
4
A Translational Pathway to Limited and Controlled Clinical Applications of Heritable Human Genome Editing
This chapter identifies the elements of a responsible translational pathway for circumstances of heritable human genome editing (HHGE) that would fall into those described in Chapter 3 for potential initial uses: (1) prospective parents for whom all children would inherit the disease-causing genotype for a serious monogenic disease and who therefore have no alternative for having genetically-related offspring unaffected by the disease (Category A); and (2) prospective parents for whom some children would inherit the disease-causing genotype for a serious monogenic disease and who have poor likelihood of success through preimplantation genetic testing (PGT) (a very small subset of couples in Category B; see Chapter 3 for further details).
Chapter 4 specifies preclinical and clinical requirements that would need to be met to enable clinical evaluation of initial proposed uses of HHGE, should a country decide to permit such uses to be considered. A pathway toward clinical use of HHGE begins with a specific proposed use and includes three major stages:
- development of a sufficient methodology and preclinical evidence of its safety and efficacy;
- decision points and required approvals; and
- clinical evaluation of a proposed use.
Each stage includes sub-components, as shown in Figure 4-1. This chapter describes these components and the requirements that would need to be met to proceed further. These pathway requirements pertain to genome
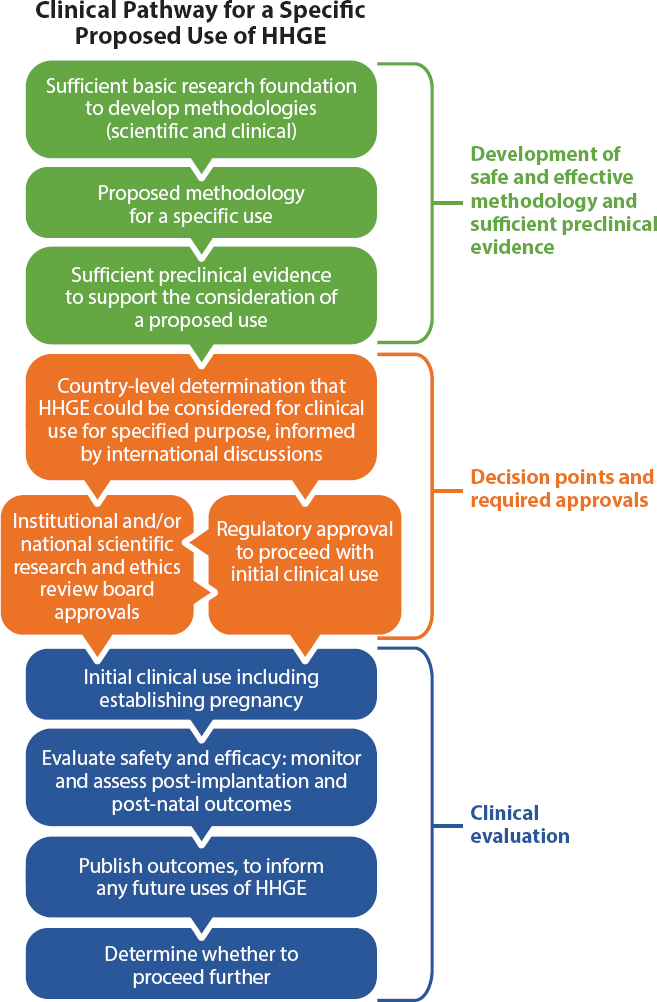
editing undertaken in human zygotes. Should in vitro–derived gametes ever be permitted as a reproductive technology, preclinical considerations for their use for HHGE are discussed later in the chapter.
As introduced in Chapter 1, important parallel processes of societal engagement also must occur throughout the pathway but are not the focus of this report.
The required elements of a responsible translational pathway are summarized in Box 4-1 and discussed in the chapter.
CONTEXT FOR ANY HERITABLE HUMAN GENOME EDITING TRANSLATIONAL PATHWAY
As noted in Chapter 3, it is not possible to describe a generic translational pathway applicable to all uses of HHGE. Any translational pathway starts with the specific proposed use, which would involve making precise changes to a targeted sequence of DNA in the context of prospective parents wishing to have a genetically-related child without a particular disease. As emphasized in Chapter 3, the proposed clinical use also needs to be one that would fall within the set of circumstances for which the Commission was able to describe a translational pathway given the current state of scientific and clinical knowledge.
For any initial uses, HHGE would represent a new technological intervention in the assisted reproductive technology (ART) clinic, with only preclinical data with which to judge safety and efficacy. There will be information relevant to safety and efficacy that could only be obtained following evaluation in humans. As a result, the preclinical and clinical standards would need to be set very high for any initial human uses.
To meet this requirement for any initial human uses, the proposed use should be to change a pathogenic genetic variant known to be responsible for the serious monogenic disease to a sequence that is common in the relevant population and that is known not to be disease-causing. The disease would also need to be one that meets the Commission’s definition of “serious” for the purpose of identifying an initial pathway toward HHGE. The Commission defines this as a life-shortening disease that causes severe morbidity or premature death.
BASIC RESEARCH FOUNDATION TO ESTABLISH SAFE AND EFFECTIVE GENOME EDITING METHODOLOGIES
As described in Chapter 2, current genome editing technologies are not sufficiently precise and specific to ensure safe and effective HHGE. Knowledge gaps remain in controlling and characterizing genome editing in human zygotes. Bringing the process of genome editing in zygotes to required levels of efficacy and safety will require substantial improvements in the editing and validation procedures themselves.
Basic Research as a Foundation to Develop Methodologies
Continued basic research is needed to expand understanding and control of genome editing in human zygotes. Continuing basic research on genome editing for purposes not linked to specific clinical uses will be very
important for issues like design of the editing reagents for maximum efficiency and specificity; methods for detecting and quantifying the broad range of outcomes at both on- and off-target sites; enhancing desired editing outcomes—for example, by favoring homology-directed repair (HDR) over non-homologous end joining (NHEJ) if introducing a double-strand break is part of the methodology; and characterizing processes in human embryos that influence editing outcomes and may differ from those in somatic and cultured cells. Accumulating evidence will help to decide to what extent cultured cells, model organisms, or other surrogates can be used to confidently predict events in human zygotes.
Key elements that will be required to develop safe and effective methodologies for HHGE include the following.
Controlling On-Target Events
The inability to control events at the genomic target site constitutes a major limitation for HHGE. The majority of disease-causing mutations would require introducing the non-disease-causing sequence by copying from a provided template or from a non-disease-causing gene copy located on the homologous chromosome. Based on limited experience, this process of HDR is not efficient in human zygotes following a double-strand break in the DNA. In zygotes and in other cell types, the more common outcome of making a double-strand break is the introduction of sequence insertions and deletions (indels) via NHEJ. The NHEJ process could result in replacing one mutation with another, the consequences of which cannot be predicted or controlled. Such products would be deleterious in almost all instances; therefore, the ratio of DNA repair by HDR to NHEJ must be increased to achieve the desired outcome with high probability. As noted in Chapter 2, both base editing and prime editing largely avoid the risks associated with making and repairing a double-strand break; and both (particularly base editing) have shown promise in embryos.
The goal of HHGE would be to generate embryos that carry only a common, non-disease-causing sequence at both alleles of a gene. Creating one non-disease-causing allele would still be effective in the case of a recessive condition, while restoring both would also eliminate carrier status. A corollary of this goal is that alteration of a pre-existing non-disease-causing allele should be avoided. Zygotes that require genome editing must be identified prior to treatment. The latter may be possible through biopsy and testing of the first and second polar bodies (see Chapter 2) or future development of efficient genome editing methodologies for multi-cellular embryos that have already been genotyped.
Minimizing Off-Target Events
The ability to reduce the frequency of unintended genetic changes and to detect such changes when they occur has progressed significantly in recent years. For CRISPR-based genome editing, testing of various guide ribonucleic acids (gRNAs) for a particular target and making modifications to both the gRNA and the Cas protein have improved specificity. Similar advances have been made for the zinc-finger nuclease (ZFN) and transcription activator–like effector nuclease (TALEN) platforms. However, there are still challenges for detecting unintended sequence changes with high confidence in embryos. Analysis of off-target events arising from genome editing can be done by whole-genome DNA sequencing; however, current whole-genome sequencing (WGS) methods are not adequate for the accurate analysis of the small amount of genetic material that can be safely extracted from blastocyst-stage embryos intended for transfer to the uterus. In addition, WGS may not capture the full range of alterations that can occur. These could include large insertions and deletions or even whole or partial chromosome losses, which are difficult to detect with WGS or with standard polymerase chain reaction–based procedures.
Minimizing Mosaicism
Preventing mosaicism requires the ability to make the desired on-target modification with very high efficiency either in the one-cell zygote with restriction of editing activity to that stage or in all cells of embryos comprised of two or more cells. If genome editing continues beyond the first cell division, different cells in an embryo may carry different sequence changes at the intended target or at off-target sites. The effect of such mosaicism is difficult to predict, but it may pose serious risks by either failing to prevent disease due to target tissues having an insufficient number of appropriately edited cells or by introducing undesired mutations—particularly large copy number variants—at the target locus or elsewhere in a fraction of cells that could result in diseases related or unrelated to the targeted disease. Mosaicism poses particular challenges to verification. For an embryo destined for transfer, only a few trophectoderm cells can be safely removed from a blastocyst for molecular analysis. No current method can determine whether all cells of an embryo intended for uterine transfer carry exactly the same edits; it is even difficult to envision one that would. This means that preclinical research must establish procedures that only very rarely lead to mosaic embryos.
Evaluating the Physiological Effects of Genome Editing of Disease Alleles
Research on the short-term and long-term physiological and functional outcomes of editing disease alleles is needed to verify that a given intended edit is sufficient to prevent the disease phenotype and to provide reassurance that significant, unanticipated health effects would be unlikely to result from the genome editing process. Useful information may be obtained from human somatic editing of the same disease allele and potentially from the use of germline editing in other mammals—for example, to alter the animal-equivalent version of the human allele in cases where the human disease phenotype is accurately reproduced.
PRECLINICAL EVIDENCE TO SUPPORT A PROPOSED USE
Extensive investigation in a variety of experimental, preclinical contexts will be required prior to any attempt to establish a human pregnancy with an edited embryo.
Proposed Methodology for a Specific Use
Developing a proposed methodology that has been independently validated to be sufficient for the proposed use is an important part of the preclinical stage. The genome editing system (e.g., the combination of a Cas protein and gRNA designed to target a specific section of DNA) would need to address the issues above: controlling on-target editing, minimizing off-target events, and avoiding the generation of mosaic embryos. For any specific clinical use, the particular reagents and processes will need to be tested carefully at the particular genomic site and in the particular context as far as possible, as described below.
Sufficient Preclinical Evidence to Support Clinical Evaluation of the Proposed Use
To undertake HHGE through the use of zygotes, the genome-editing reagents would most likely be injected directly into oocytes concomitant with sperm or into zygotes immediately after fertilization. It is possible that, as has been observed for base editing (Zhang et al., 2019), treating embryos at the two-cell stage can also be effective. If HHGE were used in circumstances in which only some embryos were likely to carry the disease-causing genotype, the criteria for a translational pathway described in Chapter 3 would require a method to ensure that no embryos without the disease-causing genotype were subjected to the process of genome editing and transfer. Genotyping the polar bodies produced as an oocyte undergoes
meiosis could identify the zygote’s genotype in circumstances in which the maternal contribution is definitive (see Chapter 2). If polar body analysis would not be sufficient to determine the zygote’s genotype, an alternate approach would be needed. One option could be editing an eight-cell or later embryo (the stage at which embryo biopsy and genotyping could be conducted). However, this would require the development of methodologies capable of effectively undertaking such editing. The goal for any of these circumstances is to produce embryos with a non-disease-causing genotype at the target sequence in all cells. Preclinical evidence for the proposed use of HHGE would need to be obtained from cultured human cells and from zygotes of model organisms before conducting preclinical experiments in human embryos.
What preclinical evidence can or should be collected depends on the genetic circumstances of the prospective parents, such as whether it is necessary to assess impacts on any non-disease-causing alleles that they might pass on. As described in Chapters 2 and 3, circumstances in which all embryos would inherit the genotype causing a serious monogenic disease include having one parent who is homozygous for an autosomal dominant disease-causing mutation or both parents homozygous for an autosomal recessive disease-causing mutation. In the latter case, no non-disease-causing allele would be present in cells of either parent or in their zygotes.
Need for Extensive Research in Cultured Human Cells and in Zygotes of Model Organisms
Each combination of a specific target gene and editing reagents would need to be evaluated, since each combination will present unique potential for on- and off-target events. To justify the design of the editing reagents proposed for potential clinical use, preclinical research in cultured human somatic cells from the prospective parents and in model organisms must include the following steps.
Assessment of Parental Genomes
Requirement: Obtain whole-genome sequences of the prospective parents using best practice protocols for investigating genetic disorders. Identify the exact sequence of the target mutation and surrounding genomic region. For a given combination of target and editing reagents, assess potential off-target sites based on these genomes.
Context: WGS is routinely used to identify new (de novo) mutations in offspring, as well as to establish the specific disease-causing genetic variant that parents with a history of genetic disease are at risk of passing on.
Examples of best practice protocols include the Deciphering Developmental Disorders study in the United Kingdom.1
Testing of Genome Editing Reagents in Cultured Parental Cells
Requirement: The following assessments need to be undertaken:
- For assessing on-target efficiency, test the editing reagents using cells from the parent(s) with the disease-causing mutation.
- For identifying sites at risk of off-target editing, test in cells from both parents.
- If a non-disease-causing allele is present in the genome of either parent, also test for any potential undesired editing of this allele.
Context: Testing in parental cells is important to allow for possible effects of genetic background on on-target and off-target outcomes. The information obtained from these assessments should be used to refine the editing reagents for efficacy at the intended target and to assess and minimize off-target mutagenesis. The cumulative frequency of off-target mutagenesis should not be significantly higher than the expected de novo mutation frequency. The cells could be primary cells from each parent, induced pluripotent stem cells (iPSCs), or embryonic stem cells (ES) derived from the parents by nuclear transfer. Because the adaptation to culture and the induction of pluripotency can both lead to the accumulation of novel mutations, testing in several independently derived lines is advisable.
Testing of Editing Reagents in Embryos of Model Organisms
Requirement: Test the efficiency of modifying the comparable target sequence in zygotes from a mammalian model organism. Use models incorporating humanized sequences—at least the sequence to be modified at the target and surrounding regions recognized by the editing reagent.
Context: Genome editing in mammalian zygotes differs from editing in somatic cells of the same species. Since some embryo-specific characteristics are likely shared among species, testing the editing reagents in zygotes of mammalian model organisms allows the characterization of the types of editing outcomes and the development of procedures to prevent and
___________________
1 See https://www.ddduk.org/intro.html. The Deciphering Developmental Disorders study is funded by the Health Innovation Challenge Fund and the Wellcome Sanger Institute to analyze genomic information from “over 12,000 undiagnosed children and adults in the U.K. with developmental disorders and their parents” in order to better understand the basis of these disorders.
assess mosaicism. Relevant contextual factors in mammalian embryos include the methods of delivering the editing reagents, which are different in zygotes compared with cells in culture, and the DNA repair mechanisms active in zygotes compared with adult cells. While processes in human zygotes may differ from those in other mammalian zygotes, information obtained from such experiments can provide guidance for further testing in the human context.
Testing in mammalian zygotes is essential for refining the editing system prior to preclinical testing in human embryos. This testing is not designed primarily to evaluate phenotypic effects of editing the target sequence in the model organism. Animals such as mice can be used to generate sequences equivalent to the human disease-causing allele, so-called humanized alleles (Zhu et al., 2019). Evidence showing that the humanized disease-causing allele can be edited to the non-disease-causing allele would be essential to have prior to the earliest clinical uses of HHGE. If the humanized sequence cannot be produced in a mammalian model organism for some reason, then this disease allele should not be selected for an initial application of HHGE.
Preclinical Testing in Human Embryos
Preclinical testing in human zygotes must be undertaken to demonstrate that the genome-editing methodology proposed for clinical use provides high levels of efficiency, specificity, and safety. No other cell type can substitute for this stage of preclinical evidence. The preclinical testing of human embryos is conducted in a laboratory, and the embryos are never used to establish a pregnancy.
Only a limited number of human zygotes are available for experimental purposes, and the Commission recognizes that many jurisdictions do not permit the creation of human embryos for research. Nonetheless, thorough validation of the genome editing process prior to clinical use would require data from human embryos. To minimize generation of embryos specifically for experimentation, zygotes created through ARTs but not used by a couple to establish a pregnancy may be donated for use in laboratory research. While such zygotes would likely lack the specific disease-causing allele(s) being targeted for the proposed use of HHGE, testing them would generate information about potential off-target editing and could provide valuable guidance regarding zygote-specific processes involved in on-target editing. This option is limited by the fact that these stored zygotes will likely be at the G2 cell cycle stage—that is, later than would be subjected to genome editing—but may still be useful for approaches that address this stage or even ones in development for two-cell embryos. In addition, most in vitro fertilization (IVF) embryos are currently stored at even later stages and may not be useful at all.
For initial uses of HHGE, there will be certain types of information that could only be obtained following human clinical use. As a result, the standards for preclinical testing must be very high, and human zygotes for preclinical assessment of the editing methodology should be obtained that contain the disease-causing mutation. The efficacy of the editing reagents on disease-causing alleles carried by the prospective male parent can be tested in zygotes produced using his sperm to fertilize donated oocytes. In the case of disease-causing alleles transmitted by the female parent, care must be taken to avoid subjecting her to multiple rounds of hormonal stimulation and oocyte collection, with their attendant risks. If she is not at further risk from the IVF process and not of advanced age, a single round of stimulation and collection might be appropriate for the purposes of generating embryos for testing. A reasonable alternative would be to recruit a sperm donor who carries the same disease-causing mutation(s) and to use his sperm in conjunction with donated oocytes to produce zygotes for testing.
After a substantial knowledge base has been gained through experimentation on human embryos, it might be possible to identify alternative cell-types that reliably allow accurate prediction of the effects of genome editing in human zygotes. In such circumstances, it might become acceptable to use these cells as a surrogate for the preclinical tests involving human embryos required in this pathway. Rigorous scientific assessment of such models and their ability to substitute for evaluation in human embryos would be critical before using such alternative cell systems as the only source of preclinical evidence for HHGE. Over time, extensive preclinical testing of the ability of a particular editing methodology to correct a variety of targeted alleles might also become considered sufficient to conclude that it was not necessary to test the correction of each specific allele in preclinical human zygotes. Ongoing and independent scientific and technical reviews to assess knowledge gained and to consider whether it may be reasonable to make changes to preclinical standards required for the earliest human uses would be crucial (see Chapter 5 for a discussion of such oversight issues).
As noted above, preclinical testing can be complicated by the particular genetic circumstances of each couple. For example, when one or both prospective parents is a compound heterozygote for a serious genetic disease, more than one disease-causing allele will be present for that disease. This could pose a challenge to the design of gRNAs as they may only be able to edit one disease-causing allele and not others. Preclinical testing will need to examine the effects of the editing reagents on both the targeted allele and any other alleles present. In such circumstances, PGT on any edited embryo intended for uterine transfer will have to determine that at least one disease-causing allele has been changed to a non-disease-causing allele that is common in that population and that the other allele is unaffected.
Preclinical testing in human embryos must include the following steps.
Characterizing Editing at the Target Site
Requirement: The efficiency of the intended edit must be very high when measured in a cohort of treated human embryos. There must be no other sequence changes induced at the target, including insertions and deletions (indels).
Context: The goal of this testing would be to guarantee that the genome-editing methodology produced sufficient numbers of high clinical grade embryos with the desired edit before moving further toward to any clinical use. For a dominant disease, both zygotic alleles would need to exhibit a non-disease-causing sequence. For a recessive disease, although the restoration of a non-disease-causing sequence in one allele would prevent the disease, the editing frequency must be high enough that a high proportion of the available embryos are so modified. This testing could be done at any multicellular stage and must include testing for large deletions, chromosome loss, and other rearrangements.
Characterizing Any Off-Target Editing
Requirement: Compare parental genomes with whole-genome sequences obtained from the edited embryos or ES cells derived from these. Targeted sequencing should also be done for any particular off-target sites identified in preclinical research. There must be no detectable editing-induced off-target sequence changes. The incidence of de novo mutations, determined in conjunction with the sequence of the biological parents, must be in the range observed for unedited embryos, with no increase in the occurrence of single-nucleotide polymorphisms, indels, copy number variants, or chromosome rearrangements.
Context: Testing would be done at the blastocyst stage to provide sufficient DNA for this analysis and because this is the stage at which embryos would be transferred in the case of actual clinical use.
Identifying off-target sites from analysis of the cultured parental cells (see the first step under “Need for Extensive Research in Cultured Human Cells and in Zygotes of Model Organisms,” above) would provide an initial indication of high-risk off-target sites in the embryo, allowing attention at this stage to focus on regions where off-target editing has previously been observed or might be anticipated to occur.
Characterizing Any Mosaicism
Requirement: All cells of the embryo must have the same on-target sequence (i.e., no mosaicism) as shown by analysis of multiple individual cells. The
sequences of the intended target and any high-risk off-target sites should be determined for each individual cell, or as many as feasible, in the early-stage embryo.
Context: If not all cells have been successfully edited, it is possible that the target organ(s) in the offspring, and/or later in the adult, will not be completely disease-free.
Characterizing Embryo Development
Requirement: The genome-edited embryos must proceed through normal development in vitro to the blastocyst stage, meeting milestones with comparable efficiency to unedited embryos. Cellular and molecular features of genome-edited embryos should be comparable to unedited embryo controls and have aneuploidy rates no higher than expected based on standard ART procedures.
Context: The goal of such testing is to ensure that the genome editing does not negatively affect normal embryo development. Developmental characterization in genome-edited embryos would be compared to expected embryo development based on what is known from the use of unedited embryos in IVF. This assessment could be continued up to the 14-day limit currently permitted for human embryo culture in many countries.
Examples of best practice protocols include those used by the Newcastle Fertility Centre at the International Life Science Centre and others in the preclinical evaluation of human embryos that had undergone mitochondrial replacement techniques (MRT). In collaboration with the Crick Institute, investigators at Newcastle analyzed the cell lineages that were present in blastocyst-stage human embryos that had undergone MRT, to ensure that all expected cell lineages were present. They also performed single-cell transcriptome analysis to check for the expected patterns of gene expression (Hyslop et al., 2016).
Only if all of these preclinical requirements are met and validated by independent expert opinion should the use of edited embryos in a clinical setting be contemplated.
Additional Consideration for Genome Editing in Which Not All Embryos Would Inherit the Disease-causing Mutation
Requirement: Development of a genome editing methodology capable of safely and efficiently editing an eight-cell or later embryo may be required.
Context: If all of the embryos that can be produced by the prospective parents will inherit the disease-causing mutation, genome editing could be
undertaken around the time of fertilization. If only some of the embryos will carry the disease-causing mutation, identifying which are affected would be necessary prior to editing. For genetic circumstances in which only the maternal genetic contribution needs to be known, identifying the oocyte genotype through polar body analysis may be sufficient. In other circumstances, in which both maternal and paternal genetic contributions need to be known, an embryo biopsy would be required. For any such envisioned uses, the preclinical development and testing of a genome editing methodology capable of safely and effectively editing embryos post-genotyping would be required.
DECISION POINTS AND REQUIRED APPROVALS
Several important approvals must be received before any clinical use of HHGE could be undertaken.
Determination That Heritable Human Genome Editing Could Be Considered for Clinical Use in a Country
As described in Chapter 1, a country must first allow the consideration of HHGE for the proposed clinical use. This decision making will not only include information on preclinical evidence of an appropriate genome-editing methodology, but also include societal engagement and input. The clinical use of HHGE remains illegal in many countries; many others have not yet established oversight systems by which HHGE would be regulated, should it be permitted. It would be important for any clinical uses of HHGE to take place only in the context of a regulated environment (see Chapter 5 for additional discussion).
Appropriate Review Board and Regulatory Approvals
A proposal to clinically evaluate a particular use of HHGE would require submission of information on the proposed disease and genomic target, preclinical evidence, and clinical protocols to relevant institutional and national advisory bodies for science and ethics. Appropriate approvals would need to be obtained as a result of these reviews. A proposal with the required supporting preclinical evidence and protocols must also be reviewed and permitted by the appropriate national regulatory authorities. Only if such approvals are obtained could the initiation of a pregnancy with edited embryos for the proposed use be undertaken.
Informed Consent from Prospective Parents
For any clinical evaluation of HHGE, the prospective parents must be informed about the procedures and projected outcomes as thoroughly as possible, and they must give their consent to the intervention. As a requirement of informed consent is that prospective parents are given detailed information about the nature and risks of HHGE, it is premature to establish specific protocols at this time. Instead, general guidelines are presented of the principles and procedures that should be considered in the informed consent process. Due to the technical nature of genome editing, this will require extensive discussions in most cases. Prospective parents would require clinical assessment and counseling, by people with no conflict of interest regarding the outcome. Counseling would need to include the presentation of all reproductive options, including the risks, benefits, and degree of unknowns associated with each, with opportunities for prospective parents to consider the implications of the choices available to them. Reproductive advice would need to cover all aspects of ARTs, including a discussion of IVF, PGT, and any interventions used for prenatal evaluation. The prospective parents would also be asked to give consent to fetal monitoring and to reasonable post-natal monitoring and assessment. Assessment and counseling would need to consider mental health as well as physical health, both in parents and in resulting offspring, and the prospective parents’ ability to care for children born. Psychological support would also need to be available throughout the consent process.
In addition to meeting standard criteria for informed consent, because HHGE would represent a novel technology without a history of clinical use, care would need to be taken for any initial human uses not to engender or to be influenced by excessive optimism. It will be essential for those leading consent discussions to have no conflict of interest regarding the outcome of HHGE and fully understand the mechanisms, procedures, and risks involved.
CLINICAL EVALUATION OF THE PROPOSED USE
Once all of the required preclinical evidence had been assembled, indicating that a suitable methodology was available, and all of the appropriate regulatory reviews and approvals had been completed, a genome-edited human embryo might be generated with the aim of establishing a pregnancy.
The required clinical elements include the following.
Identify the Genotype (for Circumstances in Which Not All Embryos Are Expected to Carry the Disease-Causing Genotype)
Requirement: Identify the genotype of human oocytes and/or embryos prior to genome editing.
Context: This element may be required to ensure that only genetically affected embryos undergo genome editing. Sufficient genotype identification could be obtained through polar body biopsy in certain circumstances; in others, an eight-cell or later embryo biopsy may be required. In such cases, a genome editing methodology capable of editing a multicellular embryo must be established during the preclinical phase.
Create Genome-Edited Human Embryos Intended for Transfer to Establish a Pregnancy
Requirement: Best practice standards for the relevant genome-editing and ARTs would need to be followed in obtaining the parental gametes and creating a genome-edited zygote.
Context: The medical center performing the creation of the zygote, introduction of the genome-editing reagents, assessment of the clinical suitability of the resulting embryos, and eventual transfer to establish a pregnancy would need to have the appropriate qualifications, experience, and demonstrated competences according to the regulatory requirements of its country and would need to adhere to professional best practice guidelines. Best practice standards for consistency and quality control would also need to be followed for all reagents and procedures.
Characterize Human Embryos Intended for Transfer
Requirement: Perform an embryo biopsy to collect cells from the trophectoderm of blastocyst-stage embryos and perform PGT to confirm the presence of the precise on-target edits, the absence of detectable off-target mutations, and no evidence of mosaicism.
Context: As detailed above, extensive preclinical evidence must demonstrate that a methodology is consistently able to deliver human embryos in which every cell has the appropriate genetic features following genome editing. As a result, a trophectoderm biopsy of an embryo intended for transfer would be expected to reliably correlate with the rest of the embryo.
Evaluation of Outcomes Including Safety and Efficacy
Should a pregnancy be established with a genome-edited human embryo, it will be important to evaluate any negative effects during the prenatal period, as well as to assess physical and psychological outcomes of any child born following HHGE. It will also be important that information on the clinical outcomes of HHGE, including any detected negative effects, be collected and assessed to inform the understanding of safety and efficacy.
Monitor a Resulting Pregnancy
Requirement: Careful monitoring of a resulting pregnancy with a genome-edited embryo is strongly recommended.
Context: Following transfer of a genome-edited embryo to establish a pregnancy, prenatal monitoring is crucial to detect any fetal abnormalities or other issues arising during the pregnancy. Should prenatal testing identify genetic or physical anomalies, counseling for the parents will be important. Such prenatal monitoring would be strongly recommended but is the choice of the mother.
Undertake Longer-term Monitoring and Follow-up
Requirement: Longer-term monitoring and follow-up of a child born following HHGE is essential, and should include:
- obtaining consent from the parents, and later from the child, for monitoring immediately after birth and at specified intervals thereafter extending into adulthood, which must be done by competent professionals and include both physical and psychological aspects; and
- using assessment tools that have been validated and standardized in an international context and, if appropriate, that have versions available across the lifespan.
Context: It is important for the health of the individual born as a result of HHGE, as well as any children that they have, that such individuals continue to be assessed for adverse genetic or health outcomes. If adverse outcomes are identified, the individual concerned should be informed of them if they so choose and offered genetic counseling.
Make Information on Decisions to Permit the Clinical Evaluation of Heritable Human Genome Editing Publicly Available
Requirement: Each country would need to make public the details of any approved applications to clinically evaluate HHGE. Information that would need to be made available includes the genetic condition for which HHGE had been allowed, the associated laboratory procedures that would be used, and the national bodies that would be providing oversight.
Context: Making such information publicly accessible would be important for ensuring transparency about any potential uses of HHGE being contemplated, the evidence base on which decisions had been made, and oversight responsibilities. However, information made available would need to protect family identity.
Evaluate Information to Inform Future Decisions about Heritable Human Genome Editing
Requirement: It would be vital to publish in peer-reviewed scientific journals the procedures involved and outcomes of any clinical evaluation of HHGE.
Context: Such information would contribute to ongoing national and international discussions on the safety and efficacy of HHGE. In conjunction with further extensive societal engagement, such information would also contribute to any decisions about whether to consider the clinical evaluation of HHGE for other uses in Categories A and B, according to the translational pathway identified in this report, whether or how to modify any of the preclinical or clinical requirements laid out in this translational pathway, or potentially whether to consider evaluating uses that would fall into other categories of potential uses described in Chapter 3.
HERITABLE HUMAN GENOME EDITING USING IN VITRO STEM CELL–DERIVED GAMETES: WHAT A POTENTIAL TRANSLATIONAL PATHWAY WOULD ENTAIL
Chapter 2 describes the prospect of genome editing in human gamete precursors by two approaches: editing gamete precursor cells, such as spermatogonial stem cells (SSCs); and editing pluripotent stem cells followed by differentiation into functional gametes in vitro (in vitro–derived gametogenesis). At present, procedures for generating functional human sperm or oocytes by these methods are not available, so this technology is not available for clinical use. It should be emphasized that methodologies for using SSCs,
(iPSCs), or nuclear transfer embryonic stem cells (ntESCs), even if safe and efficient, would need to be permitted for use in assisted reproduction in the absence of genome editing, independently of considering their use in combination with genome editing. Such approval would need to involve extensive public consultation given its societal implications. Because this technology is not yet available for approval in any clinical setting, it would be premature to describe a translational pathway that uses it to create heritable genomic changes. Nevertheless, the section below describes preclinical and clinical considerations that would be relevant to such a pathway were it ever to be feasible.
Clinical availability of in vitro–derived gametes for assisted reproduction, especially female gametes that are usually only available in small numbers, would eliminate the need for HHGE in all monogenic disease circumstances except Category A. This is because it would be possible to generate and genetically test sufficiently large numbers of embryos such that identification of those that do not have the disease-causing genotype would be practically assured. Moreover, since the availability of such unaffected embryos would no longer be limiting, it would be possible to select those of the highest clinical grade for transfer to the prospective mother.
This strategy would not be available in the context of Category A, because no in vitro stem cell–derived gametes, and therefore no embryos, could be produced that lack the disease-causing genotype. In this case, undertaking HHGE would require genome editing of patient-derived stem cells in vitro, resulting in the production of cells lacking the disease-causing genotype. Generation of functional gametes from such edited stem cells would have several advantages over genome editing in zygotes. A significant advantage arises from the fact that editing would be done in cultured cells, where methods for making and evaluating specific modifications and for analyzing genomic and epigenetic profiles are well documented, although there is no consensus on which methods are best. The per-cell editing efficiency would not have to be particularly high, because treated cells can be thoroughly characterized for on- and off-target editing to find those with only the desired changes, expanded into a pool of correctly edited cells, and then differentiated into functional gametes. Finally, mosaicism would not be an issue when a single sperm derived from an edited SSC, iPSC, or ntESC is used to fertilize an egg.
However, the use of in vitro stem cell–derived gametes could have disadvantages. The precursors of such gametes would have gone through extensive adaptation to and expansion in cell culture. During this time, de novo mutations could accumulate at levels comparable to the spontaneous germline mutation rate in vivo (Wu et al., 2015). Some cells might be selected for properties, including both genetic and epigenetic differences, that promote their ability to replicate in culture conditions, which could
have unknown effects if used clinically. Each batch of cells may acquire a unique set of mutations, unlike the more specific off-target mutations potentially induced by genome editing. It is not clear how to evaluate such genetic and epigenetic changes, were they to be unavoidable, for possible effects on embryonic, fetal, and post-natal development.
Were approaches that rely on the use of in vitro stem cell–derived gametes to be permitted for clinical use as a reproductive technology, their use for HHGE would still be subject to the same ultimate tests of safety and efficacy outlined above for genome editing in zygotes. However, there would also be some special considerations. For example, there would be no need to test the resulting embryos for mosaicism.
Before any clinical use to create a human embryo for transfer to the uterus, preclinical research for genome editing approaches using in vitro stem cell–derived gametes would include the following.
- Extensive research in human cells to develop and optimize the genome editing reagents. For editing in gamete precursor cells, such as SSCs, comparative genetic analysis with uncultured SSCs from the prospective father would be required. This would allow the development of effective editing reagents for on-target efficiency of the desired edit and absence of other on- and off-target changes. For editing in iPSCs/ntESCs that would subsequently be differentiated into functional male or female gametes, similar comparative analyses would be undertaken with unedited parental iPSCs/ntESCs to optimize the editing reagents.
- Isolation of individual cell lines and testing to characterize on- and off-target events and epigenetic profiles. Cell lines derived from iPSCs/ntESCs that had undergone genome editing would need to be examined thoroughly by WGS for mutations acquired and selected for during establishment and growth in cell culture. Selected lines would need to have only the desired edit at the intended target and no undesired modifications elsewhere in the genome as a result of the editing. Epigenetic and gene expression profiles should also be examined to better understand whether the editing reagents had affected these.
- Differentiation of the correctly edited iPSCs/ntESCs into functional gametes. As discussed in Chapter 2, protocols have been developed that permit the derivation of functional gametes from mouse pluripotent stem cells in vitro. Similar protocols are being developed in humans, but there are major challenges that still need to be overcome, not least that of ensuring normal meiosis in such cells. Continued research in this area will be of vital importance, not only for the development of in vitro–derived gametes that can be used
-
safely, but also for the insight such research will allow into human gametogenesis and abnormalities thereof associated with infertility.
- Characterization of gametes before their use in IVF. This would require comparison with parental gametes not derived from cultured cells in order to assess genetic and epigenetic properties of the edited gametes, including additional WGS to examine potential genome changes acquired during the gamete differentiation process. Variation between the transcriptomic and epigenomic properties of individual gametes could be assessed through single-cell approaches (Hermann et al., 2018).
- Testing the functionality of genome-edited gametes. The ultimate test of any gamete generated in vitro will be its ability to generate an embryo with general features that are essentially indistinguishable from an embryo generated using conventional gametes. For example, it will be necessary to demonstrate that male gametes derived from either unedited or genome-edited precursor cells in vitro are able to effectively fertilize an oocyte and that resulting embryos reach normal developmental milestones to the blastocyst stage. The genome or epigenome of a single sperm that contributed to any one embryo may not be representative of the genomes characterized in bulk produced by this method of genome editing, so characterization of genomes and epigenomes from individual embryos generated from in vitro–derived gametes will be very important. ES cell lines could be derived from such embryos to confirm by high-quality genome sequencing that such cells did not differ from parental genomes at off-target sites. Finally, conducting genetic testing on cells obtained from a blastocyst biopsy would be important in the clinical phase, prior to transfer of any embryo, at least for the intended genomic target.
CONCLUSION AND RECOMMENDATIONS
Any responsible pathway to clinical application of HHGE would need to include clear and strict criteria in technical capabilities, in the acceptable evaluation of safety and efficacy, and in oversight standards. The Commission’s recommendations set out the components that would be required for a responsible translational pathway and are provided below.
Scientific Validation and Standards for Any Proposed Use of Heritable Human Genome Editing
Evidence from preclinical research would be required to establish that HHGE may be safe enough to consider evaluating in first-in-human clinical
applications. Once the preclinical requirements set out in the translational pathway have been met, it may become appropriate to proceed with clinical interventions, subject to required approvals, informed consent, and ongoing review and monitoring. Each specific clinical use would need to be carefully considered in its own right. Even with preclinical evidence, there will still be unknowns concerning safety and efficacy that could only be revealed and resolved by the long-term monitoring of individuals born following the use of HHGE.
Recommendation 5: Before any attempt to establish a pregnancy with an embryo that has undergone genome editing, preclinical evidence must demonstrate that heritable human genome editing (HHGE) can be performed with sufficiently high efficiency and precision to be clinically useful. For any initial uses of HHGE, preclinical evidence of safety and efficacy should be based on the study of a significant cohort of edited human embryos and should demonstrate that the process has the ability to generate and select, with high accuracy, suitable numbers of embryos that:
- have the intended edit(s) and no other modification at the target(s);
- lack additional variants introduced by the editing process at off-target sites—that is, the total number of new genomic variants should not differ significantly from that found in comparable unedited embryos;
- lack evidence of mosaicism introduced by the editing process;
- are of suitable clinical grade to establish a pregnancy; and
- have aneuploidy rates no higher than expected based on standard assisted reproductive technology procedures.
Recommendation 6: Any proposal for initial clinical use of heritable human genome editing should meet the criteria for preclinical evidence set forth in Recommendation 5. A proposal for clinical use should also include plans to evaluate human embryos prior to transfer using:
- developmental milestones until the blastocyst stage comparable with standard in vitro fertilization practices; and
- a biopsy at the blastocyst stage that demonstrates
- the existence of the intended edit in all biopsied cells and no evidence of unintended edits at the target locus; and
- no evidence of additional variants introduced by the editing process at off-target sites.
If, after rigorous evaluation, a regulatory approval for embryo transfer is granted, monitoring during a resulting pregnancy and long-term follow-up of resulting children and adults is vital.
Future Developments Affecting Reproductive Options
Genome editing of gamete precursor cells or editing of pluripotent stem cells followed by in vitro differentiation into functional gametes could represent alternative methods of undertaking HHGE. However, the technologies to develop human gametes from cultured cells are still under development and are currently unavailable for clinical use.
Recommendation 7: Research should continue into the development of methods to produce functional human gametes from cultured stem cells. The ability to generate large numbers of such stem cell–derived gametes would provide a further option for prospective parents to avoid the inheritance of disease through the efficient production, testing, and selection of embryos without the disease-causing genotype. However, the use of such in vitro–derived gametes in reproductive medicine raises distinct medical, ethical, and societal issues that must be carefully evaluated, and such gametes without genome editing would need to be approved for use in assisted reproductive technology before they could be considered for clinical use of heritable human genome editing.
This page intentionally left blank.
























