
The workshops’ discussions on real-world evidence (RWE) concluded with sessions about how RWE can be used to improve different facets of the health care system, including any potential to improve health technology assessment, to transform product research and development, or to bring patients in as partners for research. Workshop participants also discussed how—based on U.S. Food and Drug Administration (FDA) input—RWE can inform regulatory decisions for biologics, drugs, and devices in the United States and abroad.
REAL-WORLD EVIDENCE TO IMPROVE HEALTH TECHNOLOGY ASSESSMENT
Health technology assessment has a problem, said Pall Jonsson. The function of the UK National Institute for Health and Care Excellence (NICE) is to understand the comparative effectiveness and comparative cost-effectiveness of new treatments compared with standard practice. However, obtaining data suited for health technology assessment is becoming increasingly difficult. For example, new treatments in orphan diseases are becoming available, and the treatments are not supported by large randomized controlled trials (RCTs) that are traditionally used in assessments. Drugs are receiving market authorization based on single-arm trials, particularly in orphan diseases or areas of unmet needs. The lack of head-to-head trial data on these products makes them difficult to assess,
he said. Some drugs are receiving accelerated approval through new regulatory mechanisms. This acceleration means the products are getting to patients quicker, but it also means the data are less mature and there is more reliance on observational data. All of these factors combined, said Jonsson, mean there is “increased uncertainty around the decisions that we have to make.”
RWE has a role to play in providing data for comparative assessments, said Jonsson. While NICE’s assessment framework traditionally relies on RCTs, real-world data (RWD) sources may provide more useful data in some cases. For example, RCTs have narrow inclusion criteria, which reduces evidence about how a product works in patients with comorbidities or with more severe diseases. RCTs are time limited, which restricts the ability to gather evidence on long-term effectiveness, particularly for patients with chronic disease. Comparators in RCTs sometimes do not reflect the local practice or clinical practice, which makes it difficult to assess real-life comparative effectiveness of a product, he said. Of course, he added, using RWE also has challenges, including limited availability of data at time of assessment, potential for bias, poor quality or missing data, and data sources that are not established for research purposes. The acceptability of RWE varies across Europe; different countries have different views on the importance and credibility of RWE and the potential value of RWE in the future (see Figure 10-1).
To address the potential use of RWE, as well as the challenges involved, the GetReal1 project was established approximately 4 years ago by the Innovative Medicines Initiative (IMI) in Europe, said Jonsson. IMI brought together stakeholders—including regulators, payers, patients, assessors, clinicians, and drug developers—to identify issues with current evidence-generation practices, and to explore how RWE might be useful for key decision makers.
The stakeholders involved in GetReal identified several pressing needs in the RWE space, said Jonsson, including
- Integrity, quality, access, and privacy protection of RWD sources;
- Guidance on RWE study design, evidence synthesis, and interpretation in decision making;
- Training and education in RWE; and
- Broader involvement of stakeholders in RWE generation and use of RWD.
A key finding of GetReal, said Jonsson, was that more attention should be paid to the “whole journey” of RWE, from designing studies to imple-
___________________
1 See http://www.imi-getreal.eu (accessed November 4, 2018).
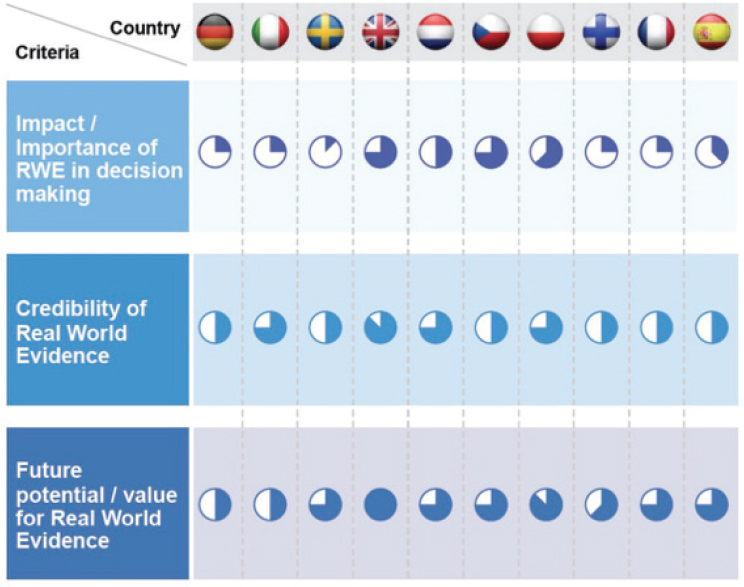
NOTE: The shaded portions of the circles indicate the value and importance placed on RWE in each country (indicated by flag across the top).
SOURCES: Jonsson presentation, July 17, 2018; Gill et al., 2016.
menting and analyzing studies. To focus on the whole journey, GetReal has received additional funding to create a sustainable, self-funded entity to continue this work in its next iteration. The next generation of GetReal will continue to drive international consensus and use of RWE in decision making, provide tools to deliver high-quality RWE, and provide the education and training required to generate and use RWE. International thought leaders within the entity will “act as ambassadors” for the use of RWE by broadly engaging with stakeholders to drive debate and facilitate uptake of best practices.
Jonsson provided three examples of how the GetReal initiative has affected the work of NICE in the past several years. Jonsson noted that NICE is a unique stakeholder in that it is not a regulator or a payer, but sits “somewhere in between” and provides guidance to a broad range of stakeholders, including health care, public health, and social care organizations. The approach that NICE takes in regard to RWE, said Jonsson,
needs to be compatible across all of these areas and all of the different stakeholder needs.
The first example of NICE progression in incorporating RWE, said Jonsson, is the development of an internal health care and data analytics team that can help identify research questions, work with data owners, analyze data, and provide quality assurance of data. This team will be supported by an external advisory group that consists of members, data owners, experts, and industry, he said. Future partnerships may include working with the UK National Health Service to facilitate access to data and to understand how NICE guidance impacts the health care system, or working with health policy agencies to implement pilot projects.
The second example Jonsson discussed is a NICE statement that incorporates lessons learned from the GetReal project. The manual describes a range of possible situations in which RWD can be used in the context of NICE guidelines, and is meant to encourage guideline developers to consider whether and when analysis of RWD could be used to support decision making. The manual “flags key areas where we think there is a role potentially for real-world data,” Jonsson said. For example, applications might include addressing the efficacy–effectiveness gap, extrapolating treatment beyond the duration of clinical trials, and understanding the impact of treatments on the health care system.
Finally, NICE’s Science Policy and Research team is prioritizing areas for methods development, said Jonsson. The team is engaging in research projects with partners in order to develop best practices for applying adjustment methods for confounding, explore the use of big data in health care decision making, and consider the use of advanced analytics and artificial intelligence for RWE analysis.
REAL-WORLD EVIDENCE TO TURN PATIENTS INTO PARTNERS
In RCTs, said Komathi Stem, founder and chief executive officer of monARC Bionetworks, patients are passive participants. Data generated by RCTs are clean, structured, and easy to analyze, but at the same time, are limited, expensive, time consuming, and not always generalizable. RWE presents an opportunity to change these dynamics, and to turn patients into partners, she said.
Stem discussed three major trends that are facilitating a shift toward a more patient-centered, real-world approach:
- First, the point of care is shifting from the clinic to the smartphone (see Figure 10-2). Smartphones can be used to track health information, conduct telemedicine visits, and facilitate communication between providers and patients. Ninety-five percent of Americans
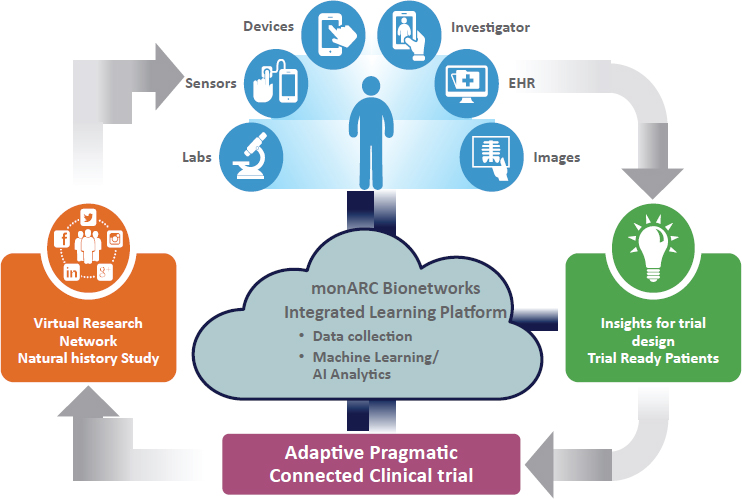
NOTE: AI = artificial intelligence; EHR = electronic health record.
SOURCE: Stem presentation, July 17, 2018.
- Second, data are abundant, but highly compartmentalized and noisy. There are data from wearables, social media, smartphones, and electronic health records (EHRs), and these data are complex and variable. However, there are issues with interoperability, and with mitigating privacy concerns and proprietary obstacles to data sharing. These issues limit the potential of what can be done with the abundant data that exist.
- Third, there is a developing shift from data ownership to data access. Ultimately, being able to access, integrate, and use a variety of data streams is what delivers value. Players in other industries—
have a cell phone, 77 percent have a smartphone, and 20 percent have online access only through a smartphone (Pew Charitable Trusts, 2018). Seventy-five percent of physicians are using mobile tools in their clinic, and 75 percent of patients are willing to use mobile tools or telemedicine (Physicians Foundation, 2018). “This is the new clinical care setting, and if we continue to develop drugs in our traditional brick-and-mortar medium, we’re going to be developing tools and drugs for a market that is completely different.”
such as Amazon, Facebook, and Apple—have begun this shift, and health care should follow suit.
In this new world, patients will be the most important partners for research, Stem said. Patients are ideal aggregators of RWD; people already own a considerable amount of data from smartphones and other technologies, and they have and can grant access to their own health records. Patients are willing to share their data—96 percent have expressed a willingness to share, provided that security is protected and that the data will be used for a trustworthy purpose, she said. Patients can provide not just RWD, but also real-world insights. Stem said that patients can help develop meaningful endpoints for research, influence the design of research, and improve the decision-making process, similar to W. Benjamin Nowell’s presentation in Chapter 7. Patients expect to be partners in this new world, said Stem, and expect to access continuous and updated information, such as personalized and dynamic drug labels on their smartphones.
To capitalize on these trends and to involve patients as partners, said Stem, monARC Bionetworks has developed an integrated RWD learning platform. monARC worked directly with patients to generate virtual research networks where patients can share data, and be engaged and recruited for research (see Figure 10-2). This system, said Stem, means that researchers can design better trials from the beginning and accelerate trial development by using data from patients and by recruiting and qualifying patients for whom data already exist in the system. Stem gave an example of an observational study on home spirometry that went from Institutional Review Board approval to published poster in 4 months.
These types of big changes to health research and practice, said Stem, will require big changes to incentives and legislation, as well as new partnerships. New incentives for data sharing across researchers and sponsors are needed, as well as incentives to develop novel endpoints. Clarity and improvement are needed in legislation about telemedicine, drug shipments, and research, she said. For example, the use of smartphones as a point of care or in research is highly limited by these laws. Finally, she said, there should be collaboration among industries such as social media, mobile devices, and artificial intelligence, and “bold partnerships” with patients to leverage the RWD and real-world insights they have to offer. Stem advocated for making processes simple so patients can participate in research, and for ensuring patients have access to or the ability to learn from the research to which they contribute. Echoing Nowell, Stem said patients have unique and invaluable insights; they “know a lot about their own condition, maybe more than we do, because they’re living it.”
REAL-WORLD EVIDENCE TO TRANSFORM RESEARCH AND DEVELOPMENT
“We are living in extraordinary times,” said Elliott Levy, head of Development at Amgen Inc. There is a confluence of new technologies and the availability of new data sources that have the potential to transform medical product development. Over the past several decades, he said, medical technologies have been developed that have incredible potential. For example, T cells can be reprogrammed to express a chimeric antigen receptor, and these CAR-T cells have been used with response rates exceeding 80 percent in patients with highly refractory malignancies. Researchers have access to unprecedented amounts of data—such as genetic code—that can be used to personalize and target therapies. These technologies and new data sources are exciting, said Levy, who cautioned that “it will be expensive” to do the research and build the infrastructure necessary to fully exploit the opportunities presented. Unfortunately, the cost of developing a new medicine has been steadily increasing over the past few decades, while revenues of pharmaceutical companies have remained flat or risen slowly. Because of this, the challenge, said Levy, will be in finding a way to exploit new technologies and new data sources, without the luxury of having new funds to invest.
Several factors increase the cost of developing drugs, Levy said. The failure rate of new drug candidates is high, with only about 1 in 10 reaching the market (DiMasi et al., 2016). Levy said this rate suggests that despite a growing understanding of human biology, the research community’s “ability to identify targets outstrips our ability to validate and confirm their importance.” The data requirements for new products are increasing, with a growing demand for active comparator data, patient-reported outcomes, and long-term safety follow-up data. Finally, the most burdensome part of drug development is the cost of collecting the data in the physician’s office, he said, a cost that continues to rise quickly.
In addition to the cost of developing new drugs, there is a significant cost to maintaining drugs that are on the market, Levy observed. There are demands for postmarket safety data, data on how products affect the health system, and further investigation into extending the product into pediatric or other special populations. The costs of developing and maintaining products present a “formidable challenge to our ability to realize the promise of new technologies and new data sources, and bring to market truly transformative medicines,” said Levy.
RWE can play a critical role in addressing this issue in a sustainable way, he said. Levy noted that traditional cost-cutting measures typically yield significant savings initially, but the savings rate diminishes over time. For example, if a company moves a data management unit to India, there
are significant initial savings, but no future decreases to this cost. The challenge is to identify ways to stop or even reverse the increase in drug development costs, and this may require “radical changes” in the design and methodology of clinical trials. Levy shared three potential approaches to reduce the cost burden of generating evidence.
One potential change is a shift to adaptive trial designs, which have the potential to increase the probability of making correct decisions with reduced sample size and cost, and on a shortened time line. In essence, adaptive designs allow researchers to “fail efficiently and succeed efficiently,” Levy said. He shared a hypothetical example of a traditional design compared to a Bayesian adaptive design (see Figure 10-3). It showed that the adaptive design needed to enroll about 20 percent fewer subjects, and saved 4 to 6 months in the time needed to reach a decision on product efficacy or futility. This type of design, said Levy, means that such decisions can be reached more quickly, and patient exposure can be limited earlier if a product is found to be ineffective. Levy noted that adaptive designs are currently used extensively in Phase I, to some extent in Phase II, but only sparingly in Phase III, where the greatest costs are incurred.
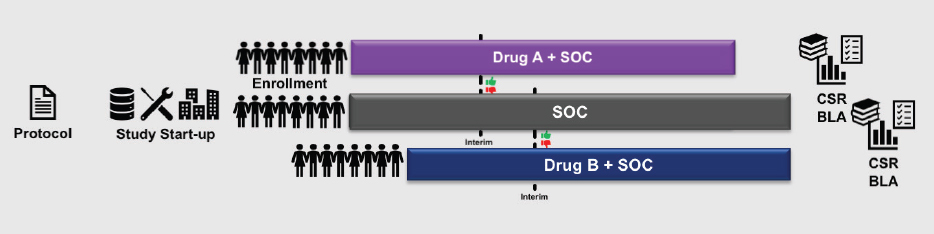
A second promising approach, said Levy, is the platform trial, a concept mentioned during the first workshop by Janet Woodcock of FDA. This design allows more than one agent to be studied in a single trial, or a single agent can be studied in multiple disease types. For example, two anticancer treatments that would normally be studied in two parallel trials (see Figure 10-4) could potentially be combined into a single platform trial that has a single master protocol, consolidated start-up procedures, and a combined comparator arm (see Figure 10-5). Consolidating two trials into one, said Levy, saves about 25 percent in terms of cost, number of patients randomized, and trial duration. A more complex platform trial (e.g., combining a larger number of experimental agents) could lead to even greater savings, he said.
The third approach for reducing costs, said Levy, is to use RWE to augment RCTs or perhaps for label expansions on existing products. RWE can be incorporated into trial design in a number of ways, he said. For example, a simplified pragmatic trial could randomize patients and collect data on baseline status, adverse events, and key endpoints, but rely on information collected in the usual course of care for all other data. By reducing the frequency of study visits and the number of laboratory assessments, this can reduce the cost per patient by about two-thirds, said Levy. In some cases, RCT evidence could be replaced entirely by RWE. If a product is already known to be safe and effective, but evidence is needed for marketing the product to a new geography or a new indication, a retrospective RWE study can use data that already exist, and reduce the time and cost significantly. Levy estimated that replacing RCTs with RWE would reduce the cost of a
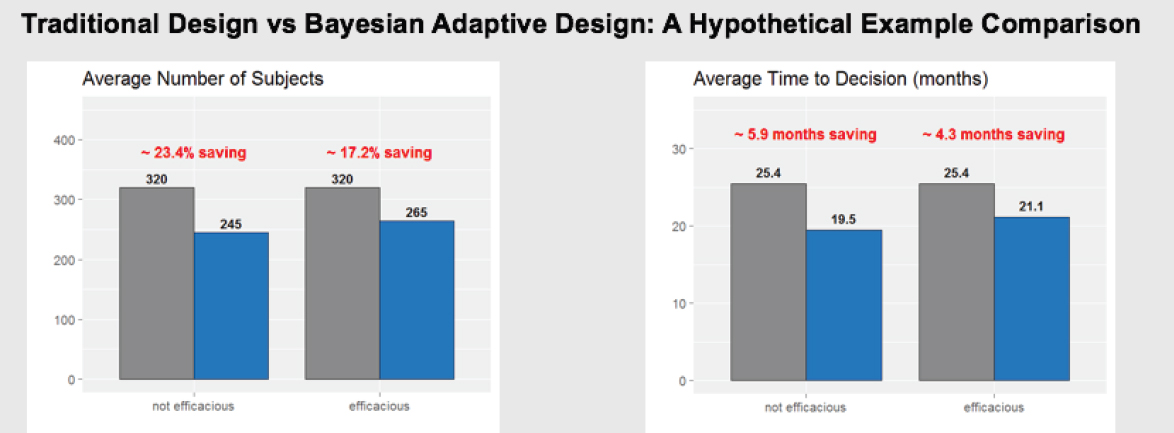
SOURCE: Levy presentation, July 17, 2018.
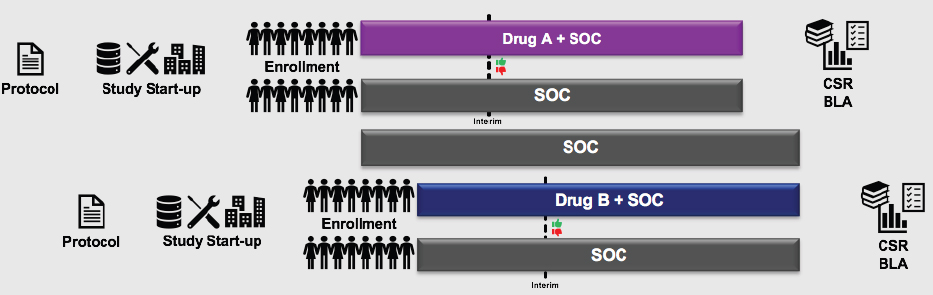
NOTE: BLA = biologics license application; CSR = clinical study report; SOC = standard of care.
SOURCE: Levy presentation, July 17, 2018.
NOTE: BLA = biologics license application; CSR = clinical study report; SOC = standard of care.
SOURCE: Levy presentation, July 17, 2018.
study by up to 90 percent and the duration of the trial by up to 80 percent. Berger added that another way to incorporate RWE is to nest trials within registries or networks where data are already being collected on a systematic basis.
While all of these approaches—adaptive design, platform trials, and using RWE to augment trials or for label expansions—can generate significant cost and time savings, said Levy, RWE has the potential for the most radical cost savings (see Table 10-1). The conventional approaches that drug development companies have pursued over the past decade for improving cost efficiency have already been fully exploited and will not yield meaningful additional gains, said Levy, so other changes will be necessary. Greater improvements in trial costs and time lines will require radical reductions in the amount of data collected specifically for the trial, instead replacing it with the RWD collected in the process of providing care, he said. Levy noted that RWE is not appropriate for every setting, and that RCTs will continue to play an important role. However, the overall cost of developing drugs could be reduced by using new trial designs and incorporating RWE whenever feasible and appropriate. Hernandez emphasized that the timing and scope of a study can greatly impact the cost of the study. A trial that aims to assess a product early in its life cycle likely requires significant data collection and must contend with many adverse events, concomitant therapies, and analysis of millions of data points. By contrast, a trial that assesses a product later in its life cycle can be more streamlined, collect fewer data, and cost significantly less.
TABLE 10-1 Hypothetical Examples of Cost Savings
| Example | Savings |
|---|---|
| Risk-based monitoring | ~5 percent of total clinical trial spend |
| Adaptive trials designs | Average saving of ~20 percent study subjects and up to 25 percent reduction in trial timeline |
| Platform trials | ~20–40 percent saving of trial costs and reduction of timeline by ~25 percent |
| Highly simplified trials | ~50 percent reduction in per-site and per-patient costs |
| Real-world evidence (in lieu of randomized controlled trial) | Up to ~90 percent reduction in total trial cost and ~75 percent reduction in trial timeline |
SOURCE: Levy presentation, July 17, 2018.
REAL-WORLD EVIDENCE TO INFORM REGULATORY DECISIONS
In this session of the third workshop, participants first heard about the European perspective on RWE, and then heard from three directors at FDA about the use of RWE for making regulatory decisions on biologics, drugs, and devices.
European Perspective
The field of RWE is active in Europe in several areas, said Alasdair Breckenridge, emeritus professor of clinical pharmacology at the University of Liverpool. Activity is still mainly in the traditional area of safety, but increasingly in dosing, drug–drug interactions, sequence of therapies, expansion to subpopulations, and new indications. A particularly interesting new use, he said, is in applying RWE-generation techniques to the data collected to meet postmarketing requirements in order to suggest new indications. Breckenridge told workshop participants about RWE work being done by three European bodies.
The UK Academy of Medical Sciences has held two workshops on the topics of RWD and RWE, he said. The first workshop was held in September 2015, and aimed to explore the acceptability of RWE in regulatory and health technology assessment decision making, to address the challenges, and to suggest practical steps to address them. The workshop brought together a number of stakeholders, including the European Medicines Agency (EMA), FDA, NICE, and the regulated industry. This workshop reached the conclusion, said Breckenridge, that the role of RWE in regulatory decision making remains to be defined. While RWE is being used, there is a need for better definitions and standards, he said. The second workshop was held in early 2018, and participants at the workshop concluded that compared with the United States, progress in Europe on RWE was limited and many of the challenges identified in 2015 remained unresolved. The workshop also explored definitions, and defined RWD as a “subset of big data relating to patient health status, delivery of routine health care, collected from a variety of sources [including] electronic health records, claims, product and disease registries, and social media.” RWE, said Breckenridge, was considered to be evidence drawn from RWD through the application of research methods. The workshop participants suggested there should be a way to define “regulatory-grade RWE.” Regulatory-grade RWE, said Breckenridge, would meet five criteria:
- Define the scientific question to be answered.
- Identify study design.
- Be specific in terms of the RWD used.
- Be rigorous in data standards and analytic methods.
- Comply with regulatory standards.
EMA, said Breckenridge, is very active in the pharmacovigilance field, routinely using RWD for safety monitoring. The EudraVigilance system, operated by EMA, received 1 million safety reports in 2016, of which 2,000 signals were detected and 48 were validated. Breckenridge discussed EMA’s involvement in two efficacy studies: the Salford Lung Studies (discussed in detail in Chapter 3) and the Phase II single-arm study on Zalmoxis, an immunotherapy for high-risk hematological malignancies, which used historical controls from a transplantation registry. Breckenridge noted that EMA gave conditional marketing authorization for this product, but asked for postauthorization efficacy and safety studies. EMA’s biggest contribution, said Breckenridge, has been its work on the Medicines Adaptive Pathways to Patients (MAPPs, previously known as Adaptive Licensing). The MAPP program is a prospectively planned, adaptive approach to give early access to important new medicines for patients with unmet needs, with lower premarket evidence requirements. The program shifts the burden of evidence from the pre- to postmarket space, and emphasizes postauthorization efficacy and safety studies. MAPP uses the existing European Union legal framework, he said, and requires the ongoing involvement of the company, regulators, health technology assessment experts, payers, and patients. As of July 2016, there were 18 accepted pilot projects, all of which included plans for the use of RWD that went beyond the traditional use of registries for pharmacovigilance. Breckenridge noted, however, that most of these projects were “vague in terms of the purpose of collecting RWD to supplement RCTs.”
IMI (discussed earlier in this chapter by Jonsson) is a public–private consortium launched in 2008, which has a budget of approximately 3 billion euros and currently supports 50 projects, said Breckenridge. He briefly outlined three IMI-sponsored projects of interest, in addition to IMI’s GetReal project, presented by Jonsson earlier in this chapter. IMI PROTECT (Pharmacoepidemiological Research Outcomes of Therapeutics by European Consortium) is a project designed to strengthen the “monitoring of benefit/risk” of treatments in Europe, with the involvement of patients and the public. The project addressed the limitations of the current methods for monitoring, and an evaluation of the project found that it had met its objectives in terms of signal detection and evaluation, and the use of routine pharmacovigilance. IMI ADAPT SMART is a project to facilitate and accelerate the availability of MAPP’s related activities. It was established in 2017 for a 2-year period to develop next-generation vaccines and medicines, to tackle Europe’s growing health care challenges, and to ensure future competitiveness of Europe’s pharmaceutical industry, he said. Finally, IMI WEB RADR (Recognising
Adverse Drug Reactions) is a project that developed a mobile application for patients and health care professionals to report suspected adverse events. Reports received from the application are compared to reports received via established reporting schemes in order to evaluate whether the application is an effective way to collect this information.
U.S. Food and Drug Administration: Drugs
FDA is “taking our obligations very seriously under 21st Century Cures,” said Jacqueline Corrigan-Curay, director of the Office of Medical Policy at FDA’s Center for Drug Evaluation and Research (CDER). This is not just because it is a requirement, she said, but “because we do want to make sure we are doing everything we can be doing to bring things efficiently to the market” while keeping strong evidence standards. RWD has been used a number of times as part of the premarket evaluation of drugs for rare diseases, said Corrigan-Curay, including examples of non-randomized, unblinded trials against historical controls (see Figure 10-6).
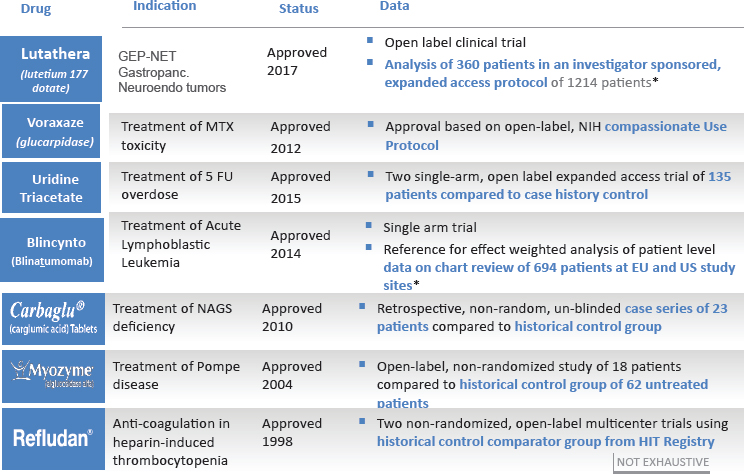
NOTES: Bolded text = examples using real-world evidence. 5 FU = 5-fluorouracil; EU = European Union; GEP-NET = gastroenteropancreatic neuroendocrine tumor; HIT = heparin-induced thrombocytopenia; MTX = methotrexate; NAGS = N-acetylglutamate synthetase; NIH = National Institutes of Health; US = United States.
SOURCES: Corrigan-Curay presentation, July 18, 2018; * data from Gökbuget et al., 2016.
RWD has also been used in the postmarket space for some limited indication expansions; for example, in 2017, FDA expanded the indications for Kalydeco to include 23 new mutations in cystic fibrosis based on clinical and in vitro data. After approval, FDA asked for postmarketing observational study using the cystic fibrosis registry, she said.
In 2013, FDA released guidance on the best practices for conducting and reporting pharmacoepidemiologic safety studies using EHR data.2 These practices can likely inform best practices in other areas of RWD analysis and RWE use, said Corrigan-Curay. For example, one goal of this guidance is to ensure that patients whose electronic health care data have been used in an outcomes analysis have actually experienced the event; it suggests practical steps such as ensuring that the code or algorithm has either been validated previously or that its predictive value was calculated, and describing the sensitivity of the outcome.
Claims data are a common source of RWD. Corrigan-Curay noted that even though use of claims data in RWE applications is well understood, systemic changes such as the switch from the International Statistical Classification of Diseases and Related Health Problems, Ninth Revision (ICD-9) to ICD-10 will create challenges for RWD-based research. ICD-10 contains 70,000 diagnoses, compared with 14,000 in ICD-9, and this transition has created some issues, she said. For example, a surveillance of hospitalizations with a diagnosis of opioid use disorder saw an uptick of about 14 percent when the transition happened; other systems observed a decrease in the likelihood of correctly reporting confounding comorbidities with the new system. In addition, published studies using different claims data sources are sometimes in conflict. Corrigan-Curay emphasized that the point was not to decide which study was correct, but rather to understand the reasons for differential results. These challenges will need to be dealt with so that data sources are valid and reliable.
To begin answering some of the questions on how to work with RWD for regulatory purposes, Corrigan-Curay discussed several demonstration projects that are being supported by FDA. One example was the One-Source3 checklist project, which supports data collection in a way that will meet the needs of both researchers and practicing clinicians. Corrigan-Curay also mentioned the promise of data networks like Sentinel or the National Patient-Centered Clinical Research Network, and the potential of also using observational data networks in the future.
___________________
2 See https://www.fda.gov/downloads/drugs/guidances/ucm243537.pdf (accessed January 4, 2019).
3 See https://www.fda.gov/ScienceResearch/SpecialTopics/RegulatoryScience/ucm574079.htm (accessed January 4, 2019).
There is a wide spectrum of potential uses of RWE in clinical studies, said Corrigan-Curay. However, she emphasized a need to be thoughtful about adopting new methodologies and data sources because “as we adapt the tools and methods of traditional trials to real-world settings, we must consider the components of such trials that are critical to obtaining valid results and minimizing bias” (Sherman et al., 2016, p. 2294). Corrigan-Curay emphasized a need to build confidence and experience in using new data streams, new technologies, and new analytic methodologies for RWE, as well as building expertise and commitments from multiple stakeholders to realize the potential of RWE. Corrigan-Curay outlined the potential future of RWE, which could include
- Research fully embedded in care settings (no data are wasted);
- Integrated/connected systems throughout the entire health care continuum with feedback loops;
- Seamless and integrated auditing and quality control mechanisms;
- Flexible and linkable on-demand data aggregation from databases/registries;
- All stakeholders engaged, including potentially increased patient engagement with mobile technologies or data capture;
- Secured and traceable access and management of data (blockchain); and/or
- RWE continuously used to support decision-making processes.
U.S. Food and Drug Administration: Biologics
The 21st Century Cures Act is a main driver of the RWD and RWE initiatives at FDA, said Steven Anderson, director of the Office of Biostatistics and Epidemiology at Center for Biologics Evaluation and Research (CBER). CBER, which regulates vaccines, blood and blood products, tissue and tissue products, and cellular and gene therapy products, has undertaken several RWE initiatives in recent years, he said.
CBER uses a number of population-based data systems to conduct RWE safety and effectiveness studies, including a system being developed specifically for CBER called the Biologics Effectiveness and Safety (BEST) program. BEST is part of the Sentinel initiative, and was launched in September 2017. One goal of BEST, said Anderson, is to build data infrastructure and tools and develop expertise to conduct queries and studies of biologic products. The second goal is to automate adverse event reporting by using methods such as machine learning and natural language processing in order to mine adverse events related to biologics from EHRs and automatically submit them to FDA. The BEST program, said Anderson, will help CBER to better meet its regulatory needs by building “better,
faster, cheaper systems” to generate evidence about safety and effectiveness of biologic products. CBER also uses the Sentinel system (described in Chapter 3) to generate evidence on biologics; CBER has conducted dozens of safety assessments and more than 100 “rapid queries” to address safety questions, as well as a pilot study on vaccine effectiveness. Anderson gave two examples of RWE studies on safety of biologic products being successfully used for label changes or regulatory action. The first was a study on immune globulins and thrombotic events, and used data from the Centers for Medicare & Medicaid Services (CMS) database (Daniel et al., 2012). The second, discussed in Chapter 3, was a Sentinel study on rotavirus vaccines and intussusception.
Anderson highlighted CBER’s use of RWE for real-time analysis. CBER uses the CMS claims data system to conduct near-real-time analysis of the annual flu vaccine and related adverse events such as Guillain-Barre syndrome, he said. One specific study that was performed in 2017–2018 was a rapid response effectiveness study of cell versus egg-based influenza vaccines. Using CMS data, FDA and CMS examined about 13 million vaccine doses, and found that cell-cultured vaccines were slightly more effective. These types of studies, said Anderson, can provide near-real-time information (within 4 to 6 weeks) to inform regulatory decisions concerning current and future influenza vaccines.
In addition to these activities, CBER is considering ways to collect patient input and information on patient preferences, said Anderson. In March 2018, the agency began soliciting proposals to collect patient input in five disease areas: sickle cell anemia, brittle diabetes, hemophilia, rheumatoid arthritis, and retinal dystrophy. CBER is also working with the Sentinel staff to develop a mobile app to collect patient input, said Anderson.
There are several challenges in using RWD for regulatory decision making, said Anderson, including bias, quality of data, missing data, and how well a patient’s exposure and outcome can be captured. In addition, there are challenges in linking different forms of data, such as data from EHRs and registries.
U.S. Food and Drug Administration: Devices
FDA’s Center for Devices and Radiological Health (CDRH) has been on “an 8-plus-year journey in our use of RWE,” said CDRH Director Jeff Shuren. He discussed three major CDRH efforts regarding RWE.
First, in 2010, the Medical Device Epidemiology Network (MDEpiNet) was established with other stakeholders from government, industry, and academia. Since its inception, MDEpiNet partners have published more than 190 studies, said Shuren. MDEpiNet has developed registries, including coordinated registry networks. These networks, he said, address the fact
that some individual registries may not contain fit-for-purpose data, but when a registry is combined with other RWD sources (e.g., claims data), it may be fit for purpose. MDEpiNet has also worked to develop active surveillance methodologies, conducted studies exploring the utility of claims and EHR data, and worked on evidence synthesis through in silico modeling and other approaches. Several dozen projects are in the MDEpiNet pipeline, Shuren said, including developing tools to move clinical data from EHRs into the Women’s Health Coordinated Registry Network4; implementing the Delta System for active surveillance in the transcatheter valve therapy registry and the cardioverter–defibrillator registry; and testing the capabilities of state-based claims.
In 2012, CDRH developed a strategy for a national system to address the limitations in the use of RWD, and to facilitate the systematic generation of RWE by a broad range of public and private entities. This effort, said Shuren, is called the National Evaluation System for health Technologies (NEST).5 NEST will soon begin a series of demonstration projects with 11 data partners, with the purpose of test driving the systems’ capabilities for addressing important device questions. At the time of the workshop, the NEST partners include approximately 150 hospitals and more than 3,000 outpatient clinics, which provide access to more than 469 million patient records. NEST has also established committees on methodology and data quality to develop standards and best practices.
CDRH’s third effort in the RWE space, said Shuren, is its involvement in the International Medical Device Regulators Forum (IMDRF). IMDRF released a series of three principle documents that focused on the infrastructure, methods, and tools for assessing the usability of registries for regulatory decision making. There is currently a study under way that aims to test these principles through research on devices for ruptured abdominal-aortic aneurysms.
CDRH has been accepting and leveraging RWE as valid scientific evidence in support of both pre- and postmarket regulatory decisions for many years, said Shuren. Since 2015, there have been at least 50 regulatory decisions for which CDRH relied on RWE, he noted, including decisions about expanded labeling and new device approval. In 2017, CDRH released final guidance on the use of RWE in regulatory decisions to provide greater clarity about when RWE is “regulatory grade.” The guidance pointed to two critical considerations in the evaluation of RWE: relevance and reliability.
Shuren gave an example of how RWE can be used in an iterative manner in the postmarket space. In 2011, FDA approved first-generation
___________________
4 For more information about the Women’s Health Coordinated Registry Network, see mdepinet.org/womens-health-crn (accessed November 2, 2018).
5 See https://nestcc.org/demo-announcement (accessed January 4, 2019).
transcatheter aortic valve replacement (TAVR), being the 42nd country to do so, and worked with the American College of Cardiology, the Society of Thoracic Surgeons, and CMS to establish a registry. At the same time, CMS issued a national coverage determination that approved coverage for the FDA-approved indication for TAVR, with the requirement that data be entered into the registry. The coverage determination also stated that if FDA expanded the indication for TAVR, coverage would automatically be expanded. In 2017, FDA approved third-generation TAVR for intermediate-risk patients 19 days after the European Union granted a CE (European conformity) mark for a similar device, said Shuren, and approved it for mitral valve-in-valve indication—the first country to do so. This represents a “really dramatic change,” he said, in FDA’s ability to obtain data to answer questions that were not previously addressable, and to do so more quickly and at a lower cost. CDRH conducted a return-on-investment analysis for decisions like this that leverage a registry, and found that it cost approximately $25 million for 20 studies to support 22 FDA decisions, compared with $127 to $134 million to conduct these studies if there had been no registry. This cost savings, he said, does not consider that a quicker time to market results in lives saved and improved quality of life for patients, as well as additional economic benefits to the companies.
The use of RWE for making regulatory decisions about devices is not a thing that is “nice to have,” it is a “need to have,” said Shuren. RCTs have inherent limitations that restrict their utility, particularly for devices. For example, when devices are used in the real world, there is a learning curve for providers who are implanting the device. This information cannot be generated from RCTs, he said. In addition, RWE can inform subgroup analysis on race, gender, and other patient characteristics. RWE is necessary for assessing these types of technologies in the real world, particularly when they are used in a broad range of patients and providers.
CDRH has long been trying to apply a total product life cycle approach for devices, said Shuren, rather than making an artificial divide into pre- and postmarket spaces. However, CDRH is not optimally structured organizationally to do this, so there is a reorganization under way. Part of the reorganization will involve the creation of a new Office of Clinical Evidence and Analysis that brings together experts on clinical trials, epidemiology, RWE, biostatistics, and surveillance. The reason for this, said Shuren, is that ultimately, the source of the clinical data is irrelevant; the true key attributes instead are whether the data are relevant and reliable.
Shuren concluded with three “lessons learned” for workshop participants to carry forward into their work with RWE:
- “Don’t let the perfect be the enemy of the good”: A single RWD source may not always be perfectly fit for purpose, but it can be
-
incredibly useful when linked with other RWD sources. Other approaches include validating an RWD source by comparing the results generated from it with a previously conducted RCT, or to conduct separate analyses with separate RWD sources to “triangulate” results; if the results match then confidence in the result is much greater.
- Consider how data are collected: Clinical research needs to be incorporated into the workflow of routine clinical practice in a systematic way that ensures consistent, high-quality data.
- Do not underestimate the value of good data scientists: Evidence generation requires expertise and appropriate methodology to turn good data into good evidence.
Discussion
After the presentations on the regulatory perspective on RWE, Breckenridge moderated a broad-ranging discussion among the panelists and workshop participants. Corrigan-Curay began by noting that there is a “chicken and egg” problem with the use of RWE in the regulatory space: companies are waiting for FDA to release guidance, and FDA is waiting for companies to come forward to inform the guidance. She said that meetings and workshops, such as this series, are enormously helpful for identifying gaps that need more discussion, as well as areas where FDA may be able to move forward. Anderson concurred and noted that one of the biggest gaps is in ensuring data quality and reliability. For example, he said, depending on the source of the data (e.g., EHRs, claims data), there can be different biases introduced and there can be issues with fully and accurately capturing the necessary information. In addition, linking the various sources of data remains an enormous challenge. Anderson noted that there needs to be a higher standard for evidence used for regulatory decision making. He said that companies should engage in conversation with FDA early and often in order to improve the likelihood that their studies will produce this type of “regulatory-grade evidence.” Shuren broadened this issue slightly, noting that evidence should be produced that is fit for purpose, whether that purpose is regulatory decision making or some other purpose such as clinical decision making. He added that FDA and other regulators do not look at RWE in a vacuum, but rather as part of the totality of the evidence from a variety of sources.
Breckenridge asked the panelists how the three Centers within FDA—CDER, CBER, and CDRH—are working together on the issue of RWE. Corrigan-Curay responded that while each Center may have different needs for RWE, communication among Centers is essential so that “we are all talking about the same thing across the agency,” including harmonizing
terminology. Shuren added that while open communication and working together are essential, it is sometimes appropriate and even necessary for different organizations within FDA to take a different approach to RWE. He said one benefit of FDA’s structure is that different parts of FDA can experiment with different approaches and the other organizations can learn from those experiences and adapt them to their own needs. The panelists noted that FDA is also working with EMA and other international regulators on the issue of RWE.
Gregory Simon asked if FDA will be moving away from solely being a “referee” of evidence produced by others to becoming a “starring player” by producing evidence, like in the Zostavax vaccine effectiveness study presented by Hector Izurieta (see Chapter 9). Corrigan-Curay responded that CDER will still primarily be recipients of evidence rather than data generators, but will try to work closely with stakeholders bringing evidence to FDA. Anderson noted that these types of studies are costly and take years to conduct, and that CBER would likely continue to “be the referee rather than [primarily generating] the data.” Shuren responded that CDRH both generates data and has built infrastructure and partnerships that enable CDRH to use RWE for regulatory decisions. For example, said Shuren, there was a clinical trial proposed by a sponsor in order to expand the indication for a device. CDRH looked at the existing evidence in the registries about the device and found that it was sufficient for expanding the indication without further study. This reduced the amount of time necessary for the expanded indication, said Shuren, from 1 or 2 years to a couple of weeks.
Mark McClellan closed the discussion by asking each of the panelists where they would choose to direct resources to best accelerate effective use of RWE. Anderson replied that if additional resources were available, he would direct them to hiring new staff to help coordinate communications among different levels and organizations at the agency. Corrigan-Curay said she would invest in demonstration and validation projects so she could better understand the best methods and approaches for turning RWD into useful, fit-for-purpose evidence. She added that in particular, there is a need for further work to ensure the accuracy and relevancy of data from EHRs. Shuren said he would use additional resources to build out the active surveillance capabilities of CDRH as well as to adopt universal device identifiers.
FINAL THOUGHTS
The third workshop concluded with McClellan offering some overarching thoughts on the series of workshops and on the decision aids that had been discussed. He noted that the decision aids were “viewed as useful”
by workshop participants, and that they captured many key considerations that could be included in designing approaches for collecting, evaluating, and using RWD and RWE. He noted that while the decision aids were basic, they could be used as a framework on which additional details could be added for specific stakeholders or applications. Workshop participants repeatedly emphasized that how one uses RWD and RWE depends on the research question, the clinical context, and the decision to be made (see Box 10-1). For example, if it is a regulatory decision for a new product, one needs the highest quality evidence available. If it is a regulatory decision for a new indication or population, the evidence can be slightly less robust, and if it is a decision about policy or insurance coverage, the evidence could be even less robust. McClellan said the decision aids could be adapted for more specific uses by individual stakeholders who are making these different types of decisions, leading to the idea that the value of RWE depends on the stakeholder and the specific questions each hopes to answer.
McClellan noted that, broadly, the goal of decision aids is to build a systematic framework for thinking about a particular topic; he highlighted the generation of evidence that is fit for purpose as an important part of utilizing RWE. The current system of evidence generation, as discussed in the first workshop, is expensive, time consuming, and cannot answer all of the questions about a product or intervention. Important questions could potentially be answered “cheaper and faster” with a systematic, validated framework for generating RWE, he said. The key to creating this framework will be clarity and specificity about when using RWE is
appropriate, and appropriate data sources and methods for different types of questions.
Improving the quality and reliability of data will also be essential for moving forward. Simon noted that improving the quality of data will be useful not just for research, but also for patient care. He said “the primary goal of improving the information that is generated in health care is to deliver better health care.” Referring to the phrase “the tail is wagging the dog,” Simon said that research is the tail, not the dog: “Actually improving people’s health is the dog, and [research] is just the tail that rides along.” Improving the data that are generated in the course of real-world care will benefit everyone, he said, and several other participants pointed to better data as a foundational principle to the increased use of RWD and RWE. He noted that many issues that were identified as making data collection more difficult are issues that affect our health care system as a whole. For example, discontinuities in the health care system and fragmentation of care affect both the collection of quality data as well as the care that people receive.
Relatedly, several workshop participants emphasized the importance of including patients and the patient perspective when thinking about the collection and use of RWE. W. Benjamin Nowell and Sharon Terry noted that patients are important sources of information for understanding the lived experience of a disease, and can provide insights about appropriate and relevant endpoints and outcomes for research. Jennifer Graff suggested that the word “patient” be incorporated into the decision aids, because while FDA and other stakeholders use evidence to make decisions, so do patients and caregivers.
McClellan closed by saying that tools such as the decision aids are “intended to help us move from case by case to more systematic and predictable opportunities.” He noted that there is growing infrastructure for RWE, from Sentinel to registries to NEST, and that FDA and other regulators were open to engaging with RWE and building the systems necessary to use RWE for decision making. These efforts, along with workshops such as this one, will contribute to the creation of “well-understood, well-curated, fit-for-purpose sources of data” that can be collected and used in routine practice, as well as the development of analytic methods and tools for turning RWD into evidence that can be used by a variety of stakeholders for making different types of decisions.
























