
2
The State of the Science
Chapter 2 provides the foundations in areas of science and medicine that are important for understanding the feasibility of heritable human genome editing (HHGE). This chapter contains substantial scientific detail; see the glossary in Appendix C for any unfamiliar terms. Part I of this chapter describes what is known about the genetics of diseases caused by mutations in a single gene—a category known as monogenic diseases. It then discusses potential reproductive options for parents at risk of passing on a disease genotype, including the use and current limitations of in vitro fertilization (IVF) in conjunction with preimplantation genetic testing (PGT) to identify any embryos that do not have the disease-causing genotype.
Part II reviews genome editing technologies and current approaches to characterizing their results. It describes what has been learned so far from genome editing in somatic cells and in early embryos. Genome editing carried out concomitant with fertilization, or in a zygote (the single cell created by fertilization), would be the most likely way in which the potential for clinical use of HHGE could currently be evaluated.
Part III discusses a technology with the potential to provide another means of preventing the inheritance of genetic disorders, as well as an alternative to zygote genome editing for undertaking HHGE: the ability to create sperm or egg cells in the laboratory from parental stem cells. At this time, further development would be required before this technology could be considered for clinical use. Even then, it would have significant scientific, ethical, and social implications. As with HHGE, the decision about
whether to make it available for clinical use would depend on much more than technological feasibility.
Part IV reviews two other areas that would be crucial components of any clinical use of HHGE. Informed consent would be needed from prospective parents, and HHGE would pose special challenges to these protocols. In addition, the long-term monitoring of any individuals born following HHGE would be important, and such monitoring would potentially span generations, raising further issues.
Part V reflects on the complexities of human genetics beyond monogenic diseases and looks ahead to other circumstances for which HHGE has been proposed. It describes what is known about the genetics of polygenic diseases, a category that includes many common diseases in which multiple genetic variants contribute to overall disease risk. And it discusses the genetics of male infertility—a special case for HHGE.
The chapter concludes by identifying key knowledge gaps that would need to be addressed before any clinical use of HHGE and provides two recommendations.
MONOGENIC DISEASES: GENETICS AND REPRODUCTIVE OPTIONS
Genetics of Monogenic Diseases
Over the past 40 years, human genetics has undergone a revolution that has enabled the systematic identification of genes underlying many human diseases (Claussnitzer et al., 2020). The scientific program started with the recombinant DNA revolution in the 1970s, which allowed the cloning and isolation of segments of the genome of any species. This led to recognition that physical and genetic maps of genomes could be unified (Wensink et al., 1974), resulting in the idea of “positional cloning.” In this paradigm, the chromosomal location of a trait-causing mutation could be determined by any of several genetic methods, and the cloned DNA segments from the section of the chromosome thought to contain the responsible mutation could then be assembled and analyzed to identify the specific gene and mutation that produce the disease or trait (Bender et al., 1983).
The development of positional cloning in humans became possible with the recognition in 1980 that there is substantial polymorphism in DNA sequence of genomes, with alternative sequences that are common in populations (Botstein et al., 1980). These alternative sequences mark a specific chromosome segment and permit the tracing of its inheritance through pedigrees or populations. The discovery of millions of these common variations allowed the development of genetic maps of the human genome, thereby permitting the systematic comparison of the inheritance
of every segment of every chromosome to the inheritance of diseases or other traits in families. For diseases caused by mutation in a single gene, this process demonstrated which chromosome segment was precisely linked with the disease or trait. From this map location, the disease gene could eventually be discovered as the gene in the mapped interval that harbors mutations specific to individuals with the disease or trait. For example, the gene in which mutations lead to cystic fibrosis (CF) was identified in 1989 (Riordan et al., 1989).
This process was greatly accelerated by the assembly of the virtually complete sequence of the human and other genomes, announced in 2001 (IHGSC, 2001, 2004), which greatly aided the discovery of human genes and facilitated the process of identifying disease-causing mutations. These efforts led to the identification of several thousand human genes in which mutation produced a disease phenotype.
Advances in DNA sequencing over the ensuing decade dramatically increased sequence production and reduced its cost by more than a million-fold. This advance led to brute-force methods of disease gene discovery in which sequencing of all ~20,000 protein-coding genes in the human genome in many unrelated patients with the same clinical disease could identify genes that are mutated more often than expected by chance, and also permitted routine establishment of clinical diagnosis of individuals with monogenic diseases.
This work collectively has led to the discovery of the genes responsible for more than 4,000 monogenic (single-gene) diseases to date, such as Duchenne muscular dystrophy, beta-thalassemia, CF, Huntington’s disease, and Tay-Sachs disease.1 As discussed in the last section of this chapter, this work has also advanced our understanding of heart disease and neurodegeneration.
Identifying the genes for monogenic diseases has had profound consequences for medicine. The ability to detect mutations in a gene has enabled clinical diagnostics—for example, early diagnosis available to women with mutations in the gene BRCA1, who are at increased risk of breast, ovarian, and other cancers. Biological understanding of disease mechanisms has enabled therapies in some cases, ranging from dietary control (patients with phenylketonuria can avoid severe brain damage by adopting a phenylalanine-restricted diet), to drugs (e.g., the ability to replace missing enzymes, as in Gaucher disease, or to mitigate the impact of mutations that cause CF), to gene-based therapies (e.g., one which delivers to cells a functional copy of a gene missing in spinal muscular atrophy [Hoy, 2019]).
___________________
1 See Online Mendelian Inheritance in Man at https://www.omim.org.
The Human Genome
Humans inherit two copies of the genome, one from their mother and one from their father. Each copy of the human genome consists of approximately 3 billion base pairs of genetic information distributed among 23 pairs of chromosomes. Of these, 22 pairs are autosomes (equivalent chromosomes inherited from each parent) and 1 pair comprises the sex chromosomes (X or Y, with females inheriting two X chromosomes and males one X and one Y chromosome). In addition to this nuclear genome, mitochondria in cells contain their own, much smaller genome, as discussed in Chapter 1.
Any two examples of the human genome have around 3 million sequence differences, many of which do not result in observable (phenotypic) effects but which reflect the degree of genetic variation in the human population. The vast majority of these differences are single nucleotide variants (SNVs), in which a single base pair in a specific location in the genome varies among people. Other differences include short insertions or deletions of DNA (indels); longer DNA segments that have been lost, added, duplicated, or transposed; and, at the largest scale, differences in chromosome numbers.
The genetic variation in the human population arises from several factors. Genetic variants originally arise as alterations to the genome sequence that arise during DNA replication or other natural processes. Each individual has an average of about 70 de novo SNVs and 6 de novo indels not present in their parents (Sasani et al., 2019). The rate of de novo mutations is increased in older men due to the high number of cell divisions during spermatogenesis and is referred to as the paternal age effect (reviewed in Cioppi et al., 2019).
Most new variants do not alter reproductive success and are unlikely to persist over time in large populations. For this reason, most common variations found in the human genome were introduced many thousands of years ago, when population sizes were small. Other variants impair reproductive success and are more rapidly eliminated from the population by negative selection. Rarely, a variant will increase reproductive success and thereby increase in frequency in a population over time due to positive selection. Lastly, occasionally a variant will have beneficial effects when it is present in a single copy (allele) but have deleterious effects when present in both alleles, resulting in balancing selection that allows a potentially deleterious variant to be maintained in the population. Over generations, the linkage of these variants to each other on a chromosome is shuffled by genetic recombination between parental chromosome pairs that occurs during the formation of gametes, thereby producing great variation in the combinations of alleles that in turn produce high phenotypic variation in populations.
Monogenic Diseases
Monogenic diseases are caused by mutation of either one or both copies (or alleles) of a single gene, typically by altering the protein-coding sequence of the gene or, less often, by altering a DNA segment that regulates the activity of the gene. The thousands of monogenic diseases vary widely in many respects, including the organ systems affected, the age of onset, and the seriousness of disease.
Some monogenic diseases are caused by dominant mutations. These diseases occur in individuals who carry one disease-causing allele and one non-disease-causing allele in the relevant gene (heterozygotes). An example is Huntington’s disease, in which a defect in the gene for a protein active in brain cells gradually causes damage to those cells through the accumulation of the abnormal protein, which leads to progressive neurological symptoms and premature death (Walker, 2007). Other examples include myotonic dystrophy and neurofibromatosis. Dominant diseases can arise because the disease-causing copy of the gene produces too little protein to allow normal function even in the presence of a normal copy of the gene (haploinsufficiency), produces an abnormal protein that interferes with the normal protein produced by the other copy of the gene (dominant negative), or causes too much activity of the normal protein (gain of function), or the abnormal protein acquires a new function, not found in the normal protein, that causes disease (neomorph). In other cases, loss of a single functioning gene copy is tolerated, but the remaining functional copy of the gene is lost in some cells during the lifetime of the individual, leading to disease manifestation restricted to the affected tissue. This is the case in some forms of familial breast and colon cancer.
In other monogenic diseases, the causative mutations are recessive. These diseases occur in individuals who carry disease-causing mutations on both alleles of a gene (mutations are homozygous if the two mutations are identical, or compound heterozygous if they are different). Recessive mutations typically cause loss of normal gene function, as occurs in CF and spinal muscular atrophy, but there are exceptions, such as sickle cell disease (SCD), in which the mutant protein acquires a deleterious function not found in the normal protein.
Still other monogenic diseases are X-linked, due to a mutation in a gene found only on the X chromosome. Males are affected if they carry a mutated allele on their single X chromosome, and females are affected if they carry a disease-causing allele on both of their X chromosomes. Some females who are carriers of the mutated allele may show signs or symptoms of the disease if there is skewed inactivation of their X chromosomes with preferential inactivation of the X chromosome without the mutation
(reviewed by Migeon, 2020). Examples include fragile X syndrome, hemophilia A, and Duchenne muscular dystrophy.
In some cases, complexities may be layered over the descriptions above. Monogenic diseases may have incomplete penetrance: only a subset of people who inherit the same disease genotype will actually have the disease. These diseases may also have variable expressivity, and people who inherit the same disease genotype may have different qualitative or quantitative manifestations of the disease. Incomplete penetrance and variable expressivity may be due to the effect of modifier genes elsewhere in the genome, only some of which have been identified. For example, the severity of SCD, caused by mutations in the gene encoding the beta chain of hemoglobin, is modified by genetic variants that affect adult expression of the gene encoding fetal hemoglobin. Disease penetrance and expressivity may also be influenced by non-genetic factors. Well-known examples include phenylketonuria in which an inherited inability to metabolize the amino acid phenylalanine can result in intellectual disability and seizures; however, the disease can be mitigated by a diet low in phenylalanine. Similarly, some immune deficiencies may have no significant clinical consequence unless an individual is exposed to a particular infectious agent such as tuberculosis or influenza.
A single gene can also have different pathogenic variants, some that are more common in particular populations and some that are rare or unique to one or a small number of families. In general, for a gene whose mutation causes a recessive disease, many different disease-causing mutations will be found in populations because there are many ways to produce loss of function mutations in a gene: these can be produced by different premature termination, splice site or frameshift mutation at many different sites along the gene, and by many different protein-altering mutations. The high diversity of these mutations may complicate editing efforts since the required editing reagents for the same gene in different cases could often be different. The same applies to dominant mutations caused by haploinsufficiency. In contrast, dominant diseases caused by gain of function mutations typically have a more restricted spectrum of disease-causing mutations because markedly increasing the activity or producing a distinct function of an encoded protein by mutation is genetically much less frequent than simply knocking out a gene’s function.
Nonetheless, some recessive mutations can dominate the allele spectrum in certain diseases. One example is SCD, in which one copy of the hemoglobin S allele can provide some protection against malaria while two mutant copies cause SCD, featuring severe morbidity and premature death (Archer et al., 2018). In this disease, most affected people in or descendent from West Africa have the same disease-causing mutation in beta-hemoglobin. Another serious red blood cell disease, thalassemia, also has relatively
frequent alleles, again owing to protection from malaria. Similarly, while more than 1,500 different loss of function mutations in the CFTR gene can cause the recessive disease CF, a specific deletion of three nucleotides in this gene comprises approximately 70 percent of all loss of function mutations in CFTR in people of northern and central European descent (European Working Group on Cystic Fibrosis Genetics, 1990), while a different mutation is enriched in people of African ancestry.
Inheritance Patterns of Monogenic Diseases
With some notable exceptions, monogenic diseases are individually very rare—with frequencies typically in the range of 1 in 10,000 to 1 in 1 million births.2 However, the thousands of rare monogenic diseases together impose a significant burden on human health. According to the World Health Organization (2019a), the global prevalence of all monogenic diseases at birth is about 1 in 100, and monogenic conditions have been reported to “collectively contribute to disease in ~0.4 percent of children and young adults” (Posey et al., 2019). In addition, as noted above, there are circumstances in which a monogenic disease is found at higher frequency in a particular population in which the heterozygous state confers an advantage, where a mutation was present in an individual whose genes were inherited by a significant proportion of a population (also known as a founder effect), or where there are high rates of consanguinity (see Chapter 3 for further discussion of circumstances in which certain monogenic diseases are found at higher frequencies).
The typical situation for the inheritance of autosomal dominant and autosomal recessive diseases is shown in Figure 2-1. For an autosomal dominant disease, if one parent is a heterozygote for the disease-causing allele, each offspring of this parent has a 50 percent chance of inheriting a disease-causing genotype and a 50 percent chance of not inheriting a disease-causing genotype. In rare circumstances in which both parents have the same autosomal dominant disease, each offspring would have a 75 percent chance of inheriting the disease-causing genotype (i.e., at least one disease-causing allele). For an autosomal recessive disease, if both parents are unaffected heterozygous carriers, each offspring would have a 25 percent chance of inheriting the disease-causing genotype (i.e., two disease-causing alleles).
There are very rare circumstances, however, in which all of a couple’s children would inherit the disease genotype, as shown in Figure 2-2. Specifically, these circumstances involve either one parent being homozygous for a dominant disease or both parents being homozygous or compound heterozygous for the same recessive disease.
___________________
2 See Online Mendelian Inheritance in Man (OMIM) at https://www.omim.org.
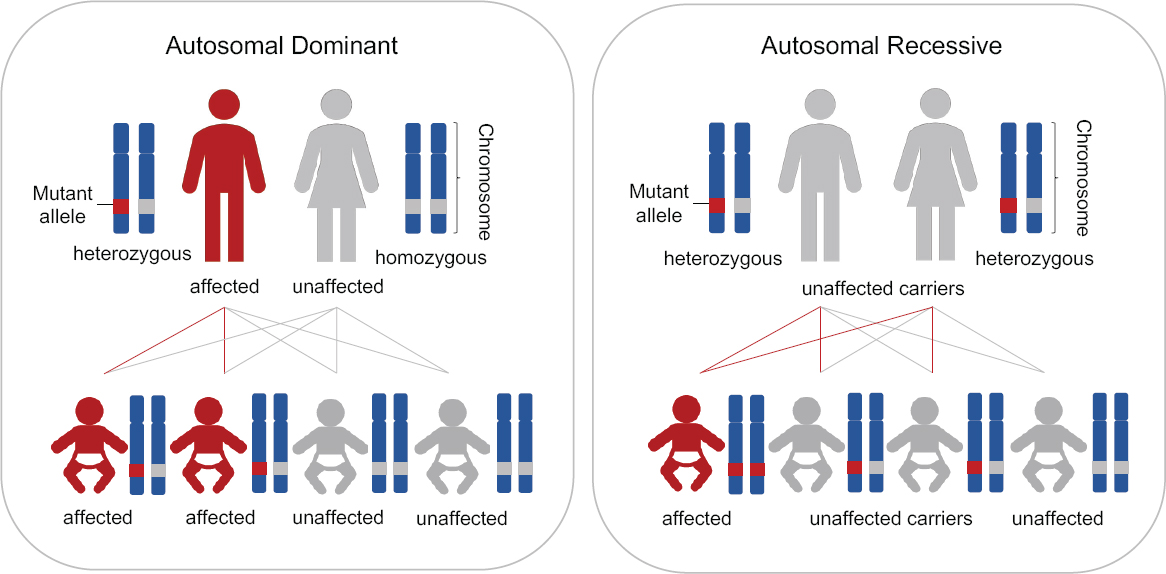
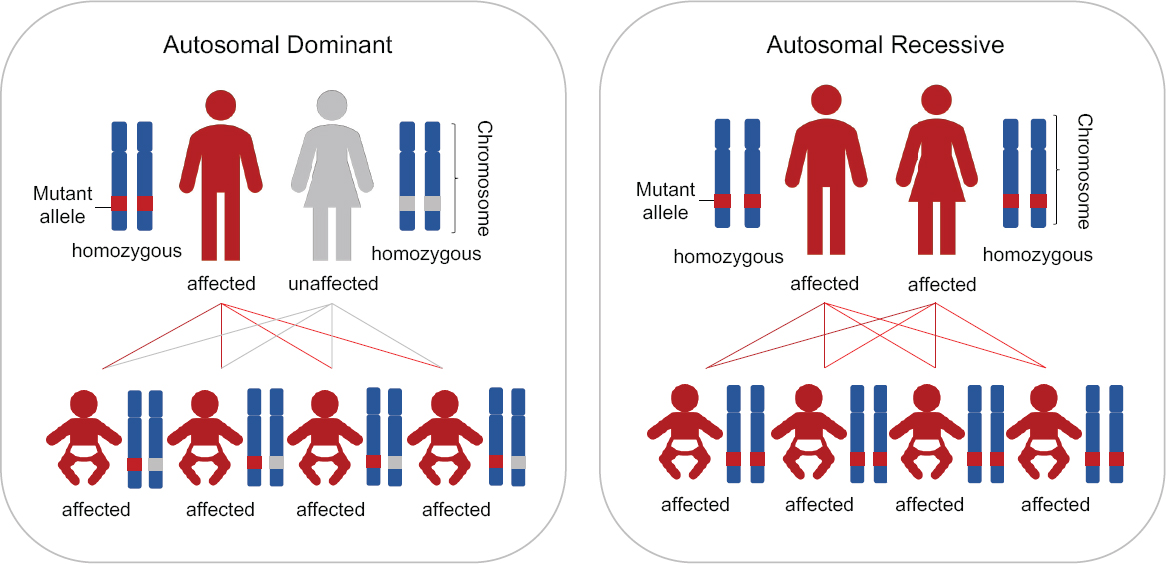
This report distinguishes between these two types of circumstances—those in which all of a couple’s children would inherit the disease-causing genotype and those in which only some children could inherit it. As discussed in the next section, these latter couples currently have various options for having children lacking the disease-causing genotype.
Current Reproductive Options for Parents at Risk of Transmitting a Monogenic Disease
Over the past 30 years, a range of options has been developed to allow prospective parents to know whether they are at high risk of having a child who will suffer from a serious genetic disease and, if so, to avoid this outcome. An understanding of these options is important in assessing the circumstances in which HHGE might meaningfully improve or expand the options already available to prospective parents. Six current options are described below. Certain options may be acceptable to some prospective parents and not to others, while the availability of a particular option to a given set of prospective parents may also be constrained by cost, access, national regulatory policies, or other factors such as religious, cultural, or personal beliefs. Of course, a proportion of genetic diseases are the result of the de novo mutations discussed above, with the precise proportion varying by disease (Acuna-Hidalgo et al., 2016). Such mutations are unpredictable; therefore, the diseases that result from them are not amenable to prevention in that first generation of offspring using preimplantation or prenatal genetic testing, or HHGE were it ever to be available.
Preconception Genetic Testing
Some prospective parents know that they are at higher risk of having a child with a serious genetic disease because one of them has a genetic disease, because they have a family history of a genetic disease, because they underwent genetic testing for a targeted set of diseases that are at higher frequency in a particular ancestry group (e.g., Finnish or Ashkenazi Jewish individuals), or as a result of population genetic screening or testing.
Other prospective parents may not have access to family history information. And many parents only learn that they are at risk when they have an affected child; this is frequently the case for the thousands of rare recessive diseases. In populations with prevalent disease-causing founder mutations and/or high levels of consanguinity, preconception testing can enable prospective parents who wish to do so to reduce the risk of having children with serious monogenic diseases.
Adoption
Adoption avoids the risk of prospective parents passing on a genetic disease because the child is not genetically-related to either parent. Some people who would like to have children find that adopting a child is a positive and fulfilling way to create a family. Others would prefer to have a child who is genetically related to them.
Gamete and Embryo Donation
Another option is to conceive a child via egg or sperm donation, depending on whether the genetic disorder is likely to be transmitted by a woman or a man. They will experience the pregnancy and birth, and the child will be genetically-related to one parent (the father in the case of egg donation, the mother when sperm donation is used). Prospective parents may also use embryo donation. As with gamete donation, they will experience the pregnancy and birth, but like adoption, neither parent will be genetically-related to the child. The large proportion of fertility patients who seek treatment using their own gametes, such as intracytoplasmic sperm injection (ICSI), in preference to treatments involving donated gametes, such as donor insemination, illustrates the value placed on having genetically-related children. Nevertheless, many fertility patients who are unable to have genetically-related children come to accept the use of donated gametes or embryos.
Prenatal Genetic Testing
Some prospective parents have a strong desire to have a child who is genetically-related to both parents—that is, conceived from their egg and sperm. In the early phase of genetic testing, prenatal screening became available as an option to avoid having a child with a serious monogenic disease and is the method of choice for some people. The prospective parents choose to conceive a child in the conventional manner, have genetic testing performed on the fetal tissue (or the placenta in the context of non-invasive prenatal testing), and have the option to terminate the pregnancy if the fetus is found to be affected by the disease.
Preimplantation Genetic Testing
In the 1990s, another option became available: IVF coupled to PGT.3 Developed in 1978, IVF made it possible to create a pregnancy by fertil-
___________________
3 PGT for monogenic diseases is usually called PGT-M. There are other types of PGT, but for the sake of simplicity in this report we use ‘PGT’ to mean ‘PGT-M’, unless otherwise stated.
izing an egg outside of the body, allowing the resulting embryo to develop for a few days, and transferring it into a woman’s uterus. PGT involves removing a few cells from an early embryo, identifying embryos that do not carry the disease genotype, and transferring one of those into the uterus (see Figure 2-3). IVF in conjunction with PGT is currently a reproductive option for many monogenic disorders. Boxes 2-1 and 2-2 discuss the processes involved, including potential harms and benefits, and current outcomes in terms of children born without a genetic disorder.
Treatment of Genetic Diseases
Finally, new options are emerging that would allow a child who is born with a serious genetic disease to be effectively treated. Our growing knowledge of the genetic basis of human disease is leading, in some cases, to therapeutics that can ameliorate or even prevent the serious effects of certain genetic diseases. Some prospective parents at risk of passing on a genetic disorder may choose to proceed to have children, depending on the effectiveness, accessibility, and affordability of the treatment options. The children with genetic disorders who are treated in such ways would remain at risk of passing on the disease to their own children.
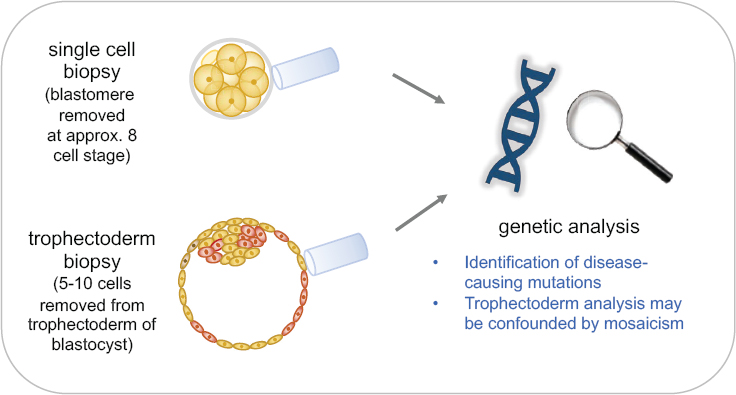
Limitations of Current Reproductive Options
IVF in conjunction with PGT offers an option for many at-risk prospective parents wanting to have a child who is genetically-related to both parents and who does not suffer from a serious genetic disease. Two limitations, however, currently keep IVF with PGT from being a complete solution. These are circumstances in which all embryos produced by a couple would carry the disease genotype, and circumstances in which a viable genetically unaffected embryo is not identified through IVF and PGT cycles. HHGE has been suggested as a possible solution to these limitations. If clinically available, HHGE could also reduce the number of ovarian stimulation cycles a woman has to undergo before having a child, which would be of particular benefit to women at greater risk of ovarian hyperstimulation syndrome and those toward the end of their reproductive years.
Couples Unable to Produce Unaffected Embryos
In extremely rare cases, couples cannot produce any unaffected embryos. For these couples, the parental genotypes guarantee that 100 percent of their embryos will carry the disease genotype (see Figure 2-2). Such couples are extremely rare, because, in the case of an autosomal recessive disorder, both partners would be affected by having the disease-causing genotype in the same gene and would need to have reached reproductive age with a health status compatible with a pregnancy. In the case of an autosomal dominant disorder, one partner would be homozygous for the disease-causing mutation and would also need to have reached reproductive age and be able to produce viable gametes and if female, be able to sustain a pregnancy. With the advent of treatments for genetic diseases, it has been proposed that the number of such couples is likely to increase in the coming decades. For such couples, HHGE would represent a major new option because it could make it possible for the first time for them to have a child genetically-related to both parents but without the disease-causing genotype.
Couples for Whom Unaffected Embryos Are Unlikely to Be Obtained by Cycles of In Vitro Fertilization in Conjunction with Preimplantation Genetic Testing
For other couples at risk of having affected offspring, some fraction of their embryos will be genetically unaffected (e.g., an average of 50 percent in the case of one parent with an autosomal dominant disease and 75 percent when both parents are heterozygous for recessive disease mutations). For such couples, PGT provides a viable option for having a genetically unaffected child. If a sufficient number of eggs can be obtained from the
female partner, it should be possible to identify and implant unaffected embryos. However, IVF followed by PGT sometimes fails to yield any unaffected high-quality embryos to transfer. Couples may choose to repeat the procedure, although some couples do not succeed even after several cycles. The current efficiency of IVF+PGT is described in Box 2-2. HHGE has been proposed as a strategy that might improve the current efficiency of IVF combined with PGT by genome editing of high-quality embryos that have the disease genotype, thus making them available for transfer (Steffann et al., 2018). Whether HHGE would provide a meaningful improvement in efficiency over existing protocols of IVF in combination with PGT is currently unclear and would depend on the extent to which it yields an increase in the number of embryos suitable for transfer.
Identifying the Genotype of a Zygote by Polar Body Genotyping
Current genome editing techniques would involve treating zygotes, at the single-cell stage, when it is not possible to determine their genotype directly without destroying the cell (see discussion in section “Heritable Genome Editing: The Use of Genome Editing in Zygotes,” below). In the case of couples who exclusively produce zygotes carrying the disease-causing genotype, genome editing could proceed on all zygotes without risk of exposing genetically unaffected embryos to the potential harm of the editing machinery without potential benefit. In contrast, when couples can produce both genetically affected and unaffected embryos, subjecting all zygotes to editing would often subject unaffected zygotes to editing.
Polar body genotyping could, in certain cases, provide a reliable way of distinguishing zygotes that do and do not have a disease-causing genotype (see Figure 2-4). Polar bodies are cells produced as an oocyte progresses through the meiotic divisions. The developing oocyte reaches a stage in which it carries four copies of each chromosome, rather than the normal two. As it proceeds through meiosis, this number is reduced to one of each (a haploid set) that is combined with one copy of each chromosome coming from the sperm upon fertilization. The reduction is accomplished by expelling two sets of chromosomes into the first polar body (PB1) at meiosis I, prior to fertilization, and one set into the second polar body (PB2) at meiosis II, after sperm entry. Both polar bodies are accessible for analysis.
PB1 contains the two copies of each particular chromosome that were inherited from either the woman’s mother or her father, selected at random. PB2 contains one copy of each of the chromosomes that were left in the zygote. Thus, analysis of the DNA in the two polar bodies reveals, by elimination, the alleles remaining in the zygote.
In the simplest case, when the woman is heterozygous for a disease-causing mutation, analysis of PB1 will show whether both copies of that mutation
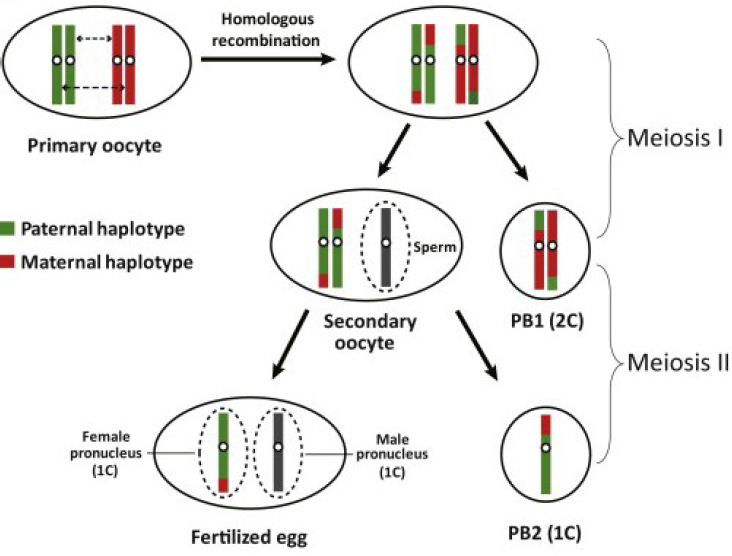
SOURCE: Reprinted with permission from Hou et al. (2013).
are present in PB1 or were retained in the oocyte. In the case of a dominant disease, if the oocyte has retained two disease-causing copies then any person that results from fertilization of that oocyte is certain to inherit that disease.
The situation is complicated by genetic recombination during meiosis I that can exchange a segment of each chromosome between parental copies prior to PB1 expulsion, in which case PB1 might show one copy of the disease-causing allele and one of the non-disease-causing allele. In this case, analysis of PB2 can resolve the issue of whether the zygote has received the disease-causing allele, since the remaining disease-causing sequence must be present either in PB2 or in the zygote. The exception to this would be if a gene conversion event has taken place that has changed the number of disease-causing alleles to one or three, but such events are rare.
In practice, determining whether PB1 carries two copies of the non-disease-causing allele is not entirely straightforward. Polymerase chain reaction (PCR)-based genotyping of PB1 is intended to detect the presence of an allele, but cannot reliably determine the number of copies present. It is possible that “allele dropout” (the failure of an allele to be detected) could cause a PB1 that is actually heterozygous to be mistakenly called
homozygous for an allele. To avoid such errors, it will be important that PB1 be genotyped with a sufficient number of flanking genetic markers to ensure that the genotype at the disease-causing locus can be inferred with a high degree of certainty.
Polar body biopsy is a common and safe technique in PGT, used to detect maternally-derived chromosomal aneuploidies and translocations in oocytes (Schenk et al., 2018). The technique is also used for preimplantation diagnosis of monogenic diseases (Griesinger et al., 2009). However, because the paternal contribution to the genetic constitution of the developing embryo cannot be diagnosed by polar body analysis, its application remains limited (Altarescu et al., 2008).
For the purposes of HHGE, secondary oocytes that were diagnosed to have a disease-causing allele (because their associated first polar bodies had been shown to be homozygous for a non-disease-causing allele) could be frozen to provide a “reserve.” These cells could potentially be used in an HHGE process, if all oocytes collected through the successive IVF attempts, and carrying at least one unaffected allele, had been used in a conventional PGT process but without success. This approach could ultimately increase the chances of a woman with autosomal or X-linked dominant disease having a healthy child without requiring a new IVF cycle.
For autosomal recessive diseases, the allele contributed by a mother heterozygous for a disease-causing allele could also be deduced by the same procedures. However, this would only allow inference that the zygote has biallelic mutations if the father had biallelic mutations.
GENOME EDITING: SCIENTIFIC BACKGROUND FOR A TRANSLATIONAL PATHWAY
The success of therapeutic genome editing depends on both the clear identification of the disease-causing DNA sequence that needs to be changed and the reliability of the technical approach to accomplishing that change without undesired consequences. In this section, the current status of genome editing methods is reviewed, and existing limitations are highlighted. The focus is on the CRISPR-Cas platform, due to its prominence in research and in developing clinical applications, while the parallel utility of other platforms—zinc-finger nucleases (ZFNs) and transcription activator–like effector nucleases (TALENs)—is acknowledged.
Genome Editing Technologies
The modern tools of genome editing have contributed to a revolution in genetics because they provide the ability to introduce with relative ease specific, desired modifications at any locus, or loci, in the chromosomes of
living cells. The idea of precisely editing the genomes of living mammalian cells dates back to the 1980s, when geneticists working with mice developed ways to use homologous recombination to introduce DNA into specific locations in the genomes of embryonic stem cells, which could then be used to create mice with desired genotypes (Capecchi, 2005; Doetschman et al., 1987). While workable for research purposes, the initial methods had very low efficiency, and the desired change was only made in a small number of the cells targeted. The secret to increasing the efficiency was the ability to introduce a targeted double-strand break at a unique, chosen target using a programmable nuclease (an enzyme that cleaves DNA). Various programmable nucleases, including mega nucleases, ZFNs, and TALENs, were successfully used (Bibikova et al., 2003; Joung and Sander, 2013). But the situation changed with a series of discoveries, over the course of two decades, culminating in the recognition that bacteria contain adaptive immune systems, called CRISPR-Cas, that are naturally programmed by ribonucleic acid (RNA) to cut specific DNA sequences and can be used to readily edit the genomes of living human cells (Doudna and Charpentier, 2014; Hsu et al., 2014; Karvelis et al., 2017).
Because of its simplicity and flexibility, the CRISPR-Cas platform has come to dominate research uses of genome editing, and it forms the basis of many preclinical studies and clinical trials (as well as applications in many animals and plants). The basic components of this platform are a Cas nuclease (Cas9 in the most widely used version) and a guide RNA (gRNA) that associate to form a complex. The gRNA usually consists of one RNA molecule (sometimes two) and provides specificity for the editing—directing the complex to a genomic DNA sequence (the target) that matches the variable portion of the gRNA. The gRNA associates with this DNA target through complementary base pairing. Typically, about 20 bases in the gRNA must match the target for effective recognition. Because of this length requirement, recognition can be quite specific, even in a complex genome like that of humans. Once a target is located, Cas9 cuts both strands of the DNA, leaving a double-strand break at that site. These breaks could be lethal to cells, but cellular mechanisms exist to repair them, providing the opportunity to change the DNA sequence at the target location (see Figure 2-5). The CRISPR-Cas system is highly flexible because the variable portion of the gRNA can be designed to match almost any desired target sequence. Although each Cas protein’s enzymatic activity is restricted to a particular short sequence next to the gRNA-determined target, called the protospacer-adjacent motif (PAM), many Cas variants, both natural and derived, recognize different PAMs, and an appropriate one can be chosen for each specific target. In addition, the Cas-induced break can be made at variable distances from the site of the desired change and still be effective. Thus, it will be quite rare for any particular target to be inaccessible due
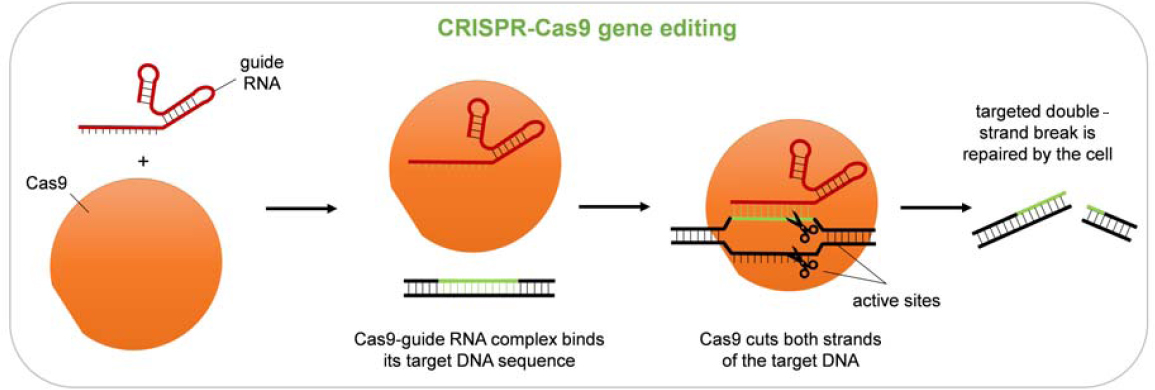
to the absence of a suitable PAM. Even in these cases, the well-developed ZFN and TALEN technologies could complement CRISPR-Cas for editing these loci.
On-Target Modifications
Genome editing technologies rely on repair mechanisms in human cells to make the desired changes in DNA. As a result, the efficiency and specificity of genomic alterations depend not only on the properties of the genome editing system introduced into cells but also on the characteristics of the cellular repair mechanisms.
Cells have several mechanisms to repair the breaks that are created, each of which has advantages and disadvantages for making intended changes. One mechanism, known as non-homologous end joining (NHEJ), simply reconnects the broken ends. This process often results in the addition or deletion of DNA sequences (indels) at the site of the break (Rouet et al., 1994) (see Figure 2-6). Such changes can disrupt the normal function of the DNA at that site if it encodes a protein, for example, or governs the expression of nearby genes. If multiple breaks are made in a single cell, DNA can undergo rearrangements that can also have consequences for gene function. Although it is sometimes possible to anticipate new sequences that will be generated by NHEJ, it is not currently possible to control the
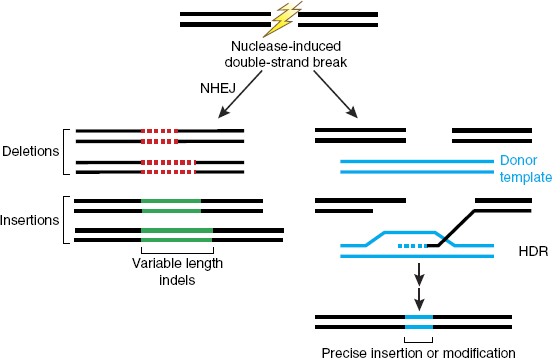
SOURCE: Sander and Joung (2014), reprinted by permission from Springer Nature.
process or to specify a particular product. Thus, NHEJ is useful when the goal is to disrupt an existing DNA sequence but not when a specific editing outcome is needed.
The other major class of processes that repair DNA breaks in cells is homology-directed repair (HDR). In this case, a related (homologous) DNA sequence is used as a template from which sequences are copied at the site of the break (see Figure 2-6). The template can already exist inside the cell—on the sister chromatid or on the other parental allele—or it can be introduced into cells along with the editing nuclease. The sequence changes introduced from a template can be as subtle as changing one or a few base pairs or can involve DNA sequence insertions or deletions of hundreds or thousands of base pairs. With both NHEJ and HDR, changes are induced specifically at the site of the break made by the editing nuclease. The overall efficiency of total modification (NHEJ plus HDR) can be very high in some circumstances, but the outcomes are difficult to control. While HDR is a more versatile and precise repair mechanism and therefore more useful for genome editing, NHEJ is the dominant repair process in most human cell types, and HDR operates efficiently only during some portions of a cell’s cycle of growth and division (Gu et al., 2020; Heyer et al., 2010; Hustedt and Durocher, 2017). The efficiency of HDR also varies widely among cell types for reasons that are not fully understood. Although non-dividing cells typically show very low levels of HDR, there is considerable variability among rapidly dividing cells of different types, and the responsible mechanistic differences have generally not been identified.
Approaches have been tried to enhance the use of the proffered DNA template by HDR. These include providing the template in various molecular formats, linking the template to the Cas9 nuclease or to the gRNA, and manipulating cellular DNA repair activities (Liu et al., 2019b). The improvements in most of these cases have been modest; HDR efficiencies do not approach 100 percent; and unintended products are still produced at some level. Encouragingly, some recent publications report improved efficiencies, including instances in mouse embryos (Gu et al., 2020). Continued research into cellular DNA repair processes will be needed to increase the efficiency and specificity of genome editing, particularly the efficiency of HDR.
Beyond small indels produced by NHEJ, larger sequence changes have been found at sites of induced double-strand breaks. These include extensive deletions (Kosicki et al., 2018), occasional insertions of DNA sequences introduced as intended HDR templates, and chromosome rearrangements. Such products are not readily detected by targeted PCR-based assays that are commonly used, so protocols must be designed explicitly to determine whether they are present.
Another strategy for precise modification that has been used experimentally is microhomology-mediated DNA insertion (Paix et al., 2017;
Sakuma et al., 2016). Because this leads to sequence additions rather than replacements, it will not be applicable for restoring common genomic sequences by introducing only one double-strand break in most cases. To adapt this approach to genetic replacements, two Cas-induced breaks would have to be made, which increases the likelihood of unwanted on- and off-target events.
It is worth noting that while NHEJ might be clinically useful for somatic genome editing, this cannot be said for HHGE, at least in its initial uses. In somatic genome editing, if an intervention derives a clinical benefit by introducing genetic alteration that breaks a gene or a regulatory element, this is acceptable even if the resulting DNA sequence is rarely if ever found in the human population because the change is limited to that tissue in that individual. However, this would not be acceptable for HHGE because the consequences of such genetic alteration in every tissue and at all stages of development could be expected to be deleterious in many cases. The change could also be inherited by future generations. For this reason, it is considered crucial for any initial uses of heritable genome editing to change a disease-causing allele to a common allele in the population that is known not to cause disease. This can only be done by HDR or other technologies that specifically change one DNA sequence into a specific desired sequence. This represents a critical issue for the future use of HHGE.
Off-Target Modifications
From the beginning of research on genome editing, concerns have been raised that at the same time that desired changes are made at the intended target sequence, changes could be introduced elsewhere in the genome. The ability to reduce the frequency of unwanted changes and the ability to detect off-target events when they occur have both progressed in recent years. For the CRISPR platform, specificity has been improved through testing various gRNAs for efficiency with a particular target and modifying both the gRNA and the Cas protein (Chen et al., 2017; Kleinstiver et al., 2016; Slaymaker et al., 2016). Similar advances have been made for the ZFN and TALEN platforms (Doyon et al., 2011; Guilinger et al., 2014). Frequencies of off-target mutagenesis below 0.01 percent at individual at-risk sites have been achieved in some cases.
Several methods exist for identifying non-target genomic sequences that may be at risk of cleavage by any particular genome editing reagent and for detecting and characterizing the off-target editing that occurs (Kim et al., 2019). Genome-wide screening using bioinformatics tools can help to identify genomic sites that are most similar to the target site and thus may be at risk of undesired editing. More useful are methods that identify sites of actual cleavage. Digenome-seq does this by cutting purified genomic
DNA and locating sites where cleavage has occurred by whole-genome sequencing (WGS) (Kim et al., 2015; Tsai and Joung, 2016). GUIDE-seq and DISCOVER-seq, by contrast, capture sites that are cleaved in living cells and subject them to DNA sequencing (Tsai and Joung, 2016; Wienert et al., 2019). Once these off-target sites are identified for a particular nuclease (particular Cas9-gRNA combination in the case of CRISPR), they can be tested by polymerase chain reaction amplification and targeted deep sequencing to see to what extent these off-target sites have been edited in any specific situation.
Unbiased WGS (see Box 2-3) can also be applied to detect off-target changes, but it has some limitations. Because all of the genomic DNA is being read, no individual site within the genome is read as many times as is the case with targeted deep sequencing. Therefore, low levels of mutagenesis can go undetected. There is an inherent error frequency in all methods of DNA sequencing, and there is a background of natural de novo mutations in cells, accumulated as cells grow and divide. Therefore, it is difficult to know which novel sequences are attributable to these effects, as opposed to genome editing. For assessing the genomes of a single cell or a few cells from early stage embryos, since only very small amounts of genomic DNA are available, the DNA must be amplified before it can be subjected to WGS using current technology. At present there is no unbiased method that can uniformly amplify all genomic sequences, although progress has been made in this direction (Chen et al., 2017; Hou et al., 2013).
Other Editing Approaches
Several genome editing systems have been developed that do not rely on the creation of double-strand breaks at the target site in DNA. The avoidance of double-strand breaks is acknowledged by many as ultimately desirable in genome editing given the unpredictability of the cellular response to these. In addition, since these approaches do not rely on HDR, they may be more effective throughout the cell cycle.
Base editing is an alternative approach that involves chemically modifying DNA bases at the desired target (see Figure 2-7). It relies on the specificity of Cas9-gRNA but uses a version that makes only a single-strand break or no break at all and is linked to a deaminase enzyme, resulting in the ultimate conversion of one base pair to another base pair at the targeted site. The tools of base editing are undergoing rapid development. Early experiments showed that sequence changes were often produced at off-target sites in DNA and even in RNA, in some cases at non-targeted sequences. Recent modifications of the base editing reagents have significantly reduced these unintended effects without significantly compromising on-target activity (Doman et al.,
2020; Gaudelli et al., 2020; Grünewald et al., 2019; Richter et al., 2020; Yu et al., 2020). In addition to concerns about potential off-target events, current base editors can make only certain types of DNA sequence changes, specifically, transition mutations (changing C to T, G to A, A to G, or T to C) but not transversions (changing A to C or T, G to C or T, C to A or G, or T to A or G). According to Rees and Liu (2018), approximately 58 percent of human disease alleles are single nucleotide variants, 62 percent of which could be reversed with current base editors. As a result, roughly one third (35 percent) of known disease mutations could potentially be addressed using
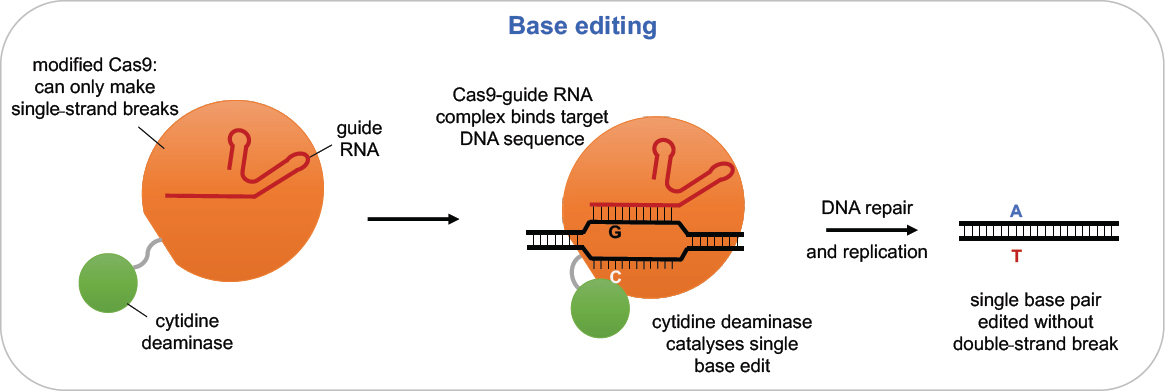
SOURCE: Rees and Liu (2018), adapted by permission from Springer Nature.
base editing technology. Nonetheless, the cytosine deaminase (C-to-T) base editor has been shown to be quite effective in human embryos, particularly at the two-cell stage (Zhang et al., 2019).
Very recently, novel base editors that induce C-to-A and C-to-G transversions have been reported (Kurt et al., 2020; Zhao et al., 2020). Currently these reagents also generate other products at the target, but they will undoubtedly be improved.4
Another recent innovation in genome editing is prime editing (Anzalone et al., 2019). This system involves modification of the gRNA that directs Cas9 to its target sequence such that the RNA also contains a repair template. The Cas9 protein is modified so that it cuts only one strand of the target DNA, in this case the strand that is not bound by gRNA. Cas9 is also linked to a reverse transcriptase enzyme that can utilize the extension on the modified gRNA to copy new sequences into the nicked strand. The provision of a template means that a much wider range of disease-causing mutations, including transitions, transversions, small insertions, and small deletions, can potentially be repaired when compared to base editing. Experience with prime editing is rapidly expanding (Sürün et al., 2020), and at least one study reports success in mouse zygotes, albeit at rather low efficiency (Liu et al., 2020).
While more research is required, both base editing and prime editing provide evidence of the flexibility of the CRISPR-Cas toolkit and the pace of ongoing development of precision genome editing methodologies. It is possible that continuing research may yield new methodologies that rapidly supersede the safety and efficacy of current editing approaches.
Non-Heritable Genome Editing: The Use of Genome Editing in Somatic Cells
One potential alternative to HHGE for the treatment of genetic diseases is somatic genome editing. This section discusses some of the relative advantages and disadvantages of somatic editing in comparison with HHGE.
The initial applications of genome editing in humans occurred in somatic cells, the cells that make up all of the cells of the body except sperm, eggs, and their precursor cells. The effects of genome editing carried
___________________
4 It has also been proposed that genome editing could be used as an alternative to MRT to prevent the transmission of mitochondrial DNA (mtDNA) disease (Reddy et al., 2015). This study used mitochondrial targeted restriction endonucleases or TALENS and showed they could be used to potentially lower mutation load. However, this procedure led to net depletion of mtDNA and thus was not suitable for oocytes with a very high level of heteroplasmic or homoplasmic mtDNA mutations. A recent paper reports the use of base editing on mtDNA (Mok et al., 2020) and may represent a new approach to addressing mitochondrial disease. A detailed analysis of mtDNA editing requires a separate study, in the context of existing treatments for mitochondrial diseases and options such as MRT.
out in somatic cells are generally limited to the individual treated and would not be transmissible to that person’s offspring. (The special circumstance of editing somatic cells that are located in an individual’s reproductive system, such as editing in the testes to treat infertility, is discussed later in this chapter.) Despite the cost that would be associated with any clinical use of HHGE and the complex social, ethical, and scientific issues that heritable genome editing raises, the potential limitations associated with somatic editing, discussed below, represent one reason that HHGE has been proposed as a theoretical alternative for parents wishing to have a genetically-related child who does not have the disease-causing genotype.
Somatic genome editing is an option for treating patients with monogenic disorders, but it remains in early stages of clinical use, and much more experience will be needed to assess its safety and efficacy. The first clinical trial, initiated in 2009, tested the safety of using ZFNs to prevent the progression to AIDS in people infected by HIV (Tebas et al., 2014); and multiple trials using ZFNs, TALENs, and CRISPR systems are currently in progress.5 With significant funding across multiple companies, somatic genome editing is likely to lead to numerous human trials in the coming decade.
The simplest targets for somatic editing are ones in which cells can be removed from a patient, treated outside the body, and returned (ex vivo genome editing) (Li et al., 2020). At present, the primary conditions that can be approached in this way are diseases resulting from mutations in HSCs. For example, promising results have been reported for patients affected with SCD and beta-thalassemia who were treated with CRISPR-Cas reagents to induce expression of fetal hemoglobin,6 although long-term follow-up will be needed before conclusions can be drawn regarding its successes and limitations. Trials are also under way using genome editing to enhance the activity of CAR T cells for cancer immunotherapy (Bailey and Maus, 2019; Stadtmauer et al., 2020).
For many other envisioned somatic therapies, the genome editing reagents will need to be delivered directly to a patient’s cells and tissues (in vivo genome editing). When a disease affects multiple organs, the challenge of delivery is magnified. Only in a few cases is the target tissue readily accessible. One favorable example is the eye, where direct injection of a viral vector carrying CRISPR-Cas reagents is feasible and is being applied for a rare retinal blindness condition.7 The liver is also relatively accessible, and ZFNs are being employed to enhance a gene addition therapy in trials targeting hemophilia and metabolic disease.8
___________________
5 See clinicaltrials.gov.
6 See, for example, clinical trial numbers NCT03745287 and NCT03655678.
7 See clinical trial NCT03872479.
8 See clinical trial numbers NCT02695160, NCT03041324, and NCT02702115.
One feature of many of the above cases is that they rely on disruption of genome sequences by NHEJ. As noted above, this pathway is more active in most cells after a double-strand break is introduced than HDR. Treatments relying on HDR are in development, but attaining therapeutically relevant efficiencies remains challenging. For quite a number of genetic conditions, a non-disease-causing allele could be created via base editing and such approaches are being pursued actively.
While somatic genome editing avoids some of the challenging issues raised by HHGE—because somatic editing involves treating existing patients who can typically consent and because the resulting genetic changes would not be passed on to subsequent generations—somatic editing has some disadvantages. First, because editing does not alter the germline, a patient receiving somatic therapy for a genetic disease could still transmit the disease-causing mutation to future children. Additionally, because only a fraction of targeted cells might be edited, eliminating cells with the disease genotype or positive selection for the edited cells might be needed to increase the fraction of stems cells that have been edited. For example, protocols for somatic editing of hematopoietic stem cells (HSCs) commonly include cytotoxic chemotherapy to eliminate native HSCs before infusion of edited cells. These treatments confer risk of harm. Somatic genome editing therapies are also likely to be very expensive, although costs are unknown and likely to vary (Rockoff, 2019).
Heritable Genome Editing: The Use of Genome Editing in Zygotes
At present, the primary approach that could be used for undertaking HHGE would involve genome editing in zygotes. Because edits introduced would be present in every cell in the body, and the resulting genetic modifications could be passed on to subsequent generations, it would be critically important to obtain the desired genetic change at the target site and ensure an absence of editing-induced changes elsewhere in the genome. There are unique challenges in characterizing the editing events in zygotes and early embryos, as well as important gaps in understanding how to precisely control genome editing in these cells.9
___________________
9 Genome editing technologies have also been adapted to affect the epigenetic state of somatic cells by altering DNA methylation (Kang et al., 2019) and histone modifications (Pulecio et al., 2017). Extensive epigenetic remodeling occurs during early development, and it is not clear whether epigenome editing would be heritable or how it would operate in zygotes and early embryos. Much more research on epigenome editing in embryos would need to be undertaken before it could be considered as an intervention for congenital imprinting disorders (Eggermann et al., 2015).
A zygote—the single, fertilized cell that results from the combination of parental gametes (the egg and sperm)—is the earliest stage of embryonic development. At first the maternal and paternal chromosomes remain in two distinct pronuclei in the cell, and then, after a round of DNA replication, they fuse to become a single nucleus. The zygote then divides into two cells, each with a nucleus containing the full complement of chromosomes from both parents. The blastocyst forms over the first week, and by day 7 consists of roughly 200 cells of 3 different types (Hardy et al., 1989; Rossant and Tam, 2017). Some cells, called the trophectoderm, are progenitor cells that will go on to form the placenta, while additional cells will go on to form the yolk sac. Approximately 10–20 cells within the inner cell mass (ICM) of the blastocyst are epiblast progenitor cells that will form the embryo proper (Niakan, 2019).
Most preclinical research on HHGE has focused on two techniques for getting the genome editing reagents (e.g., the Cas9 nuclease and gRNA, with or without a template DNA) into the zygote: (1) introducing them into an egg cell at the same time as the sperm, or (2) introducing them into the pronuclei or cytoplasm of the fertilized egg. Introducing these reagents can be done by direct mechanical injection or by electroporation, both of which have been used in human embryos without significant damage (Ma et al., 2017). At the zygote stage of development, only one copy each of the maternal and paternal chromosome sets are present, and the aim is to ensure accurate editing in these two chromosome sets while minimizing the chances of undesired consequences arising as a result of off-target events, mosaicism, or other issues. Some studies in mouse embryos have successfully used injection into two-cell embryos, where four or eight genomes are present (before or after S phase) (Gu et al., 2018; Zhang et al., 2019). This presents additional demands on the efficiency and consistency of the editing events in order to achieve uniform outcomes on all alleles and prevent mosaicism.
Another issue arises if editing is attempted in zygotes that have non-disease-causing genotypes, as would be the case if one or both parents were heterozygous for a recessive or dominant variant. To avoid making unnecessary edits in such embryos, the zygotes that require editing would have to be identified prior to treatment. The latter may be possible with polar body biopsy and genotyping (see above) or with an editing platform that can reliably edit a multicellular embryo following genotyping. This would depend on an editing protocol that can reliably edit an eight-cell embryo, the earliest point at which biopsy and genotyping is possible without harming the embryo.10
___________________
10 Were HHGE ever to be used to the extent that a long history of safe use had provided confidence that there was nothing necessarily harmful about the process of introducing editing reagents into zygotes or early embryos, it might be considered acceptable to use editing
Efficiency of Editing at the Target Site
Efficiency of genome editing refers to the ability of the editing system to make the intended edit at the target site. To be used clinically, genome editing reagents would need to exhibit high efficiency in zygotes. First, the reagents must be very effective in binding to the intended target sequence. Second, the desired sequence modification must be produced with high efficiency.
Progress has been made in deriving Cas9 proteins and gRNA designs that yield essentially complete DNA cleavage of an intended target, including in human zygotes (Lea and Niakan, 2019). However, which DNA repair pathway will be used following DNA cleavage depends on cellular characteristics—including the stage of the cell cycle, which DNA damage response components are present, other factors that influence DNA repair, and potentially genetic background. Based on very limited experience, the process of HDR is not efficient in human zygotes. The more common result of making a double-strand break is the introduction of sequence insertions and deletions (indels) via NHEJ, and larger changes can occur as well (Kosicki et al., 2018; Lea and Niakan, 2019). This could result in replacing a disease-causing mutation with another mutation, the nature of which cannot be specified in advance. While the generation of indels has proved useful in the fundamental study of gene function during human embryogenesis (Fogarty et al., 2017), it would be a very undesirable outcome in clinical uses of HHGE. Several recent and not yet peer-reviewed preprints also report significant unintended editing near the target site in human embryos, including chromosomal modifications (Alanis-Lobato et al., 2020; Liang et al., 2020; Zuccaro et al., 2020). Further fundamental characterization of the process of DNA repair in early human zygotes and the development of effective strategies to facilitate use of the HDR pathway will be required to devise safe and effective solutions.
In mice, the introduction of the genome editing reagents at the G2 stage of two-cell embryos has been shown to improve rates of HDR (Gu et al., 2018); however, it is not yet clear whether the same applies to human zygotes. As noted above, editing at this stage would demand exceptional efficiency and consistency of the editing process to avoid mosaicism and to ensure that all alleles are edited. This was not achieved in reported mouse embryo experiments (Gu et al., 2018).
___________________
protocols that only targeted disease-causing alleles to treat a group of zygotes or embryos without prior identification of ones that carried the disease-causing genotype. This would depend on the development and experimental validation of genome editing reagents that are sufficiently specific that they modify only the disease-causing allele without affecting the non-disease-causing allele and did not introduce off-target modifications.
A recent report of high efficiency HDR in human embryos by gene conversion between maternal and paternal chromosomes at the target site is promising (Ma et al., 2017), but this interpretation has been challenged (Adikusuma et al., 2018; Egli et al., 2018; Ma et al., 2018), and further experimentation will be required for validation. Indeed, further research into DNA repair mechanisms and the possibility of gene conversion events in early human embryos will be critical.
Base editing and prime editing have been shown to generate very low levels of indels. For base editors, there is a window of several nearby base pairs in the target sequence that are at risk of unintended editing (Lee et al., 2020), although progress has been made in avoiding this outcome (Huang et al., 2019; Jiang et al., 2018; Kim et al., 2017; McCann et al., 2020). Technical developments continue to advance and will help address editing efficiency and specificity. A considerable amount of evidence has accumulated on the use of base editors in embryos, including those of humans (Li et al., 2017; Liu et al., 2020; Zeng et al., 2018; Zhang et al., 2019). Newer variants of Cas9 and gRNA systems and prime editing have not yet been extensively tested in embryos (Liu et al., 2020).
Beyond the questions of efficiency and repair pathways at the target site, there is also a possible issue with having to target two different disease-causing variants in prospective parents in whom one parent is compound heterozygous for mutations in the same gene that each cause a dominant disease or in which different alleles are present in the same gene in cases of recessive disease. If it is not possible to develop a single editing reagent that targets both variants, then there are two possible editing strategies, both of which have their limitations. These would be to (1) target one variant and use PGT to ensure that the resultant embryo had not inherited the other disease-causing variant, which increases the risk that there are no viable embryos without a disease-causing genotype available for transfer; or (2) introduce two editing reagents to target the two variants, which increases the risk off-target events and the possibility of chromosomal rearrangements.
There are further complications in using HHGE to prevent the inheritance of genetic disorders caused by the expansion of repeated DNA sequences, such as Huntington’s disease. The challenge with such diseases is to reduce the number of repeats in the pathogenic copy of the gene to a non-pathogenic level, an approach that presents significant technical hurdles including the fact that there are identical triplet repeat sequences on both the other allele and elsewhere in the genome. One possible alternative editing strategy in such circumstances would be to introduce a stop codon into the disease-causing variant of the gene to prevent protein production, and therefore the pathogenic effect. Although such a strategy would prevent disease transmission, the precise DNA sequence that is produced would
introduce a loss of function mutation into the population. In a population with high rates of consanguinity or small effective population size, in future generations this mutation could cause an increased frequency of disease due to inheritance of two copies of this mutation. For this reason, introducing heritable mutations that are potentially disease-causing in future generations are unsuitable for any initial uses of HHGE.
Specificity of Editing and Minimizing Off-Target Events
The specificity of genome editing systems—the ability to restrict activity of editing reagents to the intended site in the genome and not to make edits in undesired, off-target locations—is provided largely by the sequence complementarity between the gRNA and the DNA target in the case of CRISPR-Cas systems, or the protein-DNA recognition specificity in the case of ZFNs and TALENs. Before any use of genome editing in human embryos intended for establishment of a pregnancy, careful experimentation and optimization would need to be undertaken to select the reagents that would provide the greatest specificity in the genetic context of the prospective parents. Devising genome editing tools that induce only very low levels of off-target modifications in human zygotes appears feasible but would need to be validated in each specific case.
A second critical issue in evaluating unintended sequence changes in the zygote genome is the ability to detect with high confidence whether such changes have taken place. Tools have been developed for identifying sites that are at significant risk of cleavage by any particular editing reagent, for example, Cas9-gRNA combination (see discussion in section “Genome Editing Technologies,” above). However, these methods have primarily been designed and tested in cell-free whole genomic DNA, in cultured cells, or in whole tissues or organisms, and they are not feasible in embryos, where there is limited availability of cellular DNA. Targeted sequencing of the off-target sites identified in cultured cells can be done with DNA from early stage embryos (Ma et al., 2017), but if there are sites that are uniquely at risk in zygotes, they will be missed.
Off-target sites shown to be at greater risk in somatic cell or embryonic stem cell editing with the same reagents can provide information to help guide the assessment of those sites in embryos. These experiments, however, will not be fully predictive of what occurs in a zygote, because the efficiency and specificity of editing are likely to vary with cell type (NASEM, 2017). As a result, the primary strategy for characterizing editing that has occurred in an edited embryo is WGS. To carry out whole-genome sequencing, a small number of cells are generally removed from an early embryo such as a blastocyst. Because the small amount of DNA is inadequate for processing, whole-genome amplification is undertaken
prior to sequencing. This can introduce amplification bias, including allele dropout, where some sections of genomic DNA are amplified more efficiently than others and some not represented at all. Preclinical WGS of whole-embryo DNA could be useful in identifying the zygote-specific off-target sites, and it would not be as subject to the problem of small amounts of DNA. It would also be useful to assess whether there is a particular somatic cell analysis of off-target sites that correlates well with identification of such sites in zygote editing. Another issue with sequence analysis is that it must cover the full range of likely alterations that could have occurred. This includes large insertions and deletions and even whole or partial chromosome losses that are difficult to detect with standard procedures (Kosicki et al., 2018). Current methods thus lack sufficient power to locate and characterize off-target editing in early embryos with sufficiently high confidence.
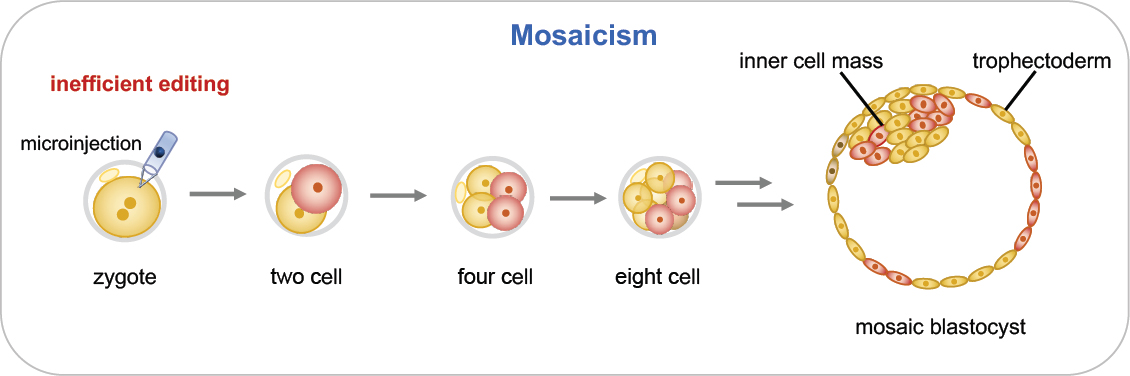
Assessment of Mosaicism
Genome editing of embryos is performed as early as possible—generally at the single-cell stage—to maximize the chance that maternal and paternal genomes have been edited before significant DNA replication and cell division take place. If editing continues beyond this stage, different cells in the embryo may carry different sequence changes at the intended target or at off-target sites. This results in mosaicism, a condition that has been commonly observed in mouse genome editing experiments (Mianné et al., 2017). Mosaicism is a serious concern because some cells in the growing embryo would have the intended sequence change while other cells would not (see Figure 2-8). Unedited cells could make a significant contribution to tissues or cell types that contribute to disease causation, thereby undermining the disease prevention strategy. In addition, editing activity that continues past the single-cell stage raises the prospect of continuing off-target mutagenesis. The effects that genetic mosaicism of this type might have on development and post-natal life are difficult to predict but could be significant.
Preventing mosaicism requires a very high efficiency of the desired on-target modification in the one-cell zygote and restriction of editing activity beyond that stage. Research in somatic cells suggests that the Cas9-gRNA complex is rather short-lived, but the lifetime of the complex in human embryos has not been well characterized. New methods to restrict the duration of active editing are needed. For example, it may be possible to reduce the time the complex remains active by fusing Cas9 to protein domains that accelerate its degradation.
Mosaicism poses particular challenges for verification that correct genome editing has occurred in a clinical context. For an embryo destined
for transfer, only one blastomere or a few trophectoderm cells can be removed for molecular analysis. On- and off-target sequence analysis of these cells does not provide information on the genotypes of the remaining cells of the embryo, including the cells in the inner cell mass that will form the embryo proper. Another method under development is analysis of DNA found in fluid within the hollow central cavity of a blastocyst or in embryo culture media (Leaver and Wells, 2020). Further research is needed to address questions such as the source of detectable cell-free DNA—whether it results from random cell loss or is lost preferentially from cells that may have other developmental anomalies (e.g., aneuploidy). The continued development of non-invasive, cell-free DNA-based techniques may provide further options for the genetic characterization of human embryos prior to clinical use. However, such cell-free methods still could not provide information on the genotype of each cell in an embryo and therefore cannot guarantee the absence of mosaicism.
No current non-destructive method can determine whether all cells in the embryo carry exactly the same edits; it is difficult even to envision a method that could do so. For this reason, it will be essential that preclinical research on human zygotes establish procedures that only very rarely lead to mosaic embryos.
Assessing Early Embryonic Development: The Epigenome and Transcriptome
A number of additional assessments are important in characterizing the impact of genome editing in zygotes. One critical issue would be whether edited zygotes proceed through subsequent steps of development in a normal fashion. Following fertilization, embryos undergo a sequence of carefully orchestrated events that include and depend on epigenetic modifications.11 Studies of preimplantation mouse embryos show that there are global changes to methylation of the maternally and paternally derived genomes, while histone proteins undergo modifications that alter nucleosome positioning and DNA accessibility to the cell’s transcriptional machinery (Eckersley-Maslin et al., 2018; Li et al., 2019; Xu and Xie, 2018). Genome organization at higher levels also occurs in early embryos, including the formation of 3-D topologies that can enable DNA sequences separated by large distances to interact (Flyamer et al., 2017). The epigenetic remodeling that takes place in human embryos is likely to be more complex than in mice, due to the inbred genome of laboratory mice and the influence of genetic variation in humans on epigenetic variation (Delahaye et al., 2018).
___________________
11 An epigenetic modification is one that can result in a change in gene expression without changing the DNA sequence of a gene.
Studies of the epigenomics of early human embryonic development are ongoing (Gao et al., 2018; Guo et al., 2014; Liu et al., 2019a; Smith et al., 2014; Wang et al., 2019; Zhou et al., 2019).
It is currently unclear whether making and repairing chromosomal breaks in zygotes, as might occur with HHGE, could have an impact on local and global DNA methylation, histone modifications, or chromatin domain organization. Preclinical research on epigenomic and transcriptomic profiles is needed to determine whether epigenomic characteristics and patterns of gene expression are altered in edited embryos compared to those that are untreated. Assessment of local chromatin dynamics in the vicinity of an edit may also be important. Research in genome-edited model organisms (e.g., mice) is likely to shed light on various molecular events requiring assessment in human embryos.
Methods for assessing some of these features at the single-cell level are available. For example, DNA methylation profiling of individual cells in the human preimplantation embryo has been reported (Zhu et al., 2018), and single-cell multi-omic approaches surveying chromatin state, nucleosome positioning, and DNA methylation are being developed (Li et al., 2018). Data emerging from stem cell–derived embryo models (Moris et al., 2020; Simunovic and Brivanlou, 2017) might also inform characterizations of early human development. Further fundamental research and refined assessment methodologies will be needed to establish whether developmental milestones, including epigenetic and transcriptomic profiles, are comparable to those of unedited human embryos.
Sources of Relevant Information to Inform Potential Development of a Translational Pathway
The evidence base that would need to be assembled for any clinical translational pathway for HHGE would need to draw on information obtained from a variety of sources.
In Vitro Systems
Most of the current information on editing of mammalian genomes comes from research in cultured cells, and these systems continue to provide valuable insights. Guidance for germline editing, at least for the foreseeable future, will come from methods for optimizing combinations of Cas9 and gRNAs, from the development of novel reagents such as base editors, and from testing various configurations of template DNAs for restoring non-disease-causing sequences. Additional methods for identifying and minimizing off-target mutations will need to be developed, including approaches for assessing the production of large insertions, deletions, and rearrangements.
Of particular importance are methods for controlling the editing outcome at the intended target, including suppressing indel formation and enhancing sequence replacement. Relevant results can be obtained from many different types of cultured cells, but perhaps most useful will be experiments in cell lines, induced pluripotent stem cells (iPSCs) and primary cells, carrying the particular disease-causing mutation. A limitation to the use of stem cells for this purpose is the fact that there is considerable variability among lines established from individual patients, and there are reports of high rates of genomic abnormalities (Henry et al., 2019).
Clinical Use of Somatic Genome Editing
Despite the fundamental differences between genome editing in somatic cells and in germline cells discussed above, clinical experience with somatic genome editing can provide some information to help inform HHGE. The differences in methodologies mean that successes or failures in the use of somatic genome editing are unlikely to have direct relevance to the prospects for HHGE in humans—the editing reagents and delivery methods used will likely differ, as will the cellular context and repair mechanisms in zygotes versus somatic cells.
Nevertheless, clinical trials of somatic editing therapies represent the use of genome editing in human primary cells, rather than in cells maintained in laboratory culture. Somatic therapy may thus be able to provide some insight into the benefits to a patient from correcting a particular disease-causing mutation, and it may also provide some data on the types and frequencies of genetic modifications observed at the intended target and at off-target locations in different cell types. Long-term monitoring of patients who receive somatic genome therapies may also reveal the extent to which there are late-appearing risks associated with the treatment, particularly if off-target mutations were created, or may help reveal whether therapeutic outcomes are influenced by the broader genomic background of patients. At the same time, populations of treated somatic cells undergo competition for growth and survival in the body, which might mask deleterious effects that could affect the development of treated embryos. Long-term follow-up of somatic therapy patients will also provide insights into compliance with long-term follow-up processes and optimization of communication and consent procedures.
Research in Zygotes of Other Mammals
While it is likely that human zygotes harbor capabilities and undergo processes that differ in significant ways from those in human somatic cells, it seems likely that human zygotes may share some of these capabilities and processes with zygotes of other organisms, particularly other mam-
mals. Germline genome editing is being employed in many animal species for the purpose of generating specific variants for research purposes or for improvement of livestock traits. In these cases, there is no need to achieve a very high efficiency of the desired edit, since multiple individual animals can be screened at many stages of development and the final examples are often obtained in subsequent generations through selective breeding. Such approaches would be unacceptable in humans.
A great deal of genome editing research has already been done in mouse embryos, but, while undeniably useful in identifying key parameters, these may not be the best model for human embryos. While human and mouse blastocysts show similar morphology, there are significant differences at later stages of embryonic development. In addition, it has been shown that the timing of expression and the early embryonic function of certain genes differ significantly between the two species (Fogarty et al., 2017; Niakan and Eggan, 2013). Recently, more attention has turned to larger mammals, including cows and pigs, in which embryo genome editing is becoming routine. Work has also been done in non-human primates, including macaques and rhesus monkeys, which are the closest equivalents to humans (Chen et al., 2015; Niu et al., 2014). In some cases, human disease alleles may have been introduced into the genomes of these organisms, creating so-called humanized genes, and these would constitute excellent models for assessing the feasibility of specific sequence repairs. However, such work in larger mammals may be more likely to give rise to ethical objections in some countries due to the relative sophistication of the animals in question.
Attempts to perfect human germline genome editing can benefit greatly from focusing research in other zygotes on the issues that are currently most troublesome. These include raising the efficiency of on-target editing and preventing unintended on-target events. This will likely require developing an understanding of how DNA repair processes operate in zygotes, how outcomes depend on the timing of introducing the editing reagents, and what formats of template DNA and delivery are most effective. Off-target events can also be addressed. The issue of embryo mosaicism is critical and cannot be addressed in somatic cells. Methods to limit editing to the one-cell stage as well as methods to analyze mosaics can be developed in model organisms before being tested in human zygotes. The issue of effects of the editing procedure per se on epigenetic programming and gene expression can also be investigated. Of course, other issues, such as off-target effects, would not reflect the vulnerabilities in the human genome.
Research in Human Zygotes
Early human embryo development is already studied for a variety of reasons, including to provide insight into infertility, implantation, and pla-
cental development, and to improve IVF technologies. Currently, little is known about how the very first cell types that emerge in a human embryo become specialized in their fate and function (Lea and Niakan, 2019) or how this might be affected by genome editing.
Before it would be appropriate to proceed with HHGE, the necessary features of reliable germline genome editing would need to be tested in human zygotes directly. These tests must demonstrate very efficient introduction of the intended sequence change, no significant level of off-target mutagenesis, and a very low probability of mosaicism, and these tests must, at least for the earliest cases, be performed for each new target and Cas nuclease/gRNA combination. Obtaining meaningful measurements of the on-target, off-target, and mosaicism rates would require analyzing a sufficiently large number of embryos.
This will present a challenge, however, because research conducted on human embryos is prohibited in some countries and is subject to stringent oversight and regulation in many others. Where research is permitted, generally only a limited number of human embryos can be obtained, and these are generally donated surplus embryos from IVF. Many human embryo genome editing studies have also used non-viable tripronuclear embryos (Li et al., 2017; Liang et al., 2015; Tang et al., 2018; Zhou et al., 2017), but their abnormal chromosome content and aberrant developmental course make them unsuitable for preclinical characterization of genome editing in human zygotes as part of a potential translational pathway to HHGE.
With respect to adverse developmental effects, it is currently prohibited to experimentally monitor human embryonic development beyond 14 days because of the culture limit that exists in many jurisdictions (Cavaliere, 2017). Synthetic embryo-like entities may be of some use here because they can be maintained beyond 14 days; however, current models do not fully reflect the cell types, environment, or developmental timings of an intact embryo (Aach et al., 2017; Moris et al., 2020; Rivron et al., 2018; Warmflash, 2017). Monitoring fetal development requires implantation to establish a pregnancy. Therefore, experiments in non-human organisms must provide assurance that genome–editing-induced adverse developmental effects are likely to be non-existent or minimal.
FUTURE ISSUES IN ASSISTED REPRODUCTION: IMPLICATIONS OF IN VITRO STEM CELL–MEDIATED GAMETOGENESIS
Rather than undertaking genome editing in zygotes, an alternative pathway for HHGE would be through genome editing of cells that are capable of forming functional male and female gametes (sperm and eggs). Genome editing is unlikely to be undertaken directly in sperm or eggs, but
several types of cells can serve as gamete precursors in which genome editing could be performed, including (i) spermatogonial stem cells (SSCs) and (ii) patient-derived pluripotent stem cells or nuclear transfer embryonic stem cells (ntESCs) that could be induced to differentiate into functional haploid gametes via in vitro gametogenesis (IVG). The technologies to develop human gametes from cultured stem cells are still under development and are currently unavailable for clinical use. Prior to any human use of such stem cell–mediated methodologies, they would need to be permitted in their own right as a component of assisted reproductive technology (ART). However, the development of in vitro stem cell–mediated gametogenesis would have significant implications for both the reproductive options that might be available to prospective parents and for HHGE.
Implications of In Vitro Stem Cell–Mediated Gametogenesis for Heritable Genome Editing
In the vast majority of cases, in vitro stem cell–mediated gametogenesis would eliminate a need for heritable genome editing as a means of preventing the transmission of monogenic diseases. For those circumstances in which a couple could produce an embryo without the disease-causing genotype, the ability to screen a large number of embryos created from male and female gametes produced from the prospective parents’ somatic cells would enable suitable embryos to be identified. This would be the case even in those circumstances in which parents have a relatively low predicted chance of producing an unaffected embryo using conventional ARTs. This technology would thus eliminate the current efficiency issues associated with PGT.
The exception that would remain would be circumstances in which all embryos that could be produced by a couple would carry the disease genotype (see Figure 2-2). For such cases, HHGE would still be the only option for producing an unaffected child genetically-related to both parents. In this circumstance, IVG offers the potential to address a number of the technical challenges associated with using current methodologies to undertake genome editing in human zygotes. Performing genome editing in cultured cells would permit very careful analysis of any edited genomes at both genetic and epigenetic levels prior to the production and use of gametes. This would have significant safety implications, since the issues of on-target editing fidelity and avoidance of off-target events could be largely settled before any gamete is considered for use in the creation of an embryo. Meanwhile, high-throughput epigenetic analyses can be employed to ensure the epigenetic stability of genome-edited cells. In addition, the use of edited gametes to generate an embryo would avoid the issue of mosaicism, since all embryonic cells would be generated from a single gamete that had previously been genome-edited.
Approaches involving the use of human gamete and precursor cells in culture for laboratory research show promise for addressing basic questions in early human development. Any future clinical use of IVG, however, raises numerous scientific and ethical issues that would require careful consideration given the potential consequences for human reproduction (Bredenoord and Hyun, 2017; Greely, 2018). What would societal attitudes be to the routine production of hundreds or even thousands of human embryos for use in research or in treatment? Research would no doubt flourish in such circumstances, but in the context of treatment, would thousands of embryos be routinely discarded, or stored indefinitely, because some patients feel that destructive embryo research is ethically unacceptable? Some practitioners might attempt to screen thousands of embryos for polygenic traits (see the next section), ranking them according to polygenic risk prior to transfer (Karavani et al., 2019). As a result, wide-ranging societal discussions would need to occur prior to any clinical use of IVG, analogous to the discussions that are required prior to clinical use of HHGE (Adashi et al., 2019).
Preclinical Research Using In Vitro Stem Cell–Derived Gametes
The current state of progress in generating gametes from stem cells cultured in vitro is important when considering the potential implications of this technology for HHGE. It is unclear at present which, if any, of these approaches might reach a stage of development at which clinical application could be considered.
Genome Editing in Spermatogonial Stem Cells
Sperm cell genomes cannot be edited directly using current technology. However, sperm cells originate from stem cells in the seminiferous epithelia of the testis, the SSCs. SSCs can be isolated from multiple species, including primates, but so far have only been maintained long term in culture from small mammals (Kubota and Brinster, 2018). Research in mice has shown that genome editing of SSCs and their subsequent transplantation into the testis results in the production of sperm with the edited genome. This method could be used to prevent human genetic disease inherited from the male lineage. Wu et al. (2015) used the CRISPR-Cas9 system to edit the genome of mouse SSCs to correct a cataract-causing mutation. They were able to identify and select SSCs carrying the desired genome editing but lacking other unwanted genomic changes or signs of epigenetic abnormalities (including abnormal genomic imprinting), and were able to produce healthy offspring after transplantation of the edited SSCs back to the mouse testis. As with zygote genome editing, for any initial human
uses to produce gametes, it would be very important to carry out investigations of the epigenetic and transcriptomic properties of embryos generated from edited SSCs. It is currently unclear whether the requirement to transplant cells into the testis would be an obstacle to clinical application or whether maturation into functional gametes could be reliably and safely accomplished in vitro.
Use of Androgenetic Haploid Embryonic Stem Cells
Research in mice also shows that androgenetic haploid embryonic stem cells (AG-haESCs) can be derived from embryos generated either by injecting sperm into oocytes from which the maternal chromosomes have been removed or by fertilizing eggs and removing the female pronucleus. These AG-haESCs, with genetic modifications to mimic the imprinted state of two paternally-imprinted genes, can be injected into oocytes to “fertilize” them, giving rise to live and fertile mice (Wang and Li, 2019). Therefore, genetic manipulation of AG-haESCs is a way of performing one-step transmission of genetic modifications in mice.
Human AG-haESCs have recently been successfully derived. These cells exhibit typical paternal imprints and can also “fertilize” human oocytes and support early embryonic development, leading to blastocysts and diploid embryonic stems cells with transcriptomes comparable to those of normal diploid embryos and embryonic stem cells, respectively, derived from ICSI (Zhang et al., 2020). Haploid embryonic stem cells thus provide a novel form of human germline stem cell that could potentially be used for editing disease-related mutations and validating the desired genotype.
Use of Induced Pluripotent Stem Cells or Nuclear Transfer Embryonic Stem Cells for In Vitro–Derived Gametogenesis
Genome editing could also be performed in patient-derived iPSCs or ntESCs, prior to these being differentiated into gametes in vitro—known as IVG (see Figure 2-9). Mouse pluripotent stem cells can be converted into cells with properties similar to primordial germ cells (called primordial germ cell–like cells, or PGCLCs) (Hayashi et al., 2011). When these are introduced into germ cell–free mouse gonads, functional spermatozoa can be produced. Further differentiation in vitro into germline stem cell–like cells and functional spermatid-like cells has also been reported (Ishikura et al., 2016; Zhou et al., 2016). These studies involve the completion of gametogenesis in vivo by gonadal transfer or in vitro by co-culture with neonatal testicular somatic cells but establish the principle that mouse stem cells could be converted into functional male gametes.
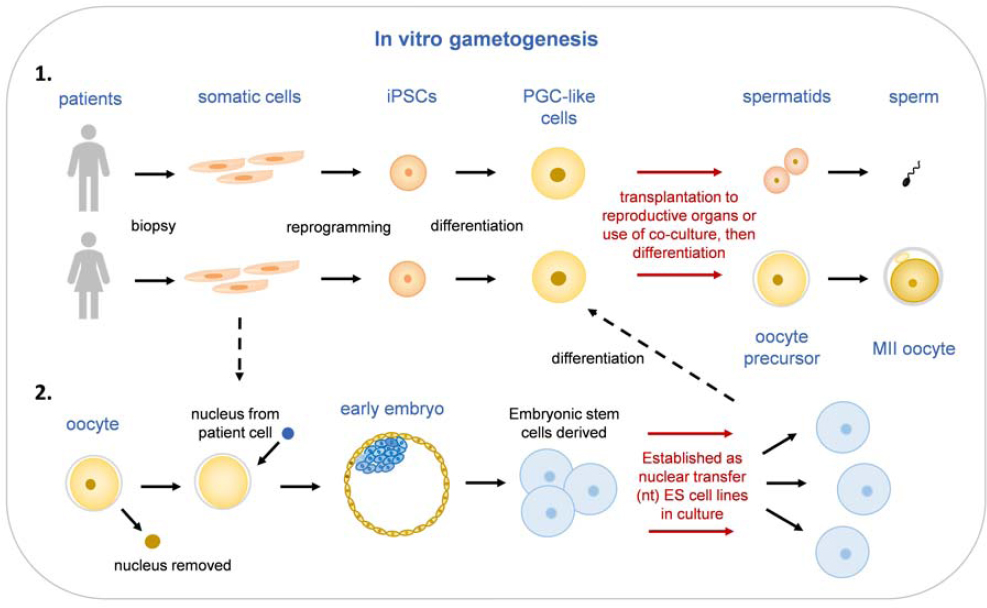
The ovary does not contain female germline stem cells analogous to SSCs that could be extracted and manipulated in cell culture, because gametogenesis is completed before birth in females. The only theoretical route to developing female gamete precursors for cultivation and genome editing is therefore through the use of IVG. Hayashi et al. (2012) reported that transplantation of PGCLCs into the ovaries of adult mice ultimately resulted in the production of fertile offspring. Morohaku et al. (2016) reported the successful in vitro maturation of mouse primordial germ cells into MII oocytes (the stage at which an oocyte would be fertilized by a sperm). Hikabe et al. (2016) reported the reconstitution of the entire process in mice, involving conversion of stem cells to PGCLCs and maturation of these PGCLCs into MII oocytes. These studies demonstrate that it is possible in mice to recapitulate the female gametogenesis pathway from pluripotent stem cells.
Studies of IVG using human cells are ongoing, but it is unclear whether it will be possible to replicate the successes reported in mice, especially the initiation and completion of meiosis. Human PGCLCs (hPGCLCs) have been derived from iPSCs (Sasaki et al., 2015) and characterized molecularly (Chen et al., 2019). One challenge to human female gamete derivation in vitro is the successful use of hPGCLCs. Yamashiro et al. (2018, 2020) reported the use of hPGCLCs to derive oogonia-like cells in a long-term culture model. However, this is not an efficient approach for the further differentiation of such in vitro–derived oogonia into primary oocytes in meiotic prophase I. Improved co-culture methods will likely be required, but the availability of fetal gonadal somatic cells of the appropriate type may be challenging. The use of such approaches to produce human sperm has not yet been achieved although in vitro reconstitution of some steps in spermatogenesis have been reported (Nagamatsu and Hiyashi, 2017; Yuan et al., 2020).
All IVG approaches would involve substantial periods of cell culture, and the adaptation to culture itself runs the risk of introducing undesired genetic or epigenetic changes. Further research would be required in mammalian models, including non-human primates, to develop this as a potential method of producing human gametes.
ADDITIONAL COMPONENTS OF ANY CLINICAL TRANSLATIONAL PATHWAY FOR HERITABLE HUMAN GENOME EDITING
In addition to the scientific and technical considerations discussed above, any potential clinical use of HHGE would entail the incorporation of detailed plans for obtaining informed consent and for monitoring the effects of genome editing. Lessons for developing such plans can be drawn
from experiences with other novel human ARTs, such as mitochondrial replacement techniques (MRT), and from current practices in reproductive medicine, although HHGE would pose additional unique challenges due to the risks associated with making heritable changes.
Informed Consent
The potential use of HHGE for monogenic diseases poses specific challenges in terms of informed and voluntary consent that are analogous to those presented by MRT. These challenges are discussed in detail in reports from the Nuffield Council on Bioethics (NCB, 2012) and the National Academies of Sciences, Engineering, and Medicine (NASEM, 2016). HHGE for other types of uses, such as for polygenic disorders (see below), would raise additional challenges.
Prospective parents, as participants in initial human uses of heritable genome editing, would need to understand the novel procedures to be employed in addition to the use of IVF and PGT, and would need to be aware of the lack of information beyond preclinical evidence on the safety and efficacy of HHGE in humans in order to weigh the potential harms, benefits, and uncertainties involved in their decision on whether or not to proceed. Prospective parents would also need to be aware of the risk that they may give birth to a seriously ill or disabled child, and of the possibility that they may be faced with the difficult decision of whether or not to terminate a pregnancy should prenatal testing identify genetic or physical anomalies. The advantages and disadvantages of alternative routes to parenthood that would avoid the transmission of genetic disease would need to be carefully discussed as part of the consent process. It will also be important to discuss with the prospective parents the pressures they may face from media attention or public interest both during pregnancy and after the birth of a child with an edited genome.
Furthermore, prospective parents would need to be informed of the importance of monitoring of the health status of such children to document outcomes of HHGE and informed that they would be asked to consent to prenatal and long-term assessment of their children who have undergone an editing procedure. Informed and voluntary consent from parents for their children to participate in monitoring would need to be obtained at each new phase of assessment until the children reach the age of consent (generally 18, though this varies by jurisdiction). This consent would need to inform parents of all the features of the assessment that may affect their willingness to allow their child to participate and also of their right to refuse or withdraw from participation without incurring any penalty to themselves or their child. Parents would also need to be informed about the purpose of the monitoring; who would be conducting evaluations; what their partici-
pation would entail, including the risks, benefits, and limitations; and the safeguards that had been put in place regarding confidentiality, anonymity, and data protection.
The children themselves would not be able to give consent to long-term monitoring at the outset, as is the case for the many longitudinal studies that exist worldwide on children’s health and development. Before they reach the age of consent, and once they are old enough to do so, children would be asked to assent, which means that they agree to participate without necessarily understanding the full significance of the assessment.
In line with guidelines from the Society for Research in Child Development (SRCD, 2007) and the report Ethical Research Involving Children (Graham et al., 2013), assent or informed consent would need to be obtained from children born following HHGE according to the children’s age and developmental level. Children would need to be informed of the nature of the assessment in a way that is appropriate for their level of understanding, and assessors would need to ensure that children understand what participation will entail. It would also need to be made clear to children that they are free to participate or not and are free to withdraw, entirely or from specific assessments, at any time without having to give a reason and without adverse consequences. Children would also need to be assured of medical confidentiality and personal privacy. The people involved in the assessment of children must be trained to recognize signs of discomfort and instructed to cease the assessment should a child become distressed.
Due to the importance of monitoring children born following HHGE, every effort would need to be made to encourage parents and children to participate in long-term follow-up. For individuals with edited genomes who continue to consent to and engage in a monitoring process into their child-bearing years, this process would provide an opportunity to invite these individuals to include any children they have in an intergenerational assessment, thus enabling the follow-up of grandchildren bearing an edited genome.
Individuals born following HHGE should be valued in the same way as any other person, and they and their parents should not be stigmatized, or discriminated against, for having undergone HHGE.
Long-Term Monitoring
Because the consequences of genome editing for children’s physical and psychological development are unknown, in order to establish whether HHGE prevents the transmission of a genetic disorder and whether there are unintended adverse and intergenerational effects it is important to assess the health of the developing fetus during pregnancy and the health
and well-being of resulting children throughout the lifespan and into the next generation, if such individuals exist. Such intergenerational monitoring would be important for evaluating the well-being of the individuals involved rather than refining HHGE technologies, since the technologies are likely to have been refined in the interim. The section below briefly reviews monitoring that has been done with other ARTs and then turns to the consideration for HHGE.
Monitoring Children Born Through Assisted Reproductive Technologies
Regarding the follow-up of children born following HHGE, the closest parallels are studies of children born through ARTs, such as IVF and ICSI, who are genetically-related to their parents; children born through ARTs using donated eggs, sperm, or embryos and who therefore lack a genetic connection to one or both parents; and children born following PGT in combination with IVF/ICSI. Although most follow-up studies have focused on childhood outcomes, a small number of studies have followed up children born by ARTs to adulthood. For example, young men conceived by ICSI have been followed up to assess their fertility, and children conceived by gamete donation have been followed up to assess their psychological well-being, relationships with their parents, and thoughts and feelings about their method of conception.
Physical and health outcomes.
There exists a substantial body of research on the physical and health outcomes of children born through ARTs involving large, representative samples, although most of this research has focused on short-term rather than longer-term outcomes. A recent, comprehensive review, largely of children born through IVF and ICSI, focused on studies of singleton children to avoid the confounding effects of multiple births (Berntsen et al., 2019). It was concluded that children born following ARTs are at some risk of adverse short-term outcomes such as low birth weight, pre-term birth, and birth defects, although these risks were described as modest. The poorer outcomes for these children appeared to result from a combination of the parents’ subfertility and specific aspects of the ART procedure, although it was difficult to disentangle the two in the absence of appropriate comparison groups.
There has been less research on the development of children born following PGT. In a cohort study of children born following PGT in Denmark (Bay et al., 2016), the level of adverse obstetric and neonatal outcomes was higher than that of spontaneous pregnancies but similar to that of pregnancies following ICSI. It appeared that the increased risk of adverse outcomes was related to the underlying parental genetic condition rather than the PGT procedure itself. Studies that followed children born after
PGT to age 2 found no differences in growth, cognitive and psychomotor development, and behavioral and health outcomes compared to ICSI and naturally conceived children. More recent studies of 5-year-olds (Heijligers et al., 2018) and 9-year-olds (Kuiper et al., 2018) produced similarly reassuring results.
Psychological outcomes.
Research on the psychological well-being of children born through ARTs dates back to the 1990s (for reviews see Golombok [2017, 2019]). These studies have shown that these children’s families are generally characterized by positive parent-child relationships and well-adjusted children, irrespective of the presence or absence of a genetic connection between one or both parents and the child. In spite of a growing trend toward greater openness, many parents do not tell their donor-conceived children about their origins, mainly because of the concern that this information would jeopardize family relationships, especially the relationship between the non-genetic parent and the child. Parents who tell their children about their biological origins when they are young generally find that their fears about the potentially negative consequences of disclosure were unfounded, and there is growing evidence that early disclosure is associated with more positive outcomes for donor-conceived children and adults. Some children and adults who are aware of their donor-conception search for information about their donor and donor siblings (genetic half-siblings born from the same donor) in order to acquire a greater understanding of who they are and how they came to be.
These findings suggest that disclosure to children born through HHGE of the circumstances of their conception at an early age is likely to be beneficial for their psychological well-being and relationships with their parents, and that persons born following HHGE may be interested in what was done to them as embryos and why. It is not known whether, or what, parents tell children born following PGT about their origins, although they may tell them that they were selected as embryos following genetic testing. However, HHGE is distinct from PGT in that it produces an alteration to the genetic make-up of their children whereas PGT does not.
Long-Term Follow-Up of Children Born Following Heritable Human Genome Editing
As with informed consent, a broad approach is described for the long-term follow-up of children born following HHGE, as the most appropriate specific assessments will be dependent upon a number of factors, including
the conditions being edited and the countries in which the editing is carried out. Comprehensive long-term follow-up would include the assessment of (i) obstetric and perinatal outcomes; (ii) genetic disorders in resulting births; (iii) other health problems in children; (iv) growth, motor, and physical development; (v) cognitive and language development including developmental delay; and (vi) psychological adjustment including mental health problems. It will be necessary to achieve a balance between the need for monitoring and the need to avoid unduly burdening the children and adults concerned.
To examine children’s growth, cognitive development, language development, and social and emotional development, assessments at key developmental milestones would be required, depending on the specific aspect of development under consideration. There are standardized tests designed for this purpose. If these assessments are to be used internationally, there is a need for versions of tests that have been translated into different languages, have been adapted to be culturally appropriate, and have normative data for the interpretation of the meaning of individual children’s scores in the context of their own language and culture (Gregoire et al., 2008). There is growing consensus that it is important, both from an ethical perspective and to increase understanding, to include children’s voices in interventions that affect them. For this reason, children affected by HHGE should be interviewed about their thoughts, feelings, and experiences, beginning in adolescence. A key issue for long-term follow-up studies of children born following initial HHGE uses is the need to minimize sample attrition in order to reduce sample bias. Challenges include the small number of children who would be born following initial uses of HHGE, the need to standardize both the genetic disorders under investigation and the genome editing undertaken, and the absence of meaningful comparison groups, all of which would restrict the reliability, validity, and generalizability of the findings.
In the United Kingdom, children conceived through MRT, the closest parallel to HHGE, are assessed from the prenatal period onward. During pregnancy, in addition to the monitoring of fetal growth and development, parents are offered amniocentesis. At birth, neonatal indicators are recorded, and infants receive routine developmental checks in the first year of life. The outcome of MRT on children born will be initially studied at 18 months (an age at which there are clear developmental milestones) and then subsequent follow-up will be conducted throughout childhood with appropriate consent from the parents and assent from the child, followed by consent in adulthood by the individual whose genome was edited (Gorman et al., 2018).
OTHER POSSIBLE USES OF HERITABLE HUMAN GENOME EDITING
The majority of this chapter has focused on the use of HHGE by prospective parents to prevent transmission of a genetic variant that causes a single-gene (monogenic) disease in order to have a genetically-related child unaffected by that disease. This section discusses the potential use of HHGE in more complex circumstances: to prevent the transmission of polygenic diseases, to affect characteristics not associated with disease, and in the special circumstance of male infertility.
Human genetics is complex. Although the distinction between monogenic and polygenic conditions is useful, the reality of human genetics is more complicated. Despite the extraordinary developments in understanding mammalian gene function, relatively little is known about the function of the majority of genes in the human and mouse genomes. This ignorance about basic biology is a reminder of the research that is still required to ground clinical interventions firmly in genomic knowledge (Brown and Lad, 2019; Oprea et al., 2018).
Charting the landscape of human genetic variation and understanding its contribution to disease will require a systematic understanding of how genomic variation reliably predisposes to disease, both monogenic and polygenic. Many genes are pleiotropic; they play a role in multiple biochemical pathways. Genes operate in functional networks, and understanding the role that genes and their variants play in such networks is a challenge that will require powerful computational tools and large-scale datasets from genomics programs, including animal models (Cacheiro et al., 2020). Monogenic and polygenic inheritable conditions might also share underlying similarities. Rare single-gene disorders often predispose to more common diseases characterized by complex inheritance, with variants underlying monogenic disease interacting to contribute to complex disease risk (Blair et al., 2013).
Polygenic Diseases
Many common diseases have significant genetic contributions, including such conditions as type 2 diabetes, rheumatoid arthritis, heart disease, schizophrenia, many cancers, and Alzheimer’s disease. Most of these diseases are polygenic, influenced by many genetic variants across many genes. These genetic variants typically have very small effects on the risk of disease (Timpson et al., 2018). Nonetheless, in some cases, particular variants in particular genes can have a comparatively large effect on the risk of a disease, although even then such variants do not completely determine
who will get a disease or how serious it will be.12 For example, individuals with a single copy of a common variant in the APOE gene, called APOE4, approximately doubles the risk of dementia in each decade after age 60 compared to individuals without this variant. Nonetheless, the absolute risk of dementia with one copy of APOE4 is only approximately 5 percent from ages 60–69, and 15–20 percent over age 80 (Rasmussen et al., 2018).
In polygenic inheritance, individual DNA variants might each alter gene function modestly, most often by altering the production of a protein, and would not individually cause the disease phenotype. For example, a genome-wide association study in 11,260 cases of schizophrenia and 24,542 controls identified 145 novel associated loci, each contributing only a small amount to the risk for developing the disease (Paradiñas et al., 2018). A genetic risk score estimate can be generated by combining the different associated loci into a single score associated with a higher or lower risk for developing the disease. However, these scores indicate only relative risk and are not deterministic. Risks for most common (polygenic) diseases depend not only on complex genetic factors, but also on a wide range of environmental influences, such as diet and lifestyle choices, and on random events whose impact is difficult to predict.
We do not have sufficiently predictive information to contemplate the use of HHGE for intervening in the many common diseases that are associated with multiple variants and complex patterns of inheritance. Editing a gene variant associated with a complex disease is likely to have only a minor effect on the risk of developing that disease, while also potentially introducing unknown effects because of other biological roles the gene may play and other genetic networks in which it may interact. Genetic variants that alter the risk for one disease frequently have effects on the risk for other diseases, often in opposite directions. Such difficulties are compounded when the editing of multiple variants is contemplated. Valuable research on genetic and non-genetic factors associated with many human diseases continues, but this knowledge is far from a stage at which it could support the use of HHGE for the prevention or reduction of risk in the case of polygenic diseases.
Complex, Non-Disease Traits
Geneticists have also identified many genetic variants associated with personal characteristics, and genome editing has been proposed as a potential way to alter these. For example, genetic variants are associated with increased muscle strength or with an improved ability to increase strength
___________________
12 Other types of muti-genic disease inheritance are possible, including digenic, in which mutations in two different genes are required for the disease (Deltas, 2018).
through training, and such traits are desirable for some people. An example is a variant in the ACTN3 gene that encodes a protein that is part of muscle fiber and is associated with muscle strength (Ma et al., 2013). Some studies have found significant associations between a particular variant of this gene and muscle strength and function, though others have not (Pickering and Kiely, 2017). It is unlikely that this variant on its own is responsible for the level of muscle strength; therefore, the outcome of genome editing for this variant would be unpredictable.
As with polygenic diseases, we have insufficient knowledge of the biological effects of the many individual genetic variants associated with complex traits. Even if it were possible to edit all the variants associated with a particular trait, it would not be possible to predict the phenotypic outcome. If the purpose of the editing were to obtain greater-than-typical function, rather than preventing a disease, this would be a form of genetic enhancement. These types of uses of HHGE would likely be very controversial, raise many additional societal and ethical concerns, and be scientifically very premature.
Treatment of Male Infertility
The circumstance of male infertility represents a unique case for which genome editing could be contemplated. For a man whose infertility has an identified monogenic genetic cause, genome editing in vivo in his reproductive tissues (e.g., testes) or in vitro in his SSCs could offer the opportunity to restore fertility. This type of use sits at the intersection of somatic and heritable genome editing. The male patient in this circumstance exists and could provide informed consent to the procedure, and the clinical intent here is to treat a condition having a negative impact on that person’s life, as with somatic genome editing. However, because the cells targeted for genome editing are reproductive, the correction of an infertility-causing mutation would produce a genetic change that is heritable. In its effect, genome editing for infertility would be a use of HHGE. Restoring fertility in a woman would be an analogous circumstance and is a theoretical possibility, but as discussed above it is likely to exist only through use of stem cell–derived gametes (IVG) and remains significantly further from potential clinical application than the ability to obtain and edit the genome of testicular SSCs.
Approximately 7 percent of men reportedly have some form of infertility (Krausz and Riera-Escamilla, 2018). This condition can arise from multiple potential causes, and the origin of many cases of infertility remains unexplained, but it has been estimated that more than 2,000 genes have transcripts specific to male germline cells (Schultz et al., 2003). Research continues to identify genes affecting fertility, but a number of single gene
mutations are associated with quantitative and qualitative spermatogenic anomalies (Ben Khelifa et al., 2011; Harbuz et al., 2011; Kasak et al., 2018; Maor-Sagie et al., 2015; Nsota Mbango et al., 2019; Okutman et al., 2015; Tenenbaum-Rakover et al., 2015; Yatsenko et al., 2015).
For men with genetic conditions in which sperm are produced but fertility is diminished (e.g., because the sperm have reduced motility), existing reproductive options such as ICSI may be able to overcome the fertility challenge and result in the creation of an embryo. On the other hand, some men possess identified genetic mutations that make them unable to produce any sperm (azoospermia) or produce sperm with significant genetic or structural abnormalities incompatible with creating a viable embryo. For this subset of people, genome editing of SSCs and these cells’ subsequent development into sperm could provide the ability to create a genetically-related child. This process may still require the use of ARTs, for example, if the SSCs are developed into sperm in culture. Or, if the genome-edited SSCs are reintroduced to the testes to develop into sperm in vivo, this intervention could provide an ability to have a genetically-related child without the use of IVF. Although infertility resulting from single gene mutations affects only a fraction of the human population, those who have identifiable genetic causes for this condition form a potential community that may be interested in access to HHGE.
CONCLUSIONS AND RECOMMENDATIONS
This chapter explored in detail the state of the science in genetics, genome editing technologies, and reproductive medicine that would be required for potential translational development of HHGE. It also explored circumstances in which HHGE has been proposed as a technology that could increase reproductive options. The chapter then discussed the current evidence for, and state of understanding of, the feasibility of these options. The key messages arising from this analysis are provided below.
Knowledge of Human Genetics Limits Potential Applications of Heritable Human Genome Editing
For monogenic diseases, the use of HHGE to change a genetic variant that causes the disease to a non-pathogenic DNA sequence that is common in the population could prevent the disease being transmitted to offspring, if it is possible to efficiently and reliably make precise genomic changes without undesired changes affecting human embryos. Many human diseases are polygenic, in which a phenotype is the result of many genetic and external factors that individually have small effects and that interact in complex ways. Current knowledge is not sufficient to use HHGE to edit variants
associated with the risk of developing complex, polygenic diseases or that affect non-disease traits.
Heritable Human Genome Editing Could Provide a Reproductive Option for Some Prospective Parents at Risk of Transmitting a Disease Genotype
In certain circumstances, HHGE would represent the sole option for a couple to have a genetically-related child who did not inherit a disease-causing genotype. These circumstances would arise when one prospective parent is homozygous for a dominant disease-causing mutation, or both prospective parents are homozygous or compound heterozygous for mutations in the same gene whose mutation causes a recessive disease.
In all other circumstances, at least some of a couple’s embryos are expected to not have a disease-causing genotype, and PGT could offer a potential solution. Demand for PGT screening for disease-causing mutations is increasing. However, PGT is not without physical and financial costs, and some couples do not achieve a live birth after one or more cycles. Genome editing might increase the number of embryos available to a couple for transfer and thus may increase PGT success rates, although improved data concerning current PGT usage and failure rates are needed to provide insight into the extent to which there are unmet needs in this area and thus potential future demand for HHGE.
Current Technologies for Heritable Human Genome Editing Are Inadequate
Genome editing of zygotes (one-cell embryos) produced by IVF is currently the most feasible approach for carrying out HHGE, but the efficiency and specificity of current embryo-editing methods are not adequate for human clinical uses. There are no procedures to adequately control the outcome of DNA repair in embryos after an editing-induced break is introduced into the genome, and current analytical methods are not sufficient to provide necessary assessment of on-target and off-target events and mosaicism in a clinical context. Genome editing methods that avoid production of a double-strand DNA break are also subject to most of these same deficiencies. Research to refine methods and understand the feasibility and limitations of genome editing in zygotes is needed. This research would necessarily involve the use of human embryos because there are differences in the DNA repair mechanisms used by different cell types and there are important species-specific differences in early development which limits the usefulness of research in model organisms and other human cell types.
Genome editing conducted in stem cells that are induced to form male, and potentially female, gametes provides a theoretical alternative approach
for HHGE. Such cells can be maintained and characterized in cell culture, but these approaches raise distinct technical, ethical, and social considerations of their own and would have to be permitted as a means of assisted reproduction in a given regulatory framework before they could be considered for use in HHGE.
Plans for Consent and Monitoring Are Needed
Any clinical use of HHGE would need to include detailed plans for obtaining informed consent and for conducting long-term monitoring of the effects of genome-editing methodologies. Established protocols and legally recognized approaches exist for obtaining informed consent from parents to participate in a clinical trial of a reproductive intervention and for obtaining consent from parents, and eventually children, to participate in future stages of monitoring and assessment. Similarly, protocols exist for the long-term monitoring and evaluation of the physical and psychological development of people born using novel ARTs. HHGE could draw on these protocols, although details would need to be adapted to the context of clinical use, and HHGE raises special considerations due to the nature of the intervention and the heritability of the genetic changes that could be made.
Gaps in Current Scientific and Technical Understanding Need to Be Addressed
Genome-editing tools are effective at making targeted DNA sequence modifications and could, in principle, be applied to HHGE. However, scientific and technical knowledge gaps would need to be addressed before any clinical use of HHGE could responsibly be considered. These gaps include the following.
Limitations in the Understanding of Human Genetics
Multiple genes influence the disease risk for the majority of human diseases and disorders, and many genetic variants identified in humans have an unknown impact on phenotype. Existing knowledge is not sufficient to predict the effects of making genetic changes in these circumstances. Even for monogenic diseases, sound evidence for a causative role of an identified genetic variant would be needed prior to genome editing. The impact of genetic background on the function of particular disease-related variants is also not well understood, and in some cases can modify the risk or clinical severity of a disease, which complicates the analysis of the potential risks and benefits of HHGE.
Limitations in the Understanding of Genome-Editing Technologies
It remains unknown to what extent the results of genome editing in model systems, such as human somatic cells and embryonic stem cells, and in other animals is predictive of the efficiency or effects of editing to correct a specific mutation in human embryos. In addition, controlling the DNA repair processes in embryos is difficult because the pathway is often dominated by complex indels produced by NHEJ. Newer variants of Cas-gRNA systems and methods such as prime editing have not yet been extensively tested in embryos.
Limitations Associated with Characterizing the Effects of Genome Editing in Human Embryos
Understanding of the effects of unintended genetic changes, both on- and off-target, on subsequent development and long-term health remains limited. Although genome editing is commonly used to create mice and other organisms that appear developmentally normal, protocols suitable for preclinical validation of human editing would need to be established. These would need to determine (a) the efficiency of achieving desired on-target edits; (b) the frequency with which undesired edits are made, including absolute and relative frequencies of NHEJ, HDR, chromosomal translocations, and large genomic deletions or duplications resulting from editing; and (c) the frequency with which mosaic embryos arise. This will likely require developing an improved understanding of how DNA repair processes operate in germline cells, zygotes, and early embryos; how outcomes depend on the timing of introducing the editing reagents; and what formats of template DNA and delivery are most effective.
Limitations Associated with the Development of Genome Editing in Stem Cell–Derived Gametes
Studies in animal models reveal that the use of genome editing in gamete precursor cells and the production of male gametes able to produce healthy embryos without a disease-causing genotype are a nearer-term prospect than the production of female gametes. The production of gametes from stem cells in vitro, if successful, would offer an alternative to HHGE in most envisaged cases. But such technology requires the same careful scientific and societal assessment needed for HHGE.
Recommendation 1: No attempt to establish a pregnancy with a human embryo that has undergone genome editing should proceed unless and until it has been clearly established that it is possible
to efficiently and reliably make precise genomic changes without undesired changes in human embryos. These criteria have not yet been met, and further research and review would be necessary to meet them.
Beyond the technical considerations, countries contemplating allowing the use of HHGE will need to address the broader issues raised by the technology. As discussed in greater detail in Chapter 5, this should include engagement with a wide range of perspectives within their jurisdictions.
Recommendation 2: Extensive societal dialogue should be undertaken before a country makes a decision on whether to permit clinical use of heritable human genome editing (HHGE). The clinical use of HHGE raises not only scientific and medical considerations but also societal and ethical issues that were beyond the Commission’s charge.




























































