
4
Catalytic Conversion of Light Alkanes
The second day of the workshop began with a plenary session featuring three presentations designed to serve as background for subsequent breakout group discussions. Jeffery Bricker, senior director of research at Honeywell UOP, formerly known as Universal Oil Products, reviewed the history and state of the art of ethane and propane dehydrogenation catalysis. Alexis Bell, the Dow Professor of Sustainable Energy at the University of California, Berkeley, and faculty senior scientist at the Lawrence Berkeley National Laboratory, described some of the lessons learned from theory and experiment about methane, ethane, and propane conversion over heterogeneous catalysts, and Shannon Stahl from the University of Wisconsin–Madison discussed the use of homogeneous catalysts to activate the carbon–hydrogen bond. Each presentation was followed by a brief discussion period.
HISTORY AND STATE OF THE ART OF ETHANE AND PROPANE DEHYDROGENATION CATALYSIS
There will be continued growth in worldwide demand for light olefins—ethylene and propylene—at a projected 4 percent compound annual growth rate, supporting investment in additional petrochemical facilities, said Jeffery Bricker. Driving this growth, he said, is demand from the expanding middle class in developing countries, particularly in China, India, and Southeast Asia. The majority of the demand for propylene, he added, will be filled by propane dehydrogenation (PDH) in North
America and methane-to-olefin (MTO) plants in China, although China is also importing an increasing amount of propane from the United States.
Bricker explained that there is a growing gap in the supply-versus-demand curve for propylene (see Figure 4-1) as a result of two factors: Steam crackers have been shifting to lighter feedstocks that produce less propylene, and flat demand for gasoline in some regions has limited the amount of propylene produced during the oil-refining process. His colleagues at UOP believe that so-called on-purpose propylene will supply one quarter of the world’s demand by 2021.
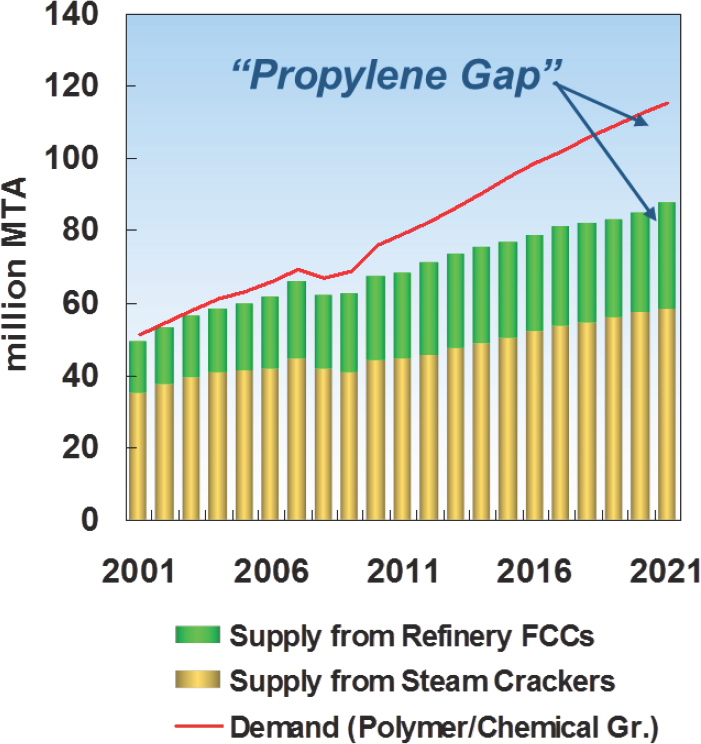
NOTE: FCC = fluid catalytic cracking; MTA = million tons per annum.
SOURCE: Bricker, 2016.
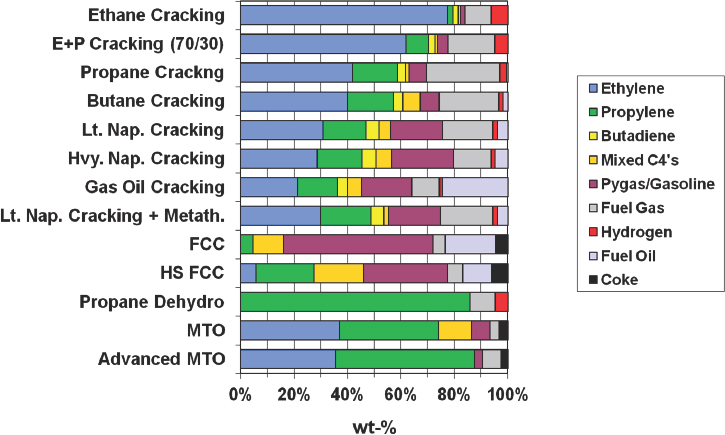
NOTE: FCC = fluid catalytic cracking; MTO = methanol to olefin.
SOURCE: Bricker, 2016.
Of all of the technologies that have been developed that produce propylene, PDH provides the highest yield (see Figure 4-2), and because of the price differential between propane and propylene, PDH economics are quite favorable and support continued investment in plant construction. Over the past few years, the price differential has been as high as $700 per ton, which Bricker said is “an unbelievable number in the petrochemical industry” and has led to the construction of plants capable of producing 1 million tons of propylene annually.
There are several challenging characteristics of PDH technology, Bricker noted, starting with the fact that the endothermic reaction is equilibrium limited, requiring temperatures that exceed 600°C and low-pressure conditions, making a reheat strategy critical. In his opinion, reheating is an area ripe for technological advancements going forward. Another feature of PDH technology is that coke formation is unavoidable, leading to a catalyst life of days and the need for frequent regeneration. As in all catalytic–process technology, he added, the process and catalyst are intertwined and cannot be separated. UOP’s Oleflex process, he noted, can use mixed feedstocks—propane plus isobutylene or n-butane—and generate a product mix of propylene plus isobutylene or butenes, the
latter of which can be converted to in-demand butadiene using other technology.
Bricker noted that World War II marked the first use of alkane dehydrogenation, and over the ensuing years several companies have steadily improved the catalysts and the processes using them. Today, two PDH technologies—UOP’s Oleflex process and the Lummus Catofin process—dominate propylene production. In total, there are now 22 PDH units in operation worldwide, and unit capacities of the newest facilities have been increasing to as large as 1 million tons per year. Since 2011, 40 new PDH units have been ordered, and although UOP has won 34 of those contracts, the company continues to improve its technology in order to remain competitive.
The Lummus Catofin technology, first developed in the 1940s, involves a cyclic reactor technology in which seven reactors go through a computer-controlled sequence of reaction, reheat, and regeneration using a catalyst developed by Clariant (see Figure 4-3). The UOP Oleflex technology uses a continuous moving bed process with a regenerator (see Figure 4-4). PDH units have four reactors, while mixed propane and butane units have three reactors. The spherical platinum-based catalyst travels several hundred meters through all of the reactors and is then regener-
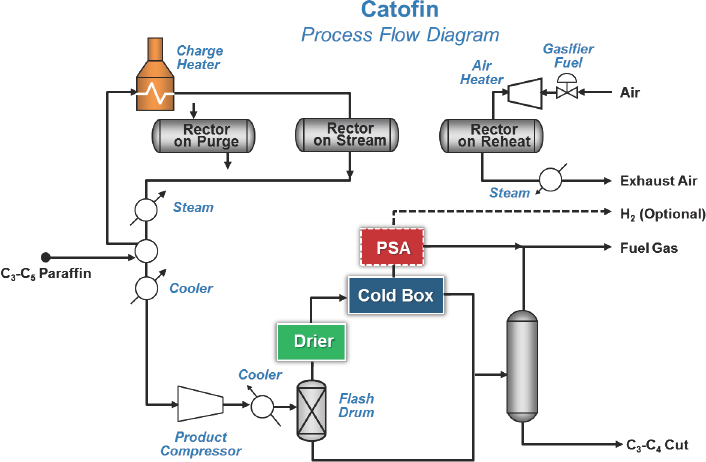
NOTE: PSA = pressure swing adsorption.
SOURCE: Bricker, 2016.
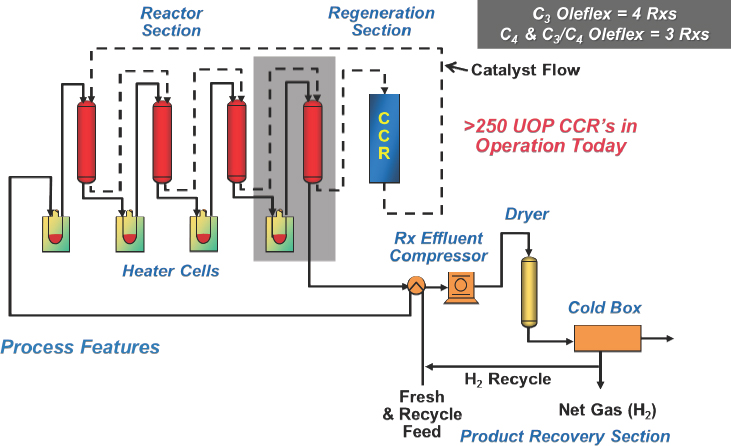
NOTE: CCR = continuous catalyst regeneration.
SOURCE: Bricker, 2016.
ated to burn off the accumulated coke. Bricker explained that the catalyst coming out of the regenerator is indistinguishable from fresh catalyst and will last several years. He noted, too, that because the chemical reaction is endothermic, heat input is critical and any yield loss results from inefficient heat transfer, not from catalyst selectivity.
Going forward, UOP researchers are working on methods for improving heat transfer and on changes that will reduce propane consumption at a constant conversion. They have already developed process improvements that will increase the yield per pass and lower both utility and capital costs. Bricker said these improvements are on fast-track development and will be commercialized soon.
With regard to ethane utilization, Bricker said that ethane steam cracking is the only process used today to produce ethylene, and ethane steam crackers produce more than 100 million metric tons of ethylene annually. Important advances have been made with two other routes to ethylene—catalytic ethane dehydrogenation and ethane oxidative dehydrogenation—but there are no commercial units that he is aware of that either use or plan to use these technologies in the near future, largely because the economics of ethane stream cracking are hard to beat. Steam crackers, explained Bricker, not only have a low cost of production and
capital cost, but the internal rate of return is projected to exceed 26 percent less than both 2016 and 2019 pricing scenarios. The main downsides to ethane steam cracking are its high energy intensity (i.e., 16 gigajoules of energy are required to produce 1 ton of ethylene) and the more than 1 ton of carbon dioxide emitted per ton of ethylene produced (Gärtner et al., 2013). Oxidative dehydrogenation, he noted, could have a much lower energy and carbon dioxide footprint if done with high selectivity and at high ethane-conversion rates, and would likely enable a continuous production process.
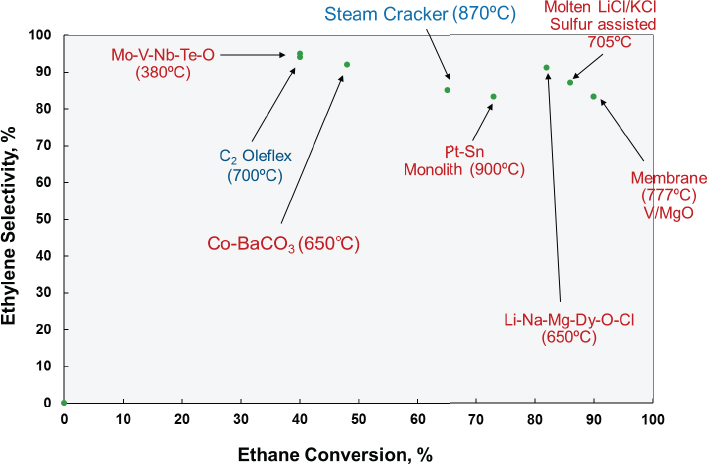
Researchers have reported a number of different oxidative dehydrogenation catalysts that do achieve high selectivity for ethylene (see Figure 4-5). In the late 1980s, for example, Bricker adapted the Oleflex process to produce ethylene from ethane, but the heating strategy required to maintain the necessary temperature across six reactors made the process uneconomical, he explained. There has also been work on novel reactor designs using oxygen-permeable membranes (Czuprat et al., 2009; Ramos et al., 2000; Rebeilleau-Dassonneville et al., 2005), hydrogen-permeable membranes (Schäfer et al., 2003), microchannel reactors, and monolith-type reactors. Hydrogen-permeable membranes, for example, can shift the equilibrium of the dehydrogenation reaction to increase conversion,
SOURCE: Bricker, 2016.
but the challenge then is to create a change in pressure across the membrane without resorting to a high-pressure regime or using a second gas to sweep hydrogen off of the membrane. The removed hydrogen could then be burned to provide some heating. Although an ionic oxygen-conducting membrane with palladium and vanadium–manganese oxide catalyst (Rebeilleau-Dassonneville et al., 2005) produced very high conversion rates and good selectivity, scaling such a system would likely be difficult, said Bricker. Also, it would probably not provide much benefit over steam cracking because of the need to run the reaction at high temperature. However, if someone could make an ion-conducting membrane that worked at 500°C rather than 777°C, such a process could prove workable at an industrial scale, he added.
A monolith reactor coated with a platinum and tin catalyst (Bodke et al., 1999; Silberova et al., 2004), similar in concept to the catalytic converter on a car, produced good results at high throughput rates. Feeding hydrogen into the reactor stream reduced carbon dioxide emissions substantially. Bricker said that at least one large chemical company tried to scale this process but without success. However, his group is now working with high-velocity reactor designs to see if it can figure out how to manage heat flow at an industrial scale.
Another interesting approach for oxidative dehydrogenation used sulfur as a mild oxidant in combination with a molten salt catalyst (Gaspar et al., 1974), which allows heat to dissipate rapidly and prevents hotspot formation that would decrease selectivity. An advantage this system offers is that coke could be removed easily from the molten catalyst. While Bricker characterized this approach as being outstanding from the catalyst perspective, he suspects that scaling a system using molten salts and sulfur is likely to be difficult.
With regard to catalytic oxidative dehydrogenation, researchers have developed upwards of 20 different systems, said Bricker. One approach he finds promising uses a molten alkali metal chloride supported on dysprosium oxide-magnesium oxide to achieve an 82 percent conversion and 91 percent selectivity to ethylene (Kumar et al., 2008). Another approach with potential uses a molybdenum-vanadium-niobium-tellurium oxide catalyst operating at the relatively low temperature of 400°C and dilution to achieve an 87 percent conversion and 84 percent selectivity (Botella et al., 2004).
Bricker concluded his presentation by noting that ethane cracking is still the most economical proven process for ethylene production, but that promising catalysts for ethane oxidative dehydrogenation are emerging. “If an overall economic process can be developed, there is a good chance that the future will include commercial ethane oxidative dehydrogenation plants,” said Bricker. For propane, PDH is now an economically attractive
technology to fill the propylene gap. In closing, he added that “catalysis still provides a tremendous lever to provide value to society for environmental reasons and by providing high-value products.”
In the discussion period that followed, Bell asked Bricker, for the benefit of those in academia and at the national laboratories, in his opinion what longer-term research should be pursued that would enable companies such as his to be more effective. Bricker replied that there will always be value in people inventing new materials, noting that almost every advance in petrochemical processes came about because of the invention of a new material. Other areas in which academia and the national laboratories could help are in developing a better fundamental understanding of catalysis and new methods of modeling catalyst structures and reaction processes.
Eric Stangland from The Dow Chemical Company remarked that just as catalysts and processes are intimately linked, so too are the separations processes. Bricker agreed with this comment, adding that each catalyst–process combination produces a different mix of byproducts that have to be separated. He noted that his company has made a significant effort aimed at improving downstream separations.
HETEROGENEOUS CATALYSIS: LESSONS LEARNED FROM EXPERIMENT AND THEORY
Chemists have developed a number of different routes by which natural gas can be converted to chemicals using catalysis (see Figure 4-6), many of which form the technological foundation for the modern chemical industry. Yet despite their successful application at an industrial scale, there are still a number of central questions common to all of these chemistries that remain unanswered, said Bell. The four questions he listed, and that he addressed in his presentation, include
- What is the rate-limiting step in the activation of methane and light alkanes?
- What factors govern the formation of coke during the conversion of methane and light alkanes?
- Can oxygenated compounds be formed directly from methane and light alkanes?
- What is on the horizon and beyond?
With regard to the conversion of ethane or propane, respectively, to ethylene or propylene plus hydrogen, it is well known that platinum is one of the better catalysts for this reaction but that it deactivates rapidly because of coke formation. Research has also shown that adding tin,
SOURCE: Bell, 2016.
gallium, indium, or other metals to create a bimetallic catalyst enhances alkene selectivity while also reducing coke formation (Feng et al., 2015; Galvita et al., 2010; Peng et al., 2012; Somodi et al., 2011, 2012; Sun et al., 2011; Wu et al., 2014a, 2014b, 2015). Several years ago, Bell and his colleagues began to look at the effect of platinum particle size and the concentration of tin on coke formation and to identify the mechanism of coke formation and its influence on platinum nanoparticles. After developing a process that enabled them to control the particle size and tin-to-platinum ratio, they were able to show that catalytic activity increased significantly when increasing the amount of tin relative to platinum while keeping the particle size constant. Activity also went up when holding the tin-to-platinum ratio constant while increasing the catalyst particle size. However, as the particle size increases, so too does the tendency to form coke.
To determine the source of the coke, Bell and his collaborators used 13C-labeled ethylene to show that coke is formed by readsorption of ethylene onto the catalyst surface. Ethylene as the source of coke was confirmed, Bell explained, by high space velocity experiments, which showed there were lower levels of coke deposition at high space velocities. Theoretical calculations showed that one of the effects of adding tin is
that it weakens the readsorption of ethylene or propylene onto platinum. High-resolution transmission electron microscopy studies showed that the amount and morphology of carbon changes with platinum particle size (Peng et al., 2012) (see Figure 4-7). These studies also confirmed theoretical predictions that carbon would grow at the steps of the catalyst particle (see Figure 4-8), and they demonstrated that carbon deposition induces step formation that then serves as additional nucleation sites for carbon formation.

NOTE: nm = nanometer.
SOURCE: Peng et al., 2012.
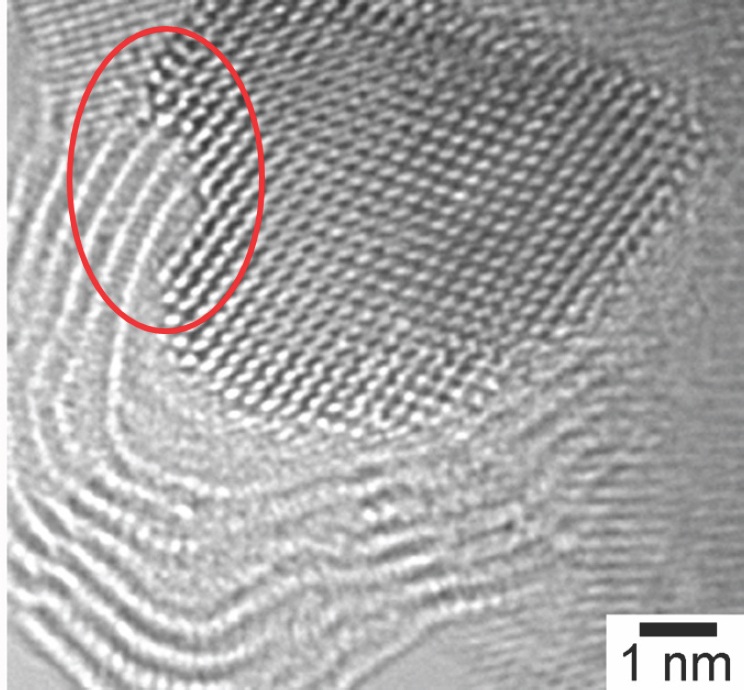
NOTE: nm = nanometer.
SOURCE: Peng et al., 2012.
Researchers have also studied oxidative dehydrogenation of ethane and propane by vanadium catalysts dispersed onto alumina. At low loading, most of the vanadium is present at isolated sites, and in that situation alkene selectivity is limited by deep oxidation of both the alkane and alkene (Zboray et al., 2009).
Summarizing what is known about the catalytic conversion of light alkanes, Bell said breaking the carbon–hydrogen bond is the rate-limiting step. Steam and dry reforming of methane follow identical kinetics, as do the thermal dehydrogenation of light alkanes and the dehydroaromatization of methane. Graphene formation, he said, is nucleated at steps on the surface of metal particles and graphene growth can cause step formation. Graphene formation, he added, is suppressed by reduc-
ing metal particle size and increasing the lattice mismatch between the graphene and the metal, and soot formation is limited by very rapid thermal quenching. With regard to oxidation of methane to methanol, the rate of this reaction is limited by catalyst reactivation, and oxidative dehydrogenation of light alkanes is limited by both primary deep oxidation of the alkane and secondary oxidation of the alkene.
Two of the best catalysts for steam reforming of methane to produce syngas, said Bell, are ruthenium and rhodium, but their low abundance and high cost make them less attractive for use in large-scale industrial processes. As a result, nickel is the catalyst used in practice. Both experiment and theory show that the turnover rate for this reaction drops slightly from ruthenium to rhodium to nickel (Jones et al., 2008). For dry reforming, the results are nearly the same, with nickel predicted to be slightly less active than ruthenium and rhodium (German and Sheintuch, 2013; Wang et al., 2007). In both cases, the initial dissociation of methane to produce a methyl group and a hydrogen atom is the critical step that determines the reaction rate. In fact, one of Bell’s colleagues demonstrated that steam reforming and dry reforming are two manifestations of the same reaction (Wei and Iglesia, 2004), and the kinetics are first order for methane and zero order for oxygen or carbon dioxide when using a nickel catalyst. It turns out, said Bell, that the kinetics of carbon accumulation, or coking, are also the same for steam and dry reforming of methane.
Given that coke formation is inevitable, the next question Bell addressed concerned where coke formation occurs. Experiments have shown that methane dissociation occurs preferentially at the steps on the catalyst surface, and the more steps present in the catalyst structure, the more carbon will form (Saadi et al., 2010; Sehested, 2006). At the same time, steps decrease the activation energy for methane dissociation, so what is good for catalytic activity is bad for coke formation, Bell explained. Experiments have also shown that carbon grows at steps as small graphene islands that eventually form graphene sheets and finally carbon nanotubes (Peng et al., 2012; Wu et al., 2016), and researchers have developed a thermodynamics-based explanation for why the steps are important and how wide they have to be to serve as nucleation sites for graphene growth (Saadi et al., 2010). These studies led to the prediction that introducing gold into the catalyst surface would inhibit this process, and indeed, experiments have shown that the presence of gold leads to smaller amounts of coke forming on the catalyst surface.
Turning to methane pyrolysis, Bell said that from a thermodynamic perspective, methane would prefer to form graphite and hydrogen rather than products such as ethylene and benzene. In fact, while molybdenum carbide nanoparticles in ZSM-5 zeolite or an iron on silica catalyst will produce some benzene and ethylene at temperatures exceeding 900 K,
coke formation occurs rapidly resulting in catalyst deactivation. However, 2 years ago, researchers at the Dalian Institute of Chemical Physics showed that a catalyst in which iron atoms are isolated from one another in a silica structure is capable of avoiding coking and produces a mixture of ethylene, benzene, and naphthalene when operated at 1,363 K and at what Bell characterized as very high space velocities (Guo et al., 2014). This catalyst, the researchers report, is stable for 60 hours, which they attribute to the isolated iron carbide sites in the catalyst’s silica framework. Bell noted that the presumed mechanism involves dissociating methane on the catalyst surface, with the resulting methyl radical then leaving the surface and reacting in the gas phase. While the chemistry is not fully understood, he suspects that these isolated iron sites are too small to nucleate the formation of graphene and that the reaction is run at such short contact time that there is rapid thermal quenching of the product gases, which would inhibit the gas-phase production of carbon.
As Tobin Marks from Northwestern University stated earlier, while the indirect conversion of methane to methanol via syngas is an important industrial process, the chemical industry would like to replace that energy-intensive process with one that directly oxidizes methane to methanol. Bell agreed that such a process would be of commercial interest, particularly if it could be run in a continuous manner. This latter feat has not been achieved yet, but researchers have shown that a copper-ZSM-5 catalyst will oxidize methane to methanol at relatively low temperatures. Catalyst reactivation, however, requires raising the temperature by several hundred degrees (Groothaert et al., 2005). Bell explained that three research teams each claim different copper structures that serve as the active catalytic site, indicating that further research to fully understand this chemistry is required.
Looking to the future, Bell listed several goals, including
- identify catalysts that operate at high temperature and are resistant to coke formation;
- identify single-site catalysts that enable the continuous conversion of methane to methanol;
- identify catalysts than can promote the oxidative dehydrogenation of alkanes to alkenes selectively; and
- understand the nature of oxygen species and what controls their activity.
During the ensuing discussion, Bala Subramaniam from the University of Kansas noted that because catalysts will increase the rate of conversion but not the equilibrium, process engineering in combination with catalyst development could be a powerful complement. Bell agreed
that process engineering has to be part of the whole package of catalyst development, and that the two do go hand in hand. He noted that when engaging in process development, it does help to know what is happening locally at the catalyst. Bricker then asked if the tin in the platinum–tin catalysts was exerting a geometric or electronic effect, and Bell replied that quantum mechanical calculations show it to be an electronic effect in addition to the geometric effect of impacting nucleation sites.
HOMOGENEOUS CATALYSIS FOR CARBON–HYDROGEN BOND ACTIVATION
Homogeneous catalysts, said Shannon Stahl, have been used in numerous major industrial processes, and he wondered if the dividing line between homogeneous and heterogeneous catalysis is meaningful or if it is an artifact of the silos that separate chemistry and chemical engineering departments at most universities. Of greatest importance in his view are the molecular processes and concepts that are operating with a given catalyst regardless of the name it is given. Indeed, there is a great deal of opportunity for cross-fertilization and mechanistic understanding that spans homogeneous catalysis, heterogeneous catalysis, and electrocatalysis.
Some of the industrially important applications of homogeneous catalysis include the Shell higher olefin process for producing linear α-olefins from ethylene, the INEOS process for making higher α-olefins from syngas, The Dow Chemical Company’s butadiene telomerization process, and Chevron Phillips’s and Sasol’s ethylene trimerization and tetramerization of ethylene processes, hydroformylation chemistry, and ethylene oxidation. Major applications for the products of these reactions are in linear low-density polyethylene (LLDPE), high-density polyethylene, detergent alcohols, and synthetic lubricants. Stahl noted that increasing 1-hexene use in LLDPE is expected to drive market growth (see Figure 4-9). Commenting on the slated construction of ethane crackers to produce ethylene, Stahl said, “If we are going to be awash in ethylene through ethane dehydrogenation, enhancing these types of processes could play an important role in terms of where we need to stimulate more activity in homogeneous catalysis.”
Hydroformylation of α-olefins and syngas to produce linear and branched aldehydes is one example of a homogeneous catalysis process carried out on a massive scale, with production exceeding 18 billion pounds per year using primarily rhodium-based catalysts that were discovered in the 1960s (Cornils et al., 1994). Liquid-phase radical chain aerobic oxidation of hydrocarbons using a cobalt-manganese-bromine catalyst discovered in 1955 produces more than 100 billion pounds per
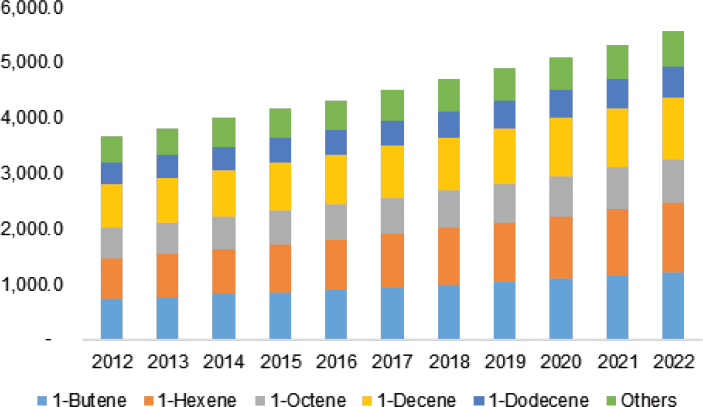
NOTE: M = million.
SOURCE: Stahl, 2016. Reproduced with permission by Grand View Research.
year of chemicals such as terephthalic acid, which is used to make the plastic polyethylene terephthalate (PET) (Tomás et al., 2013). The Wacker process, discovered in 1959 and based on an organometallic catalyst, is used to convert ethylene into more than 1 billion pounds of acetaldehyde per year (Jira, 2009). Stahl noted that while this latter reaction appears to involve oxygen transfer, water is the source of the oxygen atom in acetaldehyde and molecular oxygen merely serves as an electron acceptor and reoxidizes the reduced copper (I) to copper (II).
One of the features of homogeneous catalysis that distinguishes it from heterogeneous catalysis is that soluble molecular complexes can often activate specific carbon–hydrogen bonds, not necessarily the weakest one. In that way, organometallic chemistry can drive a different reactivity pattern relative to that of traditional oxidation methods. While this selectivity may not play a critical role in the production of many commodity chemicals, Stahl noted it is capitalized on throughout organic chemistry and in particular in pharmaceutical synthesis. In fact, most of the research on homogeneous catalysis has focused on carbon–hydrogen bond functionalization as part of complex molecule synthesis and, to a lesser extent, fine chemicals synthesis. However, selectivity is crucial for alkane functionalization, with an example of preventing over-oxidation in the partial oxidation of methane to form methanol. As an aside, Stahl said
in his opinion there has been a “brain drain” in the area of homogeneous catalysis directed toward hydrocarbon functionalization since the 1990s related to a lack of research funding from National Science Foundation (NSF) and the U.S. Department of Energy (DOE). Most of the research funding for homogeneous catalysis, he claimed, comes from the National Institutes of Health and goes toward pharmaceutical synthesis, rather than from NSF and DOE to study hydrocarbon functionalization.
The Shilov platinum chloride catalyst (Shilov and Shul’pin, 1997) is one of the more well-studied homogeneous catalysts for alkane functionalization that enables organometallic methane oxidation (see Figure 4-10). It is also possible to use oxygen as the oxidant using redox couples, and Stahl, and his collaborators have been studying that possibility in an electrocatalytic system (Gerken and Stahl, 2015; Joglekar et al., 2016).
Other uses of homogeneous catalysts include oxidative carbon–carbon coupling, which is used today to make polyimide resin from two aromatic molecules. The question, said Stahl, is whether this type of system could be used to couple two methane molecules to produce ethane, which would then undergo oxidative dehydrogenation on the same palladium catalyst to yield ethylene. In such a scheme, oxygen would act as a hydrogen acceptor in the ethane-to-ethylene reaction. Another possibility
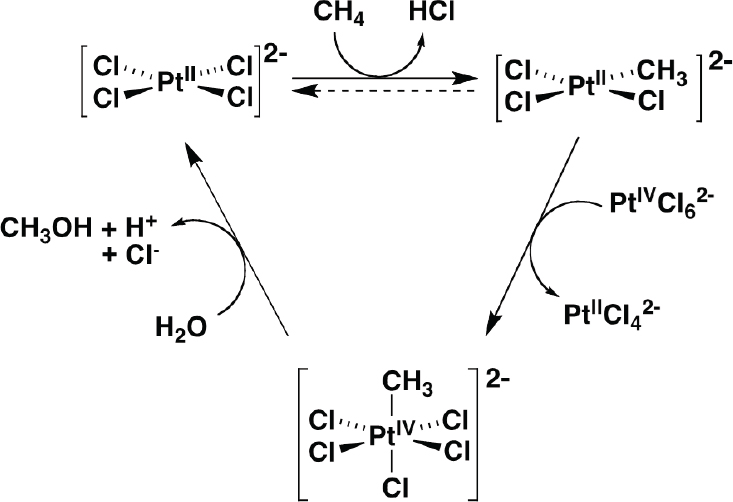
SOURCE: Stahl, 2016.
is to catalyze the non-oxidative conversion of hydrocarbons through the addition of methane to olefins (Sadow and Tilley, 2003).
Stahl noted that a team within the NSF Center for Chemical Innovation, the Center for Enabling New Technologies for Catalysis, is exploring the idea of using homogeneous catalysis for converting natural gas liquids into longer-chain hydrocarbons that could be used as liquid fuels (Goldman et al., 2006; Huang et al., 2011). They are also studying routes for making aromatics from ethylene and alkanes (Brookhart et al., 2012; Huang et al., 2011; Lyons et al., 2012). The key to developing these types of reactions, he said, is a willingness to work at elevated temperatures, which most researchers do not consider when studying homogenous catalysis because they worry about stability. This highlights the lack of sustained efforts to design homogeneous catalysts that are stable under conditions required for hydrocarbon functionalization.
During the discussion period, Matthew Neurock from the University of Minnesota noted that homogeneous catalysts offer the possibility of using analytical tools such as nuclear magnetic resonance spectroscopy to study well-defined molecules and their interactions with reactants in a way that is challenging with heterogeneous catalysts. These types of studies may not lead to an industrial process, but they can provide novel and useful information about how bonds are made, broken, and transformed through their interactions with the metal atoms in these catalysts.
WORKING GROUP SESSIONS
Following the presentations by Bricker, Bell, and Stahl, the workshop’s participants broke into four predefined working groups, each of which explored an area in which catalysis could enable novel approaches to using natural gas and natural gas liquids. The subjects discussed by the four groups included light alkanes to alkenes and dienes, light alkanes to aromatics, emerging opportunities for novel approaches to natural gas conversion, and activation of natural gas using nontraditional oxidants. As was the case with the first set of working group discussions, each group, after hearing a short introductory presentation, was asked to answer a set of questions over the course of their of deliberation. Three of the four groups addressed the questions in Box 4-1. The remaining fourth group, which discussed emerging opportunities for novel approaches to natural gas conversion, confronted a slightly different set of questions (see Box 4-2).
Following the 2-hour discussion period, each group’s designated rapporteur summarized the group’s work to the reassembled workshop participants. An open discussion followed the four reports.
Light Alkanes to Alkenes and Dienes
In her framing remarks to this working group, Angeliki Lemonidou, professor of chemical engineering and director of the Petrochemical Technology Laboratory at Aristotle University of Thessaloniki, recapped much of what earlier speakers had presented on the technologies used to convert light alkanes, primarily ethane and propane, to alkenes and dienes. She did, however, also note that while these dehydrogenation technologies are deployed on an enormous scale worldwide, there is still room for improving these processes. The three main drawbacks they suf-
fer from include coke deposition; side reactions such as hydrogenolysis,1 isomerization, and polymerization that produce unwanted byproducts; and thermodynamic limitations. Catalysis science, she said, can improve the performance of alkane dehydrogenation processes through the design of new or improved catalyst formulations and dopants based on fundamental understanding of coke formation routes and mechanism of side reactions. Research on catalysts can also aim to identify novel catalysts that replace toxic metal oxides with environmentally benign, catalytically active metals; and materials that selectively remove hydrogen from the reaction milieu by using hydrogen-permeable membranes.
With the shift from naphtha as a feedstock in the United States, butadiene production may not be able to meet future demand, so an area that Lemonidou believes merits special attention is butadiene synthesis from light alkanes. There is a desire, she said, to exploit new processes for butadiene production, and she suggested two potential routes: ethylene dimerization followed by oxidative dehydrogenation and a one-step oxidation of butane to butadiene. She also highlighted the lack of research on alternative oxidants for oxidative dehydrogenation, including the use of carbon dioxide as a mild oxidant (Ascoop et al., 2016; Koirala et al., 2015; Porosoff et al., 2015) and halogens (Upham et al., 2016).
Lemonidou concluded her remarks with her perspective on opportunities in this area. For propane to propylene, the immediate target should be to increase yield given that high selectivity is difficult with the known catalyst and using oxygen as the oxidant. For ethane to ethylene, there is immediacy for catalyst formulations that fulfill industry’s yield requirements and for scaling and testing promising catalysts under industrially relevant conditions. Current yields of butane to butadiene are low, so near-term work should focus on identifying catalysts that can boost yields to acceptable levels. The goal of all of this research on oxidative dehydrogenation, she said, should be to minimize the deep oxidation of alkanes and sequential oxidation of the resulting olefins. Approaches to achieving this goal include designing catalyst surfaces that adsorb weakly to the formed olefin, controlling active site density, and keeping the partial pressure of oxygen low, perhaps through the design of membrane-based reactors, or by decoupling the reduction and oxidation steps.
Discussion
The discussion that followed Lemonidou’s presentation, led by Tobin Marks, identified a number of impediments facing technologies for con-
___________________
1 Hydrogenolysis is a chemical reaction in which carbon–carbon bonds are cleaved by the addition of hydrogen.
verting light alkanes into alkenes and dienes. A major obstacle is that the technologies currently used are mature with acceptable selectivity, and the capital costs of implementing a new technology are considerable. For PDH, one challenge is to develop enough of a knowledge base to enable the rational design of selective and stable catalysts, which Marks added is an overarching theme for the entire workshop. Another challenge is to achieve similar selectivity but with a lower carbon footprint than oxidative dehydrogenation, and to do so with simpler reactors requiring smaller capital expenditures. For dienes, the discussion focused on direct routes from butane to butadiene.
With regard to the top two to three well-established research approaches to making alkenes and dienes, Marks reported that the group discussed identifying catalysts that would improve the carbon selectivity of oxidative dehydrogenation while maintaining acceptable turnover rates and frequencies. Alternative oxidants might be able to address this challenge, as might novel reactor designs such as circulating fluid bed and short-contact-time reactors.
The discussion on promising but higher-risk novel approaches produced a long list of ideas that Marks characterized as a good guide for developing a research program. The list included
- biological routes for syngas fermentation to butadiene;
- carbon-based catalysts, such as those using carbon nanotubes and graphene, for oxidative dehydrogenation at low temperatures;
- oxygen activation and utilization bookkeeping in oxidative dehydrogenation reactions to improve selectivity and carbon efficiency; and
- membrane reactor technology to separate hydrogen or remove products before further reaction occurs.
This group also identified a long list of new tools and scientific advances that are creating important research opportunities. This list included
- in situ observations of catalysts under a variety of dynamic reaction conditions;
- theoretical methods for better understanding spectroscopy data and mechanisms of light alkane conversion;
- tools for mechanistic studies of light alkane conversion, such as operando methods, labeling techniques, and Mossbauer and electron paramagnetic resonance spectroscopy; and
- homogeneous catalysts for conversion of light alkanes as knowledge-based platforms for developing new concepts.
With regard to the industrial requirements and environmental constraints that researchers are responsible for knowing when developing new approaches to utilizing natural gas, an overarching impediment as reported by Marks is reducing the energy requirements of any process. With oxidative dehydrogenation reactor design, safety is a critical issue given how much heat these reactions produce. The group noted that the national laboratories have facilities to test novel reactor designs safely and that researchers could collaborate with those laboratories when it comes to testing design prototypes. When incorporating oxidative dehydrogenation chemistry with other processes, or considering the use of alternative oxidants, Marks added that there is value for researchers to think about scalability and environmental viability.
According to Marks, the last constraint that researchers should consider is critically important in the real world of the chemical industry—the cost of capital. Specifically, this group suggested that lower capital costs in implementing a new technology compared with an existing technology could reduce the risk and produce a 15 to 20 percent return on investment.
Light Alkanes to Aromatics
In his opening presentation to this working group, Bruce Gates, distinguished professor of chemical engineering and materials science at the University of California, Davis, said that the conversion of propane to aromatics is less uphill thermodynamically than conversion of methane to aromatics, which had been discussed by one of the first four working groups. Various groups have reported success converting light alkanes to aromatics using acidic zeolites and zeolite-supported metals such as zinc, gallium, and molybdenum, but in his opinion these have been incompletely characterized. The metal atoms, for example, may be present as carbides or oxycarbides, or they may not be in a metallic state. It also appears, he said, that the metal atoms are both inside and outside of the zeolite pores. These catalysts, he added, coke rapidly and require frequent regeneration, which might contribute to catalyst deactivation.
The UOP CYCLAR™ process, developed by BP and UOP, produces aromatics from propane, butane, or a mixture of the two in a dehydrocyclodimerization reaction sequence (see Figure 4-11). Numerous authors, said Gates, have suggested that alkane dehydrogenation is a slow reaction catalyzed by molybdenum and that the subsequent oligomerization and cyclization are catalyzed by the acidic zeolite sites. The resulting product stream of benzene, toluene, and mixed xylenes can be recovered without an extraction unit or sent to an aromatics complex for conversion of the toluene and mixed xylenes into benzene and p-xylene. The yield of this process, said Gates, is reported to be 58 to 60 percent. Rapid catalyst
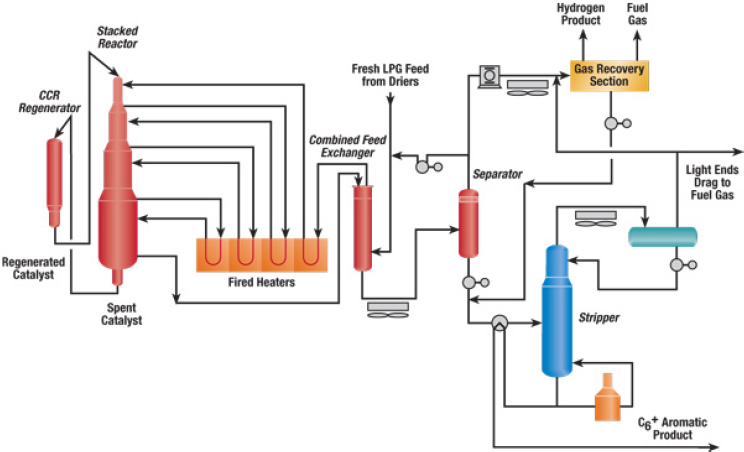
SOURCE: Bricker, 2016.
deactivation requires continuous regeneration using UOP’s moving bed reactor and catalyst regenerator design incorporated in its Oleflex process, but the robust catalyst has a substantial lifetime.
An alternative process, Chevron’s Aromax® process, uses a platinum cluster-zeolite catalyst to produce benzene and toluene, but it is more suited to converting larger alkanes, such as hexane and heptane, into aromatics. Gates noted that this reaction resembles naphtha reforming but without the acidic function in the catalyst.
Gates noted there are opportunities for discovering improved catalysts, including the use of metal-containing molecular sieves that have been shown to catalyze reactions such as hydrogenation and dehydrogenation. This is a large and growing class of catalytic materials, he explained, though many of these materials have not been well characterized and are not uniform structurally. He also explained that catalytic performance in any alkane-to-aromatic reaction scheme developed so far depends strongly on the structure of the metal-containing species. As a result, there is an opportunity for chemists to explore that structure–activity relationship with an eye on improving catalyst design through the many synthetic routes that have been developed to tune catalyst structure and other properties that influence activity. These synthetic routes include organometallic syntheses and atomic layer deposition, the latter
of which has been used to create zinc-containing catalysts that operate at atmospheric pressure and 823°C (Almutairi et al., 2012). Gates wondered if there were opportunities to use that type of approach for synthesizing well-defined catalysts containing metals such as zinc, gallium, and molybdenum in zeolites, and to create single-site catalysts.
As a conclusion to his presentation, Gates enumerated several possible directions for research. One approach would be to vary the metal or combination of metals in molecular sieves of different pore structures and sizes. Another avenue for research would be to attempt to tailor metal-containing catalytic sites on or in a molecular sieve framework, either as single sites or multi-atom clusters. He also suggested a research effort aimed at understanding the chemistry of catalyst synthesis and at relating catalytic activity, selectivity, and stability to structure using theory and spectroscopy with functioning catalysts. Gates noted the lack of, repeated frequently during the workshop, developing a deeper characterization of catalysts. He also thought it worthwhile to investigate processes that would use methane in combination with other feedstocks to produce aromatics.
Discussion
Johannes Lercher from the Pacific Northwest National Laboratory, acting as this group’s rapporteur, said the three major impediments to commercial viability of processes to convert light alkanes to aromatics are
- yield, which the group thought was surprisingly low;
- cracking, which produces methane and ethylene instead of aromatics; and
- capital costs related in large part to the product separations required.
Research approaches that could make production of aromatics from alkanes viable included varying the metal in the zeolite, which the discussion noted has been the subject of several patents involving the use of rhenium and tungsten. Other approaches would be to vary the zeolite structure and to balance the metal and acid function in the zeolite. With regard to the second of these, the working group discussed the possibility of speeding up the rate-limiting dehydrogenation step by using a gallium or zinc catalyst, but then it speculated that perhaps it was important for this step to be slow so that too much olefin did not accumulate in the zeolite pores so as to prevent higher oligomerization and runaway reactions.
Promising but higher-risk novel approaches described by this working group included the use of confinement-based catalysts to steer selec-
tivity, oxidative heating and aromatization to better manage energy use, and decoupling the dehydrogenation reaction and ring-closure reaction. With regard to this last possibility, Lercher noted there has been conflicting data in the literature so it was not clear whether this approach was a real possibility for industrial application or merely an interesting research project.
This working group discussed a long list of research opportunities, many of which, said Lercher, reiterate what other groups have proposed:
- Vary the metals, zeolites, and location of the active sites for the two reactions within the zeolites.
- Explore the chemical, structural, and mesoscopic properties of the zeolites across a wide range of structures and in a more exhaustive manner, particularly with regard to how well these structures stabilize or destabilize transitions states during dimerization and ring closure.
- Explore engineering approaches to optimize heat transfer for the endothermic reaction.
- Minimize cracking while maintaining dehydrogenation and cyclization activity, perhaps by better characterizing the role of cations in suppressing cracking.
- Determine the nature and mechanism of coke formation and devise strategies for limiting the sites at which coke is able to form or directing coke to form away from the active sites.
- Characterize the location of active sites in zeolite structures, their stability in the presence of reagents at process-relevant temperatures, and any factors that might increase the lifetime of the active sites.
- Explore other hydrothermally stable support systems beyond zeolites, such as tungsten-zirconia structures.
- Identify milder approaches to catalyst regeneration.
- Characterize the role of chemical potential on the active sites and how an active site might change as a function of the chemical potential within the reactor as a function of axial direction and reduction state.
- Develop methods for in situ operando high-temperature spectroscopy, pore size measurement, and microscopy.
- Use theoretical methods to explore mechanisms and how active sites are maintained or change over time.
Emerging Opportunities for Novel Approaches
The working group heard two short presentations, one from independent consultant Guido Pez on electrochemically mediated catalysis, and the other on biocatalysis from Mattheos Koffas, the Dorothy and Fred Chau ’71 Career Development Constellation Professor in Biocatalysis and Metabolic Engineering and professor of chemical and biological engineering and biological sciences at Rensselaer Polytechnic Institute. While electrochemical catalysis has potential as a means of converting hydrocarbons into value-added products, Pez said, one of its main limitations is the high relative cost of using electricity as a reagent to drive endothermic hydrocarbon conversion processes. Therefore, he explained, a more promising approach is to first conduct an exothermic, selective oxidation at the anode of an electrochemical system that would provide both electricity and chemicals from what has been called a “tailored” direct hydrocarbon fuel cell (Alcaide et al., 2006).
There are a number of thermodynamically feasible fuel cells for chemicals and energy cogeneration, said Pez. These include ethane plus oxygen to ethylene and water; methane coupling in the presence of oxygen to produce ethane and water or ethylene and water; and methane in the presence of oxygen to produce methanol. One group (Liu et al., 2016) has already demonstrated an ethane-to-ethylene fuel cell using a complex anode and cathode (see Figure 4-12). The published fuel cell was a 0.2 cm2 button cell, and as configured it achieved selectivity exceeding 90 percent and yields approaching 40 percent at 750°C, with only methane and trace amounts of carbon monoxide as byproducts. This system produced no carbon dioxide, so in a zero-carbon environment, it is possible to consider such as a system as a replacement for steam cracking of ethane, said Pez.
A methane to ethane and ethylene fuel cell has also been reported (Kiatkittipong et al., 2004; Quddus et al., 2010) using a lanthanum-aluminum anode and a lanthanum-strontium-manganese cathode. This system achieved 91 percent selectivity for ethane and ethylene, with the relative amount of these two products varying with temperature. At 1,273 K, this fuel cell produces ethylene almost exclusively, with only trace amounts of ethane, carbon monoxide, and carbon dioxide. There are also published reports of electrocatalytic conversion of methane to methanol (Fan, 2015; Lee and Hibino, 2011; Spinner and Mustain, 2013), but these systems required energy input.
Electrocatalysis does not have to happen solely in the context of a fuel cell. It is possible, said Pez, for electrochemistry to promote catalysis or modify catalytic activity (Katsaounis, 2010). One group has developed what it calls “non-Faradaic electroreforming” of methane to syngas that produces a dramatic decrease in reaction temperature and an increase in yield that exceeds the calculated thermodynamic equivalent (Oshima et
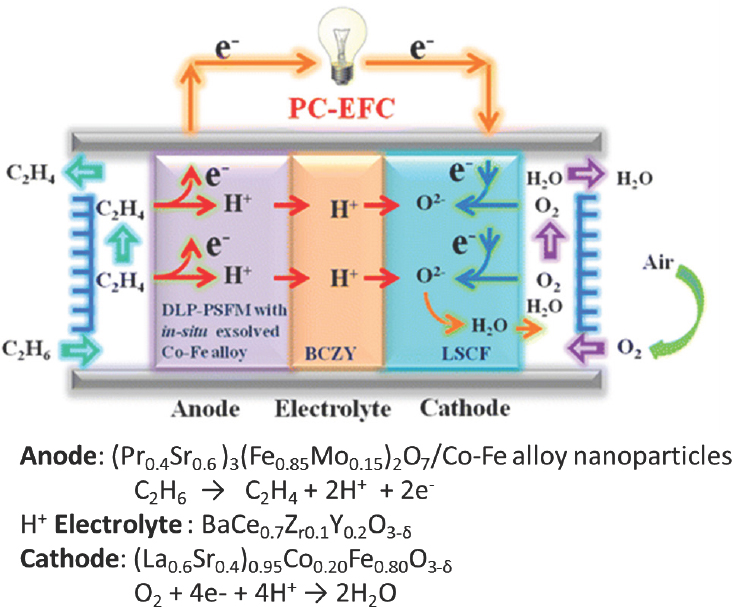
SOURCE: Liu et al., 2016.
al., 2013; Sekine et al., 2011), though with electrical efficiency of only 15 to 25 percent. Others have used spark discharge (Kado et al., 2003) or corona discharge (Marafee et al., 1997) to convert methane into ethylene, though with substantial power input.
Turning to the subject of biocatalysis, Koffas said that methane is an excellent source of carbon and energy for microorganisms known as methanotrophs, which historically have been used for producing feed-grade biomass. These bacteria are capable, he explained, of converting methane into protein, alkanes, alcohols, sugars, dicarboxylic acids, and other higher-value chemicals such as carotenoid pigments and vitamins. Currently, a plant in Norway is producing 850,000 tons per year of methanol and 10,000 tons per year of protein for animal feed from crude methane using the microorganism Methylococcus capsulatus.
One area of industrially motivated research aims to produce carotenoid pigments and antioxidants using a microorganism known as Methylomonas sp. 16a. The genome of this organism has been sequenced, said
Koffas, and this knowledge is being used to manipulate the organism’s metabolic pathways to produce different high-value carotenoids. He noted that prices for various carotenoids range from $500 per kilogram for β-carotene to $2,000 per kilogram for astaxanthin. Today, production is dominated by chemical synthesis, but researchers have engineered the organism to produce a variety of these valuable compounds and are now working to boost production to economically viable levels.
Discussion of Electrocatalysis
The discussion on electrocatalysis covered conversion of C1, C2, and C3 hydrocarbons to chemicals by use of electricity including limiting process economics. Methods covered were direct hydrocarbon fuels cells, electrically promoted catalysis, methane to syngas via electroreforming and methane to C2 hydrocarbons via electrical plasma processes.
In recapping the discussion on electrocatalysis, Monty Alger from Pennsylvania State University said that the outcome of discussion was straightforward: Researchers are working on fuel cells, others are doing work on materials development, a third group is studying electrocatalysis, and none of these groups is talking to one another, a point that he noted had been raised throughout the workshop. As far as specifics, the working group voiced interest in these electrochemical processes but the concern was that these technologies may not be viable at an industrial scale because of the difficulty in scaling the electrocatalytic systems and operating them at scale. Another barrier to commercial viability is the high expected cost of building industrial scale electrocatalytic reactors, whether they are fuel cells or systems based on non-Faradaic electrochemical modification of catalytic activity.
At one point in the discussion, Alger recounted, that it was mentioned that there has been a substantial body of work on ceramic membrane technology developed in recent years that could present an opportunity to advance an integrated solution for overall chemical conversion using electrochemical means. He noted that the opportunities that could result from merging membrane research and electrocatalysis are substantial and could lead to entirely new processes for chemical conversion. The discussion also pointed out that the drivers for fuel cell development are different than for catalysis.
Some in this working group stressed that the fuel cell community will not solve the challenges to developing industrial scale processes without collaborating with the catalyst, materials, and engineering communities. However, the group also recognized that funding is not available today for critical research on materials, such as transport membranes and mixed conductors, which will present challenges to collaboration. Others in
the group also noted that the catalysts used in these systems could be improved.
Discussion of Biocatalysis
The discussion about biocatalysis began with working group members pointing out that biocatalysis is being used in small-scale commercial processes. Calysta, for example, is converting methane into protein feed to reduce the aquaculture industry’s need for fishmeal, and Newlight Technologies has a demonstration plant that uses biocatalysis to convert methane emissions into an engineering polymer. The group also noted that, at least theoretically, anything that can be made biologically could be made from methane given enough time and money to do the necessary metabolic engineering. The resulting challenge, then, will be to select the best opportunities to pursue. However, one qualifier for that selection would be that the resulting biocatalytic process converts methane into chemicals with no carbon dioxide generation. That would be a unique outcome with a unique value proposition, Alger reported. A possibility the group mentioned was to couple biocatalysis with electrocatalysis to invent processes that convert methane to chemicals without generating carbon dioxide or water. A challenge for research in this area is addressing overall economics to be viable for long-term commercialization.
One of the biggest impediments to commercialization of biocatalytic routes is their poor yield of product. Therefore, improving yields, kinetics, reaction rates, and process costs related to separations will be critical for any commercially viable process to come out of biocatalysis research. The working group raised the possibility that the organisms developed through this research could be tainted by the “genetically modified organism” label, which could limit the ability to export products made using these organisms to certain regions of the world, and the group noted the potential challenge of having chemically trained people running biological processes. Another confounding issue for biological systems is the potential impact of natural gas impurities on the microorganisms.
One potential advantage of biosynthetic approaches to alkane modification is the possibility of making materials not currently accessible in high volumes or entirely new materials for which markets could be developed. Such systems may also be more economically viable at smaller scales than current industrial chemical processes, which could be important for utilizing stranded and flared gas. Biocatalytic systems may also have lower energy demands, though the cost of separating product from a biological reactor could negate any energy-related savings.
Activation of Natural Gas Using Nontraditional Oxidants
One of the main drivers of developing nontraditional oxidants for activating natural gas is the benefit of eliminating carbon dioxide emissions associated with electricity production, transportation, and chemical, agricultural, and other industrial processes, said Eric McFarland, professor of chemical engineering at the University of California, Santa Barbara. In the area of alkane conversion, it has so far proven impossible to partially oxidize alkanes with oxygen at high rates and low cost without producing carbon dioxide, he noted. As an example, converting methane to syngas for the production of methanol and other chemicals produces between 0.5 and 1 ton of carbon dioxide per ton of methane. Aside from the issue of carbon dioxide emissions, McFarland said there is another reason to look at alternative oxidants for hydrocarbon conversion, which is to make the best use of the chemical potential stored in the carbon–hydrogen bond.
Among the potential alternative oxidants McFarland listed were sulfur (Zhu et al., 2013), carbon dioxide (Cavani et al., 2007; Wang et al., 1999), nitrogen oxides (Cavani et al., 2007), and sulfur oxides (Hristov and Ziegler, 2003; Mukhopadhyay et al., 2005; Periana et al., 1993), though he devoted most of his remarks to the use of chlorine, bromine, and iodine. The halogens—chlorine, bromine, and iodine—are quite effective, he said, at oxidative dehydrogenation, which is why they are used as flame retardants. This has been known, said McFarland, since the late 1940s (Rust and Vaughan, 1949). In the 1960s, Shell developed a dehydrogenation process using molten iodine salts (Sanborn et al., 1968), and more recently, researchers have demonstrated the production of light olefins from methane and ethane using chlorine as the oxidant (Shalygin et al., 2013). While working with halogens presents some engineering challenges, halogen chemistry is practiced safely and profitably on massive scales, McFarland noted.
Among the benefits of converting methane to methyl halogens are that it preserves the chemical potential stored in methane’s carbon–hydrogen bond, the reaction product is easily separated from the reactants, and the hydrogen halide byproduct of the halogenation reaction also has value as an electron carrier. In fact, said McFarland, the catalytic reoxidation of the hydrogen halide by oxygen to produce the molecular halogen can be used to generate heat or electricity.
As an example of the halogen-mediated dehydrogenation reactions that he and his collaborators have explored, McFarland briefly described a process in which methane reacts with bromine at a moderate temperature of 400°C to produce methyl bromide, which is then catalytically coupled at 400°C to produce olefins, alcohols, aromatics, or ethers, depending on the catalyst. This mixture then passes over a solid metal
oxide to absorb the hydrogen bromide. The resulting solid metal bromide is regenerated with oxygen to produce metal oxide plus bromine for reuse (Lorkovic et al., 2004; Zhang et al., 2011). This reaction scheme can be used to convert ethane to ethylene, propane to propylene, and butane to butene at greater than 95 percent selectivity. The unresolved issues with these processes include the ability to regenerate the solid metal oxide, the reactive capacity of the solid metal oxide, and hydrocarbon stability over the solids. To address these and other challenges, McFarland and his collaborators changed their approach, using a molten halogen salt to generate the halogen in situ (see Figure 4-13), to control heat exchange, and to absorb and transport bromine and oxygen. This approach reduced the system complexity, he explained.
Discussion
Group rapporteur James Stevens, recently retired as the Dow Distinguished Fellow at The Dow Chemical Company, began the discussion with the comment that over the years he had seen numerous examples of methane activation with nontraditional oxidants involving the conversion of methane to methyl-X, where X is a leaving group, and that if he were to poll the workshop participants, each one could probably identify one leaving group that someone in industry or academia had tried and
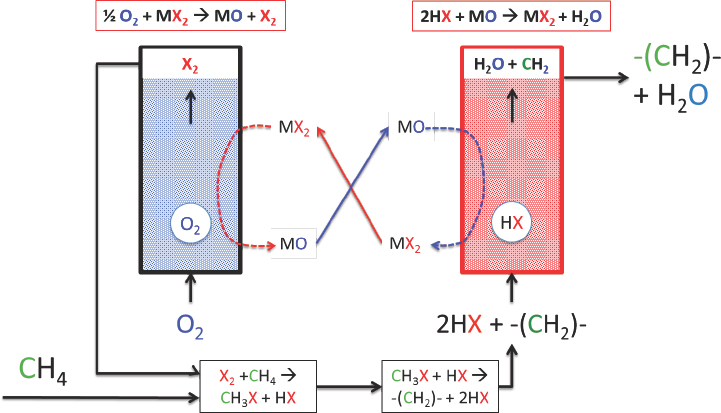
SOURCE: McFarland, 2016.
even piloted for converting methane to other hydrocarbons via this route. His point was that this group’s discussion was not so much about new science but rather about trying new ways of adapting existing science to meet specific needs. Having said that, he reported that the working group discussed a number of impediments for nontraditional oxidants to become commercially viable, including the lack of a tax on carbon dioxide emissions and the risk-aversive nature of the chemical industry with regard to new technology. The group also noted that current technology can work well in an environment where there is no penalty for emitting carbon dioxide. The group noted that dealing with corrosion issues in strong electrolyte environments raises the engineering and material demands and unknown safety issues, but these are not insurmountable if the economics become favorable. For example, the chemical industry has extensive experience in handling halogens and strong acids, and so the use of bromine to form methyl bromide and HBr, followed by coupling the methyl bromide to ethylene or other hydrocarbons, would probably not be a technical challenge for industry. There was a discussion on the use of oxygen in a cycle to form more selective oxidants, thereby moderating the selectivity. However, the relative lack of research on how to use oxygen to form more selective oxidants at industrial scales was also noted in the discussion as an obstacle to progress.
With regard to well-established research approaches for activating natural gas with nontraditional oxidants, this group pointed out that processes using halogens to make, for example, methyl chloride and methyl bromide, which would serve as intermediates to make value-added hydrocarbon products have been piloted by numerous companies. The use of bromine as a nontraditional oxidant for methane coupling has several advantages, particularly because the heat of reaction of methane with bromine is much lower than that with oxygen, while still being an exothermic reaction, which has the potential to make the bromination reaction more selective. In addition, the use of halogens as nontraditional oxidants has the potential to make product separation easier. The use of halogens to convert methane to ethylene and other hydrocarbons has not been commercialized yet, primarily because of economic rather than technical reasons. Some major drawbacks with the use of bromine include the heat management and the large bromine recycle stream that would be necessary. Ultimately, the commercial use of nontraditional oxidants is disadvantaged over current technology, as long as there is no cost for producing and releasing carbon dioxide.
There was work in the 1940s on a commercial process for reacting methane with sulfur to form carbon disulfide and hydrogen sulfide, which were then reacted to form ethylene, regenerating sulfur. This area was recently developed further by the Marks group (Zhu et al., 2013).
Sulfochlorination has also been demonstrated at scale, and again, economics are the only major impediment to commercial application. For promising but higher-risk approaches, the discussion noted that identifying new secondary regenerative and selective oxidants could have value, as could a process for selective mono–oxyhalogenation. Molten halide salt processes and solvents could offer an approach to recycle reactants and dissipate heat.
Research opportunities the group discussed as potential routes for overcoming obstacles for the use of nontraditional oxidants for natural gas included developing a more extensive knowledge base about strong electrolytes and identifying novel reactor and process materials for dealing with corrosive conditions. A better understanding of the kinetics of oxygen transfer and of multiphase reaction systems offers the possibility of improving oxidation reactions and the use of oxygen to form more selective methane oxidants and processes, while still using oxygen as the secondary oxidant. This was particularly noted for the formation of selective nitrogen-based oxidants and processes. The group also noted the lack of research on process engineering, including novel separations technologies. Each of these opportunities, the group pointed out, would be better addressed in collaborative efforts involving chemical engineers and chemists from industry, the national laboratories, and academia, which Stevens said was a common theme that others had mentioned throughout the day. This group also had a lengthy discussion, he reported, about how chemical engineering departments at U.S. universities may not be devoting enough attention today to teaching fundamental chemical engineering processes.
DISCUSSION
Alexander Orlov from the Institute for Advanced Computational Science at Stony Brook University pointed out there are two-dimensional materials, including graphene, carbides, and nitrides, that could hold promise as size-selective catalyst supports. Methods for scaling the production of such materials and research to develop those methods are lacking, which could perhaps tap into the funding opportunities associated with these materials.
Fabio Ribeiro from Purdue University said that this community can benefit from educating the public about the importance of making wise use of the nation’s natural resources, referring to shale oil and natural gas. Lercher remarked that in his opinion, the way to make the argument to those who want to leave shale gas in the ground, since it is not a renewable resource, is to point out that shale oil and natural gas, which has a lower carbon footprint, can serve as a bridge to a zero-carbon industry.
“This is not going to be the final solution, but a bridge that could last 30 to 50 years,” said Lercher. Speaking from an industrial perspective, Stevens said that while many of these opportunities could be considered “basic blocking and tackling,” it is becoming increasingly difficult to hire classically trained chemical engineers who know how to conduct these kinds of studies.
Bruce Gates turned the discussion to the subject of how to generalize the themes for how to go forward in catalysis research. Two themes that he heard repeatedly were to conduct operando characterization under more challenging conditions and to study single-site catalysts. Tobin Marks added developing more robust catalysts, and Lercher said studying carbon–hydrogen bond activation with the goal of creating more active and selective catalysts capable of operating at lower temperature. Moving forward, Mark Barteau from the University of Michigan suggested that the catalyst and process should be thought about as an integrated system. In Pez’s opinion, electrode catalysis should be a theme given the possibility of using fuel cells to make both chemicals and electricity.
Carl Mesters from Shell said in his opinion, catalysis is a tool for chemistry to make products society needs, and perhaps chemistry is taking too narrow a view of what products it can make when taking advantage of the abundance of shale gas. One such product would be graphene for electrodes, but he also suggested that the carbon in natural gas could serve as a feedstock for making strong but lightweight materials for the building industry. What catalysis science has not done is look at ways of taking the carbon in methane and turning it into materials with specific properties beyond those available today. The challenge, he said, is to broaden the opportunity space and look into what catalysis can do beyond energy and existing chemicals. Orlov noted that he and his colleagues have been studying the use of carbon to reinforce polymer composites and as soil amendments that increase productivity. “There are some unusual applications, especially if you go outside the discipline and to people at the agriculture department or in materials science for incorporating carbon into existing products,” said Orlov.
Alger voiced his support for that idea, but also cautioned that developing a new material is just the start of a process that has to include teaching the customer how to use the material and the virtues of a new material. Lercher noted that the chemical industry uses only 7 percent of the world’s methane, with the rest being burned as a fuel. As a result, it would have to develop a host of revolutionary technologies to have even a modest impact on decarbonizing the global economy.
This page intentionally left blank.


































