
5
Opportunities to Enhance the Capabilities of the Biotechnology Regulatory System
The profusion of products and the growing number of actors in the biotechnology space described in Chapter 2 present many challenges to the U.S. biotechnology regulatory system. The present chapter outlines a framework for risk analysis targeted at the types and scale of products anticipated and describes what tools, expertise, and scientific capabilities are required within and beyond the regulatory agencies in order to support oversight of future biotechnology products. The focus is not just on the regulatory process, but the broader context of presubmission and post-market activities that are an important part of the overall regulatory framework and that can provide a balanced approach to capabilities required for regulation of future biotechnology products.
As technologies and basic knowledge advance, a regulatory system should be able to adapt to new risks of future biotechnology products and also to adjust to well-established categories of products as their level and types of risk become better understood. As discussed in Chapters 2, 3, and 4, the scope, scale, complexity, and tempo of future products is expected to increase rapidly, and this increase has the potential to overwhelm the existing regulatory system. In addition, the new types of actors and new types of business models that will be involved in the development of technology and products means that the regulatory system will likely need to provide information to a broader group of stakeholders with diverse backgrounds and expertise. Finally, the possibility that some future products of biotechnology will be controversial may require substantial conversation and public debate throughout the phases of the regulatory process. A regulatory system with a greater emphasis on stratified approaches that prioritize the regulatory agencies’ familiarity with a product, the complexity of the risk assessment for the product, and the anticipated risk associated with the product (that is, proportionate oversight) could contribute to meeting the increased demands on the system.
The 2016 National Strategy for Modernizing the Regulatory System for Biotechnology Products issued by the Executive Office of the President recognizes the increased complexity of future biotechnology products and provides a strategic plan for ensuring that federal agencies can efficiently assess any risks associated with such products (EOP, 2016). It also describes several approaches to increasing public participation in the process and incorporating science-driven decision making
CONSISTENT, EFFICIENT, AND EFFECTIVE DECISION MAKING FOR FUTURE PRODUCTS OF BIOTECHNOLOGY
A key property of the U.S. biotechnology regulatory system, well articulated in the update to the Coordinated Framework, is that it effectively protects human health and the environment through a safety-evaluation process that can be understood by members of the public (EOP, 2017). As described in Chapter 3, the structure of the Coordinated Framework presents considerable flexibility for regulating future products of biotechnology but requires the agencies to appropriately apply their statutory authority. Multiple agencies may have jurisdiction over a given product, while other products may not be explicitly covered by any statute or federal agency. Either situation could lead to uncertainty in regulatory jurisdiction. The regulatory route may also be unclear at the time future products are developed. This uncertainty in regulatory jurisdiction “can make it difficult for the public to understand how the safety of biotechnology products is evaluated and create challenges for small and mid-sized businesses navigating the regulatory process” (EOP, 2017:1). As articulated in both the update to the Coordinated Framework and the National Strategy, the desired state is one in which there is a consistent, efficient, and effective decision-making framework that continues to protect human health and the environment. This section provides some properties of the risk-analysis system that will be important for meeting these goals for products anticipated in the next 5–10 years.
Preparing for Increased Scope, Scale, and Complexity of Biotechnology Products
A key theme throughout this report is the increase in scope, scale, and complexity that will accompany future biotechnology products. Scope is the new types of biotechnology products that have not yet been seen by regulators. Scale refers to the number of products as well as the number of variants of products that may interact with the regulatory system. Complexity refers to the number of traits that may be involved in a single product and the interactions between the various elements in a product. Increased scope and complexity are key components of future products that may have fewer or no comparators to nonbiotechnology products or no similar existing biotechnology products and thus little or no familiarity within the regulatory system.
Though the scale of products is likely to increase, some of this volume will be comprised of new products with a composition similar to existing biotechnology products with a history of characterization and safe use. Such products should be familiar to regulatory agencies and should have low complexity because the risk analyses for such products are well understood. The introduction of an already approved Bt protein into a new crop variety is an example. Another example of a product for which an a priori argument for familiarity and low complexity might be made is an organism that contains only a loss-of-function mutation in a gene or genes because such mutations arise spontaneously in nature. Provided the loss-of-function mutation does not create a new reading frame that encodes a novel protein, an organism with such a mutation is likely to be not complex in terms of risk analysis. A benefit of products that are familiar and not complex is the savings to regulators in terms of time and effort spent on designing and implementing risk analyses. These savings in time and resources can then be applied to devising and implementing risk analyses for products that are less familiar, more complex, or less familiar and more complex. It will be important in implementing the update to the Coordinated Framework (EOP, 2017) to make use of scientific tools to evaluate when new products can be categorized as familiar and not complex by comparison with the existing base of scientific knowledge and to apply appropriate oversight to those products (including no regulatory oversight, if appropriate) based on scientifically sound risk analyses.
Other new products—such as organisms with entirely new pathways assembled from many genes derived from multiple unrelated sources, perhaps including synthetic genes, and engineered
microbial communities planned for open-environmental release in which some community members contain engineered pathways—will pose challenges for the regulatory system because the regulatory agencies have not seen these types of products before and because the products do not have nonbiotechnology products to which they can be easily compared. Such products would be unfamiliar and have high complexity for the regulatory agencies.
Examples of products that pose new regulatory challenges are organisms engineered to contain gene drives, which are designed to introduce a trait that spreads throughout the species population. A trait could be designed to modify a species, such as one that reduces the species’ ability to transmit a disease, or to eliminate a species, which may be the case when trying to exterminate a particular disease vector from a geographical region. In the case of gene drives in insects, the same public health benefit of disease elimination could be attempted by releasing sterile males of the species (Krafsur, 1998; Benedict and Robinson, 2003), but the use of a gene drive may be more effective in reducing the population size of the target species. However, gene drives may pose new complexity for risk assessments if the speed with which the target-species population is depressed exceeds current ecological and evolutionary rates. Additional risk-assessment endpoints and pathways to those endpoints may also need to be addressed. Examples of pathways to risk-assessment endpoints could include the probability that off-target gene effects could result in an unanticipated phenotype, the probability that the gene drive could mutate and result in an unanticipated phenotype, or the changes that the system (or its mutations) could cause in a community food web. Although these examples do not represent new risk-assessment endpoints, they may require more sophisticated risk analyses, with consideration of increasingly complex interactions. As noted in Recommendation 6-3 of the National Academies report on gene drives (NASEM, 2016a:128): “To facilitate appropriate interpretation of the outcomes of an ecological risk assessment, researchers and risk assessors should collaborate early and often to design studies that will provide the information needed to evaluate risks of gene drives and reduce uncertainty to the extent possible.”
Another example of a new type of product is one that would enable the “deextinction” of a species. At the time the committee was writing its report, there were projects under way to “deextinct” the passenger pigeon and the woolly mammoth (or arguably a relative), among other animals (Biello, 2014; Callaway, 2015; Shapiro, 2015). If release to a natural ecosystem is a goal of such a project, a meaningful risk assessment should include wildlife ecologists and local experts from the area of release, including those with knowledge about migratory routes, to assist in assessing effects on the existing function and structure in the community.
An important approach for dealing with an increase in the products of biotechnology will be the increased use of stratified approaches to regulation, where new and potentially more complex risk-analysis methods will need to be developed for some products, while established risk-analysis methods can be applied or modified to address products that are familiar or that require less complex risk analysis. With this approach, new risk-analysis methods are focused on products with less familiar characteristics, more complex risk pathways, or both. Multiple criteria are usually embedded within risk analyses to ascertain if an estimated level of risk is consistent with the risk-management goals established during the problem-formulation phase of a risk assessment. In some cases, additional risk analyses may be needed to refine risk estimates, to evaluate risk-mitigation measures, or both. Criteria that could be applied to biotechnology products have also been used for risk analysis of other emerging technologies that integrate health, environmental, and life-cycle effects and occupational and socioeconomic risks, and these criteria can be weighted and rated by experts or stakeholders (Linkov et al., 2007; Tsang et al., 2014). In order to implement the appropriate rigor of risk analyses for new biotechnology products, it will be necessary to establish scientifically rigorous criteria based on factors affecting the perception of risk, the degree of uncertainty, and the magnitude of risk and nature of potential risks.
Enhancing the Responsiveness of the Regulatory System
At the time the committee was writing its report, there was no regulation, law, or statute to mandate a central review of biotechnology products or to develop an oversight system that is coordinated among agencies, minimizes gaps and redundancies in product review, provides more certainty for product developers as to the regulatory path, and embraces the principles of anticipation, participation, responsiveness, and transparency. The update to the Coordinated Framework (EOP, 2017) and the National Strategy (EOP, 2016) recognize the need for addressing these issues and provide a set of first steps for doing so. In this section, the committee provides some insights on how these topics might be addressed for the types of products that are anticipated in the next 5–10 years.
As described in Chapter 3, the statutory authorities that apply to some of the future products of biotechnology can be confusing and better coordination among the agencies would be beneficial so that risk analyses cover the impacts of biotechnology products more comprehensively in some cases or avoid duplication of data submissions in others. For example, as of 2016, genetically engineered insects were regulated by the U.S. Food and Drug Administration (FDA) under the Federal Food, Drug, and Cosmetic Act (FDCA) and environmental assessments were performed under the National Environmental Protection Act (NEPA).1 Crops with resistance to targeted insects through the insertion of genetic material from Bacillus thuringiensis were reviewed by the U.S. Department of Agriculture (USDA; under the Plant Protection Act to evaluate if the crop could be a pest) and the U.S. Environmental Protection Agency (EPA; under the Federal Insecticide, Fungicide, and Rodenticide Act [FIFRA]) to determine if the Bt toxin, the insecticide produced by the plant, would cause unreasonable adverse effects to humans and the environment); product developers of these crops also consulted with FDA (under the FDCA) before commercial release to ensure the food products derived from the engineered plant were substantially equivalent to corn products already in the marketplace. These examples underscore that developers of future products of biotechnology would benefit substantially from access to timely, consistent, and unambiguous feedback from the federal regulatory system as to whether or not a product is regulated and, if so, which agency or agencies would be response for regulatory oversight.
One possible approach would be to consider the development of a single “point of entry” as a mechanism for initiating the cross-agency cooperation that is articulated in the update to the Coordinated Framework and in the National Strategy. Box 5-2 provides an example of what such a mechanism could look like that would operate with the agencies’ existing statutory authorities. A collection of integrated resources could be maintained that provide a means for developers to initially determine if their product falls under regulation and, if so, an initial “read” on the regulatory pathway likely to be required for a future regulatory decision. A single point of entry could also provide an accessible public face for the regulatory system where interested parties can explore and understand the nature of the regulatory process. In addition, such a point of entry could be used to enable the federal agencies to decide early in the product-development cycle which authorities are relevant in cases where there have not been precedents. Throughout the process, developers would also have access to ombudsmen within each agency for additional assistance and feedback, including an opportunity to meet with the lead agency prior to a decision on a proposed oversight approach.
The concept of a single point of entry is already available for some distinct parts of the regulatory system; for example, crop developers can submit a letter to the “Am I Regulated” site2 of
___________________
1 In January 2017, FDA issued a draft guidance on mosquito-related products to clarify that its definition of nonfood regulated articles no longer included those “intended to function as pesticides by preventing, destroying, repelling, or mitigating mosquitoes for population control purposes. FDA believes that this interpretation is consistent with congressional intent and provides a rational approach for dividing responsibilities between FDA and EPA in regulating mosquito-related products” (FDA, 2017:6575).
2 Am I Regulated Under 7 CFR part 340? Available at https://www.aphis.usda.gov/aphis/ourfocus/biotechnology/am-i-regulated. Accessed January 15, 2017.
USDA’s Animal and Plant Health Inspection Service (APHIS) to find out if the agency considers their crop a regulated article. This process lets the crop developers know earlier if their crop is regulated or not, and it lets USDA know earlier what kind of crops are being developed. The concept is also a stated intent of the National Strategy. Descriptions were given in the National Strategy for multiple online resources maintained by each of EPA, FDA, and USDA, though these were not yet integrated at the time the committee’s report was written and hence product developers and other interested parties had to navigate multiple sites that reflect the complexity of the regulatory system and the agencies’ jurisdictions. There are examples from the European Union that collect together various product types into a single point of entry and provide a means for public consultation in the context of allergenicity assessment.3 A similar system for the U.S. regulatory system could provide a more easily navigated system for identifying the regulatory routes for a given product class.
It was not within the committee’s statement of task to delineate how a single point of entry could be crafted and implemented. As mentioned, such a mechanism could operate within the agencies’ statutory authorities and could range in concept from greater cooperation among the agencies in terms of sharing resources to more consistent and rigorous interagency working group collaboration. Alternatively, it could be operated by an existing coordinating unit within the executive branch or by a new agency created to be the “front door” for all biotechnology products, although the latter option would require new legislation from Congress. However it might be constructed, a key element of an effective single point of entry will be the establishment of criteria that provide guidance on the regulatory route that will be required. This guidance would not necessarily be exclusively consultative or structured through case-by-case deliberations. There are good examples of published guidance used within federal agencies that provide interested parties with relevant information, such as the content of agency website information regarding navigation through the system, and methodological guidance, such as EPA Guidelines for Ecological Risk Assessment (EPA, 1998) and FDA Guidance for Industry.4 There are clear needs for this information to be improved and continually updated, and this would be an important facet of the point-of-entry implementation. Internationally, regulatory guidance is more commonly available than in the United States; examples are the European Food Safety Authority guidance developed in response to European Union directives (EFSA, 2010, 2011a,b,c). Experience elsewhere with the use of such guidance could be considered when designing a single point of entry to be used within the Coordinated Framework.
As described in Box 5-2, the criteria for which bin a product would fall into would be based on familiarity with existing, regulated products (there should be greater certainty as to how to undertake a risk assessment with a familiar product as compared to an unfamiliar product). Additional product attributes such as the degree of confinement and/or containment (greater confinement/containment should reduce the likelihood of environmental exposure), whether it is living or nonliving (a living product may increase uncertainty and unpredictability of the assessment), and reversible or nonreversible product deployment (a nonreversible deployment may increase the complexity of risk-management measures to mitigate adverse effects) need to be considered in determining the appropriate bin for a new product (see Figure 5-2). The greater the amount and specificity of information a developer can provide for a product (including a proposed risk-analysis approach) through the single entry point, the more efficiently the agencies should be able to determine the product’s level of familiarity and the degree of complexity. The development and use of the multidimensional decision criteria for bin placement could be informed by external, independent peer review and input from interested and affected parties. Developers might be able to self-score their product as to the appropriate bin, but the ultimate determination would be an inherently govern-
___________________
3 Register of Questions. Available at http://registerofquestions.efsa.europa.eu/roqFrontend. Accessed January 15, 2017.
4 Guidance for Industry, Biotechnology Guidances. Available at http://www.fda.gov/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/ucm123631.htm. Accessed January 9, 2017.
mental decision by the appropriate regulatory authorities. The developer would be notified of the determination and provided a pointer to more information about the appropriate agency and point of contact. Consistent with the guidelines developed by the International Risk Governance Council (Renn, 2005; IRGC, 2015) and the 1996 National Research Council report Understanding Risk (NRC, 1996), the level of participation of outside experts, stakeholders, and interested and affected parties and the level of effort for both developer and the regulatory agencies would increase from the bin for familiar and not complex products through to the bin for unfamiliar and complex products. The model used by the International Risk Governance Council for managing different types of risk problems is illustrative of the degree of agency and stakeholder involvement that may be necessary depending on a product’s familiarity and complexity (Table 5-1). Thus, as complexity increases, so does a need for engaging external experts, industry stakeholders, and interested and affected parties in the dialogue.
The method and amount of public engagement for future biotechnology products would also vary according to familiarity and complexity. Products that are unfamiliar and complex could require external peer review and input from interested and affected parties. The peer review and input from parties would be facilitated by one or more of the appropriate/relevant agencies—broad agency engagement is desirable if additional, future product types are envisioned to have different regulated uses. Peer review or public engagement could be designed to protect confidential business information as needed. External peer review or external party input could be used for problem formulation and then for the subsequent draft risk assessment. Iterative risk-assessment and risk-management decision making may be appropriate based on the nature and extent of the estimated risk and associated uncertainties. Peer review and engagement by external parties on potential future products could also be initiated by the regulatory agencies based on horizon scanning. Undertaking such proactive, pilot projects will increase preparedness.
A product developer would not have to use the voluntary point of entry and could independently determine whether or not their product is regulated. If it determines the product is regulated, the developer could independently ascertain the statute(s) and agency (or agencies) appropriate for the situation and directly submit the product for review; if an incorrect determination was made, the developer could subsequently work with the regulatory agencies to route the submission to the appropriate agency. A developer could use in-house expertise, private-sector consultative legal and regulatory-science expertise, or both to provide general and product-specific guidance. At the time the committee was writing its report, this practice was common within the business community for
TABLE 5-1 Escalating Levels of Expert and Stakeholder Involvement and Effort in the Management of Different Types of Risk Problems
| Risk Problem | Simple | Complexity | Uncertainty | Ambiguity |
|---|---|---|---|---|
| Actors | Regulatory Agency Staff | Regulatory Agency Staff External Experts |
Regulatory Agency Staff External Experts Industry Stakeholders Affected Parties |
Regulatory Agency Staff External Experts Industry Stakeholders Interested and Affected Parties |
| Remedy | Statistical Risk Analysis | Probabilistic Risk Modeling | Risk Balancing with Probabilistic Risk Modeling | Risk Tradeoff Analysis and Deliberation with Risk Balancing and Probabilistic Risk Modeling |
SOURCE: Adapted from Renn (2005).
dealing with regulatory issues under the Coordinated Framework. The committee recognized from the presentations it heard from startup companies and small firms and from its deliberations that this approach is currently used, especially with those businesses with some degree of in-house experience and resources, but it is not easily or routinely used by a host of smaller enterprises entering into the biotechnology product space. Therefore, a formal structure governed through collaboration among the regulatory agencies, such as that described in Box 5-2, is an important consideration for future products of biotechnology.
Risk Analysis and Public Participation
In updating the Coordinated Framework and presenting the National Strategy, the federal agencies have taken into account the nature of biotechnology products that were visible in 2016. In looking at the products of biotechnology that are likely to emerge in the next 5–10 years, Chapter 2 describes some of the features of future products that will challenge the system and Chapter 4 articulates some of the challenges in applying the Coordinated Framework. In moving from products that are in columns B and C to those in column D of Figure 2-6, it will be important for agencies to be prepared for products that involve substantial internal complexity, complex interactions with the environment, relatively few or no comparators to nonbiotechnology products for use in risk analysis, and have little similarity with existing biotechnology products. In this section, the committee articulates some of the features of these types of products and provides possible perspectives on how risk analysis could be performed.
Natural-science evidence, social and economic evidence, and values all influence risk analyses for future biotechnology products (NRC, 1996; Thompson, 2007; Kuzma and Besley, 2008; IRGC, 2015). Given the diversity, pervasiveness, and power of new biotechnology methods and products, public concerns that have followed and are likely to continue to follow biotechnology products into the market, and increasing complexities and uncertainties associated with anticipating the human health and environmental effects of unfamiliar and unconfined releases of biotechnology products or living genetically engineered organisms, it will become increasingly important to develop oversight systems that are adaptive, iterative (learning from past experiences or new data and information, or new concerns that emerge), and engage a wider range of expertise (Wilsdon and Willis, 2004; Stirling, 2007; Meghani and Kuzma, 2011; Ramachandran et al., 2011; Marchant and Wallach, 2015).
The social-science literature suggests several middle-ground approaches to framing future conversations that could increase public confidence in the oversight of products of biotechnology. Paradigms of responsible innovation (Stilgoe et al., 2013), critical realism (Freudenburg, 1996), strong objectivity (Harding, 1996), and analytical–deliberative risk analysis (NRC, 1996) all recognize that what is available in the empirical world is useful but also that human interpretation brings meaning to that evidence and is just as crucial. These frameworks address concerns of multiple stakeholders and disciplines, consider what evidence or risk-mitigation strategies could help address those concerns, anticipate which of biotechnology products or processes should receive greater regulatory scrutiny and which should receive less, and prepare for future concerns and products by beginning the deliberations and identifying regulatory-science needs further upstream in product development (Barben et al., 2007; Kuzma et al., 2008; Guston, 2014). Life-cycle analysis of energy, water, and chemical inputs and outputs, risk–benefit analyses, the risk of doing nothing compared to alternatives, and cultural considerations (especially to disenfranchised groups) could also be part of the oversight. These approaches will be especially important for open-release, unfamiliar applications of biotechnology such as deextinction and gene drives and for other future biotechnology products that have complex interactions and risk pathways. These approaches may also be important to provide an opportunity for future governance that is science informed, public guided, and value attentive.
A common recommendation from prior National Academies reports is the need to increase public participation in the regulatory process (see, for example, NRC, 2008). As indicated already above, it is likely that future products of biotechnology could be controversial due to their complex interactions with the environment and society, and the committee anticipates that additional concern from the public will be a common feature of many future biotechnology products. Increasing public participation in the regulatory process raises the possibility of increases in agencies’ costs and inefficiencies in the overall decision-making process. Other parties may be concerned that such an approach could be fraught with complications in ensuring a balanced representation of viewpoints.
Oversight of complex and interdependent activities by their very nature requires input from multiple developers and interested and affected parties to develop and revise approaches over time. Formulating an agency approach for such complex scenarios “in secret” (bureaucratic closure) or behind closed doors with a select group of developers or interested parties (private bureaucratic learning) increases the risk of failure due to retribution from excluded participants or lack of agency capacity or statutory jurisdiction to address all the tasks needed for implementation (Moffitt, 2014). To explore these concerns, the committee considered ways additional external participation may be incorporated in those future biotechnology products that are unfamiliar and complex. The proposed uses of public participation and external peer review are generally consistent with a paradigm articulated by Moffitt (2014; see Figure 5-3). This paradigm acknowledges two dimensions in an agency’s regulatory decision making: (a) implementation independence to implementation interdependence and (b) lack of or incomplete information and understanding to full information and understanding.
In cases where a regulatory agency has high interdependence (for example, it is supporting a future voluntary, self-regulation system where the agency depends on technology developers for oversight implementation or its decision must be integrated with input from another agency) but has a high level of information, the agency could distribute information to developers that the agency depends on as well as to the public to transparently share current information strengths and limitations to develop the oversight approach (“participatory bureaucracy”). A participatory bureaucracy can increase the chance of success by exposing any information gaps and including the values of the developers and interested and affected parties in a voluntary program. When an agency has high interdependence and a low level of information, employing participatory bureaucracy can create new information by engaging input from experts, developers, and interested and affected parties. In cases where an agency has high interdependence, a lack of knowledge, and employs a closed process for making a decision, it increases the likelihood of “eroding bureaucratic administration when it prevents bureaucrats from acquiring needed expertise, from considering helpful alternatives or from learning from experience and mistakes” (Moffitt, 2014:47). Furthermore, a bureaucratic closed or private learning approach to developing and implementing an oversight approach could increase the likelihood of challenges (legal or otherwise) due to the opaque nature of decision making and the exclusion of informed input from groups (developers and interested and affected parties alike) outside the bureaucracy or outside the limited set of groups that were invited by an agency for consultation. This perspective is consistent with the findings of numerous science and technology policy scholars who have looked at biotechnology and concluded the same (Harding, 1996; Bozeman and Saretwitz, 2005; Thompson, 2007; Meghani and Kuzma, 2011; Kuzma, 2013).
One possible approach for including external input is through the use of advisory boards implemented through the Federal Advisory Committee Act (FACA) to assess strengths and limitations of alternative risk-analysis approaches by engaging developers and interested and affected parties and gaining external scientific peer review. The employment of a FACA process does not come time or cost free, and the costs of implementing such an approach could outweigh the benefits of gaining input and advice and sharing information (Balla and Wright, 2003; Box 5-3). There are also criticisms of the use of FACA groups that include allegations of privileging specific interest groups,
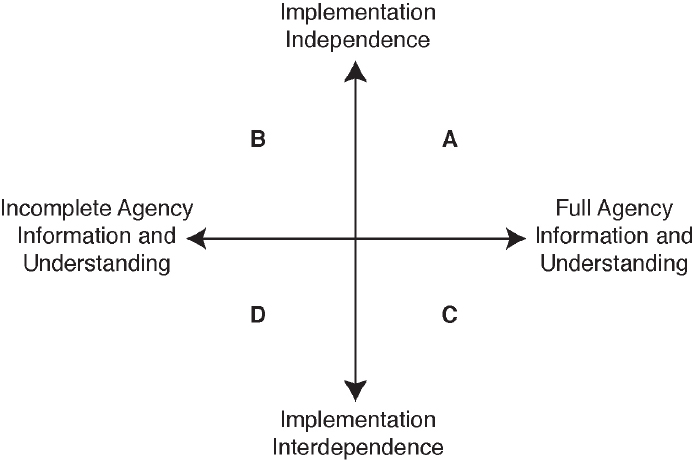
NOTES: In cases where an agency has regulatory independence and full knowledge to undertake its regulatory tasks, it could opt to make decisions “in house” and not share knowledge or decision-making logic publicly in a meaningful way (quadrant A, “bureaucratic closure”). If the agency has high independence and incomplete knowledge, it could gather information from outsiders “behind closed doors” (quadrant B, “private bureaucratic learning”). In situations where the agency has high interdependence, it could opt to share information, realizing the need to protect confidential business information, and create a process to take in and address additional information (quadrant C, “participatory bureaucratic oversight”). In cases where an agency has high interdependence but limited knowledge on an issue, it could publicly acknowledge its lack of information and initiate a public learning process to inform the future decision (quadrant D, “participatory bureaucratic learning”).
SOURCE: Adapted from Moffitt (2014).
limiting use of agency expertise, promoting secrecy rather than transparency, or driving acceptance of an agency’s position rather than receiving advice and input on an agency’s options (Moffitt, 2014). These concerns need to be addressed in an advisory process, and the committee suggests the agencies consider the program-management and conduct-of-practices principles provided in the 2008 National Research Council report Public Participation in Environmental Assessment and Decision Making, which include
- Clarity of purpose and diagnosis of context,
- Commitment to use the process to inform their actions,
- Adequate funding and staff,
- Appropriate timing in relation to decisions,
- Focus on implementation,
- Monitoring of the process and adjusting tools and techniques as needed,
- Inclusiveness of participation,
- Collaborative problem formulation and process design,
- Transparency of the process, and
- Good-faith communication.
Ginsberg (2015) also noted that an effective FACA process entails securing clear agency commitment; finding a balance between responsiveness to the agency and independence; leveraging resources through collaboration with similar FACA groups; and evaluating a FACA group’s usefulness to identify future directions or improvements.
The committee also reviewed the potential role stakeholder rulemaking and private standard setting could play in enhancing efficiency in the proposed decision-making framework. The committee concluded in some circumstances these approaches may be preferable to a FACA process or a process in which the agency independently establishes and implements a regulatory process or requirement (see Box 5-4).
TECHNICAL TOOLBOX AND CAPABILITIES FOR RISK ASSESSMENT AND REGULATORY SCIENCE
The committee synthesized information received during public meetings, webinars, and the results of National Academies reports (NRC, 2013; NASEM, 2016a,b), symposia (Drinkwater et al., 2014; Roberts et al., 2015), and relevant publications to identify gaps in risk-analyses tools and possible approaches that could be advanced to close these gaps. Addressing these gaps through a design-build-test-learn paradigm can help support development of a responsive research agenda and staffing plans for enhancing existing capacity, capability, and expertise needed for efficient and sound evaluations of future products of biotechnology. Separately, the tools and technologies used by product developers could be enhanced to ensure a higher probability of success in navigating the regulatory system. Finally, the committee identified specific needs in the area of regulatory science. The committee recognizes that the tools and techniques described here require a depth of data and analysis that may be inconsistent with the degree of risk that can be anticipated for many future products of biotechnology. Their blanket application would be inconsistent with tiered risk-assessment strategies and with the capacity available in the public and private sectors. The intent of the committee is to highlight the emergence of these approaches and their application for clarification of regulatory understanding of future products, especially when qualitative or deterministic risk analyses are uncertain as to whether they are incorporating well-characterized, worst-case assumptions to support the safety standard associated with risk-management decision criteria (see Box 4-2).
Implementation of Probabilistic Risk Analyses Associated with Future Products of Biotechnology
As discussed in Chapter 4, probabilistic risk analysis has not been widely used in the regulation of biotechnology products. However, the use of quantitative risk assessment is well established in many fields and is applied to questions of ecological, food-safety, biosecurity, and biological risk. The most common application of quantitative risk assessment for biotechnology products is for the purposes of insect-resistance management of Bt crops (Storer, 2003), but there are also examples of quantitative nontarget-species ecological risk assessment (Sears et al., 2001), dietary exposure assessment (Exponent, 2005), and endangered-species risk assessment (Peterson et al., 2006) that have been used for regulatory decision making. The Coordinated Framework would benefit from fuller implementations of probabilistic methodologies when appropriate in light of challenges to the regulatory system that are expected to occur. This section proposes the need for more probabilistic risk assessments than were conducted when the committee was writing its report. In addition, this section discusses the need to conduct risk analyses that are proportional to the quantitative risks assessed to prevent the problem of “second-order risk” (that is, the risk of missing a significant risk versus the risk of overanalyzing a negligible risk).
Biotechnology products are diverse and therefore may vary in their associated risks. That is, some biotechnology products could be used with a lower probability of risks (for example, crops genetically engineered with insect resistance or bacteria within bioreactors that are similar to engineered products already in commerce with a familiar risk profile) while other biotechnology products may have uncertain risks at greater spatial and temporal scales to consider (for example, organisms with gene drives or genetically altered bacteria released into an open environment). For
better understood products, available information from analogous systems or organisms or analyses of the published literature (for example, meta-analyses or pathway analyses) may suffice to assess associated risks. In contrast, unfamiliar products for which there is not a sufficient baseline of information may require more sophisticated quantitative analyses to estimate their associated risks. In cases of high uncertainty, data may need to be generated to be able to estimate risk with acceptable confidence.
Probabilistic approaches are summarized by recent National Academies reports and the scientific literature (for example, Suter, 2007; Warren-Hicks and Hart, 2010; NRC, 2013; NASEM, 2016a). A National Research Council report (NRC, 2013) described three principle steps in preparing a probabilistic risk assessment, which the present committee concludes are also applicable for assessing risks of biotechnology products:
- Describe uncertainty for variables with distributions (realizing all variables in a model need not require the same degree of data intensity).
- Propagate uncertainty through distributions of exposure and effects variables.
- Integrate exposure and effect estimates to calculate risk probabilities.
Example calculation methods include Monte Carlo analyses, Bayesian methods (some of which also use Monte Carlo simulations), and uncertainty bounding analyses (Warren-Hicks and Hart, 2010; NRC, 2013; NASEM, 2016a). At the time the committee was writing its report, probabilistic approaches were rarely implemented in ecological risk assessments for chemical pesticides (NRC, 2013). On the basis of the committee’s limited survey of existing risk assessments, environmental assessments, and environmental impact statements for biotechnology products, probabilistic analyses have seldom been undertaken. The committee believes that the risk analyses customarily conducted in environmental assessments and environmental impact statements required by NEPA may be inadequate to characterize the risks of certain future products of biotechnology. The committee found no statutory restriction that precludes the regulatory agencies from conducting quantitative risk assessments.
The further need for quantitative approaches for human health and environmental safety involves questions of multiple exposures, complex mixtures, and vulnerable populations, which represent broad stakeholder concerns often considered to be inadequately captured in risk analyses. A recognized need in quantitative risk assessment is improved cumulative risk assessments combining risks of aggregate exposure to mixtures that include all routes, pathways, and sources (NRC, 2009). Revision and extension of existing approaches to cumulative risk assessment will be needed to fully analyze future products of biotechnology.
Even under conditions of unfamiliarity and complexity, probabilistic risk assessments can be used to identify where information is missing. Several researchers from the Commonwealth Scientific and Industrial Research Organisation in Australia have used a combination of stakeholder, expert, and public input; Bayesian elicitation; and fault-tree analysis to develop and quantify (with uncertainty) risks from genetically engineered fish (Hayes et al., 2014) and genetically engineered insect pests (Murphy et al., 2010; Murray et al., 2016). These serve as models for both probabilistic risk analysis and public engagement in an analytical–deliberative process (NRC, 1996).
The quantitative risk analyses discussed above support the means to refine risk analyses by incorporating new data through iterative assessments and enable risk assessors, risk managers, and stakeholders to refine risk-management options as needed to meet the regulatory standard for a safety finding (NRC, 2013). An established probabilistic risk-assessment framework for a given product for a suite of use-pattern scenarios (such as those proposed in Chapter 4) also can facilitate timely updates to risk estimates based on new information and help form hypotheses for causes of unexpected risks that may emerge.
The regulatory agencies vary in the degree in which risk-assessment tiers can be implemented in concert with risk-management needs. For example, EPA’s pesticide risk-analysis approach uses risk-assessment tiers with increasing resolution based on the results of lower-tier risk assessments, the endpoints of concern, and the nature of requested use patterns. In addition, EPA can implement a higher-tier risk assessment to refine an existing assessment, based on adverse-effect information submitted through Section 6(a)2 of FIFRA (40 C.F.R. Part 159)5 and Section 8(e) of the Toxic Substances Control Act (TSCA).6 In addition, FIFRA requires EPA to reevaluate registered pesticides at least once every 15 years to ensure the existing risk analysis and regulatory decision are current with the state of the science and policy (40 C.F.R. Part 155).7 It is more difficult for USDA–APHIS to implement iterative risk analyses because the agency as of 2016 did not have authority to reassess products once they were deregulated (McHughen and Smyth, 2008).
Ecological Risk Assessment Within the Context of Future Biotechnology Products
Ecological risk assessment for future biotechnology products and their release scenarios will necessitate more emphasis on measurement and modeling of effects to populations and communities within landscapes than has been necessary with biotechnology products regulated in the 1990s and 2000s. Further challenges arise regarding the biological responses that are used to determine effects to entities of concern for ecological risk assessment (Forbes et al., 2001). The relationship between lethal and sublethal effects to individuals and the survival and reproduction of populations is a continuing uncertainty in the ecological risk-assessment process (NRC, 2013). Typical laboratory toxicity tests focus mostly on individuals through measurements of lethality, growth rate, or both and occasionally have been extended to measures more directly representative of populations (reproductive success). Field-scale studies may more fully encompass populations and communities through consideration of abundance for greater numbers of taxa (Naranjo et al., 2005). An emphasis in ecological risk assessment on individuals in and near production fields is logical and has been successful in understanding single-stressor effects within fields of genetically engineered crops as of 2016.
The environments in which some future biotechnology products will be deployed, however, will represent a dynamic temporal–spatial mosaic where multiple novel stressors with sometimes overlapping effects are being introduced at large geographic scales such as a watershed or geopolitical region and where there may be incomplete quantitative description of effects on populations. Simple approaches for lower-tier screening that may consider effects that may scale in the environment include simple functional ecology models based on life statistics for trophic–functional types to determine the magnitude of effect necessary to become evident in the ecosystem (Raybould et al., 2011) or considerations of aggregate sensitivity to species occurring within the environment (Wolt and Peterson, 2010; Wolt, 2011). These approaches, however, still place boundaries on the system to encompass limited spatial and temporal scales, thus leaving unanswered changes occurring in the contiguous landscape over time. Future products of biotechnology designed for open release in minimally managed or unmanaged environments will introduce an increasing diversity of potential environmental stressors that will necessitate improved ecological risk assessment to forecast potential effects with a view toward understanding and managing ecological services at the landscape level. The limitations of species-specific modeling and measurement in landscapes
___________________
5 Incident Reporting by Pesticide Manufacturers/Registrants. Available at https://www.epa.gov/pesticide-incidents/incident-reporting-pesticide-manufacturers-registrants. Accessed September 14, 2016.
6 Reporting a TSCA Chemical Substance Risk Notice. Available at https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/reporting-tsca-chemical-substantial-risk-notice. Accessed September 14, 2016.
7 Registration Review Process. Available at https://www.epa.gov/pesticide-reevaluation/registration-review-process. Accessed September 14, 2016.
argue for a more generalized approach focusing on functional groups and their distribution and density among elements within the landscape (Caron-Lormier et al., 2009, 2011) for certain products intended for open release in low-management environments.
To explore how ecological risk assessment might be applied, government agencies could pilot advances in ecological risk assessments and benefit analyses for open-release products expected over the next 5–10 years. Aspects to be explored could include external, independent peer review, public participation, and whether agencies’ staff will need new skills on quantitative risk-assessment practices. Risk assessors have used stakeholder and public-informed processes for broader ecological risk analyses of genetically engineered crops and fish to incorporate on-the-ground knowledge and values associated with multiple ecological and societal risk-assessment endpoints, especially in stages of problem formulation and risk-management options (Nelson et al., 2004; Kapuscinski et al., 2007). Multicriteria approaches to choose ecological indicators for risks to biodiversity and fault-tree analysis have also been applied to genetically engineered plants (Andow et al., 2013). These examples point to integration of multiple risk-assessment endpoints, modeling approaches, and societal values in the risk-assessment process. The agencies would benefit from a review of over two decades of literature on iterative and engaged methods of risk analysis for transgenic organisms.
Public–private investments in new environmental risk–benefit analytical approaches, including the identification of information needs; the development of assay methods and laboratory and field-study designs and monitoring protocols; and models (conceptual through computational) to inform risk assessments across appropriate biological, spatial, and temporal scales can also be used to address potential ecological outcomes associated with future open-release biotechnology products.
Enhancing the Capabilities, Expertise, and Tools of Regulatory Agencies
In the previous sections of this report, needed risk-analysis knowledge and technological capabilities were noted. Chapter 2 describes types of future biotechnology products, many of which will not have obvious comparators to nonbiotechnology products and in turn may require a new generation of risk-analysis approaches. Some of the use patterns for future products also highlight the need for developing spatially and temporally explicit risk-assessment capabilities. In addition, Chapter 2 points to the potential increase in the sheer number of products that may need to be assessed in the future, which highlights the need for an effective, high-throughput risk-analysis system. In Chapter 4, the need for probabilistic assessments to better interpret comparative risk assessments and management options was introduced. The above section “Consistent, Efficient, Effective Decision-Making Processes for Future Products of Biotechnology” also raises the need for assessing similarities and differences among biotechnology products and anticipates a stratified assessment process that in some cases will be highly reliant on access to existing risk-analysis data or data summaries for biotechnology products already in the market. To this end, a suite of publicly available physical and computational models and methodologies that can be accessed for risk assessments with different degrees of complexity would be helpful. Examples of sampling designs and indicators to support post-market surveillance and monitoring programs would also be beneficial.
To organize the discussion on risk-analysis tools needed for products expected in the next 5–10 years, the committee adapted categories of future research needs prepared through workshop deliberations addressing the need for a research agenda exploring the ecological implications of synthetic biology (Drinkwater et al., 2014) and a workshop and Delphi study on synthetic-biology governance (Roberts et al., 2015). The research areas identified to address gaps in risk analyses include many of those the committee sought information for in its request for information (RFI) to federal agencies (see Chapter 4 and Appendix C): comparators, off-target gene effects, and phenotypic characterizations; gene fitness, genetic stability, and horizontal gene transfer; control
of organismal traits; monitoring and surveillance; modeling and life-cycle analyses; and economic and social costs and benefits. The responses to the RFI indicate that some work is being done in these areas, but the committee thinks it is likely insufficient for the number and kinds of biotechnology products the agencies can expect to see. The committee also identified molecular characterization and standardization of risk-analysis methods and data management as areas in need of research.
Comparators, Off-Target Gene Effects, and Phenotypic Characterization
As discussed earlier in this chapter, there is a need to advance quantitative comparisons that can facilitate assessment of future biotechnology products. A key characteristic of the risk-assessment process in use at the time the committee was writing its report was comparability between biotechnology products and their nonbiotechnology counterparts. However, as noted in Chapter 4, the use of nonbiotechnology comparators is becoming more challenging. Transformations can be made in host organisms that are not well characterized, and there may not be baseline data on the nontransformed counterpart host. Furthermore, some new biotechnology products may contain only synthetic DNA, which would have no nonbiotechnology counterpart. Therefore, the idea of “comparator” may need to expand to include similar existing biotechnology products with which regulatory agencies already have experience.
Methods to quantitatively compare products will be needed for determining which bin is appropriate for a new product; selecting data from other product data sets for screening-level risk assessments or problem formulation; selecting data to use in effects or exposure analysis steps in a risk assessment; and selecting data to generate a risk characterization of a new product and/or place a characterization of a new product in context with an existing, similar product. Elements of a risk assessment are typically considered against baseline nonbiotechnology comparators to address whether, other than the intended change of the modification, the observed attributes of the transformed organism represent a substantive change relative to the comparators. The degree of uncertainty in making comparisons will need to be quantified given that different risk-assessment steps or scenarios can tolerate different levels of uncertainty at key decision points; that is, findings in risk assessments are worded in language specific to the statute under which they are being evaluated, but in every instance represent an “as safe as” determination.
Research approaches need to address questions raised by risk assessors and managers concerning comparisons that are context specific and reflect the need to assess similarity across levels of biological organization and spatial and temporal scales. Issues and questions raised in risk analyses can inform development of a research agenda. For example, products may be comparable at one level of biological organization, but not at other levels (for example, target genes and off-target genes and their expression, to protein structure and function, to biochemical function, tissue/organ function, organismal, population, and community effects). There may be variability in comparability for the same biotechnology product under different environmental conditions. Products may be comparable in terms of affecting a common physiological function, but the mechanisms by which they initiate the physiological responses could be very different. Some products may be comparable in terms of the genes being manipulated, but the commercial application of the products and their use patterns may be different. Depending on the risk-analysis question, the products may be considered comparable (that is, two open-release products initiate perturbations in the same invasive weed organism in a similar manner, but with some differences in off-target gene effects), but in the context of their environmental-effects analysis and their impact on nontarget organisms they may or may not be comparable: If one product has been deployed in Gulf Coast estuaries, how comparable will its effects be to the other product’s effects if it is intended for open release in estuaries along the U.S. mid-Atlantic coast?
Research in this area will also need to support computational approaches for estimating missing data from data available for existing, comparable products and identifying gaps in specific information that may require targeted testing. A systematic approach, taking advantage of horizon scanning, to establish the biological knowledge bases needed to inform computational similarity analyses systems and develop decision-support systems to facilitate analyses will also be needed.
In addition to comparators, research on phenotypic characterization is also needed to advance understanding of trait function and potential ecological consequences over the short and long term as well as understanding on how environmental context can affect phenotypic expression.
Gene Fitness, Genetic Stability, and Horizontal Gene Transfer
Engineered organisms that reproduce can suffer mutations that affect the physiology of the organism, leading to the potential for “instability” in the genome (engineered genetic constructs mutating in ways that could cause loss of function). In addition, many organisms can incorporate DNA from their environment, leading to the possibility of horizontal gene transfer.8 Techniques to measure these properties, including how these properties may vary with different environmental interactions, are needed. This research area includes evaluation and advancement of environmental models to assess properties; engineering for unanticipated interactions; developing standardized metrics and quantitative thresholds; and the interplay of fitness and stability, especially if an organism loses its containment mechanism.
Future approaches for risk assessment can be more streamlined, less costly, more comprehensive, and unbiased by utilizing state-of-the-art assay tools—for example, automated high-throughput biochemical assays, next-generation DNA sequencing, and advanced mass spectrometry technologies—integrated with high-capacity data storage and analytics. Rather than obtaining a targeted snapshot of single-few genomic loci via Southern blot, polymerase chain reaction, or Sanger sequencing to characterize a genetic modification, risk assessment at the molecular level should leverage recent advances in next-generation DNA sequencing and associated whole-genome sequence information to obtain an unbiased assessment of both the targeted and off-target genetic modifications in the species, whether altered by biotechnology or not (Pauwels et al., 2015). Similarly, untargeted mass spectrometry for metabolomics and proteomics is one approach for enhanced safety assessment of biotechnology products because it provides an unbiased assessment into potential pleiotropic effects derived from a modified organism (Ryals, 2016).
Control of Organismal Traits (Containment and Confinement)
Given that open-release products are deployed in dynamic environments, quantitative assessments of the safety, security, and stability of biotechnology-derived organisms should be tailored for the proper context. Metrics to test for biotechnology-derived organisms in open environments should measure
- Intrinsic biocontainment (i.e., escape frequencies into the natural environment),
- Genetic isolation (i.e., flow of horizontal gene transfer),
- Watermarking (i.e., unique sequence identifiers in the genomes of biotechnology-derived organisms), and
- Functional impact on the environment, including on nontarget organisms.
___________________
8 Horizontal gene transfer is common in nonbiotechnology organisms.
Possible areas of research include biocontainment schemes that can be adaptive to different intended applications, environmental settings, or both; establishing redundant, stacked containment approaches; and assessing the reliability of engineered reversibility.
Monitoring and Surveillance
Following completion of a premarket risk assessment, with a decision to allow the use of the products under specified conditions, there may be a need for monitoring or surveillance to evaluate specific assumptions in risk assessments, to address uncertainties in the evaluation of a risk hypothesis in an assessment, to assess the effectiveness of any required risk-mitigation measures, or all the above. In instances where products enter the marketplace through a notice to the appropriate regulatory agency, post-market monitoring or surveillance may be used to determine if future risk analyses and potential risk mitigation may be needed following use of the products (for example, cosmetics). To ensure data obtained from monitoring or surveillance address risk-management needs, designs and indicators need to be developed to directly address specific areas of uncertainties and risk hypotheses.
Examples of questions that may need to be answered through monitoring and surveillance for different types of products include the following:
- What is the current baseline of allergic responses to certain classes of cosmetics and the contribution of specific products? Has the introduction of a new class of living cosmetics increased or decreased the rate of allergic responses?
- Has the removal of contaminants by a consortium of microbes at a site met the remediation goals, has the consortium been confined or contained as planned, and is the habitat responding as predicted?
- Has the introduction of a gene drive to suppress a pest population achieved the suppression as predicted, and has the ecosystem responded as predicted with the removal/suppression of the target pest? If not, why not? Has the gene drive appeared somewhere it was not supposed to (that is, in a nontarget organism)? Has the gene drive mutated?
- Are the discharges of living engineered microorganisms into publicly owned treatment works or receiving waterbodies altering existing microbial communities in an unanticipated manner?
The sampling designs for monitoring and surveillance—for example, stratified, probability-based survey designs or fixed-site sampling programs at the national, regional, state, or watershed scale—will need to be established to address specific questions that arise for specific products or types of products. Frequency of sampling also needs to be established for addressing specific questions. Perhaps some questions concerning future biotechnology products could be integrated within existing monitoring programs (with inclusion of new indicators), while other questions may require unique monitoring programs, sampling designs, and diagnostic indicators. Although open-release products will offer little opportunity for environmental recall should unanticipated ecological effects be observed, the committee observed that environmental release in managed versus unmanaged or low-management conditions presents differences in complexity that will influence monitoring designs and potential variations in risk management.
Research to support monitoring and surveillance will be needed to assess movement and effects of specific product applications and may also be needed to provide a broad-based assessment of environmental conditions. Indicators will be needed to assess status and trends at the molecular level (for example, metagenomics) and to track changes in structural or functional attributes of ecosystems. Monitoring designs and protocols will likely be established and directed for specific
issues, but research is needed to ascertain the extent to which data sets derived with different survey features and indicators can be integrated to maximize the use of available resources.
Modeling and Life-Cycle Analyses
Both physical and computational models will be needed to help inform uncertainties in risk assessments. Physical models, such as mesocosms or controlled field studies, can provide information in specific places and time periods. For example, mesocosm experiments with GE versus wild-type Japanese medaka (Oryzias latipes) were used to assess gene flow over time in the life cycle of the fish (Pennington et al., 2010), indicating that such studies are possible and exist in the academic literature but are not routinely used in USDA or FDA assessments with live organisms. Computational models can be used to support the development of conceptual models within the problem-formulation phase of a risk assessment and to predict ecological and evolutionary responses in other places and time frames (that is, over decades rather than several years) that cannot be evaluated with a physical model. In some cases, the findings of a computational model may be needed prior to undertaking outdoor studies to help ascertain if there is an acceptable level of risk to undertake a study. Results from a computational model can provide insights for designing experiments with physical models. Optimally, collection of data through the use of physical models and computational models develops iteratively, each informing the other (NRC, 2007). Furthermore, a 2007 National Research Council report (2007:102–103) recommended
Using adaptive strategies to coordinate data collection and modeling should be a priority of decision makers and those responsible for regulatory model development and application. The interdependence of measurements and modeling needs to be fully considered as early as the conceptual model development phase. Developing adaptive strategies will benefit from the contributions of modelers, measurement experts, decision makers, and resource managers.
Research is needed to advance physical (for example, microcosms, mesocosms, and controlled field studies) and computational models to improve understanding of the ecological implications of genome engineering and to reduce uncertainties in predicting future ecoevolutionary dynamics over time frames of years to decades, which will support life-cycle analyses. Identifying gaps in current physical and computational models is needed to prioritize desired, future capabilities.
Physical Models. Microcosms, mesocosms, and controlled field studies are approaches to generate data that can reduce uncertainty in assessing potential effects across levels of biological organization, space, and time (Drinkwater et al., 2014). The 2016 National Academies report on gene drives articulated a phased testing approach for gene drives that includes research preparation, laboratory research, field research, staged environmental release, and post-release surveillance to gather information to support risk assessments and risk-mitigation measures to reduce potential nontarget effects (NASEM, 2016a). That report also provided examples of field and environmental field research for biocontrol and existing engineered organisms. External peer reviews of effects of herbicides in aquatic ecosystems (EPA, 2012b) and effects of insecticides on honey bees (EPA, 2012a) also provide insights on the design and execution of mesocosm and field studies that are intended to support ecological risk assessments. Clarity in ecosystem definition and model system design (for example, its size and composition), type of risk-assessment endpoints and responses measured (including recovery of community structure and function), and approaches to interpret and extrapolate data are important features of successful studies.
Although the need for undertaking field studies to evaluate future biotechnology products is recognized, EPA in 1992 determined that field studies or mesocosm experiments for pesticide registrations would no longer be required due to uncertainty in data interpretation and a conclusion
that the information gained from such studies did not alter risk-assessment conclusions based on data derived from laboratory studies (EPA, 2004). The agency can, however, conditionally require mesocosm and field studies for chemical pesticides9 and plant-incorporated protectants (Rose, 2007) on a case-by-case basis. USDA–APHIS uses a similar rationale for tiered testing regimes extending from the laboratory to the field. The current, limited experience in using results from physical models to inform ecological risk assessments indicates proactive research is needed on the development of study designs and risk-analysis methods for future open-release biotechnology products. Pilot efforts could be undertaken to develop and evaluate new approaches for using physical models to assess population, community, and ecosystem effects. Advances should be linked to the design-build-test-learn cycle and the scaled release of biotechnology products (from laboratory scale to small field trials, larger field trials, and eventually full-scale deployment). Consistent with this perspective, the 2016 National Academies report on gene drives (NASEM, 2016a) noted that support mechanisms for risk assessment, public engagement, and governance will be needed throughout a phased testing scheme.
Computational Models. Some future biotechnology products could be assessed with a high degree of specificity concerning spatial and temporal dimensions (for example, bacteria within bioreactors) while assessments for other biotechnology products have a more complex dimensionality to consider (for example, open-release organisms with gene drives and genetically altered bacteria consortia for open release). For new products, probabilistic risk assessments that can use information and methods available for analogous systems and organisms may be completed with a lower level of effort as compared to assessments involving unfamiliar products or products with more complex spatial and temporal use patterns. As the complexity of an assessment increases (dimensionality and number and nature of the risk-assessment endpoints), computational models to support more sophisticated quantitative analyses to estimate ecological risks and evolutionary responses will likely be required because existing assessments that provide baseline information or methods will be limited. Modeling will also support life-cycle analyses for existing and future products and can be used to help inform the socioeconomic tradeoffs associated with oversight decisions. These modeling efforts will entail integration with existing approaches to assess water and fossil-fuel utilization and other ecological goods and services.
The risk estimates and descriptions in human health and ecological risk assessments for existing biotechnology products are typically qualitative in nature; however, certain portions of an assessment may be quantitative, such as for estimates of human dietary exposure assessment or determining nontarget species sensitivity. The current assessments may provide a limited discussion of the uncertainties associated with risk estimates with the overall risk-assessment conclusion based on the perspective that assumptions used in a risk assessment will provide an adequate margin of safety. The influence these assumptions have on a quantitative estimate of risks needs clarification.
In the development cycle of future models to estimate risks of biotechnology products, the committee supports the 2007 National Research Council report (NRC, 2007:161) recommendation that model evaluation, rather than validation, be employed:
Model evaluation is the process of deciding whether and when a model is suitable for its intended purpose. This process is not a strict verification procedure but is one that builds confidence in model applications and increases the understanding of model strengths and limitations. Model evaluation is a multifaceted activity involving peer review, corroboration of results with data and other information, quality assurance and quality control checks, uncertainty and sensitivity analyses, and other activities.
___________________
9 See 40 C.F.R. Part 158.
Economic and Social Costs and Benefits
As discussed in Chapter 2, biotechnology products can have economic and social benefits, but they also frequently involve economic and social risks and tradeoffs. How important concerns about future biotechnology products are in comparison to the benefits provided depends on the social and cultural position of different communities, interpretation of evidence, context, and an individual’s and social group’s perception of risk and technologies. Research that teases out the social and economic tradeoffs involved in developing (or not developing) a biotechnology product is important for responsible decision making about technological development. However, the committee understands that social and economic research is not within the remit of every regulatory agency. Analyses that go beyond the direct health and environmental effects of biotechnology may be conducted by product developers, academe, and think-tanks. These analyses can be helpful to regulatory agencies when communicating about the possible risks and benefits involved in biotechnology products and in increasing public understanding about the science of risk assessment and the limitations of regulatory risk assessments. More research on how to consider the multiple socioeconomic, cultural, and indirect health effects of biotechnology products is needed, as these studies are not typically funded by current government programs (see Chapter 4).
Molecular Characterization as a Preliminary Assessment Tool
Molecular characterization of biotechnology products can provide important precursor information that can guide the direction and extent of human health and ecological risk assessments necessary for regulatory decisions (Corrigan-Curay et al., 2015). For instance, for the case of genome-edited plants, the use of whole-genome sequencing and/or evaluated bioinformatics models can establish the frequency of off-target mutations within the genome resulting from CRISPR-Cas9 genome editing and therefore addresses the probability for indirect downstream effects from the genome-edited product (Wolt et al., 2016). Establishing that off-target gene mutation frequencies are at or below natural mutation frequencies also indicates that nontransformed plant varieties may be appropriate comparators for genome-edited varieties. Similarly, molecular characterization can determine if CRISPR-Cas9 reagents are removed in breeding selection by establishing that transgenic elements are absent, and this can provide assurance that a gene drive has not been accidentally released as an unintended residual effect of genome engineering (Akbari et al., 2015).
More generally, advanced molecular approaches provide a possible avenue to address potential ecological risks through proper design and interdiction or elimination of poor design in biotechnology products. Such molecular characterization is critical for early screening to triage (potential) products into bins based on familiarity and/or complexity and therefore appropriately direct regulatory-science resources; this is particularly valuable as the pace of product innovation increases and stresses the regulatory system.
Standardization of Methods and Data
New approaches to conduct risk assessment will leverage state-of-the-art tools and capabilities from high-throughput and automated experimentation in genomics, metabolomics, and proteomics to site-specific and potentially national-scale monitoring programs. While cognizant of the need to establish the performance of new assay methods, the committee encourages a process for evaluating assays by determining if they are fit for their intended purpose and avoid costly and timely assay validation processes. In this regard, the committee encourages implementing an approach to establish assay performance criteria, as was being developed to evaluate bench-level and high--
throughput in vitro assays for chemical risk assessments (OECD, 2014). The use of private standard setting could provide the means to increase the efficiency of establishing performance-based assays.
Comprehensive assessment of future biotechnology products will likely generate large data sets of unprecedented size and complexity that will require state-of-the-art data storage and analytics. There will be a need to enhance existing data storage and information-technology analytical capabilities to rapidly accommodate and analyze the large data sets generated from -omics approaches to assessment. There will also be a need to establish standards under which some data sets can be made publicly available, while protecting confidential information as appropriate under federal statutes. A 2009 National Research Council report concluded with respect to advanced risk-assessment methods that “there is a need for simplified risk-assessment tools (such as databases, software packages, and other modeling resources) that would allow screening-level risk assessments and could allow communities and stakeholders to conduct assessments and thus increase stakeholder participation” (NRC, 2009:10).
In response to regulatory concerns regarding the validation and integrity of proprietary data sources used for industry data analysis and risk assessment, some shared, transparent, and publicly available resources already have been developed. Three examples are
- Allergen Online, a peer-reviewed allergen list and sequence-searchable database intended for the identification of proteins that may present a potential risk of allergenic cross-reactivity curated by the University of Nebraska.10
- The International Life Sciences Institute crop composition database, which summarizes ranges in nutrient, toxicant, and antinutrient content of crops for use in substantial-equivalence comparisons.11
- The CRISPR Genome Analysis Tool curated by Iowa State University and used for design and analysis of guide RNA for minimization of off-target genome edits.12
Given the large amounts of data that will be generated to support modeling and monitoring efforts, some degree of standardized methodologies and information systems will be required. Issues that will require attention include standardizing notation; standardizing testing procedures and assessment paradigms; characterizing the potential impacts of similar testing protocols on risk assessments; and approaches for collecting and integrating data from existing and future risk assessments and environmental impact statements, without compromising product developers’ data compensation rights when specified under a relevant statute. Another possible approach that would be enabled by common standards for data would be the creation of scientifically based, evidence-oriented “dossier” approaches for the submission of one, common scientific information and test data kit that can be used by all agencies for different purposes with different risk standards. To improve the consistency and predictability of risk assessment for future products of biotechnology, common standards for the information that is provided for different product classes as part of the assessment process could be used.
Enhancing the Capabilities and Tools of Product Developers to Enable Future Biotechnology Products to Traverse the Regulatory Path
In addition to needs in regulatory science for the regulatory agencies, there are also risk-analysis considerations and data generation that developers might employ in their design work
___________________
10 AllergenOnline. Available at http://www.allergenonline.org. Accessed January 15, 2017.
11 The International Life Sciences Institute Crop Composition Database, Version 6. Available at https://www.cropcomposition.org/query/index.html. Accessed January 15, 2017.
12 CRISPR Genome Analysis Tool. Available at http://cbc.gdcb.iastate.edu/cgat. Accessed January 15, 2017.
to optimize efficient risk analyses when a product is submitted for regulatory review. This section discusses the apparent discontinuity between basic bioscience activities and the regulatory process for biotechnology products. It then explores the development of tools that bridge the gap between fundamental biotechnology design-build-test-learn activities and the action of performing a regulatory assessment on a specific product submission. In considering options to employ approaches described below, beginning with simpler products (such as those intended to be contained and that have only one or a few deleted genes) would support developing design-build-test-learn cycles that eventually could be scaled to the potential open release into the environment of more complicated biotechnology products.
The tools used for regulatory assessments are aligned to guidance or statutes that are often not transparent to early-stage researchers or product developers. As the scale and complexity of the development process increases, failure to incorporate elements into early-stage product candidates leads to rework, delays, or abandonment of the product candidate in the regulatory process. Alternatively, being aware of criteria considered key to assessments of safe use might allow the developer to incorporate these early in a way that facilitates safety by design. Tools that bridge early research demands with anticipation of downstream regulatory requirements can increase the efficiency, predictability, and outcomes of regulatory assessments. Several examples of horizon scanning and anticipatory governance (Guston, 2014) for synthetic-biology products already exist; for example, a policy Delphi study focused on four cases of synthetic biology to outline research needs and governance issues for each using surveys, interviews, and a workshop. At the workshop, the most important research needs and governance opportunities and challenges were assessed for biomining, deextinction, Cyberplasm, and nitrogen-fixing microbes in the presence of a group of multidisciplinary scholars and practitioners coming from nongovernmental organizations, industry, academe, and government (Roberts et al., 2015). Another example of multiparty input in horizon scanning of potential future products which considered future regulatory needs was the Woodrow Wilson Center report Creating a Research Agenda for the Ecological Implications of Synthetic Biology, which identified several priority research areas (Drinkwater et al., 2014). Such workshops could serve as a model for identifying the risk-assessment tools needed in early-stage research to anticipate the downstream regulatory requirements of future biotechnology products.
A key aspect in considering these tools versus those discussed in the section “Enhancing the Capabilities, Expertise, and Tools of Regulatory Agencies” are that these are intended to be anticipatory to risk assessment. They are also tied to the technical drivers (see Chapter 2) that are enabling creation of future products. The tools here are envisioned to bridge conceptual gaps that could arise when products that fall within columns C and D of Figure 2-6 are actually placed into a regulatory framework that is underpinned by practices which might not be scalable.
The first category of tools would facilitate adoption and assessment of future products destined for open release into the environment. The key gap is biology knowledge on open-release use-pattern scenarios. The tools needed would establish systematic frameworks to enable evaluation of design or deployment concepts that have been recommended for genetic biocontainment. Such examples include modern kill-switch implementation and nutritional or genetic orthogonality. These design components need to be validated in a product-dependent framework at the point of entry to the regulatory system, but more generally they need validation as types of technologies in increasingly relevant systems. These systems might include at-scale fermentation or simulated environmental releases, up to assessment in actual limited environmental release.
Likewise, creation of proven models for microbial gene flow relevant to biotechnology products in environmental scenarios is a general need that anticipates future microbial products intended for environmental release. Establishing risk-assessment frameworks and metrics involves integrating current concepts of microbial ecology and would result in qualitative or quantitative scenarios that are important to new types of products. As a given product approaches the regulatory framework, these
models will provide insight to help ascertain the degree of oversight proportional to the risks posed by the specific product.
The tools of computational biology need to be focused on resolving questions and establishing evaluation frameworks directly relevant to increasing the probability of success in the regulatory framework. Currently such tools are heavily deployed on enabling early-stage discovery or development. The adaptation of these tools to the task of predictive modeling on environmental open-release scenarios would benefit later-stage risk assessment but can inform release scenario design, and possibly point to new opportunities to design, monitor, or enhance features of future products destined for deliberate release. The development of better in silico modeling systems for outcomes relevant to health, environment, and safety questions is important as the scale or complexity of systems advances. Access to such evaluated tools would enable developers to make better design decisions earlier in development and could also be designed to be responsive to advances in knowledge within the system’s technical domain, such as environment or health. This could enable the developer to prescreen or iterate designs to optimize desired outcomes, which is consistent with previous calls to design safety into biotechnology products as a first priority (Kapuscinski et al., 2003).
One example that illustrates a near-term opportunity is in computational assessment of allergen potential of a gene or gene product. Some current risk-assessment frameworks require detection and assessment of “stop to stop” codon hypothetical reading frames in all six frames (Young et al., 2012), some with no minimum length (EFSA, 2011b). Any hypothetical reading frame thus detected is taken through the computational analysis for allergenicity (Schein et al., 2007; Goodman et al., 2016). The increased use of computer-aided design systems for rapid design and assembly iteration represents an opportunity to incorporate a computational assessment early in the development process to minimize or eliminate the number of these hypothetical open reading frames during initial construct design in anticipation of a future regulatory assessment process (Galdzicki et al., 2014; Christen et al., 2015; Nielsen et al., 2016). Furthermore, by incorporating such a search and creating open-source tools to eliminate undesirable features in fundamental DNA design–build software, it may enable diffusion of this aspect of safety into the community of developers regardless of their size or knowledge of the downstream regulatory assessment framework (though with the caveat that if the downstream framework is based on faulty scientific assumptions or extra factors of conservatism, designers may limit their choices for product development by accepting rather than challenging the science). Likewise, in scenarios where a community of researchers has found value in creating libraries of standardized parts or standardized parts sequences, frameworks that allow routine screening of these resources, which are themselves a product of biotechnology, increase their utility in development work and enable safety by design at the earliest stages of a project.
A particular detail with regard to the intersection of gene sequence composition and regulatory assessment could impact the deployment of safeguarding concepts such as watermarking or DNA barcodes (Gibson et al., 2010; Liss et al., 2012; Iftikhar et al., 2015). As just mentioned, sequences subject to a regulatory review process are analyzed for many features, and the use of watermarking technology could introduce features that negatively affect the regulator’s analysis of the introduced genetic elements. Therefore, automated DNA design systems that could ensure optimum balance between the objective of sequence tagging and minimizing sequences of concern would be beneficial. Ease of access to such design tools which also incorporate sequence analysis schemes important to regulatory assessment could also facilitate the incorporation of such safeguarding elements by developers.
Another example of an anticipatory computational tool would be implementation of a computational framework by which novel chassis would receive a systematic taxonomic classification. The Coordinated Framework invokes genus-level classification of the host and donor in many regulatory submission documents. To the extent that such foundational information is considered essential for proper risk framing in the future, one can anticipate that developers of future products
of biotechnology, which rely on novel or orthogonal chassis, will need a scientifically sound route for establishing taxonomy.
Lastly, future products of biotechnology, particularly those in columns C and D of Figure 2-6, could require development of frameworks for rationalizing use of -omics data in anticipation of their increasing use in making risk-analysis decisions. In particular, developing nonarbitrary decision frameworks for choice of comparators will have an important impact on future risk analysis as the regulatory system is faced with increasing types of hosts and increasingly novel, engineered hosts; this will be a large undertaking and is best achieved through development of risk-analysis guidance that utilizes far-reaching engagement from an array of experts with public input. Analytical and information-technology resources that support appropriate experimental designs for using -omics data in comparative work should be enabled, including development of guidance on what is important to measure by when within a development pathway—that is, what information from the suite of -omics tools would be helpful in a consultation phase prior to submission and what information will likely be required during the submission would need to be clarified.
SUMMARY AND CONCLUSIONS
As technologies and basic knowledge advance, the regulatory system needs to be able to adapt to new risks of future biotechnology products and also to adjust to well-established categories of products as their level and types of risk become better understood. A regulatory system with a greater emphasis on stratified approaches that prioritize the regulatory agencies’ familiarity with a product, the complexity of the risk assessment for the product, and the anticipated risk associated with the product (that is, proportionate oversight) could contribute to meeting the increased demands on the system.
Conclusion 5-1: It would be beneficial to develop clear points of entry for biotechnology stakeholders that provide guidance, support, and direction to future product developers on the appropriate regulatory path for products of biotechnology on the basis of organism, product attributes, and release environment.
Given the diverse set of new actors who are likely to develop new products of biotechnology, it is important that there be a consistent approach to regulatory oversight that supports a product-based, science-driven risk assessment of consumer safety and environmental protection. Clear points of entry for biotechnology stakeholders might include a federally operated Web portal, interagency coordinating office, or targeted outreach efforts (for instance, to small business using the Small Business Administration networks). Stakeholders of interest include large industry, small- and medium-sized enterprises, the do-it-yourself biology community, direct-to-consumer entrepreneurs, nongovernmental organizations with interests in one or more classes of biotechnology products, and the public at large.
The development, use, and regular updating of guidance documents have proven effective and useful by EPA, FDA, and USDA in providing predictable pathways to market and increasing regulatory input quality. Future guidance documents will need to provide clear indications of the criteria that will be used to perform risk assessments and what processes and timelines will apply. Several such efforts were already under way at the time the committee was writing its report and were described in the update to the Coordinated Framework. Documents that bridge the different agencies and provide a more concise and unified description of the regulatory routes would be particularly helpful.
Conclusion 5-2: To be prepared for the anticipated profusion of future biotechnology products, the regulatory system should use scalable and proportional methods of risk assessment, capable of handling significant increases in the rate of biotechnology product innovation, the number of biotechnology products, the complexity of interactions, and the diversity of actors (who may have varying experience with the regulatory process).
On the basis of the information gathered as part of this study, the committee concluded that there is a strong possibility that the number of products per year that require federal oversight will increase and the complexity of future assessments for these products—and the associated level of effort required on the part of appropriate regulatory authorities—will also increase. Some future products of biotechnology will be familiar and fit into product categories for which there is already substantial experience and risk-analysis approaches are well defined and well understood. An approach that focuses the most attention and resources on developing risk-analysis methods for products that are unfamiliar and more complex in terms of risk analyses should be used.
Conclusion 5-3: Participatory governance processes are available for unfamiliar and more complex products of biotechnology, especially open-release products, to enhance input from experts, developers, and interested and affected parties early in the decision-making process.
Future biotechnology products and their use patterns will be increasingly dissimilar to existing biotechnology products and relatively well-understood applications; this is especially true for open-release products that may interact with the natural environment in increasingly complex ways. Participatory governance can help inform the development and implementation of an efficient process for identifying regulatory routes. To this end, approaches to efficiently address product-deployment cycles can engage diverse stakeholders and social and natural scientists with diverse expertise to establish a rigorous system based on research in social and natural sciences and practical policy experiences. Risk analyses for unfamiliar and more complex products will benefit from participatory governance by gaining a more complete appreciation of societal values to inform definition of risk-assessment endpoints in problem formulation, consideration of uncertainties in risk characterization, and formulation of risk-management options. Participation or peer review by independent experts will enable the strongest possible scientific judgments in performing risk analyses.
This conclusion is supported by a recommendation in the National Academies report on gene drives (NASEM, 2016a:10), which stated:
Governing authorities, including research institutions, funders, and regulators, should develop and maintain clear policies and mechanisms for how public engagement will factor into research, ecological risk assessments, and public policy decisions about gene drives. Defined mechanisms and avenues for such engagement should be built into the risk assessment and decision-making processes from the beginning.
Conclusion 5-4: Ecological risk assessment provides a methodology for more quantitative risk assessments for future biotechnology products and their release scenarios but will require more emphasis on measurement and modeling of effects on populations, communities, and ecosystems.
Comprehensive, efficient, and unbiased risk analysis requires regulatory expertise commensurate with the scale and complexity of future biotechnology products. Tools that can bridge early research demands with anticipation of downstream regulatory requirements can increase the efficiency and predictability of outcomes in regulatory assessments.
Future products of biotechnology provide many opportunities for improving risk analyses. Deficiencies in risk analyses include inadequate use of planning and problem-formulation steps in risk assessments and a resulting lack of clarity in the factors considered in selecting risk-assessment endpoints and in establishing conceptual models (that is, the rationale for determining what needs to be protected from potential harm by biotechnology products; what is the object of the protection; and what is the assumed path to harm). Increasing the quantitative nature of risk analyses required by the Coordinated Framework can, along with methods to elicit probabilities and uncertainties in the absence of empirical data (for example, Bayesian methods or fault-tree assessments), contribute to utilizing proportional efforts in risk analyses.
Conclusion 5-5: There are many opportunities for enhancing the capabilities, expertise, and tools available to regulatory agencies in areas that are likely to see increased emphasis and complexity in future products of biotechnology.
Given the nature of future biotechnology products, there are a diversity of knowledge and technological gaps in current risk-analysis approaches that if addressed on a case-by-case basis could overwhelm the capacity and capability of regulatory agencies to make efficient and sound evaluations. Risk analysis must be capable of adapting and responding to the rapid pace of technology and information. The regulatory agencies may wish to consider establishing a common risk-assessment infrastructure focused on the assessment of products designed for open release into the environment. There are unique research and risk-analysis needs for future biotechnology products, but some of the needs are similar, if not identical, to needs also faced for assessing the probability of adverse effects from other nonbiotechnology stressors. Resources in national and international programs managing these efforts could be leveraged. In addition, opportunities to establish public–private partnerships to address research needs should be explored in an open, transparent process.
Future biotechnology products will be more complex in terms of their internal and external interactions, and it is critical that the agencies involved in regulation of biotechnology develop and maintain scientific capabilities, tools, and expertise in relevant areas. Furthermore, it will be essential that the agencies stay appraised of technology trends so that they can engage in meaningful discussions with technology and product developers early in the product-development cycle, where there is often the best opportunity to affect future technologies. Determination of the key areas of scientific capability will need to adapt to the emerging technologies that underlie future products of biotechnology. On the basis of the current level of federal investments (see Chapter 4), some of the key areas of regulatory-science research for the products of biotechnology likely in the next 5–10 years include comparators, off-target gene effects, and phenotypic characterization; gene fitness, genetic stability, and horizontal gene transfer; impacts on nontarget organisms; control of organismal traits; modeling (including risk-analysis approaches under uncertainty) and life-cycle analyses; monitoring and surveillance; and economic and social costs and benefits.
Conclusion 5-6: There are many opportunities for enhancing the capabilities and tools of technology and product developers to enable future products to traverse the regulatory path.
There are substantial opportunities for the use of improved methods for scientific evaluation, risk assessment, and community engagement related to future products of biotechnology that can be applied by technology and product developers. In order to ensure that the regulatory framework is able to make use of the best available tools in performing its oversight and regulation responsibilities, it will be important to invest in those tools and make them available to regulators and product developers. Areas for consideration include stochastic methods, advances in uncertainty analysis,
better ways to integrate and interpret both qualitative and quantitative data, and communication strategies.
REFERENCES
Akbari, B.O.S., H.J. Bellen, E. Bier, S.L. Bullock, A. Burt, G.M. Church, K.R. Cook, P. Duchek, O.R. Edwards, K.M. Esvelt, V.M. Gantz, K.G. Golic, S.J. Gratz, M.M. Harrison, K.R. Hayes, A.A. James, T.C. Kaufman, J. Knoblich, H.S. Malik, K.A. Matthews, K.M. O’Connor-Giles, A.L. Parks, N. Perrimon, F. Port, S. Russell, R. Ueda, and J. Wildonger. 2015. Safeguarding gene drive experiments in the laboratory. Science 349(6251):927–929.
Andow, D., G.L. Lövei, S. Arpaia, L. Wilson, E.M. Fontes, A. Hilbeck, A. Lang, N. Van Tuat, C. Pires, E. Sujii, C. Zwahlen, A.N. Birch, D.M. Capalbo, K. Prescott, C. Omoto, and A.R. Zeilinger. 2013. An ecologically-based method for selecting ecological indicators for assessing risks to biological diversity from genetically-engineered plants. Journal of Biosafety 22(3):141–156.
Balla, S.J., and J.R. Wright. 2003. Consensual rule making and the time it takes to develop rules. Pp. 187–206 in Politics, Policy, and Organizations: Frontiers in the Scientific Study of Bureaucracy, G.A. Krause and K.J. Meier, eds. Ann Arbor: University of Michigan Press.
Barben, D., E. Fisher, C. Selin, and D.H. Guston. 2007. Anticipatory governance of nanotechnology: Foresight, engagement, and integration. Pp. 979–1000 in The Handbook of Science and Technology Studies, 3rd Ed., E. Hackett, O. Amsterdamska, M.E. Lynch, and J. Wajcman, eds. Cambridge, MA: MIT Press. Available at https://www.hks.harvard.edu/sdn/articles/files/Barben-STS_Handbook-Anticipatory_Governance_Nanotechnology-08.pdf. Accessed October 28, 2016.
Benedict, M.Q., and A.S. Robinson. 2003. The first releases of transgenic mosquitoes: An argument for the sterile insect technique. Trends in Parasitology 19(8):349–355.
Biello, D. August 29, 2014. Ancient DNA could return passenger pigeons to the sky. Scientific American. Available at https://www.scientificamerican.com/article/ancient-dna-could-return-passenger-pigeons-to-the-sky. Accessed January 8, 2017.
Bozeman, B., and D. Sarewitz. 2005. Public values and public failure in US science policy. Science and Public Policy 32(2):119–136.
Callaway, E. 2015. Mammoth genomes hold recipe for Arctic elephants. Nature 521(7550):18–19.
Caron-Lormier, G., D.A. Bohan, C. Hawes, A. Raybould, A.J. Haughton, and R.W. Humphry. 2009. How might we model an ecosystem? Ecological Modelling 220(17):1935–1949.
Caron-Lormier, G., D.A. Bohan, R. Dye, C. Hawes, R.W. Humphry, and A. Raybould. 2011. Modelling one ecosystem: The example of agro-ecosystems. Ecological Modelling 222(5):1163–1173.
Christen, M., S. Deutsch, and B. Christen. 2015. Genome calligrapher: A Web tool for refactoring bacterial genome sequences for de novo DNA synthesis. ACS Synthetic Biology 4(8):927–934.
Corrigan-Curay, J., M. O’Reilly, D.B. Kohn, P.M. Cannon, G. Bao, F.D. Bushman, D. Carroll, T. Cathomen, J.K. Joung, D. Roth, M. Sadelain, A.M. Scharenberg, C. von Kalle, F. Zhang, R. Jambou, E. Rosenthal, M. Hassani, A. Singh, and M.H. Porteus. 2015. Genome editing technologies: Defining a path to clinic. Molecular Therapy 23(5):796–806.
Drinkwater, K., T. Kuiken, S. Lightfoot, J. McNamara, and K. Oye. 2014. Creating a Research Agenda for the Ecological Implications of Synthetic Biology. MIT Center for International Studies, Cambridge, MA, and Woodrow Wilson International Center for Scholars, Washington, DC. Available at https://www.wilsoncenter.org/sites/default/files/SYNBIO_create%20an%20agenda_v4.pdf. Accessed August 10, 2016.
EFSA (European Food Safety Authority). 2010. Guidance on the environmental risk assessment of genetically modified plants. EFSA Journal 8(5):1879–1989.
EFSA. 2011a. Guidance on conducting repeated-dose 90-day oral toxicity study in rodents on whole food/feed. EFSA Journal 9(12):2438.
EFSA. 2011b. Guidance on risk assessment of food and feed from genetically modified plants. EFSA Journal 9(5):2150.
EFSA. 2011c. Guidance on the post-market environmental monitoring (PMEM) of genetically modified plants. EFSA Journal 9(8):2316.
EOP (Executive Office of the President). 2016. National Strategy for Modernizing the Regulatory System for Biotechnology Products. Available at https://obamawhitehouse.archives.gov/sites/default/files/microsites/ostp/biotech_national_strategy_final.pdf. Accessed January 31, 2017.
EOP. 2017. Modernizing the Regulatory System for Biotechnology Products: An Update to the Coordinated Framework for the Regulation of Biotechnology. Available at https://obamawhitehouse.archives.gov/sites/default/files/microsites/ostp/2017_coordinated_framework_update.pdf. Accessed January 30, 2017.
EPA (U.S. Environmental Protection Agency). 1998. Guidelines for Ecological Risk Assessment. Federal Register 63:26846–26924.
EPA. 2004. Overview of the Ecological Risk Assessment Process in the Office of Pesticide Programs, U.S. Environmental Protection Agency. Endangered and Threatened Species Effects Determinations. Available at https://www.epa.gov/sites/production/files/2014-11/documents/ecorisk-overview.pdf. Accessed September 14, 2016.
EPA. 2012a. SAP Minutes No. 2012-06: A Set of Scientific Issues Being Considered by the Environmental Protection Agency Regarding Pollinator Risk Assessment Framework. Available at https://www.regulations.gov/document?D=EPA-HQ-OPP-2012-0543-0047. Accessed September 14, 2016.
EPA. 2012b. SAP Minutes No. 2012-05: A Set of Scientific Issues Being Considered by the Environmental Protection Agency Regarding: Problem Formulation for the Reassessment of Ecological Risks from the Use of Atrazine. Available at https://www.epa.gov/sites/production/files/2015-06/documents/061212minutes.pdf. Accessed September 14, 2016.
Exponent. 2005. DEEM-FCID TM Distributional Acute Dietary Exposure Analysis Program, Version 2.03. Exponent, Washington, DC.
FDA (U.S. Food and Drug Administration). 2012. A Guide to Drug Safety Terms at FDA. FDA Consumer Health Information. November. Available at http://www.fda.gov/downloads/ForConsumers/ConsumerUpdates/UCM107976.pdf. Accessed January 9, 2017.
FDA. 2017. Regulation of Mosquito-Related Products; Draft Guidance for Industry; Availability. Federal Register 82:6574–6575.
Forbes, V.E., P. Calow, and R.M. Sibly. 2001. Are current species extrapolation models a good basis for ecological risk assessment? Environmental Toxicology and Chemistry 20(2):442–447.
Freudenburg, W.R. 1996. Strange chemistry: Environmental risk conflicts in a world of science, values, and blind spots. Pp. 11–36 in Handbook for Environmental Risk Decision Making: Values, Perceptions, and Ethics, C.R. Cothern, ed. Boca Raton, FL: Lewis Publishers.
Galdzicki, M., K.P. Clancy, E. Oberortner, M. Pocock, J.Y. Quinn, C.A. Rodriguez, N. Roehner, M.L. Wilson, L. Adam, J.C. Anderson, B.A. Bartley, J. Beal, D. Chandran, J. Chen, D. Densmore, D. Endy, R. Grünberg, J. Hallinan, N.J. Hillson, J.D. Johnson, A. Kuchinsky, M. Lux, G. Misirli, J. Peccoud, H.A. Plahar, E. Sirin, G-B. Stan, A. Villalobos, A. Wipat, J.H. Gennari, C.J. Myers, and H.M. Sauro. 2014. The Synthetic Biology Open Language (SBOL) provides a community standard for communicating designs in synthetic biology. Nature Biotechnology 32(6):545–550.
Gibson, D.G., J.I. Glass, C. Lartigue, V.N. Noskov, R.-Y. Chuang, M.A. Algire, G.A. Benders, M.G. Montague, L. Ma, M.M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N. Assad-Garcia, C. Andrews-Pfannkoch, E.A. Denisova, L. Young, Z.-Q. Qi, T.H. Segall-Shapiro, C.H. Calvey, P.P. Parmar, C.A. Hutchison, H.O. Smith, and J.C. Venter. 2010. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329(5987):52–56.
Ginsberg, W. 2015. Creating a Federal Advisory Committee in the Executive Branch. Washington, DC: Congressional Research Service.
Goodman, R.E., M. Ebisawa, F. Ferreira, H.A. Sampson, R. van Ree, S. Vieths, J.L. Baumert, B. Bohle, S. Lalithambika, J. Wise, and S.L. Taylor. 2016. AllergenOnline: A peer-reviewed, curated allergen database to assess novel food proteins for potential cross-reactivity. Molecular Nutrition and Food Research 60:1183–1198.
Guston, D.H. 2014. Understanding anticipatory governance. Social Studies of Science 44(2):218–242.
Harding, S. 1996. Rethinking standpoint epistemology: What is “strong objectivity”? Pp. 235–248 in Feminism and Science, E.F. Keller and H. Longino, eds. Oxford: Oxford University Press.
Hayes, K.R., B. Leung, R. Thresher, J.M. Dambacher, and G.R. Hosack. 2014. Meeting the challenge of quantitative risk assessment for genetic control techniques: A framework and some methods applied to the common Carp (Cyprinus carpio) in Australia. Biological Invasions 16(6):1273–1288.
Iftikhar, S., S. Khan, Z. Anwar, and M. Kamran. 2015. GenInfoGuard—a robust and distortion-free watermarking technique for genetic data. PLOS ONE 10(2):e0117717.
IRGC (International Risk Governance Council). 2015. Guidelines for Emerging Risk Governance. Lausanne, Switzerland: IRGC. Available at https://www.irgc.org/wp-content/uploads/2015/03/IRGC-Emerging-Risk-WEB-31Mar.pdf. Accessed October 17, 2016.
Kapuscinski, A.R., R.M. Goodman, S.D. Hann, L.R. Jacobs, E.E. Pullins, C.S. Johnson, J.D. Kinsey, R.L. Krall, A.G. La Viña, M.G. Mellon, and V.W. Ruttan. 2003. Making “safety first” a reality for biotechnology products. Nature Biotechnology 21:599–601.
Kapuscinski, A.R., S. Li, K.R. Hayes, and G. Dana, eds. 2007. Environmental Risk Assessment of Genetically Modified Organisms, Vol. 3. Methodologies for Transgenic Fish. Cambridge, MA: CABI. Available at https://gmoera.umn.edu/sites/gmoera.umn.edu/files/environmental_risk_assessment_volume_3.pdf. Accessed October 28, 2016.
Krafsur, E.S. 1998. Sterile insect technique for suppressing and eradicating insect populations: 55 years and counting. Journal of Agricultural Entomology 15(4):303–317.
Kuzma, J. 2006. Nanotechnology oversight and regulation—just do it. Environmental Law Reporter 36:10913–10923.
Kuzma, J. 2013. Properly paced or problematic? Examing governance of GMOs. Pp. 176–197 in Innovative Governance Models for Emerging Technologies, G. Marchant, K. Abbott, and B. Allenby, eds. Cheltenham, UK: Edward Elgar.
Kuzma, J., and J.C. Besley. 2008. Ethics of risk analysis and regulatory review: From bio- to nanotechnology. Nanoethics 2(2):149–162.
Kuzma, J., J. Romanchek, and A. Kokotovich. 2008. Upstream oversight assessment for agrifood nanotechnology: A case studies approach. Risk Analysis 28(4):1081–1098.
Lavertu, S., and D.L. Weimer. 2010. Federal advisory committees, policy expertise, and the approval of drugs and medical devices at the FDA. Journal of Public Administration Research and Theory 21:211–237.
Linkov, I., F.K. Satterstrom, J. Steevens, E. Ferguson, and R.C. Pleus. 2007. Multi-criteria decision analysis and environmental risk assessment for nanomaterials. Journal of Nanoparticle Research 9(4):543–554.
Liss, M., D. Daubert, K. Brunner, K. Kliche, U. Hammes, A. Leiherer, and R. Wagner. 2012. Embedding permanent watermarks in synthetic genes. PLOS ONE 7(8):e42465.
Marchant, G.E., and W. Wallach. 2015. Coordinating technology governance. Issues in Science and Technology 31(4):43–50.
McHughen, A., and S. Smyth. 2008. U.S. regulatory system for genetically modified [genetically modified organism (GMO), rDNA or transgenic] crop cultivars. Plant Biotechnology Journal 6(1):2–12.
Meghani, Z., and J. Kuzma. 2011. The “revolving door” between regulatory agencies and industry: A problem that requires reconceptualizing objectivity. Journal of Agricultural and Environmental Ethics 24(6):575–599.
Moffitt, S.L. 2014. Making Policy Public: Participatory Bureaucracy in American Democracy. New York: Cambridge University Press.
Murphy, B., C. Jansen, J. Murray, and P. De Barro. 2010. Risk Analysis on the Australian Release of Aedes aegypti (L.) (Diptera: Culicidae) Containing Wolbachia. Indooroopilly, Australia: CSIRO.
Murray, J.V., C.C. Jansen, and P. De Barro. 2016. Risk associated with the release of Wolbachia-infected Aedes aegypti mosquitoes into the environment in an effort to control dengue. Frontiers in Public Health 4:43.
Naranjo, S.E., G. Head, and G.P. Dively. 2005. Field studies assessing arthropod nontarget effects in Bt transgenic crops: Introduction. Environmental Entomology 34(5):1178–1180.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2016a. Gene Drives on the Horizon: Advancing Science, Navigating Uncertainty, and Aligning Research with Public Values. Washington, DC: The National Academies Press.
NASEM. 2016b. Genetically Engineered Crops: Experiences and Prospects. Washington, DC: The National Academies Press.
Nelson, K.C., G. Kibata, L. Muhammad, J. Okuro, F. Muyekho, M. Odindo, A. Ely, and J. Waquil. 2004. Problem formulation and options assessment (PFOA) for genetically modified organisms: The Kenya case study. Pp. 57–82 in Environmental Risk Assessment of Genetically Modified Organisms, Vol. 1. A Case Study of Bt Maize in Kenya, A. Hilbeck and D.A. Andow, eds. Cambridge, MA: CABI.
Nielson, A.A., B.S. Der, J. Shin, P. Vaidyanathan, V. Paralanov, E.A. Strychalski, D. Ross, D. Densmore, and C.A. Voight. 2016. Genetic circuit design automation. Science 352(6281):aac7341.
NRC (National Research Council). 1996. Understanding Risk: Informing Decisions in a Democratic Society. Washington, DC: National Academy Press.
NRC. 2007. Models in Environmental Regulatory Decision Making. Washington, DC: The National Academies Press.
NRC. 2008. Public Participation in Environmental Assessment and Decision Making. Washington, DC: The National Academies Press.
NRC. 2009. Science and Decisions: Advancing Risk Assessment. Washington, DC: The National Academies Press.
NRC. 2013. Assessing Risks to Endangered and Threatened Species from Pesticides. Washington, DC: The National Academies Press.
OECD (Organisation for Economic Co-operation and Development). 2014. Guidance Document for Describing Non-Guideline In Vitro Test Methods. Series on Testing and Assessment No. 211. Paris: OECD.
Pauwels, K., S.C. De Keersmaecker, A. De Schrijver, P. du Jardin, N.H. Roosens, and P. Herman. 2015. Next-generation sequencing as a tool for the molecular characterisation and risk assessment of genetically modified plants: Added value or not? Trends in Food Science & Technology 45(2):319–326.
Pennington, K.M., A.R. Kapuscinski, M.S. Morton, A.M. Cooper, and L.M. Miller. 2010. Full life-cycle assessment of gene flow consistent with fitness differences in transgenic and wild-type Japanese medaka fish (Oryzias latipes). Environmental Biosafety Research 9(1):41–57.
Peterson, R.K., S.J. Meyer, A.T. Wolf, J.D. Wolt, and P.M. Davis. 2006. Genetically engineered plants, endangered species, and risk: A temporal and spatial exposure assessment for Karner blue butterfly larvae and Bt maize pollen. Risk Analysis 26(3):845–858.
Ramachandran, G., S. Wolf, J. Paradise, J. Kuzma, R. Hall, E. Kokkoli, and L. Fatehi. 2011. Recommendations for oversight of nanobiotechnology: Dynamic oversight for complex and convergent technology. Journal of Nanoparticle Research 13(4):1345–1371.
Raybould, A., G. Caron-Lormier, and D.A. Bohan. 2011. Derivation and interpretation of hazard quotients to assess ecological risks from the cultivation of insect-resistant transgenic crops. Journal of Agricultural and Food Chemistry 59(11):5877–5885.
Renn, O. 2005. Risk Governance: Towards and Integrative Approach. Geneva: IRGC.
Roberts, J.P., S. Stauffer, C. Cummings, and J. Kuzma. 2015. Synthetic Biology Governance: Delphi Study Workshop Report. GES Center Report No. 2015.2. Available at https://research.ncsu.edu/ges/files/2014/04/Sloan-Workshop-Report-final-ss-081315-1.pdf. Accessed October 17, 2016.
Rose, R.I., ed. 2007. White Paper on Tier-Based Testing for the Effects of Proteinaceous Insecticidal Plant-Incorporated Protectants on Non-Target Arthropods for Regulatory Risk Assessments. Environmental Protection Agency and U.S. Department of Agriculture–Animal and Plant Health Inspection Service. Available at https://www.epa.gov/sites/production/files/2015-09/documents/tier-based-testing.pdf. Accessed January 15, 2017.
Ryals, J. 2016. Metabolomics as a High Resolution Phenotyping Tool. Webinar presentation to the National Academies of Sciences, Engineering, and Medicine Committee on Future Biotechnology Products and Opportunities to Enhance Capabilities of the Biotechnology Regulatory System, July 29.
Schein, C.H., O. Ivanciuc, and W. Braun. 2007. Bioinformatics approaches to classifying allergens and predicting cross-reactivity. Immunology and Allergy Clinics of North America 27:1–27.
Sears, M.K., R.L. Hellmich, D.E. Stanley-Horn, K.S. Oberhauser, J.M. Pleasants, H.R. Mattila, B.D. Siegfried, and G.P. Dively. 2001. Impact of Bt corn pollen on monarch butterfly populations: A risk assessment. Proceedings of the National Academy of Sciences of the United States of America 98(21):11937–11942.
Shapiro, B. 2015. How to Clone a Mammoth: The Science of De-Extinction. Princeton, NJ: Princeton University Press.
Stilgoe, J., R. Owen, and P. Macnaghten. 2013. Developing a framework for responsible innovation. Research Policy 42(9):1568–1580.
Stirling, A. 2007. Risk, precaution and science: Towards a more constructive policy debate. EMBO Reports 8(4):309–315.
Storer, N.P. 2003. A spatially explicit model simulating western corn rootworm (Coleoptera: Chrysomelidae) adaptation to insect-resistant maize. Journal of Economic Entomology 96(5):1530–1547.
Suter, G.W., II. 2007. Ecological Risk Assessment, 2nd Ed. Boca Raton, FL: CRC Press.
Thompson, P.B. 2007. Food Biotechnology in Ethical Perspective, 2nd Ed. The International Library of Environmental, Agricultural and Food Ethics Vol. 10. Dordrecht, Netherlands: Springer.
Tsang, M.P., M.E. Bates, M. Madison, and I. Linkov. 2014. Benefits and risks of emerging technologies: Integrating life cycle assessment and decision analysis to assess lumber treatment alternatives. Environmental Science & Technology 48(19):11543–11550.
Warren-Hicks, W.J., and A. Hart. 2010. Application of Uncertainty Analysis to Ecological Risks of Pesticides. Boca Raton, FL: CRC Press.
Weimer, D.L. 2006. The puzzle of private rulemaking: Expertise, flexibility, and blame avoidance in U.S. regulation. Public Administration Review 66(4):569–582.
Weimer, D.L., and L. Wilk. 2016. Allocation of indivisible life-saving goods with both intrinsic and relational quality: The new deceased-donor kidney allocation system. Administration and Society 1–30.
Wilsdon, J., and R. Willis. 2004. See-Through Science: Why Public Engagement Needs to Move Upstream. London: Demos.
Wolt, J.D. 2011. A mixture toxicity approach for environmental risk assessment of multiple insect resistance genes. Environmental Toxicology and Chemistry 30(3):763–772.
Wolt, J.D., and R.K.D. Peterson. 2010. Prospective formulation of environmental risk assessments: Probabilistic screening for Cry1A(b) maize risk to aquatic insects. Ecotoxicology and Environmental Safety 73:1182–1188.
Wolt, J.D., K. Wang, D. Sashital, and C.J. Lawrence-Dill. 2016. Achieving plant CRISPR targeting that limits off-target effects. The Plant Genome 9(3).
Young, G.J., S. Zhang, H.P. Mirsky, R.F. Cressman, B. Cong, G.S. Ladics, and C.X. Zhong. 2012. Assessment of possible allergenicity of hypothetical ORFs in common food crops using current bioinformatics guidelines and its implications for the safety assessment of GM crops. Food and Chemical Toxicology 50:3741–3751.


































