2 in HGSCs). Cells of interest include OSE and FTE.
Historically, two general approaches have been used to modify the genes of interest in the development of ovarian cancer GEMMs: (1) the expression of viral proteins (e.g., SV40 Large T-Antigen) to inactivate certain tumor suppressor proteins through direct protein–protein interactions, and (2) Cre-lox technology, in which the genes of interest are engineered to carry recognition sequences (loxP sites) for a bacteriophage enzyme called Cre-recombinase (Cre). When Cre is expressed in the desired cells, Cre-mediated recombination (i.e., the excision of DNA between two loxP sites) results in the inactivation of specific tumor-suppressor genes or the activation of specific oncogenes. Some model systems are further modified in ways that allow investigators to control the timing as well as the location of Cre expression. A third approach involves infecting p53-deficient mouse OSE cells engineered to express the avian receptor TVA with retroviruses that express various oncogenes (Orsulic et al., 2002; Xing and Orsulic, 2005, 2006).
Most of the early ovarian cancer GEMMs were based on transforming the OSE using recombinant adenovirus to express Cre-recombinase in OSE harboring engineered tumor suppressor gene and oncogene alleles. Some of these models may also inadvertently target the FTE. However, because of the new focus on the FTE as the cell of origin of many HGSCs, some of the more recent GEMMs are based on transforming the mouse oviductal epithelium (equivalent to human FTE) using the promoters of genes expressed in the FTE to drive Cre-mediated recombination of engineered tumor suppressor and oncogene alleles. Although the oviductal models of HGSC represent a significant advance, some shortcomings remain. For example, the
________________
3 Gene symbols for mice are italicized, with only the first letter in uppercase and the remaining letters in lowercase to differentiate from human genes.
Pax8 promoter drives Cre expression in other Müllerian epithelia besides the FTE (e.g., endometrium), and Ovgp1-driven expression of SV40 T-Ag does not mimic the actual genetic alterations known to play a role in HGSC pathogenesis. Furthermore, it is not clear whether any of the models faithfully reproduce the earliest events in ovarian tumorigenesis, as all of them rely on a simultaneous mutation of more than one tumor suppressor gene or oncogene, which is unlikely in humans.
While GEMMs provide a good system to examine gene functions and disease progression, patient-derived xenografts (PDXs) provide a good model system to test drug efficacy (Hasan et al., 2015). PDXs are able to recapitulate aspects of tumors found in women because they are directly transferred from the patient into the mouse. This allows the tumor in the mouse to have properties and cell proportions that are similar to those of the original tumor (Monsma et al., 2012). Therefore, PDX mice are clearly useful for clinical and co-clinical trials because they allow drugs to be tested based on a patient’s specific tumor type.
The adult laying hen is also recognized as a relevant model for human ovarian cancer, because ovarian tumors arise spontaneously in approximately 40 percent of hens around 4 years of age (King and Burdette, 2011). Ovarian tumors in hens can exhibit serous, endometrioid, mucinous, and clear cell histopathological features and express genes similar to those observed in human and mouse carcinomas including CA125; they can harbor TP53 mutations; and they can overexpress the epidermal growth factor receptor. Ovarian cancer in hens metastasizes to similar tissues with an accumulation of ascites fluid, as occurs in humans. Because of the spontaneous formation and heterogeneity of the ovarian cancers, hens provide another model system with which to study the progression of ovarian cancer and to test novel drugs in vivo for treating it.
The heterogeneity of ovarian cancers presents unique challenges to studying and treating ovarian cancer. The committee offers the following findings and conclusions:
- Ovarian cancer is not a single disease, and better diagnostic criteria, nomenclature, and classification are needed to standardize research and treatment.
- More research is needed to determine the sites of origin and the pathogenesis of each subtype because current evidence suggests that only a fraction of ovarian carcinomas originate in the ovaries.
- More research is needed to better understand the multitude of genetic alterations that characterize ovarian cancers in order to help
TABLE 2-3
Genetically Engineered Mouse Models
| Altered Genesa | Delivery Method | Targeted Cell Population(s) | Comments | References |
| HIGH-GRADE SEROUS CARCINOMA MODELS | ||||
| Brca1; Brca2; Trp53; Pten | Pax8 | FTE and endometrium | Mice also develop endometrial hyperplasia and adenocarcinoma | Perets et al., 2013 |
| Brca1; Brca2; Trp53, RbT121 | AdCre into bursa | OSE | TgK18GT121 mice allow Cre-mediated expression of the N-terminal domain of SV40 TAg in the OSE to interfere with all three Rb pocket proteins (pRB, p107, and p130) | Szabova et al., 2012 |
| Pten; Dicer ± Trp53 | MISIIR(Amhr2)-Cre | FTE and OSE | Tumors with Pten and Dicer inactivation initiate in fallopian tube stroma. Tumors with Pten, Dicer, and Trp53 inactivation form tumors in tube and ovary (ovary tumors form even if tubes are removed) | Kim et al., 2012, 2015 |
| Pten; Pik3caH1047R | AdCre into bursa | OSE | Mice also form granulosa tumors | Kinross et al., 2012 |
| SV40 T-Antigen | AdCre into bursa | OSE | TgCAG-LS-Tag allows Cre-mediated expression of SV40 Tag in the OSE. CAG is the CMV early enhancer/chicken β-actin promoter. Tumors are very poorly differentiated; treatment with estradiol decreased survival time and induced papillary architecture | Laviolette et al., 2010 |
| SV40 T-Antigen | MISIIR(Amhr2)-TAg | OSE | Female mice expressing the transgene are infertile; a subset of male transgenic mice develop Sertoli tumors | Connolly et al., 2003 |
| Trp53; Rb1 | AdCre into bursa | OSE | Other groups who used a similar approach to inactivate Trp53 and Brca1 (Quinn et al., 2009) or Trp53, Rb1 and Brca1 (Clark-Knowles et al., 2009) found that mice develop leiomyosarcomas rather than HGSCs | Flesken-Nikitin et al., 2003, 2013 |
| SV40 T-Antigen | Ovgp1-TAg | FTE | The tumor phenotype in this model is described in greater detail by Sherman-Baust et al. (2014) | Miyoshi et al., 2002; Sherman-Baust et al., 2014 |
| Altered Genesa | Delivery Method | Targeted Cell Population(s) | Comments | References |
| LOW-GRADE SEROUS CARCINOMA MODELS | ||||
| Pten; KrasG12D | MISIIR(Amhr2)-Cre | OSE | Fan et al., 2009 | |
| Pten; KrasG12D | MISIIR(Amhr2)-Cre | OSE | Mullany et al., 2011 | |
| ENDOMETRIOID CARCINOMA MODELS | ||||
| Pten; Arid1a | AdCre into bursa | OSE | Endometrioid and/or undifferentiated carcinomas arise in ~60 percent of mice within 6 months and in ~80 percent of mice within 8–9 months | Guan et al., 2014 |
| Pten; Apc ± Pik3caE545Kor Trp53 | AdCre into bursa | OSE | Tumor phenotype is more aggressive with mutant Pik3ca or Trp53 | Wu et al., 2013 |
| Pten; Apc ± Arid1a | AdCre into bursa | OSE | Tumor phenotype is less aggressive and tumors display more epithelial differentiation with Arid1a loss | Zhai et al., 2015 |
| Pten; Ctnnb1—ex3 | MISIIR(Amhr2)-Cre | OSE | Tumors are poorly differentiated | Tanwar et al., 2011 |
| Pten; Apc | AdCre into bursa | OSE | Tumors are poorly differentiated with prominent spindle component | Wu et al., 2007 |
| Pten; KrasG12D | AdCre into bursa | OSE | Kras activation alone induces endometriosis | Dinulescu et al., 2005 |
| CLEAR CELL CARCINOMA MODELS | ||||
| Pik3caH1047R; Arid1a | AdCre into bursa | OSE | Activation of Pik3ca results in OSE hyperplasia; Pik3ca activation and Arid1a inactivation results in CCC | Chandler et al., 2015 |
aGene symbols for mice are italicized, with only the first letter in uppercase and the remaining letters in lowercase to differentiate from human genes.
-
identify and prioritize promising candidates to develop for screening, prevention, and treatment methods.
- Because many ovarian cancers become drug resistant, further biological investigations are required to explore the mechanisms of drug resistance and to identify new drug targets.
- Because of the interplay between the tumor microenvironment and ovarian tumor cells, it will be important to develop treatment strategies that extend beyond targeting the tumor cell itself, with attention being placed on components within the tumor microenvironment (e.g., immune therapy and angiogenesis inhibitors).
- New cancer research models for preclinical studies need to take into account the variability among the ovarian cancer subtypes, the heterogeneity within ovarian cancer subtypes, and the different origins of these subtypes.
Abelson, S., Y. Shamai, L. Berger, K. Skorecki, and M. Tzukerman. 2013. Niche-dependent gene expression profile of intratumoral heterogeneous ovarian cancer stem cell populations. PLoS ONE 8(12):e83651.
Ahmed, A. A., D. Etemadmoghadam, J. Temple, A. G. Lynch, M. Riad, R. Sharma, C. Stewart, S. Fereday, C. Caldas, A. Defazio, D. Bowtell, and J. D. Brenton. 2010. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. Journal of Pathology 221(1):49-56.
Ahmed, N., K. T. Oliva, G. Barker, P. Hoffmann, S. Reeve, I. A. Smith, M. A. Quinn, and G. E. Rice. 2005. Proteomic tracking of serum protein isoforms as screening biomarkers of ovarian cancer. Proteomics 5(17):4625-4636.
Anglesio, M. S., M. S. Carey, M. Kobel, H. Mackay, and D. G. Huntsman. 2011. Clear cell carcinoma of the ovary: A report from the first Ovarian Clear Cell Symposium, June 24, 2010. Gynecologic Oncology 121(2):407-415.
Anglesio, M. S., S. Kommoss, M. C. Tolcher, B. Clarke, L. Galletta, et al. 2013a. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. Journal of Pathology 229(1):111-120.
Anglesio, M. S., K. C. Wiegand, N. Melnyk, C. Chow, C. Salamanca, L. M. Prentice, J. Senz, W. Yang, M. A. Spillman, D. R. Cochrane, K. Shumansky, S. P. Shah, S. E. Kalloger, and D. G. Huntsman. 2013b. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 8(9):e72162.
Asotra, S., J. Sharma, and N. Sharma. 2009. Metastatic ovarian tumor. Journal of Cytology 26(4):144-145.
Auersperg, N. 2013. Ovarian surface epithelium as a source of ovarian cancers: Unwarranted speculation or evidence-based hypothesis? Gynecologic Oncology 130(1):246-251.
Balkwill, F., K. A. Charles, and A. Mantovani. 2005. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7(3):211-217.
Barcellos-Hoff, M. H., D. Lyden, and T. C. Wang. 2013. The evolution of the cancer niche during multistage carcinogenesis. Nature Reviews: Cancer 13(7):511-518.
Barrett, C. L., C. DeBoever, K. Jepsen, C. C. Saenz, D. A. Carson, and K. A. Frazer. 2015. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proceedings of the National Academy of Sciences of the United States of America 112(23):E3050-E3057.
Bashashati, A., G. Ha, A. Tone, J. Ding, L. M. Prentice, et al. 2013. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. Journal of Pathology 231(1):21-34.
Beaufort, C. M., J. C. Helmijr, A. M. Piskorz, M. Hoogstraat, K. Ruigrok-Ritstier, N. Besselink, M. Murtaza, I. W. F. van, A. A. Heine, M. Smid, M. J. Koudijs, J. D. Brenton, E. M. Berns, and J. Helleman. 2014. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 9(9):e103988.
Belanger, M. H., L. Dolman, S. L. Arcand, Z. Shen, G. Chong, A. M. Mes-Masson, D. Provencher, and P. N. Tonin. 2015. A targeted analysis identifies a high frequency of BRCA1 and BRCA2 mutation carriers in women with ovarian cancer from a founder population. Journal of Ovarian Research 8(1):1.
Bell, D. A. 2014. Low-grade serous tumors of ovary. International Journal of Gynecological Pathology 33(4):348-356.
Benam, K. H., S. Dauth, B. Hassell, A. Herland, A. Jain, K. J. Jang, K. Karalis, H. J. Kim, L. MacQueen, R. Mahmoodian, S. Musah, Y. S. Torisawa, A. D. van der Meer, R. Villenave, M. Yadid, K. K. Parker, and D. E. Ingber. 2015. Engineered in vitro disease models. Annual Review of Pathology 10:195-262.
Berry, N. B., and S. A. Bapat. 2008. Ovarian cancer plasticity and epigenomics in the acquisition of a stem-like phenotype. Journal of Ovarian Research 1:8.
Bianco, P., P. G. Robey, and P. J. Simmons. 2008. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2(4):313-319.
Bissell, M. J., and W. C. Hines. 2011. Why don—t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nature Medicine 17(3):320-329.
Bodurka, D. C., M. T. Deavers, C. Tian, C. C. Sun, A. Malpica, R. L. Coleman, K. H. Lu, A. K. Sood, M. J. Birrer, R. Ozols, R. Baergen, R. E. Emerson, M. Steinhoff, B. Behmaram, G. Rasty, and D. M. Gershenson. 2012. Reclassification of serous ovarian carcinoma by a 2-tier system: A Gynecologic Oncology Group Study. Cancer 118(12):3087-3094.
Braicu, E. I., J. Sehouli, R. Richter, K. Pietzner, C. Denkert, and C. Fotopoulou. 2011. Role of histological type on surgical outcome and survival following radical primary tumour debulking of epithelial ovarian, fallopian tube and peritoneal cancers. British Journal of Cancer 105:1818-1824.
Brinton, L. A., L. C. Sakoda, M. E. Sherman, K. Frederiksen, S. K. Kjaer, B. I. Graubard, J. H. Olsen, and L. Mellemkjaer. 2005. Relationship of benign gynecologic diseases to subsequent risk of ovarian and uterine tumors. Cancer Epidemiology, Biomarkers and Prevention 14(12):2929-2935.
Brosens, I., and G. Benagiano. 2015. Perinatal origin of endometriosis revisited. Gynecological Endocrinology 31(6):419-421.
Butler, J. M., H. Kobayashi, and S. Rafii. 2010. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nature Reviews: Cancer 10(2):138-146.
Callahan, M. J., C. P. Crum, F. Medeiros, D. W. Kindelberger, J. A. Elvin, J. E. Garber, C. M. Feltmate, R. S. Berkowitz, and M. G. Muto. 2007. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. Journal of Clinical Oncology 25(25):3985-3990.
Campeau, P. M., W. D. Foulkes, and M. D. Tischkowitz. 2008. Hereditary breast cancer: New genetic developments, new therapeutic avenues. Human Genetics 124(1):31-42.
Cancer Genome Atlas Research Network. 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474(7353):609-615.
Castells, M., D. Milhas, C. Gandy, B. Thibault, A. Rafii, J. P. Delord, and B. Couderc. 2013. Microenvironment mesenchymal cells protect ovarian cancer cell lines from apoptosis by inhibiting XIAP inactivation. Cell Death & Disease 4:e887.
Catena, R., N. Bhattacharya, T. El Rayes, S. Wang, H. Choi, D. Gao, S. Ryu, N. Joshi, D. Bielenberg, S. B. Lee, S. A. Haukaas, K. Gravdal, O. J. Halvorsen, L. A. Akslen, R. S. Watnick, and V. Mittal. 2013. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discovery 3(5):578-589.
Chaluvally-Raghavan, P., F. Zhang, S. Pradeep, M. P. Hamilton, X. Zhao, et al. 2014. Copy number gain of hsa-miR-569 at 3q26.2 leads to loss of TP53INP1 and aggressiveness of epithelial cancers. Cancer Cell 26(6):863-879.
Chandler, R. L., J. S. Damrauer, J. R. Raab, J. C. Schisler, M. D. Wilkerson, J. P. Didion, J. Starmer, D. Serber, D. Yee, J. Xiong, D. B. Darr, F. Pardo-Manuel de Villena, W. Y. Kim, and T. Magnuson. 2015. Coexistent ARID1a-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nature Communications 6:6118.
Chen, F., X. Zhuang, L. Lin, P. Yu, Y. Wang, Y. Shi, G. Hu, and Y. Sun. 2015. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Medicine 13:45.
Cho, K. R., and I. M. Shih. 2009. Ovarian cancer. Annual Review of Pathology 4:287-313.
Clark-Knowles, K. V., M. K. Senterman, O. Collins, and B. C. Vanderhyden. 2009. Conditional inactivation of Brca1, p53 and Rb in mouse ovaries results in the development of leiomyosarcomas. PLoS ONE 4(12):e8534.
Connolly, D. C., R. Bao, A. Y. Nikitin, K. C. Stephens, T. W. Poole, X. Hua, S. S. Harris, B. C. Vanderhyden, and T. C. Hamilton. 2003. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Research 63(6):1389-1397.
Cooke, S. L., C. K. Ng, N. Melnyk, M. J. Garcia, T. Hardcastle, J. Temple, S. Langdon, D. Huntsman, and J. D. Brenton. 2010. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 29(35):4905-4913.
Cunnea, P., and E. A. Stronach. 2014. Modeling platinum sensitive and resistant high-grade serous ovarian cancer: Development and applications of experimental systems. Frontiers in Oncology 4:81.
Dehari, R., R. J. Kurman, S. Logani, and I. M. Shih. 2007. The development of high-grade serous carcinoma from atypical proliferative (borderline) serous tumors and low-grade micropapillary serous carcinoma: A morphologic and molecular genetic analysis. American Journal of Surgical Pathology 31(7):1007-1012.
Della Pepa, C., G. Tonini, D. Santini, S. Losito, C. Pisano, M. Di Napoli, S. C. Cecere, P. Gargiulo, and S. Pignata. 2015. Low grade serous ovarian carcinoma: From the molecular characterization to the best therapeutic strategy. Cancer Treatment Reviews 41(2):136-143.
DePriest, P. D., E. R. Banks, D. E. Powell, J. R. van Nagell, Jr., H. H. Gallion, L. E. Puls, J. E. Hunter, R. J. Kryscio, and M. B. Royalty. 1992. Endometrioid carcinoma of the ovary and endometriosis: The association in postmenopausal women. Gynecologic Oncology 47(1):71-75.
Dinulescu, D. M., T. A. Ince, B. J. Quade, S. A. Shafer, D. Crowley, and T. Jacks. 2005. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nature Medicine 11(1):63-70.
Domcke, S., R. Sinha, D. A. Levine, C. Sander, and N. Schultz. 2013. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature Communications 4:2126.
Easton, D. F. 1999. How many more breast cancer predisposition genes are there? Breast Cancer Research 1(1):14-17.
Edwards, S. L., R. Brough, C. J. Lord, R. Natrajan, R. Vatcheva, D. A. Levine, J. Boyd, J. S. Reis-Filho, and A. Ashworth. 2008. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451(7182):1111-1115.
Eitan, R., M. Kushnir, G. Lithwick-Yanai, M. B. David, M. Hoshen, M. Glezerman, M. Hod, G. Sabah, S. Rosenwald, and H. Levavi. 2009. Tumor microRNA expression patterns associated with resistance to platinum-based chemotherapy and survival in ovarian cancer patients. Gynecologic Oncology 114(2):253-259.
Emori, M. M., and R. Drapkin. 2014. The hormonal composition of follicular fluid and its implications for ovarian cancer pathogenesis. Reproductive Biology and Endocrinology 12:60.
Erzen, M., S. Rakar, B. Klancnik, K. Syrjanen, and B. Klancar. 2001. Endometriosis-associated ovarian carcinoma (EAOC): An entity distinct from other ovarian carcinomas as suggested by a nested case-control study. Gynecologic Oncology 83(1):100-108.
Etemadmoghadam, D., B. A. Weir, G. Au-Yeung, K. Alsop, G. Mitchell, J. George, Australian Ovarian Cancer Study Group, S. Davis, A. D. D—Andrea, K. Simpson, W. C. Hahn, and D. D. Bowtell. 2013. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proceedings of the National Academy of Sciences of the United States of America 110(48):19489-19494.
Fan, H. Y., Z. Liu, M. Paquet, J. Wang, J. P. Lydon, F. J. DeMayo, and J. S. Richards. 2009. Cell type-specific targeted mutations of Kras and Pten document proliferation arrest in granulosa cells versus oncogenic insult to ovarian surface epithelial cells. Cancer Research 69(16):6463-6472.
Ffrench, B., C. Gasch, J. J. O—Leary, and M. F. Gallagher. 2014. Developing ovarian cancer stem cell models: Laying the pipeline from discovery to clinical intervention. Molecular Cancer 13:262.
Fidler, I. J. 2003. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nature Reviews: Cancer 3(6):453-458.
Flesken-Nikitin, A., K. C. Choi, J. P. Eng, E. N. Shmidt, and A. Y. Nikitin. 2003. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Research 63(13):3459-3463.
Flesken-Nikitin, A., C. I. Hwang, C. Y. Cheng, T. V. Michurina, G. Enikolopov, and A. Y. Nikitin. 2013. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 495(7440):241-245.
Folkman, J. 2006. Angiogenesis. Annual Review of Medicine 57:1-18.
Forte, A., M. Cipollaro, and U. Galderisi. 2014. Genetic, epigenetic and stem cell alterations in endometriosis: New insights and potential therapeutic perspectives. Clinical Science (London, England: 1979) 126(2):123-138.
Fotheringham, S., K. Levanon, and R. Drapkin. 2011. Ex vivo culture of primary human fallopian tube epithelial cells. Journal of Visualized Experiments 51:e2728.
Fujii, K., Y. Yamashita, T. Yamamoto, K. Takahashi, K. Hashimoto, T. Miyata, K. Kawai, F. Kikkawa, S. Toyokuni, and T. Nagasaka. 2014. Ovarian mucinous tumors arising from mature cystic teratomas—A molecular genetic approach for understanding the cellular origin. Human Pathology 45(4):717-724.
Fukunaga, M., K. Nomura, E. Ishikawa, and S. Ushigome. 1997. Ovarian atypical endometriosis: Its close association with malignant epithelial tumours. Histopathology 30(3):249-255.
Garg, K., D. A. Levine, N. Olvera, F. Dao, M. Bisogna, A. A. Secord, A. Berchuck, E. Cerami, N. Schultz, and R. A. Soslow. 2013. BRCA1 immunohistochemistry in a molecularly characterized cohort of ovarian high-grade serous carcinomas. American Journal of Surgical Pathology 37(1):138-146.
Garrett, A. P., K. R. Lee, C. R. Colitti, M. G. Muto, R. S. Berkowitz, and S. C. Mok. 2001. k-ras mutation may be an early event in mucinous ovarian tumorigenesis. International Journal of Gynecological Pathology 20(3):244-251.
Gehlenborg, N., S. I. O—Donoghue, N. S. Baliga, A. Goesmann, M. A. Hibbs, H. Kitano, O. Kohlbacher, H. Neuweger, R. Schneider, D. Tenenbaum, and A. C. Gavin. 2010. Visualization of omics data for systems biology. Nature Methods 7(3 Suppl):S56-S68.
Gemingani, M. L., A. C. Schlaerth, F. Bogomolniy, R. R. Barakat, O. Lin, R. Soslow, E. Venkatraman, and J. Boyd. 2003. Role of KRAS and BRAF gene mutations in mucinous ovarian carcinoma. Gynecologic Oncology 90(2):378-381.
Gershenson, D. M., D. C. Bodurka, K. H. Lu, L. C. Nathan, L. Milojevic, K. K. Wong, A. Malpica, and C. C. Sun. 2015. Impact of age and primary disease site on outcome in women with low-grade serous carcinoma of the ovary or peritoneum: Results of a large single-institution registry of a rare tumor. Journal of Clinical Oncology 33(24):2675-2682.
Gilks, C. B., D. N. Ionescu, S. E. Kalloger, M. Kobel, J. Irving, B. Clarke, J. Santos, N. Le, V. Moravan, K. Swenerton, and Cheryl Brown Ovarian Cancer Outcomes Unit of the British Columbia Cancer Agency. 2008. Tumor cell type can be reproducibly diagnosed and is of independent prognostic significance in patients with maximally debulked ovarian carcinoma. Human Pathology 39(8):1239-1251.
Gourley, C., J. Farley, D. M. Provencher, S. Pignata, L. Mileshkin, P. Harter, J. Maenpaa, J. W. Kim, E. Pujaide-Lauraine, R. M. Glasspool, I. Ray-Coquard, and D. Gershenson. 2014. Gynecologic Cancer InterGroup (GCIG) consensus review for ovarian and primary peritoneal low-grade serous carcinomas. International Journal of Gynecological Cancer 24(9 Suppl 3):S9-S13.
Granot, Z., E. Henke, E. A. Comen, T. A. King, L. Norton, and R. Benezra. 2011. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20(3):300-314.
Guan, B., Y. S. Rahmanto, R. C. Wu, Y. Wang, Z. Wang, T. L. Wang, and I. M. Shih. 2014. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. Journal of the National Cancer Institute 106(7):dju146.
Hagemann, T., J. Wilson, F. Burke, H. Kulbe, N. F. Li, A. Pluddemann, K. Charles, S. Gordon, and F. R. Balkwill. 2006. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. Journal of Immunology 176(8):5023-5032.
Hanahan, D., and L. M. Coussens. 2012. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3):309-322.
Hanahan, D., and R. A. Weinberg. 2011. Hallmarks of cancer: The next generation. Cell 144(5):646-674.
Hart, W. R. 2005. Mucinous tumors of the ovary: A review. International Journal of Gynecological Pathology 24(1):4-25.
Hasan, N., A. W. Ohman, and D. M. Dinulescu. 2015. The promise and challenge of ovarian cancer models. Translational Cancer Research 4(1):14-28.
Hennessy, B. T., K. M. Timms, M. S. Carey, A. Gutin, L. A. Meyer, D. D. Flake, 2nd, V. Abkevich, J. Potter, D. Pruss, P. Glenn, Y. Li, J. Li, A. M. Gonzalez-Angulo, K. S. McCune, M. Markman, R. R. Broaddus, J. S. Lanchbury, K. H. Lu, and G. B. Mills. 2010. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. Journal of Clinical Oncology 28(22):3570-3576.
Huang, R. L., F. Gu, N. B. Kirma, J. Ruan, C. L. Chen, H. C. Wang, Y. P. Liao, C. C. Chang, M. H. Yu, J. M. Pilrose, I. M. Thompson, H. C. Huang, T. H. Huang, H. C. Lai, and K. P. Nephew. 2013. Comprehensive methylome analysis of ovarian tumors reveals hedgehog signaling pathway regulators as prognostic DNA methylation biomarkers. Epigenetics 8(6):624-634.
Ince, T. A., A. D. Sousa, M. A. Jones, J. C. Harrell, E. S. Agoston, et al. 2015. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nature Communications 6:7419.
IOM (Institute of Medicine). 2012. Evolution of translational omics: Lessons learned and the path forward. Washington, DC: The National Academies Press.
Jenison, E. L., A. G. Montag, C. T. Griffiths, W. R. Welch, P. T. Lavin, J. Greer, and R. C. Knapp. 1989. Clear cell adenocarcinoma of the ovary: A clinical analysis and comparison with serous carcinoma. Gynecologic Oncology 32(1):65-71.
Jones, S., T. L. Wang, I. M. Shih, T. L. Mao, K. Nakayama, R. Roden, R. Glas, D. Slamon, L. A. Diaz, Jr., B. Vogelstein, K. W. Kinzler, V. E. Velculescu, and N. Papadopoulos. 2010. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330(6001):228-231.
Junttila, M. R., and F. J. de Sauvage. 2013. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501(7467):346-354.
Karin, M., and F. R. Greten. 2005. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nature Reviews: Immunology 5(10):749-759.
Karnoub, A. E., A. B. Dash, A. P. Vo, A. Sullivan, M. W. Brooks, G. W. Bell, A. L. Richardson, K. Polyak, R. Tubo, and R. A. Weinberg. 2007. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449(7162):557-563.
Kashiwagi, S., Y. Izumi, T. Gohongi, Z. N. Demou, L. Xu, P. L. Huang, D. G. Buerk, L. L. Munn, R. K. Jain, and D. Fukumura. 2005. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. Journal of Clinical Investigation 115(7):1816-1827.
Kawamura, K., Y. Komohara, K. Takaishi, H. Katabuchi, and M. Takeya. 2009. Detection of M2 macrophages and colony-stimulating factor 1 expression in serous and mucinous ovarian epithelial tumors. Pathology International 59(5):300-305.
Kenny, P. A., G. Y. Lee, and M. J. Bissell. 2007. Targeting the tumor microenvironment. Frontiers in Bioscience 12:3468-3474.
Kerr, S. E., A. B. Flotte, M. J. McFalls, J. A. Vrana, K. C. Halling, and D. A. Bell. 2013. Matching maternal isodisomy in mucinous carcinomas and associated ovarian teratomas provides evidence of germ cell derivation for some mucinous ovarian tumors. American Journal of Surgical Pathology 37(8):1229-1235.
Kim, J., D. M. Coffey, C. J. Creighton, Z. Yu, S. M. Hawkins, and M. M. Matzuk. 2012. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proceedings of the National Academy of Sciences of the United States of America 109(10):3921-3926.
Kim, J., D. M. Coffey, L. Ma, and M. M. Matzuk. 2015. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 156(6):1975-1981.
Kinde, I., C. Bettegowda, Y. Wang, J. Wu, N. Agrawal, M. Shih Ie, R. Kurman, F. Dao, D. A. Levine, R. Giuntoli, R. Roden, J. R. Eshleman, J. P. Carvalho, S. K. Marie, N. Papadopoulos, K. W. Kinzler, B. Vogelstein, and L. A. Diaz, Jr. 2013. Evaluation of DNA from the papanicolaou test to detect ovarian and endometrial cancers. Science Translational Medicine 5(167):167ra4.
Kindelberger, D. W., Y. Lee, A. Miron, M. S. Hirsch, C. Feltmate, F. Medeiros, M. J. Callahan, E. O. Garner, R. W. Gordon, C. Birch, R. S. Berkowitz, M. G. Muto, and C. P. Crum. 2007. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. American Journal of Surgical Pathology 31(2):161-169.
King, M. C., J. H. Marks, J. B. Mandell, and New York Breast Cancer Study. 2003. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302(5645):643-646.
King, S. M., and J. E. Burdette. 2011. Evaluating the progenitor cells of ovarian cancer: Analysis of current animal models. BMB Reports 44(7):435-445.
Kinross, K. M., K. G. Montgomery, M. Kleinschmidt, P. Waring, I. Ivetac, et al. 2012. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. Journal of Clinical Investigation 122(2):553-557.
Klopp, A. H., E. L. Spaeth, J. L. Dembinski, W. A. Woodward, A. Munshi, R. E. Meyn, J. D. Cox, M. Andreeff, and F. C. Marini. 2007. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Research 67(24):11687-11695.
Kuchenbaecker, K. B., S. J. Ramus, J. Tyrer, A. Lee, H. C. Shen, et al. 2015. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nature Genetics 47(2):164-171.
Kuhn, E., R. J. Kurman, and I. M. Shih. 2012. Ovarian cancer is an imported disease: Fact or fiction? Current Obstetrics and Gynecology Reports 1(1):1-9.
Kurman, R. J. 2013. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Annals of Oncology 24(Suppl 10):x16-x21.
Kurman, R. J., and I. M. Shih. 2010. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. American Journal of Surgical Pathology 34(3):433-443.
Kurman, R. J., R. Vang, J. Junge, C. G. Hannibal, S. K. Kjaer, and I. M. Shih. 2011. Papillary tubal hyperplasia: The putative precursor of ovarian atypical proliferative (borderline) serous tumors, noninvasive implants, and endosalpingiosis. American Journal of Surgical Pathology 35(11):1605-1614.
Laviolette, L. A., K. Garson, E. A. Macdonald, M. K. Senterman, K. Courville, C. A. Crane, and B. C. Vanderhyden. 2010. 17beta-estradiol accelerates tumor onset and decreases survival in a transgenic mouse model of ovarian cancer. Endocrinology 151(3):929-938.
Le Blanc, K., and O. Ringden. 2007. Immunomodulation by mesenchymal stem cells and clinical experience. Journal of Internal Medicine 262(5):509-525.
Ledermann, J. A., D. Luvero, A. Shafer, D. O—Connor, G. Mangili, M. Friedlander, J. Pfisterer, M. R. Mirza, J. W. Kim, J. Alexandre, A. Oza, and J. Brown. 2014. Gynecologic Cancer InterGroup (GCIG) consensus review for mucinous ovarian carcinoma. International Journal of Gynecological Cancer 24(9 Suppl 3):S14-S19.
Lee, K. R., and R. H. Young. 2003. The distinction between primary and metastatic mucinous carcinomas of the ovary: Gross and histologic findings in 50 cases. American Journal of Surgical Pathology 27(3):281-292.
Lee, Y., F. Medeiros, D. Kindelberger, M. J. Callahan, M. G. Muto, and C. P. Crum. 2006. Advances in the recognition of tubal intraepithelial carcinoma: Applications to cancer screening and the pathogenesis of ovarian cancer. Advances in Anatomic Pathology 13(1):1-7.
Lee, Y., A. Miron, R. Drapkin, M. R. Nucci, F. Medeiros, A. Saleemuddin, J. Garber, C. Birch, H. Mou, R. W. Gordon, D. W. Cramer, F. D. McKeon, and C. P. Crum. 2007. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. Journal of Pathology 211(1):26-35.
Levanon, K., V. Ng, H. Y. Piao, Y. Zhang, M. C. Chang, M. H. Roh, D. W. Kindelberger, M. S. Hirsch, C. P. Crum, J. A. Marto, and R. Drapkin. 2010. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene 29(8):1103-1113.
Li, H., K. B. Chiappinelli, A. A. Guzzetta, H. Easwaran, R. W. Yen, R. Vatapalli, M. J. Topper, J. Luo, R. M. Connolly, N. S. Azad, V. Stearns, D. M. Pardoll, N. Davidson, P. A. Jones, D. J. Slamon, S. B. Baylin, C. A. Zahnow, and N. Ahuja. 2014. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 5(3):587-598.
Lis, R., C. Touboul, C. M. Raynaud, J. A. Malek, K. Suhre, M. Mirshahi, and A. Rafii. 2012. Mesenchymal cell interaction with ovarian cancer cells triggers pro-metastatic properties. PLoS ONE 7(5):e38340.
Malpica, A., M. T. Deavers, K. Lu, D. C. Bodurka, E. N. Atkinson, D. M. Gershenson, and E. G. Silva. 2004. Grading ovarian serous carcinoma using a two-tier system. American Journal of Surgical Pathology 28(4):496-504.
Mantovani, A., and A. Sica. 2010. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Current Opinion in Immunology 22(2):231-237.
Marquez, R. T., K. A. Baggerly, A. P. Patterson, J. Liu, R. Broaddus, M. Frumovitz, E. N. Atkinson, D. I. Smith, L. Hartmann, D. Fishman, A. Berchuck, R. Whitaker, D. M. Gershenson, G. B. Mills, R. C. Bast, Jr., and K. H. Lu. 2005. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clinical Cancer Research 11(17):6116-6126.
Martins, F. C., I. Santiago, A. Trinh, J. Xian, A. Guo, K. Sayal, M. Jimenez-Linan, S. Deen, K. Driver, M. Mack, J. Aslop, P. D. Pharoah, F. Markowetz, and J. D. Brenton. 2014. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biology 15(12):526.
Matei, D., F. Fang, C. Shen, J. Schilder, A. Arnold, Y. Zeng, W. A. Berry, T. Huang, and K. P. Nephew. 2012. Epigenetic resensitization to platinum in ovarian cancer. Cancer Research 72(9):2197-2205.
Matsumoto, T., M. Yamazaki, H. Takahashi, S. Kajita, E. Suzuki, T. Tsuruta, and M. Saegusa. 2015. Distinct beta-catenin and PIK3CA mutation profiles in endometriosis-associated ovarian endometrioid and clear cell carcinomas. American Journal of Clinical Pathology 144(3):452-463.
McConechy, M. K., M. S. Anglesio, S. E. Kalloger, W. Yang, J. Senz, C. Chow, A. Heravi-Moussavi, G. B. Morin, A. M. Mes-Masson, Australian Ovarian Cancer Study Group, M. S. Carey, J. N. McAlpine, J. S. Kwon, L. M. Prentice, N. Boyd, S. P. Shah, C. B. Gilks, and D. G. Huntsman. 2011. Subtype-specific mutation of PPP2R1A in endometrial and ovarian carcinomas. Journal of Pathology 223(5):567-573.
McConechy, M. K., J. Ding, J. Senz, W. Yang, N. Melnyk, A. A. Tone, L. M. Prentice, K. C. Wiegand, J. N. McAlpine, S. P. Shah, C. H. Lee, P. J. Goodfellow, C. B. Gilks, and D. G. Huntsman. 2014. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Modern Pathology 27(1):128-134.
McDaniel, A. S., J. N. Stall, D. H. Hovelson, A. K. Cani, C. J. Liu, S. A. Tomlins, and K. R. Cho. 2015. Next-generation sequencing of tubal intraepithelial carcinomas. JAMA Oncology 1(8):1128-1132.
Merritt, M. A., and D. W. Cramer. 2010. Molecular pathogenesis of endometrial and ovarian cancer. Cancer Biomarkers 9(1-6):287-305.
Miessen, K., S. Sharbati, R. Einspanier, and J. Schoen. 2011. Modelling the porcine oviduct epithelium: A polarized in vitro system suitable for long-term cultivation. Theriogenology 76(5):900-910.
Miles, G. D., M. Seiler, L. Rodriguez, G. Rajagopal, and G. Bhanot. 2012. Identifying microRNA/mRNA dysregulations in ovarian cancer. BMC Research Notes 5:164.
Mishra, V. K., and S. A. Johnsen. 2014. Targeted therapy of epigenomic regulatory mechanisms controlling the epithelial to mesenchymal transition during tumor progression. Cell and Tissue Research 356(3):617-630.
Mitra, A. K., C. Y. Chiang, P. Tiwari, S. Tomar, K. M. Watters, M. E. Peter, and E. Lengyel. 2015. Microenvironment-induced downregulation of miR-193b drives ovarian cancer metastasis. Oncogene (Epub ahead of print).
Miyoshi, I., K. Takahashi, Y. Kon, T. Okamura, Y. Mototani, Y. Araki, and N. Kasai. 2002. Mouse transgenic for murine oviduct-specific glycoprotein promoter-driven simian virus 40 large T-antigen: Tumor formation and its hormonal regulation. Molecular Reproduction and Development 63(2):168-176.
Monsma, D. J., N. R. Monks, D. M. Cherba, D. Dylewski, E. Eugster, H. Jahn, S. Srikanth, S. B. Scott, P. J. Richardson, R. E. Everts, A. Ishkin, Y. Nikolsky, J. H. Resau, R. Sigler, B. J. Nickoloff, and C. P. Webb. 2012. Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. Journal of Translational Medicine 10:125.
Mroue, R., and M. J. Bissell. 2013. Three-dimensional cultures of mouse mammary epithelial cells. Methods in Molecular Biology 945:221-250.
Mullany, L. K., H. Y. Fan, Z. Liu, L. D. White, A. Marshall, P. Gunaratne, M. L. Anderson, C. J. Creighton, L. Xin, M. Deavers, K. K. Wong, and J. S. Richards. 2011. Molecular and functional characteristics of ovarian surface epithelial cells transformed by KrasG12D and loss of Pten in a mouse model in vivo. Oncogene 30(32):3522-3536.
Musrap, N., and E. P. Diamandis. 2012. Revisiting the complexity of the ovarian cancer microenvironment—Clinical implications for treatment strategies. Molecular Cancer Research 10(10):1254-1264.
Mutch, D. G., and J. Prat. 2014. 2014 FIGO staging for ovarian, fallopian tube and peritoneal cancer. Gynecologic Oncology 133(3):401-404.
Myers, E. R., L. J. Havrilesky, S. L. Kulasingam, G. D. Sanders, K. E. Cline, R. N. Gray, A. Berchuck, and D. C. McCrory. 2006. Genomic tests for ovarian cancer detection and management. Evidence Report/Technology Assessment 145:1-100.
Ng, A., and N. Barker. 2015. Ovary and fimbrial stem cells: Biology, niche and cancer origins. Nature Reviews: Molecular Cell Biology 16(10):625-638.
Nieman, K. M., H. A. Kenny, C. V. Penicka, A. Ladanyi, R. Buell-Gutbrod, M. R. Zillhardt, I. L. Romero, M. S. Carey, G. B. Mills, G. S. Hotamisligil, S. D. Yamada, M. E. Peter, K. Gwin, and E. Lengyel. 2011. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature Medicine 17(11):1498-1503.
Orsini, M., A. Travaglione, and E. Capobianco. 2013. Warehousing re-annotated cancer genes for biomarker meta-analysis. Computer Methods and Programs in Biomedicine 111(1):166-180.
Orsulic, S., Y. Li, R. A. Soslow, L. A. Vitale-Cross, J. S. Gutkind, and H. E. Varmus. 2002. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 1(1):53-62.
Paget, S. 1889. The distribution of secondary growths in cancer of the breast. Lancet 1: 571-573.
Pal, T., J. Permuth-Wey, J. A. Betts, J. P. Krischer, J. Fiorica, H. Arango, J. LaPolla, M. Hoffman, M. A. Martino, K. Wakeley, G. Wilbanks, S. Nicosia, A. Cantor, and R. Sutphen. 2005. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 104(12):2807-2816.
Parker, R. L., P. B. Clement, D. J. Chercover, T. Sornarajah, and C. B. Gilks. 2004. Early recurrence of ovarian serous borderline tumor as high-grade carcinoma: A report of two cases. International Journal of Gynecological Pathology 23(3):265-272.
Pavone, M. E., and B. M. Lyttle. 2015. Endometriosis and ovarian cancer: Links, risks, and challenges faced. International Journal of Women’s Health 7:663-672.
Pennington, K. P., T. Walsh, M. I. Harrell, M. K. Lee, C. C. Pennil, M. H. Rendi, A. Thornton, B. M. Norquist, S. Casadei, A. S. Nord, K. J. Agnew, C. C. Pritchard, S. Scroggins, R. L. Garcia, M. C. King, and E. M. Swisher. 2014. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clinical Cancer Research 20(3):764-775.
Perets, R., G. A. Wyant, K. W. Muto, J. G. Bijron, B. B. Poole, K. T. Chin, J. Y. Chen, A. W. Ohman, C. D. Stepule, S. Kwak, A. M. Karst, M. S. Hirsch, S. R. Setlur, C. P. Crum, D. M. Dinulescu, and R. Drapkin. 2013. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 24(6):751-765.
Pharoah, P. D., A. Antoniou, M. Bobrow, R. L. Zimmern, D. F. Easton, and B. A. Ponder. 2002. Polygenic susceptibility to breast cancer and implications for prevention. Nature Genetics 31(1):33-36.
Piek, J. M., P. J. van Diest, R. P. Zweemer, J. W. Jansen, R. J. Poort-Keesom, F. H. Menko, J. J. Gille, A. P. Jongsma, G. Pals, P. Kenemans, and R. H. Verheijen. 2001a. Dysplastic changes in prophylactically removed fallopian tubes of women predisposed to developing ovarian cancer. Journal of Pathology 195(4):451-456.
Piek, J. M., P. J. van Diest, R. P. Zweemer, P. Kenemans, and R. H. Verheijen. 2001b. Tubal ligation and risk of ovarian cancer. Lancet 358(9284):844.
Piek, J. M., R. H. Verheijen, P. Kenemans, L. F. Massuger, H. Bulten, and P. J. van Diest. 2003. BRCA1/2-related ovarian cancers are of tubal origin: A hypothesis. Gynecologic Oncology 90(2):491.
Pinato, D. J., J. Graham, H. Gabra, and R. Sharma. 2013. Evolving concepts in the management of drug resistant ovarian cancer: Dose dense chemotherapy and the reversal of clinical platinum resistance. Cancer Treatment Reviews 39(2):153-160.
Pradeep, S., S. W. Kim, S. Y. Wu, M. Nishimura, P. Chaluvally-Raghavan, et al. 2014. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 26(1):77-91.
Przybycin, C. G., R. J. Kurman, B. M. Ronnett, I. M. Shih, and R. Vang. 2010. Are all pelvic (nonuterine) serous carcinomas of tubal origin? American Journal of Surgical Pathology 34(10):1407-1416.
Quail, D. F., and J. A. Joyce. 2013. Microenvironmental regulation of tumor progression and metastasis. Nature Medicine 19(11):1423-1437.
Quinn, B. A., T. Brake, X. Hua, K. Baxter-Jones, S. Litwin, L. H. Ellenson, and D. C. Connolly. 2009. Induction of ovarian leiomyosarcomas in mice by conditional inactivation of BRCA1 and p53. PLoS ONE 4(12):e8404.
Rechsteiner, M., A. K. Zimmermann, P. J. Wild, R. Caduff, A. von Teichman, D. Fink, H. Moch, and A. Noske. 2013. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Experimental and Molecular Pathology 95(2):235-241.
Ren, Y., Y. Cui, X. Li, B. Wang, L. Na, J. Shi, L. Wang, L. Qiu, K. Zhang, G. Liu, and Y. Xu. 2015. A co-expression network analysis reveals lncRNA abnormalities in peripheral blood in early-onset schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry 63:1-5.
Riopel, M. A., B. M. Ronnett, and R. J. Kurman. 1999. Evaluation of diagnostic criteria and behavior of ovarian intestinal-type mucinous tumors: Atypical proliferative (borderline) tumors and intraepithelial, microinvasive, invasive, and metastatic carcinomas. American Journal of Surgical Pathology 23(6):617-635.
Robey, S. S., and E. G. Silva. 1989. Epithelial hyperplasia of the fallopian tube. Its association with serous borderline tumors of the ovary. International Journal of Gynecological Pathology 8(3):214-220.
Ronnett, B. M., B. M. Shmookler, P. H. Sugarbaker, and R. J. Kurman. 1997. Pseudomyxoma peritonei: New concepts in diagnosis, origin, nomenclature, and relationship to mucinous borderline (low malignant potential) tumors of the ovary. Anatomic Pathology 2:197-226.
Roodhart, J. M., M. H. Langenberg, E. Witteveen, and E. E. Voest. 2008. The molecular basis of class side effects due to treatment with inhibitors of the VEGF/VEGFR pathway. Current Clinical Pharmacology 3(2):132-143.
Rossing, M. A., K. L. Cushing-Haugen, K. G. Wicklund, J. A. Doherty, and N. S. Weiss. 2008. Risk of epithelial ovarian cancer in relation to benign ovarian conditions and ovarian surgery. Cancer Causes and Control 19(10):1357-1364.
Ryland, G. L., S. M. Hunter, M. A. Doyle, F. Caramia, J. Li, S. M. Rowley, M. Christie, P. E. Allan, A. N. Stephens, D. D. Bowtell, Australian Ovarian Cancer Study Group, I. G. Campbell, and K. L. Gorringe. 2015. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Medicine 7(1):87.
Sainz de la Cuesta, R., J. H. Eichhorn, L. W. Rice, A. F. Fuller, Jr., N. Nikrui, and B. A. Goff. 1996. Histologic transformation of benign endometriosis to early epithelial ovarian cancer. Gynecologic Oncology 60(2):238-244.
Sakai, W., E. M. Swisher, B. Y. Karlan, M. K. Agarwal, J. Higgins, C. Friedman, E. Villegas, C. Jacquemont, D. J. Farrugia, F. J. Couch, N. Urban, and T. Taniguchi. 2008. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451(7182):1116-1120.
Sala, E., M. Y. Kataoka, A. N. Priest, A. B. Gill, M. A. McLean, I. Joubert, M. J. Graves, R. A. Crawford, M. Jimenez-Linan, H. M. Earl, C. Hodgkin, J. R. Griffiths, D. J. Lomas, and J. D. Brenton. 2012. Advanced ovarian cancer: Multiparametric MR imaging demonstrates response- and metastasis-specific effects. Radiology 263(1):149-159.
Sampson, J. A. 1927. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. American Journal of Obstetrics and Gynecology 14:422-469.
Schwarz, R. F., C. K. Ng, S. L. Cooke, S. Newman, J. Temple, A. M. Piskorz, D. Gale, K. Sayal, M. Murtaza, P. J. Baldwin, N. Rosenfeld, H. M. Earl, E. Sala, M. Jimenez-Linan, C. A. Parkinson, F. Markowetz, and J. D. Brenton. 2015. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Medicine 12(2):e1001789.
Seidman, J. D., I. Horkayne-Szakaly, M. Haiba, C. R. Boice, R. J. Kurman, and B. M. Ronnett. 2004. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. International Journal of Gynecological Pathology 23(1):41-44.
Seidman, J. D., K. R. Cho, B. M. Ronnett, and R. J. Kurman. 2011. Surface epithelial tumors of the ovary. In Blaustein’s pathology of the female genital tract, sixth edition, edited by R. J. Kurman, L. H. Ellenson, and B. M. Ronnett. New York: Springer. Pp. 679-784.
Shaw, P. A., M. Rouzbahman, E. S. Pizer, M. Pintilie, and H. Begley. 2009. Candidate serous cancer precursors in fallopian tube epithelium of BRCA1/2 mutation carriers. Modern Pathology 22(9):1133-1138.
Sherman-Baust, C. A., E. Kuhn, B. L. Valle, I. M. Shih, R. J. Kurman, T. L. Wang, T. Amano, M. S. Ko, I. Miyoshi, Y. Araki, E. Lehrmann, Y. Zhang, K. G. Becker, and P. J. Morin. 2014. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. Journal of Pathology 233(3):228-237.
Shih, I. M., P. K. Panuganti, K. T. Kuo, T. L. Mao, E. Kuhn, S. Jones, V. E. Velculescu, R. J. Kurman, and T. L. Wang. 2011. Somatic mutations of PPP2R1A in ovarian and uterine carcinomas. American Journal of Pathology 178(4):1442-1447.
Shih, K. K., L. X. Qin, E. J. Tanner, Q. Zhou, M. Bisogna, F. Dao, N. Olvera, A. Viale, R. R. Barakat, and D. A. Levine. 2011. A microRNA survival signature (MiSS) for advanced ovarian cancer. Gynecologic Oncology 121(3):444-450.
Silva, E. G., C. S. Tornos, A. Malpica, and D. M. Gershenson. 1997. Ovarian serous neoplasms of low malignant potential associated with focal areas of serous carcinoma. Modern Pathology 10(7):663-667.
Soliman, P. T., B. M. Slomovitz, R. R. Broaddus, C. C. Sun, J. C. Oh, P. J. Eifel, D. M. Gershenson, and K. H. Lu. 2004. Synchronous primary cancers of the endometrium and ovary: A single institution review of 84 cases. Gynecologic Oncology 94(2):456-462.
Storey, D. J., R. Rush, M. Stewart, T. Rye, A. Al-Nafussi, A. R. Williams, J. F. Smyth, and H. Gabra. 2008. Endometrioid epithelial ovarian cancer: 20 years of prospectively collected data from a single center. Cancer 112(10):2211-2220.
Strioga, M., S. Viswanathan, A. Darinskas, O. Slaby, and J. Michalek. 2012. Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells and Development 21(14):2724-2752.
Studeny, M., F. C. Marini, J. L. Dembinski, C. Zompetta, M. Cabreira-Hansen, B. N. Bekele, R. E. Champlin, and M. Andreeff. 2004. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. Journal of the National Cancer Institute 96(21):1593-1603.
Szabova, L., C. Yin, S. Bupp, T. M. Guerin, J. J. Schlomer, D. B. Householder, M. L. Baran, M. Yi, Y. Song, W. Sun, J. E. McDunn, P. L. Martin, T. Van Dyke, and S. Difilippantonio. 2012. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Research 72(16):4141-4153.
Szych, C., A. Staebler, D. C. Connolly, R. Wu, K. R. Cho, and B. M. Ronnett. 1999. Molecular genetic evidence supporting the clonality and appendiceal origin of pseudomyxoma peritonei in women. American Journal of Pathology 154(6):1849-1855.
Takaishi, K., Y. Komohara, H. Tashiro, H. Ohtake, T. Nakagawa, H. Katabuchi, and M. Takeya. 2010. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Science 101(10):2128-2136.
Tammela, J., J. P. Geisler, P. N. Eskew, Jr., and H. E. Geisler. 1998. Clear cell carcinoma of the ovary: Poor prognosis compared to serous carcinoma. European Journal of Gynaecological Oncology 19(5):438-440.
Tanwar, P. S., L. Zhang, T. Kaneko-Tarui, M. D. Curley, M. M. Taketo, P. Rani, D. J. Roberts, and J. M. Teixeira. 2011. Mammalian target of rapamycin is a therapeutic target for murine ovarian endometrioid adenocarcinomas with dysregulated Wnt/beta-catenin and PTEN. PLoS ONE 6(6):e20715.
Tlsty, T. D., and L. M. Coussens. 2006. Tumor stroma and regulation of cancer development. Annual Review of Pathology 1:119-150.
Tone, A. A., M. K. McConechy, W. Yang, J. Ding, S. Yip, E. Kong, K. K. Wong, D. M. Gershenson, H. Mackay, S. Shah, B. Gilks, A. V. Tinker, B. Clarke, J. N. McAlpine, and D. Huntsman. 2014. Intratumoral heterogeneity in a minority of ovarian low-grade serous carcinomas. BMC Cancer 14:982.
Toss, A., E. De Matteis, E. Rossi, L. D. Casa, A. Iannone, M. Federico, and L. Cortesi. 2013. Ovarian cancer: Can proteomics give new insights for therapy and diagnosis? International Journal of Molecular Sciences 14(4):8271-8290.
Touboul, C., F. Vidal, J. Pasquier, R. Lis, and A. Rafii. 2014. Role of mesenchymal cells in the natural history of ovarian cancer: A review. Journal of Translational Medicine 12:271.
Vang, R., A. M. Gown, T. S. Barry, D. T. Wheeler, A. Yemelyanova, J. D. Seidman, and B. M. Ronnett. 2006a. Cytokeratins 7 and 20 in primary and secondary mucinous tumors of the ovary: Analysis of coordinate immunohistochemical expression profiles and staining distribution in 179 cases. American Journal of Surgical Pathology 30(9):1130-1139.
Vang, R., A. M. Gown, L. S. Wu, T. S. Barry, D. T. Wheeler, A. Yemelyanova, J. D. Seidman, and B. M. Ronnett. 2006b. Immunohistochemical expression of CDX2 in primary ovarian mucinous tumors and metastatic mucinous carcinomas involving the ovary: Comparison with CK20 and correlation with coordinate expression of CK7. Modern Pathology 19(11):1421-1428.
Vang, R., I. M. Shih, and R. J. Kurman. 2009. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Advances in Anatomic Pathology 16(5):267-282.
Vang, R., D. A. Levine, R. A. Soslow, C. Zaloudek, I. M. Shih, and R. J. Kurman. 2015. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: A rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian Study. International Journal of Gynecological Pathology (Epub ahead of print).
Veras, E., T. L. Mao, A. Ayhan, S. Ueda, H. Lai, M. Hayran, I. M. Shih, and R. J. Kurman. 2009. Cystic and adenofibromatous clear cell carcinomas of the ovary: Distinctive tumors that differ in their pathogenesis and behavior: A clinicopathologic analysis of 122 cases. American Journal of Surgical Pathology 33(6):844-853.
Visvader, J. E., and G. J. Lindeman. 2008. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nature Reviews: Cancer 8(10):755-768.
Wang, Y., R. C. Wu, L. E. Shwartz, L. Haley, M. T. Lin, I. M. Shih, and R. J. Kurman. 2015. Clonality analysis of combined Brenner and mucinous tumours of the ovary reveals their monoclonal origin. Journal of Pathology (Epub ahead of print).
Wefers, C., L. J. Lambert, R. Torensma, and S. V. Hato. 2015. Cellular immunotherapy in ovarian cancer: Targeting the stem of recurrence. Gynecologic Oncology 137(2):335-342.
Wels, J., R. N. Kaplan, S. Rafii, and D. Lyden. 2008. Migratory neighbors and distant invaders: Tumor-associated niche cells. Genes and Development 22(5):559-574.
White, E. A., H. A. Kenny, and E. Lengyel. 2014. Three-dimensional modeling of ovarian cancer. Advanced Drug Delivery Reviews 79-80:184-192.
Wiedemeyer, W. R., J. A. Beach, and B. Y. Karlan. 2014. Reversing platinum resistance in high-grade serous ovarian carcinoma: Targeting BRCA and the homologous recombination system. Frontiers in Oncology 4:34.
Woolas, R., I. Jacobs, A. P. Davies, J. Leake, C. Brown, J. G. Grudzinskas, and D. Oram. 1994. What is the true incidence of primary fallopian tube carcinoma? International Journal of Gynecological Cancer 4(6):384-388.
Wu, R., N. Hendrix-Lucas, R. Kuick, Y. Zhai, D. R. Schwartz, A. Akyol, S. Hanash, D. E. Misek, H. Katabuchi, B. O. Williams, E. R. Fearon, and K. R. Cho. 2007. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell 11(4):321-333.
Wu, R., S. J. Baker, T. C. Hu, K. M. Norman, E. R. Fearon, and K. R. Cho. 2013. Type I to type II ovarian carcinoma progression: Mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. American Journal of Pathology 182(4):1391-1399.
Xing, D., and S. Orsulic. 2005. A genetically defined mouse ovarian carcinoma model for the molecular characterization of pathway-targeted therapy and tumor resistance. Proceedings of the National Academy of Sciences of the United States of America 102(19):6936-6941.
Xing, D., and S. Orsulic. 2006. A mouse model for the molecular characterization of BRCA1-associated ovarian carcinoma. Cancer Research 66(18):8949-8953.
Yoshikawa, H., H. Jimbo, S. Okada, K. Matsumoto, T. Onda, T. Yasugi, and Y. Taketani. 2000. Prevalence of endometriosis in ovarian cancer. Gynecologic and Obstetric Investigation 50(Suppl 1):11-17.
Zaino, R., C. Whitney, M. F. Brady, K. DeGeest, R. A. Burger, and R. E. Buller. 2001. Simultaneously detected endometrial and ovarian carcinomas—A prospective clinicopathologic study of 74 cases: A Gynecologic Oncology Group Study. Gynecologic Oncology 83(2):355-362.
Zaino, R. J., M. F. Brady, S. M. Lele, H. Michael, B. Greer, and M. A. Bookman. 2011. Advanced stage mucinous adenocarcinoma of the ovary is both rare and highly lethal: A Gynecologic Oncology Group Study. Cancer 117(3):554-562.
Zhai, Y., R. Kuick, C. Tipton, R. Wu, M. Sessine, Z. Wang, S. J. Baker, E. R. Fearon, and K. R. Cho. 2015. Arid1a inactivation in an Apc- and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. Journal of Pathology (Epub ahead of print).
Zhang, S., C. Balch, M. W. Chan, H. C. Lai, D. Matei, J. M. Schilder, P. S. Yan, T. H. Huang, and K. P. Nephew. 2008. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Research 68(11):4311-4320.
Zhang, Z., R. C. Bast, Jr., Y. Yu, J. Li, L. J. Sokoll, A. J. Rai, J. M. Rosenzweig, B. Cameron, Y. Y. Wang, X. Y. Meng, A. Berchuck, C. Van Haaften-Day, N. F. Hacker, H. W. de Bruijn, A. G. van der Zee, I. J. Jacobs, E. T. Fung, and D. W. Chan. 2004. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Research 64(16):5882-5890.
Zhu, L. Y., W. M. Zhang, X. M. Yang, L. Cui, J. Li, Y. L. Zhang, Y. H. Wang, J. P. Ao, M. Z. Ma, H. Lu, Y. Ren, S. H. Xu, G. D. Yang, W. W. Song, J. H. Wang, X. D. Zhang, R. Zhang, and Z. G. Zhang. 2015. Silencing of MICAL-L2 suppresses malignancy of ovarian cancer by inducing mesenchymal-epithelial transition. Cancer Letters 363(1):71-82.








































